K0.25Mn2O4 nanofiber microclusters as high power cathode materials for rechargeable lithium batteries†
Chaofeng
Zhang
ab,
Chuanqi
Feng
c,
Peng
Zhang
ab,
Zaiping
Guo
*ab,
Zhixin
Chen
b,
Sean
Li
d and
Huakun
Liu
a
aInstitute for Superconducting & Electronic Materials, University of Wollongong, NSW 2522, Australia. E-mail: zguo@uow.edu.au; Fax: +61 2 4221 5731
bSchool of Mechanical, Materials & Mechatronics EngineeringUniversity of Wollongong, NSW 2522, Australia
cCollege of Chemistry & Chemical Engineering, Hubei University, Wuhan 430062, China
dSchool of Materials Science and Engineering, The University of New South Wales, NSW 2052, Australia
First published on 22nd December 2011
Abstract
K0.25Mn2O4 microclusters assembled from single-crystalline nanofibers were synthesized via a hydrothermal process at different temperatures. The possibility of using these materials as cathode material for lithium ion batteries was studied for the first time. The charge/discharge results showed that the K0.25Mn2O4 nanofiber microclusters synthesized at 120 °C exhibit excellent lithium storage properties, with a high reversible capability (360 mA h g−1 at current density of 100 mA g−1) and stable lithium-ion insertion/de-insertion reversibility. The charge/discharge mechanism in lithium ion batteries was studied and proposed for the first time. During the charge process, K+ was extracted from the electrode, which made active vacated sites suitable for lithium ion intercalation and was beneficial for increasing capacity.
Introduction
During the past few decades, one of the most attractive research areas in both science and technology has been rechargeable lithium-based batteries, which are currently regarded as the power source of choice for portable devices and potential power sources for electric vehicles (EVs), hybrid electric vehicles (HEVs), etc.1–3 It is consumer demand that drives research efforts for batteries with high energy density, fast recharging times, and excellent cycling stability. Great efforts have been made to develop competitive electrode materials for rechargeable lithium batteries with superior electrochemical performance.4,5 Compared with the development of anode materials, developing cathode materials with good electrochemical performance is even more crucial to meet the requirements of the rapidly increasing portable electronics market, as well as potential applications in EVs and HEVs.6–7Lithium transition metal oxide, with the formula of LiMO2 (M: Mn, Co, and Ni),8–15 and transition metal phosphate materials (LiFePO4)16–17 have been widely investigated and reported as cathode materials, especially commercial LiCoO2. These cathode materials have limited power density (W kg−1) compared to double layer capacitors and pseudocapacitors, and therefore cannot completely meet the practical requirements for EVs and HEVs, which need both high energy density and high power density. So far, developing cathode materials with low cost, environmental friendliness, better safety, and high energy and power densities, as well as excellent cycling stability, is still a challenge.1In recent years, manganese-based materials (MnO2, LixMn2O4, etc.) have received increasing attention due to their high abundance, non-toxicity, and environmental friendliness.18–21Manganese oxides are very well known for their suitable properties as cathode materials in Li-based batteries, especially α-MnO2.22–25 Alkaline or alkaline-earth ions hosted in cathode materials can enhance their electrochemical performance, probably because they stabilize the structures of the cathode materials during the lithium insertion/extraction, as well as increasing the interlayer space, thus increasing the ion diffusion rate in materials such as NaV3O8, LiMnO4, Mg(V3O8)2, etc.26–30 On the other hand, nanosized materials and porous materials have larger surface-to-volume ratios that enhance the contact between the active material grains and the electrolyte, and shorten the ion diffusion pathways.31 It could be deduced that a cathode material with nanosize porous structure and alkaline or alkaline-earth element doping would show improved electrochemical performance.
As a member of the manganese-based materials family, cryptomelane-type K0.25Mn2O4 materials have been prepared by different approaches, which can result in different morphology, and different particle sizes and particle size distributions.32–36 Single-crystalline K0.25Mn2O4 could be suitable as a cathode material for lithium ion batteries because its potassium ions can not only maintain the structure, but also increase the ion diffusion rate.26–30 However, to the best of our knowledge, no investigations have been conducted on single-crystalline K0.25Mn2O4 as a cathode material for lithium ion batteries so far. Herein, we report the electrochemical properties of K0.25Mn2O4 microclusters for the first time and propose its reaction mechanism as cathode material in lithium ion batteries. The K0.25Mn2O4 microclusters, assembled from single-crystalline nanofibers with diameters of 10–20 nm, were prepared by a facile hydrothermal technique. The K0.25Mn2O4 prepared at 120 °C, with a high Brunauer–Emmett–Teller (BET) surface area of 84.2 m2 g−1, delivered a specific discharge capacity of 223 mA h g−1 at a current density of 300 mA g−1 and exhibited excellent cycling stability, indicating that the K0.25Mn2O4 single-crystalline nanofiber microclusters could be a very promising candidate material for rechargeable lithium batteries.
