
Acetalated-dextran as valves of mesoporous silica particles for pH responsive intracellular drug delivery
Zhe Lina,
Jizhen Lic,
Hongyan Hebd,
Huihui Kuangb,
Xuesi Chene,
Zhigang Xieb,
Xiabin Jingb and
Yubin Huang*b
aResearch and Development Center, Changchun University of Chinese Medicine, Changchun 130117, P. R. China
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China. E-mail: ybhuang@ciac.ac.cn; Fax: +86-431-85262769; Tel: +86-431-85262769
cDepartment of Organic Chemistry, College of Chemistry, Jilin University, Changchun 130023, P. R. China
dUniversity of Chinese Academy of Sciences, Beijing 100039, P. R. China
eKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
First published on 5th January 2015
Abstract
A pH-sensitive drug release system using acetalated-dextran as valves was designed to manipulate smart intracellular release of anticancer drugs. Dextran was grafted onto the exterior of MSN through a click reaction, and followed by acetalation to generate the final carriers of MSN–Dex-Ac. The hydrophobic Dex-Ac would act as valves on the MSN surface to block the entrapped drugs inside the MSN pores. While under acidic conditions mimicking the micro-environment of endosomal/lysosomal compartments, the valves could be opened by acetal hydrolysis to recover the acetalated-dextran to its hydrophilic state, resulting in fast drug release. In vitro drug release profile clearly showed that DOX release was restricted at pH 7.4 by the valves, while it was accelerated under acidic conditions. Fast endocytosis and intracellular DOX release was observed by confocal laser scanning microscopy (CLSM). Cytotoxicity evaluation showed good biocompatibility with the carriers. In vitro MTT assays revealed that the DOX-loaded particles exhibited comparable antitumor activity with free DOX towards HeLa cells.
Introduction
The construction of site-specific stimuli-responsive controlled drug delivery systems (CDDS) has been extensively investigated over the past decade, since it is an important solution for cancer chemotherapy especially for treatment of improved efficacy and reduced toxicity. The crucial problem of CDDS is how to transport an effective amount of drug with less premature release and reduced toxicity.1–6 To solve these problems, drug carriers can play the key role. Many efforts have been dedicated to design intelligent drug delivery materials to accomplish the aforementioned prerequisites, such as liposomes,7 polymeric micelles,8 polymer nanoparticles,9 dendrimers,10 inorganic–organic hybrid nanoparticles11,12 and various inorganic nanomaterials.13–15Among them, mesoporous silica nanoparticles (MSN) have attracted a lot of research attention for various controlled delivery applications due to the following unique properties: tunable particle size, controllable surface functionalization, nontoxicity and high surface area.16 In addition, MSN equipped with molecular or supramolecular nanovalves can regulate the release of loaded cargo molecules by external stimuli, such as redox,17–20 enzymatic activity,21–23 photoirradiation,24,25 magnetic actuation26 and pH.27
It is well documented that the micro environment around tumor (pH 6.5), and endosomes or lysosomes inside tumor cells (pH 5.5) are more acidic than that in blood and normal tissue (pH 7.4).28,29 This motivated us to design nanocarriers that could release therapeutic agents responding to physiopathological pH signals. To date, several kinds of pH-responsive nanovalves are explored for capping MSN, including pH-sensitive linkers,30 and polyelectrolytes.31,32 For example, Sun et al. have grafted a poly(2-diethylaminoethyl methacrylate) (PDEAEMA) shell onto the exterior surface of MSN via surface-initiated atom transfer radical polymerization (ATRP), to produce a novel nanodevice with the MSN core as the drug carrier and the pH-responsive PDEAEMA shell as a smart nanovalve. In the very recent years, some progress has been made towards designing the biocompatible nature gatekeepers like polypeptide,33,34 protein,23,35–37 chitosan,38 etc.
Dextran, a polysaccharide consisting predominantly of 1,6-glucosidic linkages, has obtained the approval from FDA and has a long history in clinical applications as a plasma expander and many kinds of commercial drug formulations.39,40 Application of dextran as a polymeric drug carrier in CDDS is developed nowadays,39,41 because dextran can be easily functionalized with biomolecules or drugs via its hydroxyl groups either by direct esterification or by the pre-introduction of spacer arms.42 Dextran itself is water-soluble, and is a natural analog to PEG to reduce the macrophage phagocytosis.43–45 Also, it could be rendered to water insoluble by modification of its hydroxyl groups with liposoluble materials like cholesterol46 and alkane.47 Recently, a simple but effective approach was proposed by Fréchet's group: dextran was modified in the presence of 2-methoxypropene and p-toluene sulfonic acid in DMSO to convert the hydroxyl groups into pH-responsive acetal groups.48–51 Acetalated dextran has two crucial properties: first, acetalation of dextran changes it to a hydrophobic material; second, hydrolysis of acetal groups under acidic conditions could regenerate hydrophilic dextran. This pH-sensitive switch mechanism is beneficial to design intelligent drug carriers.
