
Construction of g-C3N4/S-g-C3N4 metal-free isotype heterojunctions with an enhanced charge driving force and their photocatalytic performance under anoxic conditions†
Shaozheng Hua,
Lin Maa,
Fayun Lia,
Zhiping Fana,
Qiong Wanga,
Jin Baia,
Xiaoxue Kanga and
Guang Wu*b
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, Fushun 113001, China
bSchool of Chemistry and Materials Sciences, Heilongjiang University, Key Laboratory of Chemical Engineering Processes & Technology for High-efficiency Conversion (College of Heilongjiang Province), Harbin 150080, China. E-mail: guangw001@163.com
First published on 7th October 2015
Abstract
In heterojunction catalysts, the potential difference is the main driving force for efficient charge separation and transfer. The slight difference in the electronic band structure of the isotype heterojunction catalysts causes a poor driving force, leading to a dissatisfactory charge separation efficiency. In this work, g-C3N4/S-g-C3N4 metal-free isotype heterojunction catalysts (GCN–SCN) with an enhanced charge driving force were prepared by a two step calcination method. Anoxic photocatalytic degradation of RhB under visible light was used to evaluate the performance of the as-prepared g-C3N4 catalysts. The results indicate that this two step calcination method can markedly improve the charge driving force of the as-prepared isotype heterojunctions, leading to a more efficient charge-carrier migration. GCN–SCN displays the highest reaction rate constant of 0.0228 min−1, which is 3.5 and 2.9 times higher than that of GCN/SCN(1) and GCN/SCN(2) prepared by a one step calcination method. A linear relationship is observed between the VB driving force and RhB degradation rate. This paper provides a new perspective to prepare isotype heterojunctions with an improved charge driving force and photocatalytic performance.
Introduction
As a clean and green technology, photocatalytic oxidation has received more and more attention. In general, a photocatalytic process requires the participation of molecular oxygen, which can generate reactive oxygen species by capturing electrons. However, oxygen is not available in many environments, such as petroleum-contaminated aquifers, oil reservoirs and deep sediments.1 These anoxic environments would suppress the ability of traditional photocatalysts. Therefore, the design and synthesis of novel photocatalysts for the anoxic photocatalytic process is required. In recent years, graphitic carbon nitride (g-C3N4), the most stable allotrope of covalent carbon nitride, has been widely used as a new metal-free visible light photocatalyst for organic pollutant degradation,2 water reduction and oxidation,3 CO2 capture4 and organic synthesis.5 The tunable condensation degree and distinctive heptazine ring structure make g-C3N4 possess good physicochemical stability, an interesting electronic structure and a medium band gap (2.7 eV).6,7 These advantages make g-C3N4 a potential candidate for visible light photocatalysis. Besides that, g-C3N4 is composed of very common elements, C, N and H. It is easily prepared via the one-step polymerization of cheap raw materials, such as melamine,8 dicyandiamide,9 urea10 and thiourea.11 Nevertheless, the shortcomings of g-C3N4 are as obvious as its advantages, such as its low visible light utilization efficiency and rapid recombination of photogenerated electron–hole pairs. To advance this promising photocatalyst, many strategies have been used, such as metal and non-metal doping,12–15 copolymerization,16 semiconductor coupling17 and nanostructure design.18Semiconductor coupling is one of the most promising methods among those mentioned above. In the field of photocatalysis, the construction of an intimate heterojunction between two appropriate semiconductors is an effective strategy to enhance the photocatalytic performance. Several kinds of g-C3N4-based heterojunctions have been developed by coupling g-C3N4 with other types of inorganic photocatalysts, such as graphene/g-C3N4,19 TiO2/g-C3N4,20 MoS2/g-C3N4,21 CdS/g-C3N4 (ref. 22) and WO3/g-C3N4.23 However, a basic condition has to be satisfied to form the heterojunction: the energy level matching of two semiconductors. This condition limits the further development of this method.
Recently, inspired by Degussa P25 (a mixture of 75% anatase TiO2 and 25% rutile TiO2), isotype heterojunctions constructed between two different crystal phases of a single substance have been developed.24–26 The slight difference in the electronic band structure between two different crystal phases of a single substance enables the formation of an isotype junction at their crystal interfaces. In general, the band gap structure of g-C3N4 can be simply tuned from 2.4 to 2.8 eV by using different precursors. Coupling two components of g-C3N4 with a well-matched band structure forming a g-C3N4/g-C3N4 isotype heterojunction can provide an alternative novel pathway to address the intrinsic drawbacks of g-C3N4 to enhance the photocatalytic performance without relying on extra semiconductors. Zhang et al. prepared a g-C3N4/S-doped g-C3N4 heterojunction photocatalyst using trithiocyanuric acid and dicyandiamide as raw materials.24 The obtained heterojunction was demonstrated to promote charge separation which arises from the band offsets, leading to a significant enhancement in the photocatalytic hydrogen production. Dong et al. prepared a g-C3N4/g-C3N4 isotype heterojunction using urea and thiourea as raw materials.25,26 They suggested that the lifetime of the charge carriers was prolonged due to the band offsets in this isotype heterojunction catalyst, resulting in an improved photocatalytic NO removal ability under visible light irradiation.
All of these reported g-C3N4 based isotype heterojunctions are prepared using different precursors. However, the potential difference between the two components of the heterojunctions is the main driving force for efficient charge separation and transfer.24,27,28 The slight difference in the electronic band structure of these isotype heterojunction catalysts causes the poor driving force, leading to a dissatisfactory charge separation efficiency. It is known that the calcination temperature is the key factor to determine the polycondensation degree. Besides that, polycondensation degree of different precursors at the same calcination temperature is also different, thus leading to a different band structure. Therefore, compared with a one-step calcination method, the two-step calcination method (520 °C for 2 h at a rate of 5 °C min−1 and then 550 °C for 4 h at a rate of 10 °C min−1) can make different precursors exhibit the polycondensation degree with a greater difference, which can promote the charge driving force and improve the photocatalytic performance. Here, we report a g-C3N4/S-g-C3N4 metal-free isotype heterojunction catalyst with an enhanced charge driving force prepared by a two step calcination method. The charge driving force can be increased by more than two times by this two step calcination method, leading to a more efficient charge-carrier migration. Anoxic photocatalytic degradation of RhB under visible light was used to evaluate the performance of the as-prepared g-C3N4 catalysts.
Experimental
Preparation and characterization
All chemicals used in this study were of analytical grade and used without further treatment. 6 g of urea or thiourea was calcined at 520 °C for 2 h (at a rate of 5 °C min−1). The prepared catalyst is referred to as GCN(1) or SCN(1). 6 g of urea or thiourea was calcined at 550 °C for 4 h (at a rate of 10 °C min−1). The prepared catalyst is referred to as GCN(2) or SCN(2). 6 g of thiourea was dissolved into 20 ml deionized water to form a solution. 1 g of as-prepared GCN(1) was added into the above solution and ultrasound-treated for an additional 60 min. After that, the mixture was stirred vigorously for another 2 h to achieve a homogeneous suspension. The obtained suspension was heated to 100 °C to remove the water. The solid product was dried at 100 °C in an oven, followed by milling and annealing at 550 °C for 4 h (at a rate of 10 °C min−1). The obtained product was GCN–SCN. Neat SCN was prepared by the two-step annealing of thiourea: 520 °C for 2 h (at a rate of 5 °C min−1) and then 550 °C for 4 h (at a rate of 10 °C min−1). For comparison, GCN was prepared following the same procedure as in the synthesis of GCN–SCN but in the absence of thiourea.In order to confirm the effect of this two step calcination method on the promotion of the charge driving force, g-C3N4/S-g-C3N4 isotype heterojunctions were prepared by the one step calcination method. 6 g of thiourea and 6 g of urea were dissolved into 30 ml water. This solution was then dried at 100 °C to remove the water and get the molecular composite precursors. The precursors were calcined by two different methods, 520 °C for 2 h at a heating rate of 5 °C min−1 and 550 °C for 4 h at a heating rate of 10 °C min−1. The resultant products were denoted as GCN/SCN(1) and GCN/SCN(2), respectively.
The XRD patterns of the prepared samples were recorded on a Rigaku D/max-2400 instrument using Cu-Kα radiation (λ = 1.54 Å). The scan rate, step size, voltage and current were 0.05° min−1, 0.01°, 40 kV and 30 mA, respectively. Elemental analysis was performed with a vario EL cube from Elementar Analysensysteme GmbH. UV-Vis spectroscopy was carried out on a JASCO V-550 model UV-Vis spectrophotometer using BaSO4 as the reflectance sample. The XPS measurements were performed on a Thermo Escalab 250 XPS system with Al Kα radiation as the excitation source. The binding energies were calibrated by referencing the C 1s peak (284.6 eV) to reduce the sample charge effect. Nitrogen adsorption was measured at −196 °C on a Micromeritics 2010 analyser. All the samples were degassed at 393 K prior to the measurement. The BET surface area (SBET) was calculated based on the adsorption isotherm. The photoluminescence (PL) spectra were measured at room temperature with a fluorospectrophotometer (FP-6300) using a Xe lamp as the excitation source. The electrochemical impedance spectra (EIS) were recorded using an EIS spectrometer (EC-Lab SP-150, BioLogic Science Instruments) in a three electrode cell by applying a 10 mV alternative signal versus the reference electrode (SCE) over a frequency range of 1 MHz to 100 mHz. The cyclic voltammograms were measured in a 0.1 M KCl solution containing 2.5 mM K3[Fe(CN)6]/K4[Fe(CN)6] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a redox probe at a scanning rate of 20 mV s−1 in the same three electrode cell as the EIS measurement.
:1) as a redox probe at a scanning rate of 20 mV s−1 in the same three electrode cell as the EIS measurement.