Experimental
Sample preparation
All chemicals, including manganese(II) acetate tetrahydrate (MnAc2·4H2O) 99%, oxone monopersulfate compound (triple salt 2KHSO5·KHSO4·K2SO4), and potassium nitrate (KNO3), were bought from Sigma-Aldrich Co. Ltd. All reagents were used without any further purification. Nanofiber microclusters of cryptomelane-type manganese oxide (K0.25Mn2O4) were prepared via a template-free, one-step hydrothermal method.36 Five representative samples, designated as KMO-80, KMO-100, KMO-110, KMO-120, and KMO-130 were synthesized at different hydrothermal temperatures of 80, 100, 110, 120, and 130 °C, respectively. The synthesis procedure employed oxone and potassium nitrate as the oxidant, while manganese(II) acetate tetrahydrate was selected to build the oxide network. In one typical synthesis, 0.2 g of manganese(II) acetate tetrahydrate and 0.165 g of potassium nitrate were dissolved in 5 mL de-ionized water, and the resulting solution was labeled as solution A. Solution B was prepared by dissolving 1.5 g of oxone in 10 mL of double de-ionized water. After the mixing of the two solutions, the resultant mixture was then transferred to an autoclave and kept in an oven at 80 °C for 20 h. After the autoclave was cooled down, the product was separated and washed by centrifugation with de-ionized water several times, and then dried at 60 °C under vacuum conditions.Sample characterization
The microstructures of the as-prepared samples were characterized by X-ray diffraction (XRD; GBC MMA diffractometer) with Cu-Kα radiation and a graphite monochromator at a scanning rate of 2° min−1 in the range of 10–70°. Raman analysis was performed with a Jobin Yvon HR800 Raman spectrometer. X-Ray photoelectron spectroscopy (XPS) experiments were carried out on a VG Scientific ESCALAB 2201XL instrument using Al-Kα X-ray radiation. The morphologies of the samples were investigated by field emission scanning electron microscopy (FE-SEM; JEOL JSM-7500FA). The Brunauer–Emmett–Teller (BET) surface areas of the synthesized materials were measured by a NOVA 1000 high speed gas sorption analyser (Quantachrome Corporation, USA). Before BET measurements, the samples were degassed in vacuum at 100 °C for at least 12 h. Transmission electron microscope (TEM) images were collected on a JEOL 2011 200 kV instrument, with a JEOL Energy Dispersive X-ray Spectroscopy (EDS) detector and a JEOL EDS software analysis system. The working electrodes were prepared by mixing 75 wt% as-prepared K0.25Mn2O4 active materials with 15 wt% carbon black and 10 wt% polyvinylidene difluoride (PVdF, Sigma-Aldrich) binder in N-methyl-2-pyrrolidinone (NMP, Sigma-Aldrich, anhydrous, 99.5%) to form a homogeneous slurry, which was then uniformly pasted onto aluminium foil. The prepared working electrodes were dried in a vacuum oven at 100 °C over 12 h and were then ready for assembling into a test cell after pressing. Electrochemical cells (CR2032 coin type) using the K0.25Mn2O4 as working electrode, Li foil as the counter electrode, a microporous polypropylene film as the separator, and 1 M LiPF6 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) as the electrolyte were assembled in an Ar-filled glove box (H2O, O2 < 0.1 ppm, Mbraun, Unilab, USA). The cells were galvanostatically charged and discharged over a voltage range of 1.5–4.2 V versusLi/Li+ at different constant current densities, based on the weight of the active materials, on a Land CT2001A cycler. Cyclic voltammetry was conducted using a three-electrode system with Li foil as the counter electrode and reference electrode on a CHI660C electrochemical workstation.
:1 (v/v) mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) as the electrolyte were assembled in an Ar-filled glove box (H2O, O2 < 0.1 ppm, Mbraun, Unilab, USA). The cells were galvanostatically charged and discharged over a voltage range of 1.5–4.2 V versusLi/Li+ at different constant current densities, based on the weight of the active materials, on a Land CT2001A cycler. Cyclic voltammetry was conducted using a three-electrode system with Li foil as the counter electrode and reference electrode on a CHI660C electrochemical workstation.