Inspired by the previous works, we for the first time designed an MSN–acetalated dextran (MSN–Dex-Ac) hybrid drug carrier system for intracellular drug delivery. Dextran was grafted onto the exterior of MSN through click reaction firstly, followed by the acetal reaction to generate the final carriers of MSN–Dex-Ac. Under neutral environment like in blood circulation, the hydrophobic Dex-Ac would collapse on MSN surface acting as valves to block the entrapped drugs inside MSN pores. While under acidic conditions mimicking the micro-environment of endosomal/lysosomal compartments, the valves could be opened by acetal hydrolysis to recover the acetalated-dextran to its hydrophilic state, resulting in fast drug release. In our experiments, doxorubicin (DOX), as the model drugs, could be efficiently locked in the pores by the collapsed hydrophobic dextran chain during the circulation process. Once DOX-loaded nanoparticles were internalized by cancer cells, DOX would be efficiently released into the cytosol due to the hydrophilic conversion of dextran in response to an acidic stimulus (Scheme 1). The hybrid particles of MSN–Dex-Ac provided promising vehicles for smart drug delivery.
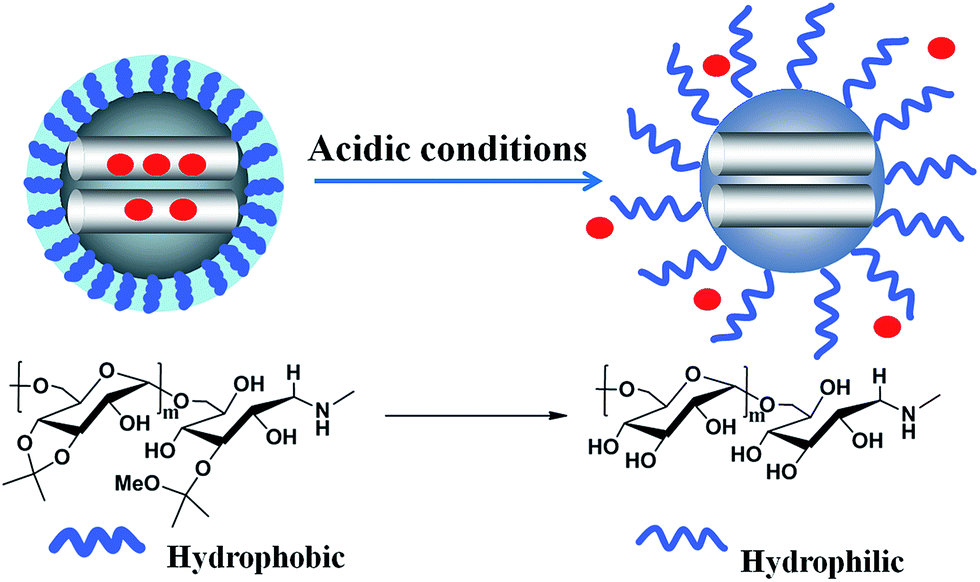 | ||
| Scheme 1 Schematic illustration of intracellular microenvironment triggered drug release for DOX loaded MSN–Dex-Ac@DOX particles. | ||
Experiment
Materials
Dextran (Dex, Mn = 6 kDa) was purchased from Sigma-Aldrich without further purification. Propargylamine (98%, Sigma), (3-chloropropyl)trimethoxysilane (Sigma), sodium azide (Aladdin), tetraethyl orthosilicate (TEOS, Aladdin), n-cetyltrimethylammonium bromide (CTAB, YiLi fine chemicals Co. Ltd. Beijing), sodium cyanoborohydride (95%, Sigma), 2-methoxypropene (Energy Chemical), p-toluenesulfonic acid (Energy Chemical) were used as received. Toluene was purified by distillation from sodium with benzophenone. DMSO and N,N-dimethylformamide (DMF) was dried over calcium hydride for 48 h and then distilled before use. Doxorubicin hydrochloride (DOX·HCl) was purchased from Zhejiang Hisun Pharmaceutical Co. Ltd. 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich. Other reagents were commercially available and used as received.Characterizations
Transmission electron microscopy (TEM) studies were performed on a JEM-1011 electron microscope operating at an acceleration voltage of 100 kV. Fourier transform infrared spectrophotometer (FT-IR) spectra were recorded on a Bruker Vertex70 Win-IR instrument. X-ray measurements were performed on a Bruker D8 FOCUS Powder X-ray diffractometer using Cu Kα radiation. N2 adsorption–desorption isotherms were recorded on a Micromeritics ASAP 2020M automated sorption analyzer. The samples were degassed at 150 °C for 5 hours. Thermogravimetric analyses were carried out on a PerkinElmer Pyris Diamond TG/DTA Analyzer, using an oxidant atmosphere (air) with a heating program consisting of a dynamic segment (10 °C min−1) from 373 to 1073 K. Particle size and zeta potential measurements were conducted on a Malvern Zetasizer Nano ZS, samples were dissolved in de-ionized water. The UV spectrum was determined using a UV-Vis spectrophotometer (UV-2450PC, Shimadzu). 1H NMR spectra were characterized on a Bruker AV 300M spectrometer in CDCl3 and DMSO-d6 at 25 °C. Chemical shifts were given in parts per million with respect to tetramethylsilane (TMS) as an internal reference.Synthesis of α-alkyne dextran
Briefly, Dex (5.00 g, 0.0833 mmol) was dissolved in acetate buffer (0.01 M, pH = 5.0) in a flask followed by adding propargylamide (5.71 mL, 8.33 mmol) and sodium cyanoborohydride (5.23 g, 8.33 mmol). Then, pH of the solution was adjusted to 5.0 using 1.0 M of HCl and the mixture was allowed to react at 50 °C for 96 h. After that, the crude reaction mixture was concentrated and then dialyzed against deionized water for 96 h (MWCO = 3500) and freeze-dried to obtain the product (4.76 g, yield: 95.2%).Synthesis of azido-functionalized particles MSN–N3
The organic compound of 3-azidopropyltrimethoxysilane (AzPTMS) was first synthesized. 3-Chloropropyltriethoxysilane (10.00 mL, 54.4 mmol), sodium azide (14.00 g, 217.6 mmol) and 100 mL of dry DMF were heated to 90 °C under a N2 atmosphere for 4 h. The mixture was concentrated under reduced pressure, and then poured into 100 mL of cold diethyl ether to remove the precipitated salts. After that, the mixture was concentrated again and the residual oil was dried under reduced pressure to get the liquid product of AzPTMS.Then, CTAB (1 g) was dissolved in 480 mL of deionized water. NaOH solution (2.00 M, 3.50 mL) was added to CTAB solution, followed by heating to 80 °C and vigorous stirring. TEOS (5.00 mL) was then introduced dropwisely to the above solution and the mixture was maintained at 80 °C for 2 h to produce a white precipitate. This solid crude product of MSN was collected, washed with water and ethanol, and dried in air.
This obtained MSN (900 mg) were suspended in 50 mL of dry toluene containing 1 mL of AzPTMS. The solution was stirred at 90 °C under nitrogen for 24 h. The product was recovered by centrifugation and washed with ethanol. To remove the surfactant template (CTAB), the obtained particles were dispersed in 300 mL of ethanol solution containing 3 g of NH4NO3 and refluxed at 80 °C for 24 h. The final particles were separated by centrifugation (9000 rpm, 10 min) and extensively washed with methanol. After dried under vacuum, MSN–N3 particles was finally obtained.
Dextran modification on MSN (MSN–Dex)
Dextran was grafted onto the surface of MSN through a click reaction as follows: MSN–N3 (200 mg), α-alkyne Dex (1.2 g, 0.2 mmol) and CuSO4·5H2O (50 mg, 0.2 mmol) were dissolved in 20 mL of dried DMSO. The mixture was stirred for 10 min and degassed by freeze–thaw in a Schlenk flask. Then, under the frozen state, sodium ascorbate (38.8 mg, 0.2 mmol) was added. Thereafter, another two cycles of freeze–thaw were conducted. The flask was placed in an oil bath at 60 °C for 72 h. The reaction was terminated by contact with air and the particles were isolated by centrifugation, extensively washed with DMSO and H2O, and dried under vacuum to obtain the intermediate particles of MSN–Dex.Acetal modification on MSN–Dex particles (MSN–Dex-Ac)
MSN–Dex (100 mg), p-toluenesulfonic acid (2.8 mg) were dissolved in anhydrous DMSO, followed by addition of 2-methoxypropene (150 μL). The reaction was allowed to proceed for 18 h at 20 °C and quenched by addition of triethylamine. The particles were isolated by centrifugation, extensively washed with DMSO, acetone and H2O (pH = 7.4) in order. After lysophilization, the MSN–Dex-Ac particles were finally obtained.To track the acetal reaction, dextran itself was acetalated using the similar method.
Preparation of DOX-loaded particles
Doxorubicin (DOX) was used as a model drug for in vitro drug loading and release. Briefly, DOX·HCl (20.00 mg) and 3-time excess of triethylamine were dissolved in DMSO (0.50 mL) and stirred for 0.5 h. This DOX solution was added to THF (10 mL) solution containing MSN–Dex-Ac (60 mg). Then, deionized water (50 mL) was added dropwisely to the above mixture and stirred for an additional 2 h. After that, THF was removed by rotate evaporator, and the mixture was dialyzed against deionized water for 48 h (MWCO = 3500) and freeze-dried. The drug loaded particles were named as MSN–Dex-Ac@DOX. To determine the loading amount of DOX into particles, DOX loaded in particles was extracted with DMF. After removing the blank particles by centrifugation, the UV absorbance of the supernatant at 480 nm was measured. Drug loading content was obtained according to a standard curve obtained from DOX/DMF solutions at a series of DOX concentrations.Two control groups of MSN–Dex and MSN–N3 were also loaded with DOX in the same way and named as MSN–Dex@DOX and MSN@DOX, respectively.
In vitro DOX release
In vitro drug release behavior of freeze-dried DOX-loaded particles of MSN–Dex-Ac@DOX, MSN–Dex@DOX and MSN@DOX were investigated in buffer solution at pH 7.4, 6.5 and 5.0, respectively. Briefly, DOX-loaded particles were suspended in 5 mL of phosphate buffer (pH 7.4 or 6.5) or acetate buffer (pH 5.0) solutions (1 mg mL−1) and transferred into a dialysis bag (MWCO = 3500). The bag was immersed in 25 mL of corresponding buffer at 37 °C with continuous shaking (90 rpm). At predetermined time, 3 mL of release medium was taken out for UV-Vis measurement and an equal volume of fresh buffer was replenished. The amount of released DOX was determined from the absorbance at 480 nm with the help of a calibration curve of DOX in the same buffer.Intracellular uptaken and drug release
The cellular uptake and intracellular release behaviors of DOX-loaded particles were determined by confocal laser scanning microscopy (CLSM) toward HeLa cells (a human cervical cancer cells). Hela cells were cultured in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C under 5% CO2. Cells were split in 6-well culture plates with a sterile coverslip in each well at a density of 5 × 104 cells per well and cultured for 24 h. Then, after the culture medium were removed, cells were exposed to free DOX or MSN–Dex-Ac@DOX at a final DOX concentration of 10 μg mL−1 for 0.5 h, 2 h or 4 h at 37 °C. Thereafter, the culture medium was removed and cells were washed three times with ice-cold PBS and fixed with 4% formaldehyde for 30 min at room temperature. After the nucleus were stained with DAPI for 20 min, CLSM images of was observed through Olympus FV1000 confocal laser scanning microscope (CLSM) imaging system (Japan).In vitro cytotoxicity of the blank particles and drug-loaded particles
The biocompatibility of blank particles (MSN–Dex-Ac and MSN–N3) was investigated by MTT assay against L929 normal cells (a mouse fibroblasts cell line), which were cultured in Dulbecco's modified Eagle's medium (DMEM, GIBCO) supplemented with 10% heat-inactivated fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C under 5% CO2. Cells were split in 96-well culture plates at a density of 6 × 103 cells per well in100 μL DMEM. After incubated to adhere for 24 h, cells were treated with a series of concentrations of particles solutions and cultured for further 48 h. Then, 20 μL of MTT solution in PBS (5 mg mL−1) was added to each well and cells were incubated for additional 4 h at 37 °C. After that, the medium was replaced by 150 μL of DMSO to dissolve the MTT formazan crystals. Finally, the plates were shaken for 10 min, and the absorbance of formazan product was measured at 492 nm using a Bio-Rad 680 microplate reader.The anti-cancer activity of DOX-loaded particles and free DOX were assessed with MTT assay against HeLa cells. Cells were split in 96-well culture plates at a density of 6 × 103 cells per well in 100 μL DMEM. After incubated to adhere for 24 h, cells were treated with DOX-loaded particles suspension or free DOX solution with a series of DOX concentrations from 8 mg mL−1 to 0.0128 mg mL−1 and cultured for further 48 h. Then, 20 μL of MTT solution in PBS (5 mg mL−1) was added to each well and cells were incubated for additional 4 h at 37 °C. After that, the medium was replaced by 150 μL of DMSO to dissolve the MTT formazan crystals. Finally, the plates were shaken for 10 min, and the absorbance of formazan product was measured at 492 nm using a Bio-Rad 680 microplate reader.
Result and discussion
Synthesis of MSN–Dex-Ac
The drug carriers based on MSN with pH-sensitive acetalated dextran as caps was prepared as described in Scheme 2. For easy and efficient modification, parent MSN with azide groups on the surface (MSN–N3) and dextran with a terminal α-alkyne group were designed and synthesized. Through the click reaction, dextran could be covalently grafted onto the surfaces of MSN to obtain MSN–Dex. Further acetalation of dextran on MSN easily generated the pH-sensitive MSN named as MSN–Dex-Ac. This modification introduced a solubility switch which rendered dextran insoluble in water while degraded to the original hydrophilic state under mildly acidic conditions. Because of the hydrophobic nature, the acetal-derivatized dextrans would collapse on the surface of MSN under neutral condition and act as the valves or caps to block the entrance of MSN pores to prevent leakage of the entrapped drugs inside pores. However under acidic condition, hydrolysis of the acetal structures would recover the water-soluble state of dextrans. The stretching out behavior of dextrans would switch on the valves to MSN pores to release the loaded drugs rapidly. | ||
| Scheme 2 Synthesis routes of the hybrid drug carriers of MSN–Dex-Ac. | ||
Parent particles with azide groups on the surface are obviously necessary (Scheme 2A). The silica nanoparticles produced by one-pot method were treated with AzPTMS52 in toluene to introduce azide groups.53 TEM images (Fig. 1A) showed that the obtained MSN–N3 particles were uniform spherical ones with a mean diameter of approximately 100 nm. The characteristic diffraction peaks of MCM-41 type of particles of (100), (110), (200) and (210) in the XRD profile (Fig. 2) clearly indicated that MSN–N3 with a mesoporous nature was successfully prepared.54 In addition, FT-IR spectrum showed the strong adsorption peak at 2100 cm−1, which could be assigned to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN stretching vibration in azide groups (Fig. 3a).55
NN stretching vibration in azide groups (Fig. 3a).55
 | ||
| Fig. 1 TEM images of MSN–N3 (A) and MSN–Dex (B). | ||
 | ||
| Fig. 2 Low-angle X-ray diffraction (XRD) pattern of MSN–N3 (a) and MSN–Dex powders (b). | ||
 | ||
| Fig. 3 The FT-IR spectra of MSN–N3 (a), MSN–Dex (b) and MSN–Dex-Ac (c). | ||
On the other hand, α-alkyne dextran was synthesized and confirmed by 1H NMR (Fig. 4). The characteristic resonances of the anomeric proton peaks of the reducing end group at 6.7 and 6.3 ppm disappeared, suggesting the successful synthesis of the desired α-alkyne dextran.56
 | ||
| Fig. 4 1H NMR spectra of dextran and α-alkyne dextran in DMSO-d6. | ||
We know that acetal structure is pH sensitive. In order to avoid the possible degradation of acetal-derivatized dextran during the subsequent reactions, we grafted α-alkyne dextran to particles by click reaction to synthesize MSN–Dex at first. The acetalation of grafted dextran was carried out at the final step (Scheme 2B).
TEM image of MSN–Dex was shown in Fig. 1B. MSN–Dex still remained uniform spherical morphologies after click reaction. In comparison with the parent particles of MSN–N3 (Fig. 1A), the clear polymer nanoshell around the nanoparticle is visible. The XPS pattern of MSN–Dex still clearly exhibited the characteristic diffraction pattern of hexagonal MCM-41-type mesoporous silicas (Fig. 2). However, the diffraction peaks are weaker than the corresponding ones in MSN–N3, owing to the capping effect of dextran. In FT-IR spectrum (Fig. 3), MSN–Dex showed an increase of the adsorption peak at 2930 cm−1, indexing the dissymmetry stretching vibration of C–H groups in dextran. In addition, strengthened peaks at 3400 cm−1 were assigned to –OH stretching vibration of intermolecular hydrogen bonding, also owing to the introduction of dextran. The thermogravimetric analysis of MSN was performed. The difference of weight loss between MSN–N3 and MSN–Dex was 8 wt%, which should be mainly from grafted dextran (Fig. 5). Combining the TEM, XPS, FT-IR and TGA results, we can deduce that dextran was successfully grafted on the surface of MSN.
 | ||
| Fig. 5 The thermogravimetric analysis of MSN–N3 (a) and MSN–Dex (b). | ||
An important aspect of this study focused on the acetalation reaction. The acetalation method we used was according to the previous literature with some modification.47,53 In the acetalation reaction, two different acetal structures (cyclic and acyclic forms) could be generated and attached to dextran. The ratio of cyclic to acyclic forms and the total number of them affect the degradation time of the acetal-derivatized dextran on MSN. In the previous literature,51 the ratio of cyclic to acyclic forms under different reaction conditions has been described for details. In our paper, we grafted α-alkyne dextran to particles by click reaction to synthesize MSN–Dex at first, followed by the acetalation of grafted dextran. For inorganic particles, it's difficult to track the reaction explicitly and directly. To solve this problem, we conducted the acetalation for free dextran as a model reaction. In the 1H NMR spectrum of this acetalized dextran (Fig. 6), chemical shifts at 5.13, 4.92, and 4.15–3.60 ppm can be attributed to the anomeric and ring protons of dextran, and peaks at 1.5 ppm (peak 7), 3.25 ppm (peak 8) can be assigned to –CH3 of cyclic and acyclic acetal forms, respectively. The results were in good agreement with the literatures and indicated the successfully modification of dextran. Based on these results, MSN–Dex-Ac was then prepared under the same conditions of the above model reaction.
 | ||
| Fig. 6 1H NMR spectrum of acetalated dextran in CDCl3. | ||
It is necessary to address that the acetalation of dextran will affect the dispersibility of the hybrid particles, but not to the extent of causing fast aggregation. The reason might lie in that the acetalation of dextran was not complete. The particles still keep well-dispersion for few hours. After that, only slight aggregation could be observed. We found that, dispersibility could be improved by modifying the surface of the particles with hydrophilic polymers (like PEG) in our subsequent works.
The structure of MSN–Dex-Ac was well proved by FT-IR analysis (Fig. 3). Comparing to MSN–Dex, the key feature to note was the significant decrease of the –OH peak at around 3400 cm−1, indicating the extent of reaction for hydroxyl groups during the acetalation process. IR spectrum also appeared some expected peaks at 1460 cm−1, 1375 cm−1 and 2990 cm−1 which were assigned to dissymmetry bending vibration, symmetry bending vibration and stretching vibration of –CH– in –CH3 introduced by acetalation reaction, respectively.57
In vitro DOX loading and pH triggered release
DOX is a widely used antineoplastic drug in the treatment of many solid tumors. In the current study, DOX was used as the model drug and loaded into particle pores by means of solvent evaporation. In the organic solvent, acetal-derivatized dextran on the surface of the particles was soluble, leaving the entrance of MSN pores open. DOX could enter the nanopores of MSN through free diffusion. After solvent evaporation and dispersion in water, the hydrophobic acetal-derivatized dextran would collapse on the surface of MSN to block the pores, serving as the caps to lock the entrapped DOX in the pores. In order to evaluate effect of the Dex-Ac caps, bare particles of MSN–N3 were also loaded with DOX in the same way. DOX loading content of MSN–Dex-Ac@DOX was calculated to be 22.58 wt%, which was higher than that of MSN@DOX (14.97 wt%). The introduced Dex-Ac caps on MSN surface clearly increased the drug loading efficiency and enhanced the stability of the drug-loading system.DOX release behavior from MSN–Dex-Ac@DOX was investigated in buffer solution at pH 7.4, 6.5 and 5.0, mimicking the pH in blood or normal tissue, solid tumor and late endosome, respectively. MSN@DOX was also tested as a control. As shown in Fig. 7, DOX release rates from both DOX-loaded particles were pH dependent and increased with the decrease of pH. Compared with that of MSN@DOX, DOX release rate from MSN–Dex-Ac@DOX was slower at pH 7.4. After 48 h, the released DOX from MSN–Dex-Ac@DOX and MSN@DOX were about 15% and 24% of the loaded DOX, respectively. The acetal-derivatized dextran as caps significantly restricted DOX release from MSN–Dex-Ac@DOX in neutral condition. While upon decreasing the pH to 5.0, the released amount from MSN–Dex-Ac@DOX after 48 h increased to 45%, greater than that of MSN@DOX (38%). It is easy to understand that the better solubility of DOX in acidic solutions increased the release of loaded DOX in MSN pores. At the same time, the Dex-Ac caps was degraded to be water soluble under acidic condition, switching on the valves of MSN pores to enable much more amount of DOX released. The above results suggested that the drug could be efficiently confined in the pores using acetal-derivatized dextran as caps during blood circulation, and fast released upon the acid-trigger in cancer cells. This character made the MSN–Dex-Ac@DOX effective to be a controlled drug delivery system.
