
Highly percolated poly(vinyl alcohol) and bacterial nanocellulose synthesized in situ by physical-crosslinking: exploiting polymer synergies for biomedical nanocomposites
 a
a
Abstract
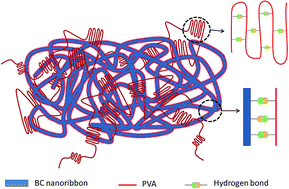
Bacterial cellulose (BC) grown from a culture medium in the presence of water-soluble poly(vinyl alcohol) (PVA) produced an assemblage that was used as precursor for the synthesis of biocompatible nanocomposites. Physical crosslinking via cyclic freezing and thawing of the formed hydrogel facilitated retention of PVA matrix upon composite separation and purification. The composites displayed a porous architecture within the PVA matrix and an excellent compressive strength as a result of the synergism between BC and PVA. BC largely improved the thermo-mechanical performance as well as moisture and dimensional stability of the systems while PVA imparted optical transparency and extensibility. Compared to the respective reference sample (BC-free material), elastic modulus increments of 40, 98 and 510% were measured for PVA-based nanocomposites loaded with BC at 10, 20 and 30% levels, respectively. Likewise, the corresponding strength at break were 30, 77 and 104% higher. The results indicate an exceptional reinforcing effect endowed by the three-dimensional network structure that was formed in situ upon BC biosynthesis in the presence of PVA and also suggest a large percolation within the matrix. BC is relatively inexpensive, can produce scaffolds of given shapes and with high strength and acts as an excellent reinforcing element that promotes cell proliferation. Taken these properties together, BC and BC/PVA composites are promising materials in biomedical engineering.
 Please wait while we load your content...
Please wait while we load your content...