Results and discussion
The K0.25Mn2O4 is composed of an octahedral molecular sieve (OMS), which is built from double chains of edge-sharing MnO6 octahedra, forming (2 × 2) + (1 × 1) tunnel structures, with potassium ions situated in the large (2 × 2) tunnels, as shown in Fig. 1. The structural features of these K0.25Mn2O4 samples were confirmed by X-ray diffraction (XRD) patterns, as shown in Fig. 2, with all peaks labeled, which demonstrates that all the reflections of these samples are in good agreement with the standard pattern of the pure tetragonal cryptomelane phase (JCPDS card file 29-1020). No other phases or impurities were detected, even though the microstructures were synthesized at a slightly lower temperature (80, 100, and 110 °C) than the temperature used in the previous study (120 °C).36 As can be observed, the KMO-80 sample exhibits weaker and broader XRD peaks compared to samples synthesized at higher temperatures. The XRD peaks become sharper and more intense with increasing synthesis temperature, suggesting enhancement of the crystallinity.29 However, the XRD peak of KMO-130 is weaker than that of KMO-120, probably because the higher temperature has destroyed the morphological structure of the material. The chemical compositions of the as-prepared products were analyzed by using inductively coupled plasma-atomic emission spectroscopy (ICP-AES). These samples give similar molar ratios of K:Mn, which are between 0.121 and 0.126. The ratios of K:Mn are very close to the ratio for cryptomelane with a half occupancy of the tunnel sides, K0.125MnO2.36
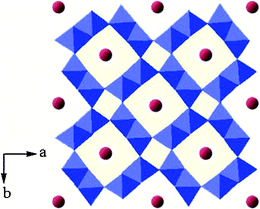 | ||
| Fig. 1 Structure of cryptomelane-type manganese oxide K0.25Mn2O4, with K and MnO6 octahedra shown as red balls and blue polyhedra, respectively. | ||
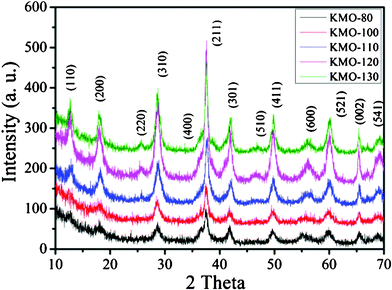 | ||
| Fig. 2 Powder XRD patterns of different K0.25Mn2O4 nanomaterials. | ||
Scanning electron microscope (SEM) images (Fig. 3) show that the four samples (KMO-80, KMO-100, KMO-110, and KMO-120) have uniform morphology over large domains. The KMO-80 and KMO-100 samples exhibit uniform microspherical morphology (Fig. 3a and 3b). When the synthesis temperature was increased to 110 °C, circular or oval holes (Fig. 3c) began to appear in the uniform spherical microstructures. On further increasing the hydrothermal temperature to 120 °C, the circular or oval holes were enlarged and hollow microstructures formed. Fig. S1 (ESI†) shows high resolution SEM (HRSEM) images of these four samples. Compared to the images of KMO-80 (Fig. S1a†), KMO-100 (Fig. S1b†), and KMO-110 (Fig. S1c†), the nanofibers in KMO-120 (Fig. S1d†) are more straight, which is probably due to their higher crystallinity. The uniform hollow microstructures, with diameters of less than 5 μm, are assembled from a complex arrangement of long nanoscale fibers with diameters from 10–20 nm (inset of Fig. S1d† and Fig. 3d). Diverse microspheres, with non-uniform structures were found in the images of KMO-130 (Fig. S2†).
 | ||
| Fig. 3 SEM images of different K0.25Mn2O4 nanomaterials: (a) KMO-80; (b) KMO-100; (c) KMO-110; (d) KMO-120. The insets of (a), (b), and (c) show enlargements of representative structures, respectively. | ||
A transmission electron microscope (TEM) image (Fig. 4a) of the as-prepared KMO-120 sample shows an individual sphere which is composed of numerous fibers. Fig. 4b and c contain high resolution TEM (HRTEM) images, showing that the diameters of these nanofibers are in the range of 10–20 nm, The inset of Fig. 4c shows the lattice fringes of a selected nanofiber with an interplanar distance of 0.49 nm, which is in good agreement with the d-spacing of (200) planes of the cryptomelane structure (JCPDS 29-1020). Fig. 4d shows the corresponding selected area electron diffraction (SAED) pattern from the nanofiber, which is recorded from the [0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1] zone axis. The white spots verify the single crystalline nature of the K0.25Mn2O4 nanofibers and show that the crystal grows along [211]*. These single crystal nanofibers facilitate the transportation of Li ions.
1] zone axis. The white spots verify the single crystalline nature of the K0.25Mn2O4 nanofibers and show that the crystal grows along [211]*. These single crystal nanofibers facilitate the transportation of Li ions.
 | ||
| Fig. 4 (a, b) TEM images of KMO-120; (c) HRTEM image of the nanoscale fibers; the inset shows the lattice fringe spacing for the area indicated by the small rectangle; (d) the corresponding selected area electron diffraction pattern. | ||
The electrochemical performance of the as-prepared K0.25Mn2O4 microclusters as cathode materials for lithium ion batteries was investigated. Fig. 5a presents the first cycle charge–discharge profiles for the different K0.25Mn2O4 electrodes at 100 mA g−1 at room temperature. KMO-80 delivers the lowest discharge capacity of 246 mA h g−1, whereas KMO-120 exhibits the best performance for Li insertion, with a capacity of 360 mA h g−1. To evaluate the cycling performance of different samples, electrochemical tests up to 100 cycles were performed at a current density of 100 mA g−1 (Fig. 5b). After 100 cycles, the discharge capacities of KMO-100, KMO-110, and KMO-120 are 99, 125, and 160 mA h g−1, which are about 40%, 60%, and 80% of their second discharge capacities, respectively. The capacity of KMO-80 drops rapidly and is only 126 mA h g−1 after 51 cycles, which is only 60% of its second discharge capacity. The capacity of KMO-130 drops quickly and is about 134 mA h g−1 after 67 cycles, which is far less than that of KMO-120. The high temperature has destroyed the uniform structure of the microclusters, which results in the agglomeration of nanofibers. Therefore, the contact area between electrolyte and active materials is decreased. Generally, the performance of electrode materials is very strongly influenced by the morphology, purity, crystalline phase, and porosity of the structure.37 Zhou et al. prepared single-crystalline nanowires of LiMn2O4, which showed capacity of around 100 mA h g−1 after 100 cycles at a 5 A g−1 rate.38 Zhao et al. synthesized single-crystalline LiMn2O4 nanotubes using β-MnO2 nanotubes as a self-sacrifice template, presenting superior high-rate capability and cycling stability (with 70% of its initial capacity retained after 1500 cycles at the 5 C rate).8 As one can see from Fig. 5a and b, KMO-120 exhibits the best electrochemical performance and cycling stability, probably due to two reasons: the single crystalline nature of its nanofibers29,37,38 (shown in Fig. 4d), and its larger surface area, which is 84.2 m2 g−1 (71.7, 68.2, and 67.6 m2 g−1 for KMO-110, KMO-100, and KMO-80, respectively), as estimated by the BET method. For the above-mentioned reasons, all of the following work is based on KMO-120 unless otherwise noted.
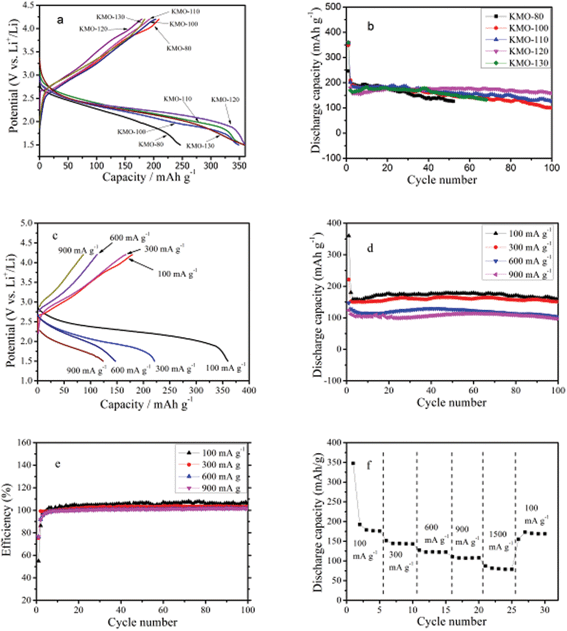 | ||
| Fig. 5 (a) First cycle discharge–charge profiles and (b) cycling performance for KMO-80, KMO-100, KMO-110, KMO-120, and KMO-130 at current density of 100 mA g−1. Electrochemical measurements on the K0.25Mn2O4 electrode (KMO-120): (c) first cycle charge–discharge profiles at various current densities for K0.25Mn2O4 electrode; (d) cycling performance of the K0.25Mn2O4 electrode at various current densities between 1.5 and 4.2 V vs.Li/Li+; (e) coulombic efficiency of the K0.25Mn2O4 electrode at various current densities between 1.5 and 4.2 V vs.Li/Li+; (f) rate capability of the K0.25Mn2O4 electrode at various current densities between 1.5 and 4.2 V vs.Li/Li+. | ||
The results of typical cyclic voltammetry (CV) measurements on KMO-120 (assigned as K0.25Mn2O4) are displayed in Fig. S3a.† There is a strong cathodic peak at about 2.22 V vs.Li/Li+ during the first cycle of the CV scan, and the peak shifts to higher potential in the following cycles. Similar effects have been observed previously.39–41 The reason for such a phenomenon is not exactly clear; however, it can be conjectured that some sort of local restructuring or ‘activation’ takes place in the material during the initial lithium intercalation. The peak in the cathodic sweep is ascribed to the lithium insertion process in the K0.25Mn2O4 electrode. One wide peak at about 3.0 V vs.Li/Li+ appeared in the anodic sweep, indicating the behavior of lithium ions during extraction from the host electrode material. Therefore, the possible electrochemical reaction mechanism of lithium ions with K0.25Mn2O4 in lithium ion batteries can be tentatively expressed in the following eqn (1):29
| K0.25Mn2O4 + xLi + xe− ↔ LixK0.25Mn2O4 | (1) |
Fig. 5c shows the charge–discharge voltage profiles for the first cycle of the K0.25Mn2O4 electrode, which were collected at various current densities from 100 to 900 mA g−1. The discharge capacities of the K0.25Mn2O4 cathode at 100, 300, 600, and 900 mA g−1 are 360, 221, 147, and 124 mA h g−1, respectively. The capacities and the discharge potentials decrease as the current density increases, due to the internal resistance and polarization. The cycling performance of the sample was investigated up to 100 cycles at different current densities from 100 to 900 mA g−1 (Fig. 5d). After 100 discharge–charge cycles, the capacities of the sample cycled at 100, 300, 600, and 900 mA g−1 were measured to be 160, 150, 102, and 96 mA h g−1, respectively. The relevant coulombic efficiencies (Fig. 5e) of the K0.25Mn2O4 electrodes at different current densities remain around 100% after several cycles, indicating the good reversibility. In order to investigate possible application in high power density devices, the rate capability of the K0.25Mn2O4 nanomaterial electrode was studied (Fig. 5f). The discharge capacities at 100, 300, 600, and 900 mA g−1 are approximately 175, 151, 121, and 107 mA h g−1, respectively, and the capacity is still 80 mA h g−1 even at the 1500 mA g−1 rate. When the current density is reduced to 100 mA h g−1 after the rate performance testing, the K0.25Mn2O4 cell can provide the formerly measured value (169 mA h g−1) again, indicating its good reversibility and high rate capability.
Fig. S3b† shows the number of Li ions inserted per formula unit during the first charge–discharge measurement at various current densities from 100 to 900 mA g−1. At the current density of 100 mA g−1, approximately 2.4 Li+ ions per formula unit were intercalated into the host material, while only 1.2 Li+ ions per formula unit can be de-intercalated from the material during charging. When the current density was increased to 300, 600, and 900 mA g−1, the numbers of Li+ ions intercalated were around 1.5, 1.0, and 0.8 per formula unit, while the numbers of Li+ ions extracted were about 1.1, 0.7, and 0.6 per formula unit, respectively.
As can be seen from Fig. 5d, the capacities of the K0.25Mn2O4 electrodes kept rising at the initial stage, except for the first few cycles, which is similar to the phenomenon that occurs in the NaV6O15 electrode.29 It is assumed that the potassium ions in the tunnels were de-intercalated during the charging process in the initial stage and then more lithium ions could be intercalated into the K0.25Mn2O4 electrode in the following discharge process. Thus, the amount of K+ in the K0.25Mn2O4 electrode before and after the charge–discharge process is significant for investigating the behavior of K+ in the charge–discharge procedure.
To determine the change in the number of K+ ions and the reversible insertion and extraction behavior of lithium ions in the K0.25Mn2O4 nanofibers, X-ray photoelectron spectroscopy (XPS) was carried out to investigate the surface concentrations of the corresponding elements and the chemical states of the species in the K0.25Mn2O4. Fig. 6a shows the O1s and Mn2p spectra for the K0.25Mn2O4 electrode before and after charge–discharge cycling. There are three distinctive binding energies in the O1s spectrum of fresh K0.25Mn2O4 electrode: 531.08, 531.58, and 533.38 eV. The binding energy at 531.08 eV is assigned to the lattice oxygen (O2−); the medium binding energy (531.58 eV) is ascribed to the surface absorbed oxygen (O2−, O−), OH groups, or oxygen vacancies; and the binding energy of 533.38 eV is probably due to the absorbed molecular water from air during sample transportation.42–47 As indicated in Fig. 6a, a freshly prepared electrode typically shows two Mn2p peaks, with binding energies of 642.38 eV for Mn2p3/2 and 654.18 eV for Mn2p1/2, and with a spin-energy separation of 11.8 eV, indicating the presence of tetravalent Mn and trivalent Mn.46–48 After 100 charge–discharge cycles at 100 mA g−1, the K0.25Mn2O4 cell was disassembled and washed in ethylene carbonate (EC) in an Ar-filled glove box before being investigated by XPS. As one can see from Fig. 6a, the XPS spectra of Mn2p show that the binding energies of the K0.25Mn2O4 electrode after 100 charge–discharge cycles are 641.68 eV for Mn2p3/2 and 653.38 eV for Mn2p1/2, which are shifted to lower binding energies in comparison with the fresh electrode. The shift towards lower binding energies suggests that the electron densities of Mn atoms have increased, which indicates an increase in low-valence Mn species.49–51 This is consistent with the decrease in intensity of the oxygen low binding energy peak (531.08 eV, corresponding to O2−) and the increase of the oxygen medium binding energy peak (531.58 eV, corresponding to O− or O2−) to keep the valence state balanced. Fig. 6b shows the core level K2p peak recorded from the fresh K0.25Mn2O4 electrode and the same electrode after electrochemical measurements, as well as the relative ratio of K:Mn. The amount of potassium element is reduced, and the ratio of K:Mn was reduced from the initial value of 0.313 to 0.068 after the electrochemical measurements, indicating that approximately 80% of the K+ ions were extracted during the charge process. For the freshly prepared electrode, the main sharp line at 292.5 eV for K2p3/2, accompanied by the satellite line at 295.3 eV for K2p1/2, is attributed to the K–O group.52–54 After 100 cycles, the binding energies of K are shifted to 293.9 and 296.3 eV. The positive binding energy shift indicates a decrease in the electron density of potassium due to the extraction of potassium from K0.25Mn2O4, in which there is a stronger interaction between potassium and lattice oxygen.52–57 Since the potassium was extracted from the K0.25Mn2O4 host materials, eqn (1), which describes the reversible insertion/extraction behavior of lithium, should be amended as:
| K0.25Mn2O4 + xLi + xe− → LixK0.25Mn2O4 (First discharge process) | (2) |
| LixK0.25Mn2O4 − yLi − mK − (y + m)e− → Lix−yK0.25−mMn2O4 (y < x, m < 0.25) (First charge process) | (3) |
| Lix−yK0.25−mMn2O4 (y < x, m < 0.25) + zLi + ze− ↔ Lix−y+zK0.25−mMn2O4 (y < x, m < 0.25) (Reversible electrochemical reaction) | (4) |
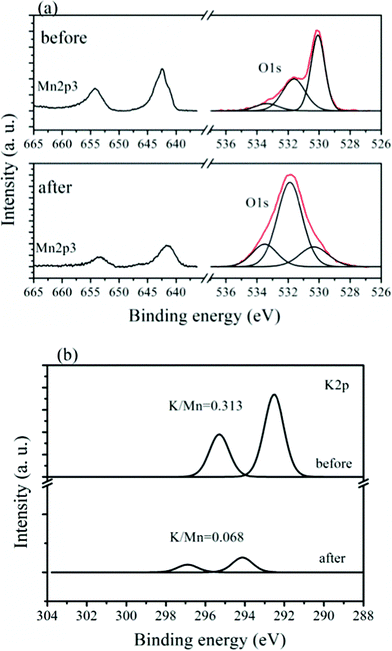 | ||
| Fig. 6 XPS spectra of the (a) Mn2p and O1s, and (b) the K2p regions for the K0.25Mn2O4 electrode before and after 100 cycles of charge–discharge at a current density of 100 mA g−1. | ||
The Li+ was intercalated into the K0.25Mn2O4 electrode during the first discharge, and then a certain number of Li+ ions, together with some K+ ions, were de-intercalated from the cathode in the first charge process. Since K+ was extracted from the electrode, some vacated active sites appeared and became available for Li+ in the following discharge process, resulting in the increasing capacity. In addition, there may be chemical ion exchange between K and Li due to the presence of excess Li in the electrolyte. The intercalated Li and K already there in the pristine form were electrochemically removed upon charge, which is supported by the increase in capacity (Fig. 5d) and the coulombic efficiencies above 100%, as shown in Fig. 5e (calculated from the charge capacity/discharge capacity). After 100 cycles of charge–discharge, there are a certain amount of Li+ ions in the electrode, which is consistent with the Raman results (Fig. S4†). However, the irreversible Li+ in the LixK0.25Mn2O4 reduces the trend towards increasing Li+ per formula unit in the following cycles, as is shown in Fig. 5d (where the capacity slightly increases in the initial stage, but exhibits capacity dropping in the first several cycles). With stable charge–discharge cycling at the current density of 100 mA g−1, the amount of lithium which could be electrochemically intercalated into the K0.25Mn2O4 electrode was about 1.1 Li+ per formula unit. Even when cycled at the current density of 900 mA g−1, there was around 0.66 Li+ per formula unit inserted into the hostK0.25Mn2O4 electrode after 100 cycles, corresponding to 96 mA h g−1.
Conclusions
The electrochemical performance of cryptomelane-type K0.25Mn2O4 nanofiber microclusters is reported for the first time. Cryptomelane-type K0.25Mn2O4 microclusters assembled from nanofibers show excellent electrochemical performance. At a current density of 100 mA g−1, the K0.25Mn2O4 electrode exhibited a reversible discharge capacity as high as 160 mA h g−1 for up to 100 cycles. Even when measured at high current densities of 300, 600, and 900 mA g−1, it was able to deliver high discharge capacities (around 150, 102, and 96 mA h g−1, respectively) after 100 cycles of charge–discharge. The high rate capability and excellent cycling performance of the K0.25Mn2O4 electrode can be attributed to its unique nanostructures, single-crystalline nature, and the presence of K+, which helps to stabilize the structures and favor lithium ion diffusion. During the charge process, K+ was extracted from the structure, which made active vacated sites suitable for lithium ion intercalation. Considering the excellent rate capability, environmental friendliness, and facile fabrication method, K0.25Mn2O4 microclusters assembled from single-crystalline nanofibers could be a potential cathode material for lithium rechargeable batteries.Acknowledgements
Financial support provided by the Australian Research Council (ARC) through an ARC Discovery project (DP1094261) is gratefully acknowledged. The authors would like to thank Mr. Guodong Du, Dr Shulei Chou, and Dr Jun Liu at the University of Wollongong for their assistance in using laboratory equipment or valuable remarks.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Berger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271 CrossRef CAS.
- G. Derrien, J. Hassoun, S. Panero and B. Scrosati, Adv. Mater., 2007, 19, 2336 CrossRef CAS.
- Y. G. Wang, H. Q. Li and Y. Y. Xia, Adv. Mater., 2006, 18, 2619 CrossRef CAS.
- J. Kim and A. Manthiram, Nature, 1997, 390, 265 CrossRef CAS.
- H. Zhou, D. Li, M. Hibino and I. Honma, Angew. Chem., Int. Ed., 2005, 44, 797 CrossRef CAS.
- Y.-L. Ding, J. Xie, G.-S. Cao, T.-J. Zhu, H.-M. Yu and X.-B. Zhao, Adv. Funct. Mater., 2011, 21, 348 CrossRef CAS.
- J. Y. Luo, H. M. Xiong and Y. Y. Xia, J. Phys. Chem. C, 2008, 112, 12051 CAS.
- M. Okubo, E. Hosono, J. Kim, M. Enomoto, N. Kojima, T. Kudo, H. Zhou and I. Honma, J. Am. Chem. Soc., 2007, 129, 7444 CrossRef CAS.
- V. Subramanian, C. L. Chen, H. S. Chou and G. T. K. Fey, J. Mater. Chem., 2001, 11, 3348 RSC.
- K. Mukai, J. Sugiyama, Y. Ikedo, P. L. Russo, D. Andreica, A. Amato, K. Ariyoshi and T. Ohzuku, J. Power Sources, 2009, 189, 665 CrossRef CAS.
- W. S. Yoon, K. Y. Chung, J. McBreen, D. A. Fischer and X. Q. Yang, J. Power Sources, 2006, 163, 234 CrossRef CAS.
- D. Liu, Y. Liu, A. Pan, K. P. Nagle, G. T. Seidler, Y.-H. Jeong and G. Cao, J. Phys. Chem. C, 2011, 115, 4959 CAS.
- L. Mai, L. Xu, C. Han, X. Xu, Y. Luo, S. Zhao and Y. Zhao, Nano Lett., 2010, 10, 4750 CrossRef CAS.
- Y. G. Wang, Y. R. Wang, E. Hosono, K. X. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461 CrossRef CAS.
- A. Yamada, H. Koizumi, S.-I. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Nat. Mater., 2006, 5, 357 CrossRef CAS.
- F. Cheng, J. Zhao, W. Song, C. Li, H. Ma, J. Chen and P. Shen, Inorg. Chem., 2006, 45, 2038 CrossRef CAS.
- H. Fang, L. Li, Y. Yang, G. Yan and G. Li, J. Power Sources, 2008, 184, 494 CrossRef CAS.
- G. Du, J. Wang, Z. Guo, Z. Chen and H. Liu, Mater. Lett., 2011, 65, 1319 CrossRef CAS.
- F. Jiao, J. Bao, A. H. Hill and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 9711 CrossRef CAS.
- M. H. Rossouw, D. C. Liles, M. M. Thackeray, W. I. F. David and S. Hull, Mater. Res. Bull., 1991, 27, 221 CrossRef.
- C. S. Johnson, D. W. Dees, M. F. Mansuetto, M. M. Thackeray, D. R. Vissers, D. Argyriou, C.-K. Loong and L. Christensen, J. Power Sources, 1997, 68, 570 CrossRef CAS.
- P. Botkovitz, P. Deniard, M. Tournoux and R. Brec, J. Power Sources, 1993, 44, 657 CrossRef CAS.
- L. I. Hill, A. Verbaere and D. Guyomard, J. Power Sources, 2003, 119–121, 226 CrossRef CAS.
- R. Chen and M. S. Whittingham, J. Electrochem. Soc., 1997, 144, L64 CrossRef CAS.
- D. Ahn, I. Yoo, Y.-M. Koo, N. Shin, J. Kim and T. J. Shin, J. Mater. Chem., 2011, 21, 5282 RSC.
- M. E. Spahr, P. Novák, W. Scheifele, O. Haas and R. Nesper, J. Electrochem. Soc., 1998, 145, 421 CrossRef CAS.
- H. Liu, Y. Wang, L. Li, K. Wang, E. Hosono and H. Zhou, J. Mater. Chem., 2009, 19, 7885 RSC.
- G. Pistoia, M. Pasquali, G. Wang and L. Li, J. Electrochem. Soc., 1990, 137, 2365 CrossRef CAS.
- M. M. Rahman, J.-Z. Wang, M. F. Hassan, S. Chou, Z. Chen and H. K. Liu, Energy Environ. Sci., 2011, 4, 952 CAS.
- H. Huang, S. Sithambaram, C.-H. Chen, C. K. Kithongo, L. Xu, A. Iyer, H. F. Garces and S. L. Suib, Chem. Mater., 2010, 22, 3664 CrossRef CAS.
- B. Hu, C.-H. Chen, S. J. Frueh, L. Jin, R. Joesten and S. L. Suib, J. Phys. Chem. C, 2010, 114, 9835 CAS.
- N. N. Opembe, C. K. King'ondu, A. E. Espinal, C.-H. Chen, E. K. Nyutu, V. M. Crisostomo and S. L. Suib, J. Phys. Chem. C, 2010, 114, 14417 CAS.
- T. Gao, M. Glerup, F. Krumeich, R. Nesper, H. Fjellvåg and P. Norby, J. Phys. Chem. C, 2008, 112, 13134 CAS.
- H. M. Galindo, Y. Carvajal, E. Njagi, R. A. Ristau and S. L. Suib, Langmuir, 2010, 26, 13677 CrossRef CAS.
- H.-W. Lee, P. Muralidharan, R. Ruffo, C. M. Mari, Y. Cui and D. K. Kim, Nano Lett., 2010, 10, 3852 CrossRef CAS.
- E. Hosono, T. Kudo, I. Honma, H. Matsuda and H. Zhou, Nano Lett., 2009, 9, 1045 CrossRef CAS.
- A. Esmanski and G. A. Ozin, Adv. Funct. Mater., 2009, 19, 1999 CrossRef CAS.
- M. Green, E. Fielder, B. Scrosati, M. Wachtler and J. S. Moreno, Electrochem. Solid-State Lett., 2003, 6, A75 CrossRef CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS.
- L. Yu, G. Diao, F. Ye, M. Sun, J. Zhou, Y. Li and Y. Liu, Catal. Lett., 2011, 141, 111 CrossRef CAS.
- V. P. Santos, M. F. R. Pereira, J. J. M. Órfão and J. L. Figueiredo, Appl. Catal., B, 2010, 99, 353 CrossRef CAS.
- L. Sun, Q. Cao, B. Hu, J. Li, J. Hao, G. Jing and X. Tang, Appl. Catal., A, 2011, 393, 323 CrossRef CAS.
- S. Kim, H.-B. Pyo, S. H. Ko, C. S. Ah, A. Kim and W.-J. Kim, Langmuir, 2010, 26, 7355 CrossRef CAS.
- X. Yang, J. Han, Z. Du, H. Yuan, F. Jin and Y. Wu, Catal. Commun., 2010, 11, 643 CrossRef CAS.
- T. Gao, P. Norby, F. Krumeich, H. Okamoto, R. Nesper and H. Fjellvåg, J. Phys. Chem. C, 2010, 114, 922 CAS.
- X. Tang, Y. Li, J. Chen, Y. Xu and W. Shen, Microporous Mesoporous Mater., 2007, 103, 250 CrossRef CAS.
- R. Hu, C. Yan, L. Xie, Y. Cheng and D. Wang, Int. J. Hydrogen Energy, 2011, 36, 64 CrossRef CAS.
- X.-S. Liu, Z.-N. Jin, J.-Q. Lu, X.-X. Wang and M.-F. Luo, Chem. Eng. J., 2010, 162, 151 CrossRef CAS.
- J. Ge, L. Zhuo, F. Yang, B. Tang, L. Wu and C. Tung, J. Phys. Chem. B, 2006, 110, 17854 CrossRef CAS.
- Y. Joseph, G. Ketteler, C. Kuhrs, W. Ranke, W. Weiss and R. Schlögl, Phys. Chem. Chem. Phys., 2001, 3, 4141 RSC.
- K. Jirátová, J. Mikulová, J. Klempa, T. Grygar, Z. Bastl and F. Kovanda, Appl. Catal., A, 2009, 361, 106 CrossRef.
- L. Xue, C. Zhang, H. He and Y. Teraoka, Catal. Today, 2007, 126, 449 CrossRef CAS.
- A. Miyakoshi, A. Ueno and M. Ichikawa, Appl. Catal., A, 2001, 219, 249 CrossRef CAS.
- M. Xiang, D. Li, J. Zou, W. Li, Y. Sun and X. She, J. Nat. Gas Chem., 2010, 19, 151 CrossRef CAS.
- A. Caballero, J. P. Espinós, A. Fernández, L. Soriano and A. R. González-Elipe, Surf. Sci., 1996, 364, 253 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00510c |
| This journal is © The Royal Society of Chemistry 2012 |