 | ||
| Fig. 7 In vitro DOX release in buffer solutions at pH 7.4, 6.5 and 5.0, at 37 °C: (A) MSN@DOX, (B) MSN–Dex-Ac@DOX. | ||
Intracellular drug release
Cellular uptake and intracellular drug release behavior of MSN–Dex-Ac@DOX were investigated by CLSM in HeLa cells with free DOX as controls. The CLSM images clearly revealed the internalization of MSN–Dex-Ac@DOX, with obvious red fluorescence visible in cells after 0.5 h incubation, mainly located in cytoplasm (Fig. 8D). Differently, when cells were incubated with free DOX, the fluorescence was detected only in the nuclei of cells (Fig. 8A), which was consistent with the reported literature.58 The above CLSM results implied that free DOX entered cells through fast diffusing, while MSN–Dex-Ac@DOX might be uptaken through the endocytosis mechanism.59 The red fluorescence became stronger after a prolonged incubation time of 2 h and 4 h in both free DOX (Fig. 8B and 8C) and MSN–Dex-Ac@DOX (Fig. 8E and 8F) groups. As expected, in MSN–Dex-Ac@DOX group, the fluorescence gradually appeared in the nuclei area instead of the perinuclei, most likely resulting from the diffusion of released DOX from the particles into nuclei. All the above results declared that DOX molecules could release from the DOX-loaded particles of MSN–Dex-Ac@DOX under the acidic environment of the lysosomes and diffuse to the nucleus eventually. With prolonging the incubation time, the drug caused cell inhibition, making the CLSM observation very difficult. Nevertheless, according to the above results, we believe that the released DOX from the particles into nuclei was comparable to the free DOX and would have similar anti-cancer effect to induce the cell inhibition. | ||
| Fig. 8 CLSM-images of HeLa cells incubated with free DOX for 0.5 (A), 2 (B) and 4 h (C), and with DOX-loaded particles MSN–Dex-Ac@DOX for 0.5 (D), 2 (E) and 4 h (F). For each row, images from left to right were: the cells with nucleus stained with DAPI (blue), with DOX (red) fluorescence and overlaid images. Bar = 30 μm. | ||
Cytotoxicity assay
Cytotoxicity of the two blank particles MSN–N3 and MSN–Dex-Ac against L929 cells were tested by MTT assay. The results presented in Fig. 9A showed that the cell viability cultured with these particles were all above 80% even at the highest concentrations of 1.0 mg mL−1, demonstrating the high safety as drug carriers. | ||
| Fig. 9 Cytotoxicity analysis: (A) cell viability of L929 cells after 48 h incubation at 37 °C; (B–D) cell viability of HeLa cells after 12 h, 24 h and 48 h incubation at 37 °C. | ||
The anti-cancer activity of DOX-loaded nanoparticles (MSN–Dex-Ac@DOX, MSN@DOX) as well as free DOX against HeLa cancer cells were also investigated by MTT assay. The results revealed that cytotoxicity of the samples were all time dependent, increasing with prolonged culture time (Fig. 9). Among these, MSN–Dex-Ac@DOX and MSN@DOX exhibited almost the same cytotoxicity after incubation of 12 h, 24 h and 48 h. For the investigation time of 12 h and 24 h, as evidenced by the in vitro DOX release studies above, both the MSN nanoparticles were taken up by cells through endocytosis and concentrated within acidic endosomes and lysosomes (pH 5.0–5.5), where MSN–Dex-Ac@DOX and MSN@DOX exhibited the similar DOX release rate. As for the investigation time of 48 h, although more drugs released from MSN–Dex-Ac@DOX, the concentration of DOX was already high enough to cause cell apoptosis. Therefore, similar cytotoxicity at the highest concentration of DOX was reasonable. The most important, MSN–Dex-Ac@DOX possessed efficient anti-cancer activity comparable to free DOX. With the help of MSN–Dex-Ac@DOX to restrict drug release under blood circulation, the DOX-loaded particles can play an enormous role to ensure higher drug concentration in tumor site. Although it is a time-consuming process, the steadily increased drug accumulation via MSN–Dex-Ac@DOX would finally catch up the diffusion of free DOX in tumor cells to obtain a comparable and sustainable anti-cancer effect.
Conclusions
In summary, the current work demonstrated an efficient CDDS for pH-sensitive drug release using acid-degradable acetalated-dextran as valves of MSN. The hydrophobic acetalated-dextrans were attached onto the surface of particles to block drug molecules inside the mesopores. The in vitro DOX release data indicated that the introduction of polymer valves resulted in a restricted drug release in a simulated blood circulation condition. The DOX-loaded particles could be uptaken via endocytosis by cells and specifically released DOX intracellularly under the help of hydrophilic conversion of dextran to open the valves, resulting in comparable antitumor activity towards HeLa cells in a time-dependent manner.Acknowledgements
The authors would like to thank the financial support from National Natural Science Foundation of China (no. 51321062, 51273194 and 21174143), the Ministry of Science and Technology of China (973 Project, no. 2009CB930102; 863 Project, no. 2012AA021900), “100 Talents Program” of the Chinese Academy of Sciences (no. KGCX2-YW-802), and Jilin Provincial Science and Technology Department (no. 20100588).Notes and references
- H. Xiao, L. Yan, Y. Zhang, R. Qi, W. Li, R. Wang, S. Liu, Y. Huang, Y. Li and X. Jing, Chem. Commun., 2012, 48, 10730–10732 RSC.
- H. Xiao, H. Song, Q. Yang, H. Cai, R. Qi, L. Yan, S. Liu, Y. Zheng, Y. Huang, T. Liu and X. Jing, Biomaterials, 2012, 33, 6507–6519 CrossRef CAS PubMed.
- H. Xiao, H. Song, Y. Zhang, R. Qi, R. Wang, Z. Xie, Y. Huang, Y. Li, Y. Wu and X. Jing, Biomaterials, 2012, 33, 8657–8669 CrossRef CAS PubMed.
- H. Xiao, J. F. Stefanick, X. Jia, X. Jing, T. Kiziltepe, Y. Zhang and B. Bilgicer, Chem. Commun., 2013, 49, 4809–4811 RSC.
- W. H. D. Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- M. J. Vicent and R. Duncan, Trends Biotechnol., 2006, 24, 39–47 CrossRef CAS PubMed.
- H.-Y. Lian, M. Hu, C.-H. Liu, Y. Yamauchi and K. C. W. Wu, Chem. Commun., 2012, 48, 5151–5153 RSC.
- E. R. Gillies and J. M. J. Fréchet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS.
- B. P. Bastakoti, Y.-C. Hsu, S.-H. Liao, K. C. W. Wu, M. Inoue, S.-i. Yusa, K. Nakashima and Y. Yamauchi, Chem.–Asian J., 2013, 8, 1301–1305 CrossRef CAS PubMed.
- B. P. Bastakoti, K. C. W. Wu, M. Inoue, S.-i. Yusa, K. Nakashima and Y. Yamauchi, Chem.–Eur. J., 2013, 19, 4812–4817 CrossRef CAS PubMed.
- S. D. Brown, P. Nativo, J.-A. Smith, D. Stirling, P. R. Edwards, B. Venugopal, D. J. Flint, J. A. Plumb, D. Graham and N. J. Wheate, J. Am. Chem. Soc., 2010, 132, 4678–4684 CrossRef CAS PubMed.
- C.-Y. Hong, Y. Yamauchi and K. C. W. Wu, Chem. Lett., 2011, 40, 408–409 CrossRef CAS.
- L. Wang, C.-H. Liu, Y. Nemoto, N. Fukata, K. C. W. Wu and Y. Yamauchi, RSC Adv., 2012, 2, 4608–4611 RSC.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- J. Lee, H. Kim, S. Kim, H. Lee, J. Kim, N. Kim, H. J. Park, E. K. Choi, J. S. Lee and C. Kim, J. Mater. Chem., 2012, 22, 14061–14067 RSC.
- Q. Zhang, F. Liu, K. T. Nguyen, X. Ma, X. Wang, B. Xing and Y. Zhao, Adv. Funct. Mater., 2012, 22, 5144–5156 CrossRef CAS.
- C.-L. Zhu, X.-Y. Song, W.-H. Zhou, H.-H. Yang, Y.-H. Wen and X.-R. Wang, J. Mater. Chem., 2009, 19, 7765–7770 RSC.
- K. Patel, S. Angelos, W. R. Dichtel, A. Coskun, Y.-W. Yang, J. I. Zink and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 2382–2383 CrossRef CAS PubMed.
- C. Park, H. Kim, S. Kim and C. Kim, J. Am. Chem. Soc., 2009, 131, 16614–16615 CrossRef CAS PubMed.
- A. Schlossbauer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef CAS PubMed.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS PubMed.
- Q. Lin, Q. Huang, C. Li, C. Bao, Z. Liu, F. Li and L. Zhu, J. Am. Chem. Soc., 2010, 132, 10645–10647 CrossRef CAS PubMed.
- C. R. Thomas, D. P. Ferris, J.-H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J.-S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo and G. Zhu, J. Am. Chem. Soc., 2011, 133, 8778–8781 CrossRef CAS PubMed.
- H. Tang, J. Guo, Y. Sun, B. Chang, Q. Ren and W. Yang, Int. J. Pharm., 2011, 421, 388–396 CrossRef CAS PubMed.
- T. Chen, N. Yang and J. Fu, Chem. Commun., 2013, 49, 6555–6557 RSC.
- E. Aznar, M. D. Marcos, R. N. Martínez-Máñez, F. I. Sancenón, J. Soto, P. Amorós and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS PubMed.
- L. Yuan, Q. Tang, D. Yang, J. Z. Zhang, F. Zhang and J. Hu, J. Phys. Chem. C, 2011, 115, 9926–9932 CAS.
- J.-T. Sun, Z.-Q. Yu, C.-Y. Hong and C.-Y. Pan, Macromol. Rapid Commun., 2012, 33, 811–818 CrossRef CAS PubMed.
- X. Zhang, M. Oulad-Abdelghani, A. N. Zelkin, Y. Wang, Y. Haîkel, D. Mainard, J.-C. Voegel, F. Caruso and N. Benkirane-Jessel, Biomaterials, 2010, 31, 1699–1706 CrossRef CAS PubMed.
- S. Yu, G. Wu, X. Gu, J. Wang, Y. Wang, H. Gao and J. Ma, Colloids Surf., B, 2013, 103, 15–22 CrossRef CAS PubMed.
- R. Guillet-Nicolas, A. Popat, J. L. Bridot, G. Monteith, S. Z. Qiao and F. Kleitz, Angew. Chem., Int. Ed., 2013, 52, 2318–2322 CrossRef CAS PubMed.
- M. Xue and G. H. Findenegg, Langmuir, 2012, 28, 17578–17584 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, B. Zhang and D. Xu, Adv. Healthcare Mater., 2012, 1, 321–325 CrossRef CAS PubMed.
- F. Chen and Y. Zhu, Microporous Mesoporous Mater., 2012, 150, 83–89 CrossRef CAS PubMed.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- S. R. Van Tomme and W. E. Hennink, Expert Rev. Med. Devices, 2007, 4, 147–164 CrossRef CAS PubMed.
- E. Osterberg, K. Bergstrom, K. Holmberg, T. P. Schuman, J. A. Riggs, N. L. Burns, J. M. Van Alstine and J. M. Harris, J. Biomed. Mater. Res., 1995, 29, 741–747 CrossRef CAS PubMed.
- R. Mehvar, J. Controlled Release, 2000, 69, 1–25 CrossRef CAS.
- Y. L. Zhang, X. W. Dou and T. Jin, eXPRESS Polym. Lett., 2010, 4, 599–610 CrossRef CAS.
- Z. Zhang, J. Ding, X. Chen, C. Xiao, C. He, X. Zhuang, L. Chen and X. Chen, Polym. Chem., 2013, 4, 3265–3271 RSC.
- E.-K. Lim, E. Jang, K. Lee, S. Haam and Y.-M. Huh, Pharmaceutics, 2013, 5, 294–317 CrossRef CAS PubMed.
- Y.-I. L. Jeong, K.-D. Chung and K. Choi, Arch. Pharmacal Res., 2011, 34, 159–167 CrossRef CAS PubMed.
- S. C. Abeylath and M. M. Amiji, Bioorg. Med. Chem., 2011, 19, 6167–6173 CrossRef CAS PubMed.
- J. A. Cohen, T. T. Beaudette, J. L. Cohen, K. E. Broaders, E. M. Bachelder and J. M. J. Fréchet, Adv. Mater., 2010, 22, 3593–3597 CrossRef CAS PubMed.
- J. L. Cohen, S. Schubert, P. R. Wich, L. Cui, J. A. Cohen, J. L. Mynar and J. M. J. Fréchet, Bioconjugate Chem., 2011, 22, 1056–1065 CrossRef CAS PubMed.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- E. M. Bachelder, T. T. Beaudette, K. E. Broaders, J. Dashe and J. M. J. Fréchet, J. Am. Chem. Soc., 2008, 130, 10494–10495 CrossRef CAS PubMed.
- J. Nakazawa and T. D. P. Stack, J. Am. Chem. Soc., 2008, 130, 14360–14361 CrossRef CAS PubMed.
- C. Chen, J. Geng, F. Pu, X. Yang, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2011, 50, 882–886 CrossRef CAS PubMed.
- F. Carniato, C. Bisio, G. Paul, G. Gatti, L. Bertinetti, S. Coluccia and L. Marchese, J. Mater. Chem., 2010, 20, 5504–5509 RSC.
- A. Schlossbauer, D. Schaffert, J. Kecht, E. Wagner and T. Bein, J. Am. Chem. Soc., 2008, 130, 12558–12559 CrossRef CAS PubMed.
- C. Schatz, S. Louguet, J.-F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS PubMed.
- H. T. T. Duong, F. Hughes, S. Sagnella, M. Kavallaris, A. Macmillan, R. Whan, J. Hook, T. P. Davis and C. Boyer, Mol. Pharm., 2012, 9, 3046–3061 CrossRef CAS PubMed.
- N. Nasongkla, E. Bey, J. Ren, H. Ai, C. Khemtong, J. S. Guthi, S.-F. Chin, A. D. Sherry, D. A. Boothman and J. Gao, Nano Lett., 2006, 6, 2427–2430 CrossRef CAS PubMed.
- W. Zhang, Y. Li, L. Liu, Q. Sun, X. Shuai, W. Zhu and Y. Chen, Biomacromolecules, 2010, 11, 1331–1338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |