
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron porphyrin-catalyzed bromination of unactivated C–H bonds: inhibition of oxygen rebound by redox-inactive metal ions†
Yiran
Xu‡
a,
Peng
Wu‡
b,
Duanfeng
Xie
a,
Yue
Cui
a,
Yuheng
Zhang
a,
Binju
Wang
 *c and
Mian
Guo
*a
*c and
Mian
Guo
*a
aCollege of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, P. R. China
bState Key Laboratory of High-Efficiency Utilization of Coal and Green Chemical Engineering, School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan 750021, P. R. China
cKey Laboratory of Physical Chemistry of Solid Surfaces and Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China
First published on 22nd March 2025
Abstract
Heme-containing oxygenases have been known to catalyze the oxidation of unactivated C–H bonds. In most cases, hydroxylated compounds (alcohols) are the predominant products of the oxygen rebound pathway. Alternatively, non-hydroxylated products can be obtained under conditions when the oxygen rebound pathway is inhibited. However, biomimetic oxidative functionalization reactions catalyzed by synthetic iron porphyrin complexes have yet to be explored owing to the fast oxygen rebound step. In this study, metal bromide LiBr was introduced into the iron porphyrin-catalyzed oxidation of hydrocarbons, such as cycloalkanes, linear alkanes and benzyl compounds. In all the cases, brominated products were the sole products, indicating that the oxygen rebound pathway was completely inhibited in the presence of LiBr. Mechanistic studies combined with theoretical calculations revealed that the active intermediate iron(IV)–oxo porphyrin π-cation radical species interacted with lithium ions, which significantly inhibited the oxygen rebound pathway. As a result, a carbocation intermediate was formed and was responsible for the formation of brominated products. This carbocation mechanism is reminiscent of the P450 OleTJE and CYP19A1 enzymatic systems, in which the oxygen rebound pathway is inhibited and desaturated products are obtained. These results demonstrate that the redox-inactive metal ion acting as a Lewis acid was capable of tuning the reactivity of high-valent metal-oxo species from the oxygen rebound to the non-oxygen rebound pathway, providing potential application to produce versatile organic compounds stemming from simple hydrocarbons.
1. Introduction
Heme-containing enzymes, such as cytochrome P450 (CYP450), have been known to activate inert C–H bonds in the presence of an oxidant.1–7 It has been shown that the iron(IV)–oxo porphyrin π-cation radical species (compound I, Cpd I) is the active intermediate responsible for C–H bond activation to form the oxygenated product.8–13 In most cases, Cpd I abstracts one hydrogen atom from the hydrocarbon substrate, affording a corresponding Fe(IV)–OH porphyrin species and a carbon radical.3,14–18 Then, the –OH moiety will rebound to the incipient carbon radical to generate an hydroxylated product (R-OH) and a ferric porphyrin precursor (Scheme 1a). This critical step is defined as the oxygen rebound mechanism, which has been demonstrated as the general mechanism for both heme and non-heme oxygenases and related biomimetic systems.19–27 However, under certain conditions, the oxygen rebound pathway may be inhibited, whereby the carbon radical may undergo a non-oxygen rebound pathway to give an alternative functionalized product other than the hydroxylated product.24 For example, desaturation reactions forming a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond were found in some native P450 enzymes,28 while a decarboxylation reaction and aromatization reaction were found in cytochrome P450 OleTJE and steroid aromatase CYP19A1 catalyzed oxidation reactions, respectively.29–32 The expanded reactivities of heme proteins provide potential application to produce versatile organic compounds, such as fine chemicals and biofuels, through oxidative functionalization of simple hydrocarbons.33–36
C double bond were found in some native P450 enzymes,28 while a decarboxylation reaction and aromatization reaction were found in cytochrome P450 OleTJE and steroid aromatase CYP19A1 catalyzed oxidation reactions, respectively.29–32 The expanded reactivities of heme proteins provide potential application to produce versatile organic compounds, such as fine chemicals and biofuels, through oxidative functionalization of simple hydrocarbons.33–36
 | ||
| Scheme 1 Metalloporphyrin-catalyzed oxidation of hydrocarbons. | ||
Inspired by this intriguing reactivity, many efforts have been devoted to mimic the catalytic performances of heme enzymes using synthetic metalloporphyrin complexes.37–39 For instance, Groves and co-workers developed a series of reactions involving the oxidative functionalization of hydrocarbons catalyzed by manganese porphyrin complexes, such as chlorination, fluorination, azidation and isocyanation reactions, in the presence of an oxidant and X− nucleophiles (X− = Cl−, F−, N3− or NCO−).40–43 They found that the nucleophiles were able to rebound to the incipient carbon radical instead of oxygen when a strong electron donor was used as the axial ligand, which significantly suppressed the oxygen rebound step in manganese porphyrin systems (Scheme 1b).37 However, non-oxygen rebound reactions could be hardly achieved under similar conditions in iron porphyrin systems because the oxygen rebound rate is much faster and the carbon radical lifetime is too short for other transformations.44–46 Common methods such as those relying on the electronic properties of the equatorial porphyrin ligand and axial ligand are insufficient to inhibit the oxygen rebound reaction in the iron case.3 Indeed, the radical lifetimes in iron porphyrin-catalyzed reactions are in the order of tens to hundreds of picoseconds, while reactions catalyzed by manganese porphyrins exhibited radical lifetimes in the nanosecond regime.45,47 Therefore, the use of an earth-abundant and environmentally benign iron-based porphyrin catalyst in the catalytic oxidative functionalization (non-hydroxylation) of hydrocarbons still remains undeveloped.48
Although the intrinsic factors controlling the oxygen rebound pathway versus the non-oxygen rebound pathway are still elusive, some mechanistic studies have shown that the relative redox potential between the Fe(IV)–O porphyrin intermediate and carbon radical species is crucial for the selectivity.15,49 Groves et al. found that halogenation selectivity was influenced by the electronic properties of the porphyrin ligand in manganese porphyrin-catalyzed reactions.50 Goldberg et al. demonstrated that the rate constant of the oxygen rebound step exhibited a linear correlation with the redox potential of carbon radicals.20,23,51–53 Moreover, very recently Houk et al. revealed that lower hinderance around the Fe(IV)–OH porphyrin species could facilitate the oxygen rebound step.54 Conversely, a large steric effect may inhibit the oxygen rebound step. Taken together, if one factor is able to simultaneously tune the redox potential and increase the hinderance of iron-oxo species, the oxygen rebound step may be significantly inhibited. Consequently, the oxidative functionalization of hydrocarbons catalyzed by an iron porphyrin complex through a non-oxygen rebound pathway could be achieved.
Redox-inactive metal ions play crucial roles in various enzymes. One of the most representative examples of such ions is the calcium ion from the Mn4O5Ca cluster in photosystem II, which promotes O–O bond formation for O2 evolution in the water oxidation reaction.55–58 In biomimetic systems, these redox-inactive metal ions, acting as Lewis acids, have been proven to significantly increase the redox potential of high-valent metal oxygen species, resulting in an enhanced reactivity or even different reaction pathways.59–68 Furthermore, it was found that the binding of metal ions with the oxygen atom of the metal-oxo species through Lewis acid–base pair interaction would remarkably increase the hinderance of the metal-oxo species.68 Therefore, it can be proposed that the introduction of metal salts, such as metal bromides (MBr), into iron porphyrin-catalyzed oxidation reactions may inhibit the oxygen rebound step owing to the Lewis acid effect of the M+ cation, and then, the carbon radical species could undergo other transformations to react with Br−, affording the desired brominated product R–Br instead of the hydroxylated product R–OH (Scheme 1c).
Herein, we examined a series of metal bromides in the oxidation of unactivated C–H bonds catalyzed by the iron porphyrin complex. It was found that the addition of LiBr could significantly inhibit the oxygen rebound pathway in the iron porphyrin system. As a result, the brominated compound was the sole product without the formation of an alcohol or ketone (Scheme 1c). The substrate scope showed that this strategy could be applied for the bromination of various simple hydrocarbons, such as cycloalkanes, linear alkanes, and benzyl compounds. In all the cases, brominated products were the only detectable products with satisfactory turnover numbers (TON: 10–45). To the best of our knowledge, this is the first reported example of the selective bromination of simple hydrocarbons through biomimetic oxidative functionalization catalyzed by an iron porphyrin complex. Furthermore, mechanistic studies combined with theoretical calculations revealed that two molecules of LiBr bonded with the high-valent iron-oxo intermediate, which significantly inhibited the oxygen rebound pathway. Experimental data and density functional theory (DFT) calculations also suggested that a carbocation intermediate formed from the incipient carbon radical was involved in the reaction in the presence of LiBr. The carbocation intermediate then reacted with a bromide anion to afford the final brominated product with high selectivity. This unprecedented reaction pathway catalyzed by the synthetic iron porphyrin complex is totally different from manganese porphyrin-catalyzed halogenation reactions but similar to the reaction mechanism of certain native P450 enzymes (vide supra). The key role of Li+ in tuning the reaction pathway was also investigated in this work.
2. Results and discussion
2.1 Reaction development
|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal Bromide | TONb | Selectivityc (%) | ||
| –Br | –OH | –one | |||
| a Reaction conditions were as follows: cyclohexane (1.0 mmol), [Fe(TDFPP)Cl] (2.0 μmol), PhIO (0.2 mmol), MBr (0.25 mmol) and 2.0 mL of solvent CH3CN, r.t., argon atmosphere, and 12 h. b TON was based on the amount of [Fe(TDFPP)Cl] using n-dodecane as the internal standard. c Selectivity was calculated as the percentage of TON(–Br)/[TON(–Br) + TON(–OH) + TON(–one)]. | |||||
| 1 | None | 0 | 4 | 3 | 0 |
| 2 | LiBr | 45 | — | — | >99 |
| 3 | NaBr | 11 | 4 | 2 | 64 |
| 4 | KBr | 2 | 5 | 3 | 20 |
| 5 | RbBr | 7 | 5 | 2 | 50 |
| 6 | CsBr | 6 | 5 | 2 | 47 |
| 7 | MgBr2 | 1 | — | 0.5 | 67 |
| 8 | CaBr2 | 3 | — | 3 | 50 |
| 9 | BaBr2 | 5 | 4 | 3 | 41 |
| 10 | YBr3 | — | — | — | — |
| 11 | LaBr3 | — | — | — | — |

