
Recent advances in perovskite air electrode materials for protonic solid oxide electrochemical cells
Cancan
Peng
,
Xu
Han
 ,
Sebete
Mabaleha
,
Philip
Kwong
,
Yao
Zheng
* and
Xiaoyong
Xu
*
,
Sebete
Mabaleha
,
Philip
Kwong
,
Yao
Zheng
* and
Xiaoyong
Xu
*
School of Chemical Engineering, The University of Adelaide, Adelaide, South Australia 5005, Australia. E-mail: yao.zheng01@adelaide.edu.au; xiaoyong.xu@adelaide.edu.au
First published on 10th April 2025
Abstract
Intermediate-temperature proton-conducting solid oxide cells (P-SOCs) have emerged as a promising technology for power generation and hydrogen production. They have gained significant attention due to their lower operating temperature, higher efficiency, better safety and durability and simplified water management over conventional high-temperature oxygen-conducting solid oxide cells (O-SOCs). However, the performance of P-SOC air electrodes is hindered by the sluggish kinetics of oxygen reduction and evolution reactions, necessitating efficient conductivities of H+, O2−, and e−. Despite critical advancements, the search for optimal air electrode materials remains challenging. This review provides a comprehensive overview of recent advancements in perovskite materials for P-SOC air electrodes, covering fundamental mechanisms, material development, theoretical modeling, and practical applications. It highlights key progress in reaction kinetics, structure–property relationships, and modification strategies across widely studied perovskite-based systems. Particular emphasis is placed on understanding the correlation between structural characteristics and the electrochemical activity and stability of electrodes, which is essential for the rational design of high-performance, durable P-SOC materials. Additionally, advanced methodologies and mechanistic insights into newly developed air electrode materials are explored, with a focus on the role of theoretical simulations, including artificial intelligence (AI)-driven machine learning (ML) techniques. Finally, perspectives are provided on the future development of high-performance P-SOC air electrodes for industrial applications.
Broader contextAs the climate and environmental crisis deepens, the development of clean energy sources has become increasingly critical. Intermediate-temperature proton-conducting solid oxide cells (P-SOCs) show immense potential for power generation and hydrogen production due to their favorable thermodynamics, efficient kinetics, high energy efficiency, low carbon emissions, high tolerance to fuel impurities, modularity, and reversible operation capabilities. In contrast, commercially available oxygen-ion SOCs (O-SOCs) typically operate at temperatures exceeding 750 °C, where the extreme conditions accelerate material degradation and pose significant reliability challenges. To address these issues, researchers are increasingly focusing on P-SOCs, which operate at reduced temperatures of approximately 400–600 °C, enabled by their higher proton conductivity and lower activation energy. Despite these advances, several challenges persist. Even at 500 °C, the air electrode performance of P-SOCs lags that of O-SOCs due to substantial polarization resistance associated with redox reactions at the air electrode. Among potential air electrode materials, perovskite oxides emerge as promising candidates due to their unique electronic structure, superior catalytic properties for oxygen reactions, high thermal stability, and relative ease of synthesis. This review explores cutting-edge P-SOC technologies utilizing perovskite air electrode materials and provides a detailed outlook on future research directions. More broadly, this review provides a valuable reference for designing advanced air electrodes for next-generation P-SOCs. |
1. Introduction
Amid the urgent push toward carbon neutrality and the global transition to sustainable development, electrochemical energy conversion and storage technologies have gained substantial attention.1 Their unique capability to integrate renewable energy sources into existing infrastructures offers a powerful solution for reducing reliance on fossil fuels and supporting a resilient energy ecosystem.2 Among these electrochemical technologies, solid oxide fuel cells (SOFCs) and solid oxide electrolysis cells (SOECs) hold particular promise due to their rapid reaction kinetics, high round-trip efficiency, fuel flexibility, and cost-effectiveness in terms of materials.3 However, the commercialization of their conventional versions, that rely on oxygen-ion conducting electrolytes, faces substantial challenges associated with high operation temperature, high operational costs, and low reliability detrimental to sustainability. Significant efforts have, consequently, been dedicated to enhancing their sustainability through improvement of cell durability and simplification of management system on oxygen-ion-based SOFCs/SOECs (O-SOFCs/SOECs) by reducing the operating temperatures to manageable levels (400–600 °C).4,5 However, achieving these lower temperatures without compromising efficiency and performance remains challenging, as lower temperatures can hinder reaction kinetics and reduce ionic conductivity. Proton (H+) migration is believed to be much faster and has significantly lower activation energy (0.4–0.6 eV) than O2− (>1 eV) as the smaller size and mass of the H+ allow it easily hop in the lattice.6,7 Thus, protonic solid oxide electrochemical cells (P-SOCs) hold a greater promise for lower temperature operation.P-SOCs is a sustainable hydrogen production and power generation technology that integrates economy, efficiency, and safety (Fig. 1(a)). Compared to low-temperature proton exchange membrane systems (PEM, Fig. 1(b1)) which typically operate at 50–80 °C, P-SOCs provide superior thermodynamic performance—including improved catalytic activity, reaction kinetics, and efficiency. Thermal energy compensation (TΔS) for total energy (ΔH) in P-SOC systems lead to lower electrical energy (ΔG) requirements compared to low-temperature catalysts, as shown by eqn (1).8–10
| ΔH = ΔG + TΔS | (1) |
 | ||
| Fig. 1 (a) Schematic of steam electrocatalysis and power generation in P-SOCs. (b) Principal schemes of (b1) PEMs, (b2) O-SOCs, and (b3) P-SOCs. (c) Comparison among low-temperature PEM, intermediate-temperature P-SOCs, and high-temperature O-SOCs. | ||
Unlike PEM water electrolyzers, P-SOCs facilitate direct production of pure, dry hydrogen in electrolysis mode without a need for external gas purification and dehumidification, thereby making it substantially cost effective. Furthermore, the application of low-temperature PEM systems is constrained by the high cost of platinum group metal catalysts and the stringent raw material requirements, including specific levels of acidity, conductivity, and resistivity. Compared with O-SOFCs (Fig. 1(b2)), water generation at the P-SOFC (Fig. 1(b3)) prevents fuel dilution which contributes to enhanced efficiency and better fuel utilization. High temperatures, according to Stefan–Boltzmann law, increase thermal radiation losses and impose stricter requirements on the cells, interconnectors, and glass sealant to withstand high thermal stresses.11 Consequently, it is economically imprudent to employ them amid their potential to decreases hydrogen production efficiency, shorten device lifespans, and accelerate degradation, in addition.12,13 The “intermediate-temperature” P-SOCs with their fairly manageable temperatures are, therefore, better alternatives for efficient hydrogen generation via water electrolysis and power production through fuel cell mode (Fig. 1(c)).14
A typical P-SOC includes a dense electrolyte that enables proton conduction and is sandwiched between two porous electrodes: an air electrode and a fuel electrode. The electrolyte supports efficient proton transport, while the porous electrodes fulfil specific roles: the air electrode assists in oxygen reduction and evolution reactions (ORR/OER), and the fuel electrode is involved in the oxidation and reduction of fuel (H2).15 It is imperative to develop solutions that are cost-effective, reliable, and long-lasting for P-SOC technology to be effectively applied, the solutions that include finding catalytic materials with high feed conversion and stability at typical elevated reaction temperatures. Consequently, advances through material design, e.g., modifying material structure/composition alongside SOC technology design are essential and key to the technology.16 The simultaneous involvement of O2−, H+, and e− in reactions at the air electrode necessitates the corresponding conductivities for all these three charge carriers. However, materials with this unique triple-conductive capability is rare, highlighting their development an imperative.17,18 Engineering of air electrodes for P-SOCs is highly complex as they play a crucial role in determining overall performance. This complexity has drawn significant attention to their development. The sluggish kinetics of the proton-involved ORR and OER at the air electrode within the range of 400 °C to 700 °C severely limit the electrochemical performance and energy efficiency.5,19 This effectively necessitates development of more electrocatalytically active and stable materials, currently a major challenge, for sustainable future of P-SOCs.20,21
Researchers have dedicated considerable effort to develop advanced air electrode materials for P-SOCs. Metal oxide electrodes (e.g., LiCoO2) are cost-effective and chemically stable but suffer from low electronic conductivity and phase instability in humid or CO2-rich environments, limiting their long-term performance. Precious metal electrodes (e.g., Pt, Ru) offer exceptional catalytic activity and high electronic conductivity, ensuring fast reaction kinetics, but their high cost, limited availability, and susceptibility to sintering make them less viable for large-scale applications.5,8 In contrast, perovskite oxides are the dominant choice for air electrodes, owing to their distinctive electronic structure, catalytic properties in oxygen-related reactions, high thermal stability, and ease of raw material synthesis. Over time, studies on perovskite air electrodes have covered various categories, including oxide materials exhibiting dual O2−/e− conductivity, composites that combine proton conductors with dual O2−/e− conductors, and newly developed single-phase materials that achieve triple H+/O2−/e− conductivity. Herein, the recent progress and challenges associated with perovskite air electrode materials for P-SOCs are systematically reviewed. This review aims to provide an in-depth examination of the recent advancements and ongoing challenges in perovskite air electrode materials for P-SOCs. The state-of-the-art research is categorized into several representative air electrode material types—(Ba/Sr/Pr)MO3−δ-type, LnBaM2O5+δ-type, and Ln2NiO4+δ-type—with modification methods spanning bulk structures, interfaces, and composites (Fig. 2). Moreover, the application of advanced computational techniques, particularly artificial intelligence (AI)-driven machine learning (ML), is explored for materials design and theoretical explanation. Commercial considerations and future research directions for P-SOCs air electrodes are also discussed, providing a comprehensive perspective on advancing P-SOCs technology.
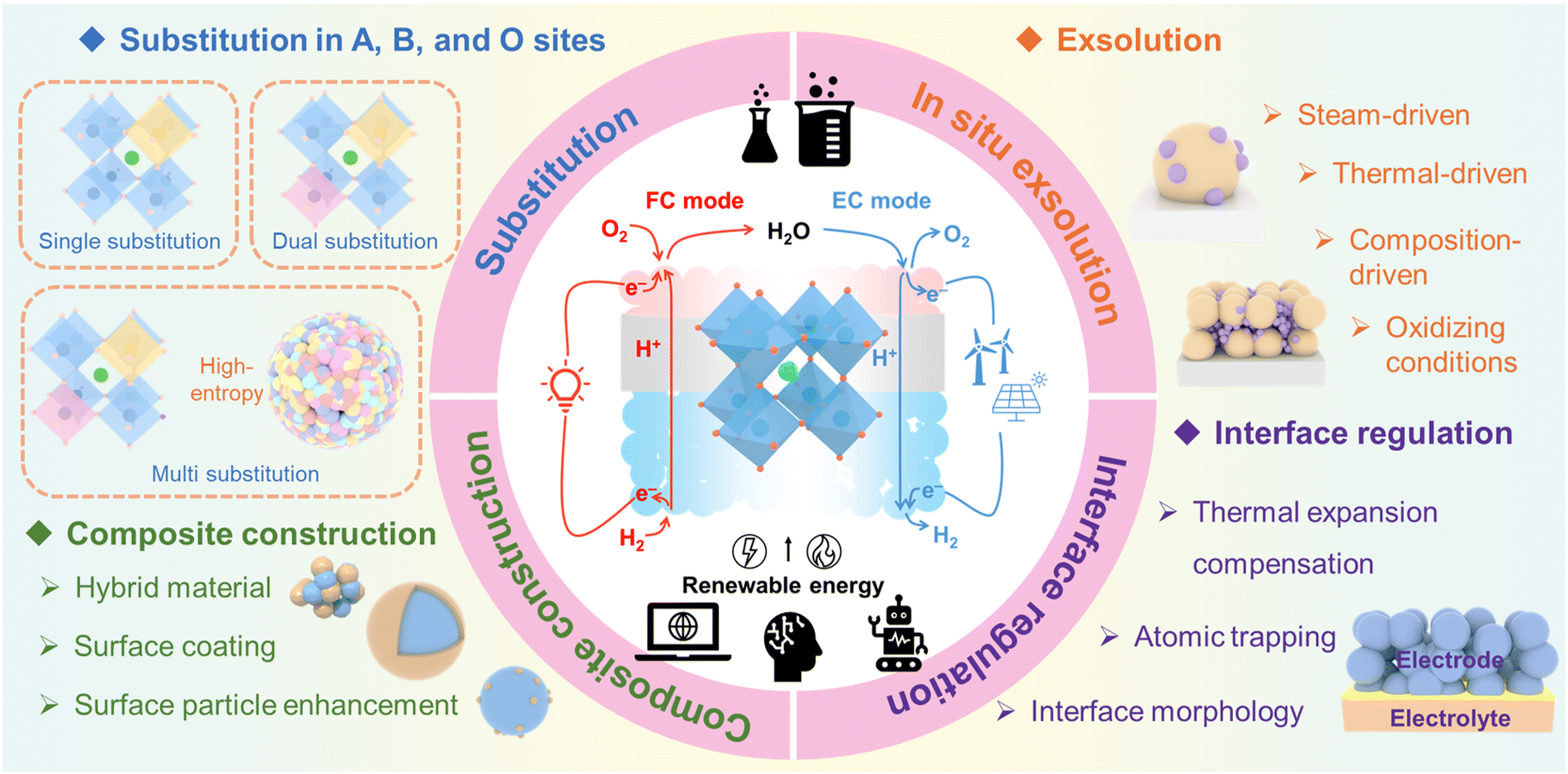 | ||
| Fig. 2 Typical modifications of perovskite air electrode materials. | ||
2. Perovskite air electrode materials for P-SOCs
P-SOCs represent a significant advancement in the fields of hydrogen production and energy conversion, operating efficiently in both fuel cell (FC) and electrolysis cell (EC) modes. Their operational versatility makes them crucial technologies in sustainable energy systems, leading to great interest in recent years. This section discusses the electrochemical processes involved in hydrogen generation and power generation and then analyzes the role and requirements of perovskite air electrode materials and the corresponding evaluation methods.2.1. Fundamentals of protonic solid oxide electrochemical cells
A typical P-SOC single cell features a sandwich-like structure with the configuration of air electrode/proton-conducting electrolyte/fuel electrode (Fig. 3(a)). The electrolyte functions as a dense electron-blocking layer, preventing direct combustion between the fuel (H2) and air (primarily O2) while facilitating rapid proton transport.22,23 The fuel electrode materials comprising of metal (commonly Ni) and electrolyte component is where H2 generated or consumed.24 The air electrode serves as a site for ORR or OER.25 During typical FC operation, electricity is generated through the electrochemical conversion of hydrogen (supplied as fuel on the fuel electrode) which serves as the proton source. Oxygen (from the air) in the air electrode reacts with the proton in the present of electrons (ORR) to form water (eqn (2) – FC mode). In EC mode, steam undergoes electrolysis to produce H2 by flowing it through the air electrode for OER (eqn (2) – EC mode).26 When voltage is applied, water decomposes into H+ and O2, releasing the electrons. H+ then migrates through the electrolyte to generate dry H2 which is then swept off the reaction chamber using inert gas, e.g., N2 or Ar, while electrons flow through the external circuit to the fuel electrode.27,28Eqn (2) and (3) show reactions at both electrodes and Fig. 3(a) exhibits their simple schematic versions.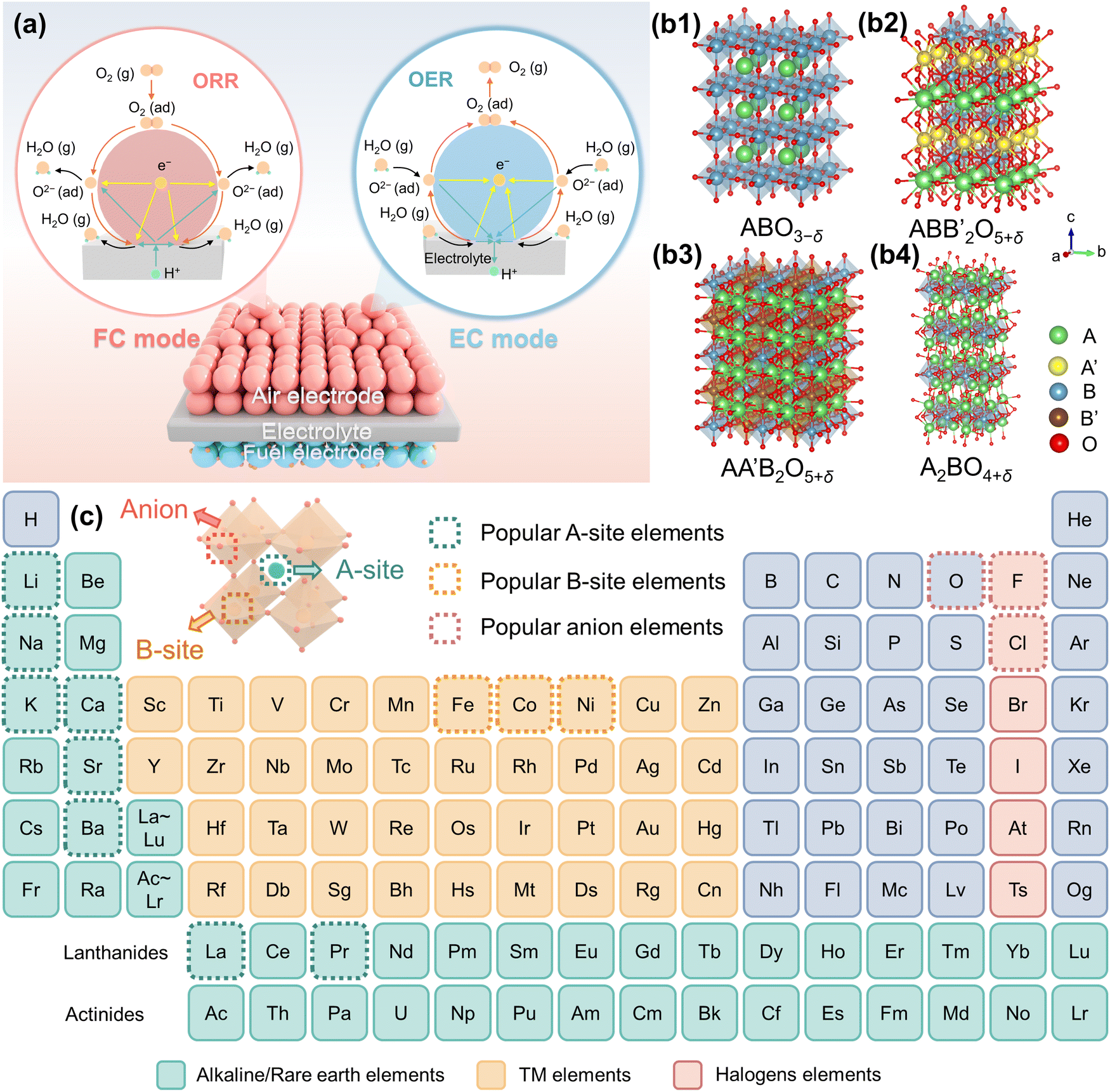 | ||
| Fig. 3 Fundamentals of perovskite air electrode materials in P-SOCs. (a) Schematic of typical P-SOCs and reactions at the air electrode: ORR reaction and OER reaction (as exemplified by triple-conducting material). (b) Structure of perovskite: (b1) simple perovskite, (b2) and (b3) double perovskite, and (b4) Ruddlesden–Popper phase. (c) Typical elements in perovskite. | ||
Air electrode:
 | (2) |
Fuel electrode:
 | (3) |
In recent years, new reversible P-SOCs have emerged that can convert fuel into electricity in FC mode and water into hydrogen in EC mode.29 Unlike prevalent batteries, which face limitations such as self-discharge and high storage costs, the reversibility of reversible P-SOCs potentially enables long-term energy storage by coping with seasonal energy storage adjustments.30 It operates electrolytically when additional baseload power is needed to supplement solar and wind.31 Since chemical fuels can be stored indefinitely or used immediately as renewable feedstocks in various industrial applications, converting renewable electricity into chemical fuels, aiding the transition to a fully sustainable energy economy.9 The operational reversibility enables efficient energy storage during periods of renewable electricity oversupply and supports smooth electricity generation, helping to balance supply and demand while expanding the potential for large-scale SOC applications.
2.2. Key properties of perovskites for air electrodes
Essentially, the electrocatalytic process at air electrode of P-SOCs encompasses both the OER and ORR along with hydration reaction, involving the transport of O2−, H+, e−. By virtue of multi-conducting materials, the active sites of the proton-involved ORR and OER can cover the entire electrode surface instead of being confined to the electrode/electrolyte interface. Perovskites and perovskite-like structures dominate multiphase conducting materials, sensibly making them widely utilized as air electrodes.32–34Simple perovskite (ABO3−δ, Fig. 3(b1)), such as BaCoO3−δ (BCO), BaFeO3−δ (BFO), and SrCoO3−δ (SCO),39–41 feature rare earth metal or alkaline-earth metal (A site) typically coordinating with oxygen in a 12-fold cuboctahedral arrangement within BO6 octahedra.42 TMs (B site) are generally found in sixfold coordination within these octahedra. Double perovskite (A′AB2O5+δ and A2BB′O5+δ, Fig. 3(b2) and (b3)), e.g., PrBaCo2O5+δ (PBC), PrBa0.8Sr0.2Co2O5+δ (PBSC), and PrBa0.8Ca0.2Co1.95Pd0.05O5+δ9,43 consist of alternatively stacked layers in the form of AO|BO2|A′O|BO2|. A is typically a lanthanide ion (+3), e.g., Pr, Nd, Sm, or Gd or alkaline-earth ion (Ba or Sr), and B is often a first-row TM ion or a mixture thereof. The RP phase has general formula: An+1BnO3n+1, where n denotes the number of perovskite-like octahedral layers (Fig. 3(b4)), exemplary materials being La2NiO4, La3Co2O7, and La4Ni3O10,44–46 They exhibit a unique layered structure in which a sequence of (ABO3)n perovskite layers is interleaved with rock-salt-type AO layers. A2BO4+δ represents the simplest RP structure. The perovskite structure is flexible, adaptable, and tunable. By modifying their properties, e.g., type, composition, phase structures, and synthesis methods, electronic conductivity, ionic conductivity, pore morphology etc., can be altered and subsequently, catalytic performance.36,37
 | ||
| Fig. 4 Charge conduction mechanism (red spheres: O2−; yellow spheres: e−; green spheres: H+). (a) e− conduction mechanism: small-polaron hopping mechanism. O2− conduction mechanisms: (b) vacancy diffusion mechanism and (c) oxygen interstitial diffusion mechanism. H+ uptake mechanism: (d) hydration mechanism, (e) hydrogeneration mechanism, and (f) new-type hydration mechanism; H+ transfer mechanism: (g) vehicle mechanism, (h) Grotthuss mechanism, and (i) H+ reorientation and hopping for Grotthuss mechanism. | ||
In perovskite air electrodes for oxidizing atmospheres, O2− diffusion is the primary mode of mass transport. This process is facilitated by O2− vacancies 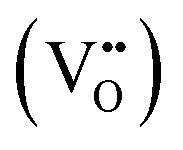 formed through acceptor substitution which create channels for transport. As a result, the mechanism is conveniently referred to as vacancy diffusion (Fig. 4(b)).57,58 The symbol, “˙˙”, represents a net double negative charge while “×” represents a neutral charge. On the surface, these
formed through acceptor substitution which create channels for transport. As a result, the mechanism is conveniently referred to as vacancy diffusion (Fig. 4(b)).57,58 The symbol, “˙˙”, represents a net double negative charge while “×” represents a neutral charge. On the surface, these  capture adsorbed oxygen (O2) into lattice oxygen (
capture adsorbed oxygen (O2) into lattice oxygen ( ) and release holes (h˙), which then combine with electrons from the fuel electrode to complete the electrical circuit (eqn (4)). In single-structure perovskites, oxygen ion transport via the vacancy diffusion mechanism whereas some layered and spinel perovskites utilize the oxygen interstitial diffusion mechanism.46,59–61 Oxygen ions, in the latter process, migrate through the interstitial spaces between lattice sites (Fig. 4(c)).55,62
) and release holes (h˙), which then combine with electrons from the fuel electrode to complete the electrical circuit (eqn (4)). In single-structure perovskites, oxygen ion transport via the vacancy diffusion mechanism whereas some layered and spinel perovskites utilize the oxygen interstitial diffusion mechanism.46,59–61 Oxygen ions, in the latter process, migrate through the interstitial spaces between lattice sites (Fig. 4(c)).55,62
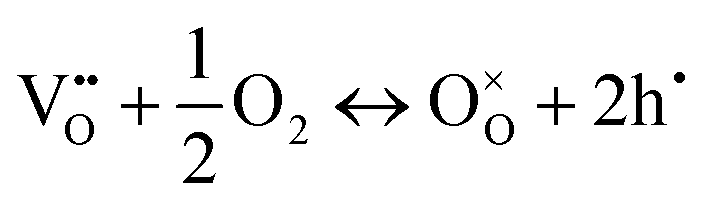 | (4) |
Since protons are not native to oxide lattices, proton uptake and conduction in perovskites heavily relies on hydroxide defects.63 Three mechanisms have been proposed for proton uptake: hydration mechanism, the hydrogeneration mechanism, and a novel hydration mechanism recently introduced in the literature.20,64,65 The Kröger–Vink notation describes the primary hydration mechanism, where H2O decompose into hydroxide ions (OH˙) and H+, with OH˙ filling 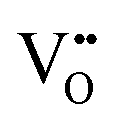 and H+ bonding with
and H+ bonding with  (eqn (5) and Fig. 4(d)). It is predominant when hole concentration is low.66–68 Hydrogenation mechanism, occurs when oxides have abundant h˙, involving a redox reaction that incorporates H+ and releases O2, bypassing the need for
(eqn (5) and Fig. 4(d)). It is predominant when hole concentration is low.66–68 Hydrogenation mechanism, occurs when oxides have abundant h˙, involving a redox reaction that incorporates H+ and releases O2, bypassing the need for  (eqn (6) and Fig. 4(e)).69–71 Recently proposed hydration mechanism based on La0.7Sr0.3MnO2.95 (LSM) suggests that water absorption and desorption are linked to the oxidation of manganese cations
(eqn (6) and Fig. 4(e)).69–71 Recently proposed hydration mechanism based on La0.7Sr0.3MnO2.95 (LSM) suggests that water absorption and desorption are linked to the oxidation of manganese cations 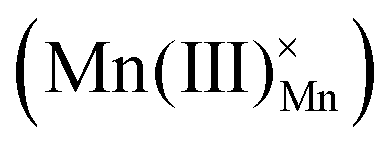 and the occupation of oxygen holes
and the occupation of oxygen holes  by H+ (eqn (7) and Fig. 4(f)).72 It further suggests that oxides with significant oxygen vacancy carriers and mixed valence states, like LSM, are effective for proton conduction, making them suitable for applications requiring triple-conducting materials.72 This theory has since been applied to a wider range of perovskite materials, including those containing cobalt cations, iron oxide ions, and others. Since water adsorption is an exothermic reaction,73,74 proton conduction becomes the dominant mechanism at lower temperatures, leading to a reduced operating temperature for P-SOCs compared to O-SOCs.
by H+ (eqn (7) and Fig. 4(f)).72 It further suggests that oxides with significant oxygen vacancy carriers and mixed valence states, like LSM, are effective for proton conduction, making them suitable for applications requiring triple-conducting materials.72 This theory has since been applied to a wider range of perovskite materials, including those containing cobalt cations, iron oxide ions, and others. Since water adsorption is an exothermic reaction,73,74 proton conduction becomes the dominant mechanism at lower temperatures, leading to a reduced operating temperature for P-SOCs compared to O-SOCs.
 | (5) |
 | (6) |
 | (7) |
Proton transfer in perovskite can occur through the vehicle and Grotthuss mechanisms, both identified by Kreuer et al.68,75,76 In the vehicle mechanism (Fig. 4(g)), protons move with the O2−, a process constrained by the diffusion rate of O2−, leading to higher activation energy because of the limited mobility of O2− ions.77,78 Conversely, the Grotthuss mechanism (Fig. 4(h)), which is more commonly accepted, exhibits lower activation energy (<0.4 eV) and involves protons hopping between O2− ions through the alternation of OH˙ bonds. This process consists of two steps: (1) reorientation of the OH˙ group to reduce the energy barrier for H+ transfer, and (2) H+ hopping between O2− ions, aided by protonic defects (Fig. 4(i)).77,79 In P-SOCs air electrodes, the reaction kinetics are dictated by the balance between electronic and ionic conductivity. High electronic conductivity facilitates the rapid movement of electrons, ensuring that redox reactions occur efficiently at the electrode–electrolyte interface. However, excessive electronic conductivity can reduce the concentration of mobile ionic species, limiting the transport of O2− or H+. Conversely, high ionic conductivity is essential for sustaining continuous ion diffusion within the electrode bulk, but it may come at the cost of reduced electronic charge transfer, leading to increased polarization resistance and sluggish electrode kinetics.80,81 However, optimizing one property often compromises the other. For example, 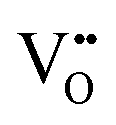 accelerate ionic transport but can deteriorate the electronic conductivity and weaken structural stability. Therefore, air electrode design is not solely about maximizing one type of conductivity but rather achieving an optimal balance between ionic and electronic transport properties to enhance overall performance.
accelerate ionic transport but can deteriorate the electronic conductivity and weaken structural stability. Therefore, air electrode design is not solely about maximizing one type of conductivity but rather achieving an optimal balance between ionic and electronic transport properties to enhance overall performance.
2.3. Performance-influencing factors
Effective operation of air electrodes relies on several complex sub-steps including the adsorption/desorption of reactants, generation of active ions, and ion transport within the electrode. High-performance air electrodes should possess both superior ion transfer capabilities and surface catalytic activity to improve reaction kinetics.82 Key factors influencing performance include oxygen vacancies,83 hydration abilities,84 oxygen/proton diffusion capabilities,85 and surface oxygen ORR and OER activities.86–89 Optimal performance in P-SOCs requires a balance among these various factors which this section looks into, based on recent advances.Catalytic activity of perovskite influences generation of active ions thus influencing the concentration of charges in system.63,103–105 The electronic structure is considered crucial for catalytic activity involve the occupancy of the eg orbitals in surface TMs and the energy levels of the O p-band center.106,107 To enable rapid surface oxygen-exchange kinetics and unimpeded transport of dissociated oxygen species, to controlling the redox activity of lattice oxygen in perovskites is crucial.108 This control can generally be achieved through several strategies: substituting A-site with lower-valence cations, inducing oxygen non-stoichiometry, and replacing B-site cations with more electronegative elements.109–113 The concentration of  affects the adsorption process of reactants in the proton-involved ORR and OER, thereby affecting the efficiency of the catalytic reaction. For instance,
affects the adsorption process of reactants in the proton-involved ORR and OER, thereby affecting the efficiency of the catalytic reaction. For instance,  concentration connected to B-site cations influences proton uptake in oxides that rely on a hydration mechanism. The micro/nano-structure of perovskite, including its surface area and exposed lattice planes, directly influences surface reaction activity.105 Lattice planes exhibit higher activity like (110) for PrO1.8, high surface area, and large pores, for example, improve the activity through increasing active sites dispersion, reducing the distance for bulk ionic diffusion, etc.114–116 To advance the industrialization of P-SOCs, it is, therefore, imperative to develop electrodes with excellent triple-phase conductivity and highly electrocatalytic activity for both ORR and OER.117,118
concentration connected to B-site cations influences proton uptake in oxides that rely on a hydration mechanism. The micro/nano-structure of perovskite, including its surface area and exposed lattice planes, directly influences surface reaction activity.105 Lattice planes exhibit higher activity like (110) for PrO1.8, high surface area, and large pores, for example, improve the activity through increasing active sites dispersion, reducing the distance for bulk ionic diffusion, etc.114–116 To advance the industrialization of P-SOCs, it is, therefore, imperative to develop electrodes with excellent triple-phase conductivity and highly electrocatalytic activity for both ORR and OER.117,118
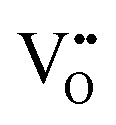 , thereby impeding proton conduction. Additionally, hydration enthalpies differ based on bond distances and local environments, influencing H+ absorption and conduction. Lower hydration enthalpies generally favor enhanced H+ conduction contrary to high enthalpies.
, thereby impeding proton conduction. Additionally, hydration enthalpies differ based on bond distances and local environments, influencing H+ absorption and conduction. Lower hydration enthalpies generally favor enhanced H+ conduction contrary to high enthalpies.
In addition to charge-carrier mobility which is influenced by structure of perovskites, structural stability including chemical and thermodynamic stability is significate parameters. Based on the findings of Anthony F. et al., the tolerance factor (t), calculated from the ionic radii of A-site (rA), B-site (rB), and O site ions (rO) (eqn (8)), accurately estimates stability of perovskite structures.122,123
 | (8) |
Perovskite oxides exhibit structural stability within the range of 0.75 < t < 1 with deviations causing varying degrees of lattice distortion.124,125 Besides radii, the ordering of cations is also important. A-site ordering improves oxygen transport and hydration, as evidenced by materials like GdBaMn2O6−δ and LaBaCo2O6−δ,126–128 while B-site ordering and oxygen-vacancy ordering are detrimental to proton conductivity and hydration.91,129,130
Structural defects such as vacancies, dislocations, stacking faults, and grain boundaries play a crucial role in determining the electrochemical performance of perovskite air electrodes. 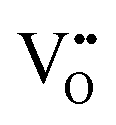 , h˙, and protonic defects are particularly important in enhancing ionic and electronic conductivity, as they facilitate O2−/H+ diffusion and e− transfer through the electrode bulk. Dislocations, which occur due to lattice mismatches or strain during synthesis and operation, can alter charge carrier mobility and introduce localized stress points that may accelerate material degradation.131 As an effective strain carrier, stacking faults can directly introduce strain fields and significantly affect the centre position of the d-band, thereby changing the interaction of surface adsorbates. Grain boundaries serve as pathways for ion transport but can also act as barriers that increase resistance, depending on their structure and chemical composition. However, excessive defects formation can lead to crystal lattice distortion and phase instability, negatively impacting electrode durability. Therefore, an ideal perovskite structure features symmetry cubic with a moderate defect concentration, and ordered A-site occupancy, while maintaining disorder in the B-site and anion sublattices to maximize overall conductivity.132
, h˙, and protonic defects are particularly important in enhancing ionic and electronic conductivity, as they facilitate O2−/H+ diffusion and e− transfer through the electrode bulk. Dislocations, which occur due to lattice mismatches or strain during synthesis and operation, can alter charge carrier mobility and introduce localized stress points that may accelerate material degradation.131 As an effective strain carrier, stacking faults can directly introduce strain fields and significantly affect the centre position of the d-band, thereby changing the interaction of surface adsorbates. Grain boundaries serve as pathways for ion transport but can also act as barriers that increase resistance, depending on their structure and chemical composition. However, excessive defects formation can lead to crystal lattice distortion and phase instability, negatively impacting electrode durability. Therefore, an ideal perovskite structure features symmetry cubic with a moderate defect concentration, and ordered A-site occupancy, while maintaining disorder in the B-site and anion sublattices to maximize overall conductivity.132
2.4. Evaluation methods
The development of new technologies, such as P-SOCs, typically follows a conventional progression from lab-scale research to pilot plant trials and, eventually to industrial-scale production.16 The initial lab-scale phase is crucial, as the reliability and accuracy of the experimental data gathered at this stage heavily influence the subsequent stages of technology development. For successful scaling-up and industrialization of sustainable P-SOCs technology, it is essential to have all necessary information well documented and understood. This section gives an overview of electrochemical methods and characterizations based on recent developments, and the recommended research path for air electrodes is summarized in Fig. 5. | ||
| Fig. 5 Recommended research paths for air electrode materials. | ||
| ASR = (A × Rp)/2 | (9) |
 | ||
| Fig. 6 Recent advances in evaluation methods of air electrode materials. (a) EIS curves, (b) DRT, and (c) kH and DH of PBSCF and W infiltrated PBSCF. Reproduced with permission.150 Copyright 2024, Wiley-VCH. (d) I–V curves of FC and EC modes. Reproduced with permission.151 Copyright 2024, Wiley-VCH. (e) Scheme for the H2 permeation test with the proton-conducting membrane. Reproduced with permission.85 Copyright 2023, Wiley-VCH. (f) Schematic of ToF-SIMS measurement. Reproduced with permission.152 Copyright 2021, Wiley-VCH. (g) XPS spectra of O 1s. Reproduced with permission.150 Copyright 2024, Wiley-VCH. | ||
Distribution of relaxation time (DRT) analysis can be performed based on impedance spectra by deconvoluting the EIS response to distinguish overlapping electrode processes and activation energy (Ea) is then determined from the Arrhenius plots (Fig. 6(b)).153,154 Catalytic activity test is typically performed in dry air or humidified air. Typically, electronic conductivity dominates because the mobility of e− is several orders of magnitude greater than that of H+ and O2−.155,156 The conductive mechanism of the electrode material can be inferred from its conductivity behavior (Fig. 6(c)). For instance, the conductivity of SSNCF material initially rises and then declines with increasing temperature, peaking at 375 °C, which suggests a transition in its conductive mechanism from p-type to n-type around this temperature. Electrical conductivity relaxation (ECR) measurements, utilizing a 4-probe DC conductivity method, are employed to investigate the chemical diffusivities and surface exchange of H+ and O2−, as well as electrical conductivity.83,157 Chemical diffusion coefficient (Dchem) and surface exchange coefficient (kchem) are derived by fitting the transient conductivity data to the solution of Fick's second law using a non-linear least-squares fitting approach, based on the Levenberg–Marquardt algorithm.157Dchem is calculated from the transient conductivity behavior in response to changes in partial pressure. However, for samples with low proton concentrations, the accuracy of ECR measurements may be limited.31 To isolate the conductivity attributed solely to surface protons, a common approach involves calculating the difference between conductivities measured in wet and dry atmospheres.158 It is crucial to ensure that the volume conductivity of the target materials remains unaffected by atmospheric conditions.159
Electrochemical performance of air electrode materials in SOCs is typically accessed using single cell configuration featuring air electrode|BaZr0.1Ce0.7Y0.1Yb0.1O3−δ (BZCYYb)|Ni-BZCYYb. In P-SOFC, H2 serves as the fuel on the anode side, while air acts as the oxidant on the cathode side. P-SOEC utilizes steam as the feedstock for H2 production, on the contrary. The evaluation of electrochemical properties involves measuring polarization resistance (Rp) from EIS test, determining peak power densities (PPDs) and current densities from current density–voltage (I–V) curve, and assessing faradaic efficiency (FE) for hydrogen production (Fig. 6(d)). Additionally, air electrode stability tests are performed to monitor any performance degradation. Tests in different atmospheres such as CO2 or steam are also conducted to assess the chemical stability of the electrode materials.160 Perovskite air electrodes in P-SOCs are vulnerable to humidity, which can lead to surface hydroxylation, phase decomposition, and a reduction in oxygen ion conductivity, particularly in Ba-containing perovskites. Similarly, CO2 exposure can cause carbonate formation (BaCO3, SrCO3), which blocks active sites, lowers oxygen vacancy concentration, and degrades ionic transport, especially at moderate to high temperatures (500–800 °C). Currently, to enhance overall catalytic efficiency for practical production, researchers are exploring large-area cells configurations, such as 12 × 12 cm2 planar cells and tubular cells, in addition to traditional button cells.161–163
For protonic conductivity, Shao et al. pioneered measuring the H2 permeation flux through the Pd|Dense Sr2Sc0.1Nb0.1Co1.5Fe0.3O6−δ (SSNCF, air electrode)|Pd membrane.165 Based on this, hydrogen permeation membrane technique was developed. The SSNCF powder materials are directly compressed, followed by sintering in air into dense pellets, H2 and N2 are fed into one side of the chamber, respectively. During the experiments, the hydrogen permeation was detected with gas chromatography (GC) (Fig. 6(e)).85 Time-of-flight secondary ion mass spectrometry (ToF-SIMS) is widely used to quantify proton kinetic properties by analyzing the distribution of deuterium/hydrogen (D/H) isotope concentrations in a quenched air electrode (Fig. 6(f)).152 For instance, Ren et al. combined ToF-SIMS and isotope exchange diffusion profile (IEDP) method to observe stronger distribution of D signal in BaCo0.4Fe0.4Zr0.1Y0.1O2.9−δF0.1 (BCFZYF) sample than undoped.166 However, its implementation can be challenging due to high costs and the limited availability of specialized equipment. Additionally, the presence of a hydroxide layer on the air electrode surface can lead to an uneven distribution of the measured ion signals. Protonic conductivity of P-SOCs system via vehicle mechanism at low-temperature can be evaluated with a “sandwich-type” membrane conductivity test designed in PEM design. Zhou et al. placed a layer of the oxide between two Nafion layers, each in contact with a gas diffusion layer (GDL) electrode that features a standard polymer electrolyte membrane fuel cells Pt/C catalyst-coated membrane under 75 °C.167 Proton conduction in the Nafion membrane occurs through hydronium ions (H3O+), and the conductivity can be calculated using the formula σ (S cm−1) = (h (cm))/(R (ohm) × S (cm2)), where h represents the thickness of the oxide layer, R is the resistance, and S denotes the cross-sectional area through which H3O+ migrate.168–170 The thickness of oxide layer is determined using SEM analysis. This reflects proton conducting via vehicle mechanism, as the Grotthuss mechanism does not involve an H2O vehicle.
Proton conduction relies on the introduction of protons into the crystal lattice via hydration reactions. Therefore, beyond directly measuring the protons conductivity of air electrode materials, it is important to investigate their hydration properties, including hydration amount, enthalpy, and entropy. The proton adsorption state at elevated temperatures can be preserved by quenching from high to room temperature of H2O-temperature-programmed desorption (TPD).167In situ H2O-TPD has been employed to assess the hydration levels of materials like (Ba,Sr,La)(Fe,Co,Zn,Y)O3−δ, (La,Sr)(Co,Mn,Ni)O3, and PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF), in both dry and humidified condition, as a function of temperature and pH2O. The hydration ability can also be accessed with Fourier transform infrared spectroscopy (FTIR), which sensitively detects adsorbed water and OH˙, OH˙ characteristic peak being detected between 3400 cm−1 and 3800 cm−1.171,172 For water uptake with special characteristics such as the absorption of water with proton defect results in a noticeable increase in sample weight, thermogravimetric (TG) analysis is employed to measure the proton defect concentration following water uptake.73
The oxygen conduction abilities of air electrode can be measured by oxygen permeation testing which involves measuring O2 permeation flux detected by GC and ToF-SIMS.85 Additionally, the oxygen adsorption behavior can be analysis via O2-TPD. Chen et al.151 detected the oxygen desorption properties using O2-TPD over a temperature range of 50 °C to 800 °C. As temperature increases, the active cations undergo thermal reduction, leading to a decrease in their oxidation state and the concurrent release of oxygen. The optimized air electrode materials exhibit a lower initial desorption temperature (303.9 °C), suggesting enhanced surface oxygen exchange kinetics and migration rates within the perovskite lattice. Additionally, charge transfer within the oxide bulk, along with the formation of H+ carriers, is often linked to modifications in electronic structure. X-ray photoelectron spectrum (XPS) is a valuable tool for analyzing surface chemical state while X-ray absorption spectroscopy (XAS) provides powerful, element-selective insights into the oxidation states of TMs.173–175 For example, Zhou et al. conducted operando hard XAS to observe changes in both the geometric and electronic structures of the air electrodes.167
Oxygen vacancies are essential in influencing the catalytic activity of air electrodes, as they function both as active sites for electrochemical reactions and as pathways for ionic transport.176 Considering the importance of oxygen vacancies, a series of advanced methods have been employed to detect the existence of oxygen vacancies, concentration, and homogeneity.177 TG analysis and Iodometric titration178 is carried out to determine the oxygen nonstoichiometry (δ), while XPS is conducted to analyze the chemical state of oxygen in air electrode materials. In the study of Liu et al., O 1s XPS was fitted in four peaks: 529 eV, 530.6 eV, 531.5 eV, and 532.8 eV, corresponding to lattice oxygen (Olatt), superoxidative oxygen species (O22−/O−), adsorbed oxygen/hydroxide (O2/OH−), and adsorbed water (H2O), respectively (Fig. 6(g)).179 The oxygen vacancy density is directly related to the amount of O22−/O−. Raman spectroscopy is used to investigate the vibrational modes which results in shifts in the Raman spectrum or appearance of new peak.180,181 Additionally, positron annihilation lifetime spectroscopy (PALS) is carried out to detect oxygen vacancies concentrations based on the positron lifetime.182 And electron paramagnetic resonance (EPR) test can detect peak intensity for oxygen vacancies by comparing the g-factor.183 Soft X-ray adsorption spectroscopy (sXAS) can be used to investigate the electronic structures of the O K-edge, which are highly sensitive to oxygen vacancy concentration, electron concentration and hybridized states.184 Resonant inelastic X-ray scattering (RIXS) can complement XAS measurements by providing detailed information about electron occupancy in d-orbitals and the extent of metal–oxygen hybridization.185 Scanning transmission electron microscopy (STEM) is employed to analyze the structure of nanomaterials, offering direct atomic-level imaging. Mao et al. utilized STEM to examine the atomic scale structure of RuO2 and identified defects on the materials surface.186 The stability of air electrode material is assessed using TG methods, along with comparative analyses of morphology, structure, and composition before and after operation. The thermal expansion coefficient (TEC) of air electrode is confirmed by determining lattice parameters through temperature-dependent XRD testing. Advanced in situ characterization techniques, such as in situ XRD, are employed to monitor the dynamic changes occurring during the operation of the air electrode.187,188
3. BaMO3−δ-based perovskite air electrode
Among the diverse family of perovskite oxides, Ba-based simple perovskite BaMO3−δ (M = Co, Fe) stands out as a particularly attractive option for air electrode in P-SOCs. The larger ionic radii of Ba (1.61 Å) compared to Sr (1.44 Å) and La (1.36 Å), along with its lower electronegativity (Ba: 0.89; Sr: 0.95; Ca: 1.0; La: 1.1; Pr: 1.13, according to Pauling electronegativity), significantly lowers the activation energy required for O2− migration within the crystal lattice.91,189 This characteristic simultaneously enhances the overall basicity of the oxide ions, promoting adequate conductivity for both oxygen ions and protons. Thus, placing Ba at the A site effectively ensures the transport of carriers. Additionally, its abundant reserves and significantly low cost of Ba (∼1029 USD mt−1) compared to La (∼2621 USD mt−1), Sr (∼8830 USD mt−1), Ca (∼3353 USD mt−1), and Pr (∼73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 779 USD mt−1) place it among best candidate for A-site in sustainable industrialization P-SOCs (Prices as of March 2025).91,190–192 Redox-active TMs like Co and Fe are commonly employed at its B-site for adequate electronic conductivity and enhanced catalytic activity. This chapter gives a comprehensive overview of typical BaMO3−δ-type perovskites used as high-performance air electrode materials (Fig. 7, performance summarized in Table 1).
779 USD mt−1) place it among best candidate for A-site in sustainable industrialization P-SOCs (Prices as of March 2025).91,190–192 Redox-active TMs like Co and Fe are commonly employed at its B-site for adequate electronic conductivity and enhanced catalytic activity. This chapter gives a comprehensive overview of typical BaMO3−δ-type perovskites used as high-performance air electrode materials (Fig. 7, performance summarized in Table 1).
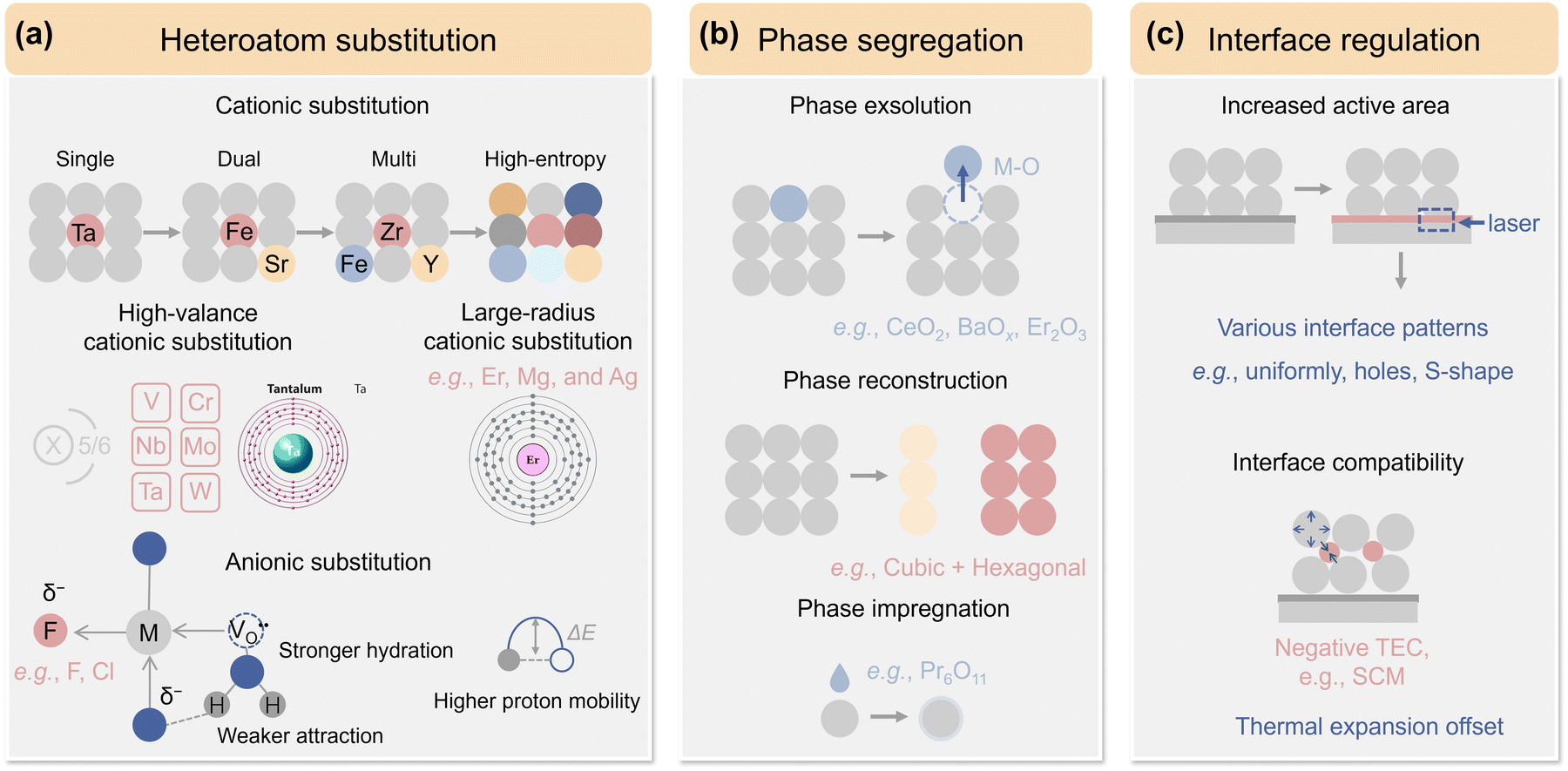 | ||
| Fig. 7 Schematic of recent advances in BaMO3−δ-type air electrode materials, including (a) heteroatom substitution, (b) phase segregation, and (c) interface regulation. | ||
| Air electrode | ASR (Ω cm2) at 3% H2O in symmetrical cell | Single cell | Ref. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 700 °C | 650 °C | 600 °C | 550 °C | 500 °C | 450 °C | Electrolyte thickness (μm) | Temperature (°C) | PPD (mW cm−2) | Current density at 1.3 V (mA cm−2) | Stability (h) | ||
| BCO–PBC–LSCF: BaCoO3−δ–Pr1−xBaxCoO3−δ–(La0.6Sr0.4)0.95Co0.2Fe0.8O3−δ; PBCC–BCO: PrBa0.8Ca0.2Co2O5+δ–BaCoO3−δ; BCO–LSCF: BaCoO3−δ–La0.6Sr0.4Co0.2Fe0.8O3−δ; BCO–BPCFY: BaCoO3−δ–Ba0.9Pr0.1Co0.7Fe0.2Y0.1O3−δ; BCT: BaCo0.8Ta0.2O3−δ; BCFT: BaCo0.7Fe0.2Ta0.1O3−δ; BSTC: BaSc0.1Ta0.1Co0.8O3−δ; BCFZnY: BaCo0.4Fe0.4Zn0.1Y0.1O3−δ; PBCF: Pr0.5Ba0.5Co0.7Fe0.3O3−δ; HE-FMCNC: Fe0.6Mn0.6Co0.6Ni0.6Cr0.6O4; HE-PLNBSCC: Pr1/6La1/6Nd1/6Ba1/6Sr1/6Ca1/6CoO3−δ; HE-PBSLCC: Pr0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ; HE-BCFZSP: BaCo0.2Fe0.2Zr0.2Sn0.2Pr0.2O3−δ; HE-BCZGZrY: BaCo0.2Zn0.2Ga0.2Zr0.2Y0.2O3−δ; BCCY: BaCo0.7(Ce0.8Y0.2)0.3O3−δ; BC1.5MN: Ba2Co1.5Mo0.25Nb0.25O6−δ; HE-CBSLCC: Ce0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ; BSCF–BZCY: Ba0.5Sr0.5Co0.8Fe0.2O3−δ–BaZr0.65Ce0.2Y0.15O3; BSCF–BZCYYb: Ba0.5Sr0.5Co0.8Fe0.2O3−δ–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ; BSCFS: Ba0.5Sr0.5Co0.8Fe0.1Sc0.1O3−δ; LBSCF: La0.1Ba0.4Sr0.5Co0.8Fe0.2O3−δ; BSCFP: Ba0.5Sr0.5(Co0.8Fe0.2)0.95P0.05O3−δ; BSCFF: Ba0.5Sr0.5Co0.8Fe0.2O2.9−δF0.1; BSCFF: Ba0.5Sr0.5Co0.8Fe0.2O2.9−δCl0.1; BSCFE–Er2O3: Ba0.5Sr0.5Co0.72Fe0.18Er0.09O3−δ–Er2O3; C/H-BSCF: Ba0.5Sr0.5Co0.8Fe0.2O3−δ–Ba4Sr4(Co0.8Fe0.2)4O16−δ; N-BCFZYNF: Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05F0.1O2.9−δ; BCFZYN: Ba0.95(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05O3−δ; Ag-BCFZY: Ba0.95Ag0.05Co0.4Fe0.4Zr0.1Y0.1O3−δ; BCFZY: BaCo0.4Fe0.4Zr0.1Y0.1O3−δ; BCF–30Pr: BaCe0.2Fe0.8O3−δ–Pr6O11; BCF36: BaCe0.36Fe0.64O3−δ; S-BCFZY: self-recoverable BaCo0.4Fe0.4Zr0.1Y0.1O3−δ; L-BCFZY: laser-engineering BaCo0.4Fe0.4Zr0.1Y0.1O3−δ; SCM–BCFZY: Sm0.85Cu0.15MnO3−δ–offset BaCo0.4Fe0.4Zr0.1Y0.1O3−δ; BCYF: BaCe0.16Y0.04Fe0.8O3−δ; BFZ: Ba0.875Fe0.875Zr0.125O3−δ; BLFZ0.95: Ba0.95La0.05(Fe0.8Zn0.2)0.95O3−δ; BFCS: BaFe0.6Ce0.2Sc0.2O3−δ; 2Ru–BCF: Ru–anchor BaCe0.125Fe0.875O3−δ; PBSCF: PrBa0.5Sr0.5Co1.5Fe0.5O5+δ; PBCsC: PrBa0.9Cs0.1Co2O5+δ; 16F-PBSCF: PrBa0.5Sr0.5Co1.5Fe0.5O5.84−δF0.16; PBCCHf0.1: PrBa0.8Ca0.2Co1.9Hf0.1O5+δ; PBCFN: PrBaCo1.6Fe0.2Nb0.2O5+δ@PrBaCo1.6Fe0.2Nb0.2−xO5+δ; BZC@PBCZ: BaZrO3@PrBaCo1.92Zr0.08O5+δ; DP-BGCO: BaGdCo2O6−δ; BSCFW@PBSCF: (Ba/Sr)(Co/Fe/W)O3−δ@PrBa0.5Sr0.5Co1.5Fe0.5O5+δ; PCO@PBC: Pr0.1Ce0.9O2+δ@PrBaCo2O5+δ; PFC@PBSCF: Pr0.9Fe0.7Co0.3O3@PrBa0.5Sr0.5Co1.5Fe0.5O5+δ; PCNCFO: Pr0.2Ce0.2Ni0.2Co0.2Fe0.2Ox; PLD-PBSCF: pulsed laser deposition-PrBa0.5Sr0.5Co1.5Fe0.5O5+δ; PBCHf10: PrBaCo1.9Hf0.1O5+δ; PNBFZ: Pr0.8Nd0.2BaFe1.9Zn0.1O5+δ; LSN: La1.2Sr0.8NiO4+δ; PNCu: Pr2Ni0.8Cu0.2O4+δ; NSTF: NaySrzTiuFe1−uO3−δ; NSTF0.3@SC: Na0.3Sr0.7Ti0.1Fe0.9O3−δ@SrCoO3−δ; SCN–PSCN: SrCo0.5Nb0.5O3−δ@PrSrCo1.8Nb0.2O6−δ; SCFN: Sr0.9Ce0.1Fe0.8Ni0.2O3−δ; PNC: PrNi0.5Co0.5O3−δ; PNC–LSCF: Pr2Ni0.5Co0.5O4−δ–La0.6Sr0.4Co0.2Fe0.8O3−δ. | ||||||||||||
| BaCoO3−δ-based air electrode in P-SOCs | ||||||||||||
| BCO–PBC–LSCF | 0.06 | 0.15 | 0.25 | 0.45 | 0.95 | 10 | 550 | 620 | 850 | 300 h (600 °C at −1 A cm−2) | 193 | |
| PBCC–BCO | 10 | 550 | 660 | 690 | 1500 h (600 °C at −1 A cm−2) | 194 | ||||||
| BCO–LSCF | 0.04 | 0.07 | 0.16 | 0.35 | 0.84 | 10 | 500 | 410 | 500 | 1150 h (550 °C at −1 A cm−2) | 195 | |
| BCO–BPCFY | 70 | 700 | 750 | 85 h (700 °C at 0.7 V) | 196 | |||||||
| Ba0.95CoO3 | 15 | 700 | 1155 | 197 | ||||||||
| BCT | 0.12 | 0.23 | 0.39 | 0.65 | 1.2 | 10 | 450 | 450 | 250 | 357 h (550 °C at −1 A cm−2) | 132 | |
| BCFT | 24 | 700 | 1272 | 120 h (700 °C at 0.84 V) | 198 | |||||||
| BSTC | 0.08 | 0.15 | 0.27 | 0.5 | 1.3 | 6.5 | 199 | |||||
| BCFZnY450 | 11.8 | 450 | 320 | 200 | ||||||||
| PBCF | 6 | 600 | 1120 | 1790 | 100 h (600 °C at 0.7 V) | 201 | ||||||
| HE-FMCNC | 0.057 | 0.03 | 10 | 600 | 453 | 120 h (600 °C at 0.2 A cm−2) | 202 | |||||
| HE-PLNBSCC | 6.5 | 500 | 660 | 1100 | 200 h (500 °C at −0.5 A cm−2, >10% H2O) | 203 | ||||||
| HE-PBSLCC | 0.06 | 0.12 | 0.26 | 0.75 | 2.13 | 10 | 500 | 400 | 280 | 500 h (600 °C at −0.5 A cm−2) | 204 | |
| HE-BCFZSP | 0.15 | 8 | 600 | 680 | 920 | 120 h (600 °C at −1.3 A cm−2) | 205 | |||||
| HE-BCZGZrY | 6.5 | 600 | 287 | 100 h (600 °C) | 206 | |||||||
| BCCY | 0.051 | 0.11 | 0.20 | 0.50 | 1.49 | 16.1 | 450 | 187 | 800 h (550 °C) | 207 | ||
| BC1.5MN | 0.07 | 0.13 | 0.24 | 0.4 | 1 | 3 | 10 | 550 | 540 | 680 | 1100 h (550 °C at 0.4 A cm−2) | 208 |
| HE-CBSLCC | 0.039 | 0.088 | 0.23 | 0.6 | 1.65 | 6 | 550 | 1140 | 840 | 200 h (600 °C) | 209 | |
| BaCoO3−δ derivatives in P-SOCs | ||||||||||||
| BSCF–BZCY | 0.16 | 700 | 418 | 210 | ||||||||
| BSCFS600 | 15 | 600 | 566 | 100 h (600 °C at 0.2 A cm−2) | 211 | |||||||
| LBSCF | 0.04 | 0.42 | 20 | 600 | 350 | 870 | 211 | |||||
| BSCFP | 20 | 500 | 425 | 401 | 240 h (550 °C) | 212 | ||||||
| BSCFF | 0.06 | 0.12 | 0.25 | 0.54 | 1.41 | 16 | 500 | 302 | 190 | 70 h (550 °C at −0.295 A cm−2) | 213 | |
| BSCFC | 0.10 | 0.23 | 0.53 | 1.37 | 4.37 | 16 | 650 | 770 | 485 | 213 | ||
| BSCFE–Er2O3 | 7 | 500 | 525 | 564 | 300 h (550 °C at 0.2 A cm−2) | 179 | ||||||
| C/H-BSCF | 0.11 | 0.26 | 0.68 | 2.13 | 8 | 500 | 1000 | 1240 | 500 h (600 °C at −2 A cm−2) | 214 | ||
| N-BCFZYNF | 0.076 | 0.165 | 0.407 | 1.338 | 4.783 | 11 | 500 | 374 | 345 | 80 h (550 °C at −0.5 A cm−2) | 151 | |
| BCFZYN | 20 | 550 | 540 | 400 h (550 °C at 0.2 A cm−2) | 215 | |||||||
| Ag-BCFZY | 0.06 | 0.107 | 0.242 | 0.5 | 10 | 500 | 398 | 600 h (500 °C at 0.75 V) | 216 | |||
| KBCFZY | 0.43 | 0.75 | 7 | 550 | 770 | 600 h (600 °C at −4 A cm−2 and 0.5 A cm−2) | 217 | |||||
| BCFZY | 0.71 | 20 | 500 | 131 | 218 | |||||||
| S-BCFZY | 90 | 600 | 350 | 60 h (600 °C at 0.45 A cm−2) | 218 | |||||||
| L-BCFZY | 0.1 | 0.17 | 0.28 | 20 | 550 | 900 | 100 h (600 °C at 0.2 A cm−2) | 219 | ||||
| SCM–BCFZY | 0.35 | 14 | 600 | 655 | 220 | |||||||
| BaFeO3−δ-based air electrode in P-SOCs | ||||||||||||
| BCF–30Pr | 0.039 | 16 | 700 | 1406 | 100 h (700 °C at 0.3 A cm−2) | 221 | ||||||
| BCF36 | 20 | 600 | 550 | 100 h (600 °C at 0.6 V) | 222 | |||||||
| BCYF | 15.8 | 450 | 151 | 450 h (550 °C at 0.2 A cm−2) | 160 | |||||||
| BFZ | 0.06 | 0.25 | 0.5 | 1.2 | 3 | 6 | 500 | 670 | 230 h (600 °C at 1.2 A cm−2) | 34 | ||
| BLFZ0.95 | 0.04 | 0.07 | 0.1 | 10 | 600 | 620 | 250 h (600 °C at 0.9 V) | 223 | ||||
| BFCS | 8 | 550 | 330 | 610 | 120 h (600 °C at 0.8 V) | 224 | ||||||
| 2Ru–BCF | 0.033 | 0.2 | 0.48 | 1 | 7 | 700 | 1780 | 200 h (55 °C at 0.7 V) | 225 | |||
| LnBaM2O5+δ-based air electrode in P-SOCs | ||||||||||||
| PBSCF | 15.7 | 500 | 420 | 200 h (600 °C at 0.6 V) | 152 | |||||||
| PBCsC | 0.162 | 0.275 | 0.531 | 1.526 | 8 | 500 | 410 | 310 | 200 h (650 °C at 0.5 A cm−2) | 226 | ||
| 16F-PBSCF | 0.2 | 0.49 | 1.2 | 8 | 550 | 300 | 227 | |||||
| PBCIn | 0.06 | 0.13 | 0.24 | 0.52 | 1.47 | 10 | 500 | 480 | 310 | 200 h (re P-SOC) | 228 | |
| PBCCHf0.1 | 0.068 | 0.107 | 0.269 | 0.546 | 7 | 550 | 673 | 836 | 200 h (600 °C at ±0.5 A cm−2) | 229 | ||
| PBCFN | 0.08 | 0.24 | 0.8 | 10 | 600 | 723 | 1036 | 200 h (650 °C at ±0.5 A cm−2) | 230 | |||
| BZC@PBCZ | 12 | 600 | 551 | 140 h (600 °C at 0.55 A cm−2) | 231 | |||||||
| DP–BGCO | 0.13 | 0.22 | 0.4 | 0.1 | 20 | 550 | 386.4 | 600 h (600 °C at 0.5 V) | 232 | |||
| BSCFW@PBSCF | 0.24 | 0.36 | 0.80 | 10 | 550 | 570 | 240 h (650 °C at 0.7 V) | 150 | ||||
| PCO@PBC | 0.06 | 0.1 | 0.3 | 10 | 600 | 870 | 2090 | 100 h (650 °C at ±0.5 A cm−2) | 233 | |||
| PFC@PBSCF | 0.25 | 0.81 | 1.88 | 10 | 550 | 640 | 100 h (650 °C at 0.5 A cm−2) | 234 | ||||
| PCNCFO | 0.04 | 0.09 | 0.18 | 10 | 600 | 910 | 100 h (650 °C at 0.5 A cm−2) | 235 | ||||
| PLD–PBSCF | 7 | 350 | 500 | 250 | 300 h (500 °C at 1 A cm−2 and −1 A cm−2) | 236 | ||||||
| PBCHf10 | 0.187 | 0.374 | 0.933 | 7 | 450 | 600 | 400 | 500 h (600 °C at −0.5 A cm−2) | 237 | |||
| PNBFZ | 16 | 550 | 401 | 24 h (600 °C at 0.7 V) | 238 | |||||||
| Others | ||||||||||||
| PNCu | 26 | 650 | 280 | 239 | ||||||||
| NSTF | 20 | 600 | 807 | 167 | ||||||||
| NSTF0.3@SC | 22.5 | 600 | 960 | 167 | ||||||||
| SCN–PSCN | 8 | 550 | 600 | 750 | 120 h (600 °C at 0.85 V) | 240 | ||||||
| SCFN | 0.2 | 0.57 | 2.05 | 26 | 500 | 225 | 179 | 290 h (550 °C at 0.8 V) | 171 | |||
| PNC | 16 | 450 | 640 | 1600 | 200 h (600 °C at 0.75 V) | 241 | ||||||
| PNC–LSCF | 30 | 550 | 662 | 200 h (700 °C at 0.8 A cm−2) | 242 | |||||||
3.1. BaCoO3−δ-based series
Co, as a B-site cation, is particularly effective for reactions involving electron exchange, such as OER and ORR. This is due to the strong hybridization between the metal 3d and O 2p orbitals,32,243 as well as the close values of their intra-atomic exchange energy and crystal electric field splitting energy, which leads to small energy differences between their spin states and oxidation states.244–247 The unique properties combined with the aforementioned of Ba collectively make BaCoO3−δ (BCO) an exceptional choice for P-SOCs.193,194,248However, BCO's hexagonal structure of BCO restricts charge carrier mobility within the lattice,249–251 which limit its effectiveness as an independent air electrode.132,252 It is, consequently, utilized as a reinforcement material, functioning as a modified layer to enhance catalytic activities as well as air electrode durability as in reported in situ studies using PrBa0.8Ca0.2Co2O5+δ, Ba0.9Pr0.1Co0.7Fe0.2Y0.1O3−δ, and La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF).193–196 The BCO coatings additionally mitigate toxic effects of Cr from interconnectors owing to their ability to reduce Cr accumulation on the electrode surface.253 Recent research efforts have increasingly focused on the bulk structural regulation of BCO for its ultimate autonomy. Atomic substitution techniques have been widely employed to modify their air electrodes phase structure, evolving from single-element substitutions (like Pr, La, Nb, Zn, Sc, etc.)254–262 to double and then multi-element modifications (Fig. 7(a)).197,259,263,264 Notably, heteroatomic doping with high-valence cations like Nb5+, Ta5+, or Mo6+ at the B-site has proven particularly effective. For instance, Kim et al. developed cubic BaCo0.8Ta0.2O3−δ (BCT) by incorporating Ta5+, which has highly corrosion-resistance, stable valence state, and a lower electronegativity (Ta: 1.5; Nb: 1.6; Mo: 2.16).132 The inclusion of Ta5+ stabilized the high-symmetry cubic perovskite structure, significantly enhancing phase stability, conductivity, and catalytic activity for both OER and ORR (Fig. 8(a)). Thus, BCT air electrode achieved remarkable PPD of 2.26 W cm−2 at 650 °C and electrolysis current density of 1.1 A cm−2 at 1.3 V and 550 °C in P-SOC. Combining two dopants on BCO-type electrodes, like Fe–Ta,198,264,265 Sm–Zr,263 and Co–Zr,266 creates synergistic effects that enhance electrical, ionic, and catalytic properties to levels superior to single-element doping. Inspired by dual-doping strategy observed in SrCoO3 for O-SOCs,267 Kim et al. developed Sc–Ta co-doped BaSc0.1Ta0.1Co0.8O3−δ (BSTC), which stabilizes the cubic perovskite structure.199 Dual-doping strategy can lead to reduced TEC, improved water uptake, hydration properties, proton transport capabilities, and higher resistance to CO2 (Fig. 8(b)). The BSTC air electrodes in P-SOCs achieved PPD of 3.15 W cm−2 and 2.25 W cm−2 at 650 °C and 600 °C in FC mode, and a current density of 4.21 A cm−2 at 1.3 V and 650 °C in EC mode. Additionally, Sr–Fe co-doped Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) inspired from O-SOCs has seen widespread practical implementation in P-SOCs, which is discussed in detail in Section 3.2. Multi-element doped materials,200,201 represented by BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) inspired by electrolyte materials, demonstrate exceptional electrochemical potential and serve as the foundation for numerous research studies. A detailed exploration of BCFZY is presented in Section 3.2. In the design of multi-element substitution strategy, high-entropy design has emerged as a significant topic.202,268–270 High-entropy perovskite oxides (HEPOs) that contain five or more elements in nearly equimolar ratios exhibit unique properties due to complexity of their localized environments and expected broad adjustability afforded by the “cocktail” effect.271 Unlike traditional enthalpy-driven phase separation during multi-doping processes,272,273 HEPOs are stabilized by configurational entropy. In addition, they display a range of unique properties that differ from those of individual oxides including unique crystal structures, enhanced thermal and electronic characteristics, superior catalytic activity, improved electrochemical performance, efficient ionic transport, and distinct thermal expansion behaviors.203,274–278 He et al. developed A-site HEPO Pr0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (HE-PBSLCC), that effectively combined high conductivity and low TEC of Pr, larger ionic radius of La3+, and chemical stability of Ca2+. This composition led to reduced TEC (23.8 × 10−6 K−1) and effective phase segregation suppress.187 HE-PBSLCC achieved a PPD of 1.51 W cm−2 and a current density of 2.68 A cm−2 at 1.3 V at 650 °C, while operational lifetimes exceeding 270-h FC and 500-h EC opeartion.187 Sun et al. constructed B-site HEPO BaCo0.2Fe0.2Zr0.2Sn0.2Pr0.2O3−δ, demonstrating integrated triple-phase conduction, along with exceptional structural stability in high concentration steam.205,206 In addition to bulk structure modification, atomic substitution can also facilitate the formation of multiple phases within a single bulk grain which is called surface segregation or exsolution. Improper t of certain elements in perovskites can lead to phase separation, resulting in the formation of two or more distinct phases with varying elemental compositions. The self-assembly strategy for creating hybrid electrodes ensures a more homogeneous phases distribution and improved phase contact compared to straightforward physical mixing, thereby reducing energy barriers for charges conduction.207 Song et al. introduced a novel self-assembled BaCo0.7(Ce0.8Y0.2)0.3O3−δ electrode for P-SOFC, incorporating Ce and Y cations into the B-site of the BCO base.207 This material undergoes self-assembly into nanocomposite consisting of mixed H+/e− phase, mixed O2−/e− phase, and the BCO phase during calcination. Enhanced electrochemical activity and ionic transport were achieved due to the synergistic effects of the distinct phases. The composite electrode Ba2Co1.5Mo0.25Nb0.25O6−δ (BC1.5MN), as reported by He et al., undergoes elegant decomposition into BCO and double-perovskite Ba2−xCo1.5−xMo0.5Nb0.5O6−δ (DP-BCMN).208 This transformation enables the electrode to achieve a PPD of 1.17 W cm−2 and a current density of 2.04 A cm−2 at 1.3 V and 650 °C. A-Site HEPO Ce0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (HE-CBSLCC), developed by He et al.,209 self-assembles into a three-phase heterostructure under specific processing conditions. This structure includes a deficient Ce0.2−yBa0.2Sr0.2−xLa0.2−xCa0.2CoO3−δ (CD-CBSLCC), along with CeO2, and La0.5Sr0.5CoO3−δ (LSC) nano-catalysts. Oxygen reduction occurs across the entire air electrode surface, with water formation occurring predominantly at or near CD-CBSLCC. CeO2 phase can either donate or accept H+, thereby facilitating the ORR and OER kinetics in reversible P-SOCs. The electrode achieves a PPD of 1.66 W cm−2 and a current density of 1.76 A cm−2 at 1.3 V and 600 °C. They also exhibit excellent operational stability, with 200 h in FC mode, 200 h in EC mode, and 548 h in reversible cycling at 550 °C.
 | ||
| Fig. 8 Recent advances in BaMO3−δ-type air electrode materials. (a) XRD patterns of BCT. Reproduced with permission.132 Copyright 2023, Royal Society of Chemistry. (b) Tolerance factors of bimetal-doped BMM’C. Reproduced with permission.199 Copyright 2024, Wiley-VCH. (c) O-site substitution with F/Cl of BSCFF. Reproduced with permission.213 Copyright 2023, Elsevier. (d) Schematic of phase content-controlled hybrid electrode. Reproduced with permission.214 Copyright 2024, Springer Nature. (e) Reaction mechanism on the surface of ABCFZY electrode in humidified air. Reproduced with permission. Copyright 2024, Royal Society of Chemistry.216 (f) Schematic of the laser ablating process and SEM micrographs of holes and S-shape. Reproduced with permission.219 Copyright 2024, American Chemical Society. (g) Mechanism of the SCM limited reactive bonding “glue”. Reproduced with permission.220 Copyright 2024, Wiley-VCH. | ||
3.2. Eye-catching BaCoO3−δ derivatives
Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) is the center of attention as air electrode material for intermediate temperature O-SOCs.279,280 In 2004, BSCF made its debut as a cathode for O-SOFCs, achieving remarkable PPDs of 1.01 W cm−2 at 600 °C and 402 mW cm−2 at 500 °C.281,282 With mixed conductivity of oxygen ions and a reported proton exchange k value of 3.85 × 10−6 cm s−1 at 500 °C in 0.21 atm pO2, the cubic perovskite BSCF has been identified as air electrode candidate for P-SOCs, too.158,281 To extend three-phase boundaries and minimize the TEC mismatch between the electrode and electrolyte, BSCF has been composited with various electrolyte materials including BaCe0.7Zr0.1Y0.2O3−δ,283 BaCe0.9Y0.1O3,284 BaCe0.8Zr0.1Y0.1O3−δ,285 BaCe0.4Zr0.4Y0.2O3−δ,286 BaZr0.65Ce0.20Y0.15O3,210 and BZCYYb.287 However, this kind of composite design may compromise the conductivity of air electrode.Due to the differences in the working principles of P-SOCs and O-SOCs—specifically, that in O-SOCs, water vapor is generated and consumed on the fuel electrode side288,289—the electrode materials for the two types of cells are not fully interchangeable.290 BSCF as P-SOCs air electrode, for example, degrades in high-concentration steam environments.291 To tackle this problem, atomic substitution (like Sc, La, P) has been attempted to BSCF to stabilize its structure.211,212,292 Besides cation regulation, anion (O-site) substitution has recently emerged as an effective method for tailoring perovskite air electrode (Fig. 7(a)).293–295 Anion doping involves substituting lattice oxygen or occupying oxygen vacancies with anions, typically at a stoichiometric ratio of 0.1 to 0.2, thereby modifying the basicity, valence balance, electronegativity, and band structure. To date, halogens (F− and Cl−) have been used as anionic dopants.293,296–298 Halogen doping can reduce the basicity of perovskite oxides without compromising their proton conductivity, thus enhancing their chemical stability against acidic gases like CO2.293,294,299 Halogen doping can alleviate F− diffusion from the electrolyte.300 By introducing halogen, the oxygen vacancy concentration is decreased to maintain electron neutrality, with halogen potentially occupying these vacancies, further lowering vacancy concentration.22,293,297 The fluorinated Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCFF) air electrode, for instance at 650 °C, achieved a PPD of 977 mW cm−2 and a current density of 950 mA cm−2 at 1.3 V.213 Chen et al. further compared the effects of F− and Cl− (Fig. 8(c)), noting that although both have similar valence states, they differ significantly in electronegativity (χF: 3.98 > χO: 3.44 > χCl: 3.16) and ionic radii (F−: 1.33 Å < O2−: 1.40 Å < Cl−: 1.81 Å).213 The higher electronegativity of F− enhances its electron-attracting ability, thereby increasing the polarity of the M–O bonds. In P-SOCs, the fluorinated BSCFF air electrode outperformed chlorine Ba0.5Sr0.5Co0.8Fe0.2O2.9−δCl0.1 (BSCFC) air electrode in terms of both Dchem and kchem. The fluorinated BSCFF also exhibited superior electrochemical performance in both FC (977 mW cm−2vs. 770 mW cm−2 for chlorine BSCFC at 650 °C) and EC (950 mA cm−2vs. 485 mA cm−2 for chlorine BSCFC at 1.3 V and 650 °C).
Recent research indicates that exceeding material tolerance limits through atomic substitution can form self-assembled multi-phase nanocomposites within a single grain. By carefully controlling concentration of high-valence cations (like Mo6+ and W6+) or large-radius cations (like Er, Mg, and Ag), it is possible to customize electrical conductivity, catalytic activity, and structural stability within composite air electrode (Fig. 7(a)).301,302 Liu et al., for instance, introduced Er into BSCF, which lead to the formation of a nanocomposite featuring predominant cubic perovskite phase (Ba0.5Sr0.5Co0.72Fe0.18Er0.09O3−δ), along with a minor Er2O3 component.179 The Er2O3 phase acts as a rapid H+ transport channel, thereby facilitating kinetics of both ORR and OER. While the self-assembly strategy effectively creates strong interactions between hybrid phases, the dynamically changing composition and inconsistent elemental composition render concerns about durability. Liu et al. efficiently controlled the phase contents of hybrid material by adjusting the stoichiometric ratio of A-site and B-site elements, while ensuring the consistency of elemental composition (Fig. 8(d)). The resulting Ba1.5Sr1.5Co1.6Fe0.4O7−δ consisted of 57.26 wt.% cubic Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) and 42.74 wt.% hexagonal Ba4Sr4(Co0.8Fe0.2)4O16−δ (H-BSCF).214 This hybrid perovskite integrated the superior oxygen activation and conductivity of cubic phase and strong hydration reaction and abundant 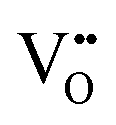 of hexagonal phase. The strengthened interaction between the two phases enhanced the structural and chemical stability.
of hexagonal phase. The strengthened interaction between the two phases enhanced the structural and chemical stability.
Since 2013, BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) has garnered significant attention from researchers, particularly after O’Hayre's group proposed its application in P-SOFC.103,266 Numerous studies have since been published exploring the chemistry and electrochemistry of BCFZY based on typical compositing (BCFZY–BaZr0.5Y0.5O3 and BCFZY–NiO)303,304 and substitution (Zn,305 Mg,200,306 Sc,307 Y3+,308 F,166,213 and A-site-deficient) strategies. Recent investigations on BCFZY increasingly advocate for a dual-functional strategy that combines both bulk and interfacial modifications instead of focusing in one aspect.195,215,309–311 Chen et al. involved Ni, F co-doped Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05F0.1O2.9−δ (N-BCFZYNE), resulting in precipitation of NiO as a surface catalyst.151 The higher electronegativity of F− compared to O2− enhances the polarity of  bonds, facilitating H2O adsorption on
bonds, facilitating H2O adsorption on  and leading to the formation of additional protonic defects. The F− introduction decreases the negative charge on lattice oxygen, which in turn weakens the O⋯H interaction, thereby lowering the barrier for proton diffusion. The in situ generated metal oxide catalyst demonstrated enhanced surface reaction activity. This approach achieved improvement both internally (via highly reactive metal–oxygen bonds) and externally (via enabled nanoscale catalysis). Secondary phase nanoparticles (NPs) exsolution can occur under various conditions including water vapor, reducing/oxidizing atmospheres, high temperatures, and electric fields.215,312 Kim et al. designed steam-induced Ag metal nanocatalysts that functions solely as an electronic conductor, without engaging in proton uptake and diffusion.216 Building on this, through steam-introduced strategy, Park et al. introduced exsolved BaOx species in alkali metal ions (Li+, Na+, K+)-doped BCFZY, which served as active sites for ORR/OER (Fig. 8(e)).217 The driving force for Ba dissolution arises from lattice strain caused by differences in ionic radius and defect interactions, while BaOx is thermodynamically favored due to its negative Gibbs free energy. In addition to water-induced exsolution, treating Ba0.95Ag0.5Co0.8Nb0.1Ta0.1O3−δ in a reductive atmosphere313 and employing voltage-driven exsolution in Ln0.2Ba0.8Co0.7Fe0.3O3−δ314,315 were also effective in generating NPs catalyst on the surfaces.
and leading to the formation of additional protonic defects. The F− introduction decreases the negative charge on lattice oxygen, which in turn weakens the O⋯H interaction, thereby lowering the barrier for proton diffusion. The in situ generated metal oxide catalyst demonstrated enhanced surface reaction activity. This approach achieved improvement both internally (via highly reactive metal–oxygen bonds) and externally (via enabled nanoscale catalysis). Secondary phase nanoparticles (NPs) exsolution can occur under various conditions including water vapor, reducing/oxidizing atmospheres, high temperatures, and electric fields.215,312 Kim et al. designed steam-induced Ag metal nanocatalysts that functions solely as an electronic conductor, without engaging in proton uptake and diffusion.216 Building on this, through steam-introduced strategy, Park et al. introduced exsolved BaOx species in alkali metal ions (Li+, Na+, K+)-doped BCFZY, which served as active sites for ORR/OER (Fig. 8(e)).217 The driving force for Ba dissolution arises from lattice strain caused by differences in ionic radius and defect interactions, while BaOx is thermodynamically favored due to its negative Gibbs free energy. In addition to water-induced exsolution, treating Ba0.95Ag0.5Co0.8Nb0.1Ta0.1O3−δ in a reductive atmosphere313 and employing voltage-driven exsolution in Ln0.2Ba0.8Co0.7Fe0.3O3−δ314,315 were also effective in generating NPs catalyst on the surfaces.
Exsolved NPs like Co, Fe, and Ni may slowly dissolve back into the perovskite lattice at elevated temperatures and oxidizing environment.316,317 Inspired by this, Wang et al. developed a self-recoverable BCFZY (S-BCFZY) electrode for P-SOFCs, featuring a reversible exsolution/dissolution mechanism based on the incorporation of NPs.218 Under cathodic conditions, the weak Co–O and Fe–O bonds effectively lower the energy barrier for oxygen release, promoting the generation of  and H+.218 In anodic conditions, conversely, these weakened TM–O bonds facilitate the segregation of TM atoms (Co0 and Fe0) to the perovskite surface, leading to NPs formation and enhanced electrocatalytic activity.318,319 This multifunctional modification, spanning from the bulk to the surface, positively influences the catalytic process. However, significant differences in the elemental composition of various phases can create thermal strains that are difficult to mitigate and can impair both catalytic activity and stability. Additionally, elemental migration may lead to confusion regarding the actual composition of the hybrid electrode, particularly under high-temperature operational conditions. The exsolution process may inadvertently induce structural modifications in the parent oxide, potentially resulting in the development of RP phases or unwanted cation precipitation.320 Therefore, the long-term effectiveness of this approach must be thoroughly assessed.
and H+.218 In anodic conditions, conversely, these weakened TM–O bonds facilitate the segregation of TM atoms (Co0 and Fe0) to the perovskite surface, leading to NPs formation and enhanced electrocatalytic activity.318,319 This multifunctional modification, spanning from the bulk to the surface, positively influences the catalytic process. However, significant differences in the elemental composition of various phases can create thermal strains that are difficult to mitigate and can impair both catalytic activity and stability. Additionally, elemental migration may lead to confusion regarding the actual composition of the hybrid electrode, particularly under high-temperature operational conditions. The exsolution process may inadvertently induce structural modifications in the parent oxide, potentially resulting in the development of RP phases or unwanted cation precipitation.320 Therefore, the long-term effectiveness of this approach must be thoroughly assessed.
Modifying air electrodes involves not only the electrode materials themselves but also the electrode/electrolyte interface (Fig. 7(c)).321,322 The blunt surface of perovskite-type proton-conducting electrolytes contributes to interfacial resistances, leading to significant discrepancies in the ASR compared to theoretical values.323,324 To alleviate interface problem, Zhou et al. developed a novel picosecond laser ablation technique that precisely modifies the electrolyte surface (Fig. 8(f)).219 This method enables uniform removal of the electrolyte's top layer and creates various patterns, such as cross, S-shape, and hole designs, which increase the effective surface area and establish strong bonding anchors between the BCFZY electrode and BZCYYb electrolyte (Fig. 8(f)). Another interface-controlled approach is to improve the compatibility between electrode materials and electrolytes, especially thermomechanical compatibility.170,325 Yu et al. employed negative thermal expansion oxide Sm0.85Cu0.15MnO3−δ (SCM with TEC of −5.93 × 10−6 K−1) to offset the thermal expansion of BCFZY.220 SCM can also strengthen electrode structure and adhesion, and provide acceptable oxygen–reduction–reaction activity (Fig. 8(g)). Thus, this strategy achieves a PPD of 1.455 W cm−2 at 700 °C.
3.3. BaFeO3−δ-based perovskite
Given the significant differences in TEC between cobalt-based electrodes and electrolytes (e.g., BSCF: 24.7 × 10−6 K−1; BCFZY: 22.9 × 10−6 K−1; BZCYYb: 10.8 × 10−6 K−1; BZCY: 9.3 × 10−6 K−1),199,207,326 along with the easy evaporation of Co at elevated temperatures and economic considerations (Co: 66.5 USD kg−1; Fe: 6.7 USD kg−1),190 Fe emerges as a viable alternative.160,309,327–329 Fe shares comparable reducibility with Co and, unlike Co-based perovskites, Fe-based perovskites are more easily hydrated and demonstrate improved chemical and thermal stability. The TEC of Fe-based air electrode materials shows enhanced compatibility with proton-conducting electrolytes.330 Among all Fe-based perovskite, BaFeO3 (BFO) exhibit superior H+ conductivity compared to placing Sr or La at A-site.32 However, undoped BFO crystallizes in a hexagonal system with low symmetry, poor oxygen ion conductivity, limited tolerance to CO2 and H2O, thereby hindering its effectiveness as a high-performance air electrode.331–333 To mitigate these challenges, doping (e.g., La, Zn, Sm, Cl, and F) and compositing (e.g., Sm0.2Ce0.8O2−δ–Ba0.5Sr0.5Fe0.8Sb0.2O3−δ) strategies are frequently employed.332,334–343 Recently, Lu et al. combined the two modifications to develop a BaCe0.2Fe0.8O3−δ–Pr6O11 (BCF–30Pr) composite cathode for P-SOFC through impregnation technique.221 The incorporation of Pr6O11 increased the specific surface area of electrode, reduced the Fe3+ content, and enhanced the concentration of on the BCF surface. These changes improved both the ORR activity and H+ conduction, leading to a PPD of 1406 mW cm−2 at 700 °C.
on the BCF surface. These changes improved both the ORR activity and H+ conduction, leading to a PPD of 1406 mW cm−2 at 700 °C.
Inspired by the design of BCFZY, Ce, Zr and Y are also favored dopants in BFO phase.223,344–346 The Ce-doped BaCe0.36Fe0.64O3−δ (BCF36) developed by Tong et al. features a cubic BFO phase with high O2−/e− conductivity and an orthorhombic BaCeO3 phase with H+ transfer, thus achieving a PPD of 1525 mW cm−2 at 700 °C.222 By tailoring the Ce/Y ratio in bimetal-doped Ba(Ce0.8Y0.2)xFe1−xO3−δ (BCYF), a O2−/e− conducting single-phase transforms into a triple-conducting multi-phase composite.347 The optimized BCYF nanocomposite displayed improved e− conductivity, ORR activity, thermo-mechanical compatibility, and CO2 tolerance, owing to the strong interaction between O2−/e− and H+/e− conducting phases, as well as the optimized dual-phase composition. The BaCe0.16Y0.04Fe0.8O3−δ cathode demonstrates 450 h durable operation at 550 °C for P-SOFC. Based on BFO, Wang et al. combined A- and B-site co-substitution to construct Ba0.875Fe0.875Zr0.125O3−δ (BFZ) with the formation of  and
and  while retaining structural, which can catch up power outputs of state-of-the-art Co-based BCFZY with a PPD of 0.67 W cm−2 at 500 °C and operational stability of 230 h at 600 °C.22,34 In the context of BFO-based perovskites used in reversible P-SOCs, Wang et al. developed BaFe0.6Ce0.2Sc0.2O3−δ (BFCS) perovskite characterized by optimized Fe 3d-eg orbital occupancy, a higher concentration of
while retaining structural, which can catch up power outputs of state-of-the-art Co-based BCFZY with a PPD of 0.67 W cm−2 at 500 °C and operational stability of 230 h at 600 °C.22,34 In the context of BFO-based perovskites used in reversible P-SOCs, Wang et al. developed BaFe0.6Ce0.2Sc0.2O3−δ (BFCS) perovskite characterized by optimized Fe 3d-eg orbital occupancy, a higher concentration of  , and enhanced Fe4+–O2− interactions which promotes the activation and mobility of lattice oxygen.224 The BFCS air electrode for P-SOC achieves encouraging output performance in both EC (1.55 W cm−2) and FC (−2.96 A cm−2 at 1.3 V) modes at 700 °C. Single-atom catalysis, an emerging technology, has recently been utilized in Fe-based air electrodes. Zhao et al. developed a novel air electrode (2Ru-BCF) by integrating single-atom Ru rivets onto BaCe0.125Fe0.875O3−δ (BCF).225 The Ru atoms in this structure adopt a distinctive 4-coordinate Ru–O–Fe configuration, which not only promotes reverse hydrogen spillover but also functions as an active site for P-ORR. This optimized 2Ru–BCF cathode (2 wt% Ru) delivers a remarkable PPD of 1.78 W cm−2 at 700 °C and demonstrates stable operation over 200 h.
, and enhanced Fe4+–O2− interactions which promotes the activation and mobility of lattice oxygen.224 The BFCS air electrode for P-SOC achieves encouraging output performance in both EC (1.55 W cm−2) and FC (−2.96 A cm−2 at 1.3 V) modes at 700 °C. Single-atom catalysis, an emerging technology, has recently been utilized in Fe-based air electrodes. Zhao et al. developed a novel air electrode (2Ru-BCF) by integrating single-atom Ru rivets onto BaCe0.125Fe0.875O3−δ (BCF).225 The Ru atoms in this structure adopt a distinctive 4-coordinate Ru–O–Fe configuration, which not only promotes reverse hydrogen spillover but also functions as an active site for P-ORR. This optimized 2Ru–BCF cathode (2 wt% Ru) delivers a remarkable PPD of 1.78 W cm−2 at 700 °C and demonstrates stable operation over 200 h.
4. LnBaM2O5+δ-based perovskite
LnBaM2O5+δ-based double perovskites feature A-sites occupied by 4f lanthanide and alkaline earth cation Ba. The B-sites host abundant TM cations, including Mn, Fe, Co, and Ni. These materials exhibit enhanced electrochemical performance due to their higher Dchem and kchem (performance summarized in Table 1).348,349 The atomic level of the separation of cations with an alternative layer structure happens due to the large difference in the A or B sites for the two different cations with equal amounts. This chapter offers an in-depth review of typical LnBaM2O5+δ-type perovskites employed as high-performance materials for air electrodes (summarized as Fig. 9). | ||
| Fig. 9 Schematic of recent advances in LnBaM2O5+δ-type air electrode materials, including (a) heteroatom substitution, (b) phase segregation, (c) phase infiltration, (d) atomic trapping, and (e) interface regulation. | ||
4.1. LnBaCo2O5+δ-based perovskite
The presence of Co on the B-sites has proven advantageous for both OER and ORR processes. LnBaCo2O5+δ-type double perovskite oxides including NdBaFe2−xCoxO5+δ,350 NdBa0.5Sr0.5Co1.5Fe0.5O5+δ,43,351 and Ba1−xGd0.8La0.2+xCo2O6−δ352,353 with triple-conducting capability, have been demonstrated as effective air electrodes for P-SOCs. Among cobalt-based double perovskites, incorporation of Pr2+/Pr3+ in the A site lead to favorable TECs, high conductivity, and strong catalytic activity for OER and ORR reactions.354,355 Thus, PrBaCo2O5+δ (PBC) system is widely studied as air electrode material,84,231,335,356–366e.g., famous PrBa0.5Sr0.5Co2−xFexO5+δ developed by Choi et al., which can achieve PPD exceed 500 mW cm−2 at 500 °C and long-term durability under CO2.84,152 Doping with aliovalent cations including Zn, Ca, Sr, Fe, Ca, and Pd in PBC has been explored to enhance oxygen transport, match TECs with electrolyte, and improve stability.335,367–371 Xu et al. introduced a low-Lewis-acid-strength Cs+-doped PrBa0.9Cs0.1Co2O5+δ (PBCsC) electrode (Fig. 9(a)).226 A-Site dopants with lower acidity facilitate easier H+ uptake (Fig. 10(a)). The polarization of ionic Lewis acid strength at the A site shifts electrons pairs, leading to increased and surface exchange kinetics (Fig. 10(b)).372 PBCsC electrode demonstrates excellent performance in both FC mode (1.66 W cm−2) and EC mode (−2.85 A cm−2 at 1.3 V) at 650 °C. Regarding anion substitution, the high electronegativity of F weakens the bond strength between transition-metal 3d orbitals and O 2p states (metal–oxygen bonds) in perovskites, thereby enhancing oxygen surface exchange and diffusion processes.227,343 The PrBa0.5Sr0.5Co1.5Fe0.5O5.84−δF0.16 electrode for P-SOFC exhibits an improved PPD of 510 mW cm−2 at 600 °C.227 The long-term durability of perovskite electrodes is significantly influenced by the alkaline earth cations (e.g., Ba and Sr) surface segregation,373 which stems from composition, temperature, acidity or alkalinity, oxygen/steam partial pressure, and polarization current (Fig. 9(c)).374 The resultant insulating impurities impede the transfer of electrons and ions in proton-involved ORR and OER processes.374–376 Researchers explored various strategies to mitigate the negative impacts of segregation, represented by phase structure engineering.18,26,313 Du et al. utilized In3+, which has a large ionic radius (In3+ = 0.8 Å, Co3+ = 0.545 Å, and Co4+ = 0.53 Å) and is recognized for enhancing sinterability and stability in proton-conducting electrolytes,377–379 to develop a PrBaCo1.9In0.1O5+δ (PBCIn) air electrode.228 The composition-driven formation of composite phases in the PBCIn electrode consists of a primary phase of deficient PrBa0.95Co1.85In0.09O5+δ and a secondary cubic perovskite phase BaCo0.85In0.15O3−δ.228 The synergistic effect of these two phases enables PBIn to show 2.25 W cm−2 in FC mode, −4.41 A cm−2 at 1.3 V in EC mode at 700 °C and 210 h durability under 600 °C. High-valence cation Hf4+-doped PrBa0.8Ca0.2Co1.9Hf0.1O5+δ (PBCCHf0.1) naturally is regarded as double perovskite backbone, PrBa0.8−xCa0.2Co1.9Hf0.1−xO5+δ (PBCCHf0.1−x), with nano-sized BaHfO3 forming on the surface (Fig. 10(c)).229,380,381 The in situ exsolution of BaHfO3 NPs induces additional oxygen vacancies on the surface of Ba- and Hf-deficient PBCCHf0.1−x. Xu et al. demonstrated that Fe and Nb co-doped PBC form a heterostructure under steam-driven conditions (Fig. 10(d)), featuring in situ exsolved Nb-deficient PrBaCo1.6Fe0.2Nb0.2−xO5+δ (Nb-deficient PBCFN) on a double perovskite backbone, PrBaCo1.6Fe0.2Nb0.2O5+δ (Nb-rich PBCFN).230 The combination of in situ formed Nb-deficient NPs and the Nb-rich parent perovskite significantly faster surface exchange process, enhancing both catalytic activity (PPD of 1.059 W cm−2 and current density of 2.148 A cm−2 at 1.3 V at 650 °C) and durability (200 h). In situ exsolution of BaZrO3 NPs from the host PrBaCo1.92Zr0.08O5+δ (BZO@PBCZ) occurs under oxidizing conditions, facilitating the liberation Ba and Zr cations and forming proton transfer channels (Fig. 10(e)),231 Thereby accelerating the oxygen reduction catalytic activity. Cation-nonstoichiometric Ba1+xGd1−xCo2O6−δ spontaneously grows into the double perovskite BaGdCo2O6−δ (BGCO), anchored by BCO NPs under thermal-driven conditions (Fig. 10(f)).232 The synergy between the two components leads to exceptional performance, with the mixed O2−/e−-conducting perovskite-derived oxide exhibiting superior catalytic activity for the ORR, while the double perovskite structure enhances bulk H+ conductivity, thus increasing the number of available reaction sites.
and surface exchange kinetics (Fig. 10(b)).372 PBCsC electrode demonstrates excellent performance in both FC mode (1.66 W cm−2) and EC mode (−2.85 A cm−2 at 1.3 V) at 650 °C. Regarding anion substitution, the high electronegativity of F weakens the bond strength between transition-metal 3d orbitals and O 2p states (metal–oxygen bonds) in perovskites, thereby enhancing oxygen surface exchange and diffusion processes.227,343 The PrBa0.5Sr0.5Co1.5Fe0.5O5.84−δF0.16 electrode for P-SOFC exhibits an improved PPD of 510 mW cm−2 at 600 °C.227 The long-term durability of perovskite electrodes is significantly influenced by the alkaline earth cations (e.g., Ba and Sr) surface segregation,373 which stems from composition, temperature, acidity or alkalinity, oxygen/steam partial pressure, and polarization current (Fig. 9(c)).374 The resultant insulating impurities impede the transfer of electrons and ions in proton-involved ORR and OER processes.374–376 Researchers explored various strategies to mitigate the negative impacts of segregation, represented by phase structure engineering.18,26,313 Du et al. utilized In3+, which has a large ionic radius (In3+ = 0.8 Å, Co3+ = 0.545 Å, and Co4+ = 0.53 Å) and is recognized for enhancing sinterability and stability in proton-conducting electrolytes,377–379 to develop a PrBaCo1.9In0.1O5+δ (PBCIn) air electrode.228 The composition-driven formation of composite phases in the PBCIn electrode consists of a primary phase of deficient PrBa0.95Co1.85In0.09O5+δ and a secondary cubic perovskite phase BaCo0.85In0.15O3−δ.228 The synergistic effect of these two phases enables PBIn to show 2.25 W cm−2 in FC mode, −4.41 A cm−2 at 1.3 V in EC mode at 700 °C and 210 h durability under 600 °C. High-valence cation Hf4+-doped PrBa0.8Ca0.2Co1.9Hf0.1O5+δ (PBCCHf0.1) naturally is regarded as double perovskite backbone, PrBa0.8−xCa0.2Co1.9Hf0.1−xO5+δ (PBCCHf0.1−x), with nano-sized BaHfO3 forming on the surface (Fig. 10(c)).229,380,381 The in situ exsolution of BaHfO3 NPs induces additional oxygen vacancies on the surface of Ba- and Hf-deficient PBCCHf0.1−x. Xu et al. demonstrated that Fe and Nb co-doped PBC form a heterostructure under steam-driven conditions (Fig. 10(d)), featuring in situ exsolved Nb-deficient PrBaCo1.6Fe0.2Nb0.2−xO5+δ (Nb-deficient PBCFN) on a double perovskite backbone, PrBaCo1.6Fe0.2Nb0.2O5+δ (Nb-rich PBCFN).230 The combination of in situ formed Nb-deficient NPs and the Nb-rich parent perovskite significantly faster surface exchange process, enhancing both catalytic activity (PPD of 1.059 W cm−2 and current density of 2.148 A cm−2 at 1.3 V at 650 °C) and durability (200 h). In situ exsolution of BaZrO3 NPs from the host PrBaCo1.92Zr0.08O5+δ (BZO@PBCZ) occurs under oxidizing conditions, facilitating the liberation Ba and Zr cations and forming proton transfer channels (Fig. 10(e)),231 Thereby accelerating the oxygen reduction catalytic activity. Cation-nonstoichiometric Ba1+xGd1−xCo2O6−δ spontaneously grows into the double perovskite BaGdCo2O6−δ (BGCO), anchored by BCO NPs under thermal-driven conditions (Fig. 10(f)).232 The synergy between the two components leads to exceptional performance, with the mixed O2−/e−-conducting perovskite-derived oxide exhibiting superior catalytic activity for the ORR, while the double perovskite structure enhances bulk H+ conductivity, thus increasing the number of available reaction sites.
 | ||
| Fig. 10 Recent advances in LnBaCo2O5+δ-type air electrode materials. (a) Structure of PBCsC, (b) comparisons of different oxygen species contents of PBS and PBCsC. Reproduced with permission.226 Copyright 2023, American Chemical Society. (c) Schematic of Hf-doped PBCCHf0.1. Reproduced with permission.229 Copyright 2024, Elsevier. (d) Schematic of PBCFN. Reproduced with permission.230 Copyright 2022, Wiley-VCH. (e) Schematic of BZO@PBC. Reproduced with permission.231 Copyright 2022, Wiley-VCH. (f) Schematic of BGCO–BCO hybrid catalyst. Reproduced with permission.232 Copyright 2024, Wiley-VCH. (g) Schematic of PFC@PBSCF in the presence of contaminants (Cr and steam). Reproduced with permission.234 Copyright 2022, Wiley-VCH. (h) Schematic for the fabrication procedures of PCNCFO–PBC. Reproduced with permission.235 Copyright 2024, Wiley-VCH. (i) Structural evolution during reverse atom capture process. Reproduced with permission.150 Copyright 2024, Wiley-VCH. (j) Mechanism underlying the thermo-mechanical enhancement of TEC gradient formation. Reproduced with permission.382 Copyright 2024, Wiley-VCH. (k) Cross-sectional SEM image of PBSCF/BZCYYb composite interlayer. Reproduced with permission.236 Copyright 2024, Wiley-VCH. | ||
Surface decoration of nano-catalysts, typically on the order of tens of nanometers, on cathodes through a cost-effective infiltration process has garnered considerable attention (Fig. 9(c)).383,384 The porous skeleton facilitates significant electronic and ionic conductivity and the infiltrated coating enhances both catalytic activity and durability, e.g., Gd0.1Ce0.9O2−δ-infiltrated PBC.385 Pei et al. incorporated a fluorite-based Pr0.1Ce0.9O2+δ (PCeO) catalyst coating on PBC skeleton, demonstrating a much-reduced polarization resistance (reduced 58%), improved performance (PPD of 1.21 W cm−2 at 650 °C), and Ba segregation inhibition.233 The infiltration method promotes a well-mixed combination of raw materials at the atomic level, allowing for lower synthesis temperatures, and extends the length of the three-phase boundaries,386–388 where the electron, ion, and gas phases converge. For instance, at 700 °C, the Pr0.9Fe0.7Co0.3O3 (PFC) coating formed on PBSCF base after infiltration resulted in substantial performance improvements and markedly poisoning (including Cr and steam) tolerance (Fig. 10(g)).234,389,390 Besides creating a surface coating, the infiltrated solution may interact with the perovskite matrix. Gao et al. designed a multi-cationic oxide nano-catalyst, Pr0.2Ce0.2Ni0.2Co0.2Fe0.2Ox (PCNCFO), anchored on the surface of the PBC electrode via infiltration (Fig. 10(h)).235 The cerium oxide in the PCNCFO coating reacts dynamically with Ba segregated from the PBC, forming BaCeO3, which stabilizes the PBC phase structure, enhances proton conduction and transfer, and accelerate oxygen surface exchange. Consequently, the PCNCFO-coated PBC cathode achieved a high PPD of 1.31 W cm−2 at 650 °C.
Recently, an innovative method involving atomic trapping at high temperatures has been effective in modifying the surface chemistry of perovskites to tackle segregation.391–393 In the (La0.6Sr0.4)0.95Co0.2Fe0.8O3−δ perovskite, strontium atoms can be selectively extracted using an acidic MoO4 trap, forming inactive SrMoO4, which enhances durability.394 Building on this atomic capture approach, Zhao et al. introduced liquid-phase dispersible (NH4)10W12O41 onto the PBSCF surface, allowing for the capture of segregated barium and strontium cations from the PBSCF matrix (Fig. 9(d)).150 This process results in the formation of a heterostructure (Ba/Sr)(Co/Fe/W)O3−δ (BSCFW)@PBSCF (Fig. 10(i)). The novel P-SOFC cathode achieved remarkable performance, attaining a PPD of 1.32 W cm−2 at 650 °C, alongside impressive long-term durability for 240 h.
Several methods exist for modifying the bulk structure of PBC-type air electrode materials including morphology engineering,395 heterostructure construction,234,396–398 compositing,399–402 and microstructure design.203,271,403 However, the large TECs of cobalt-based PBC materials (10.8 × 10−6 K−1 for BZCYYb, 23.7 × 10−6 K−1 for PBSCF) restrict their practical application by leading to mechanical incompatibility with electrolyte and poor electrolyte/electrode interface adhesion.400,404 Negative thermal expansion materials represented by Y2W3O12 and Sm0.85Cu0.15MnO3−δ are added to the air electrode to offset the thermal expansion.170,220,325 However, inadequate mechanical mixing and poor physical contact between powders can result in delamination due to the significant variation in the thermal expansion coefficient of the electrode material and added powders. Additionally, the use of negative thermal expansion materials is constrained by phase transitions triggered by temperature or pressure changes, as well as its low stiffness and hygroscopic nature. To address these challenges, Gao et al. proposed composite electrodes with a TEC gradient.382 The transition phase, generated in situ due to topological atomic effects, helps bridge the thermal behavior gap and maintains thermo-mechanical stability. This approach also ensures chemical stability by preventing water vapor infiltration into the electrode. In this recently published study, the formation of the transitional phases, BaWO4 and Y10W8O21, at the interface between PrBa(Co0.7Fe0.3)2O5−δ (PBCF) and Y2W3O12 contributes to the development of TEC gradient electrode (Fig. 10(j)). And the transition phase captures the A-site element Ba in the electrolyte layer, thereby optimizing the electrolyte–electrode interface.
The abrupt transition from a dense electrolyte to a porous electrode creates discontinuous reaction pathways and oxygen vacancies, impeding charge transfer and the transport of gas and ionic phases, leading to a high polarization resistance at electrolyte/electrode interface. This weak adhesion, structural and chemical discontinuities, and limited contact points, exacerbates stagnation at the interface and restricts effectiveness of electrode at low operating temperatures.84,132,405,406 Therefore, interface engineering is essential to address the challenges of efficient operation in P-SOCs at intermediate to low temperatures by ensuring a seamless transition between the electrolyte and electrode. Choi et al. introduced a mono-grain PBSCF/BZCYYb composite interlayer, which enabled an even lower operating temperature of 350 °C.236,407 This composite interlayer was fabricated using pulsed laser deposition (PLD), with triple-conducting oxide PBSCF and proton-conducting oxide BZCYYb co-deposited onto a dense BZCYYb electrolyte (Fig. 10(i)). The resulting mono-grain composite interlayer features a quasi-2D thin layer with an alternately arranged, phase-separated structure, which extends the electrode/electrolyte interface by approximately 13.7 times compared to a reference sample with a conventional interface between a porous electrode and a dense electrolyte (Fig. 10(k)). The composite also exhibits a higher  concentration, about twice that of the PBSCF bulk powder electrode, due to charge modulation reactions at the heterointerface. This unique structure facilitates reaction pathways through vertically aligned oxygen vacancies, significantly reducing both ohmic and polarization resistances. This mono-grain composite interlayer in a fuel electrode-supported configuration achieved a PPD of 0.50 W cm−2 and a current density of 0.25 A cm−2 at 1.3 V at 350 °C.
concentration, about twice that of the PBSCF bulk powder electrode, due to charge modulation reactions at the heterointerface. This unique structure facilitates reaction pathways through vertically aligned oxygen vacancies, significantly reducing both ohmic and polarization resistances. This mono-grain composite interlayer in a fuel electrode-supported configuration achieved a PPD of 0.50 W cm−2 and a current density of 0.25 A cm−2 at 1.3 V at 350 °C.
4.2. LnBaFe2O5+δ-based perovskite
Despite their advantageous properties for electrochemical water splitting and power generation, cobalt-based perovskites exhibit drawbacks such as cobalt toxicity, high cost, and large TEC. Recent studies have highlighted several Fe-based double perovskites that show promising performance,408–410 including LaBaFe2O5+δ,411 PrBaFe2O5+δ,412 GdBaFe2O5+δ,413 NdBaFe2O5+δ,414 and SmBaFe2O5+δ.411 Rational substitution strategies have been employed to enhance oxygen vacancy concentrations in LnBaFe2O5+δ, for example, doping the A-site with Sr415 and Ca,416 and substituting the B-site with W,417 Ta,418 Nb,419 and Mo.420 Pr0.8Nd0.2BaFe1.9Zn0.1O5+δ (PNBFZ)238 developed by Teketel et al., demonstrates improved electrical conductivities, lower TEC (13.9 × 10−6 K−1), and higher PPD of 401 mW cm−2 at 550 °C. BCO have been incorporated to construct composites.421 However, compared to cobalt-based materials, layered iron-based perovskite air electrode materials exhibit slightly lower performance, and their potential requires further investigation.5. Other representative perovskite
In addition to barium-containing perovskite materials, various other perovskite materials have seen significant advancements in recent years. This chapter focuses on lanthanide-rich perovskite structures and strontium-based perovskite materials, inspired by O-SOCs, to highlight their recent developments in intermediate-temperature P-SOCs air electrodes (summarized in Table 1).5.1. Ln2NiO4+δ series
The nickel-based perovskite represented by RP-type oxide Ln2NiO4+δ (where Ln = La, Pr, and Nd) possesses a unique K2NiF4 structure, characterized by the alternating arrangement of perovskite and Ln–O rock salt layers along the c-axis. The presence of excess interstitial oxygen in the Ln–O layer serves as an ionic carrier, resulting additional electron holes,422 which contribute to enhanced electrocatalytic capacity and enhanced surface oxygen-exchange kinetics.423–425 Grimaud et al. were pioneers in investigating the hydration and electrochemical behaviors of La2NiO4+δ (LNO), Pr2−xSrxNiO4+δ, and other related compounds.63 Their work established a direct connection between the layered perovskite structure and triple-conducting behavior, making the RP phase Ln2NiO4+δ promising candidates for P-SOCs.426–428La3+ in the A site with the larger ionic radius (La3+: 1.36 Å; Ca2+: 1.00 Å; Pr3+: 0.99 Å) can effectively lower B–O bond strength for easier O2− migration in the crystal lattice, which is beneficial for ensuring sufficient O2− conductivity at reduced temperatures.429–432 Unlike typical perovskite structures, where protons reside and migrate on regular O2−, layered perovskite (RP), exampled by LNO, may contain oxygen interstitials that allow protons to locate and migrate. However, the performance of LNO-based electrodes is limited by their low electronic conductivity and excessive alkaline earth metals. To improve this, various doping and compositing strategies have been employed.433,434 Sr-substituted La1.2Sr0.8NiO4+δ (LSN), for instance, achieved resistance of 0.15 Ω cm2 in symmetric cell and a PPD of 460 mW cm−2 at 700 °C.435 Zhong et al. reported that the water content in both pristine LNO and La2−xAxNiO4+δ (where A = Ca, Sr, Ba) rises with the level and basicity of the dopant increase (Fig. 11(a) and (b)).427,436 Wang et al. improved electronic conductivity by partially substituted Ni with Cu in La1.5Sr0.5NiO4+δ, increasing it from 74 S cm−1 to 171 S cm−1 at 800 °C. This enhancement was primarily attributed to the contraction of Ni–O bonds and the oxidation of Ni2+ to Ni3+. Cu, Zn co-doped LSN demonstrates a high electrolysis current density of 1.5 A cm−2 (1.3 V) at 600 °C under high humidity (60%) in air. A self-assembled composite of La0.6Sr0.4FeO3−δ–LSN, developed by Yang et al., exhibited low polarization resistances of 0.055 Ω cm2 at 700 °C in O2.437
 | ||
| Fig. 11 Recent advances in Ln2NiO4+δ, SrMO3−δ, and PrMO3−δ-type air electrode materials. Proton concentrations per formula unit (pfu) of (a) Ln2NiO4+δ, and (b) La2NiO4+δ and La1.6A0.4Ni4+δ (A = Ca, Sr, Ba). Reproduced with permission.427 Copyright 2022, Wiley-VCH. (c) Schematic of H3O+ diffusion in NSTF. Reproduced with permission.167 Copyright 2024, Wiley-VCH. (d) Schematic fabrication process. (e) Peeling strength of cathode-electrolyte interface. (f) AFM for electrolyte surfaces. Reproduced with permission.241 Copyright 2022, Springer Nature. | ||
The Pr2NiO4+δ (PrN)-based phases have been attempted in P-SOCs systems. Grimaud et al. found that PrN can function as a triple-conducting conducting oxide with low Rp when combined with a proton conductor like BaCe0.9Y0.1O3−δ (BCY10).63 In their subsequent work, they developed the BCY10-PrN composite to reduce TEC and optimize electrochemical properties through microstructure tuning.438 To improve insufficient adhesion of mixed systems, Tarutin et al. developed Cu-doped Pr2Ni0.8Cu0.2O4+δ, which demonstrates a PPD of ∼340 mW cm−2 at 750 °C.239 Nevertheless, the phase stability of PrN requires further improvement due to the decomposition of PrN into PrNiO3 and PrO2, leading to mixed ionic oxides formation at the electrode/electrolyte interface.439 The electrocatalytic,424 oxygen transport,440 and electrochemical properties441,442 of Nd2NiO4+δ (NNO) have been extensively documented in the literature.443 Luebber et al. utilized NNO to fabricate micro-tubular SOFC. Ba-doped Nd1.9Ba0.1NiO4+δ demonstrates lower polarization resistances, reaching as low as 1.7 Ω cm2 at 700 °C.444 Additionally, the incorporation of Sr in Nd2−xSrxNiO4+δ alters the oxidation state of Ni from Ni2+ to Ni3+, thereby influencing its magnetic properties.445 However, NNO undergoes a tetragonal to orthorhombic phase transition at 610 °C, which necessitates careful consideration of its application in intermedium-temperature catalysis.446
5.2. SrMO3−δ-based perovskite
SrMO3−δ with alkaline earth element Sr at A site is used as catalytic activity enhancement materials. SrCoO3−δ (SCO), for example, was infiltrated on Na0.3Sr0.7Ti0.1Fe0.9O3−δ (NSCF0.3@SC) thus achieving an improved PPD of 966 mW cm−2 at 600 °C.167 Zhu et al. decorated SrCo0.5Nb0.5O3−δ (SCN) NPs on the surface of PrSrCo1.8Nb0.2O6−δ (PSCN).240 This SCN NPs decoration significantly reduces the energy barriers associated with surface oxygen and vapor dissociation. Thus, SCN-PSCN composite electrode manifests a PPD of 1.30 W cm−2 and a current density of 1.91 A cm−2 at 1.3 V at 650 °C. However, Sr-containing electrodes often degrade due to Sr segregation, which results from the reactivity of Sr with steam. This segregation problem is particularly pronounced in EC mode, where higher steam concentrations exacerbate the issue.194,447 To mitigate it, various methods have been proposed, including doping with Ca,448 however, these have shown limited effectiveness. A more promising approach is transforming unstable Sr-containing single phases into stable hybrid materials by leveraging the segregation phenomenon through careful structure regulation.449–451 Song et al. synthesized a Sr0.9Ce0.1Fe0.8Ni0.2O3−δ (SCFN)-based nanocomposite electrode, featuring tetragonal and RP phases with surface-enriched CeO2 and NiO NPs.171 The RP phase in SCFN enhances hydration and H+ conduction, while the nanoscale NiO and CeO2 phases facilitate oxygen surface exchange and transfer of O2− ions from the RP or NiO surfaces to the primary phase. Zhou et al. developed a self-assembling two-phase Na-doped SrTi0.1Fe0.9O3−δ cathode for P-SOFCs, featuring a NaySrzTiuFe1−uO3−δ (NSTF) perovskite main phase and nanosized β-NaFeO2 (NF) through thermal-induced phase exsolution (Fig. 11(c)).167 Unlike most air electrode materials that exhibit a negative dependence on pH2O, the NSTF/NF nanocomposite cathode demonstrates a positive pH2O dependence in SOFC applications. This is attributed to enhanced H+ uptake and the potential development of a H3O+ transport pathway at the NSTF and NF phases interface, resulting in a new quadruple-conducting (H3O+/H+/O2−/e−) cathode that yields a PPD of 0.807 W cm−2 at 600 °C.Extrinsic Cr and S impurities at the surface represent another source of degradation in strontium-based perovskites.452 The accumulation of electronically and ionically insulating contaminants—such as Cr2O3, SO2, and secondary compounds like SrCrO4 and SrSO4—on the perovskite surface during SOFCs operation, primarily from vaporized Cr originated from metal interconnects and S-based species in the gas flow, ultimately degrades catalytic performance.453–458 Several strategies have been explored to mitigate the detrimental effects of Cr, including: (i) the use of Cr getters,459 (ii) coating of the interconnects,460 and (iii) surface modifications that demonstrate high resistance to Cr poisoning.383,461,462 However, the issue of Cr and S poisoning of Sr-based materials in P-SOCs requires further investigation.
5.3. PrMO3−δ-based perovskite
PrNi0.5Co0.5O3−δ (PNC), belonging to the PrMO3 family, is recognized as an effective triple-conducting air electrode material for P-SOCs due to its high proton conductivity. PNC is inspired by PrCoO3 (PCO) and lanthanide nickelates, combined the advantages of segregation mitigation and excellent tolerance to high steam vapor and varying oxygen partial pressures.463–466 Ding et al. demonstrated that appropriate nickel substitution at the B-sites of the PNC perovskite can lower the migration barrier for proton conduction, and easily introduce proton defects through hydration reactions.172 This enhancement in proton conduction, combined with the triple-conducting capabilities of PNC, facilitates both WOR and ORR, thereby improving the electrochemical performance during self-sustained and reversible operations at reduced temperatures. To broaden the application of PNC in low-temperature P-SOCs, addressing the issue of poor contact at the electrode/electrolyte interface is crucial. A straightforward acid treatment developed by Bian et al. can effectively rejuvenate the surface of high-temperature annealed electrolytes (Fig. 11(d)).241 This process fosters reactive bonding between the air electrode PNC and the electrolyte BZCYYb, leading to enhanced electrochemical performance and stability (Fig. 11(e) and (f)). Exceptional performance in P-SOFC is achieved at temperatures as low as 350 °C, with PPDs reaching 1.6 W cm−2 at 600 °C, 650 mW cm−2 at 450 °C, and 300 mW cm−2 at 350 °C. However, Pr is classified as a critical material by the European Commission and its large-scale use both unsustainable and costly.467 Additionally, When PNC is combined with the proton-conducting electrolyte BZCY, it demonstrates low tolerance to H2O and CO2.468 Therefore, more research is needed to improve their compatibility. Yao et al., for example, impregnated Pr2Ni0.5Co0.5O4−δ, which consists of a PNC perovskite phase and a PrO2 phase, onto LSCF base.242 The PNC perovskite phase facilitates rapid proton transport, while the PrO2 nanoparticles, characterized by a high concentration of oxygen vacancies, enhance both oxygen adsorption and transfer. The modified electrode achieving a PPD of 1857 mW cm−2 at 700 °C, and exhibit durability for 200 h.In addition to alkaline earth and rare earth element-based air electrodes, alkali metal-based materials are also being investigated for solid oxide cell development. For example, LiCoO2, a widely used cathode material in lithium-ion batteries, has achieved a PPD of 0.86 W cm−2 at 800 °C in O-SOFCs, benefiting from coordinated lithium volatilization and anion doping.469 Given the extensive adoption of lithium-ion technology, integrating recycled lithium-ion battery materials into solid oxide cell research presents a cost-effective approach. Moreover, the high-temperature operating environment efficiently removes volatile impurities, enhancing the feasibility of repurposing recycled materials for solid oxide cell applications.
6. Theoretical insights and computational design
Theoretical calculations are a powerful research tool in the field of SOCs including synthesis of perovskite and their application as air electrode materials. The complex compositions of perovskites render the exploration and development of efficient air electrode materials both time-consuming and challenging, which inevitably drove a search for more efficient tools. Theoretical calculations—especially high-throughput screening and machine learning—inherently became leading tools for the role thanks to their reliability and adaptability. The operational temperature of approximately 600 °C complicates direct observation of the air electrode's functioning SOCs which reduce their reliability there and beyond. Theoretical calculations effectively simulate operating conditions of the electrode and elucidate the underlying microscopic principles at play (summarized in Fig. 12). | ||
| Fig. 12 Application of theoretical calculations in the development of perovskite air electrodes. | ||
6.1. Computational approaches for high-performance design
Traditional approaches of discovering and developing new materials typically depend on empirical experiments and trial-and-error techniques, which can be time-consuming, costly, and often constrained in their scope. In contrast, simulations can integrate computational chemistry, molecular orbital theory, and experimental methods to effectively guide the discovery and optimization of air electrodes.117,470By leveraging theoretical calculations, researchers can systematically explore material properties and behaviors, thereby streamlining the design process and identifying optimal candidates more efficiently. In the study conducted by Mao et al., the optimization of bimetal-doped SCO was guided by DFT calculations focusing on two key parameters: the formation energy and the migration barrier of oxygen vacancies.267 A lot of 13 different TMs was evaluated as potential dopants with working resulting in 91 possible combinations for bimetal-doped SCO to identify the most promising electrode candidate. Ciucci and colleagues have utilized ab initio simulations, molecular orbital insights to design A- and B-site co-substituted BFO materials.34 By combining calculated lattice constants, substitutional defect formation energy Eform, oxygen vacancy formation energy 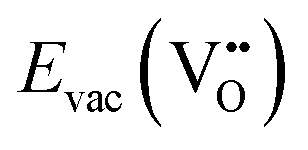 and hydration energy Ehydr, they concluded a potential Ba0.875Fe0.875Zr0.125O3−δ candidate (Fig. 13(a)).34 And as validated in experiments, the designed cathode can achieve impressive PPDs of 0.67 W cm−2, 1.28 W cm−2, and 2.04 W cm−2 at 500 °C, 600 °C, and 700 °C, respectively, and outstanding stability exceeding 200 h even in high-steam environments. Simulation-driven strategies enhance the potential for innovative material development while reducing time, manpower, and economic expenditures. Researchers can streamline the experimental process, focus resources on the most promising candidates of high effective catalyst, and avoid redundant or less effective experiments. The high-throughput computational methods employed by Luo et al. facilitated the screening of 932 materials based on key parameters such as
and hydration energy Ehydr, they concluded a potential Ba0.875Fe0.875Zr0.125O3−δ candidate (Fig. 13(a)).34 And as validated in experiments, the designed cathode can achieve impressive PPDs of 0.67 W cm−2, 1.28 W cm−2, and 2.04 W cm−2 at 500 °C, 600 °C, and 700 °C, respectively, and outstanding stability exceeding 200 h even in high-steam environments. Simulation-driven strategies enhance the potential for innovative material development while reducing time, manpower, and economic expenditures. Researchers can streamline the experimental process, focus resources on the most promising candidates of high effective catalyst, and avoid redundant or less effective experiments. The high-throughput computational methods employed by Luo et al. facilitated the screening of 932 materials based on key parameters such as  formation energy, hydration energy, and adsorption energy, thereby expediting the discovery and optimization of proton conductors.471 Very recently, Hu et al. used high-throughput calculations combined with data-driven decomposition analysis to forecast essential properties for 4455 unique perovskite oxides, focusing on their thermodynamic stability and decomposition tendencies crucial for various applications (Fig. 13(b)).237
formation energy, hydration energy, and adsorption energy, thereby expediting the discovery and optimization of proton conductors.471 Very recently, Hu et al. used high-throughput calculations combined with data-driven decomposition analysis to forecast essential properties for 4455 unique perovskite oxides, focusing on their thermodynamic stability and decomposition tendencies crucial for various applications (Fig. 13(b)).237
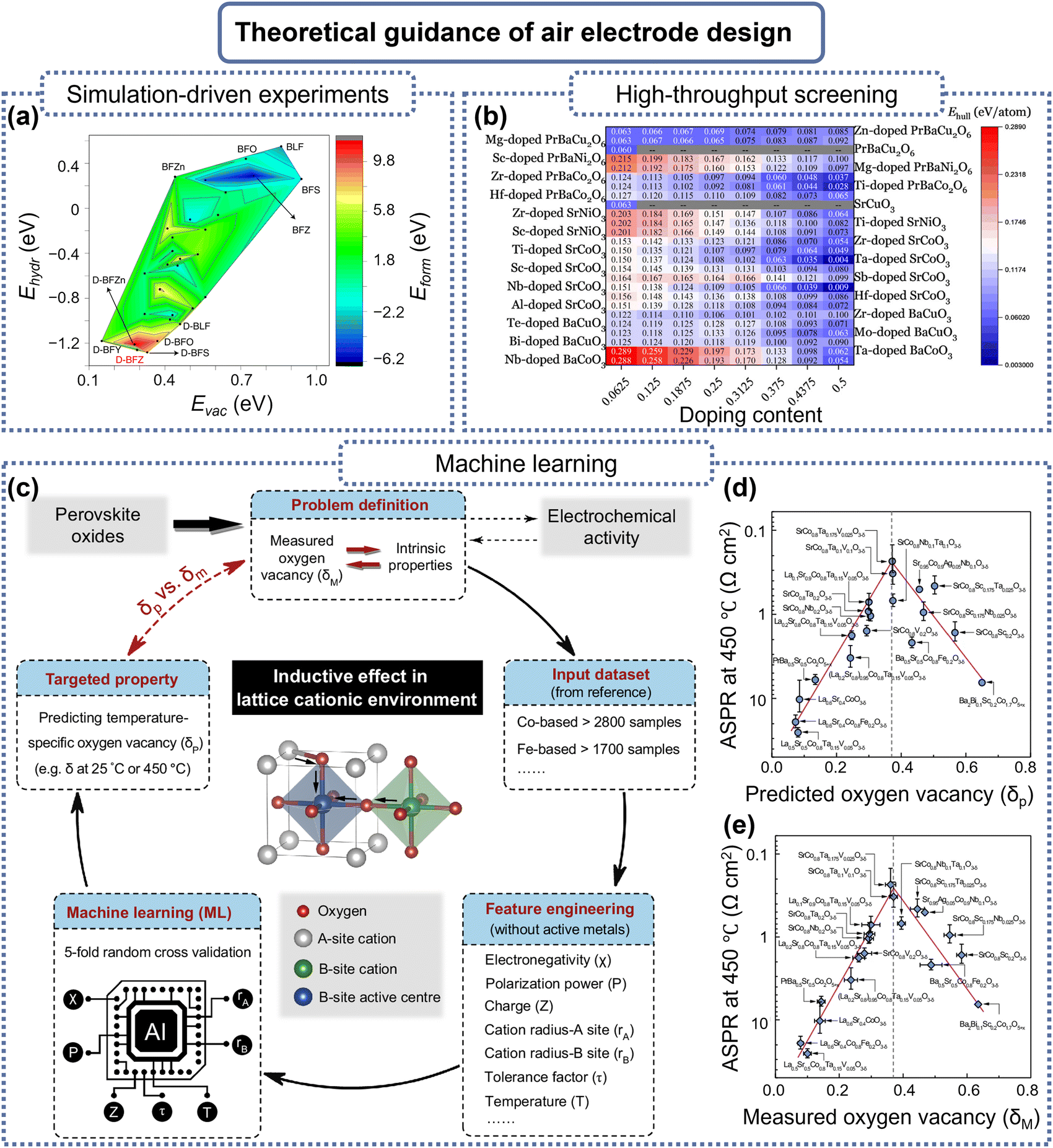 | ||
| Fig. 13 (a) Plot of computed Eform against Evac and Ehydr for BFO and its derivative materials (dots). Reproduced with permission.34 Copyright 2022, Springer Nature. (b) Promising perovskite candidates with exceptional thermodynamic stability. Reproduced with permission.237 Copyright 2024, Royal Society of Chemistry. (c) Workflow of oxygen vacancies prediction, and (d) and (e) the measured and predicted ASR. Reproduced with permission.81 Copyright 2024, Springer Nature. | ||
Since the advent of artificial intelligence (AI) and the increasing amount of experimental data in material synthesis, machine learning (ML) techniques have become the corner stone of new material discoveries, accelerating the design, prediction of property, and optimization.372 ML involves the use of computational algorithms and statistical models to analyze extensive datasets, enabling the extraction of meaningful patterns and facilitating predictions or optimizations based on these insights.472–474 Consequently, it is able to speed up the discoveries and guides experimental work towards the most promising candidates materials at relatively short time and low budge.475–479 ML models to date have been used to identify the critical factors affecting H+/O2−/e− conductivities through feature selection and engineering techniques. In 2021, an XGBoost model was constructed to efficiently and accurately predict the conductivities of 97625 perovskite oxides under wet (3% H2O) hydrogen and wet air environments.480 In hydration abilities analysis, Wang et al. employed a random forest (RF) model to identify efficient mixed proton–electron conductive oxides by predicting the hydrated proton concentration (HPC) of 3200 A1−xA1xB1−yB1yO3 perovskite oxides.481 Through this RF approach, they successfully predicted the HPC of these perovskite oxides, enabling the selection of highly efficient mixed proton–electron conductive materials. Recent studies also indicate that ML models can effectively predict the catalytic activity of air electrode materials in solid oxide cells, particularly regarding resistance to oxygen reduction (Rp), power density, energy barriers, oxygen p-band centers, and vacancy formation energies, often calculated via DFT.482–485 Integrating ML with DFT has been found enhancing efficiency and accuracy of these calculations, facilitating faster material innovation.486 Ni et al. introduced ionic Lewis acid strength (ISA) as an effective physical descriptor to accelerate the research of ORR activity.372 Their model successfully predicted the Rp of 6871 oxides, demonstrating the best fit among eight different regression models.
The important score assigned to each descriptor revealed that A/B-site ionic Lewis acid (AISA/BISA) significantly influences model predictions, suggesting a strong correlation with intrinsic ORR activity. An active learning strategy can be used to predict H2 production in high-entropy oxides, enabling a more rational design of high-entropy materials rather than relying solely on experience and intuition.487 A very recent study by Li et al. shows ML techniques have been used by to quantify the relationship between oxygen vacancy concentrations and catalytic activity, based on 235 cobalt-based and 200 iron-based perovskite catalysts (Fig. 13(c)).81 The temperature-dependent variation in oxygen vacancy concentrations in both Co-based and Fe-based perovskites can be accurately predicted from well-established elemental properties. The oxygen electrocatalytic activity of perovskite oxides exhibit a “volcano” dependence on oxygen vacancies concentrations, where the optimal oxygen vacancy concentration for electrocatalysis changes with the operating temperatures (Fig. 13(d) and (e)). This study offers an efficient approach with oxygen vacancy-activity relations to efficiently predict perovskite for electrocatalysis operated across a broad temperature range.
Additionally, ML models have been developed to predict the thermodynamic phase stability of perovskite oxides by estimating their energy above the convex hull (Ehull) under realistic conditions.488 The reliability of these models was confirmed through comparisons with DFT results and validation against newly generated compounds.489 Furthermore, EIS data has been used to train neural networks, which successfully forecast long-term performance degradation. The challenges, however remain as shown by Hagen et al., who found weak correlations in degradation assessments based on a comprehensive dataset of SOC tests, highlighting the need for more extensive data to enhance statistical significance in ML applications for durability predictions.490 ML holds great promise for developing high-efficiency perovskite air electrode materials for P-SOCs. However, challenges such as data availability, complexity of materials data, and the need for effective feature engineering hinder its application. Balancing multiple target properties like catalytic activity and stability is also difficult due to trade-offs between performance and degradation. Despite these obstacles, leveraging big data and advanced computing can enhance efficiency and accuracy of ML in materials research. Overcoming these challenges requires collaboration, better data standards, and improved model interpretability.
6.2. Mechanistic understanding from simulations
Due to the challenges of direct observation, theoretical calculations have emerged as a powerful tool for elucidating the microscopic mechanisms of efficient air electrodes. These mechanisms primarily involve reactions related to protons and oxygen ions. By analyzing the adsorption, transfer, and reaction processes of charge carriers, researchers can establish a connection between structure and performance, thereby guiding the optimization of air electrode materials. Hydration reactions involve proton incorporation and interactions with lattice oxygen, which is regarded as water adsorption and proton transport. Li et al. evaluated the impact of doped boron (B) on surface chemical properties by calculating water adsorption energies.491 They found that the B-modified Pr4Ni3O10+δ (0.5B-PN) surface had a water adsorption energy of −1.4 eV, compared to −0.9 eV for the PN surface, reflecting a 55.6% improvement in hydration ability due to the surface modification with dispersed B. Lu et al. calculated hydration energies for different cells by inserting H2O into oxygen vacancies of extracted cells in parent perovskite structures (Fig. 14(a)).183 BaCo0.4Fe0.4Nb0.2O3−δ (BCFN) showed the highest hydration energy which is indicative of strong affinity for H2O molecules, the performance partly attributed to Nb5+ doping. For F-doped BCFZY examined by Ren et al., the reduction in lattice size was confirmed through cell structure analysis, and hydration properties were illustrated by the adsorption energies of H2O on Fe–VO–Co sites and integrated crystal-orbital Hamilton populations (iCOHPs, Fig. 14(b)).166 By simulating the electronic structure and potential energy surfaces of perovskites, proton conduction mechanisms can be provided. Liu et al. used climbing image nudged elastic band (CINEB) calculations to show higher energy barrier (Eb) for proton migration for hopping pathway than rotation pathway in perovskites, identifying hopping as the rate-limiting step (Fig. 14(c)).179 They found Er-doping in BSCF lowering Eb for the hopping pathway by 0.13 eV. Er2O3, with its simpler proton hopping process, achieves an even lower Eb of 0.35 eV and a greater hopping distance compared to Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) and Ba0.5Sr0.5(Co0.8Fe0.2)0.9Er0.1O3−δ (BSCFE), indicating that Er2O3 markedly improves proton migration in composite electrodes. Beyond Eb, factors such as the O⋯H bonds formation energy and distance are also crucial for predicting proton conduction. Chen et al. reported higher positive formation energy for O⋯H bonds in Ba0.5Sr0.5Co0.8Fe0.2O2.9−δF0.1 (BSCFF) than Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF), suggesting more robust bonds formation with oxygen by the proton in BSCF (Fig. 14(d)).213 This stronger bonding results in a more challenging hopping process for protons, following the Grotthus mechanism. Moreover, fluorinated oxides like BSCFF exhibit longer O⋯H distances, which implies easier disruption of these bonds and a reduced energy barrier for proton displacement. To analyze proton diffusion pathways and conductivity,492 first-principles molecular dynamics (FPMD) simulations at 1000 K were employed by Kim et al.199 These simulations showed more rapidly protons diffusion in BaSc0.1Ta0.1Co0.8O3−δ (BSTC) compared to BaCoO3−δ (BCO) as evidenced by a higher mean squared displacement (MSD) for BSTC (Fig. 14(e)). This enhanced diffusion is linked to hydroxide rotation, proton transfer, and trapping phenomena. In terms of diffusion pathways, BSTC establishes a continuous 3D proton transport network within 100 ps, whereas BCO develops a more limited 2D network (Fig. 14(f)). The proton conductivity of BSTC measured at 4.94 × 10−4 S cm−1 using the Nernst–Einstein relation, indicating its suitability for P-SOCs air electrodes. Furthermore, negative crystal orbital Hamiltonian populations (–COHPs) reveal that O⋯H bonds in BSTC are weaker than those in BCO with integrated –COHP values of 3.87 eV bond−1 for BSTC and 3.95 eV bond−1 for BCO. This suggests that BSTC facilitates more efficient proton transfer due to its weaker O⋯H bonds and decomposition in the O 2p and H 1s orbitals. | ||
| Fig. 14 Recent advances in theoretical explanation for efficient air electrode materials. (a) Hydration energies of the BCFN, BCFNS, and BCFS (inset: optimized structural models for the hydrated samples of BCFN). Reproduced with permission.166 Copyright 2023, Elsevier. (b) M–O iCOHP value comparison for BCFZY(F). Reproduced with permission.164 Copyright 2022, Elsevier. (c) Energy profile for proton hopping and rotation in BSCF, BSCFE, and Er2O3. Reproduced with permission.179 Copyright 2023, Wiley-VCH. (d) O⋯H formation energy and distance for BSCF(F). Reproduced with permission.213 Copyright 2023, Elsevier. (e) MSD of a hydrogen atom and (f) time-dependent evolution of H+ diffusion pathways in BSTC and BCO. Reproduced with permission.199 Copyright 2024, Wiley-VCH. (g) PDOS of the O 2p and Co 3d orbitals of PBCC and PBSLCC. Reproduced with permission.493 Copyright 2023, Elsevier. (h) Oxygen vacancy formation energy in electrodes SBCF-2L and SBCF-5L. Reproduced with permission.494 Copyright 2024, Wiley-VCH. (i) Energy barrier of cubic C-BSCF (100) and hexagonal H-BSCF (0001) surfaces. Reproduced with permission.214 Copyright 2024, Springer Nature. | ||
Proton-involved oxygen reactions dominate the kinetics of air electrode. He et al. utilized DFT calculations to investigate how A-site entropy tuning affects the performance of perovskite materials for OER and ORR.212,493 The study compared high-entropy Pr0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (PBSLCC) with binary PrBaCo2O5+δ (PBC) and ternary Pr0.8Ba0.8Ca0.4Co2O5+δ (PBCC), focusing on key descriptors such as the O p-band center,36,107,495 formation energies, and oxygen adsorption. The projected density of states (PDOS) of PBCC and PBSLCC was displayed to verify the strong hybridization of cations in the B-site Co ion with oxygen (Fig. 14(g)).493 The O p-band center PBSLCC shifted closer to the Fermi level (−3.29 eV) compared to PBC and PBCC. The oxygen vacancy formation energies (Evac) in PBSLCC (0.97 eV) is lower compared to PBC (1.35 eV) and PBCC (1.31 eV).496 Oxygen adsorption energies (ΔEads)studies revealed that PBSLCC exhibits weaker oxygen bonds and better kinetics, in line with Sabatier's principle.497,498 Overall, the research confirms that A-site entropy tuning significantly enhances OER and ORR performance by optimizing the activity of oxygen reactions.
formation energies, and oxygen adsorption. The projected density of states (PDOS) of PBCC and PBSLCC was displayed to verify the strong hybridization of cations in the B-site Co ion with oxygen (Fig. 14(g)).493 The O p-band center PBSLCC shifted closer to the Fermi level (−3.29 eV) compared to PBC and PBCC. The oxygen vacancy formation energies (Evac) in PBSLCC (0.97 eV) is lower compared to PBC (1.35 eV) and PBCC (1.31 eV).496 Oxygen adsorption energies (ΔEads)studies revealed that PBSLCC exhibits weaker oxygen bonds and better kinetics, in line with Sabatier's principle.497,498 Overall, the research confirms that A-site entropy tuning significantly enhances OER and ORR performance by optimizing the activity of oxygen reactions.
Oxygen vacancies play a crucial role in affecting ion transfer and hydration properties of perovskite oxides. To assess the hydration and oxygen ion transfer capabilities of air electrode materials, researchers examine the formation energy of oxygen vacancies in different materials and lattice sites. Liu et al. showed that the Er-doping notably reduces Evac and hydration energy (Ehdr) by 0.12 eV and 0.22 eV, respectively.179 This doping strategy thus enhances hydration ability of Ba0.5Sr0.5Co0.72Fe0.18Er0.09O3−δ–Er2O3 composite air electrode. In a separate study, Sun et al. used DFT calculations to evaluate the oxygen vacancies formation energy in Sm0.8Ba1.2Co0.8Fe1.2O5+δ (SBCF-5L) and SmBaCo0.8Fe1.2O5+δ (SBCF-2L).494 he vacancies for SBCF-5L are more easily formed in the –[SmO1−x]– layer, with formation energies of 0.289 eV and 0.314 eV for the –[SmO1−x]– and perovskite layers, respectively. In contrast, SBCF-2L shows values of 0.217 eV and 0.291 eV (Fig. 14(h)). When vacancies are present in the –[SmO1−x]– layer of SBCF-5L, the formation energy in the perovskite layer drops, facilitating enhanced three-dimensional oxygen ion conduction. This results in a higher initial concentration of oxygen vacancies in SBCF-5L compared to SBCF-2L, making SBCF-5L more efficient for oxygen ion transport. In Chen et al.'s work, the oxygen vacancy formation and PDOS of Fe-substituted Nd0.8Sr1.2Ni1−xFexO4±δ was predicted by DFT.499 The oxygen vacancy formation energy was calculated as 4.34 eV for Nd0.8Sr1.2NiO4±δ (NSN) and 4.19 eV for Nd0.8Sr1.2Ni0.7Fe0.3O4±δ (NSNF), showing that Fe doping reduces this energy compared to other perovskite oxides. Fe 3d electrons are closer to the Fermi level compared to Ni 3d, indicating improved catalytic activity with Fe doping. Lower oxygen vacancy formation energy, coupled with accelerated charge transfer through Fe–O–Ni bridges, suggests enhanced catalytic activity for ORR and OER in Fe-doped NSN.500,501
In perovskite oxides, electronic conduction often occurs via polaron hopping. To understand factors influencing overall electronic conductivity, density-of-states (DOS) and band alignment simulations are employed. Yu et al. analyzed DOS plots for Sr3Fe1.8Nb0.2O7−δ (SFN) and Sr2.8Fe1.8Nb0.2O7−δ (D-SFN) to understand the effects of Nb doping and A-site cation defects.502 These plots show that the conduction band near the Fermi level is mainly influenced by Fe 3d orbitals while the valence band comes from O 2p orbitals. In D-SFN, there are more unoccupied states near the valence band which suggests better charge transfer. The addition of Sr defects increases Fe valence states, enhancing the overlap between Fe 3d and O 2p orbitals. This stronger Fe–O interaction improves electron conduction in D-SFN. Additionally, DOS analysis was performed by Zhou et al. to analyze different active sites of PrBa0.8Ca0.2Co2O5+δ–BaCoO3−δ (PBCC–BCO) composite air electrode.194 Their findings showed that Co majority spin states at PBCC (010) lose more electrons compared to BaCoO3 (110), which is linked to differences in coordination. Specifically, BaCoO3 (110) benefits from electron-donating Ba, which creates an electron-rich region at the Co sites, enhancing interactions with oxygen-containing intermediates. This increased interaction facilitates water deprotonation but limits oxygen desorption on BaCoO3 (110).
The OER process on the air electrode surface involves several steps: H2O adsorption (*H2O, * indicates the adsorbed state), oxygen evolution (*H2O → *OH + *H → *O + *H → *O → *O2), and *O2 desorption, whereas the ORR process is essentially the reverse of OER process.30,194,503 Combining calculating adsorption energies, reaction intermediates, and activation barriers, OER and ORR pathways can be simulated at the atomic level. This approach helps identify the rate-determining steps and optimize the catalytic activity of air electrodes. Liu et al. investigated the energy barriers for OER and ORR on cubic Ba0.5Sr0.5Co0.8Fe0.2O3−δ (C-BSCF) and hexagonal Ba4Sr4(Co0.8Fe0.2)4O16−δ (H-BSCF) surfaces to understand their mechanisms in P-SOCs air electrodes.214 They determined the most stable surface configurations by evaluating various A- and B-site arrangements and surface terminations. The results revealed that C-BSCF has a higher energy barrier of 3.25 eV for OER mainly due to limited proton conduction whereas H-BSCF requires a lower barrier of 1.15 eV, highlighting its superior proton migration capability. Conversely, H-BSCF faces a 2.03 eV barrier for oxygen desorption while C-BSCF performs better in this regard. The hybrid Ba1.5Sr1.5Co1.6Fe0.4O7−δ (C/H-BSCF) electrode combines the advantages of both phases, with initial water decomposition occurring on the cubic phase and further decomposition and oxygen desorption on the hexagonal phase. For ORR, C-BSCF shows a more favorable oxygen reduction process with a significant advantage over H-BSCF which has lower energy barriers for oxygen adsorption and desorption (Fig. 14(i)). Overall, the C/H-BSCF hybrid electrode benefits from enhanced ORR and OER activities due to the synergistic effects of combining cubic and hexagonal phases. When examining complex hybrid air electrodes, it is often highly effective to investigate the microscopic mechanisms of oxygen reactions with theoretical calculations. In composite air electrode PBCC–BCO constructed by Zhou et al., the H2O adsorption and oxygen evolution are favored on BCO with ΔG values of 0.099 eV (H2O adsorption) and 0.62 eV (oxygen evolution) for BCO, compared to 1.794 eV (H2O adsorption) and 1.055 eV (oxygen evolution) for PBCC at 600 °C.194 The PBCC surface, on the other hand, is more favorable for O2 formation and desorption (1.307 eV for BCO vs. −0.823 eV for PBCC at 600 °C). Combined with DOS, the proposed OER sequence for the PBCC–BCO air electrode involves initial H2O absorption on the BCO (110) surface, where it dissociated into *OH and *H. The *OH intermediate is then further dissociated into *O and *H on BCO (110), and finally, O2 is formed and desorbed on PBCC (010). This sequence highlights the combined effects of rapid H2O dissociation on BCO NPs and fast oxygen desorption on PBCC in enhancing OER activity.
7. Pre-commercialization considerations
Renewable energy sources now contribute more than 29% of global electricity generation, with countries like Australia reaching levels as high as 35%.504 Several large-scale hydrogen plants have recently been launched worldwide by corporations including European Energy in Denmark,505 TotalEnergies in French,506 and German Aerospace Center in Germany.507 By integrating electrolysis with renewable energy sources, fuels and chemicals production can move away from fossil fuels dependence, achieving the development of a fully renewable energy system. P-SOCs offer a reliable and affordable solution to incorporate green energy into the current energy infrastructure. This chapter outlines key factors to address before P-SOCs technology can be fully commercialized.7.1. Scaling up from lab to industry
Lab-scale P-SOCs are generally designed with compact sizes, for instance, O’Hayre, Shao, and Duan et al. report active areas of just around 1 cm2 for their P-SOCs.9,81 This small scale is suitable for laboratory research, where studies focus on optimizing electrode materials, cell structure, and performance parameters under controlled conditions. However, for practical applications, P-SOCs must be significantly larger to meet the energy demands of real-world systems (102 W for electronic devices and 104 W for electronic vehicles). Application-oriented P-SOCs typically adopt a planar or tubular design to increase surface area and enhance efficiency. Currently, numerous studies have explored larger-scale P-SOCs (as shown in Fig. 15(a) and Table 2) including 12 × 12 cm2 planar designs and 8 cm2 tubular configurations.161,162 However, despite these increased sizes, the total output power remains insufficient to operate electronic devices. The production capacity of a P-SOC plant is directly proportional to the total active area. Expanding the active area at the individual cell level and assembling multiple cells into stacks are the two most straightforward methods to increase output.508,509 Scaling up requires addressing challenges such as maintaining mechanical integrity, achieving uniform gas distribution, and managing thermal stresses across a larger area. In particular, the design must ensure durability and stable performance under operating conditions that differ from the controlled lab environment (Fig. 15(b)), making factors like material selection, sealing methods, and thermal management crucial to the development of commercial-scale P-SOCs systems.510,511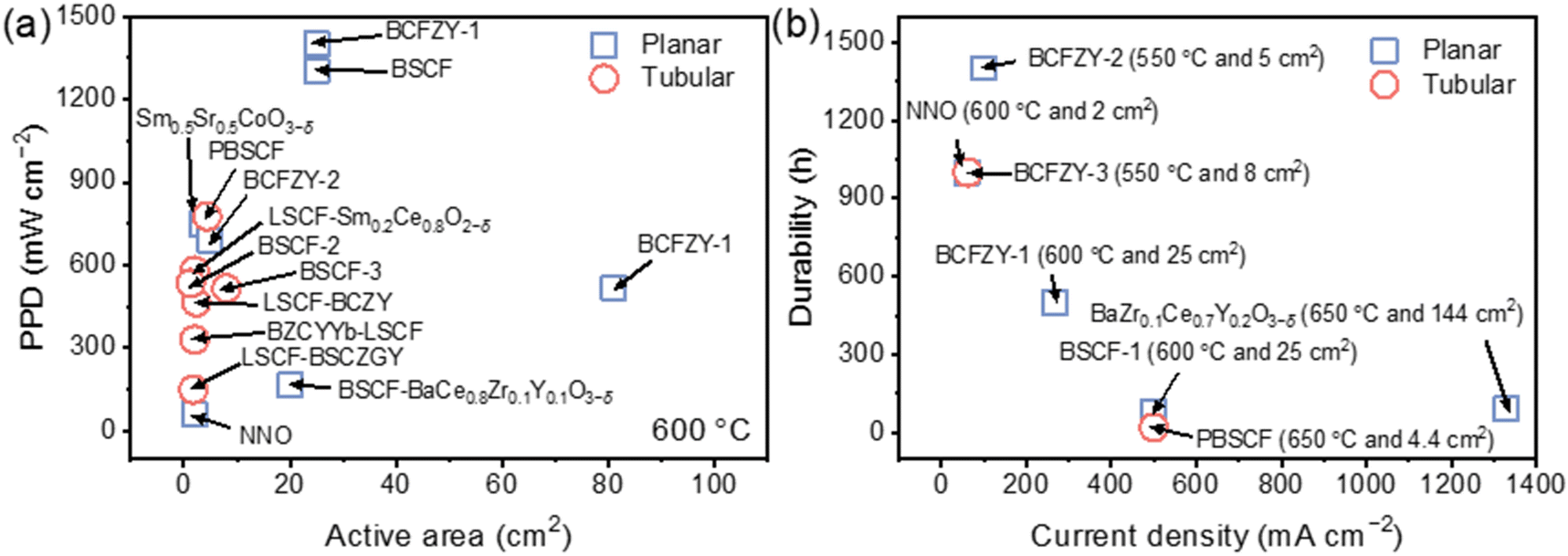 | ||
| Fig. 15 Large-area P-SOCs: (a) PPDs at 600 °C and (b) durability (data detailed in Table 2). | ||
| Air electrode | Area (cm2) | PPD at 600 °C (mW cm−2) | Current density at 1.3 V (mA cm−2) | Stability (h) | Ref. |
|---|---|---|---|---|---|
| Planar | |||||
| BaCe0.2Zr0.7Y0.1O3−δ | 135 | 400 h (ASR) | 512 | ||
| BaZr0.1Ce0.7Y0.2O3−δ | 144 | 90 h (650 °C and 1330 mA cm−2) | 162 | ||
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ–BaCe0.8Zr0.1Y0.1O3−δ (BSCF–BCZY) | 20 | 165 | 3000 h (600 °C, 170 mA cm−2 and −100 mA cm−2) | 285 | |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF-1) | 25 | 1302 | 80 h (550 °C and 500 mA cm−2) | 513 | |
| BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-1) | 81 | 518 | 514 | ||
| BCFZY-1 | 25 | 1400 | 500 h (550 °C and 267 mA cm−2) | 514 | |
| BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-2) | 5 | 690 | 1400 h (550 °C and 100 mA cm−2) | 515 | |
| Nd2NiO4+δ (NNO) | 2.01 | 60 | 1000 h (600 °C at 60 mA cm−2) | 516 | |
| Sm0.5Sr0.5CoO3−δ (SSC) | 3.33 | 747 | 517 | ||
| Tubular | |||||
| BaZr0.1Ce0.7Y0.1Yb0.1O3−δ–La0.6Sr0.4Co0.2Fe0.8O3−δ (BZCYYb–LSCF) | 2 | 331 | 518 | ||
| La0.6Sr0.4Co0.2Fe0.8O3−δ–Ba0.5Sr0.5Ce0.6Zr0.2Gd0.1Y0.1O3−δ (LSCF–BSCZGY) | 1.79 | 150 | 96 h (700 °C and 0.7 V) | 519 | |
| BCFZY-3 | 8 | 517 | 915 | 1000 h (550 °C and 0.062 A cm−2) | 161 |
| La0.6Sr0.4Co0.2Fe0.8O3−δ–Sm0.2Ce0.8O2−δ (LSCF–Sm0.2Ce0.8O2−δ) | 2 | 580 | 520 | ||
| La0.6Sr0.4Co0.2Fe0.8O3−δ–BaCe0.7Zr0.1Y0.2O3−δ (LSCF–BCZY) | 2.3 | 465 | 70 h (600 °C and 0.7 V) | 521 | |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF-2) | 1.29 | 534 | 359 | ||
| PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) | 4.4 | 776 | 20 h (650 °C and 0.5 A cm−2) | 522 | |
7.2. Key challenges for commercialization
As an air electrode material progresses toward practical applications, the criteria for evaluating its performance shift accordingly. Several practical considerations must be addressed, spanning technical, economic, and market-related factors. A primary concern is production scalability, which requires the development of cost-effective synthesis methods and dependable supply chains for essential raw materials such as Ba and Co. The traditional approaches for fabricating perovskite electrode materials include molten salt synthesis, hydrothermal reaction, sol–gel synthesis, and solid-state method. Each method plays a crucial role in determining phase purity, microstructure, defect concentration, and ionic/electronic transport properties, which directly impact electrode performance in P-SOCs. The hydrothermal method, performed under low-temperature (∼150–300 °C) and high-pressure conditions, enables fine morphology control and low-defect-density perovskites, which enhance proton and oxygen ion transport, but scalability remains a challenge due to high-pressure requirements.523,524 Molten salt synthesis is an emerging technique that allows for the formation of highly crystalline, defect-engineered perovskites at around 800 °C by utilizing molten salts as a reaction medium to promote controlled grain growth and enhanced ionic diffusion, which improves oxygen vacancy mobility and catalytic activity.525 The sol–gel method, operating at a relatively low calcination temperature (∼600–900 °C), facilitates the synthesis of nanostructured perovskites with a high surface area and uniform dopant distribution while preserving porosity and enhancing gas diffusion. These characteristics contribute to improved catalytic activity and ionic conductivity. However, potential issues such as organic solvent impurities, phase separation require careful management, unavailability and high price of the corresponding salts.526 Conversely, the solid-state reaction method, the most cost-effective and scalable approach for large-scale perovskite production, involves high-temperature calcination (∼1000–1400 °C) of mixed solid precursors, producing highly crystalline and thermodynamically stable perovskites with superior bulk electronic conductivity. However, this approach often results in large grain sizes with a low surface area, leading to non-uniform dopant distribution that may necessitate additional grinding or doping strategies for optimization.527 Additionally, advanced electrode structuring techniques such as freeze-casting, sacrificial templating, and 3D printing can help create highly porous, hierarchical structures that enhance both electronic and ionic transport without compromising mechanical integrity. Moreover, roll-to-roll processing and screen-printing techniques offer promising routes for integrating perovskite electrodes into commercial-scale P-SOCs with high reproducibility and cost efficiency.528,529 By implementing these strategies, the transition from research to commercialization can be accelerated. Achieving competitive costs is crucial, necessitating reductions in manufacturing expenses through process optimization, material efficiency, and economies of scale.Performance improvement also remains essential, particularly in enhancing outputs, efficiency, and lifespan, to ensure that P-SOCs can meet the rigorous demands of high-performance applications like electric vehicles and energy storage systems. In addition to technical and economic considerations, practical deployment of P-SOCs requires attention to delivery logistics, safety, and recycling strategies.530–532 Efficient delivery systems ensure that both raw materials and finished products reach their destinations securely and cost-effectively, with protective packaging, optimized shipping routes, and local distribution centers to minimize damage and costs. Safety is also critical, especially given the high operating temperatures and reactivity of materials in P-SOCs; ensuring thermal stability, reliable sealing, and resistance to environmental stress are essential for operational integrity. Finally, recycling strategies are needed to recover valuable materials like Co from spent cells, reducing reliance on raw material extraction and supporting sustainable production. Together, these considerations enable P-SOC technology to be deployed safely, sustainably, and at scale.
8. Challenges and prospects
In the intermediate-to-low temperature range, state-of-the-art P-SOCs continue to exhibit suboptimal performance compared to conventional O-SOCs.533 Air electrodes for P-SOCs have garnered significant interest due to their high polarization resistance, yet no truly successful air electrode material has emerged, unlike the established LSM and LSCF for O-SOCs. A review of the latest advancements in P-SOC air electrodes shows that the BCO series achieves the lowest ASR (Fig. 16(a) and (b)), with LnBaM2O6+δ double perovskite materials following closely (Fig. 16(c)). In contrast, Fe-based materials typically demonstrate less remarkable performance (Fig. 16(d)). To minimize the resistance of P-SOCs device and enhance both power output and hydrogen production efficiency, the BCO series may emerge as a key focus in future research efforts (Fig. 16).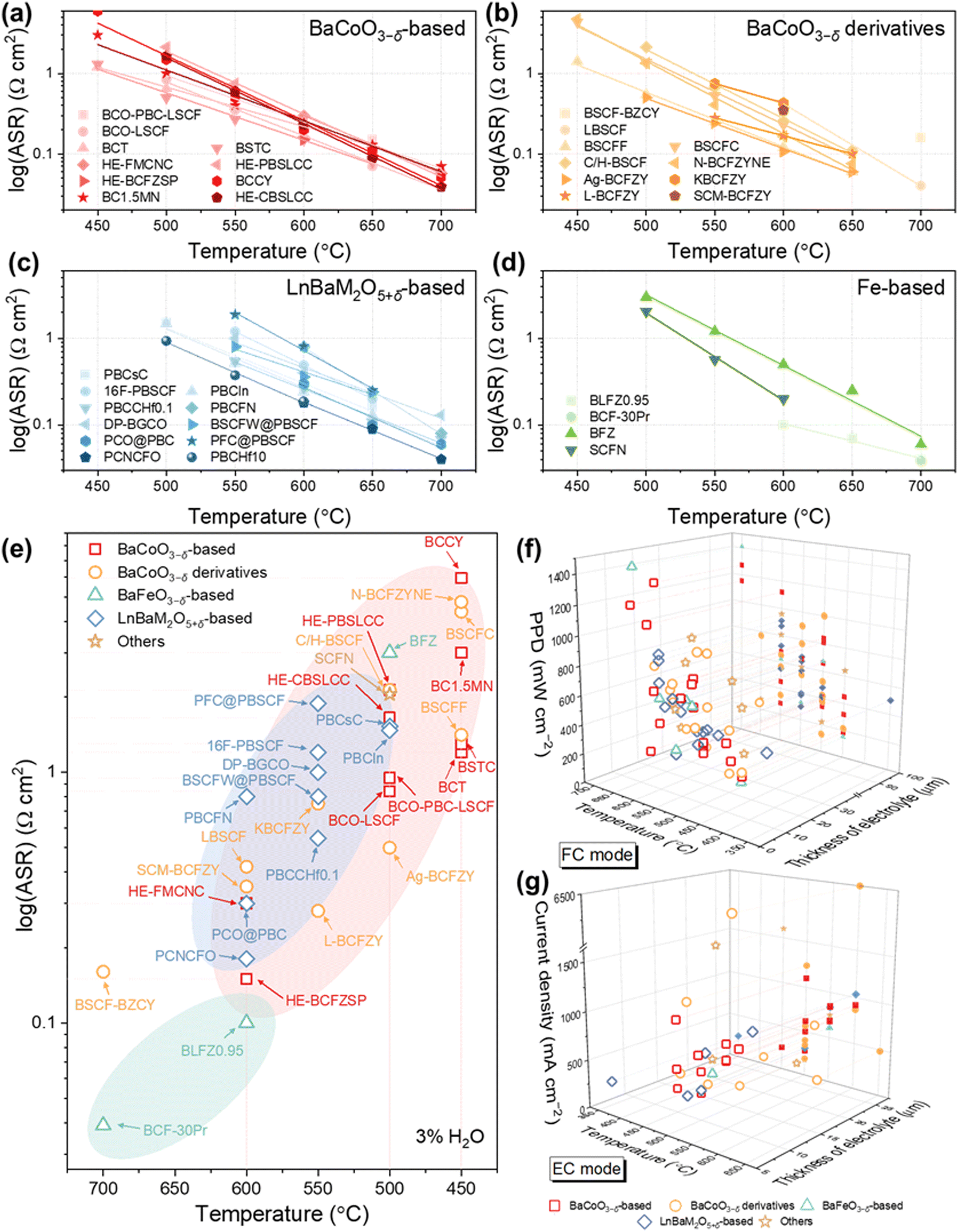 | ||
| Fig. 16 Comparison of ASR in symmetrical cell configuration of advanced air electrodes for P-SOCs at 3% H2O: (a) BaCoO3−δ-based perovskites, (b) BaCoO3−δ-derivated perovskites, (c) LnBaM2O6+δ-based perovskites, and (d) Fe-based perovskites. Summary of electrochemical properties of advanced perovskite air electrode materials (detailed data summarized in Table 1): (e) ASR of lowest working temperature in symmetrical cells configuration, (f) PPDs in fuel cell (FC) mode, and (g) current densities in electrolysis cell (EC) mode. | ||
Lowering operational temperatures is crucial for sustainability, as it reduces costs, enhances market feasibility, and opens the door for portable and transportable applications. Although P-SOCs operate at lower temperatures than O-SOCs—down to 350 °C—their performance remains insufficient for practical applications (Fig. 16(e)–(g)).236 At low temperatures, the primary challenge is typically the slow kinetics of ORR and OER. Current research often focuses on modifying existing air electrodes to enhance proton conduction, resulting in numerous variations and studies but limited genuine innovation. As operating temperatures continue to decrease, combining insights from low-temperature catalytic systems, such as PEM and anion exchange membranes, could offer valuable inspiration. For instance, low-temperature catalysis often achieves performance optimization through controlled crystal orientation and complex surface interface design. Furthermore, the reduced operating temperature enables direct observation of microevolution within the P-SOCs system. Advanced characterization techniques, including in situ TEM, can provide insights into the underlying mechanisms, thereby aiding and expediting the development of advanced air electrode materials.
Ensuring stable operation is a key aspect of optimizing both power generation and hydrogen production in P-SOCs, directly impacting efficiency, reliability, and system lifespan. Current treatment methods for air electrode materials like segregation, can raise concerns about durability. The failure of P-SOCs during operation may also arise from interface issues stemming from the thermal expansion differences between the electrode and the electrolyte. Improving the electrolyte/electrode interface is essential for enhancing air electrode. Given the complexity of the composition and elements in perovskite air electrode materials, employing AI-driven design can significantly speed up the material development process. Moreover, the P-SOC system is strongly influenced by physical factors like temperature and pressure. Finite element analysis can be applied to assess and forecast the performance of P-SOC devices in real-world operating environments.534
Lab-scale experiments are crucial for developing new technologies as they provide essential data for subsequent pilot and industrial-scale investigations. The reliability of this experimental data is vital as it guides future research directions, especially in the development of SOC technology for electrochemical energy storage and conversion which is key for achieving a net-zero carbon society. However, the complexity of lab-scale testing involving multiple parameters such as cell configuration, performance measurement strategies, and temperature distribution, can obscure true cell performance and affect power output. Discrepancies in results often arise from the omission of important parameter information in reports, leading to significant variances in literature. To facilitate reliable comparisons in future research, it is recommended that detailed information on various parameters including cell size, surface area ratios, sealing methods, and flow rates, be consistently provided.163 Additionally, the extra heat produced due to irreducible internal resistance during testing may lead to actual operating temperatures significantly exceeding the recorded values. This discrepancy underscores the need for more precisely defined experimental conditions.
Despite extensive research, commercial implementation of P-SOCs systems still has not materialized because of insufficient power outputs and high cost.34 Various considerations must be addressed during the laboratory stage to effectively align with commercial goals including the costs and availability of raw materials, expanded cells area and electrolyte sealing, safety and lifespan during storage and transportation, as well as the recyclability and environmental impact of waste P-SOCs.535,536 Consequently, it is advisable, in the early phases of research and development, to choose air electrode materials that are low-cost, safe, and environmentally friendly. This approach ensures that the materials are not only functional but also viable for future commercial applications.
Author contributions
C. P. and X. X. contributed to the central idea and conducted the draft. S. M. collected resources. P. K., Y. Z., and X. X. contributed to the writing and editing of the document.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. P. gratefully acknowledge financial support provided by University of Adelaide research scholarship. Open Access publishing facilitated by The University of Adelaide – The University of Adelaide agreement via the Council of Australian University Librarians. Y. Z., X. X. acknowledges funding from the Australian Research Council (LP210301397).References
- X. Yang, C. P. Nielsen, S. Song and M. B. McElroy, Nat. Energy, 2022, 7, 955–965 CrossRef CAS.
- I. Cho, J. Yun, B. Seong, J. Kim, S. H. Choi, H.-I. Ji and S. Choi, J. Energy Chem., 2024, 88, 1–9 CrossRef CAS.
- X. Xu, X. Han, Y. Zheng, W. Zhou, K. Davey and S.-Z. Qiao, Chem Catal., 2023, 3, 100794 CrossRef CAS.
- S. Tao and J. T. Irvine, Chem. Rev., 2004, 4, 83–95 CAS.
- I. Jang, S. A. C. Juliana, J. O. Crawford, Y. J. Cho, S. Parvin, D. A. Gonzalez-Casamachin, J. Baltrusaitis, R. P. Lively and E. Nikolla, Chem. Rev., 2024, 124, 8233–8306 CrossRef CAS PubMed.
- C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Phys. Chem. Chem. Phys., 2008, 10, 4623–4635 RSC.
- A. Peters, C. Korte, D. Hesse, N. Zakharov and J. Janek, Solid State Ionics, 2007, 178, 67–76 CrossRef CAS.
- L. Bi, S. Boulfrad and E. Traversa, Chem. Soc. Rev., 2014, 43, 8255–8270 RSC.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O’Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS.
- S. D. Ebbesen, S. H. Jensen, A. Hauch and M. B. Mogensen, Chem. Rev., 2014, 114, 10697–10734 CrossRef CAS PubMed.
- C. Tien, Trans. Phenom. Plasma, 1969, 5, 253–324 Search PubMed.
- E. Tezel, A. Whitten, G. Yarema, R. Denecke, J.-S. McEwen and E. Nikolla, ACS Catal., 2022, 12, 11456–11471 CrossRef CAS.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- C. Gunathilake, I. Soliman, D. Panthi, P. Tandler, O. Fatani, N. A. Ghulamullah, D. Marasinghe, M. Farhath, T. Madhujith and K. Conrad, Chem. Soc. Rev., 2024, 53, 10900–10969 RSC.
- J. Cao, Y. Ji and Z. Shao, Energy Environ. Sci., 2022, 15, 2200–2232 RSC.
- Z. Luo, Z. Wang, T. Zhu, Y. Song, Z. Lin, S. Jiang, Z. Zhu and Z. Shao, Energy Environ. Sci., 2024, 17, 6873–6896 RSC.
- Z. Shao and M. O. Tadé, Chem. Soc. Rev., 2016, 37, 1568 Search PubMed.
- J. Cao, Y. Ji and Z. Shao, Chem. Soc. Rev., 2024, 53, 450–501 RSC.
- F. Liu, D. Ding and C. Duan, Adv. Sci., 2023, 10, 2206478 CrossRef CAS PubMed.
- N. Wang, C. Tang, L. Du, R. Zhu, L. Xing, Z. Song, B. Yuan, L. Zhao, Y. Aoki and S. Ye, Adv. Energy Mater., 2022, 12, 2201882 CrossRef CAS.
- M. Wang, C. Su, Z. Zhu, H. Wang and L. Ge, Composites, Part B, 2022, 238, 109881 CrossRef CAS.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O’Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- Z. Wang, Z. Luo, H. Xu, T. Zhu, D. Guan, Z. Lin, T.-S. Chan, Y.-C. Huang, Z. Hu, S. P. Jiang and Z. Shao, Adv. Funct. Mater., 2024, 2402716, DOI:10.1002/adfm.202402716.
- C. Tang, Y. Yao, N. Wang, X. Zhang, F. Zheng, L. Du, D. Luo, Y. Aoki and S. Ye, InfoMat, 2024, 6, e12515 CrossRef CAS.
- L. Wang, R. Merkle and J. Maier, J. Electrochem. Soc., 2010, 157, B1802 CrossRef CAS.
- Y. Wang, Y. Ling, B. Wang, G. Zhai, G. Yang, Z. Shao, R. Xiao and T. Li, Energy Environ. Sci., 2023, 16, 5721–5770 RSC.
- P. Kazempoor and R. J. Braun, Int. J. Hydrogen Energy, 2014, 39, 2669–2684 CrossRef CAS.
- F. Gallucci, F. Chiaravalloti, S. Tosti, E. Drioli and A. Basile, Int. J. Hydrogen Energy, 2007, 32, 1837–1845 CrossRef CAS.
- Z. Wang, X. Zhou, L. Liu, H. Chen, X. Qian, F. He, Y. Sheng and X. Jiang, J. Chin. Ceram. Soc., 2023, 51, 2689–2699 CAS.
- C. Duan, J. Huang, N. Sullivan and R. O'Hayre, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- B. Yuan, N. Wang, C. Tang, L. Meng, L. Du, Q. Su, Y. Aoki and S. Ye, Nano Energy, 2024, 122, 109306 CrossRef CAS.
- M. Papac, V. Stevanovic, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed.
- W. Zhang, M. Liu, X. Gu, Y. Shi, Z. Deng and N. Cai, Chem. Rev., 2023, 123, 7119–7192 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, J. Wang, Y. Song, M. J. Robson, A. Seong, M. Yang, Z. Zhang, A. Belotti, J. Liu, G. Kim, J. Lim, Z. Shao and F. Ciucci, Nat. Catal., 2022, 5, 777–787 CrossRef CAS.
- T. Cooper, J. R. Scheffe, M. E. Galvez, R. Jacot, G. Patzke and A. Steinfeld, Energy Technol., 2015, 3, 1130–1142 CrossRef CAS.
- R. Jacobs, T. Mayeshiba, J. Booske and D. Morgan, Adv. Energy Mater., 2018, 8, 1702708 CrossRef.
- N. Ramadass, Mater. Sci. Eng., B, 1978, 36, 231–239 CrossRef CAS.
- Y. Song, X. Zhang, K. Xie, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1902033 CrossRef CAS PubMed.
- J. Jing, J. Pang, L. Chen, H. Zhang, Z. Lei and Z. Yang, Chem. Eng. J., 2022, 429, 132314 CrossRef CAS.
- K. Zhu, H. Liu, X. Li, Q. Li, J. Wang, X. Zhu and W. Yang, Electrochim. Acta, 2017, 241, 433–439 CrossRef CAS.
- Q. Zhang, J. Ding and M. He, J. Phys. Chem. Solids, 2017, 108, 76–81 CrossRef CAS.
- C. S. Choi and E. Prince, Acta Crystallogr., 1972, 28, 2857–2862 CrossRef CAS.
- J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu and G. Kim, ChemSusChem, 2014, 7, 2811–2815 CrossRef CAS PubMed.
- P. Ding, W. Li, H. Zhao, C. Wu, L. Zhao, B. Dong and S. Wang, JPhys Mater., 2021, 4, 022002 CrossRef CAS.
- A. Kushima, D. Parfitt, A. Chroneos, B. Yildiz, J. A. Kilner and R. W. Grimes, Phys. Chem. Chem. Phys., 2011, 13, 2242–2249 RSC.
- Y. Huan, S. Chen, R. Zeng, T. Wei, D. Dong, X. Hu and Y. Huang, Adv. Energy Mater., 2019, 9, 1901573 CrossRef.
- O. F. Schirmer, J. Phys.: Condens. Matter, 2006, 18, R667–R704 CrossRef CAS.
- A. J. Rettie, W. D. Chemelewski, D. Emin and C. B. Mullins, J. Phys. Chem. Lett., 2016, 7, 471–479 CrossRef CAS PubMed.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2011, 23, 1607–1617 CrossRef CAS.
- L. Tai, M. Nasrallah, H. Anderson, D. Sparlin and S. Sehlin, Solid State Ionics, 1995, 76, 259–271 CrossRef CAS.
- R. Raffaelle, H. U. Anderson, D. M. Sparlin and P. E. Parris, Phys. Rev. Lett., 1990, 65, 1383–1386 CrossRef CAS PubMed.
- X. Liu, H. Zhao, J. Yang, Y. Li, T. Chen, X. Lu, W. Ding and F. Li, J. Membr. Sci., 2011, 383, 235–240 CrossRef CAS.
- S. Kirkpatrick, Rev. Mod. Phys., 1973, 45, 574–588 CrossRef.
- D. Kim, S. Miyoshi, T. Tsuchiya and S. Yamaguchi, Solid State Ionics, 2014, 262, 875–878 CrossRef CAS.
- A. S. Foster, A. L. Shluger and R. M. Nieminen, Phys. Rev. Lett., 2002, 89, 225901 CrossRef CAS PubMed.
- L. J. Duckers, Phys. Lett., 1978, 67A, 93–94 CrossRef.
- Y. Ji and Y. Luo, J. Am. Chem. Soc., 2016, 138, 15896–15902 CrossRef CAS PubMed.
- C. L. Fu, M. Krčmar, G. S. Painter and X.-Q. Chen, Phys. Rev. Mater., 2007, 99, 225502 CAS.
- J. Cao, C. Su, Y. Ji, G. Yang and Z. Shao, J. Energy Chem., 2021, 57, 406–427 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Solid State Ionics, 2000, 135, 709–712 CrossRef CAS.
- C. Munnings, S. Skinner, G. Amow, P. Whitfield and I. Davidson, Solid State Ionics, 2005, 176, 1895–1901 CrossRef CAS.
- A. Chroneos, D. Parfitt, J. A. Kilner and R. W. Grimes, J. Mater. Chem., 2010, 20, 266–270 RSC.
- A. Grimaud, F. Mauvy, J. M. Bassat, S. Fourcade, L. Rocheron, M. Marrony and J. C. Grenier, J. Electrochem. Soc., 2012, 159, B683–B694 CrossRef CAS.
- Y. Yamazaki, P. Babilo and S. M. Haile, Chem. Mater., 2008, 20, 6352–6357 CrossRef CAS.
- I. Zvonareva, X.-Z. Fu, D. Medvedev and Z. Shao, Energy Environ. Sci., 2022, 15, 439–465 RSC.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- K. D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1999, 125, 285–302 CrossRef CAS.
- H. Uchida, N. Maeda and H. Iwahara, Solid State Ionics, 1983, 11, 117–124 CrossRef CAS.
- D. Poetzsch, R. Merkle and J. Maier, Faraday Discuss., 2015, 182, 129–143 RSC.
- D. Poetzsch, R. Merkle and J. Maier, Adv. Funct. Mater., 2015, 25, 1542–1557 CrossRef CAS.
- N. Wang, S. Hinokuma, T. Ina, H. Toriumi, M. Katayama, Y. Inada, C. Zhu, H. Habazaki and Y. Aoki, Chem. Mater., 2019, 31, 8383–8393 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- T. Scherban, R. Villeneuve, L. Abello and G. Lucazeau, Solid State Ionics, 1993, 61, 93–98 CrossRef CAS.
- T. Ueki and M. Watanabe, Macromolecules, 2008, 41, 3739 CrossRef CAS.
- K. D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed. Engl., 1982, 21, 208–209 CrossRef.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, Renewable Sustainable Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- P. Ramaswamy, N. E. Wong and G. K. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- G. Geneste, Solid State Ionics, 2018, 323, 172–202 CrossRef CAS.
- M. Li, M. Zhao, F. Li, W. Zhou, V. K. Peterson, X. Xu, Z. Shao, I. Gentle and Z. Zhu, Nat. Commun., 2017, 8, 13990 CrossRef CAS PubMed.
- Z. Li, X. Mao, D. Feng, M. Li, X. Xu, Y. Luo, L. Zhuang, R. Lin, T. Zhu, F. Liang, Z. Huang, D. Liu, Z. Yan, A. Du, Z. Shao and Z. Zhu, Nat. Commun., 2024, 15, 9318 CrossRef CAS PubMed.
- H. Dai, Y. Yin, X. Li, C. Ma, Z. Chen, M. Hua and L. Bi, Sustainable Mater. Technol., 2022, 32, e00409 CrossRef CAS.
- R. Ren, Z. Wang, X. Meng, X. Wang, C. Xu, J. Qiao, W. Sun and K. Sun, ACS Appl. Energy Mater., 2020, 3, 4914–4922 CrossRef CAS.
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS.
- Y. Yin, D. Xiao, S. Wu, E. H. Da'as, Y. Gu and L. Bi, Susmat, 2023, 3, 697–708 CrossRef CAS.
- Y. Yin, Y. Zhou, Y. Gu and L. Bi, J. Adv. Ceram., 2023, 12, 587–597 CrossRef CAS.
- Y. Lin, R. Ran, Y. Zheng, Z. Shao, W. Jin, N. Xu and J. Ahn, J. Power Sources, 2008, 180, 15–22 CrossRef CAS.
- E. Fabbri, D. Pergolesi, S. Licoccia and E. Traversa, Solid State Ionics, 2010, 181, 1043–1051 CrossRef CAS.
- Y. Yamazaki, F. Blanc, Y. Okuyama, L. Buannic, J. C. Lucio-Vega, C. P. Grey and S. M. Haile, Nat. Mater., 2013, 12, 647–651 CrossRef CAS PubMed.
- M. Yu, Q. Feng, Z. Liu, P. Zhang, X. Zhu and S. Mu, Crystals, 2024, 14, 225 CrossRef CAS.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef.
- K. Bae, D. Y. Jang, H. J. Choi, D. Kim, J. Hong, B. K. Kim, J. H. Lee, J. W. Son and J. H. Shim, Nat. Commun., 2017, 8, 14553 CrossRef CAS PubMed.
- Z. Wang, W. Yang, S. P. Shafi, L. Bi, Z. Wang, R. Peng, C. Xia, W. Liu and Y. Lu, J. Mater. Chem. A, 2015, 3, 8405–8412 RSC.
- E. Fabbri, I. Markus, L. Bi, D. Pergolesi and E. Traversa, Solid State Ionics, 2011, 202, 30–35 CrossRef CAS.
- Z. Tao, L. Bi, Z. Zhu and W. Liu, J. Power Sources, 2009, 194, 801–804 CrossRef CAS.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Energy Environ. Sci., 2011, 4, 4984 RSC.
- J. Choi, B. Kim, S.-H. Song and J.-S. Park, Int. J. Hydrogen Energy, 2016, 41, 9619–9626 CrossRef CAS.
- J. Dailly, G. Taillades, M. Ancelin, P. Pers and M. Marrony, J. Power Sources, 2017, 361, 221–226 CrossRef CAS.
- D. Kim, T. K. Lee, S. Han, Y. Jung, D. G. Lee, M. Choi and W. Lee, Mater, Today Energy, 2023, 36, 101365 CAS.
- H. Zhu, S. Ricote, C. Duan, R. P. O’Hayre, D. S. Tsvetkov and R. J. Kee, J. Electrochem. Soc., 2018, 165, F581–F588 CrossRef CAS.
- D. K. Lim, C. J. Park, M. B. Choi, C. N. Park and S. J. Song, Int. J. Hydrogen Energy, 2010, 35, 10624–10629 CrossRef CAS.
- H. Zhu and R. J. Kee, Int. J. Hydrogen Energy, 2016, 41, 2931–2943 CrossRef CAS.
- C. Duan, D. Hook, Y. Chen, J. Tong and R. O'Hayre, Energy Environ. Sci., 2017, 10, 176–182 RSC.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Adv. Mater., 2012, 24, 195–208 CrossRef CAS PubMed.
- D. Chen, C. Chen, Z. M. Baiyee, Z. Shao and F. Ciucci, Chem. Rev., 2015, 115, 9869–9921 CrossRef CAS PubMed.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- Y.-L. Lee, J. Kleis, J. Rossmeisl, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2011, 4, 3966 RSC.
- E. D. Wachsman and K. T. Lee, Science, 2011, 334, 935–939 CrossRef CAS PubMed.
- D. N. Mueller, M. L. Machala, H. Bluhm and W. C. Chueh, Nat. Commun., 2015, 6, 6097 CrossRef CAS PubMed.
- J. Suntivich, W. T. Hong, Y.-L. Lee, J. M. Rondinelli, W. Yang, J. B. Goodenough, B. Dabrowski, J. W. Freeland and Y. Shao-Horn, J. Phys. Chem. C, 2014, 118, 1856–1863 CrossRef CAS.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- W. T. Hong, K. A. Stoerzinger, B. Moritz, T. P. Devereaux, W. Yang and Y. Shao-Horn, J. Phys. Chem. C, 2015, 119, 2063–2072 CrossRef CAS.
- K. Li, Y. Liang, J. Zhang, B. Yu and L. Jia, Int. J. Hydrogen Energy, 2024, 91, 858–866 CrossRef CAS.
- R. Peng, T. Wu, W. Liu, X. Liu and G. Meng, J. Mater. Chem., 2010, 20, 6218 RSC.
- E. Fabbri, D. Pergolesi and E. Traversa, Sci. Technol. Adv. Mater., 2010, 11, 044301 CrossRef PubMed.
- R. Majee, Q. A. Islam, S. Mondal and S. Bhattacharyya, Chem. Sci., 2020, 11, 10180–10189 RSC.
- Y. Yuan, R. K. Patel, S. Banik, T. B. Reta, R. S. Bisht, D. D. Fong, S. Sankaranarayanan and S. Ramanathan, Chem. Rev., 2024, 124, 9733–9784 CrossRef CAS PubMed.
- H. Tellez Lozano, J. Druce, S. J. Cooper and J. A. Kilner, Sci. Technol. Adv. Mater., 2017, 18, 977–986 CrossRef PubMed.
- H. Hayashi, H. Inaba, M. Matsuyama, N. G. Lan, M. Dokiya and H. Tagawa, Solid State Ionics, 1999, 122, 1–15 CrossRef CAS.
- J. A. Kilner and R. J. Brook, Solid State Ionics, 1982, 6, 237–252 CrossRef CAS.
- M. S. Islam, Solid State Ionics, 2002, 154–155, 75–85 CrossRef CAS.
- A. F. Sammells, R. L. Cook, J. H. White, J. J. Osborne and R. C. MacDuff, Solid State Ionics, 1992, 52, 111–123 CrossRef CAS.
- M. A. Peña and L. G. Fierro, Chem. Rev., 2001, 101, 1981–2017 CrossRef PubMed.
- L. Liang, L. Wencong and C. Nianyi, J. Phys. Chem. Solids, 2004, 65, 855–860 CrossRef.
- H. Yokokawa, N. Sakai, T. Kawada and M. Dokiya, J. Solid State Chem., 1991, 94, 106–120 CrossRef CAS.
- A. A. Taskin, A. N. Lavrov and Y. Ando, Appl. Phys. Lett., 2005, 86, 091910 CrossRef.
- C. Bernuy-Lopez, L. Rioja-Monllor, T. Nakamura, S. Ricote, R. O'Hayre, K. Amezawa, M. A. Einarsrud and T. Grande, Materials, 2018, 11, 196 CrossRef PubMed.
- D. Parfitt, A. Chroneos, A. Tarancón and J. A. Kilner, J. Mater. Chem., 2011, 21, 2183–2186 RSC.
- A. Arulraj, F. Goutenoire, M. Tabellout, O. Bohnke and P. Lacorre, Chem. Mater., 2002, 14, 2492–2498 CrossRef CAS.
- I. Animitsa, N. Tarasova and Y. Filinkova, Solid State Ionics, 2012, 207, 29–37 CrossRef CAS.
- J. C. Ruiz-Morales, J. Canales-Vázquez, C. Savaniu, D. Marrero-López, W. Zhou and J. T. Irvine, Nature, 2006, 439, 568–571 CrossRef CAS PubMed.
- J. H. Kim, D. Kim, S. Ahn, K. J. Kim, S. Jeon, D.-K. Lim, J. K. Kim, U. Kim, H.-N. Im, B. Koo, K. T. Lee and W. Jung, Energy Environ. Sci., 2023, 16, 3803–3814 RSC.
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC.
- J. Wang, B. Wang, G. Huangfu, H. Zhang and Y. Guo, Acta Mater., 2024, 120520 Search PubMed.
- N. Ran, W. Qiu, E. Song, Y. Wang, X. Zhao, Z. Liu and J. Liu, Chem. Mater., 2020, 32, 1224–1234 CrossRef CAS.
- J. Bielecki, S. F. Parker, L. Mazzei, L. Börjesson and M. Karlsson, J. Mater. Chem. A, 2016, 4, 1224–1232 RSC.
- K. Saito, K. Umeda, K. Fujii, K. Mori and M. Yashima, J. Mater. Chem. A, 2024, 12, 13310–13319 RSC.
- M. T. Greiner, L. Chai, M. G. Helander, W. M. Tang and Z. H. Lu, Adv. Funct. Mater., 2012, 22, 4557–4568 CrossRef CAS.
- Z. Sherafat, M. H. Paydar, I. Antunes, N. Nasani, A. D. Brandão and D. P. Fagg, Electrochim. Acta, 2015, 165, 443–449 CrossRef CAS.
- N. Sakai, Solid State Ionics, 2004, 175, 387–391 CrossRef CAS.
- N. Sakai, K. Yamaji, T. Horita, Y. P. Xiong, H. Kishimoto and H. Yokokawa, J. Electrochem. Soc., 2003, 150, A689–A694 CrossRef CAS.
- R. R. Liu, S. H. Kim, S. Taniguchi, T. Oshima, Y. Shiratori, K. Ito and K. Sasaki, J. Power Sources, 2011, 196, 7090–7096 CrossRef CAS.
- S. Ricote, G. Caboche, C. Estournès and N. Bonanos, J. Nanomater., 2008, 2008, 354258–354262 CrossRef.
- D. Hashimoto, D. Han and T. Uda, Solid State Ionics, 2014, 262, 687–690 CrossRef CAS.
- A. Grimaud, J. M. Bassat, F. Mauvy, M. Pollet, A. Wattiaux, M. Marrony and J. C. Grenier, J. Mater. Chem. A, 2014, 2, 3594 RSC.
- X. Xu, J. Zhao, M. Li, L. Zhuang, J. Zhang, S. Aruliah, F. Liang, H. Wang and Z. Zhu, Composites, Part B, 2019, 178, 107491 CrossRef CAS.
- W. Zhou, B. An, R. Ran and Z. Shao, J. Electrochem. Soc., 2009, 156, B884–B890 CrossRef CAS.
- J. Tallgren, O. Himanen, M. Bianco, J. Mikkola, O. Thomann, M. Rautanen, J. Kiviaho and J. Van herle, Fuel Cells, 2019, 19, 570–577 CrossRef CAS.
- W. Preis, E. Bucher and W. Sitte, Fuel Cells, 2012, 12, 543–549 CrossRef CAS.
- S. Zhao, W. Ma, W. Wang, Y. Huang, J. Wang, S. Wang, Z. Shu, B. He and L. Zhao, Adv. Mater., 2024, 36, 2405052 CrossRef CAS PubMed.
- X. Chen, N. Yu, Y. Song, T. Liu, H. Xu, D. Guan, Z. Li, W. H. Huang, Z. Shao, F. Ciucci and M. Ni, Adv. Mater., 2024, 36, 2403998 CrossRef CAS PubMed.
- A. Seong, J. Kim, D. Jeong, S. Sengodan, M. Liu, S. Choi and G. Kim, Adv. Sci., 2021, 8, 2004099 CrossRef CAS PubMed.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- C. Mänken, D. Schäfer and R.-A. Eichel, J. Electrochem. Soc., 2024, 171, 114501 CrossRef.
- J.-I. Jung, S. T. Misture and D. D. Edwards, Solid State Ionics, 2010, 181, 1287–1293 CrossRef CAS.
- S. Wang, Solid State Ionics, 2003, 159, 71–78 CrossRef CAS.
- J. Song, S. Zhu, D. Ning and H. J. Bouwmeester, J. Mater. Chem. A, 2021, 9, 974–989 RSC.
- Y. Chen, T. Hong, P. Wang, K. Brinkman, J. Tong and J. Cheng, J. Power Sources, 2019, 440, 227122 CrossRef CAS.
- J. Gu, L. Jiang, S. A. Ismail, H. Guo and D. Han, Adv. Mater. Interfaces, 2023, 10, 2201764 CrossRef CAS.
- D. Zou, Y. Yi, Y. Song, D. Guan, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 5381–5390 RSC.
- Y.-D. Kim, C. Meisel, I.-H. Kim, C. Herradón, P. Rand, J. Yang, N. P. Sullivan and R. O'Hayre, J. Power Sources, 2025, 625, 235700 CrossRef CAS.
- Q. Wang, T. Luo, Y. Tong, M. Dai, X.-Y. Miao, S. Ricote, Z. Zhan and M. Chen, Sep. Purif. Technol., 2022, 295, 121301 CrossRef CAS.
- S. Guo, L. Jiang, Y. Li, P. Zhong, S. A. Ismail, T. Norby and D. Han, Adv. Funct. Mater., 2024, 34, 2304729 CrossRef CAS.
- U. Tariq, M. Z. Khan, O. Gohar, Z. U. Din Babar, F. Ali, R. A. Malik, I. A. Starostina, Samia, J. Rehman, I. Hussain, M. Saleem, A. Ghaffar, M. A. Marwat, K. Zheng, M. Motola and M. B. Hanif, J. Power Sources, 2024, 613, 234910 CrossRef CAS.
- C. Zhou, J. Sunarso, Y. Song, J. Dai, J. Zhang, B. Gu, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 13265–13274 RSC.
- R. Ren, X. Yu, Z. Wang, C. Xu, T. Song, W. Sun, J. Qiao and K. Sun, Appl. Catal., B, 2022, 317, 121759 CrossRef CAS.
- C. Zhou, X. Wang, D. Liu, M. Fei, J. Dai, D. Guan, Z. Hu, L. Zhang, Y. Wang, W. Wang, R. O'Hayre, S. P. Jiang, W. Zhou, M. Liu and Z. Shao, Energy Environ. Mater., 2024, 7, e12660 CrossRef CAS.
- S. Paul, S.-J. Choi and H. J. Kim, Energy Fuels, 2020, 34, 10067–10077 CrossRef CAS.
- N. N. Krishnan, N. M. H. Duong, A. Konovalova, J. H. Jang, H. S. Park, H. J. Kim, A. Roznowska, A. Michalak and D. Henkensmeier, J. Membr. Sci., 2020, 614, 118494 CrossRef CAS.
- Y. Zhang, B. Chen, D. Guan, M. Xu, R. Ran, M. Ni, W. Zhou, R. O'Hayre and Z. Shao, Nature, 2021, 591, 246–251 CrossRef CAS PubMed.
- Y. Song, J. Liu, Y. Wang, D. Guan, A. Seong, M. Liang, M. J. Robson, X. Xiong, Z. Zhang, G. Kim, Z. Shao and F. Ciucci, Adv. Energy Mater., 2021, 11, 2101899 CrossRef CAS.
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang and Y. Zhang, Nat. Commun., 2020, 11, 1907 CrossRef CAS PubMed.
- X. Wu, Y. Guo, Z. Sun, F. Xie, D. Guan, J. Dai, F. Yu, Z. Hu, Y.-C. Huang and C.-W. Pao, Nat. Commun., 2021, 12, 660 CrossRef CAS PubMed.
- D. Guan, G. Ryu, Z. Hu, J. Zhou, C.-L. Dong, Y.-C. Huang, K. Zhang, Y. Zhong, A. C. Komarek and M. Zhu, Nat. Commun., 2020, 11, 3376 CrossRef CAS PubMed.
- S. Agrestini, K. Chen, C.-Y. Kuo, L. Zhao, H.-J. Lin, C.-T. Chen, A. Rogalev, P. Ohresser, T.-S. Chan and S.-C. Weng, Phys. Rev. B, 2019, 100, 014443 CrossRef CAS.
- K. Ye, K. Li, Y. Lu, Z. Guo, N. Ni, H. Liu, Y. Huang, H. Ji and P. Wang, TrAC, Trends Anal. Chem., 2019, 116, 102–108 CrossRef CAS.
- Y.-M. Kim, J. He, M. D. Biegalski, H. Ambaye, V. Lauter, H. M. Christen, S. T. Pantelides, S. J. Pennycook, S. V. Kalinin and A. Y. Borisevich, Nat. Mater., 2012, 11, 888–894 CrossRef CAS PubMed.
- Z. F. Zhao, Z. X. Xue, Q. Q. Xiong, Y. Q. Zhang, X. S. Hu, H. Z. Chi, H. Y. Qin, Y. J. Yuan and H. L. Ni, Sustainable Mater. Technol., 2021, 30, e00357 CrossRef CAS.
- Z. Liu, Y. Lin, H. Nie, D. Liu, Y. Li, X. Zhao, T. Li, G. Yang, Y. Sun, Y. Zhu, W. Wang, R. Ran, W. Zhou and Z. Shao, Adv. Funct. Mater., 2024, 34, 2311140 CrossRef CAS.
- L. A. Lyon, C. D. Keating, A. P. Fox, B. E. Baker, L. He, S. R. Nicewarner, S. P. Mulvaney and M. J. Natan, Anal. Chem., 1998, 70, 341–362 CrossRef PubMed.
- X. Li, K. Blinn, D. Chen and M. Liu, Electrochem. Energy Rep., 2018, 1, 433–459 CrossRef CAS.
- Y. Song, J. Min, Y. Guo, R. Li, G. Zou, M. Li, Y. Zang, W. Feng, X. Yao, T. Liu, X. Zhang, J. Yu, Q. Liu, P. Zhang, R. Yu, X. Cao, J. Zhu, K. Dong, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2024, 63, e202313361 CrossRef CAS PubMed.
- C. Lu, R. Ren, Z. Zhu, G. Pan, G. Wang, C. Xu, J. Qiao, W. Sun, Q. Huang, H. Liang, Z. Wang and K. Sun, Chem. Eng. J., 2023, 472, 144878 CrossRef CAS.
- J. W. Smith and R. J. Saykally, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- F. M. de Groot, M. W. Haverkort, H. Elnaggar, A. Juhin, K.-J. Zhou and P. Glatzel, Nat. Rev. Methods Primers, 2024, 4, 45 CrossRef CAS.
- H. Over, Y. D. Kim, A. Seitsonen, S. Wendt, E. Lundgren, M. Schmid, P. Varga, A. Morgante and G. Ertl, Science, 2000, 287, 1474–1476 CrossRef CAS PubMed.
- F. He, Y. Zhou, T. Hu, Y. Xu, M. Hou, F. Zhu, D. Liu, H. Zhang, K. Xu, M. Liu and Y. Chen, Adv. Mater., 2023, 35, 2209469 CrossRef CAS PubMed.
- Z. Cheng, J.-H. Wang, Y. Choi, L. Yang, M.-C. Lin and M. Liu, Energy Environ. Sci., 2011, 4, 4380–4409 RSC.
- L. Pauling, J. Chem. Educ., 1992, 69, 519–521 CrossRef CAS.
- https://www.metal.com/price .
- https://en.institut-seltene-erden.de/current-prices-of-strategicmetals/ .
- R. Shannon, Acta Crystallogr., 1976, 32, 751–767 CrossRef.
- Y. Niu, Y. Zhou, W. Zhang, Y. Zhang, C. Evans, Z. Luo, N. Kane, Y. Ding, Y. Chen, X. Guo, W. Lv and M. Liu, Adv. Energy Mater., 2022, 12, 2103783 CrossRef CAS.
- Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao, L. Soule, Y. Niu, Y. Ding, H. Ding, D. Ding and M. Liu, ACS Energy Lett., 2021, 6, 1511–1520 CrossRef CAS.
- Y. Zhou, W. Zhang, N. Kane, Z. Luo, K. Pei, K. Sasaki, Y. Choi, Y. Chen, D. Ding and M. Liu, Adv. Funct. Mater., 2021, 31, 2105386 CrossRef CAS.
- H. Zheng, X. Zhou, X. Wang, Z. Zhao, Y. Wang, C. Xie, Y. Wang, H. Li and X. Ding, J. Power Sources, 2024, 597, 234164 CrossRef CAS.
- K. Shi, Y. Yin, Z. Tang, S. Yu and Q. Zhang, Ceram. Int., 2022, 48, 13024–13031 CrossRef CAS.
- S. Peng, S. Lei, S. Wen, G. Weng, K. Ouyang, Z. Yin, J. Xue and H. Wang, Int. J. Hydrogen Energy, 2023, 48, 22209–22219 CrossRef CAS.
- D. Kim, I. Jeong, S. Ahn, S. Oh, H. N. Im, H. Bae, S. J. Song, C. W. Lee, W. Jung and K. T. Lee, Adv. Energy Mater., 2024, 14, 2304059 CrossRef CAS.
- X. Wang, W. Li, C. Zhou, M. Xu, Z. Hu, C.-W. Pao, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2023, 15, 1339–1347 CrossRef CAS PubMed.
- M. Fu, W. Hu, H. Tong, X. Ling, L. Tan, F. Chen and Z. Tao, J. Adv. Ceram., 2024, 13, 63–72 CrossRef CAS.
- Y. Xu, X. Xu and L. Bi, J. Adv. Ceram., 2022, 11, 794–804 CrossRef CAS.
- Z. Liu, Z. Tang, Y. Song, G. Yang, W. Qian, M. Yang, Y. Zhu, R. Ran, W. Wang, W. Zhou and Z. Shao, Nano-Micro Lett., 2022, 14, 217 CrossRef CAS PubMed.
- F. He, Y. Zhou, T. Hu, Y. Xu, M. Hou, F. Zhu, D. Liu, H. Zhang, K. Xu, M. Liu and Y. Chen, Adv. Mater., 2023, 35, 2209469 CrossRef CAS PubMed.
- J. Sun, R. Ren, H. Yue, W. Cui, G. Wang, C. Xu, J. Qiao, W. Sun, K. Sun and Z. Wang, Chin. Chem. Lett., 2023, 34, 107776 CrossRef CAS.
- C. Yang, J. Li, S. Hu, J. Pu and B. Chi, Ceram. Int., 2023, 49, 38331–38338 CrossRef CAS.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Joule, 2019, 3, 2842–2853 CrossRef CAS.
- F. He, S. Liu, T. Wu, M. Yang, W. Li, G. Yang, F. Zhu, H. Zhang, K. Pei, Y. Chen, W. Zhou and Z. Shao, Adv. Funct. Mater., 2022, 32, 2206756 CrossRef CAS.
- F. He, M. Hou, D. Liu, Y. Ding, K. Sasaki, Y. Choi, S. Guo, D. Han, Y. Liu, M. Liu and Y. Chen, Energy Environ. Sci., 2024, 17, 3898–3907 RSC.
- J. Seo, H.-W. Kim, J. H. Yu and H. J. Park, J. Korean Ceram. Soc., 2022, 59, 217–223 CrossRef CAS.
- X. Yang, Z. Wang, G. Li, Y. Zhou, C. Sun and L. Bi, Ceram. Int., 2024, 50, 40375–40383 CrossRef CAS.
- Z. Liu, D. Cheng, Y. Zhu, M. Liang, M. Yang, G. Yang, R. Ran, W. Wang, W. Zhou and Z. Shao, Chem. Eng. J., 2022, 450, 137787 CrossRef CAS.
- X. Chen, N. Yu, I. T. Bello, D. Guan, Z. Li, T. Liu, T. Liu, Z. Shao and M. Ni, Energy Storage Mater., 2023, 63, 103056 CrossRef.
- Z. Liu, Y. Bai, H. Sun, D. Guan, W. Li, W.-H. Huang, C.-W. Pao, Z. Hu, G. Yang, Y. Zhu, R. Ran, W. Zhou and Z. Shao, Nat. Commun., 2024, 15, 472 CrossRef CAS PubMed.
- M. Liang, Y. Zhu, Y. Song, D. Guan, Z. Luo, G. Yang, S. P. Jiang, W. Zhou, R. Ran and Z. Shao, Adv. Mater., 2022, 34, 2106379 CrossRef CAS PubMed.
- J. H. Kim, J. Hong, D.-K. Lim, S. Ahn, J. Kim, J. K. Kim, D. Oh, S. Jeon, S.-J. Song and W. Jung, Energy Environ. Sci., 2022, 15, 1097–1105 RSC.
- K. Park, M. Saqib, H. Lee, D. Shin, M. Jo, K. M. Park, M. Hamayun, S. H. Kim, S. Kim, K.-S. Lee, R. O’Hayre, M. Choi, S.-J. Song and J.-Y. Park, Energy Environ. Sci., 2024, 17, 1175–1188 RSC.
- Y. Wang, Z. Wang, K. Yang, J. Liu, Y. Song, J. Li, Z. Hu, M. J. Robson, Z. Zhang, Y. Tian, S. Xu, Y. Lu, H. M. Law, F. Liu, Q. Chen, Z. Yang and F. Ciucci, Adv. Funct. Mater., 2024, 34, 2404846 CrossRef CAS.
- T. Zhou, H. Huang, Y. Meng, J. Conrad, M. Zou, Z. Zhao, K. S. Brinkman and J. Tong, ACS Energy Lett., 2024, 9, 4557–4563 CrossRef CAS.
- Z. Yu, L. Ge, Q. Ni, Y. Zheng, H. Chen, X. Zhou, Y. Mi, B. Shi, X. Yu, B. Wu, L. Bi and Y. Zhu, Adv. Funct. Mater., 2024, 34, 2309698 CrossRef CAS.
- X. Lu, Z. Yang, J. Zhang, X. Zhao, J. Chen, W. Liu, Y. Zhao and Y. Li, J. Power Sources, 2024, 599, 234233 CrossRef CAS.
- H. Tong, M. Fu, Y. Yang, F. Chen and Z. Tao, Adv. Funct. Mater., 2022, 32, 2209695 CrossRef CAS.
- Q. Ye, H. Ye, Z. Ma, H. Lin, B. Zhao, G. Yang, F. Dong, M. Ni, Z. Lin and S. Zhang, Small, 2024, 20, 2307900 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, Y. Xiao, Y. Zhang, X. Wang, F. Wang and T. He, Small, 2024, 20, 2312148 CrossRef CAS PubMed.
- S. Zhao, W. Ma, B. He, Y. Ling, Y. Huang, F. Hu, Z. Shu and L. Zhao, Adv. Mater., 2025, e2501387, DOI:10.1002/adma.202501387.
- Y. Xu, K. Xu, F. Zhu, F. He, H. Zhang, C. Fang, Y. Liu, Y. Zhou, Y. Choi and Y. Chen, ACS Energy Lett., 2023, 8, 4145–4155 CrossRef CAS.
- Y. Xu, Y. Huang, Y. Guo, F. Hu, J. Xu, W. Zhou, Z. Yang, J. Sun, B. He and L. Zhao, Sep. Purif. Technol., 2022, 290, 120844 CrossRef CAS.
- Z. Du, K. Xu, F. Zhu, Y. Xu, F. He, H. Gao, W. Gong, Y. Choi and Y. Chen, Adv. Funct. Mater., 2024, 34, 2409188 CrossRef CAS.
- K. Xu, H. Zhang, Y. Xu, D. Liu, F. Zhu, F. He, Y. Liu, H. Wang and Y. Chen, Adv. Powder Mater., 2024, 3, 100187 CrossRef.
- K. Xu, H. Zhang, Y. Xu, F. He, Y. Zhou, Y. Pan, J. Ma, B. Zhao, W. Yuan, Y. Chen and M. Liu, Adv. Funct. Mater., 2022, 32, 2110998 CrossRef CAS.
- X. Zhang, R. Song, D. Huan, K. Zhu, X. Li, H. Han, C. Xia, R. Peng and Y. Lu, Small, 2022, 18, 2205190 CrossRef CAS PubMed.
- X. Li, Z. Jin, C. Wang, R. Peng, Y. Zha, J. Cao, Y. Ji and Z. Shao, Adv. Energy Mater., 2024, 14, 2400319 CrossRef CAS.
- K. Pei, S. Luo, F. He, J. Arbiol, Y. Xu, F. Zhu, Y. Wang and Y. Chen, Appl. Catal., B, 2023, 330, 122601 CrossRef CAS.
- H. Zhang, K. Xu, F. He, Y. Zhou, K. Sasaki, B. Zhao, Y. Choi, M. Liu and Y. Chen, Adv. Energy Mater., 2022, 12, 2200761 CrossRef CAS.
- H. Gao, F. He, F. Zhu, J. Xia, Z. Du, Y. Huang, L. Zhu and Y. Chen, Adv. Funct. Mater., 2024, 34, 2401747 CrossRef CAS.
- M. Choi, D. Kim, T. K. Lee, J. Lee, H. S. Yoo and W. Lee, Adv. Energy Mater., 2025, 15, 2400124 CrossRef CAS.
- X. Hu, Y. Zhou, Z. Luo, H. Li, N. Shi, Z. Liu, W. Zhang, W. Wang, Y. Ding and M. Liu, Energy Environ. Sci., 2024, 17, 9335 RSC.
- B. S. Teketel, B. A. Beshiwork, X. Luo, D. Tian, S. Zhu, H. G. Desta, Q. Yang, Y. Chen and B. Lin, Ceram. Int., 2022, 48, 37232–37241 CrossRef CAS.
- A. P. Tarutin, Y. G. Lyagaeva, A. I. Vylkov, M. Y. Gorshkov, G. K. Vdovin and D. A. Medvedev, J. Mater. Sci. Technol., 2021, 93, 157–168 CrossRef CAS.
- K. Zhu, L. Zhang, N. Shi, B. Qiu, X. Hu, D. Huan, C. Xia, R. Peng and Y. Lu, ACS Nano, 2024, 18, 5141–5151 CrossRef CAS PubMed.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Nature, 2022, 604, 479–485 CrossRef CAS PubMed.
- P. Yao, J. Zhang, Q. Qiu, Y. Zhao, F. Yu and Y. Li, Adv. Energy Mater., 2024, 2403335, DOI:10.1002/aenm.202403335.
- J. Hwang, R. R. Rao, L. Giordano, Y. Katayama, Y. Yu and Y. Shao-Horn, Science, 2017, 358, 751–756 CrossRef CAS PubMed.
- M. Medarde, C. Dallera, M. Grioni, J. Voigt, A. Podlesnyak, E. Pomjakushina, K. Conder, T. Neisius, O. Tjernberg and S. N. Barilo, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 054424 CrossRef.
- C. Frontera, J. L. García-Muñoz, A. E. Carrillo, C. Ritter, D. Martín y Marero and A. Caneiro, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 184428 CrossRef.
- E. Marelli, J. Lyu, M. Morin, M. Lemenager, T. Shang, N. S. Yuzbasi, D. Aegerter, J. Huang, N. D. Daffe, A. H. Clark, D. Sheptyakov, T. Graule, M. Nachtegaal, E. Pomjakushina, T. J. Schmidt, M. Krack, E. Fabbri and M. Medarde, EES Catal., 2024, 2, 335–350 RSC.
- A. J. Barón-González, C. Frontera, J. L. García-Muñoz, J. Blasco and C. Ritter, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 054427 CrossRef.
- Y. Song, Y. Yi, R. Ran, W. Zhou and W. Wang, Small, 2024, e2406627, DOI:10.1002/smll.202406627.
- V. Cascos, R. Martínez-Coronado and J. A. Alonso, Int. J. Hydrogen Energy, 2014, 39, 14349–14354 CrossRef CAS.
- I. Jeong, S. J. Jeong, B.-H. Yun, J.-W. Lee, C.-W. Lee, W. Jung and K. T. Lee, NPG Asia Mater., 2022, 14, 53 CrossRef CAS.
- M. Li, W. Zhou and Z. Zhu, ChemElectroChem, 2015, 2, 1331–1338 CrossRef CAS.
- Z. Xu, J. Yu and W. Wang, Membranes, 2022, 12, 831 CrossRef CAS PubMed.
- K. Pei, Y. Zhou, K. Xu, Z. He, Y. Chen, W. Zhang, S. Yoo, B. Zhao, W. Yuan, M. Liu and Y. Chen, Nano Energy, 2020, 72, 104704 CrossRef CAS.
- T. Hibino, A. Hashimoto, M. Suzuki and M. Sano, J. Electrochem. Soc., 2002, 149, A1503 CrossRef CAS.
- S. Svarcova, Solid State Ionics, 2008, 178, 1787–1791 CrossRef CAS.
- W. Zhou, J. Sunarso, J. Motuzas, F. Liang, Z. Chen, L. Ge, S. Liu, A. Julbe and Z. Zhu, Chem. Mater., 2011, 23, 1618–1624 CrossRef CAS.
- T. Ishihara, S. Fukui, H. Nishiguchi and Y. Takita, J. Electrochem. Soc., 2002, 149, A823 CrossRef CAS.
- T. Ishihara, Proc. Vol., 2001, 2001-16, 439–448 CrossRef CAS.
- T. Ishihara, S. Fukui, H. Nishiguchi and Y. Takita, Solid State Ionics, 2002, 152–153, 609–613 CrossRef CAS.
- Y. Niu, Y. Kong, C. Sun, X. Song, N. Zhang and K. Sun, ChemistrySelect, 2019, 4, 10851–10855 CrossRef CAS.
- Z. Zhao, H. Qi, S. Tang, C. Zhang, X. Wang, M. Cheng and Z. Shao, Int. J. Hydrogen Energy, 2021, 46, 36012–36022 CrossRef CAS.
- B. Liu, J. Sunarso, Y. Zhang, G. Yang, W. Zhou and Z. Shao, ChemElectroChem, 2018, 5, 785–792 CrossRef CAS.
- X. Wang, T. Zhang, H. Qi and B. Tu, Mater. Lett., 2024, 355, 135513 CrossRef CAS.
- X. Yang, X. Han, T. He and Y. Du, ECS Trans., 2017, 78, 543 CrossRef CAS.
- K. Xie, J. Zhou and G. Meng, J. Alloys Compd., 2010, 506, L8–L11 CrossRef CAS.
- M. Shang, J. Tong and R. O'Hayre, RSC Adv., 2013, 3, 15769 RSC.
- X. Mao, Z. Li, M. Li, X. Xu, C. Yan, Z. Zhu and A. Du, J. Am. Chem. Soc., 2021, 143, 9507–9514 CrossRef CAS PubMed.
- R. Guo and T. He, ACS Mater. Lett., 2022, 4, 1646–1652 CrossRef CAS.
- E. Y. Y. Pikalova, E. G. G. Kalinina, N. S. S. Pikalova and E. A. A. Filonova, Materials, 2022, 15, 8783 CrossRef CAS PubMed.
- S. Oh, D. Kim, H. J. Ryu and K. T. Lee, Adv. Funct. Mater., 2023, 34, 2311426 CrossRef.
- Q. Wang, X. Liu, D. He and D. Wang, Mater. Today, 2023, 70, 218–236 CrossRef CAS.
- J. Cheng, A. Navrotsky, X.-D. Zhou and H. U. Anderson, Chem. Mater., 2005, 17, 2197 CrossRef CAS.
- R. Witte, A. Sarkar, R. Kruk, B. Eggert, R. A. Brand, H. Wende and H. Hahn, Phys. Rev. Mater., 2019, 3, 034406 CrossRef CAS.
- S.-W. Cheong and M. Mostovoy, Nat. Mater., 2007, 6, 13–20 CrossRef CAS PubMed.
- J. R. Mawdsley and T. R. Krause, Appl. Catal., A, 2008, 334, 311–320 CrossRef CAS.
- J. Dąbrowa, A. Olszewska, A. Falkenstein, C. Schwab, M. Szymczak, M. Zajusz, M. Moździerz, A. Mikuła, K. Zielińska, K. Berent, T. Czeppe, M. Martin and K. Świerczek, J. Mater. Chem. A, 2020, 8, 24455–24468 RSC.
- Y. Yang, H. Bao, H. Ni, X. Ou, S. Wang, B. Lin, P. Feng and Y. Ling, J. Power Sources, 2021, 482, 228959 CrossRef CAS.
- Y. Shi, N. Ni, Q. Ding and X. Zhao, J. Mater. Chem. A, 2022, 10, 2256–2270 RSC.
- W. Zhou, R. Ran and Z. Shao, J. Power Sources, 2009, 192, 231–246 CrossRef CAS.
- M. Marrony, M. Ancelin, G. Lefevre and J. Dailly, Solid State Ionics, 2015, 275, 97–100 CrossRef CAS.
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed.
- Z. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. Zhan and S. A. Barnett, Nature, 2005, 435, 795–798 CrossRef CAS PubMed.
- B. Lin, H. Ding, Y. Dong, S. Wang, X. Zhang, D. Fang and G. Meng, J. Power Sources, 2009, 186, 58–61 CrossRef CAS.
- G. Taillades, P. Pers, P. Batocchi and M. Taillades, ECS Trans., 2013, 57, 1289 CrossRef.
- J. Dailly, M. Ancelin and M. Marrony, Solid State Ionics, 2017, 306, 69–75 CrossRef CAS.
- T. Yang, A. Shaula, D. Pukazhselvan, D. Ramasamy, J. Deng, E. L. da Silva, R. Duarte and J. A. Saraiva, Appl. Surf. Sci., 2017, 424, 82–86 CrossRef CAS.
- G. Taillades, P. Pers, V. Mao and M. Taillades, Int. J. Hydrogen Energy, 2016, 41, 12330–12336 CrossRef CAS.
- J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim and G. Kim, Nano Energy, 2018, 44, 121–126 CrossRef CAS.
- L. Lei, J. Zhang, Z. Yuan, J. Liu, M. Ni and F. Chen, Adv. Funct. Mater., 2019, 29, 1903805 CrossRef.
- M. Choi, S. J. Kim and W. Lee, Ceram. Int., 2021, 47, 7790–7797 CrossRef CAS.
- N. Bausá, C. Solís, R. Strandbakke and J. M. Serra, Solid State Ionics, 2017, 306, 62–68 CrossRef.
- Y. Xie, N. Shi, X. Hu, K. Zhu, R. Peng, C. Xia and M. Chen, J. Electrochem. Soc., 2023, 170, 024513 CrossRef CAS.
- Z. Zhang, Y. Zhu, Y. Zhong, W. Zhou and Z. Shao, Adv. Energy Mater., 2017, 7, 1700242 CrossRef.
- J. Zhu, G. Liu, Z. Liu, Z. Chu, W. Jin and N. Xu, Adv. Mater., 2016, 28, 3511–3515 CrossRef CAS PubMed.
- Y. Li, Y. Li, Y. Wan, Y. Xie, J. Zhu, H. Pan, X. Zheng and C. Xia, Adv. Energy Mater., 2018, 9, 1803156 CrossRef.
- C. Yang, Y. Tian, C. Yang, G. Kim, J. Pu and B. Chi, Adv. Sci., 2023, 10, 2304224 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhu, Y. Gu, H. Chen, Y. Zheng and L. Ge, Ceram. Int., 2020, 46, 22787–22796 CrossRef CAS.
- C. Yang, Y. Tian, J. Pu and B. Chi, ACS Sustainable Chem. Eng., 2022, 10, 1047–1058 CrossRef CAS.
- Y. Wang, H. Wang, T. Liu, F. Chen and C. Xia, Electrochem. Commun., 2013, 28, 87–90 CrossRef.
- Y. Xie, N. Shi, D. Huan, W. Tan, J. Zhu, X. Zheng, H. Pan, R. Peng and C. Xia, ChemSusChem, 2018, 11, 3423–3430 CrossRef CAS PubMed.
- J. F. Shin, W. Xu, M. Zanella, K. Dawson, S. N. Savvin, J. B. Claridge and M. J. Rosseinsky, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- A. Demont, R. Sayers, M. A. Tsiamtsouri, S. Romani, P. A. Chater, H. Niu, C. Marti-Gastaldo, Z. Xu, Z. Deng, Y. Breard, M. F. Thomas, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2013, 135, 10114–10123 CrossRef CAS PubMed.
- Z. He, J. Nie, K. Liu, K. S. Ganesh, M. Akbar, C. Xia, X. Wang, W. Dong, J. Huang and B. Wang, Int. J. Hydrogen Energy, 2021, 46, 9799–9808 CrossRef CAS.
- H. Lee, H. Jung, C. Kim, S. Kim, I. Jang, H. Yoon, U. Paik and T. Song, ACS Appl. Energy Mater., 2021, 4, 11564–11573 CrossRef CAS.
- L. Li, Y. Fan, C. Zhou, J. Zhou, M. Wang, L. Zhou, L. Zhang and J.-Q. Wang, J. Phys. Chem. C, 2023, 127, 19107–19114 CrossRef CAS.
- M. Liang, Y. Song, D. Liu, L. Xu, G. Yang, W. Wang, W. Zhou, R. Ran, M. Xu and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS.
- R. Song, X. Zhang, D. Huan, X. Li, N. Shi, C. Xia, R. Peng and Y. Lu, Int. J. Hydrogen Energy, 2023, 48, 32943–32954 CrossRef CAS.
- J. H. Duffy, H. W. Abernathy and K. S. Brinkman, J. Mater. Chem. A, 2023, 11, 8929–8938 RSC.
- N. Wang, S. Hinokuma, T. Ina, C. Zhu, H. Habazaki and Y. Aoki, J. Mater. Chem. A, 2020, 8, 11043–11055 RSC.
- T. Zhu, H. E. Troiani, L. V. Mogni, M. Han and S. A. Barnett, Joule, 2018, 2, 478–496 CrossRef CAS.
- L. Rioja-Monllor, C. Bernuy-Lopez, M.-L. Fontaine, T. Grande and M.-A. Einarsrud, J. Mater. Chem. A, 2019, 7, 8609–8619 RSC.
- S. Liu, K. T. Chuang and J.-L. Luo, ACS Catal., 2015, 6, 760–768 CrossRef.
- J. H. Kim, J. K. Kim, H. G. Seo, D. K. Lim, S. J. Jeong, J. Seo, J. Kim and W. Jung, Adv. Funct. Mater., 2020, 30, 2001326 CrossRef CAS.
- W. Zhang, H. Wang, X. Chen, X. Liu and J. Meng, Chem. Eng. J., 2022, 446, 136934 CrossRef CAS.
- X. Zhang, W. Zhang, L. Zhang, J. Meng, F. Meng, X. Liu and J. Meng, Electrochim. Acta, 2017, 258, 1096–1105 CrossRef CAS.
- H. Qi, F. Xia, T. Yang, W. Li, W. Li, L. Ma, G. Collins, W. Shi, H. Tian, S. Hu, T. Thomas, E. M. Sabolsky, J. Zondlo, R. Hart, H. Finklea, G. A. Hackett and X. Liu, J. Electrochem. Soc., 2020, 167, 024510 CrossRef CAS.
- L. Adijanto, V. B. Padmanabhan, R. J. Gorte and J. M. Vohs, J. Electrochem. Soc., 2012, 159, F751–F756 CrossRef CAS.
- J. H. Kim, J. K. Kim, J. Liu, A. Curcio, J. S. Jang, I. D. Kim, F. Ciucci and W. Jung, ACS Nano, 2021, 15, 81–110 CrossRef CAS PubMed.
- J. Wang, D. Kalaev, J. Yang, I. Waluyo, A. Hunt, J. T. Sadowski, H. L. Tuller and B. Yildiz, J. Am. Chem. Soc., 2023, 145, 1714–1727 CrossRef CAS PubMed.
- Y. S. Chung, T. Kim, T. H. Shin, H. Yoon, S. Park, N. M. Sammes, W. B. Kim and J. S. Chung, J. Mater. Chem. A, 2017, 5, 6437–6446 RSC.
- S. He, Prog. Nat. Sci.: Mater. Int., 2021, 31, 341–372 CrossRef CAS.
- C.-C. Chueh, A. Bertei and C. Nicolella, J. Power Sources, 2019, 437, 226888 CrossRef CAS.
- K. Chen, Int. J. Hydrogen Energy, 2011, 36, 10541–10549 CrossRef CAS.
- Y. Wang, W. Li, L. Ma, W. Li and X. Liu, J. Mater. Sci. Technol., 2020, 55, 35–55 CrossRef CAS.
- P. Lin, X. Xu, S. Yu, Y. Yin, M. Andersson and L. Bi, J. Power Sources, 2022, 551, 232073 CrossRef CAS.
- Y. Wan, Y. Xing, Y. Li, D. Huan and C. Xia, J. Power Sources, 2018, 402, 363–372 CrossRef CAS.
- P. L. Knöchel, P. J. Keenan, C. Loho, C. Reitz, R. Witte, K. S. Knight, A. J. Wright, H. Hahn, P. R. Slater and O. Clemens, J. Mater. Chem. A, 2016, 4, 3415–3430 RSC.
- Y. Xia, X. Xu, Y. Teng, H. Lv, Z. Jin, D. Wang, R. Peng and W. Liu, Ceram. Int., 2020, 46, 25453–25459 CrossRef CAS.
- X. Xu, H. Wang, J. Ma, W. Liu, X. Wang, M. Fronzi and L. Bi, J. Mater. Chem. A, 2019, 7, 18792–18798 RSC.
- T. Hu, F. Zhu, J. Xia, F. He, Z. Du, Y. Zhou, Y. Liu, H. Wang and Y. Chen, Adv. Funct. Mater., 2023, 33, 2305567 CrossRef CAS.
- H. Wang, C. Tablet, A. Feldhoff and J. Caro, Adv. Mater., 2005, 17, 1785–1788 CrossRef CAS.
- Z. Wei, Z. Li, Z. Wang, Y. Zhao, J. Wang and J. Chai, Int. J. Hydrogen Energy, 2022, 47, 13490–13501 CrossRef CAS.
- J. Ma, F. Zhu, Y. Pan, H. Zhang, K. Xu, Y. Wang and Y. Chen, Sep. Purif. Technol., 2022, 288, 120657 CrossRef CAS.
- J. Jing, Z. Lei, Z. Wu, Z. Wang, H. Yu, Z. Yang and S. Peng, J. Eur. Ceram. Soc., 2022, 42, 6566–6573 CrossRef CAS.
- T. Hu, Y. Xu, K. Xu, F. Zhu and Y. Chen, SusMat, 2023, 3, 91–101 CrossRef CAS.
- Y. Lu, N. Mushtaq, M. A. K. Y. Shah, M. S. Irshad, S. Rauf, M. Yousaf and B. Zhu, Ceram. Int., 2023, 49, 2811–2820 CrossRef CAS.
- Y. Xiao, D. Bao, Z. Wang, Y. Wang and T. He, Int. J. Hydrogen Energy, 2024, 57, 880–888 CrossRef CAS.
- J. Jing, Z. Lei, Z. Zheng, H. Wang, Z. Yang and S. Peng, Int. J. Hydrogen Energy, 2022, 47, 35449–35457 CrossRef CAS.
- J. Wang, Z. Li, H. Zang, Y. Sun, Y. Zhao, Z. Wang, Z. Zhu, Z. Wei and Q. Zheng, Int. J. Hydrogen Energy, 2022, 47, 9395–9407 CrossRef CAS.
- C. Zhang and H. Zhao, J. Mater. Chem., 2012, 22, 18387–18394 RSC.
- J. Jing, Z. Lei, Z. Zheng, H. Wang, P. Zhang, Z. Wang, H. Xu and Z. Yang, Int. J. Hydrogen Energy, 2023, 48, 9037–9045 CrossRef CAS.
- L. Zhang, S. Yang and S. Zhang, J. Power Sources, 2019, 440, 227125 CrossRef CAS.
- J. Liu, Z. Jin, L. Miao, J. Ding, H. Tang, Z. Gong, R. Peng and W. Liu, Int. J. Hydrogen Energy, 2019, 44, 11079–11087 CrossRef CAS.
- C. Yang, J. Li, A. Hu, J. Pu and B. Chi, Trans. Tianjin Univ., 2023, 29, 444–452 CrossRef CAS.
- Z. Wei, Z. Li, Z. Wang, Y. Zhao, J. Wang and J. Chai, Int. J. Hydrogen Energy, 2022, 47, 13490–13501 CrossRef CAS.
- J. Ma, Y. Xu, F. Zhu and Y. Chen, Sustainable Mater. Technol., 2024, 40, e00888 CrossRef CAS.
- D. Zou, Y. Yi, Y. Song, D. Guan, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 5381–5390 RSC.
- J.-H. Kim and A. Manthiram, J. Mater. Chem. A, 2015, 3, 24195–24210 RSC.
- S. Yoo, J. Y. Shin and G. Kim, J. Mater. Chem., 2011, 21, 439–443 RSC.
- J. Jiang, Y. Zhang, X. Yang, Y. Shen and T. He, ACS Appl. Energy Mater., 2020, 4, 134–145 CrossRef.
- G.-M. Park, K. Park, M. Jo, M. Asif, Y. Bae, S.-H. Kim, A. K. Azad, S.-J. Song and J.-Y. Park, J. Alloys Compd., 2023, 968, 171987 CrossRef CAS.
- E. Vollestad, R. Strandbakke, M. Tarach, D. Catalan-Martinez, M. L. Fontaine, D. Beeaff, D. R. Clark, J. M. Serra and T. Norby, Nat. Mater., 2019, 18, 752–759 CrossRef PubMed.
- H. Zheng, F. Han, N. Sata and R. Costa, J. Energy Chem., 2023, 86, 437–446 CrossRef CAS.
- Y. Gu, Y. Zhang, Y. Zheng, H. Chen, L. Ge and L. Guo, Appl. Catal., B, 2019, 257, 117868 CrossRef CAS.
- C. Ferchaud, J.-C. Grenier, Y. Zhang-Steenwinkel, M. M. A. van Tuel, F. P. F. van Berkel and J.-M. Bassat, J. Power Sources, 2011, 196, 1872–1879 CrossRef CAS.
- L. Jiang, F. Li, T. Wei, R. Zeng and Y. Huang, Electrochim. Acta, 2014, 133, 364–372 CrossRef CAS.
- L. Zhao, J. Shen, B. He, F. Chen and C. Xia, Int. J. Hydrogen Energy, 2011, 36, 3658–3665 CrossRef CAS.
- H. Ding, Y. Xie and X. Xue, J. Power Sources, 2011, 196, 2602–2607 CrossRef CAS.
- L. Zhu, R. O'Hayre and N. P. Sullivan, Int. J. Hydrogen Energy, 2021, 46, 27784–27792 CrossRef CAS.
- C. Zhu, X. Liu, C. Yi, D. Yan and W. Su, J. Power Sources, 2008, 185, 193–196 CrossRef CAS.
- J. Li, L. Jia, B. Chi, J. Pu, J. Li and S. Wang, Int. J. Hydrogen Energy, 2019, 44, 31525–31530 CrossRef CAS.
- J. L. García-Muñoz, C. Frontera, A. Llobet, A. E. Carrillo, A. Caneiro, M. A. G. Aranda, M. Respaud, C. Ritter and E. Dooryee, J. Magn. Magn. Mater., 2004, 272–276, 1762–1763 CrossRef.
- K. Zhang, L. Ge, R. Ran, Z. Shao and S. Liu, Acta Mater., 2008, 56, 4876–4889 CrossRef CAS.
- G. Kim, S. Wang, A. J. Jacobson, Z. Yuan, W. Donner, C. L. Chen, L. Reimus, P. Brodersen and C. A. Mims, Appl. Phys. Lett., 2006, 88, 024103 CrossRef.
- H. Gu, G. Yang, Y. Hu, M. Liang, S. Chen, R. Ran, M. Xu, W. Wang, W. Zhou and Z. Shao, Int. J. Hydrogen Energy, 2020, 45, 25996–26004 CrossRef CAS.
- G. Kim, S. Wang, A. J. Jacobson, L. Reimus, P. Brodersen and C. A. Mims, J. Mater. Chem., 2007, 17, 2500 RSC.
- P. Winiarz, E. A. Sroczyk, A. Brzoza-Kos, P. Czaja, K. Kapusta and K. Świerczek, Acta Mater., 2024, 277, 120186 CrossRef CAS.
- Y. Xie, N. Shi, S. Ricote and M. Chen, Chem. Eng. J., 2024, 492, 151939 CrossRef CAS.
- U. Anjum, M. Agarwal, T. S. Khan and M. A. Haider, ECS Trans., 2017, 78, 499–506 CrossRef CAS.
- Z. Du, F. He, H. Gao, Y. Xu, F. Zhu, K. Xu, J. Xia, H. Zhang, Y. Huang, Y. Liu and Y. Chen, Energy Storage Mater., 2024, 68, 103345 CrossRef.
- U. Anjum, S. Vashishtha, N. Sinha and M. A. Haider, Solid State Ionics, 2015, 280, 24–29 CrossRef CAS.
- S. Zhai, H. Xie, P. Cui, D. Guan, J. Wang, S. Zhao, B. Chen, Y. Song, Z. Shao and M. Ni, Nat. Energy, 2022, 7, 866–875 CrossRef CAS.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- D. Kim, A. Hunt, I. Waluyo and B. Yildiz, J. Mater. Chem. A, 2023, 11, 7299–7313 RSC.
- S. Koohfar, M. Ghasemi, T. Hafen, G. Dimitrakopoulos, D. Kim, J. Pike, S. Elangovan, E. D. Gomez and B. Yildiz, Nat. Commun., 2023, 14, 7203 CrossRef CAS PubMed.
- J. Pan, Y. Ye, M. Zhou, X. Sun and Y. Chen, Energy Fuels, 2022, 36, 12253–12260 CrossRef CAS.
- W. Sun, Z. Shi, M. Liu, L. Bi and W. Liu, Adv. Funct. Mater., 2014, 24, 5695–5702 CrossRef CAS.
- L. Bi, S. Zhang, L. Zhang, Z. Tao, H. Wang and W. Liu, Int. J. Hydrogen Energy, 2009, 34, 2421–2425 CrossRef CAS.
- Y. Ma, L. Zhang, K. Zhu, B. Zhang, R. Peng, C. Xia and L. Huang, Nano Res., 2023, 17, 407–415 CrossRef.
- R. Murphy, Y. Zhou, L. Zhang, L. Soule, W. Zhang, Y. Chen and M. Liu, Adv. Funct. Mater., 2020, 30, 2002265 CrossRef CAS.
- T. Nagai, W. Ito and T. Sakon, Solid State Ionics, 2007, 177, 3433–3444 CrossRef CAS.
- Y. Gao, M. Fu, S. Zhao, Z. Hou, K. Liu, X. Deng and Z. Tao, Adv. Funct. Mater., 2025, 35, 2416625 CrossRef CAS.
- Y. Niu, Y. Zhou, W. Lv, Y. Chen, Y. Zhang, W. Zhang, Z. Luo, N. Kane, Y. Ding, L. Soule, Y. Liu, W. He and M. Liu, Adv. Funct. Mater., 2021, 31, 2100034 CrossRef CAS.
- Y. Chen, Y. Choi, S. Yoo, Y. Ding, R. Yan, K. Pei, C. Qu, L. Zhang, I. Chang, B. Zhao, Y. Zhang, H. Chen, Y. Chen, C. Yang, B. deGlee, R. Murphy, J. Liu and M. Liu, Joule, 2018, 2, 938–949 CrossRef CAS.
- G. Li, Y. Zhang, Y. Ling, B. He, J. Xu and L. Zhao, Int. J. Hydrogen Energy, 2016, 41, 5074–5083 CrossRef CAS.
- D. Ding, W. Zhu, J. Gao and C. Xia, J. Power Sources, 2008, 179, 177–185 CrossRef CAS.
- Z. Zhan, D. M. Bierschenk, J. S. Cronin and S. A. Barnett, Energy Environ. Sci., 2011, 4, 3951 RSC.
- Y. Namgung, J. Hong, A. Kumar, D.-K. Lim and S.-J. Song, Appl. Catal., B, 2020, 267, 118374 CrossRef CAS.
- J. A. Schuler, H. Yokokawa, C. F. Calderone, Q. Jeangros, Z. Wuillemin and A. Hessler-Wyser, J. Power Sources, 2012, 201, 112–120 CrossRef CAS.
- L. Zhou, J. H. Mason, W. Li and X. Liu, Renewable Sustainable Energy Rev., 2020, 134, 110320 CrossRef CAS.
- B. Wei, M. Schroeder and M. Martin, ACS Appl. Mater. Interfaces, 2018, 10, 8621–8629 CrossRef CAS PubMed.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- W. Lee, J. W. Han, Y. Chen, Z. Cai and B. Yildiz, J. Am. Chem. Soc., 2013, 135, 7909–7925 CrossRef CAS PubMed.
- Z. Zhuang, Y. Li, R. Yu, L. Xia, J. Yang, Z. Lang, J. Zhu, J. Huang, J. Wang, Y. Wang, L. Fan, J. Wu, Y. Zhao, D. Wang and Y. Li, Nat. Catal., 2022, 5, 300–310 CrossRef CAS.
- W. Wu, H. Ding, Y. Zhang, Y. Ding, P. Katiyar, P. K. Majumdar, T. He and D. Ding, Adv. Sci., 2018, 5, 1800360 CrossRef PubMed.
- F. Liu, H. Deng, D. Diercks, P. Kumar, M. H. A. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, L. Fang, D. Chen, B. Liu and C. Duan, Nat. Energy, 2023, 8, 1145–1157 CrossRef CAS.
- D. Chen, R. Ran and Z. Shao, J. Power Sources, 2010, 195, 4667–4675 CrossRef CAS.
- S. Im, J.-H. Lee and H.-I. Ji, J. Korean Ceram. Soc., 2021, 58, 351–358 CrossRef CAS.
- A. Seong, D. Jeong, M. Kim, S. Choi and G. Kim, J. Power Sources, 2022, 530, 231241 CrossRef CAS.
- S. J. Yang, W. Chang, H. J. Jeong, D. H. Kim and J. H. Shim, Int. J. Energy Res., 2021, 46, 6553–6561 CrossRef.
- J. S. Park, H. J. Choi, G. D. Han, J. Koo, E. H. Kang, D. H. Kim, K. Bae and J. H. Shim, J. Power Sources, 2021, 482, 229043 CrossRef CAS.
- H. S. Park, H. J. Jeong, K.-H. Kim, W. Chang, Y. S. Kim, Y. S. Choi and J. H. Shim, Appl. Surf. Sci., 2023, 612, 155812 CrossRef CAS.
- W. Tang, H. Ding, W. Bian, W. Wu, W. Li, X. Liu, J. Y. Gomez, C. Y. Regalado Vera, M. Zhou and D. Ding, J. Mater. Chem. A, 2020, 8, 14600–14608 RSC.
- M. Choi, J. Paik, D. Kim, D. Woo, J. Lee, S. J. Kim, J. Lee and W. Lee, Energy Environ. Sci., 2021, 14, 6476–6483 RSC.
- S. Bang, J. Lee and W. Lee, J. Power Sources, 2023, 553, 232290 CrossRef CAS.
- J. Lee, S. Hwang, M. Ahn, M. Choi, S. Han, D. Byun and W. Lee, J. Mater. Chem. A, 2019, 7, 21120–21127 RSC.
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 RSC.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- L. Wang, K. A. Stoerzinger, L. Chang, J. Zhao, Y. Li, C. S. Tang, X. Yin, M. E. Bowden, Z. Yang, H. Guo, L. You, R. Guo, J. Wang, K. Ibrahim, J. Chen, A. Rusydi, J. Wang, S. A. Chambers and Y. Du, Adv. Funct. Mater., 2018, 28, 1803712 CrossRef.
- D. Chen, F. Wang, H. Shi, R. Ran and Z. Shao, Electrochim. Acta, 2012, 78, 466–474 CrossRef CAS.
- H. Ding and X. Xue, J. Power Sources, 2010, 195, 7038–7041 CrossRef CAS.
- H. Ding and X. Xue, J. Power Sources, 2010, 195, 4139–4142 CrossRef CAS.
- Q. Yang, M. Chen, B. S. Teketel, D. Tian, Y. Ding, X. Lu, S. Zhu, Y. Chen and B. Lin, J. Alloys Compd., 2022, 918, 165368 CrossRef CAS.
- C. Wen, K. Chen, D. Guo, W. Yang, S. Gao, C. Lu, B. Niu and B. Wang, Solid State Ionics, 2022, 386, 116048 CrossRef CAS.
- L. Wang, P. Xie, L. Bian, X. Liu and K. Chou, Catal. Today, 2018, 318, 132–136 CrossRef CAS.
- B. Zhang, Y. Wan, Z. Hua, K. Tang and C. Xia, ACS Appl. Energy Mater., 2021, 4, 8401–8409 CrossRef CAS.
- C. Liu, F. Wang, Y. Ni, S. Wang, B. Qian, Q. Ni, Y. Zheng, H. Chen and L. Ge, Int. J. Hydrogen Energy, 2023, 48, 9812–9822 CrossRef CAS.
- X. Mao, W. Wang and G. Ma, Ceram. Int., 2015, 41, 10276–10280 CrossRef CAS.
- H.-X. Zhang, J.-X. Yang, P.-F. Wang, C.-G. Yao, X.-D. Yu and F.-N. Shi, Solid State Ionics, 2023, 391, 116144 CrossRef CAS.
- M. Zhang, Z. Du, Z. Sun and H. Zhao, J. Mater. Chem. A, 2023, 11, 21645–21654 RSC.
- A. Montenegro-Hernandez, J. Vega-Castillo, L. Mogni and A. Caneiro, Int. J. Hydrogen Energy, 2011, 36, 15704–15714 CrossRef CAS.
- A. Tarancón, S. J. Skinner, R. J. Chater, F. Hernández-Ramírez and J. A. Kilner, J. Mater. Chem., 2007, 17, 3175 RSC.
- J. M. Bassat, P. Odier, A. Villesuzanne, C. Marin and M. Pouchard, Solid State Ionics, 2004, 167, 341–347 CrossRef CAS.
- D. Zhu, J. Yu, Y. Zhang, J. Liu, Y. Ouyang, J. Yu, Z. Liu, W. Fan, X. Bai, N. Wang, E. Bu and C. Zhu, Int. J. Hydrogen Energy, 2024, 92, 1401–1408 CrossRef CAS.
- A. P. Tarutin, J. G. Lyagaeva, D. A. Medvedev, L. Bi and A. A. Yaremchenko, J. Mater. Chem. A, 2021, 9, 154–195 RSC.
- P. Zhong, K. Toyoura, L. Jiang, L. Chen, S. A. Ismail, N. Hatada, T. Norby and D. Han, Adv. Energy Mater., 2022, 12, 2200392 CrossRef CAS.
- M. Yashima, M. Enoki, T. Wakita, R. Ali, Y. Matsushita, F. Izumi and T. Ishihara, J. Am. Chem. Soc., 2008, 130, 2762–2763 CrossRef CAS PubMed.
- P. Kumar, N. K. Singh, G. Gupta and P. Singh, RSC Adv., 2016, 6, 22094–22102 RSC.
- P. Kumar, P. Jena, P. K. Patro, R. K. Lenka, A. S. K. Sinha, P. Singh and R. K. Singh, ACS Appl. Mater. Interfaces, 2019, 11, 24659–24667 CrossRef CAS PubMed.
- Y. Watanabe, Y. Hiruma, H. Nagata and T. Takenaka, Ceram. Int., 2008, 34, 761–764 CrossRef CAS.
- K. Koyama, T. Tange, T. Saito and K. Mizuno, Phys. B, 2000, 281–282, 909–911 CrossRef.
- Z. Wang, X. Miao, X. Zhu, S. Guo, D. Han, X. Ye and Z. Wen, J. Power Sources, 2024, 597, 234141 CrossRef CAS.
- Y. Ling, T. Guo, Y. Guo, Y. Yang, Y. Tian, X. Wang, X. Ou and P. Feng, J. Adv. Ceram., 2021, 10, 1052–1060 CrossRef CAS.
- S. Yang, Y. Wen, J. Zhang, Y. Lu, X. Ye and Z. Wen, Electrochim. Acta, 2018, 267, 269–277 CrossRef CAS.
- Q. Cao, R. Qiu, L. Lei, W. Zhang, S. Deng, J. Chen, L. He and J. Xu, ACS Appl. Energy Mater., 2022, 5, 11213–11222 CrossRef CAS.
- Z. Yang, J. Zhang, B. Wu, X. Zhao, X. Lu, Y. Zhao and Y. Li, Ceram. Int., 2023, 49, 25381–25388 CrossRef CAS.
- P. Batocchi, F. Mauvy, S. Fourcade and M. Parco, Electrochim. Acta, 2014, 145, 1–10 CrossRef CAS.
- M. Laguna-Bercero, H. Monzón, A. Larrea and V. Orera, J. Mater. Chem. A, 2016, 4, 1446–1453 RSC.
- C. Lalanne, F. Mauvy, E. Siebert, M. L. Fontaine, J. M. Bassat, F. Ansart, P. Stevens and J. C. Grenier, J. Eur. Ceram. Soc., 2007, 27, 4195–4198 CrossRef CAS.
- E. Boehm, J. Bassat, P. Dordor, F. Mauvy, J. Grenier and P. Stevens, Solid State Ionics, 2005, 176, 2717–2725 CrossRef CAS.
- C. Lalanne, G. Prosperi, J. Bassat, F. Mauvy, S. Fourcade, P. Stevens, M. Zahid, S. Diethelm, J. Vanherle and J. Grenier, J. Power Sources, 2008, 185, 1218–1224 CrossRef CAS.
- J. Wan, J. Goodenough and J. Zhu, Solid State Ionics, 2007, 178, 281–286 CrossRef CAS.
- A. P. Tarutin, M. Y. Gorshkov, I. N. Bainov, G. K. Vdovin, A. I. Vylkov, J. G. Lyagaeva and D. A. Medvedev, Ceram. Int., 2020, 46, 24355–24364 CrossRef CAS.
- A. P. Khandale, J. D. Punde and S. S. Bhoga, J. Solid State Electrochem., 2012, 17, 617–626 CrossRef.
- T. Nakamura, K. Yashiro, K. Sato and J. Mizusaki, Solid State Ionics, 2010, 181, 402–411 CrossRef CAS.
- H. G. Seo, H. Kim, W. Jung and H. L. Tuller, Appl. Catal., B, 2024, 355, 124172 CrossRef CAS.
- S. Choi, S. Park, J. Shin and G. Kim, J. Mater. Chem. A, 2015, 3, 6088–6095 RSC.
- S. Ingavale, M. Gopalakrishnan, C. M. Enoch, C. Pornrungroj, M. Rittiruam, S. Praserthdam, A. Somwangthanaroj, K. Nootong, R. Pornprasertsuk and S. Kheawhom, Small, 2024, 20, 2308443 CrossRef CAS PubMed.
- Y. Zhu, Q. Lin, Z. Hu, Y. Chen, Y. Yin, H. A. Tahini, H. J. Lin, C. T. Chen, X. Zhang, Z. Shao and H. Wang, Small, 2020, 16, 2001204 CrossRef CAS PubMed.
- X. Xu, Y. Pan, L. Ge, Y. Chen, X. Mao, D. Guan, M. Li, Y. Zhong, Z. Hu, V. K. Peterson, M. Saunders, C. T. Chen, H. Zhang, R. Ran, A. Du, H. Wang, S. P. Jiang, W. Zhou and Z. Shao, Small, 2021, 17, 2101573 CrossRef CAS PubMed.
- M. B. Liu and B. Yildiz, Chem. Mater., 2024, 36, 10571–10582 CrossRef CAS.
- H. G. Seo, A. Staerz, D. S. Kim, D. Klotz, C. Nicollet, M. Xu, J. M. LeBeau and H. L. Tuller, Energy Environ. Sci., 2022, 15, 4038–4047 RSC.
- H. G. Seo, A. Staerz, D. S. Kim, J. M. LeBeau and H. L. Tuller, Adv. Mater., 2023, 35, 2208182 CrossRef CAS PubMed.
- Y. Chen, S. Yoo, X. Li, D. Ding, K. Pei, D. Chen, Y. Ding, B. Zhao, R. Murphy, B. deGlee, J. Liu and M. Liu, Nano Energy, 2018, 47, 474–480 CrossRef CAS.
- M. Perz, E. Bucher, C. Gspan, J. Waldhäusl, F. Hofer and W. Sitte, Solid State Ionics, 2016, 288, 22–27 CrossRef CAS.
- S. P. Jiang, J. P. Zhang and X. G. Zheng, J. Eur. Ceram. Soc., 2002, 22, 361–373 CrossRef CAS.
- Y. Zhen, Solid State Ionics, 2008, 179, 1459–1464 CrossRef.
- J. Hong, S. J. Heo, A. N. Aphale, B. Hu and P. Singh, J. Electrochem. Soc., 2019, 166, F59–F65 CrossRef CAS.
- B.-K. Park, R.-H. Song, S.-B. Lee, T.-H. Lim, S.-J. Park, W. Jung and J.-W. Lee, J. Power Sources, 2017, 348, 40–47 CrossRef CAS.
- J. Huang, Q. Liu, S. P. Jiang, L. Zhao, N. Ai, X. Wang, Y. Shao, C. Guan, H. Fang, Y. Luo and K. Chen, Appl. Catal., B, 2023, 321, 122080 CrossRef CAS.
- H. A. Ishfaq, M. Z. Khan, Y. M. Shirke, S. Qamar, A. Hussain, M. T. Mehran, R.-H. Song and M. Saleem, Appl. Catal., B, 2023, 323, 122178 CrossRef CAS.
- Y. Chen, W. Zhou, D. Ding, M. Liu, F. Ciucci, M. Tade and Z. Shao, Adv. Energy Mater., 2015, 5, 1500537 CrossRef.
- Y. Li, W. Zhang, Y. Zheng, J. Chen, B. Yu, Y. Chen and M. Liu, Chem. Soc. Rev., 2017, 46, 6345–6378 RSC.
- Y. Chen, Y. Choi, S. Yoo, Y. Ding, R. Yan, K. Pei, C. Qu, L. Zhang, I. Chang and B. Zhao, Joule, 2018, 2, 938–949 CrossRef CAS.
- S. J. Kim, T. Akbay, J. Matsuda, A. Takagaki and T. Ishihara, ACS Appl. Energy Mater., 2019, 2, 1210–1220 CrossRef CAS.
- R. Zhang and G. Azimi, Ind. Eng. Chem. Res., 2023, 62, 13117–13132 CrossRef CAS.
- M. S. I. Sozal, W. Tang, S. Das, W. Li, A. Durygin, V. Drozd, C. Zhang, B. Jafarizadeh, C. Wang and A. Agarwal, Int. J. Hydrogen Energy, 2022, 47, 21817–21827 CrossRef CAS.
- Z. Zhang, C. Yao, H. Zhang, Y. Sun, B. Xia, W. Liu, J. Ding, L. Zhang, X. Lang and K. Cai, ACS Sustainable Chem. Eng., 2025, 13, 3234–3246 CrossRef CAS.
- G. Pacchioni, Nat. Rev. Mater., 2022, 7, 751 CrossRef CAS.
- Z. Luo, X. Hu, Y. Zhou, Y. Ding, W. Zhang, T. Li and M. Liu, Adv. Mater., 2024, 36, 2311159 CrossRef CAS PubMed.
- J. Wei, X. Chu, X. Y. Sun, K. Xu, H. X. Deng, J. Chen, Z. Wei and M. Lei, InfoMat, 2019, 1, 338–358 CrossRef CAS.
- G. H. Gu, C. Choi, Y. Lee, A. B. Situmorang, J. Noh, Y. H. Kim and Y. Jung, Adv. Mater., 2020, 32, 1907865 CrossRef CAS PubMed.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- Y. Chen, Y. Huang, T. Cheng and W. A. Goddard, 3rd, J. Am. Chem. Soc., 2019, 141, 11651–11657 CrossRef CAS PubMed.
- K. H. Tu, H. Huang, S. Lee, W. Lee, Z. Sun, A. Alexander-Katz and C. A. Ross, Adv. Mater., 2020, 32, 2005713 CrossRef PubMed.
- C. Rao and Y. Liu, Comput. Mater. Sci., 2020, 184, 109850 CrossRef.
- K. Choudhary and B. DeCost, npj Comput. Mater., 2021, 7, 185 CrossRef.
- J. Schmidt, M. R. G. Marques, S. Botti and M. A. L. Marques, npj Comput. Mater., 2019, 5, 83 CrossRef.
- A. Chen, X. Zhang and Z. Zhou, InfoMat, 2020, 2, 553–576 CrossRef CAS.
- N. Wang, B. Yuan, C. Tang, L. Du, R. Zhu, Y. Aoki, W. Wang, L. Xing and S. Ye, Adv. Mater., 2022, 34, 2203446 CrossRef CAS PubMed.
- A. M. Deml, V. Stevanović, C. L. Muhich, C. B. Musgrave and R. O'Hayre, Energy Environ. Sci., 2014, 7, 1996 RSC.
- R. B. Wexler, G. S. Gautam, E. B. Stechel and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 13212–13227 CrossRef CAS PubMed.
- Y.-L. Lee, J. Kleis, J. Rossmeisl and D. Morgan, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224101 CrossRef.
- M. T. Curnan and J. R. Kitchin, J. Phys. Chem. C, 2014, 118, 28776–28790 CrossRef CAS.
- W. Li, R. Jacobs and D. Morgan, Comput. Mater. Sci., 2018, 150, 454–463 CrossRef CAS.
- S. Nie, Y. Xiang, L. Wu, G. Lin, Q. Liu, S. Chu and X. Wang, J. Am. Chem. Soc., 2024, 146, 29325–29334 CrossRef CAS PubMed.
- A. Talapatra, B. P. Uberuaga, C. R. Stanek and G. Pilania, Chem. Mater., 2021, 33, 845–858 CrossRef CAS.
- K. Yang, J. Liu, Y. Wang, X. Shi, J. Wang, Q. Lu, F. Ciucci and Z. Yang, J. Mater. Chem. A, 2022, 10, 23683–23690 RSC.
- A. K. Padinjarethil, S. Pollok and A. Hagen, Fuel Cells, 2021, 21, 566–576 CrossRef CAS.
- X. Li, Z. Chen, D. Huan, B. Qiu, K. Zhu, Z. Qi, H. Liu, C. Xia, R. Peng and Y. Lu, ACS Mater. Lett., 2023, 5, 2896–2905 CrossRef CAS.
- K. Saito and M. Yashima, Nat. Commun., 2023, 14, 7466 CrossRef PubMed.
- F. He, F. Zhu, D. Liu, Y. Zhou, K. Sasaki, Y. Choi, M. Liu and Y. Chen, Mater. Today, 2023, 63, 89–98 CrossRef CAS.
- Z. Sun, Z. Shen, Z. Du, Y. Zhang, Y. Gong, M. Zhang, K. Wang, K. Świerczek, J. Zeng and H. Zhao, Adv. Funct. Mater., 2024, 34, 2403312 CrossRef CAS.
- R. Jacobs, J. Hwang, Y. Shao-Horn and D. Morgan, Chem. Mater., 2019, 31, 785–797 CrossRef CAS.
- L. Tang, Y. Yang, H. Guo, Y. Wang, M. Wang, Z. Liu, G. Yang, X. Fu, Y. Luo, C. Jiang, Y. Zhao, Z. Shao and Y. Sun, Adv. Funct. Mater., 2022, 32, 2112157 CrossRef CAS.
- Y. Choi, M. C. Lin and M. Liu, J. Power Sources, 2010, 195, 1441–1445 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, 146 CrossRef PubMed.
- T. Chen, G. Zhang, K. Liu, C. Wang, G. Zheng, Z. Huang, N. Sun, L. Xu, J. Zhou, Y. Zhou and S. Wang, Adv. Funct. Mater., 2024, 34, 2316485 CrossRef CAS.
- A. P. Tarutin, J. G. Lyagaeva, D. A. Medvedev, L. Bi and A. A. Yaremchenko, J. Mater. Chem. A, 2021, 9, 154–195 RSC.
- R. K. Lenka, P. K. Patro, V. Patel, L. Muhmood and T. Mahata, J. Alloys Compd., 2021, 860, 158490 CrossRef CAS.
- N. Yu, I. T. Bello, X. Chen, T. Liu, Z. Li, Y. Song and M. Ni, Nano-Micro Lett., 2024, 16, 177 CrossRef CAS PubMed.
- X. Li, Z. Chen, Y. Yang, D. Huan, H. Su, K. Zhu, N. Shi, Z. Qi, X. Zheng, H. Pan, Z. Zhan, C. Xia, R. Peng, S. Wei and Y. Lu, Appl. Catal., B, 2022, 316, 121627 CrossRef CAS.
- Q. Hassan, P. Viktor, T. J. Al-Musawi, B. M. Ali, S. Algburi, H. M. Alzoubi, A. K. Al-Jiboory, A. Z. Sameen, H. M. Salman and M. Jaszczur, Renewable Energy Focus, 2024, 48, 100545 CrossRef.
- https://www.renews.biz/96664/european-energy-opens-first-green-hydrogen-plant/ .
- https://energynews.pro/en/totalenergies-and-morocco-launch-green-hydrogen-production-project-for-the-european-market/ .
- https://www.offshore-energy.biz/germany-opens-test-field-for-hydrogen-fuel-cell-propulsion-systems/ .
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- M. Tsuchiya, B.-K. Lai and S. Ramanathan, Nat. Nanotechnol., 2011, 6, 282–286 CrossRef CAS PubMed.
- S. Robinson, A. Manerbino, W. Grover Coors and N. Sullivan, Fuel Cells, 2013, 13, 584–591 CrossRef CAS.
- M. Li, J. Wang, Z. Chen, X. Qian, C. Sun, D. Gan, K. Xiong, M. Rao, C. Chen and X. Li, Energies, 2024, 17, 1005 CrossRef CAS.
- S. Pirou, Q. Wang, P. Khajavi, X. Georgolamprou, S. Ricote, M. Chen and R. Kiebach, Int. J. Hydrogen Energy, 2022, 47, 6745–6754 CrossRef CAS.
- H. An, H.-W. Lee, B.-K. Kim, J.-W. Son, K. J. Yoon, H. Kim, D. Shin, H.-I. Ji and J.-H. Lee, Nat. Energy, 2018, 3, 870–875 CrossRef CAS.
- R. J. Braun, A. Dubois, K. Ferguson, C. Duan, C. Karakaya, R. J. Kee, H. Zhu, N. P. Sullivan, E. Tang and M. Pastula, ECS Trans., 2019, 91, 997 CrossRef CAS.
- L. Q. Le, C. H. Hernandez, M. H. Rodriguez, L. Zhu, C. Duan, H. Ding, R. P. O'Hayre and N. P. Sullivan, J. Power Sources, 2021, 482, 228868 CrossRef CAS.
- J. Dailly and M. Marrony, J. Power Sources, 2013, 240, 323–327 CrossRef CAS.
- S. Nien, C. Hsu, C. Chang and B. Hwang, Fuel Cells, 2011, 11, 178–183 CrossRef CAS.
- A. R. Hanifi, N. K. Sandhu, T. H. Etsell, J.-L. Luo and P. Sarkar, J. Power Sources, 2017, 341, 264–269 CrossRef CAS.
- T. Amiri, K. Singh, N. K. Sandhu, A. R. Hanifi, T. H. Etsell, J.-L. Luo, V. Thangadurai and P. Sarkar, J. Electrochem. Soc., 2018, 165, F764 CrossRef CAS.
- C. Chen, Y. Dong, L. Li, Z. Wang, M. Liu, B. H. Rainwater and Y. Bai, J. Electroanal. Chem., 2019, 844, 49–57 CrossRef CAS.
- D. Cao, M. Zhou, X. Yan, Z. Liu and J. Liu, Electrochem. Commun., 2021, 125, 106986 CrossRef CAS.
- Y. Pan, H. Zhang, K. Xu, Y. Zhou, B. Zhao, W. Yuan, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Appl. Catal., B, 2022, 306, 121071 CrossRef CAS.
- R. I. Walton, Chem. – Eur. J., 2020, 26, 9041–9069 CrossRef CAS PubMed.
- K.-Y. Park, Y. Seo, K. B. Kim, S.-J. Song, B. Park and J.-Y. Park, J. Alloys Compd., 2015, 639, 435–444 CrossRef CAS.
- L. Vradman, E. Friedland, J. Zana, R. Vidruk-Nehemya and M. Herskowitz, J. Mater. Sci., 2017, 52, 11383–11390 CrossRef CAS.
- D. Navas, S. Fuentes, A. Castro-Alvarez and E. Chavez-Angel, Gels, 2021, 7, 275 CrossRef CAS PubMed.
- S. Sharma, V. Singh, R. Kotnala and R. K. Dwivedi, J. Mater. Sci.: Mater. Electron., 2014, 25, 1915–1921 CrossRef CAS.
- R. R. Suresh, M. Lakshmanakumar, J. Arockia Jayalatha, K. Rajan, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappan, J. Mater. Sci., 2021, 56, 8951–9006 CrossRef CAS.
- X. Gao, S. Zhang, P. Wang, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Chem. Soc. Rev., 2024, 53, 1552–1591 RSC.
- G. Liu, W. Zhao, Z. Li, Z. Xia, C. Jiang, J. Kupecki, S. Pang, Z. Deng and X. Li, Energy Convers. Manage., 2022, 255, 115318 CrossRef CAS.
- G. Liu, Z. Wang, X. Liu, J. Kupecki, D. Zhao, B. Jin, Z. Wang and X. Li, J. Cleaner Prod., 2023, 425, 139000 CrossRef CAS.
- M. Mogensen, M. Chen, H. Frandsen, C. Graves, J. Hansen, K. Hansen, A. Hauch, T. Jacobsen, S. Jensen and T. Skafte, Clean Energy, 2019, 3, 175–201 CrossRef.
- G. Yang, C. Su, H. Shi, Y. Zhu, Y. Song, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 15169–15194 CrossRef CAS.
- D. Nath, Ankit, D. R. Neog and S. S. Gautam, Arch. Comput. Methods Eng., 2024, 1–40 CAS.
- B. Timurkutluk and E. Ucar, J. Power Sources, 2025, 625, 235715 CrossRef CAS.
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2023, 6, e12441 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |