
Room-temperature magnetism in the crystal of a 1,6-heptadiyne derivative and its processable polymer†
Manyu
Chen
a,
Guangze
Hu
 a,
Zuping
Xiong
a,
Haoyuan
Hu
a,
Jing Zhi
Sun
*a,
Haoke
Zhang
*ab and
Ben Zhong
Tang
*ac
a,
Zuping
Xiong
a,
Haoyuan
Hu
a,
Jing Zhi
Sun
*a,
Haoke
Zhang
*ab and
Ben Zhong
Tang
*ac
aMOE Key Laboratory of Macromolecules Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310058, China. E-mail: sunjz@zju.edu.cn; zhanghaoke@zju.edu.cn
bHangzhou Global Scientific and Technological Innovation Centre, Zhejiang University, Hangzhou 311215, China
cShenzhen Institute of Molecular Aggregate Science and Engineering, School of Science and Engineering, The Chinese University of Hong Kong, Shenzhen 518172, China. E-mail: tangbenz@cuhk.edu.cn
First published on 13th February 2025
Abstract
Room-temperature organic magnetic materials have long been a sought-after but challenging topic. Besides the reported organic-based magnets including pure organic radicals, charge-transfer salts, and coordination polymers, we report a novel and alternative approach to fabricating purely organic/polymeric magnets based on the crystal of a 4-substituted 1,6-diyne (M1) and its polymer (P1). Both the white M1 crystal and the black P1 powder samples exhibit room-temperature magnetism. The saturation magnetization of P1 is about 0.25 emu g−1 and its Curie temperature is higher than 400 K. After repeated recrystallization of M1 and precipitation of P1 to thoroughly remove the metal-catalyst residues, the room-temperature magnetism of M1 and P1 is tentatively assigned to the stable radicals in the solid samples. The results demonstrated in this work suggest an unprecedented strategy to obtain room-temperature organic magnets.
Introduction
Magnetic materials have irreplaceable applications in many important areas such as information storage, quantum computing and spin sensors. Characterized by the traits of low density, easy processing and tailorable chemical structure, organic/polymeric magnets have attracted increasing attention in both academic and industrial fields.1–7 Although the prediction of the existence of magnetic exchange interaction between π-electron spins in aromatic and olefinic free radicals was proposed by McConnell as early as 1963,8 the first magnetic polymer (poly-BIPO) was reported by Ovchinnikov and Spector et al. in 1987,9 and the discovery of the first pure organic ferromagnet (p-nitrophenyl nitronyl nitroxide crystal) was reported by Takahashi and Turek et al. in 1991.10 These pioneering works led to the development of this particular research field, and a series of magnetic organic compounds (e.g., nitroxide radicals, phenoxy radicals, and ammonium sulphate radicals) and polymers (e.g., polyaryl–methylene, fullerene C60 polymers, and poly(9,10-anthracene-acetylene)) emerged and were documented.11–20Despite these significant advances, most of the organic/polymer magnets have Curie temperatures far below room temperature and low stability, making their practical application difficult. In recent years, researchers have explored a variety of strategies to obtain room-temperature magnetic organic/polymeric materials with high Curie temperatures and high stability. For example, in 2018, Huang and coworkers obtained a room-temperature ferromagnet by ultrasonic treatment and low-temperature annealing of naphthalene.21 In 2022, Ma and colleagues reported a solvothermal approach for the preparation of a room-temperature ferromagnet from perylene diimide aggregates with a Curie temperature over 400 K.22 In recent years, two-dimensional and framework-based constructions have been adopted for the design and synthesis of organic/polymeric magnetic materials to enhance the thermal and structural stability of free radical species.23–30 For example, in 2012, Kulszewicz-Bajer et al. reported magnetic spin interaction in polymer aromatic amines by adjusting the π-conjugated system.18 In 2018, Yoo and Baek et al. reported highly stable free radicals achieved through the efficient self-polymerization of a tetracyano-quinodimethane monomer.23 In 2019, Wu and colleagues described the synthesis of 1,3,5-triazine-linked porous organic radical frameworks via thermal or triflic acid-assisted polymerization of cyano-containing stable radical monomers.24 Due to the magnetic radicals being coupled with each other through the 1,3,5-triazine connector, the polymers exhibited spontaneous magnetization or super-paramagnetism at room temperature.
As a theoretical prediction, polyacetylene with a polyene backbone may exhibit magnetic behaviour.31,32 In fact, electron spin resonance (ESR) experimental data showed that the concentration of paramagnetic centres in non-doped trans-polyacetylene films was about 3 × 1019 spin per g, but such a spin density and very low stability could not afford evident magnetism in intrinsic polyacetylene.32 Inspired by the pioneer work on the magnetic polymer (poly-BIPO), a series of polyacetylene and polyyne derivatives modified with stable radicals were prepared, increasing the concentration of radicals by several orders of magnitude. The magnetic behaviour suggested the presence of spin-glass.33–40 Generally, the magnetism of these polymers originates from the pendant polyradicals because they accumulate radical molecules along one conjugated polymer chain. To date, magnetic materials based on conjugated polymers including polyacetylenes still face three problems: (1) extrinsic magnetic properties, where spin exchange originates from radical modifiers rather than the conjugated main chain; (2) poor stability, as magnetism disappears after exposure to air for a period of time; and (3) low Curie temperature, with magnetic response being lost at ambient temperature. In this work, we demonstrate the room temperature magnetism of the powders of a polymer (P1) and the crystalline monomer of 4,4-bis-methoxycarbonyl-1,6-heptadiyne (M1). P1 exhibited a Curie temperature >400 K. The magnetic property is retained in the dark and at ambient temperature for months. In addition, the free radicals are generated in the polymer's backbone rather than introduced through the modification of the polymer with stable radical side chains.
Results and discussion
The chemical structures of the 1,6-diyne and its polymer, and the synthetic route are shown in Scheme 1. White crystals of the monomer (M1, 4,4-bis-methoxycarbonyl-1,6-heptadiyne) were obtained after recrystallization of the primary product (Scheme S1†). The polymerization of M1 followed the procedure of metathesis cyclopolymerization, as described in the literature.41–43 Under optimized reaction conditions, the poly(1,6-heptadiyne) derivative P1 was obtained in good yield using the transition metal catalyst MoCl5 in ultra-dry 1,2-dichloroethane (DCE) under a nitrogen atmosphere, with the optimization processes described in the Experimental section and the ESI (Table S1†). The data on the structural characterization of the monomer and polymer are also presented in the Experimental section and the ESI (Scheme S1, Table S1, Fig. S1 and S2†).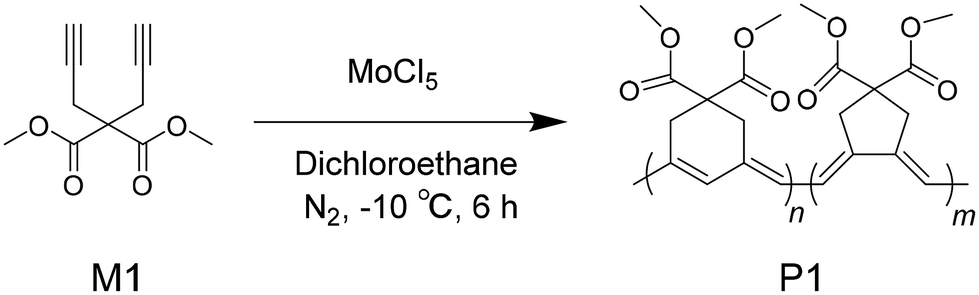 | ||
| Scheme 1 Synthetic route to poly(4,4-bismethoxycarbonyl-1,6-heptadiyne) (P1). | ||
According to the characterization data, the expected monomer and polymer were successfully derived. A noticeable experimental phenomenon was that the clear solution of M1 turned into a dark red solution during the polymerization reaction. After purification, black powders of P1 were obtained (inset of Fig. 1 and videos in the ESI†). The UV-visible absorption spectra of M1 and P1 are displayed in Fig. 1. In tetrahydrofuran (THF) solution, M1 shows no absorption in the visible spectral region, while the absorption spectrum of P1 features a very broad band ranging from 300 to 640 nm with a broad peak at around 490 nm.
 | ||
| Fig. 1 UV-visible absorption spectra of M1 and P1 in THF solution (1 × 10−5 mol L−1). Inset photographs: right, M1 and P1 in THF solutions; bottom, powders of M1 and P1 adsorbed onto the surface of magnetic stir bars. | ||
A fortuitous observation was that M1 crystals could be attracted by the magnetic stir bar (inset photograph in Fig. 1). After eliminating the possibility of electrostatic action, this observation was attributed to a magnetic effect, though it was weak. It is interesting that the magnetic property was also observed for the polymer product P1. During the purification of P1 using the dissolution and precipitation technique, it was observed that the black P1 powders were attracted by and closely adsorbed onto the surface of the magnetic stir bar (inset in Fig. 1), and the magnetized powders could be adsorbed by a pair of iron scissors (Video 2, ESI†). This phenomenon was observed in repeated dissolution and precipitation operations. After ruling out the leakage of magnet components from the stir bar by repeating the polymerization experiment, we ensured that the black powders are magnetic.
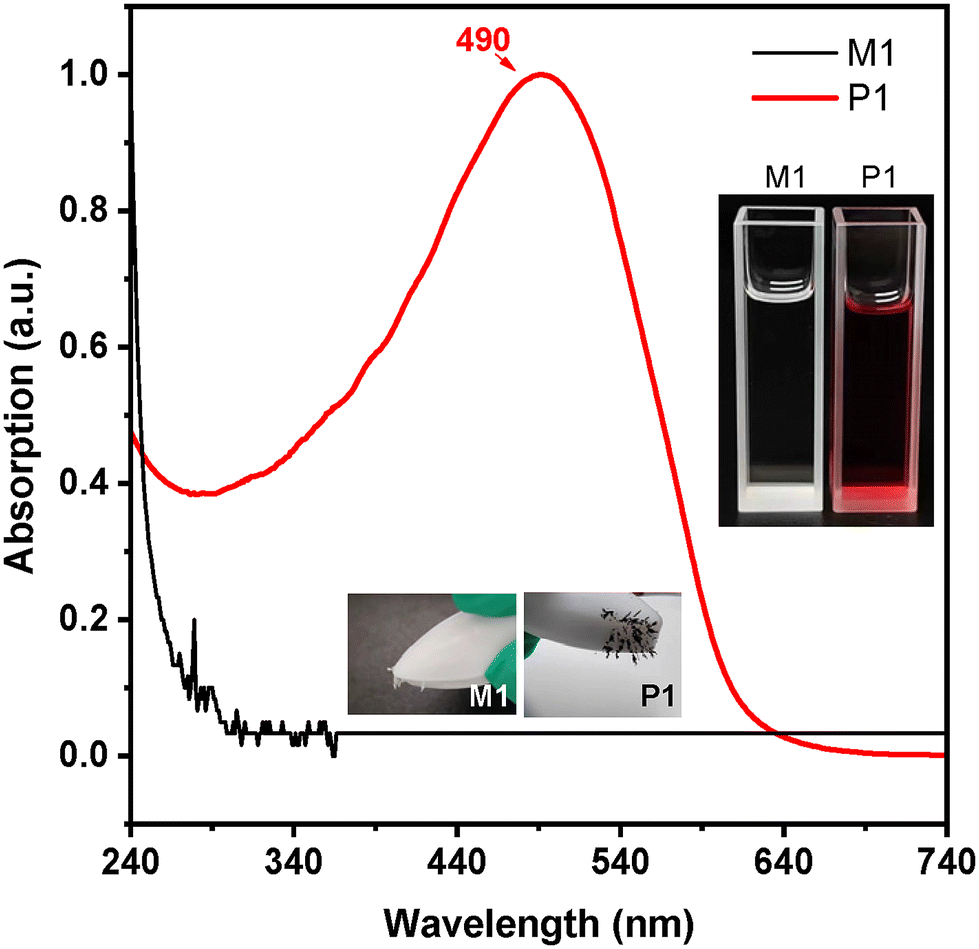
Since the magnetic behaviour usually originates from the free radical species, the electron paramagnetic resonance (EPR) spectra of the crystals of M1 and powders of P1 were recorded, and the results are displayed in Fig. 2. For M1, the EPR signals are weak but authentic (Fig. 2A). The g factor is 2.0062, which is larger than that of a free electron (2.0023) and can be tentatively assigned to the organic carbon free radicals. The wide and asymmetric peaks suggest that the free radicals are in a slowly relaxing and anisotropic environment, consistent with their localization in anisotropic and stable crystalline arrays. For P1, a g factor of 2.0025 is recorded (Fig. 2B), and the resonance signals are relatively strong and largely symmetric as compared with M1. It means that the radicals in P1 powders are in a slowly relaxing and isotropic environment, with lower rigidity and higher activity than those in the crystals of M1. These features are consistent with the fact that P1 is an amorphous solid with a conjugated main chain. The g values of M1 and P1 are much smaller than those of magnetic metals, implying that the magnetic behaviour does not result from metal species. Due to the generally poor stability of substances containing free radicals, we recorded the UV absorption spectra of the solution of the sample placed in air for two weeks (Fig. S5†) and found only slight changes compared to the freshly prepared sample, indicating that P1 has good air stability. Furthermore, we found that although P1 shows broad ultraviolet absorption, it does not exhibit fluorescence emission behaviour (Fig. S6 and S7†), which demonstrates its feasibility as a photothermal material.
 | ||
| Fig. 2 Electron paramagnetic resonance (EPR) spectra of M1 (A) and P1 (B) powders. | ||
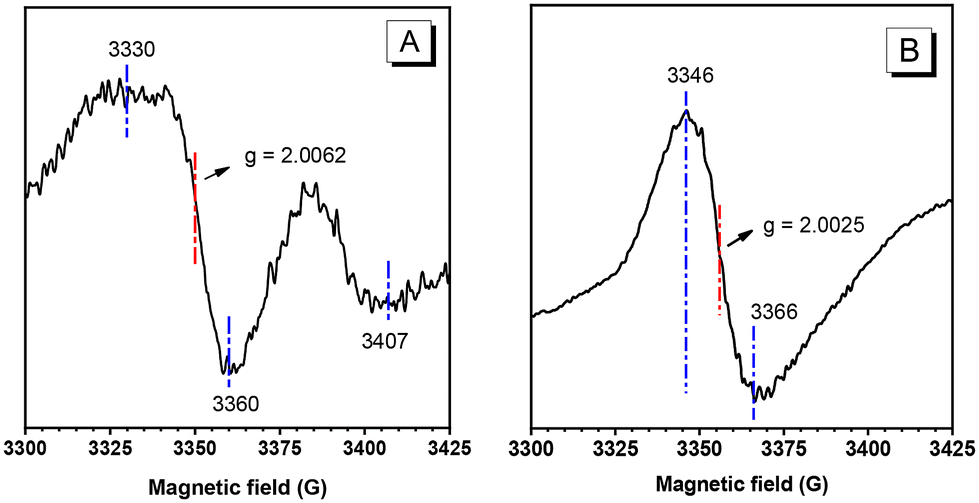
To study the magnetism of M1 and P1, magnetization–magnetic field (M–H) curves were recorded at 300 K, and the data are shown in Fig. 3. Typical magnetic hysteresis loops were obtained for both M1 and P1. For M1, the saturation magnetization was around 0.025 emu g−1 at 300 K, indicating very weak magnetic properties. For P1, the saturation magnetization at 300 K was 0.25 emu g−1, which is 10 times higher than that of M1. This value is about 66 times greater than that of 1,3,5-trizaine-linked porous organic radical frameworks (3.8 × 10−3 emu g−1) measured at room temperature.24 Meanwhile, the temperature of P1 is higher than that of the recently reported amorphous polymerized TCNQ framework (36 K).30 In addition, P1 obtained from the polymerization reaction has better thermal stability compared to M1 (Fig. S8†). The temperatures at which the samples lose 5% of their weight are 121 and 247 °C for M1 and P1, respectively, suggesting that P1 is stable in the temperature range used for magnetic property measurements. Moreover, P1 is soluble in some organic solvents such as THF and DCM, thus allowing it to be processed via solution casting and spin coating techniques to fabricate self-supporting solid films, as shown in the photographs in Fig. S4.† Accordingly, the magnetic properties of P1 were carefully investigated. As demonstrated in Fig. S3,† the M–H curve reaches saturation magnetization at 7600 Oe, with a coercive field of 133.7 Oe at 300 K. The residual magnetism (Mr) and coercivity (Hc) of P1 are 0.01 emu g−1 and 33.9 Oe, respectively.
 | ||
| Fig. 3 The magnetization–magnetic field (M–H) curves of M1 crystals (upper) and P1 powders (lower) measured at 300 K. The saturation magnetization values for M1 and P1 are 0.025 and 0.25 emu g−1, respectively. The insets show the magnified sections of the curves at around zero field. | ||
Since metal catalysts containing Mo and Sn were used in the polymer preparation, Mo-based compound residues (e.g., molybdenum oxide) may result in a weak magnetic behaviour. To eliminate the interference of metal impurities, the synthesized polymer was purified by performing repeated precipitation treatments multiple times, and the content of molybdenum in P1 was found to be lower than 0.01%, as revealed by the data from inductively coupled plasma optical emission spectrometry (ICP-OES, Table S2†). Such a low Mo content in P1 is not enough to cause the magnetization phenomenon, as shown in Fig. 3 and the inset of Fig. 1. Furthermore, both molybdenum chloride and molybdenum oxide show diamagnetism properties (Fig. S9†). Therefore, the results indicate that the magnetization phenomenon originates from P1 rather than metal-catalyst residues.
Furthermore, the content of iron, cobalt, and nickel in P1 was determined by ICP, and the data are summarized in Table S2,† with an iron content of 58 ppm, a cobalt content of 0.1 ppm, and a nickel content of 1 ppm. To further eliminate the influence of metal impurities, we conducted X-ray photoelectron spectroscopy (XPS) and high-temperature ablation experiments. No characteristic signal of iron was detected in XPS analysis (Fig. S10 and S11†). After heating P1 at 500 °C for 1 h, the hysteresis curve of the residual material was recorded (Fig. S12†), which revealed that the ferromagnetism disappeared while diamagnetism was observed, indirectly proving that the magnetism does not originate from inorganic metal impurities.
It is significant that the magnetic property could be tested at 300 K, indicating that the Curie temperature (Tc) of P1 must be higher than room temperature. Then, we measured the magnetic susceptibility and Curie temperature of P1 using a superconducting quantum interference device (SQUID) and a physical property measurement system (PPMS) in the temperature range of 2–300 K under an external field of 0.5 T. The experimental results are shown in Fig. 4 and S13.† The temperature dependence of magnetic susceptibility (χ) suggests that P1 is a typical magnetic material, with the maximum value appearing at about 206 K, indicating the best matching between the spin-alignment and the applied field at this temperature (Fig. S13(a)†).
 | ||
| Fig. 4 Temperature-dependent magnetic properties of P1 powders: plots showing the variations in ZFC and FC magnetization intensity of P1 with temperature, measured at an applied magnetic field of 100 Oe. | ||
The temperature dependence of the product of magnetic susceptibility and temperature (χT) exhibits a good linear relationship with a monotonous increase, and the χT value at 300 K exceeds 90 emu g−1 T−1, which is reasonable for weakly coupled radicals (Fig. S13(b)†). Fig. 4 displays the temperature dependence of magnetization under zero-field-cooled (ZFC) and field-cooled (FC) conditions in a magnetic field of 100 Oe. A bifurcation point between the plots of ZFC and FC can be seen at 400 K, indicating that the Tc value of the magnetic P1 powders should be no lower than 400 K, which is rarely observed for organic polymeric magnetic materials.
Experimental
Materials
Dimethyl malonate was purchased from Bidepharm; propargyl bromide was purchased from Macklin; sodium hydride (NaH), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride, 4-dimethyl-aminopyridine (DMAP), ultra-dry tetrahydrofuran (THF) and ultra-dry 1,2-dichloroethane (DCE) were purchased from J&K Scientific; molybdenum chloride (MoCl5) was purchased from Sigma-Aldrich; and tetra-n-butyltin (n-Bu4Sn) was purchased from Alfa Aesar. Anhydrous methanol (MeOH) and potassium hydroxide (KOH), ammonium chloride (NH4Cl) and anhydrous sodium sulfate (Na2SO4) were purchased from Sinopharm. All chemicals were used directly without further purification.Instruments
The molecular weight (Mw and Mn) and polydispersity (Mw/Mn) of the polymer were estimated in THF using a gel permeation chromatography (GPC, PL-GPC-50, Waters Corporation, Milford, CT, USA) system, calibrated with a set of monodisperse polystyrene standards covering molecular weights from 103 to 107. FTIR spectra were recorded on a VECTOR 22 spectrometer (Bruker Corporation, Billerica, MA, USA). 1H NMR spectra were recorded on AVANCE III 400 spectrometers (Bruker Corporation, Billerica, MA, USA), and tetramethylsilane (TMS) was used as an internal standard. UV-vis absorption spectra were recorded on a Varian CARY 100 Bio UV-vis spectrometer (Agilent Technologies Inc., Santa Clara, CA, USA). Free radical signals were analyzed using a Bruker's Brooke EMXplus-9.5/12 device (Bruker EMXplus EPR Spectrometer). The room temperature magnetization curve was recorded using a VersaLab system (Quantum Design, USA). The variable temperature magnetic susceptibility curve and ZFC–FC curve were recorded using a PPMS-9 system (Quantum Design, USA).Polymer preparation
In a glove box, 0.01 mol of catalyst (MoCl5) was weighed into a polymerization tube; then, 0.02 mol of tetrabutyltin was added using a micro-injector after the catalyst was added, followed by the addition of 1 mL of DCE solvent. In another polymerization tube, 0.5 mol of monomer was weighed, and 1 mL of ultra-dry DCE was added to dissolve the monomer. Then, the monomer solution was transferred to the polymerization tube where the catalyst had been activated for 15 min. At the end of the reaction, at the preset temperature and time, 1 mL of methanol was added to terminate the reaction. The solution was then dropped into a large amount of methanol through a glass dropper and left to precipitate overnight. The resultant product was filtered using a sand core funnel. It was then purified through repeated dissolving and precipitation operations no fewer than 6 times The precipitate was dried under vacuum to a constant weight, and finally a black powder (P1) was obtained. The synthetic procedures for the monomer and the structure characterization data of the monomer and polymer are included in the ESI.†Conclusions
The crystals of 4,4-bis-methoxycarbonyl-1,6-heptadiyne (M1) and its polymer (P1) show distinct magnetism and stable electron paramagnetic signals at ambient temperature. The results of magnetic measurements indicate that both M1 and P1 exhibit room-temperature magnetism. P1 powders exhibit a Curie temperature of at least 400 K and a saturation magnetization of 0.25 emu g−1. The interference from the metal-catalyst residues has been fully removed, and the magnetism of P1 can be tentatively ascribed to the intrinsic radicals in the conjugated polymer chains. In addition to its pronounced magnetism, P1 exhibits solution-processability, a unique property of polymer materials. The findings of this work imply that room-temperature magnetism could be achieved through rational polymer design, furnishing an alternative approach to fabricating pure organic polymeric materials with room-temperature magnetism.Author contributions
Monomer and polymer synthesis, structural characterization, property investigation and manuscript preparation were performed by M. C. and G. H.; fluorescent detection and graphical abstract design were performed by Z. X. and H. H.; data analyses and treatments were directed by H. Z.; and guidance on manuscript writing, review and editing was provided by J. Z. S. and B. Z. T.Data availability
The data supporting this article are available in the manuscript and the ESI.† Additional data can be obtained from the authors upon request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 22071215).References
- R. M. White, Science, 1985, 229, 11–15, DOI:10.1126/science.229.4708.11
.
- J. S. Miller, Adv. Mater., 1994, 6, 322–324, DOI:10.1002/adma.19940060416
- M. Kamachi, J. Macromol. Sci., Polym. Rev., 2002, C42, 541–561, DOI:10.1081/MC-120015990
- A. Rajca, Chem. – Eur. J., 2002, 8, 4834–4841, DOI:10.1002/1521-3765(20021104)
- S. J. Blundell and F. L. Pratt, J. Phys.: Condens. Matter, 2004, 16, R771, DOI:10.1088/0953-8984/16/24/R03
- P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895–8999, 10.1039/C3CS60257E
- J. S. Miller, Pramana, 2006, 67, 1–16, DOI:10.1007/s12043-006-0032-y
- H. M. Mcconnell, J. Chem. Phys., 1963, 39, 1910, DOI:10.1063/1.1734562
- Y. V. Korshak, T. V. Medvedeva, A. A. Ovchinnikov and V. N. Spector, Nature, 1987, 326, 370–372, DOI:10.1038/326370a0
- M. Takahashi, P. Turek, Y. Nakazawa, M. Tamura, K. Nozawa, D. Shiomi, M. Ishikawa and M. Kinoshita, Phys. Rev. Lett., 1991, 67, 746–748, DOI:10.1103/PhysRevLett.67.746
- E. Coronado, C. Giménez-Saiz, C. J. Gómez-García, F. M. Romero and A. Tarazón, J. Mater. Chem., 2008, 18, 929–934, 10.1039/b715739h
- C. Paulsen, J. Souletie and P. Rey, J. Magn. Magn. Mater., 2001, 226, 1964–1966, DOI:10.1016/S0304-8853(01)00005-1
- F. M. Romero, R. Ziessel, M. Drillon, J. L. Tholence, C. Paulsen, N. Kyritsakas and J. Fisher, Adv. Mater., 1996, 8, 826, DOI:10.1002/adma.19960081013
- T. Kaneko, K. Iwamura, R. Nishikawa, M. Teraguchi and T. Aoki, Polymer, 2014, 55, 1097–1102, DOI:10.1016/j.polymer.2014.01.019
- M. Mitani, D. Yamaki, Y. Yoshioka and K. Yamaguchi, J. Chem. Phys., 1999, 111, 2283–2294, DOI:10.1063/1.479499
- H. Nishide, Adv. Mater., 1995, 7, 937, DOI:10.1002/adma.19950071116
- T. Kaneko, T. Matsubara and T. Aoki, Chem. Mater., 2002, 14, 3898–3906, DOI:10.1021/cm020317t
- E. Dobrzyńska, M. Jouni, P. Gawryś, S. Gambarelli, J. Mouesca, D. Djurado, L. Dubois, I. Wielgus, V. Maurel and I. Kulszewicz-Bajer, J. Phys. Chem. B, 2012, 116, 14968–14978, DOI:10.1021/jp309935a
- T. L. Makarova, B. Sundqvist, R. Höhne, P. Esquinazi, Y. Kopelevich, P. Scharff, V. A. Davydov, L. S. Kashevarova and A. V. Rakhmanina, Nature, 2001, 413, 716–718, DOI:10.1038/35099527
- G. Z. Magda, X. Jin, I. Hagymási, P. Vancsó, Z. Osváth, P. Nemes-Incze, C. Hwang, L. P. Biró and L. Tapasztó, Nature, 2014, 514, 608–611, DOI:10.1038/nature13831
- X. L. Wu, R. S. Wang, J. Cheng, G. H. Zhong, X. J. Chen, Y. Gao and Z. B. Huang, Carbon, 2018, 136, 125–129, DOI:10.1016/j.carbon.2018.04.078
- Q. Jiang, J. Zhang, Z. Mao, Y. Yao, D. Zhao, Y. Jia, D. Hu and Y. Ma, Adv. Mater., 2022, 34, 2108103, DOI:10.1002/adma.202108103
- J. Mahmood, J. Park, D. Shin, H. Choi, J. Seo, J. Yoo and J. Baek, Chem, 2018, 4, 2357–2369, DOI:10.1016/j.chempr.2018.07.006
- H. Phan, T. S. Herng, D. Wang, X. Li, W. Zeng, J. Ding, K. P. Loh, A. T. Shen Wee and J. Wu, Chem, 2019, 5, 1223–1234, DOI:10.1016/j.chempr.2019.02.024
- S. Wu, M. Li, H. Phan, D. Wang, T. S. Herng, J. Ding, Z. Lu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8007–8011, DOI:10.1002/anie.201801998
- J. Mahmood and J. B. Baek, Chem, 2019, 5, 1012–1014, DOI:10.1016/j.chempr.2019.04.004
- K. Sakaushi and M. Antonietti, Acc. Chem. Res., 2015, 48, 1591–1600, DOI:10.1021/acs.accounts.5b00010
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676, DOI:10.1126/science.aan0202
- K. S. Burch, D. Mandrus and J. Park, Nature, 2018, 563, 47–52, DOI:10.1038/s41586-018-0631-z
- M. Wei, K. Song, Y. Yang, Q. Huang, Y. Tian, X. Hao and W. Qin, Adv. Mater., 2020, 32, 2003293, DOI:10.1002/adma.202003293
- W. P. Su, J. R. Schrieffer and A. J. Heeger, Phys. Rev. Lett., 1979, 42, 1698–1701, DOI:10.1103/PhysRevLett.42.1698
- H. Fukutome, A. Takahashi and M. Ozaki, Chem. Phys. Lett., 1987, 133, 34–38, DOI:10.1016/0009-2614(87)80048-9
- S. Ramasesha and B. Sinha, Chem. Phys. Lett., 1991, 179, 379–384, DOI:10.1016/0009-2614(91)85170-2
- H. Nishide, N. Yoshioka, T. Kaneko and E. Tsuchida, Macromolecules, 1990, 23, 4487–4488, DOI:10.1021/ma00222a028
- A. Fujii, T. Ishida, N. Koga and H. Iwamura, Macromolecules, 1991, 24, 1077–1082, DOI:10.1021/ma00005a016
- R. Saf, K. Hummel, K. Gatterer and H. P. Fritzer, Polym. Bull., 1992, 28, 395–402, DOI:10.1007/BF00297331
- H. Nishide, T. Kaneko, N. Yoshioka, H. Akiyama, M. Igarashi and E. Tsuchida, Macromolecules, 1993, 26, 4567–4571, DOI:10.1021/ma00069a023
- H. Murata, D. Miyajima, R. Takada and H. Nishide, Polym. J., 2005, 37, 818–825, DOI:10.1295/polymj.37.818
- T. Itoh, Y. Jinbo, K. Hirai and H. Tomioka, J. Am. Chem. Soc., 2005, 127, 1650–1651, DOI:10.1021/ja0455345
- M. F. Beristain, M. F. Jimenez-Solomon, A. Ortega, R. Escudero, E. Muñoz, Y. Maekawa, H. Koshikawa and T. Ogawa, Mater. Chem. Phys., 2012, 136, 1116–1123, DOI:10.1016/j.matchemphys.2012.08.062
- Y. S. Gal, S. H. Jin and S. K. Choi, J. Mol. Catal. A: Chem., 2004, 213, 115–121, DOI:10.1016/j.molcata.2003.10.055
- M. G. Mayershofer, O. Nuyken and M. R. Buchmeiser, Macromolecules, 2006, 39, 3484–3493, DOI:10.1021/ma052510p
- F. Yang, Z. Zhang, M. Chen, H. Zhang, J. Zhang and J. Z. Sun, Polym. Chem., 2022, 13, 6492–6499, 10.1039/D2PY01145J
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01459f |
| This journal is © The Royal Society of Chemistry 2025 |