Preliminary studies showed that the presence of LiBr could significantly inhibit the oxygen rebound pathway, leading to the selective bromination of CHA. We thus investigated the influence of the amount of LiBr on the selectivity. As expected, the selectivity of bromocyclohexane increased as the amount of LiBr increased (ESI, Fig. S2†). Once the loading of LiBr was larger than that of PhIO, bromocyclohexane was obtained as the sole product. These results indicated that a larger amount of LiBr may facilitate the binding of the Lewis acid to the high-valent iron-oxo intermediate, which inhibited the oxygen rebound reaction pathway. However, a large excess amount of LiBr could only slightly increase the TON without changing the selectivity. Therefore, 125 eq. of LiBr was chosen for the subsequent bromination reactions.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Solvent | TON-Brb | Selectivityc (%) |
| a Reaction conditions were as follows: cyclohexane (1.0 mmol), catalyst (0.2 μmol), oxidant (2.0 mmol), LiBr (0.25 mmol) and 2.0 mL of solvent, r.t., argon atmosphere, and 12 h. b TON was based on the amount of catalyst using n-dodecane as the internal standard. c Selectivity was calculated as the percentage of TON(–Br)/[TON(–Br) + TON(–OH) + TON(-one)]. | |||||
| 1 | [Fe(TMFPP)Cl] | PhIO | CH3CN | 10 | >99 |
| 2 | [Fe(TPFPP)Cl] | PhIO | CH3CN | 9 | >99 |
| 3 | [Fe(TDCPP)Cl] | PhIO | CH3CN | 11 | >99 |
| 4 | [Fe(TMP)Cl] | PhIO | CH3CN | — | — |
| 5 | [Fe(TDFPP)Cl] | NaClO | CH3CN | 22 | >99 |
| 6 | [Fe(TDFPP)Cl] | PhIO | EtOAc | — | — |
| 7 | [Fe(TDFPP)Cl] | PhIO | Acetone | — | — |
| 8 | [Fe(TDFPP)Cl] | PhIO | n PrCN | — | — |
| 9 | [Fe(TDFPP)Cl] | PhIO | PhCF3 | 4 | 17 |

Other terminal oxidants, such as sodium hypochlorite (NaClO), are often used in the formation of high-valent iron-oxo porphyrin species and related catalyzed oxidation reactions.13,73–76 In our system, when PhIO was replaced by NaClO, bromocyclohexane was obtained as the sole product in the presence of LiBr (Table 2, entry 5), but the TON was decreased. The better performance of PhIO in terms of the TON might be ascribed to its poor solubility, which prevented overoxidation of the catalyst.37 It should be noted that no chlorocyclohexane was observed when NaClO was used as the oxidant in the presence of LiBr, while chlorocyclohexane was the major product obtained in the manganese porphyrin-catalyzed oxidation of hydrocarbons when using NaClO without LiBr.43 The same selectivity obtained when using different oxidants suggested that identical high-valent iron-oxo intermediate interactions with LiBr may be involved in the mechanism (vide infra).
When the polar solvent acetonitrile was replaced by a less polar solvent, such as ethyl acetate or acetone (Table 2, entries 6 and 7), no reaction occurred under these conditions. Furthermore, extending the carbon chain of acetonitrile to butyronitrile (nPrCN) totally shut down the reaction (Table 2, entry 8). Use of the special solvent trifluoromethyl benzene (PhCF3) (Table 2, entry 9), which was previously used to generate Cpd I at room temperature, afforded bromocyclohexane with a poor TON and selectivity.77 Finally, the control experiments indicated that no product was formed in the absence of the iron porphyrin catalyst or oxidant (ESI, Table S1†). Taken together, the best reaction conditions were optimized as follows: catalyst [Fe(TDFPP)Cl], oxidant PhIO (100 eq.), bromide source LiBr (125 eq.), substrate (500 eq.) and solvent CH3CN at room temperature.
|
|
|||
|---|---|---|---|
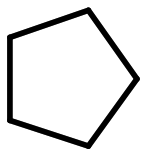
| Entry | Substrate | Productb | TONc |
| a Reaction conditions were as follows: substrate (1.0 mmol), [Fe(TDFPP)Cl] (2.0 μmol), PhIO (0.2 mmol), LiBr (0.25 mmol)and 2.0 mL of solvent CH3CN, r.t., argon atmosphere, and 12 h. b In all cases, only brominated products were obtained. c TON was based on the amount of [Fe(TDFPP)Cl] using n-dodecane as the internal standard. | |||
| 1 |
|

|
13 |
| 2 |

|

|
26 |
| 3 |
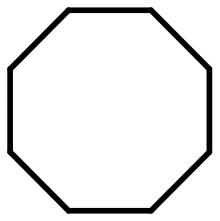
|

|
35 |
| 4 |

|

|
20 |
| 5 |

|

|
8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:8 :8:8 |
| 6 |
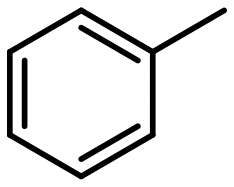
|

