
Water-induced self-assembly of an amphiphilic perylene bisimide dyad into vesicles, fibers, coils, and rings†
Mutsumi
Ogasawara
a,
Xu
Lin
a,
Hiroki
Kurata
a,
Hayato
Ouchi
a,
Mitsuaki
Yamauchi
a,
Tomonori
Ohba
 b,
Takashi
Kajitani
cd,
Takanori
Fukushima
c,
Munenori
Numata
e,
Rie
Nogami
e,
Bimalendu
Adhikari
a and
Shiki
Yagai
*af
b,
Takashi
Kajitani
cd,
Takanori
Fukushima
c,
Munenori
Numata
e,
Rie
Nogami
e,
Bimalendu
Adhikari
a and
Shiki
Yagai
*af
aDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: yagai@faculty.chiba-u.jp
bDepartment of Chemistry, Graduate School of Science, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
cLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
dRIKEN SPring-8 Center, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
eDepartment of Biomolecular Chemistry, Graduate School of Life and Environmental Sciences, Kyoto Prefectural University, Japan
fInstitute for Global Prominent Research (IGPR), Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
First published on 27th November 2017
Abstract
Control over the self-assembly pathways of small functional molecules is a current trend in supramolecular chemistry, as a variety of metastable self-assemblies with nanostructures distinct from those of thermodynamically stable assemblies can be formed as kinetic products. If such kinetically formed assemblies can be trapped under non-equilibrium conditions, diverse self-organized structures beyond the immediate molecular design may be accessible. The self-assembly of specifically designed amphiphilic π-conjugated molecules in water represents a promising strategy to realize such conditions, wherein strong hydrophobic molecular interactions play an important role. Based on the alkyl-tethered covalent perylene bisimide (PBI) dyad scaffold that can potentially aggregate into one-dimensional supramolecular polymers, herein we demonstrate that an amphiphilic molecular design can open up new self-assembly pathways, which can potentially afford distinct nanostructures in aqueous media. We synthesized amphiphilic PBI dyads that contain one PBI unit functionalized with hydrophobic branched alkyl chains, and one PBI unit functionalized with hydrophilic branched alkyl chains. By changing the composition ratio of the THF/water mixtures, we obtained one-dimensional fibrous and vesicular aggregates as a result of increasing hydrophobic effects. Increasing the temperature of the THF/water mixtures in which fibrous aggregates are preferentially formed subdued the aggregation to entropy control and resulted in the formation of coil- and ring-shaped kinetic nanoaggregates.
Introduction
Control over the self-assembly of π-conjugated molecules enables the generation of functional nanomaterials with bespoke morphologies and properties.1 Despite the same molecular design, identical functional molecules can be organized into different supramolecular structures in solution via different self-assembly pathways that are accessible by the formation of metastable assemblies, which can be controlled by using different preparation protocols.2,3 Establishing non-equilibrium conditions in solution is especially important for this approach, as transiently formed metastable assemblies can thus be converted into kinetically trapped assemblies.2b Upon changing the conditions, kinetically trapped assemblies can be reconverted into metastable species that eventually transform into thermodynamically stable species.One rational approach to such non-equilibrium conditions is the self-assembly of amphiphilic molecules in aqueous media,4 wherein strong solvophobic interactions between π-conjugated units and hydrophobic chains enthalpically drive molecular aggregation.5 In contrast, hydrophilic chains, e.g. oligo(ethylene glycol)s, which are frequently employed in the design of amphiphilic molecules,6 can entropically contribute to aggregation by releasing hydrogen-bonded water molecules upon the aggregation of amphiphilic molecules. In this situation, the noncovalent interaction that enthalpically drives aggregation does not necessarily dominate the assembly process, and hence unique opportunities arise in the design of specific aggregates, where molecular arrangements are not thermodynamically stabilized.
This approach to construct functional nanoaggregates has been applied to many π-conjugated systems,6 but has been rarely used to control the supramolecular structures of perylene bisimide (PBI) dyes,7 which are among the most frequently exploited supramolecular building blocks.8 Würthner and co-workers have recently demonstrated experimentally that the self-assembly of bolaamphiphilic PBI in pure water is entropically driven and that the enthalpic contribution braced by π–π interactions increases upon gradual addition of the organic co-solvent THF.9 However, examples of PBI aggregates whose nanostructures can be controlled by changing the entropic contribution in aqueous media remain scarce. As a compensation of their exceptionally strong and π–π stacking interactions in organic and aqueous media, most of the supramolecular structures in PBI dye aggregates are one-dimensional (1D).7 Several research groups have recently contributed to the self-assembly of amphiphilic PBI dyes into non-1D aggregates in aqueous systems.10–12 For example, Würthner and co-workers have reported the formation of vesicular aggregates upon co-aggregation of two amphiphilic PBI dyes with distinct configurations.10 Hirch et al. have prepared cylindrical micelles by hybridizing hydrophobic PBI and hydrophilic calixarene.11 Furthermore, Rybtchinski and co-workers have incorporated hydrophobic interactions between metal–ligand complexes to regulate the self-assembly of PBI, which resulted in the formation of spiral nanoaggregates.12
Previously reported PBI dyads 113 and 214 represent a simple but rational molecular design to enhance the capability of PBI dyes to aggregate one-dimensionally. In these systems, two PBI moieties with swallow-tailed alkyl (STalkyl) or oligo (ethylene glycol) chains (STOEG)15 are connected via a (CH2)7 spacer (Fig. 1). This simple covalent dimerization strongly enforces isodesmic aggregation of the PBI chromophores (Kiso = 1.0 × 106 M−1 in pure THF),14 while 3D agglomeration into insoluble precipitates is effectively avoided by a boat-like conformation of the dyad.13 As a result, this dyad stacks into 1D aggregates that eventually form a gel-like state in nonpolar organic solvents (1) or THF/water mixtures (2). After the successful enhancement of the 1D aggregation capability of PBI dyes, we were interested in how the self-assembly of this PBI dyad in aqueous media may be affected upon increasing its amphiphilic character. For this purpose, we synthesized amphiphilic PBI dyad 3, where one PBI unit was functionalized with a hydrophobic STalkyl moiety, while the other PBI unit contained a hydrophilic STOEG unit (Fig. 1). According to the previous studies,13,14 we chose the (CH2)7 chain as the spacer in order to avoid strong aggregation that induces precipitation (observed for (CH2)5) and to avoid intramolecular folding (observed for (CH2)9). We anticipated that increasing the entropic contribution by changing the aggregation conditions could deter the enthalpy-driven aggregation of the dyad, thus providing an opportunity to access different aggregate structures.
 | ||
| Fig. 1 Chemical structures of the hydrophobic (1), bolaamphiphilic (2), and amphiphilic (3) PBI dyads used in this study. | ||
Results and discussion
Self-assembly in THF/water mixtures
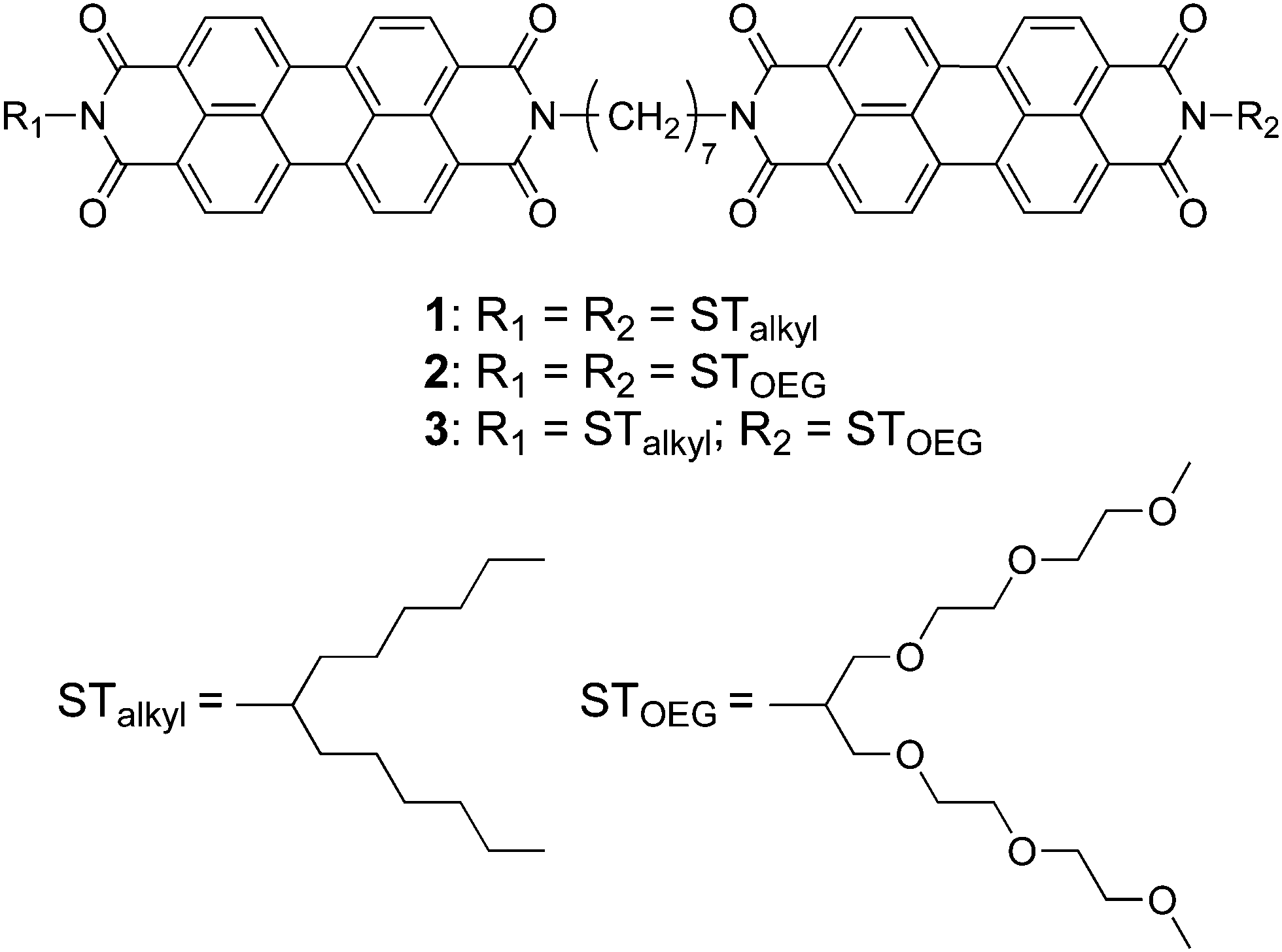
When dissolved in THF, PBI dyad 3 exists in a monomer/aggregate equilibrium with unfolded conformations, which is reflected in the reversible concentration-dependent absorption spectral change typically observed for the aggregation of PBI dyes driven by π–π stacking (Fig. 2a). When the molar fraction of the aggregated molecules (αagg) estimated from the concentration-dependent spectra measured at 20 °C was plotted as a function of concentration, a sigmoidal transition was observed, indicating an isodesmic aggregation process (Fig. 2b).16 By fitting the transition curve with an isodesmic model, an association constant (Kiso) of 1.5 × 105 M−1 was estimated for 3 in THF.1b,17 The number-averaged degree of polymerization (DPN) was estimated to be 2.6 for c = 1.0 × 10−4 M at 20 °C. In comparison, the amphiphilic PBI monoad is fully monomeric, even at c = 1.0 × 10−4 M at 20 °C (Fig. S1, ESI†), as covalent dimerization enforces intermolecular interactions. Reflecting the low DPN, atomic force microscopy (AFM) images of 3 spin-coated from THF solution showed only ill-defined nanostructures (Fig. S2, ESI†).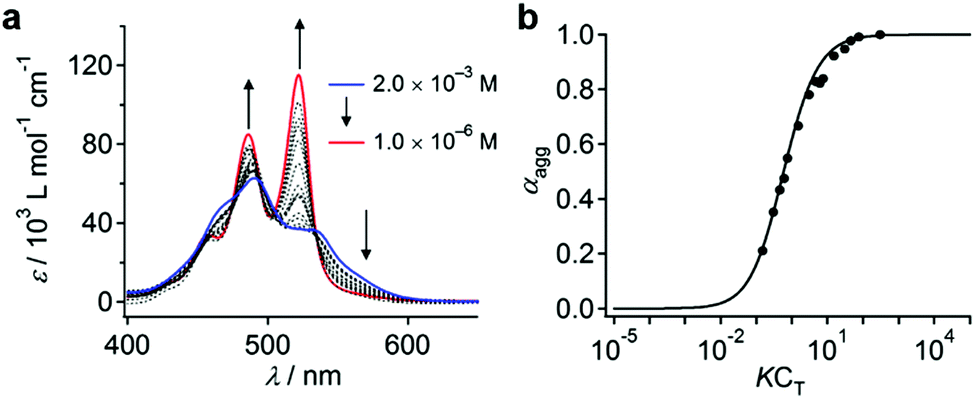 | ||
| Fig. 2 (a) Concentration-dependent UV/vis absorption spectra of 3 in THF (c = 2.0 × 10−6 to 1.0 × 10−6 M) at 20 °C. (b) A plot of concentration-dependent molar fraction of aggregates (αagg) calculated from the apparent ε at λ = 520 nm. A solid line was obtained by fitting the plot to the isodesmic model. For the calculation of αagg, see Materials and Methods. | ||
To investigate how thermodynamic parameters in the aggregation of 3 change in the presence of water, temperature-dependent UV/vis absorption spectra were measured in pure THF and in THF/water mixtures (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v) at concentrations ranging from 5.0 × 10−6 M to 1.2 × 10−5 M. Upon heating the solutions, spectral changes suggesting a transformation from the aggregated state to the monomeric state were observed for all solutions (Fig. S3, ESI†). The temperature dependence of αagg could be fitted using a thermal isodesmic aggregation model,16 from which aggregation constants (Kiso) were obtained (Fig. S4a and b and Table S1, ESI†). At each concentration, a more than two-fold increase of Kiso was noted in the presence of water. Plotting lnKiso as a function of T−1 (van’t Hoff plot) delivered standard enthalpy (ΔH°) and entropy (ΔS°) values of −44 kJ mol−1 and −51 J mol−1 K−1, respectively, for pure THF solutions, and −42 kJ mol−1 and −38 J mol−1 K−1 for THF/water mixtures (90:10, v/v) (Fig. S4c, d and Table S2, ESI†). The negative ΔH° and negative ΔS° values indicate that the aggregation is enthalpy-driven in both media. Interestingly, the presence of water did not influence the ΔH° values significantly, but substantially decreased the ΔS° values. This result indicates that the entropic penalty associated with the aggregation of the dyad molecules in aqueous medium is compensated by the release of water molecules that are confined in the STOEG unit.
:10, v/v) at concentrations ranging from 5.0 × 10−6 M to 1.2 × 10−5 M. Upon heating the solutions, spectral changes suggesting a transformation from the aggregated state to the monomeric state were observed for all solutions (Fig. S3, ESI†). The temperature dependence of αagg could be fitted using a thermal isodesmic aggregation model,16 from which aggregation constants (Kiso) were obtained (Fig. S4a and b and Table S1, ESI†). At each concentration, a more than two-fold increase of Kiso was noted in the presence of water. Plotting lnKiso as a function of T−1 (van’t Hoff plot) delivered standard enthalpy (ΔH°) and entropy (ΔS°) values of −44 kJ mol−1 and −51 J mol−1 K−1, respectively, for pure THF solutions, and −42 kJ mol−1 and −38 J mol−1 K−1 for THF/water mixtures (90:10, v/v) (Fig. S4c, d and Table S2, ESI†). The negative ΔH° and negative ΔS° values indicate that the aggregation is enthalpy-driven in both media. Interestingly, the presence of water did not influence the ΔH° values significantly, but substantially decreased the ΔS° values. This result indicates that the entropic penalty associated with the aggregation of the dyad molecules in aqueous medium is compensated by the release of water molecules that are confined in the STOEG unit.
Vesicles
When water was added to the THF solution of 3, the solution color changed from orange to pink, before turning to red within a few minutes (path A in Fig. 3a). Time-lapse UV/vis absorption measurements showed that this color transition was due to a decrease in the absorption intensity of the shoulder band at 500–550 nm relative to the absorption maximum at 474 nm (Fig. S5a, ESI†). Upon the addition of an equal amount of water (THF/water = 50:50, v/v), the spectral transition was completed within 5 min (Fig. S5b, ESI†). The absorption intensity of the resulting red solution decreased upon increasing the amount of water added, suggesting the formation of more tightly π–π stacked aggregates and/or much more extended aggregates in the aqueous environment (Fig. 3b and c). Solutions containing 30–40 vol% water were stable for several hours, before eventually suffering from precipitation, while those containing 10–20 vol% water remained homogeneous for several months. In contrast, solutions containing 50–80 vol% water were very unstable, and precipitation occurred within 30 min (Fig. 3d). This observation suggests the initial formation of kinetic species (pink solution) upon the addition of water, and a subsequent transformation into red insoluble aggregates.
 | ||
| Fig. 3 (a) Sample preparation protocol for 3 in THF/water mixtures and schematic illustration of the characteristic solution colors and states. (b) UV/vis absorption spectra of 3 (c = 5.0 × 10−5 M) in THF/water mixtures with different mixing ratios. (c) The dependence of the molar extinction coefficient (ε) at 491 nm on the water content. (d) Photographs showing time-course changes of 3 in THF/water mixtures. | ||
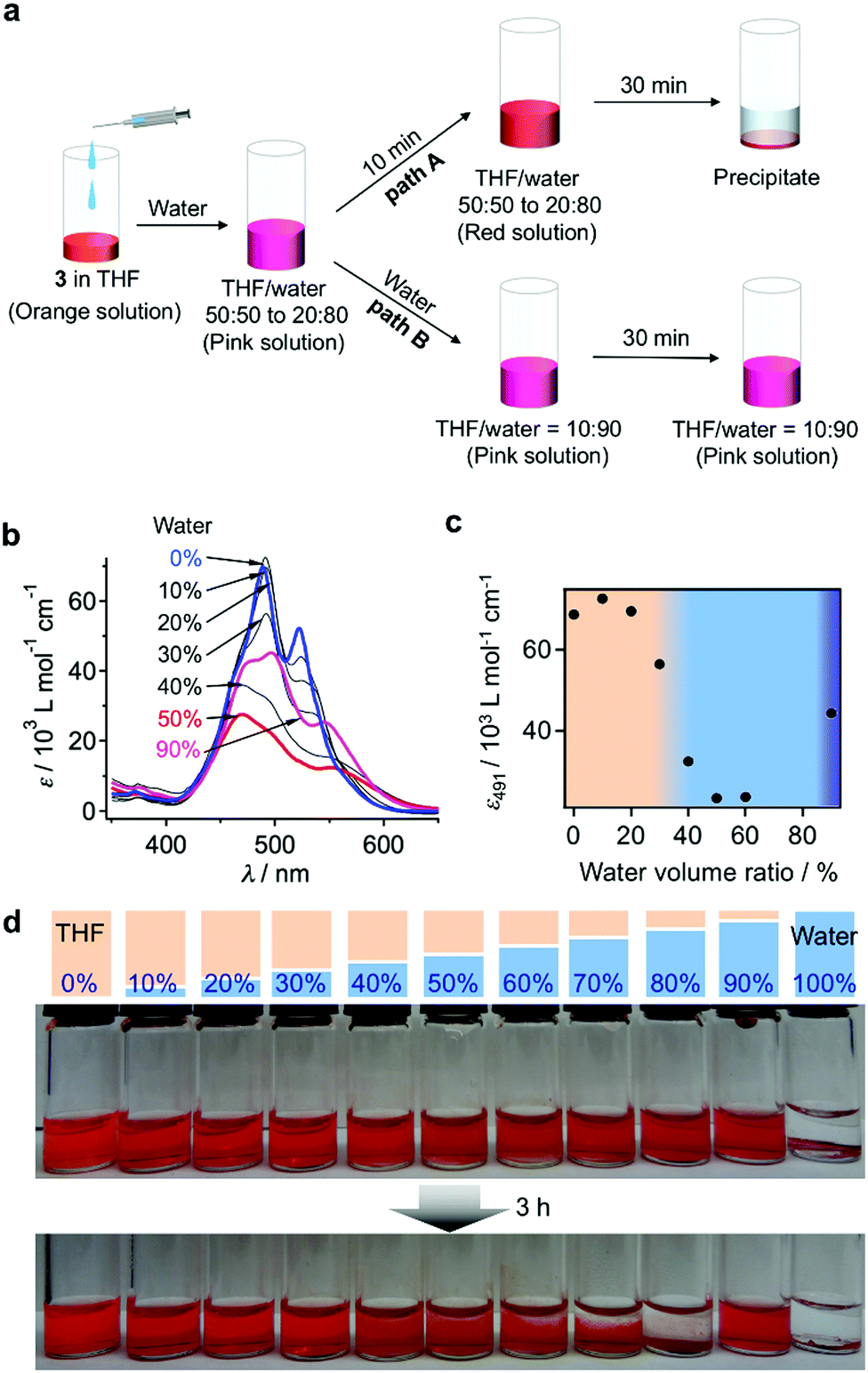
The pink kinetic aggregates were converted into stable species by adding an excess of water (THF/water = 10:90, v/v; path B in Fig. 3a). Dynamic light scattering (DLS) measurements showed that the resulting dispersion contained particles with hydrodynamic diameters (DH) of 130–470 nm (Fig. 4a), which were stable for at least one month. Interestingly, for these aggregates, an unexpected increase of the molar extinction coefficient of the PBI chromophore was observed as shown in Fig. 3b (εmax = 2.8 × 104 M−1 cm−1 in THF/water = 50:50, v/v; εmax = 4.5 × 104 M−1 cm−1 in THF/water = 10:90, v/v), which indicates the formation of other aggregates, wherein the chromophores are more loosely packed. These aggregates displayed an irreversible decrease in the ε value of the PBI chromophore upon increasing the temperature from 20–60 °C (Fig. S6, ESI†), while DLS measurements suggested a decrease in the aggregate size (Fig. S7, ESI†). This observation indicates that intermolecular interactions within the aggregates are enhanced at elevated temperatures, which is a characteristic of entropy-driven aggregation.
 | ||
| Fig. 4 (a) The DLS-derived size distribution of 3 (c = 1.8 × 10−4 M) in THF/water = 10:90 (v/v). (b and c) TEM images of vesicular aggregates of 3 in THF/water = 10:90 (v/v; unstained sample). (d) The schematic model of the vesicular aggregates of 3. (e) The XRD pattern of dried vesicular aggregates of 3 (the inset shows the bilayered packing model that would explain the XRD pattern). | ||
For the aforementioned aggregates that are stabilized in THF/water = 10:90 (v/v) mixtures, confocal laser scanning microscopy (CLSM), scanning electron microscopy (SEM), and atomic force microscopy (AFM) measurements implied a particulate morphology with sizes over hundreds of nanometers (Fig. S8, ESI†). Transmission electron microscopy (TEM) measurements revealed that these particulate aggregates consist of hollow vesicular nanostructures (Fig. 4b and c)9 with a wall thickness of 5.3–7.8 nm, which corresponds to one to two times the extended molecular length of 3 (4.2 nm). On the basis of these results, we propose a bilayered structure featuring interdigitated STalkyl units (Fig. 4d).18 X-ray diffraction (XRD) measurements of a dried sample of these vesicular aggregates showed merely two peaks at d = 2.49 and 0.34 nm (Fig. 4e), but not the multiple peaks that are typically associated with multilamellar structures. The former peak, which is broadened, may be attributed to a scattering peak from the monomeric units, while the latter should correspond to diffraction from the stacked PBI π-planes. It is worth noting that vesicular aggregates could not be prepared by direct dissolution of 3 in pure water due to their limited solubility, while those formed in THF/water = 10:90 (v/v) mixtures remained stable after removing the THF by dialysis (Fig. S10, ESI†). Hence, the formation of vesicles can improve the solubility in water similar to folded proteins under biological conditions.
Vesicle-to-fiber transformation
The red solutions obtained by adding moderate amounts of water to the THF solutions of 3 are very unstable (path A in Fig. 3a), and eventually afford a precipitate that consists of entangled micrometer-length fibers (Fig. 5a). TEM measurements revealed a width of 100–200 nm for these fibers (Fig. 5b). The results of an XRD analysis suggested a hexagonal columnar internal order with a lattice parameter a = 3.21 nm (Fig. 5c). The periodicity of the π–π stacked PBI π-planes was observed in the form of a very intense peak at d = 0.34 nm.19 Taking into account the extended conformation of 3 in THF as evident from the concentration-dependent absorption change (Fig. S1, ESI†), these molecules with an amphiphilic PBI dyad should stack on top of each other with a rotational offset to form double-stranded helical PBI stacks that are decorated with hydrophobic STalkyl and hydrophilic STOEG units, respectively. Considering the lattice parameter a, which is shorter than the extended molecular length of 3 (∼4.2 nm), the STalkyl and STOEG chains most likely extend parallel to the columnar axis (Fig. 5d).20 According to the aforementioned concentration-dependent UV-vis changes observed in pure THF as well as an aqueous THF system, the possibility of intramolecular folding followed by π–π stacked aggregation into a columnar structure can be excluded.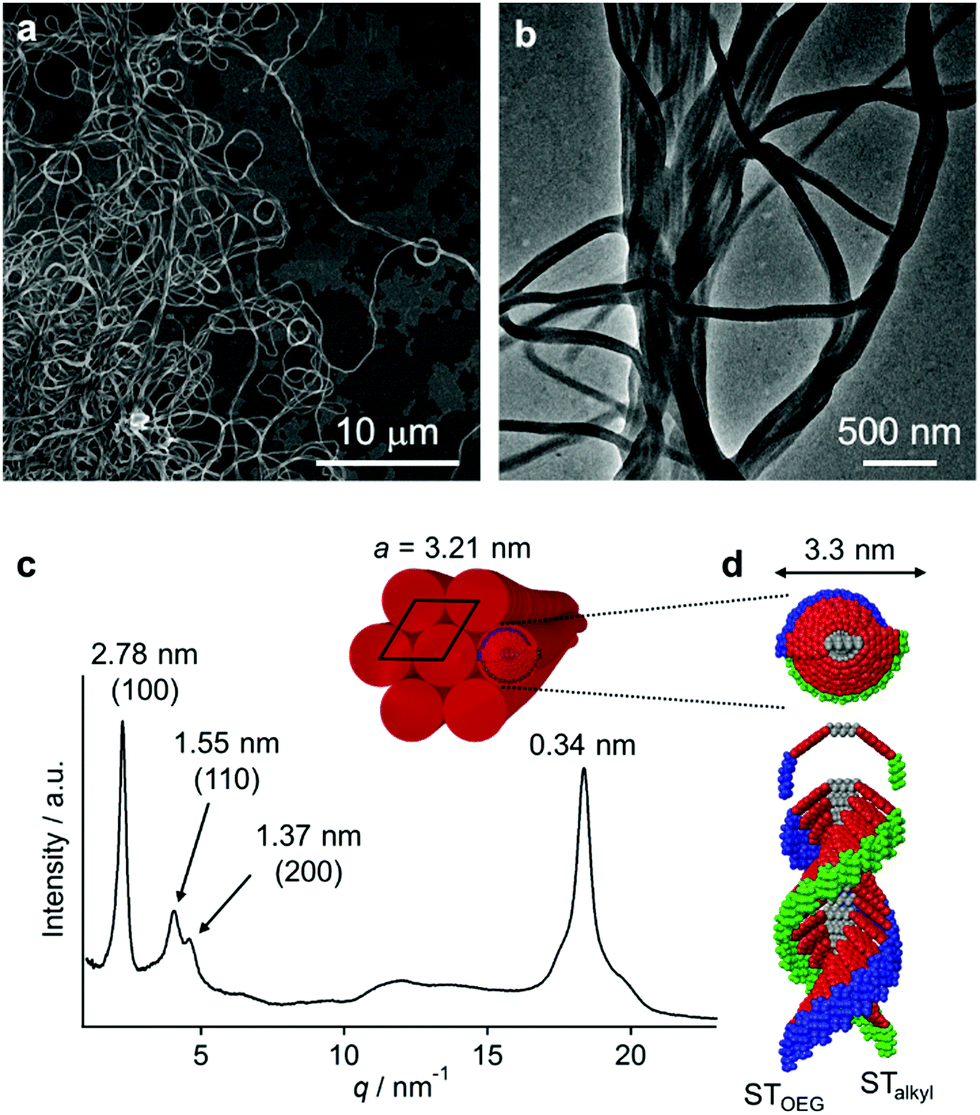 | ||
| Fig. 5 (a and b) SEM (a) and TEM images (b) of nanofibers of 3 (c = 5.0 × 10−5 M) obtained from THF/water = 50:50 (v/v) solutions. (c) The XRD pattern of nanofibers (values in parentheses denote Miller indices). (d) A schematic model for the hexagonal molecular packing of helically stacked 3. | ||
The different aggregate structures of 3 in THF/water mixtures with varying contents of water prompted us to investigate the potential transformation of vesicular to fibrous aggregates. For this purpose, appropriate amounts of THF were added to the vesicular solution of 3, and the transition kinetics were examined by UV/vis absorption spectroscopy. When THF was added to a THF/water = 10:90 (v/v) solution of 3 to afford a final THF/water = 35:65 ratio (c = 5.6 × 10−5 M), the spectral transition proceeded with first-order kinetics and a rate constant of k = 4.2 × 10−4 s−1 at 20 °C (Fig. 6a and b). This transition was accompanied by an increase of the average DH from 300 nm to 630 nm, as evident from DLS measurements (Fig. 6b). The transition accelerated with increasing temperature (Fig. 6c) and the Arrhenius plot showed a linear relationship between lnk and T−1, from which an activation energy (Ea) of 137 kJ mol−1 at 20 °C was estimated (Fig. 6d). Decreasing the amount of added THF resulted in an increase of the transition time (Fig. 6b and Fig. S11, ESI†).
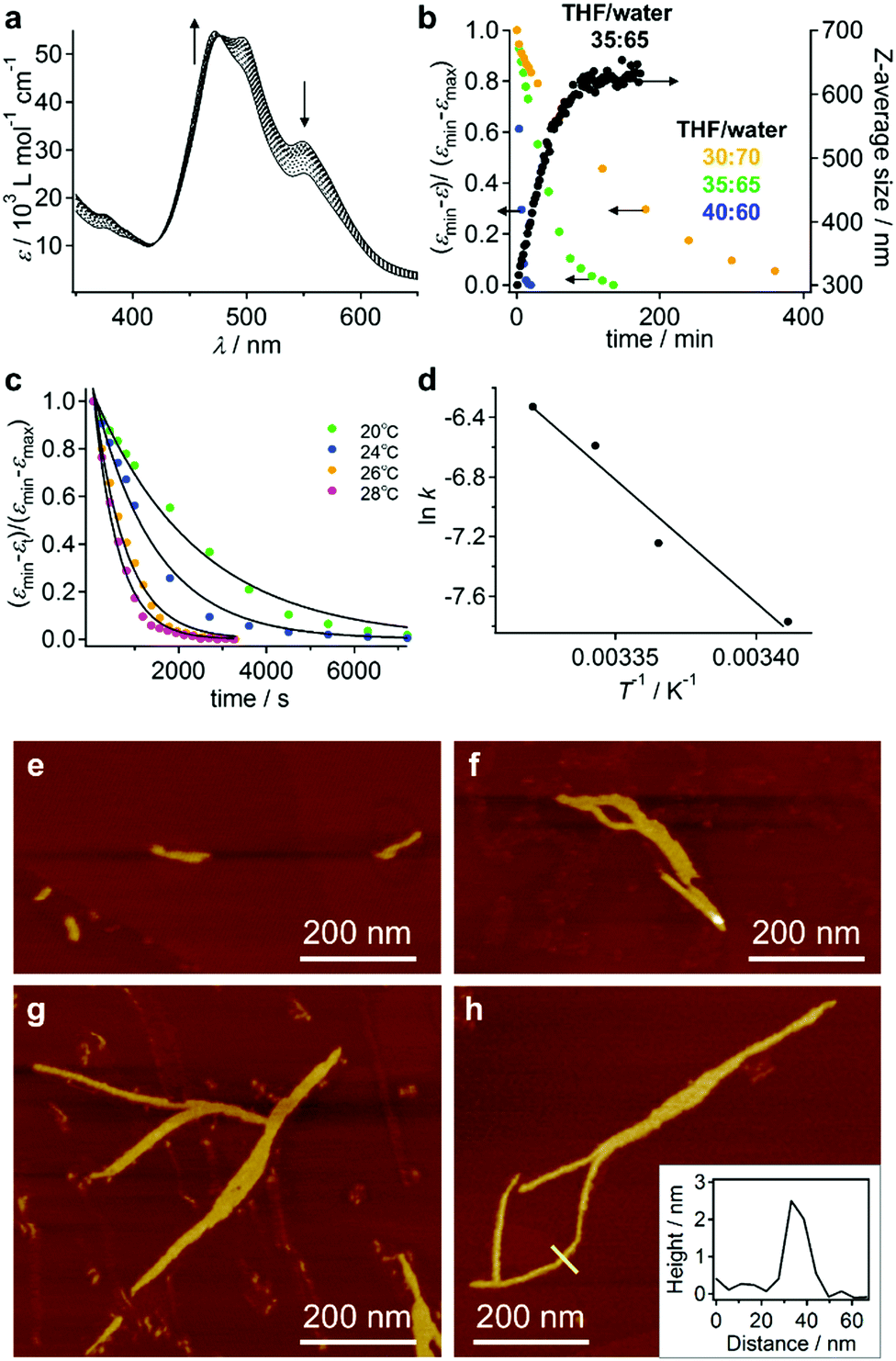 | ||
| Fig. 6 (a) Time-dependent UV/vis absorption spectra (t = 1–90 min) of 3 (c = 5.6 × 10−5 M) in THF/water = 35:65 (v/v) mixtures prepared by the addition of THF to vesicular solutions (THF/water = 10:90, v/v). (b) Time-dependent change of ε (normalized) at λ = 494 nm for different THF/water ratios (yellow dots: 30:70, v/v; green dots: 35:65, v/v; blue dots: 40:60, v/v). For the THF/water = 35:65 (v/v) solutions, the time-dependent change of the z-average size in the DLS measurements is also shown (black dots). (c) The time-dependent change of the THF/water = 35:65 (v/v) solutions at 20 °C (green dots), 24 °C (blue dots), 26 °C (yellow dots) and 28 °C (pink dots). Solid curves represent nonlinear regression analyses using a first-order kinetics model. (d) An Arrhenius plot for the kinetic data shown in (c). (e–h) AFM images for the time-dependent growth of nanofibers of 3 in THF/water = 35:65 (v/v) mixtures upon the addition of THF to THF/water = 10:90 (v/v) mixtures that contain vesicular aggregates (c = 7.7 × 10−5 M). Samples were taken at t = 20 s (e), t = 5 min (f), t = 30 min (g), and t = 60 min (h) after the addition of THF. The inset in (h) shows a cross-section analysis along the yellow line. | ||
The aforementioned transformation was also monitored by AFM. At t = 20 s after the addition of THF, short fibers (length ≈ 100 nm) were observed in addition to the ellipsoidal aggregates that presumably originated from the vesicular aggregates (Fig. 6e). These fibers elongated with time, and after t = 60 min, longer fibers (length ≈ 800 nm) were observed (Fig. 6f–h). The thickness of these fibers was relatively uniform (∼2.5 nm; cf. inset in Fig. 6h), suggesting that elementary stacks of 3 were observed by AFM. After t = 3 h, these fibers precipitated as micrometer-length fibers. The smaller thickness value of these fibers in comparison with the diameter of the helical columnar structure as shown Fig. 5d is most likely due to deformation upon interacting with the graphite surface.
Nanocoils and nanorings
Without the addition of excess water, the entropy contribution to the aggregation can be increased by increasing the temperature.9 Irrespective of the presence of precipitates, heating a solution of 3 in a THF/water = 50:50 (v/v) mixture over 90 °C led to phase separation, that is, into a homogeneous THF solution phase of 3 and an aqueous phase due to dehydration of the THF molecules (step i; Fig. 7a).21 This biphasic system was homogenized upon cooling to 80 °C and shaking (step ii), before becoming turbid below 60 °C and eventually affording a precipitate (step iii). The results of SEM (Fig. 7b and Fig. S12, ESI†), TEM (Fig. 7c), and CLSM (Fig. S13, ESI†) measurements revealed that the precipitates were composed mainly of agglomerated coil-shaped nanostructures (nanocoils) that exhibit outer diameters of 300–500 nm and inner diameters of 50–200 nm. In addition to these nanocoils, nanofibers were observed, which may possibly be ascribed to local gradients in the solvent composition and/or temperature (vide infra). AFM measurements indicated a height of ∼100 nm for these nanocoils (Fig. 7d–f), and TEM images suggest that they consist of several loops of tape-like fibers (Fig. 7c). The XRD analysis of the precipitates displayed a very broad diffusion halo centered at ∼1.3 nm, which covers sharp diffraction patterns derived from the coexisting nanofibers with a hexagonal lattice (Fig. 7g). The presence of the diffusion halo suggests that the nanocoils lack a well-defined internal molecular order (Fig. 7h). This is further supported by a microscopic UV/vis absorption analysis of nanocoils (Fig. S14, ESI†), which revealed a vibronically less-structured absorption band of the PBI chromophores.
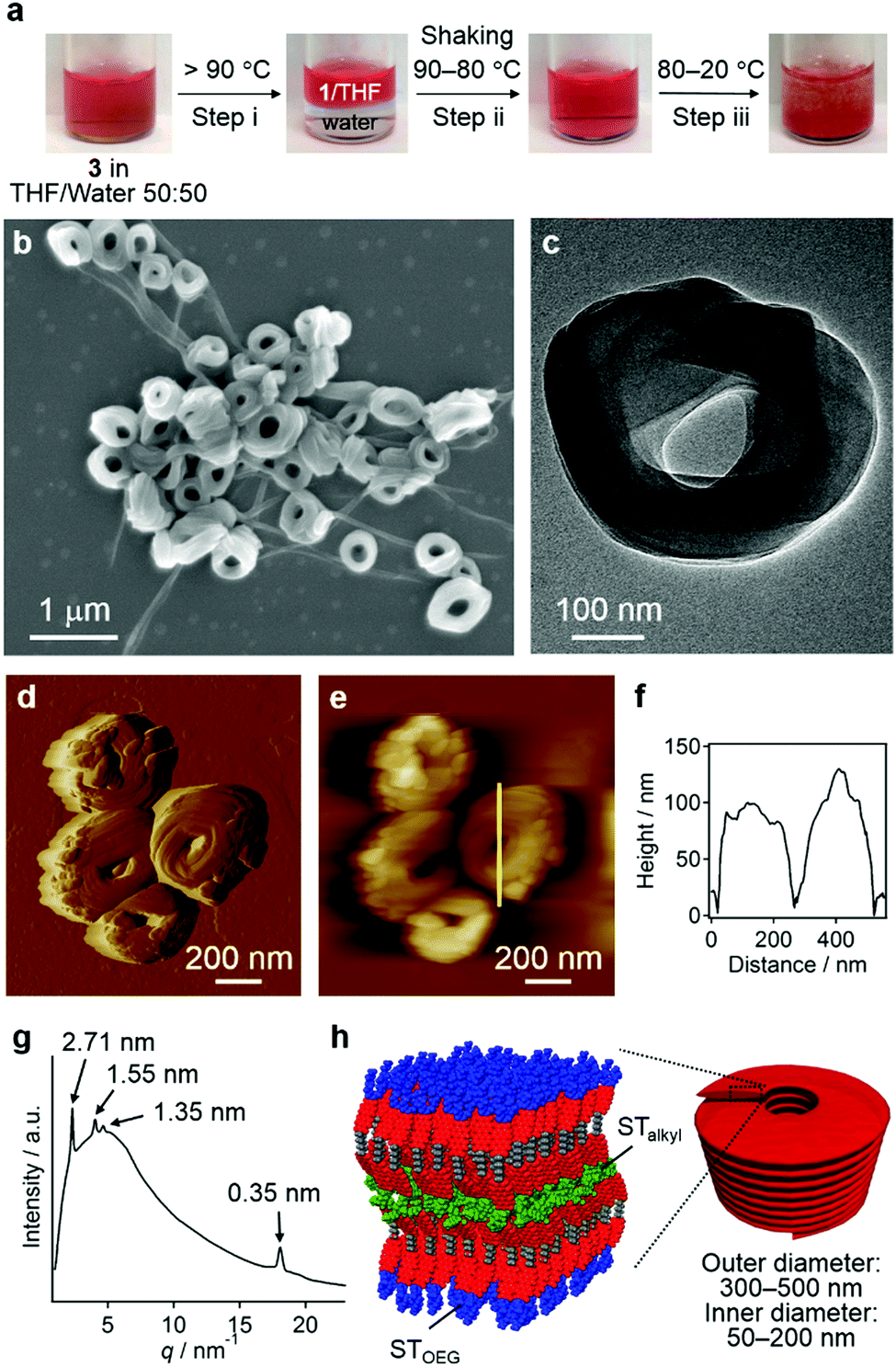 | ||
| Fig. 7 (a) Preparation protocol for nanocoils of 3. Heating a solution of 3 in THF/water = 50:50 (v/v) mixtures over 90 °C leads to phase separation into an organic phase that contains THF and 3 and an aqueous phase. Shaking the phase-separated system upon cooling to 80 °C results in a homogeneous phase, which affords precipitates upon subsequent cooling to 20 °C. (b) SEM, (c) TEM, and (d) AFM error amplitude images, as well as (e) the AFM height image of the precipitated nanocoils. (f) Cross-sectional analysis along the yellow line in (e). (g) The XRD pattern obtained for the precipitated nanocoils (values in parenthesis denote Miller indices). Arrows indicate diffraction patterns from nanofibers. (h) Schematic representation of molecular packing in the nanocoils. | ||
To understand the mechanism of the formation of these nanocoils, we removed aliquots from the solution during step iii as shown in Fig. 7a at different temperatures, which were spin-coated onto a silicon substrate and examined by SEM. Interestingly, the sample taken at 80 °C contained vesicular aggregates similar to those found in THF/water = 10:90 (v/v) at room temperature (Fig. S15a, ESI†), which corroborates the notion that vesicular aggregates were predominantly formed under conditions with large entropic contributions. The sample taken at 60 °C already contained both nanocoils and nanofibers that grew from the vesicular aggregates (Fig. S15b, ESI†). It should be noted that when this homogeneous solution was kept at 60 °C overnight, only nanofibers were observed (Fig. S16, ESI†). Accordingly, nanocoils represent metastable aggregates at 60 °C, which can be converted into thermodynamically stable nanofibers over time at higher temperatures.
The formation of nanofibers in addition to nanocoils may reflect a local gradient in the solvent composition and/or temperature in step iii of Fig. 7a.1b This obstacle can be circumvented by carefully controlling the diffusion of the solvent and temperature by employing microfluidic conditions that mix the two phases in a laminar flow. One remarkable feature of the diffusion kinetics is that the diffusion time depends on the square of the diffusion length, which means in principle that mass and thermal diffusion in a typical microflow channel (diffusion length ≈ 100 μm) occurs 104 times faster than that in a typical vial (diffusion length ≈ 1 cm). Accordingly, we expected self-assembly events to start simultaneously during rapid solution mixing and temperature fluctuation under such microflow conditions.22
In our microflow experiments, THF solutions of 3 (c = 1.0 × 10−4 M) were injected from the central channel of a typical cross-type microchannel setup, while water was injected from the two inlets from a lateral flow (Fig. 8a; for details, see the Supporting Information). Prior to mixing with the central THF solution, the two lateral water streams were heated to 100 °C using Peltier devices. The resulting solution was directly cast onto a silicon substrate and dried under an N2 flow. SEM images of the thus obtained samples revealed that these solutions contained almost exclusively circular nanostructures with outer diameters of 180–240 nm and inner diameters of 50–80 nm (Fig. 8b and Fig. S17, ESI†), most of which seem to be closed (nanorings23) rather than open-ended structures (nanocoils). AFM measurements revealed a uniform thickness (∼20 nm) for these structures, thus corroborating the formation of nanorings (Fig. 8c, inset). In the present system, the simultaneous mixing of THF solutions with hot water (∼100 °C) induced an effective aggregation of 3 in moderately hot solutions. The resulting π–π stacked aggregates transformed spontaneously into nanorings in the down-stream region. It is therefore conceivable that all monomers initiate their self-assembly events at the same spatial point and that π–π stacked aggregates always experience the same chemical environment during the organization processes.
 | ||
| Fig. 8 (a) Schematic illustration of a microflow device used for the preparation of nanorings of 3, which are formed at higher temperatures. (b) The SEM image of the nanorings of 3 prepared by using this microflow device. (c and d) AFM height (c) and phase (d) images of the thus prepared nanorings of 3 [the inset in (c) shows the cross-sectional analysis along the yellow line]. | ||
Discussion
In their entirety, the combined experimental results demonstrate that amphiphilic PBI dyad 3 can be organized into distinct nanostructures by changing the solvent composition and temperature (Fig. 9a). In the THF/water = 50:50 (v/v) mixture at 20 °C, STOEG units of 3 interact less with water molecules through hydrogen bonding, due to competing interactions between THF and the STOEG units (Fig. 9b). Under these conditions, π–π stacking interactions enthalpically drive aggregation, and an optimal stacking arrangement to maximize enthalpic release leads to a helical stacking of dyad molecules in columnar aggregates that bundle in a hexagonal fashion into extended nanofibers. In contrast, in THF/water = 10:90 (v/v) mixtures, i.e., a very water-rich environment, STOEG units are very well hydrated, which decreases the entropy of the water molecules (Fig. 9c). The aggregation of dyad molecules of 3 is thus accompanied by the release of a large amount of water molecules to decrease the steric demand around the STOEG units. Accordingly, this process should be an entropically favored process, and enthalpic contributions might not be required to drive the overall aggregation. This consideration does not contradict the non-optimal π–π stacking arrangement of the PBI units as evident from the absorption spectrum of the vesicular solution (Fig. 3b). Non-optimal, i.e., non-helical, stacking of PBI units in addition to solvophobic interactions of STalkyl units allows the dyad molecules to assemble into 2D lamellar structures, which leads to the formation of vesicular aggregates. The addition of THF to the vesicular solution diminishes the entropic advantage by interacting with water, thus disfavoring the entropy-driven assembly relative to an enthalpy-driven one, wherein strong solvophobic interactions between the PBI units become dominant. As an appreciable concentration dependence was not observed during this vesicle-to-fiber conversion experiment, we could not clearly distinguish whether this conversion proceeds through a monomeric state (off-pathway) or occurs directly without forming monomers (on-pathway).3c
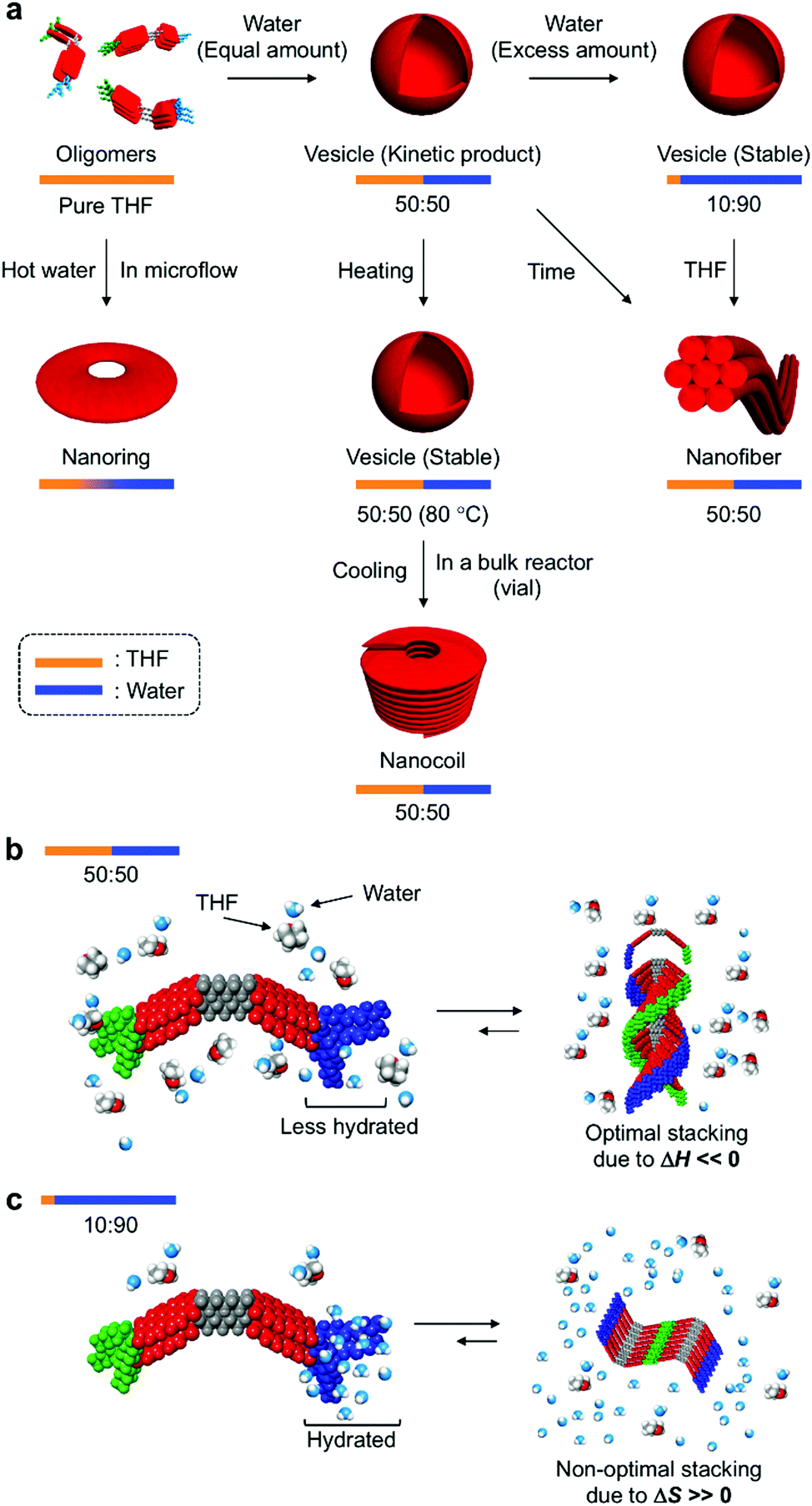 | ||
| Fig. 9 (a) Schematic illustration of the self-assembly of 3 into nanoaggregates with distinct morphologies depending on the THF/water ratio of the solvent and the temperature. (b and c) Distinct aggregation of 3 for THF/water ratios of 50:50 (v/v) (b) and 10:90 (v/v) (c), which is due to the different contributions of ΔH and ΔS. | ||
When aggregation of 3 is initiated in a THF/water = 50:50 (v/v) mixture at a high temperature (∼80 °C), the entropy gain is large on account of the dehydration of the STOEG units. Accordingly, the self-assembly becomes entropy-driven again, and a concomitant decrease of the enthalpy contribution results in a loss of helical stacking of the dyad even in this medium, which allows the formation of vesicular aggregates. Decreasing the temperature gradually increases the enthalpy contribution to promote the 1D growth of aggregates by π–π stacking between the PBI units. However, this process competes with strong segregated, i.e., amphiphilic, interactions between the dehydrated hydrophilic STOEG and hydrophobic STalkyl units, and this interplay between solvophobic interactions may initiate the unique formation of different curved nanoaggregates, which leads to the predominant formation of nanocoils in the vial, and nanorings under microflow conditions. The generation of curvature due to macroscopic forces induced by shaking the vial is clearly excluded by the nanoring formation under microflow conditions. The formation of different nanostructures in the bulk solution and the microfluidic phase can be rationalized in terms of the difference in temperature gradient upon cooling. In bulk reactors (e.g. a vial), the temperature decreases from ∼80 °C to room temperature within minutes. Under such high-temperature conditions, kinetically formed closed nanostructures, i.e., nanorings, exist in equilibrium with monomeric species, so that they can further elongate to form nanocoils. In contrast, under microflow conditions, the solution temperature drops rapidly within seconds, which effectively traps the kinetically formed nanorings.
Conclusions
We have demonstrated that bestowing amphiphilic properties onto specifically designed perylene bisimide (PBI) dyads, which can inherently self-assemble into 1D supramolecular polymers, can provide new self-assembly pathways in THF/water mixtures. In media composed of equal parts of THF and water, vesicular aggregates are kinetically formed. However, these vesicular aggregates eventually transform into thermodynamically stable nanofibers composed of hexagonally packed helical supramolecular polymers. Although this kinetic trap is not deep when using a solvent with comparable contents of THF and water, the addition of water stabilizes the vesicular aggregates by increasing the entropy contribution. Additionally, an increase in the entropy contribution by increasing the temperature of the medium that is composed of equal volumes of THF and water opens up a new self-assembly pathway leading to the assembly of the PBI dyad molecules into unique coil-shaped nanostructures. In summary, this work may represent a highly valuable design paradigm for functional molecules that can produce diverse nanostructures with distinct functionality. Achieving similar levels of complex nanoarchitectures in non-aqueous systems is intriguing with respect to organic nano-electronics and photonics, wherein peculiar electronic and optical properties are expected to arise from exotic nanostructures. However, without strong solvophobic interactions and entropic contributions that surpass optimal molecular interactions to release large enthalpic energies, more complicated molecular designs are required, and these remain to be addressed.24Author contributions
S. Y. initiated the project. M. O., X. L., H. K. and S. Y. synthesized the molecules, and designed and performed the self-assembly experiments. T. O. obtained TEM images. T. K. and T. F. performed XRD measurements. M. N. and R. N. performed the microflow experiments. H. O., M. Y. and B. A. supported and discussed the experiments. M. O., B. A. and S. Y. interpreted the data and also wrote the manuscript. All authors read and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the KAKENHI grant (26102010 for S. Y. and 26102008 for T. F.) and the Grant-in-Aid for Scientific Research on Innovative Areas “π-Figuration” (26102001) of the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT). Synchrotron radiation XRD experiments were carried out at the BL45XU beamline of SPring-8 with the approval of the RIKEN SPring-8 Center (proposals 20140056 and 20150068). T. F. appreciates the financial support by “Dynamic Alliance for Open Innovation Bridging Human, Environment and Materials” from MEXT. M. N. appreciates the partial financial support from the Grant-in-Aid for Scientific Research (B) (15H03532).Notes and references
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed; (b) Z. Chen, A. Lohr, C. R. Saha-Möller and F. Würthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC; (c) D. González-Rodríguez and A. P. H. J. Schenning, Chem. Mater., 2011, 23, 310–325 CrossRef; (d) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed; (e) L. Zhang, X. Wang, T. Wang and M. Liu, Small, 2015, 11, 1025–1038 CrossRef CAS PubMed; (f) S. Yagai, Bull. Chem. Soc. Jpn., 2015, 88, 28–58 CrossRef.
- (a) P. A. Korevaar, T. F. A. De Greef and E. W. Meijer, Chem. Mater., 2014, 26, 576–586 CrossRef CAS; (b) A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- (a) Y. Tidhar, H. Weissman, S. G. Wolf, A. Gulino and B. Rybtchinski, Chem. – Eur. J., 2011, 17, 6068–6075 CrossRef CAS PubMed; (b) I. D. Tevis, L. C. Palmer, D. J. Herman, I. P. Murray, D. A. Stone and S. I. Stupp, J. Am. Chem. Soc., 2011, 133, 16486–16494 CrossRef CAS PubMed; (c) P. A. Korevaar, S. J. George, A. J. Markvoort, M. M. J. Smulders, P. A. J. Hilbers, A. P. H. J. Schenning, T. F. A. De Greef and E. W. Meijer, Nature, 2012, 481, 492–496 CrossRef CAS PubMed; (d) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013 CAS; (e) D. Görl, X. Zhang, V. Stepanenko and F. Würthner, Nat. Commun., 2015, 6, 7009 CrossRef PubMed; (f) A. Aliprandi, M. Mauro and L. De Cola, Nat. Chem., 2015, 8, 10–15 CrossRef PubMed; (g) T. Fukui, S. Kawai, S. Fujinuma, Y. Matsushita, T. Yasuda, T. Sakurai, S. Seki, M. Takeuchi and K. Sugiyasu, Nat. Chem., 2016, 9, 493–499 CrossRef PubMed.
- (a) G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed; (b) P. Besenius, G. Portale, P. H. H. Bomans, H. M. Janssen, A. R. A. Palmans and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17888–17893 CrossRef CAS PubMed; (c) D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed; (d) C. Rest, M. J. Mayoral and G. Fernández, Int. J. Mol. Sci., 2013, 14, 1541–1565 CrossRef CAS PubMed; (e) E. Krieg, M. M. C. Bastings, P. Besenius and B. Rybtchinski, Chem. Rev., 2016, 116, 2414–2477 CrossRef CAS PubMed.
- B. Widom, P. Bhimalapuram and K. Koga, Phys. Chem. Chem. Phys., 2003, 5, 3085 RSC.
- (a) A. P. H. J. Schenning, A. F. M. Kilbinger, F. Biscarini, M. Cavallini, H. J. Cooper, P. J. Derrick, W. J. Feast, R. Lazzaroni, P. Lecle, L. A. Mcdonell, E. W. Meijer and S. C. J. Meskers, J. Am. Chem. Soc., 2002, 124, 1269–1275 CrossRef CAS PubMed; (b) J. P. Hill, W. Jin, A. Kosaka, T. Fukushima, H. Ichihara, T. Shimomura, K. Ito, T. Hashizume, N. Ishii and T. Aida, Science, 2004, 304, 1481–1483 CrossRef CAS PubMed; (c) J. K. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2006, 45, 7195–7198 CrossRef CAS PubMed; (d) W. S. Li, Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, H. Masunaga, S. Sasaki, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 8886–8887 CrossRef CAS PubMed; (e) T. Sakurai, K. Shi, H. Sato, K. Tashiro, A. Osuka, A. Saeki, S. Seki, S. Tagawa, S. Sasaki, H. Masunaga, K. Osaka, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 13812–13813 CrossRef CAS PubMed; (f) H. Maeda, Y. Ito, Y. Haketa, N. Eifuku, E. Lee, M. Lee, T. Hashishin and K. Kaneko, Chem. – Eur. J., 2009, 15, 3706–3719 CrossRef CAS PubMed; (g) F. García and L. Sánchez, Chem. – Eur. J., 2010, 16, 3138–3146 CrossRef PubMed; (h) T. Hirose, K. Higashiguchi and K. Matsuda, Chem. – Asian J., 2011, 6, 1057–1063 CrossRef CAS PubMed; (i) Z. Huang, S.-K. Kang, M. Banno, T. Yamaguchi, D. Lee, C. Seok, E. Yashima and M. Lee, Science, 2012, 337, 1521–1526 CrossRef CAS PubMed; (j) L. Chen, K. S. Mali, S. R. Puniredd, M. Baumgarten, K. Parvez, W. Pisula, S. De Feyter and K. Müllen, J. Am. Chem. Soc., 2013, 135, 13531–13537 CrossRef CAS PubMed; (k) M. C. Yeh, Y. Lou Su, M. C. Tzeng, C. W. Ong, T. Kajitani, H. Enozawa, M. Takata, Y. Koizumi, A. Saeki, S. Seki and T. Fukushima, Angew. Chem., Int. Ed., 2013, 52, 1031–1034 CrossRef CAS PubMed; (l) M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekemans, A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336–343 CrossRef CAS PubMed; (m) A. S. Weingarten, R. V. Kazantsev, L. C. Palmer, M. McClendon, A. R. Koltonow, A. P. S. Samuel, D. J. Kiebala, M. R. Wasielewski and S. I. Stupp, Nat. Chem., 2014, 6, 964–970 CrossRef CAS PubMed; (n) C. Rest, M. J. Mayoral, K. Fucke, J. Schellheimer, V. Stepanenko and G. Fernández, Angew. Chem., Int. Ed., 2014, 53, 700–705 CrossRef CAS PubMed; (o) P. Wei, T. R. Cook, X. Yan, F. Huang and P. J. Stang, J. Am. Chem. Soc., 2014, 136, 15497–15500 CrossRef CAS PubMed; (p) K. Higashiguchi, G. Taira, J. I. Kitai, T. Hirose and K. Matsuda, J. Am. Chem. Soc., 2015, 137, 2722–2729 CrossRef CAS PubMed; (q) D. J. Van Dijken, J. Chen, M. C. A. Stuart, L. Hou and B. L. Feringa, J. Am. Chem. Soc., 2016, 138, 660–669 CrossRef CAS PubMed; (r) S. Ghosh, D. S. Philips, A. Saeki and A. Ajayaghosh, Adv. Mater., 2017, 29, 1605408 CrossRef PubMed; (s) Z. Chen, Y. Liu, W. Wagner, V. Stepanenko, X. Ren, S. Ogi and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 5729–5733 CrossRef CAS PubMed; (t) K. Jalani, S. Dhiman, A. Jain and S. J. George, Chem. Sci., 2017, 8, 6030–6036 RSC.
- (a) A. Ustinov, H. Weissman, E. Shirman, I. Pinkas, X. Zuo and B. Rybtchinski, J. Am. Chem. Soc., 2011, 133, 16201–16211 CrossRef CAS PubMed; (b) X. Zhang, D. Görl, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 1270–1274 CrossRef CAS PubMed.
- (a) F. Würthner, Chem. Commun., 2004, 1564–1579 RSC; (b) H. Langhals and J. Gold, Helv. Chim. Acta, 2005, 88, 2832–2836 CrossRef CAS; (c) T. Seki, X. Lin and S. Yagai, Asian J. Org. Chem., 2013, 2, 708–724 CrossRef CAS; (d) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- D. Görl and F. Würthner, Angew. Chem., Int. Ed., 2016, 55, 12094–12098 CrossRef PubMed.
- (a) X. Zhang, Z. Chen and F. Würthner, J. Am. Chem. Soc., 2007, 129, 4886–4887 CrossRef CAS PubMed; (b) X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS PubMed.
- (a) K. Rosenlehner, B. Schade, C. Böttcher, C. M. Jäger, T. Clark, F. W. Heinemann and A. Hirsch, Chem. – Eur. J., 2010, 16, 9544–9554 CrossRef CAS PubMed; (b) F. Rodler, B. Schade, S. Backes, F. Hampel, C. Böttcher, T. Clark and A. Hirsch, J. Am. Chem. Soc., 2015, 137, 3308–3317 CrossRef CAS PubMed.
- E. Kossoy, H. Weissman and B. Rybtchinski, Chem. – Eur. J., 2015, 21, 166–176 CrossRef CAS PubMed.
- X. Lin, M. Hirono, T. Seki, H. Kurata, T. Karatsu, A. Kitamura, D. Kuzuhara, H. Yamada, T. Ohba, A. Saeki, S. Seki and S. Yagai, Chem. – Eur. J., 2013, 19, 6561–6565 CrossRef CAS PubMed.
- X. Lin, H. Kurata, D. D. Prabhu, M. Yamauchi, T. Ohba and S. Yagai, Chem. Commun., 2017, 53, 168–171 RSC.
- A. Wicklein, A. Lang, M. Muth and M. Thelakkat, J. Am. Chem. Soc., 2009, 131, 14442–14453 CrossRef CAS PubMed.
- (a) R. B. Martin, Chem. Rev., 1996, 96, 3043–3064 CrossRef CAS PubMed; (b) M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. De Greef, P. Van Der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362–367 CrossRef CAS PubMed.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845 Search PubMed.
- The C–H stretching vibrations of the vesicular aggregates are almost identical to those of nanofibers and nanocoils (Fig. S9, ESI†). Therefore, the conformation of the ST alkyl chains is not important to provide different morphologies: (a) W. Jin, Y. Yamamoto, T. Fukushima, N. Ishii, J. Kim, K. Kato, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 9434–9440 CrossRef CAS PubMed; (b) J. S. Valera, R. Sánchez-Naya, F. J. Ramírez, J. L. Zafra, R. Gómez, J. Casado and L. Sánchez, Chem. – Eur. J., 2017, 23, 11141–11146 CrossRef CAS PubMed.
- (a) F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem. – Eur. J., 2001, 7, 2245–2253 CrossRef; (b) S. Yagai, M. Usui, T. Seki, H. Murayama, Y. Kikkawa, S. Uemura, T. Karatsu, A. Kitamura, A. Asano and S. Seki, J. Am. Chem. Soc., 2012, 134, 7983–7994 CrossRef CAS PubMed.
- C. Roche, H.-J. Sun, P. Leowanawat, F. Araoka, B. E. Partridge, M. Peterca, D. A. Wilson, M. E. Prendergast, P. A. Heiney, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and V. Percec, Nat. Chem., 2015, 8, 80–89 CrossRef PubMed.
- I. M. Smallwood, Solvent Recovery Handbook, Blackwell Science, Oxford, 2nd edn, 2002 Search PubMed.
- M. Numata, Chem. – Asian J., 2015, 10, 2574–2588 CrossRef CAS PubMed.
- (a) D. J. Pochan, Z. Chen, H. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS PubMed; (b) J. Kim, E. Lee, Z. Huang and M. Lee, J. Am. Chem. Soc., 2006, 128, 14022–14023 CrossRef CAS PubMed; (c) H. Huang, B. Chung, J. Jung, H. W. Park and T. Chang, Angew. Chem., Int. Ed., 2009, 48, 4594–4597 CrossRef CAS PubMed; (d) I. S. Park, Y. R. Yoon, M. Jung, K. Kim, S. Park, S. Shin, Y. B. Lim and M. Lee, Chem. – Asian J., 2011, 6, 452–458 CrossRef CAS PubMed; (e) S. Tu, S. H. Kim, J. Joseph, D. A. Modarelli and J. R. Parquette, J. Am. Chem. Soc., 2011, 133, 19125–19130 CrossRef CAS PubMed.
- (a) S. Yagai, S. Kubota, H. Saito, K. Unoike, T. Karatsu, A. Kitamura, A. Ajayaghosh, M. Kanesato and Y. Kikkawa, J. Am. Chem. Soc., 2009, 131, 5408–5410 CrossRef CAS PubMed; (b) S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205–18208 CrossRef CAS PubMed; (c) S. Yagai, Y. Goto, X. Lin, T. Karatsu, A. Kitamura, D. Kuzuhara, H. Yamada, Y. Kikkawa, A. Saeki and S. Seki, Angew. Chem., Int. Ed., 2012, 51, 6643–6647 CrossRef CAS PubMed; (d) M. J. Hollamby, K. Aratsu, B. R. Pauw, S. E. Rogers, A. J. Smith, M. Yamauchi, X. Lin and S. Yagai, Angew. Chem., Int. Ed., 2016, 55, 9890–9893 CrossRef CAS PubMed; (e) B. Adhikari, Y. Yamada, M. Yamauchi, K. Wakita, X. Lin, K. Aratsu, T. Ohba, T. Karatsu, M. Hollamby, N. Shimizu, H. Takagi, R. Haruki, S. Adachi and S. Yagai, Nat. Commun., 2017, 8, 15254 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of 3, experimental details, additional UV/vis spectra and AFM, SEM, and CLSM images. See DOI: 10.1039/c7qm00494j |
| This journal is © the Partner Organisations 2018 |