Photocatalytic reaction
RhB was selected as the model compound to evaluate the anoxic photocatalytic performance of the prepared g-C3N4 based catalysts in an aqueous solution under visible light irradiation. A total of 0.05 g of catalyst was dispersed in 200 ml of an aqueous solution containing RhB (10 ppm) in an ultrasound generator for 10 min. The suspension was transferred to a self-designed glass reactor and stirred for 30 min in the dark to achieve adsorption equilibrium. N2 was continuously bubbled through the solution to remove O2 during the reaction process. In the photoreaction under visible light irradiation, the suspension was exposed to a 250 W high-pressure sodium lamp with main emissions in the 400–800 nm range, and air was bubbled at 130 ml min−1 through the solution. The UV light portion of the sodium lamp was filtered by a 0.5 M NaNO2 solution. All the runs were conducted at ambient pressure and 30 °C. At specific time intervals, 4 ml of the suspension was removed and immediately centrifuged to separate the liquid samples from the solid catalyst. The RhB concentrations before and after the reaction were measured using a UV-Vis spectrophotometer at a wavelength of 550 nm.Results and discussion
Fig. 1 presents the XRD patterns of as-prepared GCN, SCN and GCN–SCN. The XRD patterns confirmed the formation of graphitic carbon nitride. For GCN, the peak at 13.1° corresponds to the in-plane structural packing motif of tri-s-triazine units, which is indexed as the (100) peak. The distance is calculated as d = 0.675 nm, corresponding to the hole-to-hole distance in the nitride pores. The peak at 27.6° corresponds to the interlayer stacking of aromatic segments with a distance of 0.323 nm, which is indexed as the (002) peak. From the Fig. 1 insert, it can be seen that the diffraction angle of the (002) peak for GCN (27.6°) is higher than that of SCN (27.3°), due to the interlayer distance of GCN (0.323 nm) being smaller than that of SCN (0.326 nm). This is probably due to the additional motif of the oxygen in the urea that facilitates the condensation process and shortens the interlayer distance.9 Further observation indicates that the diffraction angle of the (002) peak for GCN–SCN (27.5°) is located between GCN and SCN, confirming the formation of an isotype heterojunction. | ||
| Fig. 1 XRD patterns of the as-prepared GCN, SCN and GCN–SCN. | ||
Generally, a high specific surface area (SBET) and large pore volume is significant to the enhancement of the performance of a catalyst. As presented in Table 1, GCN exhibits the highest SBET of 14.5 m2 g−1, whereas SCN and GCN–SCN have an SBET of 8.6 and 12.4 m2 g−1. This hints that the additional oxygen in the urea is beneficial for enlarging the surface area of g-C3N4, presumably because of the formation of CO2 during the condensation which suppresses the advance of the grain boundary.9 No essential difference in the SBET among the three as-prepared isotype heterojunctions is observed, indicating that the preparation method does not influence their SBET. For the pore volume, a similar trend is shown. The C/N ratio is 0.68 for GCN, which is lower than that of SCN (0.71). This result confirms that the condensation process can be influenced by different precursors, which is consistent with the XRD results. For the three as-prepared isotype heterojunctions, the C/N ratio of GCN–SCN is 0.66, much smaller than that of GCN/SCN(1) and GCN/SCN(2) (0.69 and 0.70). This hints that the preparation method can influence the condensation degree of the as-prepared isotype heterojunctions.
| Sample | SBET (m2 g−1) | Pore volume (cm3 g−1) | C/N ratio | Band gap (eV) | k (min−1) |
|---|---|---|---|---|---|
| GCN | 14.5 | 0.06 | 0.68 | 2.72 | 0.0024 |
| SCN | 8.6 | 0.03 | 0.71 | 2.54 | 0.0035 |
| GCN–SCN | 12.4 | 0.05 | 0.66 | 2.60 | 0.0228 |
| GCN/SCN(1) | 11.6 | 0.04 | 0.69 | 2.55 | 0.0064 |
| GCN/SCN(2) | 12.9 | 0.04 | 0.70 | 2.46 | 0.0080 |
UV-Vis spectra were used to investigate the optical properties and electronic band structure of the as-prepared g-C3N4 catalysts. As shown in Fig. 2, all the samples feature an intrinsic semiconductor-like absorption. The intensity of the absorbance of SCN is higher than that of GCN in the full spectrum, accompanying the absorption edge shift from 455 for GCN to 477 for SCN. The band gaps are estimated from the tangent lines in the plots of the square root of the Kubelka–Munk function as a function of the photon energy (Fig. 2 insert).29 The results show that the band gap energy for GCN and SCN is 2.72 and 2.54 eV, respectively. The remarkable difference in the band gap energy between GCN and SCN provides great potential for the design of a g-C3N4 based isotype heterojunction with a well-matched band structure. It is noted that a broad absorption peak from 450 nm to 600 nm is observed for the SCN. Wang et al. obtained a similar result and suggested that sulfur was most likely doped into the crystal lattice of g-C3N4, creating more defects.30 Wang et al. simulated the total and partial DOS of the pure and S-doped g-C3N4 using first principle calculations and suggested that the doped S atom influenced the distribution of C and N atoms in the lattice, leading to hybrid p orbitals among the C, N and S atoms and causing a broad absorption peak at 450–600 nm. The band gap energy of GCN–SCN is located between GCN and SCN (2.60 eV), which further confirmed the electronic coupling of these two components in the GCN–SCN heterojunction. In Fig. S1 and S2,† the band gaps for GCN(1), GCN(2), SCN(1) and SCN(2) are 2.66, 2.65, 2.55 and 2.46 eV, which are different from GCN and SCN. This confirms that the calcination method can determine the condensation degree, which significantly influences the band gap structure of g-C3N4. The band gap energies of GCN/SCN(1) and GCN/SCN(2) are also located between their components (2.58 and 2.57 eV, Table 1), confirming the formation of heterojunction catalysts.
 | ||
| Fig. 2 UV-Vis spectra of the as-prepared GCN, SCN and GCN–SCN. | ||
To confirm the structure of g-C3N4 and further identify the chemical state of the sulfur element, the g-C3N4 based catalysts were characterized using XPS. In Fig. 3a and b, the spectra of the as-prepared g-C3N4 based catalysts in both the N 1s and C 1s regions can be fitted with two contributions. In the C 1s region (Fig. 3a), two components are located at 284.6 and 288 eV for GCN and SCN. The sharp peak around 284.6 eV is attributed to the pure graphitic species in the CN matrix. The peak with a binding energy of 288 eV indicates the presence of sp2 C atoms bonded to the aliphatic amine (–NH2 or –NH–) in the aromatic rings.31 In Fig. 3b, the main N 1s peak at a binding energy of 398.5 eV can be assigned to sp2 hybridized nitrogen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C), thus confirming the presence of sp2 bonded graphitic carbon nitride. The peak at the higher binding energy of 400.5 eV is attributed to tertiary nitrogen (N–(C)3) groups.11 The spectra of GCN–SCN in the C 1s and N 1s regions do not exhibit a shift in the binding energy. In the S 2p region (Fig. 3c), no S species is found in the GCN. The binding energy is located at 163.9 eV for SCN. According to the literature reported by Chen et al., this binding energy is assigned to doping sulfur in the nitrogen position to form a S–C bond.32 For GCN–SCN, no obvious shift in the binding energy is observed in the S 2p region, suggesting that the formation of the GCN–SCN heterojunction does not influence the chemical state of sulfur.
N–C), thus confirming the presence of sp2 bonded graphitic carbon nitride. The peak at the higher binding energy of 400.5 eV is attributed to tertiary nitrogen (N–(C)3) groups.11 The spectra of GCN–SCN in the C 1s and N 1s regions do not exhibit a shift in the binding energy. In the S 2p region (Fig. 3c), no S species is found in the GCN. The binding energy is located at 163.9 eV for SCN. According to the literature reported by Chen et al., this binding energy is assigned to doping sulfur in the nitrogen position to form a S–C bond.32 For GCN–SCN, no obvious shift in the binding energy is observed in the S 2p region, suggesting that the formation of the GCN–SCN heterojunction does not influence the chemical state of sulfur.
 | ||
| Fig. 3 XPS spectra of the as-prepared g-C3N4 catalysts in the region of C 1s (a), N 1s (b) and S 2p (c). | ||
The band structures of GCN and SCN were further investigated by VB XPS (Fig. 4). The VB maxima of GCN and SCN are shown to be 1.52 and 1.08 eV, respectively.27,33 The VB potential of GCN is more positive than that of SCN. Combined with the band gap energies obtained from UV-Vis spectra, the CB position of GCN and SCN are calculated to be −1.2 and −1.46 eV, indicating that the CB potential of SCN is more negative than that of GCN. On the basis of VB and CB levels of GCN and SCN, a band structure diagram for the GCN–SCN isotype heterojunction can be drawn as shown in Fig. 5. Once GCN and SCN are electronically coupled together, the band alignment between the two kinds of g-C3N4 materials results in the formation of a heterojunction with a well-matched band structure. Upon visible-light irradiation, the photogenerated electrons tend to transfer rapidly from SCN to GCN driven by the CB offset of 0.26 eV, whereas the photogenerated holes transfer from GCN to SCN driven by the VB offset of 0.44 eV. The potential difference is the main driving force for the efficient charge separation and transfer. These two charge transfer processes are beneficial for overcoming the high dissociation barrier of the Frenkel exciton and stabilizing electrons and holes. The redistribution of electrons on one side of the heterojunction (GCN) and holes on the opposite side (SCN) could establish steady internal electric fields, which reduces the electron–hole pair recombination. As the photogenerated electrons and holes are spatially separated into two components, the charge recombination is drastically inhibited, which is of great benefit for enhancing the photocatalytic activity. In addition, with the effective separation of electron–hole pairs, the lifetime of the photogenerated charge carriers is expected to be prolonged. The prolonged lifetime allows fast charge transfer to the reactive substrates on the photocatalyst surface, promoting the photocatalysis reaction.27,28 As shown in Fig. S3 and S4,† the VB maxima of GCN(1), SCN(1), GCN(2) and SCN(2) are 1.54, 1.40, 1.58 and 1.38 eV, respectively. Accordingly, the CB position of GCN(1), SCN(1), GCN(2) and SCN(2) are calculated to be −1.12, −1.15, −1.07 and −1.08 eV. Thus the heterojunction catalysts GCN/SCN(1) and GCN/SCN(2) with a well-matched band structure are also obtained.
 | ||
| Fig. 4 VB XPS of the as-prepared GCN and SCN. | ||
 | ||
| Fig. 5 The schematic illustration of electron–hole separation and transport at the GCN–SCN heterojunction interface: EC is the contact electric field for the two components; EB is the potential barrier in the interfacial depletion layer; E1 and E2 are the internal electric fields induced by the redistribution of the spatial charges in GCN and SCN, respectively. | ||
EIS and PL spectra are very useful tools for characterizing the charge-carrier migration, and these spectra were employed to further confirm the interfacial charge transfer effect of the as-prepared g-C3N4 catalysts. As shown in Fig. 6a, the as-prepared heterojunction catalysts exhibit a decreased arc radius compared to that of GCN and SCN. In general, the radius of the arc in the EIS spectra reflects the reaction rate on the surface of the electrode.34 The reduced arc radius indicates the diminished resistance of the working electrodes, suggesting a decrease in the solid state interface layer resistance and the charge transfer resistance across the solid–liquid junction on the surface between the GCN and SCN.35,36 GCN–SCN shows the smallest arc radius. Since the radius of the arc on the EIS spectra reflects the migration rate occurring at the surface, it suggests that a more effective separation of the photogenerated electron–hole pairs and a faster interfacial charge transfer occurs on the GCN–SCN surface.36,37 In general, at a lower PL intensity, the separation rate of the photogenerated electron–hole pairs is higher. In Fig. 6b, the PL intensity follows the sequence: GCN–SCN < GCN/SCN(2) < GCN/SCN(1) < SCN < GCN. This order in the degree of PL quenching is consistent with the arc radius shown in the EIS spectra, confirming the efficient transfer of photoinduced electrons and holes in GCN–SCN.
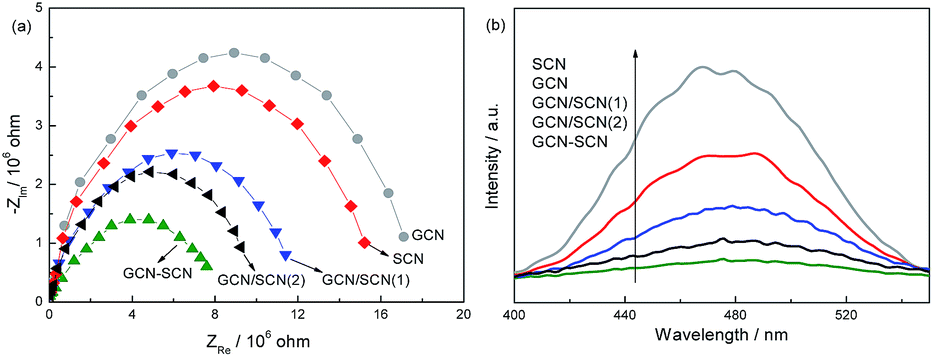 | ||
| Fig. 6 EIS and PL spectra of the as-prepared g-C3N4 catalysts. | ||
The I–t curve is provided to compare the carrier separation ability of as-prepared heterojunction catalysts (Fig. 7). The decay of the photocurrent indicates that partial h+ arriving at the catalyst surface does not capture e− from the electrolyte but accumulates at the surface or recombines with electrons from the conduction band. After the recombination of the excessive h+, the generation and transfer of electron–hole pairs reach equilibrium, and a stable photocurrent is formed. The photocurrent value of GCN–SCN is obviously higher than that of GCN/SCN(1) and GCN/SCN(2), which can be attributed to the more efficient separation of photogenerated electron–hole pairs. This is consistent with the PL and EIS results.
 | ||
| Fig. 7 The photocurrent responses of the as-prepared metal-free isotype heterojunction catalysts. | ||
The anoxic photocatalytic performance of the as-prepared catalysts was evaluated by studying the degradation of RhB, which has positively charged organic end groups, in the absence of oxygen (Fig. 8). The pH of the RhB solutions is 6.1 during the anoxic photodegradation process. Due to the high recombination rate of the electrons and holes, GCN and SCN show low degradation rates (∼23% and 32%) and reaction rate constants (0.0024 and 0.0035 min−1), as shown in Table 1. For GCN/SCN(1) and GCN/SCN(2), the activities (reaction rate constants) increase to 50% (0.0064 min−1) and 58% (0.008 min−1). GCN–SCN displays the highest anoxic photocatalytic performance, 92% and 0.0228 min−1, which is 3.5 and 2.9 times higher than that of GCN/SCN(1) and GCN/SCN(2). The RhB photocatalytic degradation performance of the as-prepared heterojunction catalysts was compared in the presence of oxygen (Fig. S5†). It was shown that the presence of oxygen does not remarkably promote the activity of the as-prepared heterojunction catalysts, indicating that molecular oxygen is not necessary for the photocatalytic oxidation of RhB in the current system.
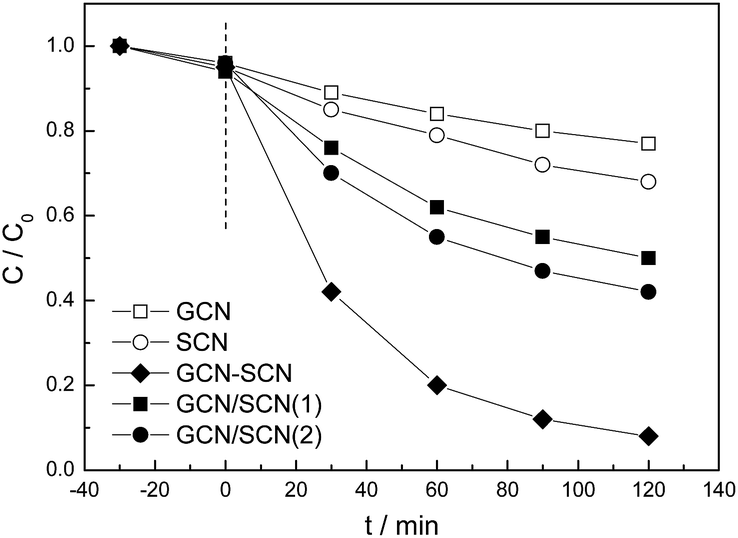 | ||
| Fig. 8 Anoxic photocatalytic degradation performance of RhB over the as-prepared g-C3N4 catalysts under visible light. | ||
From the UV-Vis and VB XPS results, it is calculated that the VB driving force for GCN–SCN, GCN/SCN(1) and GCN/SCN(2) is 0.44, 0.14 and 0.2 eV, respectively. Accordingly, the CB driving force for GCN–SCN, GCN/SCN(1) and GCN/SCN(2) is 0.26, 0.03 and 0.01 eV, respectively. Fig. 9 shows the relationship between the charge driving force and the RhB degradation rate of the as-prepared metal-free isotype heterojunction catalysts. Interestingly, a linear relationship is observed between the VB driving force and RhB degradation rate. The larger the VB driving force, the higher the RhB degradation rate. GCN–SCN with an enhanced charge driving force shows a much higher RhB degradation rate than that of GCN/SCN(1) and GCN/SCN(2). A nonlinear relationship is shown in Fig. 9 between the CB driving force and the RhB degradation rate. It is known that the VB and CB driving forces determine the migration rate of the photogenerated holes and electrons, respectively. This indicates that the photogenerated holes are responsible for the photocatalytic oxidation of RhB in the current system. This is consistent with the reaction results. In the photocatalytic process, photogenerated electrons can trap molecular oxygen to form the reactive oxygen species.38 The reaction results indicate that molecular oxygen is not necessary for the photocatalytic oxidation of RhB in the current system. Thus it is reasonable that a nonlinear relationship is observed between the CB driving force and the RhB degradation rate. In summary, a linear relationship is observed between the VB driving force and the RhB degradation rate. GCN–SCN with an enhanced charge driving force prepared by a two step calcination method shows a much higher RhB degradation rate than that of GCN/SCN(1) and GCN/SCN(2). This paper provides a new perspective to prepare isotype heterojunctions with an increased charge driving force and photocatalytic performance.
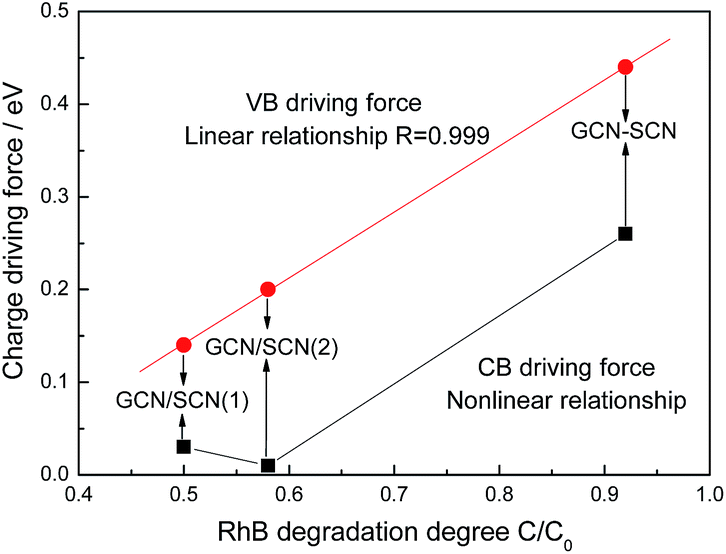 | ||
| Fig. 9 The relationship between the charge driving force and RhB degradation degree of the as-prepared metal-free isotype heterojunction catalysts. | ||
In order to determine the role of electrons and holes in the photodegradation process, EDTA-2Na and KBrO3 are used as the hole (h+) and electron (e−) scavengers, respectively.39–41 Fig. 10 shows the influence of various scavengers on the visible light photocatalytic activity of the as-prepared heterojunction catalysts. With the addition of EDTA-2Na, the RhB degradation rate for GCN–SCN, GCN/SCN(2), and GCN/SCN(1) decreased from 92%, 58% and 50% to 36%, 20% and 17%, respectively. This indicates that photogenerated holes are the main active species for RhB degradation. When KBrO3 was added, the RhB degradation rate for GCN–SCN, GCN/SCN(2) and GCN/SCN(1) did not decrease but increased to 96%, 72% and 66%, respectively. This is probably due to the fact that the addition of KBrO3 to trap the electrons can promote the separation rate of e−/h+ pairs, leading to an increased photocatalytic performance.
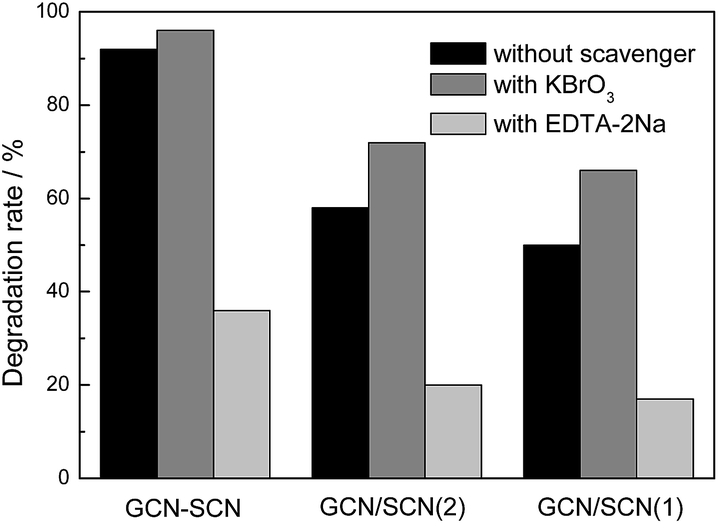 | ||
| Fig. 10 The influence of various scavengers on the visible light photocatalytic activity of the as-prepared heterojunction catalysts. | ||
Conclusions
GCN–SCN, a g-C3N4/S-g-C3N4 metal-free isotype heterojunction catalyst, was prepared by a two step calcination method. Compared with the isotype heterojunction catalysts prepared by a one step calcination method, GCN/SCN(1) and GCN/SCN(2), the charge driving force of GCN–SCN is increased by more than two times, leading to a more efficient charge-carrier migration. The results of RhB anoxic photocatalytic degradation indicate that GCN–SCN displays the highest reaction rate constant of 0.0228 min−1, which is 3.5 and 2.9 times higher than that of GCN/SCN(1) and GCN/SCN(2). Molecular oxygen is not necessary, and photogenerated holes are responsible for the photocatalytic oxidation of RhB in the current system. A linear relationship is observed between the VB driving force and the RhB degradation rate. This paper provides a new perspective to prepare isotype heterojunctions with an increased charge driving force and photocatalytic performance.Acknowledgements
This work was supported by the Science & Technology Research Foundation of Heilongjiang Province Education Bureau of China (No. 12541626), the Postdoctoral Fund of Heilongjiang province of China (No. LBH-Z14208), the Education Department of Liaoning Province (No. L2014145), the Environmental Science and Engineering Innovation Team of Liaoning Shihua University ([2014]-11), and the Students’ Innovation Fund Project of China.Notes and references
- C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206 RSC.
- S. Z. Hu, F. Y. Li, Z. P. Fan, F. Wang, Y. F. Zhao and Z. B. Lv, Dalton Trans., 2015, 44, 1084 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- Q. F. Deng, L. Liu, X. Z. Lin, G. H. Du, Y. P. Liu and Z. Y. Yuan, Chem. Eng. J., 2012, 203, 63 CrossRef CAS PubMed.
- M. B. Ansari, H. L. Jin, M. N. Parvin and S. E. Park, Catal. Today, 2012, 185, 211 CrossRef CAS PubMed.
- G. Liu, L. Z. Wang, H. G. Yang, H. M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831 RSC.
- D. Q. Zhang, G. S. Li and J. C. Yu, J. Mater. Chem., 2010, 20, 4529 RSC.
- Y. J. Wang, Z. X. Wang, S. Muhammad and J. He, CrystEngComm, 2012, 14, 5065 RSC.
- G. G. Zhang, J. S. Zhang, M. W. Zhang and X. C. Wang, J. Mater. Chem., 2012, 22, 8083 RSC.
- F. Dong, L. W. Wu, Y. J. Sun, M. Fu, Z. B. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171 RSC.
- Y. W. Zhang, J. H. Liu, G. Wu and W. Chen, Nanoscale, 2012, 4, 5300 RSC.
- Y. J. Zhang, T. Mori, J. H. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294 CrossRef CAS PubMed.
- Y. Y. Bu, Z. Y. Chen and W. B. Li, Appl. Catal., B, 2014, 144, 622 CrossRef CAS PubMed.
- C. Chang, Y. Fu, M. Hu, C. Y. Wang, G. Q. Shan and L. Y. Zhu, Appl. Catal., B, 2013, 142–143, 553 CrossRef CAS PubMed.
- J. D. Hong, X. Y. Xia, Y. S. Wang and R. Xu, J. Mater. Chem., 2012, 22, 15006 RSC.
- J. S. Zhang, X. F. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Z. Fu, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2010, 49, 441 CrossRef CAS PubMed.
- C. S. Pan, J. Xu, Y. J. Wang, D. Li and Y. F. Zhu, Adv. Funct. Mater., 2012, 22, 1518 CrossRef CAS PubMed.
- X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe and K. Domen, J. Am. Chem. Soc., 2009, 131, 1680 CrossRef CAS PubMed.
- G. Z. Liao, S. Chen, X. Quan, H. T. Yu and H. M. Zhao, J. Mater. Chem., 2012, 22, 2721 RSC.
- K. Sridharan, E. Jang and T. J. Park, Appl. Catal., B, 2013, 142–143, 718 CrossRef CAS PubMed.
- L. Ge, C. C. Han, X. L. Xiao and L. L. Guo, Int. J. Hydrogen Energy, 2013, 38, 6960 CrossRef CAS PubMed.
- M. L. Lu, Z. X. Pei, S. X. Weng, W. H. Feng, Z. B. Fang, Z. Y. Zheng, M. L. Huang and P. Liu, Phys. Chem. Chem. Phys., 2014, 16, 21280 RSC.
- M. Yang, S. Z. Hu, F. Y. Li, Z. P. Fan, F. Wang, D. Liu and J. Z. Gui, Ceram. Interfaces, 2014, 40, 11963 CrossRef CAS PubMed.
- J. S. Zhang, M. W. Zhang, R. Q. Sun and X. C. Wang, Angew. Chem., Int. Ed., 2012, 51, 10145 CrossRef CAS PubMed.
- F. Dong, Z. W. Zhao, T. Xiong, Z. L. Ni, W. D. Zhang, Y. J. Sun and W. K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392 CAS.
- F. Dong, Z. L. Ni, P. D. Li and Z. B. Wu, New J. Chem., 2015, 39, 4737 RSC.
- X. Wang, Q. Xu, M. Li, S. Shen, X. Wang, Y. Wang, Z. Feng, J. Shi, H. Han and C. Li, Angew. Chem., Int. Ed., 2012, 51, 13089 CrossRef CAS PubMed.
- J. Hou, C. Yang, Z. Wang, W. Zhou, S. Jiao and H. Zhu, Appl. Catal., B, 2013, 142–143, 504 CrossRef CAS PubMed.
- Y. I. Kim, S. J. Atherton, E. S. Brigham and T. E. Mallouk, J. Phys. Chem., 1993, 97, 11802 CrossRef CAS.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. G. Yu, Appl. Catal., B, 2015, 176, 44 CrossRef PubMed.
- W. Lei, D. Portehault, R. Dimova and M. Antoniettit, J. Am. Chem. Soc., 2011, 133, 7121 CrossRef CAS PubMed.
- G. Liu, P. Niu, C. H. Sun, S. C. Smith, Z. G. Chen, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2010, 132, 11642 CrossRef CAS PubMed.
- S. Chu, Y. Wang, Y. Guo, J. Feng, C. Wang, W. Luo, X. Fan and Z. G. Zou, ACS Catal., 2013, 3, 912 CrossRef CAS.
- Y. Xu, H. Xu, L. Wang, J. Yan, H. Li, Y. Song, L. Huang and G. Cai, Dalton Trans., 2013, 42, 7604 RSC.
- B. L. He, B. Dong and H. L. Li, Electrochem. Commun., 2007, 9, 425 CrossRef CAS PubMed.
- S. He, L. Ma, H. Wang, L. Zhang, Y. Zhang and G. Wu, RSC Adv., 2015, 5, 31947–31953 RSC.
- Q. W. Huang, S. Q. Tian, D. W. Zeng, X. X. Wang, W. L. Song, Y. Y. Li, W. Xiao and C. S. Xie, ACS Catal., 2013, 3, 1477 CrossRef CAS.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- S. Q. Zhang, Y. X. Yang, Y. N. Guo, W. Guo, M. Wang, Y. H. Guo and M. X. Huo, J. Hazard. Mater., 2013, 261, 235 CrossRef CAS PubMed.
- M. Muneera and D. Bahnemannb, Appl. Catal., B, 2002, 36, 95 CrossRef.
- W. Liu, S. F. Chen, W. Zhao and S. J. Zhang, J. Hazard. Mater., 2009, 164, 154 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra15611d |
| This journal is © The Royal Society of Chemistry 2015 |