|
3 |
| 7 |

|

|
10 |
| 8 |

|
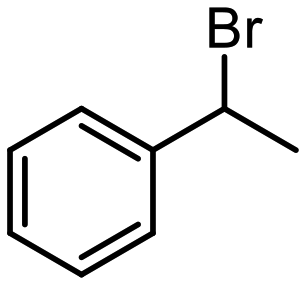
|
14 |
| 9 |
|

|
12 |
| 10 |

|
|
10:3 |
| 11 |

|

|
20:6 |
Moreover, the bromination of mono-bromo cyclopentane was also achieved under these conditions, affording 1,2-dibromocyclopentane and 1,3-dibromocyclopentane with TONs of 20 and 6, respectively (Table 3, entry 11). These results indicated that the reaction system developed in this work is capable of enabling the further bromination of brominated hydrocarbons.
2.2 Mechanistic studies
Although the bromination reaction catalyzed by the synthetic iron porphyrin complex is unprecedented, the halogenation of C–H bonds catalyzed by heme enzyme has already been discovered with the chloroperoxidase (CPO) system. Similar to the CYP450 system, the high-valent iron–oxo intermediate Cpd I was originally formed in the active site of CPO, followed by combination with a halide anion to generate the active species [FeIII(OX)(Por)].48,79–82 The substrate then reacted with this intermediate to afford the halogenated product (Scheme 2a). If the bromination reaction developed in this work went through a similar reaction pathway to CPO, the selectivity for bromination should occur irrespective of the metal ions. However, in our system, the selectivity for bromination was sensitive to the metal ion used (Table 1). Therefore, the CPO-like reaction pathway could be excluded in this work. | ||
| Scheme 2 Possible mechanisms of the metalloporphyrin-catalyzed functionalization of C–H bonds. | ||
Another possible reaction route for bromination is through a bromo radical as the bromination source, which may be derived from the oxidation of the Br− anion by the oxidant PhIO. Cyclic voltammetry (CV) analysis of LiBr and n-Bu4N+Br− (TBABr) showed an oxidized peak for TBABr at 0.59 V (vs. Fc/Fc+), whereas no oxidized peak was observed for LiBr in the same scanning range (ESI, Fig. S3†). This result indicated that LiBr was more difficult to be oxidized than TBABr in CH3CN. Moreover, DFT calculations showed that the dissociation of LiBr into Li+ and Br− in CH3CN was highly endothermic (ΔG ≈ 65.7 kcal mol−1), while the dissociation energy of LiBr in water was calculated as −124.5 kcal mol−1. The CV data combined with the results from the DFT calculations suggested that LiBr was more likely to be present as an independent molecule in CH3CN other than its ionic form. However, the oxidation of the Br− anion to bromo radical is unfavored in CH3CN. Therefore, the bromo radical as the bromination source could be excluded in this work. To further study the mechanism of the iron porphyrin-catalyzed bromination reactions, especially the effect of metal ions on selectivity, kinetic studies, characterization of the reaction intermediate, radical trapping experiments and DFT calculations were then performed.
 | ||
| Fig. 1 Mechanistic studies of bromination reactions catalyzed by [Fe(TDFPP)Cl]. (a) Competitive kinetic isotope experiments with cyclohexane or toluene as the substrate. (b) cis-Decalin as the diagnostic substrate. (c) The standard reaction under air. | ||
 | ||
| Fig. 2 UV–vis spectra of [FeIV(OLi)(TDFPP)] (red line) and [FeIV(O)(TDFPP)] (blue dashed line) generated through the reaction of [Fe(TDFPP)Cl] (0.05 mM) and PhIO (0.4 mM) in the presence or absence of LiBr in CH3CN at 263 K. Inset shows the ESI-MS spectrum of the iron(IV)–oxo species generated in the bromination reaction under standard conditions. m/z = 835.0992: [FeIV(OLi)(TDFPP)]+; m/z = 870.0702: [FeIV(OLi)(TDFPP)˙Cl]+. | ||
Previous studies have proved that the Cpd II species is a weak oxidant, which has difficultly in activating inert C–H bonds with a BDE larger than 90 kcal mol−1.3,14,86,99 However, the Cpd I species is a much stronger oxidant and can react with cyclohexane, octane or benzene.3,11 Therefore, we propose that the Cpd II species bound with Li+ were originally generated in the reaction solution and then converted to the Li+ bound Cpd I species through disproportionation. The latter one then abstracted the H atom of the substrate to form the incipient carbon radical for the following bromination reaction.
In both cases of manganese porphyrin and non-heme biomimetic systems, radical species with a relatively long lifetime were essential for the following radical escape and were then captured by radical scavengers. However, the lifetime of the incipient carbon radical was too short to be directly detected. In this regard, diagnostic substrates, which form radicals that undergo changes in their stereochemistry or structure after hydrogen abstraction, provide a powerful tool to study the intermediate radical and corresponding mechanism.44 Therefore, cis-decalin was used as the diagnostic substrate to probe the reaction pathway. The rearrangement rate constant of 9-cis-decalyl radical derived from cis-decalin was determined as 1 × 108 s−1.45 If a long-lived radical was involved in the reaction, trans-brominated decalin would be obtained through radical rearrangement.45,103 However, in the bromination reaction of cis-decalin under standard conditions, no trans-brominated decalin was observed. The configuration retained cis-brominated decalin as the only brominated product (Fig. 1b). This result demonstrated that the lifetime of the incipient radical derived from cis-decalin must be much shorter than 1 × 108 s−1, which means it would not be able to escape from the radical cage to react with the bromide source.45 Moreover, when the bromination of CHA was carried out under air, a similar TON to that of bromocyclohexane with high selectivity was obtained (Fig. 1c). The absence of a cyclohexanone product under aerobic conditions further confirmed that the incipient radical could not escape from the cage and then be captured by dioxygen. Thus, taken together, the radical escape mechanism shown in Scheme 2b could be unambiguously excluded in this work.
To further unveil the reaction mechanism, especially the key role of LiBr in the selectivity of the C–H bond bromination, DFT calculations were performed for the reactions of CHA and p-chlorotoluene (PCT). First, we examined the reaction of the active intermediate [FeIV(O)(TDFPP)˙]+ (Cpd I) and CHA without LiBr. Consistent with our experimental data, the reaction afforded the hydroxylation of CHA, with a barrier of 13.5 kcal mol−1 (Fig. S7†). Then, 1–3 molecules of LiBr were added into the Cpd I species to elucidate the effect of the LiBr.
If one molecule of LiBr interacted with Cpd I species, our calculations show that the H-atom transfer (HAT) from CHA to Cpd I and the following oxygen rebound process via a single step would still be favored, suggesting that a single LiBr molecule interacting with Cpd I would not be sufficient to inhibit the oxygen rebound for bromination reactions (Fig. S8†). Therefore, we next tested the case of two molecules of LiBr interacting with Cpd I species. Fig. 3 shows the DFT-calculated energy profile of the bromination of CHA in the presence of two molecules of LiBr. In the optimized structure of the starting reactant complex A (RCA), two LiBr molecules are ligated to the oxo moiety of Cpd I via a lithium ion (Fig. 3b). Similar to the hydroxylation reaction, the bromination reaction was also initiated by the HAT of CHA with Cpd I in the transition state A1 (Fig. 3a, TSA1). This process was coupled with a proton transfer to the neighboring Br−, leading to the formation of an HBr molecule, the binding of Fe(III)–OH species with Li+ and the cyclohexene intermediate in the intermediate complex A1 (Fig. 3a, ICA1). This step involved a barrier of 14.7 kcal mol−1 (Fig. 3a, RCA → TSA1), consistent with our KIE results in which HAT was involved in the rate-determining step.
 | ||
| Fig. 3 (a) Gibbs free energy profile (kcal mol−1) for the bromination of CHA by Cpd I with two LiBr molecules. (b) Key structures on the doublet involved in the reactions. Key distances (in angstroms) and spin population are given. Some atoms are omitted for clarity. | ||
To further understand the detailed process from TSA1 to ICA1, population and orbit analyses were performed (ESI, Fig. S9 and S10†). Along the reaction coordinate, it was found that the spin population of the CHA moiety initially increased to 0.66 and then decreased to zero. Concurrently, the spin population of the iron-oxo moiety and porphyrin dropped to 1.09 and zero, respectively. These observations indicate that the desaturated intermediate cyclohexene was formed through two states: the incipient carbon radical and subsequent carbocation. Firstly, the HAT of CHA begins to form the carbon radical state (spin population from 0 to 0.66), followed by fast electron transfer to the iron-oxo species to form the carbocation state (spin population from 0.66 to 0). The carbocation state is also highly unstable and donates its proton to the neighboring Br− simultaneously, affording ICA1 (Fig. 3a). The large downhill energy change from TSA1 to ICA1 further confirmed that the transformation of the incipient radical state was very fast, which was then not able to escape from the radical cage, as discovered in the radical trapping experiments (vide supra).
After the formation of ICA1, it was found that nascent HBr can add to the double bond of cyclohexene via TSA2 to form the final brominated product (Fig. 3a). In the optimized structure of TSA2, the Br− moiety of HBr was strongly coordinated to the adjacent Li+ (ESI, Fig. S11†), suggesting this addition step was significantly promoted by the nearby Li+. This addition step had a barrier of 15.6 kcal mol−1 (Fig. 3a, ICA1 → TSA2). Indeed, the addition of HBr to cyclohexene without Li+ required quite a high barrier of 20.3 kcal mol−1 (ESI, Fig. S12†).
For comparison, we also investigated the oxidation of CHA with three molecules of LiBr. Slightly different from the case of two LiBr molecules, the bromination with three LiBr molecules proceeded through a concerted hydride transfer, which was coupled with the attack by a neighboring Br− on the carbocation, forming a brominated product via a single reaction step (ESI, Fig. S13†). However, the reaction involved a much higher barrier of 22.5 kcal mol−1, suggesting that the ligation of more Li+ to the iron-oxo group could, to some extent, reduce the reactivity of Cpd I for HAT. Therefore, we propose that selective bromination promoted by the binding of two molecules of LiBr with Cpd I species is preferred.
To further confirm the carbocation mechanism of the bromination promoted by LiBr, DFT calculations were also carried out on the bromination of PCT with 2 LiBr, in which the alkene intermediate could not be formed owing to the benzyl structure. As shown in Fig. 4a, the hydride transfer from PCT to Cpd I afforded a relatively stable carbocation intermediate ICB1. This step experienced a barrier of 15.1 kcal mol−1 (Fig. 4a, RCB → ICB1). Similarly, the spin population of the PCT moiety rose to 0.79 and then fell to 0.13 along the reaction coordinate (ESI, Fig. S24†). Concurrently, the charge on the PCT moiety increased to 0.65. These changes indicate that the process involved a hybrid transfer mechanism through an incipient carbon radical state, eventually resulting in the formation of a relatively stable benzyl cation. After the formation of ICB1, we considered two possible routes: One was oxygen rebound to form the hydroxylated product, which had a barrier of 4.3 kcal mol−1 (Fig. 4a, red lines, ICB1 → PCBa); and an alternative pathway, in which the transfer of Br− to the substrate cation led to the brominated product (Fig. 4a, black lines, ICB1 → PCB), which involved a much smaller barrier of 2.4 kcal mol−1. This latter result was consistent with our experiment, in which the brominated product was the sole product in the reaction of PCT (Table 3, entry 7).
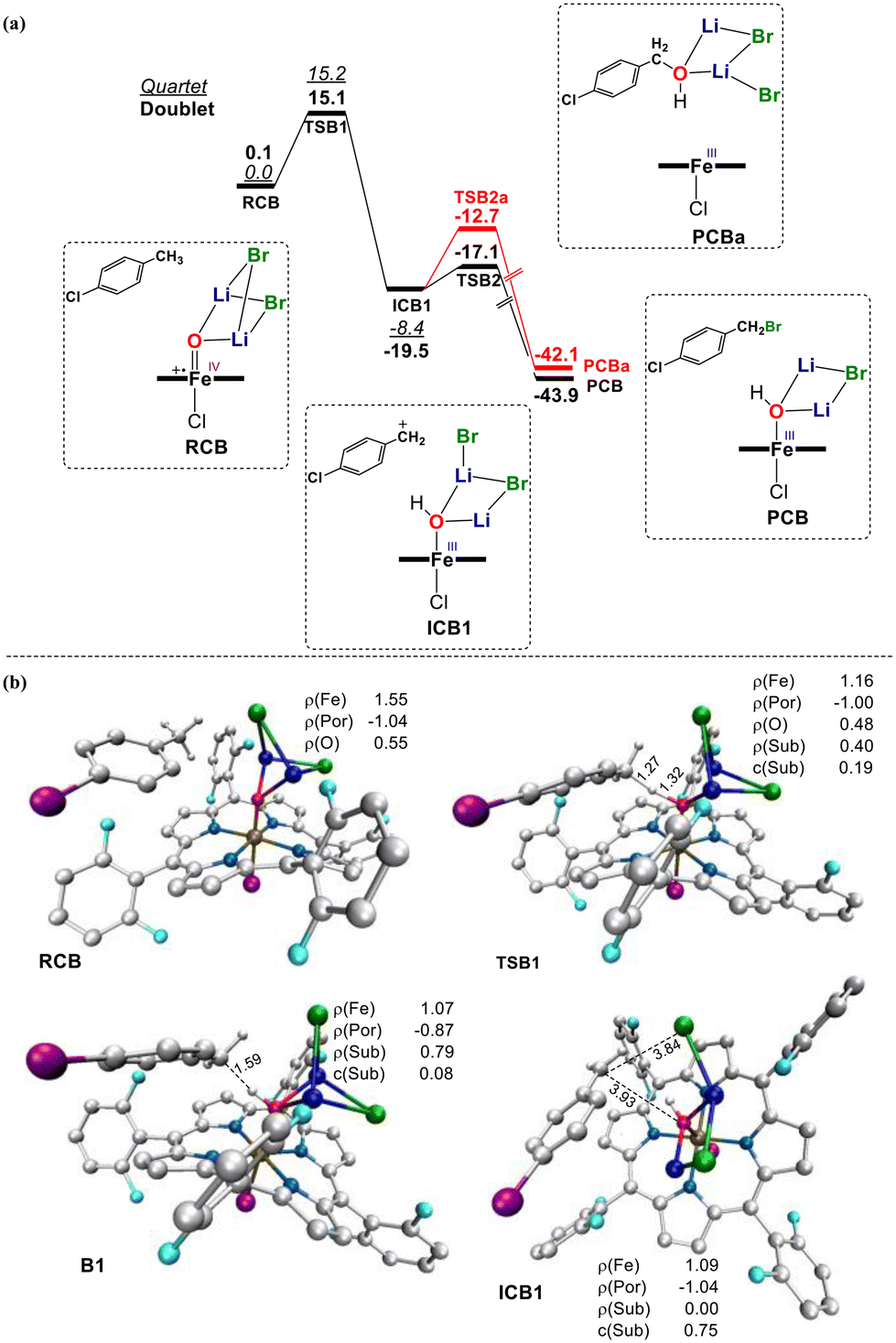 | ||
| Fig. 4 (a) Gibbs free energy profile (kcal mol−1) for the bromination of PCT by Cpd I with two LiBr molecules. (b) Key structures on the doublet involved in the reactions. Key distances (in angstroms) and spin population are given. Some atoms are omitted for clarity. | ||
To verify that Cpd I rather than the Cpd II species were responsible for the bromination in the presence of LiBr, we conducted calculations on the CHA oxidation involving Cpd II interacting with LiBr. Our calculations show that the oxygen rebound step could not be inhibited in this case. Both the hydroxylated product and bromination product could be formed when Cpd II was the reactive intermediate even when LiBr was employed in the reaction (ESI, Fig. S15 and S16†). Therefore, Cpd II as the active species for bromination reactions could be ruled out.
Interestingly, when indane was used as the substrate, the desaturated compound indene was the only product obtained under the standard conditions (with LiBr, Fig. 5a and ESI, Fig. S17†). However, the hydroxylated compound indanol was the only product obtained in the oxidation of indane without LiBr (Fig. 5a). This result clearly indicated that the alkene was indeed an intermediate in the catalytic bromination when using substrates containing a β-H bond in the presence of LiBr. Further bromination of indene was not observed because the addition of in situ HBr to indene was unfavored, as suggested by the DFT calculations (ESI, Fig. S18–S20†).
 | ||
| Fig. 5 (a) Indane as a substrate to investigate the intermediate of the catalytic oxidation reactions; (b) determination of LiOH generated in the reaction under standard conditions. | ||
Although the carbocation intermediate was too unstable to be characterized in the catalytic reactions, the trapping of the desaturated product alkene demonstrated that the carbocation route should be involved in the reaction mechanism, as previously discovered for native CYP450 enzymes. For example, in the oxidation of cyclopropyl derivatives by CYP2B1 and the oxidation of valproic acid by CYP450, the formation of a desaturated product was considered to occur through the carbocation reaction pathway.28,105 Therefore, trapping of the alkene intermediate in the reaction of indane further supported the carbocation route as being involved in the bromination when LiBr was employed. Besides, this result further excluded the involvement of the bromo radical in the present work.
In addition to the facile electron transfer promoted by Li+, the steric hinderance of the iron-oxo species is another key factor for the oxygen rebound step. Obviously, binding with 2 molecules of LiBr will greatly increase the steric hinderance of the iron-oxo species. Indeed, the crystal structure of the Sc3+-bound [FeIV(O)(TMC)] complex clearly showed the bulky hinderance around the iron-oxo moiety.68 Therefore, the oxygen rebound step was more difficult when LiBr was introduced in this work, leading to the selective bromination reaction.
After the formation of the brominated product, the OH group that was not rebound to the radical still binds with iron(III) porphyrin (Fig. 3a, PCA and Fig. 4a, PCB). However, the next catalytic turnover requires a vacant coordination site of the ferric porphyrin complex to launch the reaction. Therefore, we propose that LiOH precipitates from the solution after the reaction. The pH titration experiments of the reaction residue under standard conditions showed that the amount of LiOH generated in the reaction was approximately equal to the amount of bromocyclohexane (Fig. 5b). Therefore, the formation of LiOH precipitate may be another driving force for the catalytic bromination reaction.
 | ||
| Scheme 3 Proposed mechanism. | ||
3. Conclusions
In summary, we developed a method for biomimetic selective bromination of unactivated C–H bonds catalyzed by an iron porphyrin complex with LiBr, in which the oxygen rebound step was completely inhibited owing to the interaction of iron-oxo species with Li+. This method can be applied to the bromination of various hydrocarbons, such as cycloalkanes, linear alkanes, benzyl compounds and adamantane. Combined mechanistic studies and DFT calculations revealed a carbocation route instead of oxygen rebound pathway. This novel mechanism is different from the halogenation by chloroperoxidase or a manganese porphyrin complex. The key roles of Li+ were concluded as follows:(1) Binding with Li+ can greatly elevate the redox potential of iron-oxo species, resulting in fast electron transfer from the incipient carbon radical to the iron-oxo species through an MCET mechanism, which gives the carbocation intermediate/state responsible for the formation of the brominated product.
(2) Binding with Li+ significantly increases the steric hinderance of the iron-oxo species, in which case, oxygen rebound is inaccessible.
The use of the iron porphyrin catalyst to achieve oxidative functionalization is a big challenge in the biomimetic field because of the fast oxygen rebound step. This work presents a convenient strategy to inhibit the oxygen rebound step by introducing redox-inactive metal ions, which can guide efforts to produce versatile organic compounds from simple hydrocarbons. The development of other biomimetic functionalization reactions of hydrocarbons by changing the anion of metal salts and the extensive characterization of the metal-ion-bound iron-oxo species are ongoing in our laboratory.
Author contributions
All the authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NFSC) (22171216), the National Key R&D Program of China (2021YFA1500100) and Fundamental Research Funds for Central Universities (2042023kf0128) for Prof. Mian Guo; NSFC (22201222, 22122305) and the National Key R&D Program of China (2022YFA1505100) for Prof. Binju Wang; and NSFC (22403055) for Dr Peng Wu. The authors thank Prof. Weizhen Wei and Core Research Facilities of Wuhan University for the support of ESI-MS spectroscopies.References
- M. Guo, T. Corona, K. Ray and W. Nam, Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- K. Dutta Dubey and S. Shaik, Cytochrome P450-The Wonderful Nanomachine Revealed through Dynamic Simulations of the Catalytic Cycle, Acc. Chem. Res., 2019, 52, 389–399 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- T. L. Poulos, Heme Enzyme Structure and Function, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- W. Nam, Guest Editorial: Dioxygen Activation by Metalloenzymes and Models, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes, Chem. Rev., 2005, 105, 2279–2328 CrossRef CAS PubMed.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Heme-Containing Oxygenases, Chem. Rev., 1996, 96, 2841–2887 CrossRef CAS PubMed.
- Z. Wang, S. Shaik and B. Wang, Conformational Motion of Ferredoxin Enables Efficient Electron Transfer to Heme in the Full-Length P450TT, J. Am. Chem. Soc., 2021, 143, 1005–1016 CrossRef CAS PubMed.
- E. L. Onderko, A. Silakov, T. H. Yosca and M. T. Green, Characterization of a Selenocysteine-Ligated P450 Compound I Reveals Direct Link between Electron Donation and Reactivity, Nat. Chem., 2017, 9, 623–628 CrossRef CAS PubMed.
- C. M. Krest, A. Silakov, J. Rittle, T. H. Yosca, E. L. Onderko, J. C. Calixto and M. T. Green, Significantly Shorter Fe-S Bond in Cytochrome P450-I is Consistent with Greater Reactivity Relative to Chloroperoxidase, Nat. Chem., 2015, 7, 696–702 CrossRef CAS PubMed.
- X. Wang, S. Peter, M. Kinne, M. Hofrichter and J. T. Groves, Detection and Kinetic Characterization of a Highly Reactive Heme–Thiolate Peroxygenase Compound I, J. Am. Chem. Soc., 2012, 134, 12897–12900 CrossRef CAS PubMed.
- J. Rittle and M. T. Green, Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics, Science, 2010, 330, 933–937 CrossRef CAS PubMed.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, High-Valent Iron-Porphyrin Complexes Related to Peroxidase and Cytochrome P-450, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef CAS.
- R. Gupta, X.-X. Li, Y. Lee, M. S. Seo, Y.-M. Lee, S. Yanagisawa, M. Kubo, R. Sarangi, K.-B. Cho, S. Fukuzumi and W. Nam, Heme Compound II Models in Chemoselectivity and Disproportionation Reactions, Chem. Sci., 2022, 13, 5707–5717 RSC.
- P. C. E. Moody and E. L. Raven, The Nature and Reactivity of Ferryl Heme in Compounds I and II, Acc. Chem. Res., 2018, 51, 427–435 CrossRef CAS PubMed.
- T. H. Yosca, J. Rittle, C. M. Krest, E. L. Onderko, A. Silakov, J. C. Calixto, R. K. Behan and M. T. Green, Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450, Science, 2013, 342, 825–829 CrossRef CAS PubMed.
- K. Jayaraj, A. Gold, R. N. Austin, D. Mandon, R. Weiss, J. Terner, E. Bill, M. Muether and A. X. Trautwein, Compound I and II Analogs of a Chlorin, J. Am. Chem. Soc., 1995, 117, 9079–9080 CrossRef CAS.
- S. Shaik, W. Lai, H. Chen and Y. Wang, The Valence Bond Way: Reactivity Patterns of Cytochrome P450 Enzymes and Synthetic Analogs, Acc. Chem. Res., 2010, 43, 1154–1165 CrossRef CAS PubMed.
- J. Thomas, T. Mokkawes, L. Senft, A. Dey, J. B. Gordon, I. Ivanovic-Burmazovic, S. P. de Visser and D. P. Goldberg, Axial Ligation Impedes Proton-Coupled Electron-Transfer Reactivity of a Synthetic Compound-I Analogue, J. Am. Chem. Soc., 2024, 146, 12338–12354 CrossRef CAS PubMed.
- E. F. Gerard, V. Yadav, D. P. Goldberg and S. P. de Visser, What Drives Radical Halogenation versus Hydroxylation in Mononuclear Nonheme Iron Complexes? A Combined Experimental and Computational Study, J. Am. Chem. Soc., 2022, 144, 10752–10767 CrossRef CAS PubMed.
- K. Bleher, P. Comba, D. Faltermeier, A. Gupta, M. Kerscher, S. Krieg, B. Martin, G. Velmurugan and S. Yang, Non-Heme-Iron-Mediated Selective Halogenation of Unactivated Carbon-Hydrogen Bonds, Chem. – Eur. J., 2022, 28, e202103452 CrossRef CAS PubMed.
- J. Han, L. Tan, Y. Wan, G. Li and S. N. Anderson, C(sp(3))-H Oxidation and Chlorination Catalysed by a Bioinspired Pincer Iron(III) Complex, Dalton Trans., 2022, 51, 11620–11624 RSC.
- J. P. T. Zaragoza, T. H. Yosca, M. A. Siegler, P. Moenne-Loccoz, M. T. Green and D. P. Goldberg, Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex, J. Am. Chem. Soc., 2017, 139, 13640–13643 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Beyond Ferryl-Mediated Hydroxylation: 40 years of the Rebound Mechanism and C-H Activation, J. Biol. Inorg. Chem., 2017, 22, 185–207 CrossRef CAS PubMed.
- K.-B. Cho, H. Hirao, S. Shaik and W. Nam, To Rebound or Dissociate? This is the Mechanistic Question in C-H Hydroxylation by Heme and Nonheme Metal-Oxo Complexes, Chem. Soc. Rev., 2016, 45, 1197–1210 RSC.
- O. Planas, M. Clemancey, J. M. Latour, A. Company and M. Costas, Structural Modeling of Iron Halogenases: Synthesis and Reactivity of Halide-Iron(IV)-Oxo Compounds, Chem. Commun., 2014, 50, 10887–10890 RSC.
- P. Comba and S. Wunderlich, Iron-Catalyzed Halogenation of Alkanes: Modeling of Nonheme Halogenases by Experiment and DFT Calculations, Chem. – Eur. J., 2010, 16, 7293–7399 CrossRef CAS PubMed.
- A. E. Rettie, A. W. Rettenmeier, W. H. Howald and T. A. Baille, Cytochrome P-450—Catalyzed Formation of Δ4-VPA, a Toxic Metabolite of Valproic Acid, Science, 1987, 235, 890–893 CrossRef CAS PubMed.
- S. Yadav, S. Shaik and K. D. Dubey, Decarboxylation and Protonation Enigma in the H85Q Mutant of Cytochrome P450OleT, J. Phys. Chem. B, 2023, 127, 2927–2933 CrossRef CAS PubMed.
- J. L. Grant, C. H. Hsieh and T. M. Makris, Decarboxylation of Fatty Acids to Terminal Alkenes by Cytochrome P450 Compound I, J. Am. Chem. Soc., 2015, 137, 4940–4943 CrossRef CAS PubMed.
- F. K. Yoshimoto and F. P. Guengerich, Mechanism of the Third Oxidative Step in the Conversion of Androgens to Estrogens by Cytochrome P450 19A1 Steroid Aromatase, J. Am. Chem. Soc., 2014, 136, 15016–15025 CrossRef CAS PubMed.
- M. A. Rude, T. S. Baron, S. Brubaker, M. Alibhai, S. B. D. Cardayre and A. Schirmer, Terminal Olefin (1-Alkene) Biosynthesis by a Novel P450 Fatty Acid Decarboxylase from Jeotgalicoccus Species, Appl. Environ. Microbiol., 2011, 77, 1718–1727 CrossRef CAS PubMed.
- C. E. Wise, J. L. Grant, J. A. Amaya, S. C. Ratigan, C. H. Hsieh, O. M. Manley and T. M. Makris, Divergent Mechanisms of Iron-Containing Enzymes for Hydrocarbon Biosynthesis, J. Biol. Inorg. Chem., 2017, 22, 221–235 CrossRef CAS PubMed.
- J.-B. Wang, R. Lonsdale and M. T. Reetz, Exploring Substrate Scope and Stereoselectivity of P450 Peroxygenase OleTJE in Olefin-Forming Oxidative Decarboxylation, Chem. Commun., 2016, 52, 8131–8133 RSC.
- C. H. Hsieh and T. M. Makris, Expanding the Substrate Scope and Reactivity of Cytochrome P450 OleT, Biochem. Biophys. Res. Commun., 2016, 476, 462–466 CrossRef CAS PubMed.
- A. Dennig, M. Kuhn, S. Tassoti, A. Thiessenhusen, S. Gilch, T. Bulter, T. Haas, M. Hall and K. Faber, Oxidative Decarboxylation of Short-Chain Fatty Acids to 1-Alkenes, Angew. Chem., Int. Ed., 2015, 54, 8819–8822 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Manganese Catalyzed C-H Halogenation, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- M. Costas, Selective C-H Oxidation Catalyzed by Metalloporphyrins, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS.
- C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Selective Functionalisation of Saturated C-H Bonds with Metalloporphyrin Catalysts, Chem. Soc. Rev., 2011, 40, 1950–1975 RSC.
- X. Huang, T. Zhuang, P. A. Kates, H. Gao, X. Chen and J. T. Groves, Alkyl Isocyanates via Manganese-Catalyzed C-H Activation for the Preparation of Substituted Ureas, J. Am. Chem. Soc., 2017, 139, 15407–15413 CrossRef CAS PubMed.
- X. Huang, T. M. Bergsten and J. T. Groves, Manganese-Catalyzed Late-Stage Aliphatic C-H Azidation, J. Am. Chem. Soc., 2015, 137, 5300–5303 CrossRef CAS PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard III and J. T. Groves, Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- W. Liu and J. T. Groves, Manganese Porphyrins Catalyze Selective C-H Bond Halogenations, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed.
- C. F. McQueen and J. T. Groves, A Reevaluation of Iron Binding by Mycobactin J, J. Biol. Inorg. Chem., 2018, 23, 995–1007 CrossRef CAS PubMed.
- W. Liu, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Probing the C-O Bond-Formation Step in Metalloporphyrin-Catalyzed C-H Oxygenation Reactions, ACS Catal., 2017, 7, 4182–4188 CrossRef CAS.
- K. Auclair, Z. Hu, D. M. Little, P. R. O. de Montellano and J. T. Groves, Revisiting the Mechanism of P450 Enzymes with the Radical Clocks Norcarane and Spiro[2,5]octane, J. Am. Chem. Soc., 2002, 124, 6020–6027 CrossRef CAS PubMed.
- K. Omura, Y. Aiba, K. Suzuki, S. Ariyasu, H. Sugimoto and O. Shoji, A P450 Harboring Manganese Protoporphyrin IX Generates a Manganese Analogue of Compound I by Activating Dioxygen, ACS Catal., 2022, 12, 11108–11117 CrossRef CAS.
- J. P. Biswas, S. Guin and D. Maiti, High Valent 3d Metal-Oxo Mediated C-H Halogenation: Biomimetic Approaches, Coord. Chem. Rev., 2020, 408, 213174 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Selective C-H Halogenation with a Highly Fluorinated Manganese Porphyrin, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef CAS PubMed.
- G. W. Farley, M. A. Siegler and D. P. Goldberg, Halogen Transfer to Carbon Radicals by High-Valent Iron Chloride and Iron Fluoride Corroles, Inorg. Chem., 2021, 60, 17288–17302 CrossRef CAS PubMed.
- D. C. Cummins, J. G. Alvarado, J. P. T. Zaragoza, M. Q. E. Mubarak, Y.-T. Lin, S. P. de Visser and D. P. Goldberg, Hydroxyl Transfer to Carbon Radicals by Mn(OH) vs Fe(OH) Corrole Complexes, Inorg. Chem., 2020, 59, 16053–16064 CrossRef CAS PubMed.
- V. Yadav, J. B. Gordon, M. A. Siegler and D. P. Goldberg, Dioxygen-Derived Nonheme Mononuclear FeIII(OH) Complex and Its Reactivity with Carbon Radicals, J. Am. Chem. Soc., 2019, 141, 10148–10153 CrossRef CAS PubMed.
- Y. Zhang, C. Cao, Y. She, Y. F. Yang and K. N. Houk, Molecular Dynamics of Iron Porphyrin-Catalyzed C-H Hydroxylation of Ethylbenzene, J. Am. Chem. Soc., 2023, 145, 14446–14455 CrossRef CAS PubMed.
- H. Li, Y. Nakajima, E. Nango, S. Owada, D. Yamada, K. Hashimoto, F. Luo, R. Tanaka, F. Akita, K. Kato, J. Kang, Y. Saitoh, S. Kishi, H. Yu, N. Matsubara, H. Fujii, M. Sugahara, M. Suzuki, T. Masuda, T. Kimura, T. N. Thao, S. Yonekura, L. J. Yu, T. Tosha, K. Tono, Y. Joti, T. Hatsui, M. Yabashi, M. Kubo, S. Iwata, H. Isobe, K. Yamaguchi, M. Suga and J. R. Shen, Oxygen-Evolving Photosystem II Structures During S1-S2-S3 Transitions, Nature, 2024, 626, 670–677 CrossRef CAS PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Native Structure of Photosystem II at 1.95 Å Resolution Viewed by Femtosecond X-Ray Pulses, Nature, 2015, 517, 99–103 CrossRef CAS PubMed.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Molecular Catalysts for Water Oxidation, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- J. Yano and V. Yachandra, Mn4Ca Cluster in Photosynthesis: Where and How Water is Oxidized to Dioxygen, Chem. Rev., 2014, 114, 4175–4205 CrossRef CAS PubMed.
- Y. Liu and T. C. Lau, Activation of Metal Oxo and Nitrido Complexes by Lewis Acids, J. Am. Chem. Soc., 2019, 141, 3755–3766 CrossRef CAS PubMed.
- A. J. Jasniewski and L. Que Jr., Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes, Chem. Rev., 2018, 118, 2554–2592 CrossRef CAS PubMed.
- S. Hong, F. F. Pfaff, E. Kwon, Y. Wang, M.-S. Seo, E. Bill, K. Ray and W. Nam, Spectroscopic Capture and Reactivity of a Low-Spin Cobalt(IV)-Oxo Complex Stabilized by Binding Redox-Inactive Metal Ions, Angew. Chem., Int. Ed., 2014, 53, 10403–10407 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Redox-Inactive Metal Ions Modulate the Reactivity and Oxygen Release of Mononuclear Non-Haem Iron(III)-Peroxo Complexes, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- H. Yoon, Y.-M. Lee, X. Wu, K.-B. Cho, R. Sarangi, W. Nam and S. Fukuzumi, Enhanced Electron-Transfer Reactivity of Nonheme Manganese(IV)-Oxo Complexes by Binding Scandium Ions, J. Am. Chem. Soc., 2013, 135, 9186–9194 CrossRef CAS PubMed.
- P. Leeladee, R. A. Baglia, K. A. Prokop, R. Latifi, S. P. de Visser and D. P. Goldberg, Valence Tautomerism in a High-Valent Manganese-Oxo Porphyrinoid Complex Induced by a Lewis Acid, J. Am. Chem. Soc., 2012, 134, 10397–10400 CrossRef CAS PubMed.
- J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Metal Ion Effect on the Switch of Mechanism from Direct Oxygen Transfer to Metal Ion-Coupled Electron Transfer in the Sulfoxidation of Thioanisoles by a Non-Heme Iron(IV)−Oxo Complex, J. Am. Chem. Soc., 2011, 133, 5236–5239 CrossRef CAS PubMed.
- Y. Morimoto, H. Kotani, J. Park, Y.-M. Lee, W. Nam and S. Fukuzumi, Metal Ion-Coupled Electron Transfer of a Nonheme Oxoiron(IV) Complex: Remarkable Enhancement of Electron-Transfer Rates by Sc3+, J. Am. Chem. Soc., 2011, 133, 403–405 CrossRef CAS PubMed.
- S. Fukuzumi, Y. Morimoto, H. Kotani, P. Naumov, Y.-M. Lee and W. Nam, Crystal Structure of Metal Ion-Bound Oxoiron(IV) Complex and Implications for Biological Electron Transfer, Nat. Chem., 2010, 2, 756–759 CrossRef CAS PubMed.
- Abbreviations used: TDFPP, meso-tetrakis(2,6-difluorophenyl)porphinato dianion; TMFPP, meso-tetrakis(2-fluorophenyl)porphinato dianion; TPFPP, meso-tetrakis(pentafluorophenyl)porphinato dianion; TDCPP, meso-tetrakis(2,6-dichlorophenyl)porphinato dianion; TMP, meso-tetramesithylporphinato dianion.
- J. T. Groves and G. A. McClusky, Aliphatic Hydroxylation via Oxygen Rebound. Oxygen Transfer Catalyzed by Iron, J. Am. Chem. Soc., 1976, 98, 859–861 CrossRef CAS.
- J. T. Groves and M. Van der Puy, Stereospecific Aliphatic Hydroxylation by an Iron-Based Oxidant, J. Am. Chem. Soc., 1974, 96, 5274–5275 CrossRef CAS.
- J. T. Groves and T. E. Nemo, Aliphatic Hydroxylation Catalyzed byIron Porphyrin Complexes, J. Am. Chem. Soc., 1983, 105, 6243–6248 CrossRef CAS.
- M. Guo, Y.-M. Lee, S. Fukuzumi and W. Nam, Biomimetic Metal-Oxidant Adducts as Active Oxidants in Oxidation Reactions, Coord. Chem. Rev., 2021, 435, 213807 CrossRef CAS.
- S. Yokota and H. Fujii, Critical Factors in Determining the Heterolytic versus Homolytic Bond Cleavage of Terminal Oxidants by Iron(III) Porphyrin Complexes, J. Am. Chem. Soc., 2018, 140, 5127–5137 CrossRef CAS PubMed.
- Z. Cong, T. Kurahashi and H. Fujii, Oxidation of Chloride and Subsequent Chlorination of Organic Compounds by Oxoiron(IV) Porphyrin π-Cation Radicals, Angew. Chem., Int. Ed., 2011, 50, 9935–9939 CrossRef CAS PubMed.
- K. Czarnecki, L. M. Proniewicz, H. Fujii and J. R. Kincaid, Resonance Raman Spectrum of a 2A1u Ferryl Porphyrin π-Cation Radical, J. Am. Chem. Soc., 1996, 118, 4680–4685 CrossRef CAS.
- Y. Morimoto, Y. Shimaoka, Y. Ishimizu, H. Fujii and S. Itoh, Direct Observation of Primary C-H Bond Oxidation by an Oxido-Iron(IV) Porphyrin π-Radical Cation Complex in a Fluorinated Carbon Solvent, Angew. Chem., Int. Ed., 2019, 58, 10863–10866 CrossRef CAS PubMed.
- Y. R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, New York, 2002 Search PubMed.
- J. H. Dawson and M. Sono, Cytochrome P-450 and Chloroperoxidase: Thiolate-Ligated Heme Enzymes. Spectroscopic Determination of Their Active Site Structures and Mechanistic Implications of Thiolate Ligation, Chem. Rev., 1987, 87, 1255–1276 CrossRef CAS.
- W. Lai, H. Chen and S. Shaik, What Kinds of Ferryl Species Exist for Compound II of Chloroperoxidase? A Dialog of Theory with Experiment, J. Phys. Chem. B, 2009, 113, 7912–7917 CrossRef CAS PubMed.
- H. Chen, H. Hirao, E. Derat, I. Schlichting and S. Shaik, Quantum Mechanical/Molecular Mechanical Study on the Mechanisms of Compound I Formation in the Catalytic Cycle of Chloroperoxidase: an Overview on Heme Enzymes, J. Phys. Chem. B, 2008, 112, 9490–9500 CrossRef CAS PubMed.
- K. Kuhnel, E. Derat, J. Terner, S. Shaik and I. Schlichting, Structure and Quantum Chemical Characterization of Chloroperoxidase Compound 0, a Common Reaction Intermediate of Diverse Heme Enzymes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 99–104 CrossRef PubMed.
- D. Mandal, D. Mallick and S. Shaik, Kinetic Isotope Effect Determination Probes the Spin of the Transition State, Its Stereochemistry, and Its Ligand Sphere in Hydrogen Abstraction Reactions of Oxoiron(IV) Complexes, Acc. Chem. Res., 2018, 51, 107–117 CrossRef CAS PubMed.
- D. Mallick and S. Shaik, Kinetic Isotope Effect Probes the Reactive Spin State, As Well As the Geometric Feature and Constitution of the Transition State during H-Abstraction by Heme Compound II Complexes, J. Am. Chem. Soc., 2017, 139, 11451–11459 CrossRef CAS PubMed.
- Z. Cong, H. Kinemuchi, T. Kurahashi and H. Fujii, Factors Affecting Hydrogen-Tunneling Contribution in Hydroxylation Reactions Promoted by Oxoiron(IV) Porphyrin π-Cation Radical Complexes, Inorg. Chem., 2014, 53, 10632–10641 CrossRef CAS PubMed.
- Y. J. Jeong, Y. Kang, A.-R. Han, Y.-M. Lee, H. Kotani, S. Fukuzumi and W. Nam, Hydrogen Atom Abstraction and Hydride Transfer Reactions by Iron(IV)-Oxo Porphyrins, Angew. Chem., Int. Ed., 2008, 47, 7321–7324 CrossRef CAS PubMed.
- J. Li, B. Cheng, X. Shu, Z. Xu, C. Li and H. Huo, Enantioselective Alkylation of α-Amino C(sp3)−H Bonds via Photoredox and Nickel Catalysis, Nat. Catal., 2024, 7, 889–899 CrossRef CAS.
- L. Huan, X. Shu, W. Zu, D. Zhong and H. Huo, Asymmetric Benzylic C(sp3)-H Acylation via Dual Nickel and Photoredox Catalysis, Nat. Commun., 2021, 12, 3536 CrossRef CAS PubMed.
- Z. Gong, L. Wang, Y. Xu, D. Xie, X. Qi, W. Nam and M. Guo, Enhanced Reactivities of Iron(IV)-Oxo Porphyrin Species in Oxidation Reactions Promoted by Intramolecular Hydrogen-Bonding, Adv. Sci., 2024, 11, e2310333 CrossRef PubMed.
- L. Ji, A. Franke, M. Brindell, M. Oszajca, A. Zahl and R. van Eldik, Combined Experimental and Theoretical Study on the Reactivity of Compounds I and II in Horseradish Peroxidase Biomimetics, Chem. – Eur. J., 2014, 20, 14437–14450 CrossRef CAS PubMed.
- C. Fertinger, N. Hessenauer-Ilicheva, A. Franke and R. van Eldik, Direct Comparison of the Reactivity of Model Complexes for Compounds 0, I, and II in Oxygenation, Hydrogen-Abstraction, and Hydride-Transfer Processes, Chem. – Eur. J., 2009, 15, 13435–13440 CrossRef CAS PubMed.
- S. N. Dhuri, M. S. Seo, Y.-M. Lee, H. Hirao, Y. Wang, W. Nam and S. Shaik, Experiment and Theory Reveal the Fundamental Difference between Two-State and Single-State Reactivity Patterns in Nonheme FeIV=O versus RuIV=O Oxidants, Angew. Chem., Int. Ed., 2008, 47, 3356–3359 CrossRef CAS PubMed.
- W. Nam, High-Valent Iron(IV)-Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- W. Nam, M. H. Lim and S.-Y. Oh, Effect of Anionic Axial Ligands on the Formation of Oxoiron(IV) Porphyrin Intermediates, Inorg. Chem., 2000, 39, 5572–5575 CrossRef CAS PubMed.
- We have found that the Lewis-acid such as the redox-inactive metal ions could promote the disproportionation of Cpd II to form Cpd I species. The details and mechanism about the disproportionation is another ongoing project in our labrotary.
- K. Nishikawa, Y. Honda and H. Fujii, Spectroscopic Evidence for Acid-Catalyzed Disproportionation Reaction of Oxoiron(IV) Porphyrin to Oxoiron(IV) Porphyrin π-Cation Radical and Iron(III) Porphyrin, J. Am. Chem. Soc., 2020, 142, 4980–4984 CrossRef CAS PubMed.
- M. Oszajca, A. Drzewiecka-Matuszek, A. Franke, D. Rutkowska-Zbik, M. Brindell, M. Witko, G. Stochel and R. van Eldik, Mechanistic Insight into Peroxo-Shunt Formation of Biomimetic Models for Compound II, Their Reactivity toward Organic Substrates, and the Influence of N-Methylimidazole Axial Ligation, Chem. – Eur. J., 2014, 20, 2328–2343 CrossRef CAS PubMed.
- Z. Pan and M. Newcomb, Acid-Catalyzed Disproportionation of Oxoiron(IV) Porphyrins to Give Oxoiron(IV) Porphyrin Radical Cations, Inorg. Chem. Commun., 2011, 14, 968–970 CrossRef CAS PubMed.
- M. A. Ehudin, D. A. Quist and K. D. Karlin, Enhanced Rates of C-H Bond Cleavage by a Hydrogen-Bonded Synthetic Heme High-Valent Iron(IV) Oxo Complex, J. Am. Chem. Soc., 2019, 141, 12558–12569 CrossRef CAS PubMed.
- M. Guo, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, Highly Reactive Manganese(IV)-Oxo Porphyrins Showing Temperature-Dependent Reversed Electronic Effect in C-H Bond Activation Reactions, J. Am. Chem. Soc., 2019, 141, 12187–12191 CrossRef CAS PubMed.
- S. H. Bae, X.-X. Li, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, Tunneling Controls the Reaction Pathway in the Deformylation of Aldehydes by a Nonheme Iron(III)-Hydroperoxo Complex: Hydrogen Atom Abstraction versus Nucleophilic Addition, J. Am. Chem. Soc., 2019, 141, 7675–7679 CrossRef CAS PubMed.
- E. Kwon, K.-B. Cho, S. Hong and W. Nam, Mechanistic Insight into the Hydroxylation of Alkanes by a Nonheme Iron(V)-Oxo Complex, Chem. Commun., 2014, 50, 5572–5575 RSC.
- To the opposite, only configuration retained product trans-brominated decalin would be obtained when trans-decalin was used, since no radical rearrangement can occur in this case. The details about product analysis in the reaction of decalin was elucidated in ESI, Fig. S5.†.
- D. Zhu, M. J. Seo, H. Ikeda and D. E. Cane, Genome Mining in Streptomyces. Discovery of an Unprecedented P450-Catalyzed Oxidative Rearrangement That Is the Final Step in the Biosynthesis of Pentalenolactone, J. Am. Chem. Soc., 2011, 133, 2128–2131 CrossRef CAS PubMed.
- P. R. O. de Montellano, Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes, Chem. Rev., 2010, 110, 932–948 CrossRef PubMed.
- J. Park, Y. Morimoto, Y.-M. Lee, Y. You, W. Nam and S. Fukuzumi, Scandium Ion-Enhanced Oxidative Dimerization and N-Demethylation of N,N-Dimethylanilines by a Non-Heme Iron(IV)-Oxo Complex, Inorg. Chem., 2011, 50, 11612–11622 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, instrumentation, experimental section, theoretical studies, control experiments, Table S1, Figures S1–S20 and appendix. See DOI: https://doi.org/10.1039/d5qi00508f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |