
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect seawater electrolysis for green hydrogen production: electrode designs, cell configurations, and system integrations
Lizhen
Wu†
a,
Yifan
Xu†
 b,
Qing
Wang
a,
Xiaohong
Zou
a,
Zhefei
Pan
*cd,
Michael K. H.
Leung
*b and
Liang
An
*ae
b,
Qing
Wang
a,
Xiaohong
Zou
a,
Zhefei
Pan
*cd,
Michael K. H.
Leung
*b and
Liang
An
*ae
aDepartment of Mechanical Engineering, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China. E-mail: liang.an@polyu.edu.hk
bAbility R&D Energy Research Centre, School of Energy and Environment, City University of Hong Kong, Kowloon, Hong Kong SAR, China. E-mail: mkh.leung@cityu.edu.hk
cKey Laboratory of Low-grade Energy Utilization Technologies and Systems (Chongqing University), Ministry of Education, Chongqing, 400030, China. E-mail: zhefei.pan@cqu.edu.cn
dInstitute of Engineering Thermophysics, Chongqing University, Chongqing 400030, China
eResearch Institute for Smart Energy, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong SAR, China
First published on 28th April 2025
Abstract
Direct seawater electrolysis (DSE) is a promising technology for sustainable hydrogen production, utilizing abundant marine resources. However, industrialization of DSE faces significant long-term stability challenges due to the complex composition of seawater, which contains various ions and microorganisms that can lead to both chemical and physical degradation of the electrolysis system. For instance, the presence of chloride ions (Cl−) hinders the desired oxygen evolution reaction (OER) because competing chlorine evolution reactions (CER) occur and adversely impact electrode materials, resulting in low system efficiency and poor longevity. To enhance long-term stability of DSE, researchers are investigating robust electrocatalysts and advanced surface modifications that improve protection against corrosive environments and enhance selectivity. Innovative electrode designs are also being developed to manage bubble transport and decrease precipitation. Additionally, the design of electrolysis cells, such as bipolar membrane cells, offers a viable solution by minimizing Cl− transport and corrosive environment. With an increasing number of offshore renewable energy projects, the integration of effective DSE technologies in the offshore environment is critical. This review provides the state-of-the-art of electrodes, cells and systems, contributing to the development of DSE for long-term stable operation.
Broader contextDirect seawater electrolysis (DSE) is emerging as a critical technology for sustainable hydrogen production, leveraging the abundant supply of seawater to address global energy demands and reduce reliance on fossil fuels. Despite its potential, the widespread application of DSE faces significant challenges due to the complex chemical composition of seawater, particularly the presence of chloride ions (Cl−), which can lead to competing reactions like chlorine evolution that degrade the performance of electrocatalysts. Ongoing research focuses on developing robust electrocatalysts, advanced surface modifications, and innovative electrode designs, including novel cell configurations, to enhance the resilience and efficiency of electrolysis systems. As offshore renewable energy projects increase, integrating DSE technologies is vital for sustainable hydrogen production and achieving environmental sustainability goals, paving the way for a more resilient and renewable energy future. |
1 Introduction
The serious challenges faced globally with increasing energy and environmental risks have prompted a profound shift in the energy mix from traditional fossil energy sources to emerging carbon-free energy alternatives.1,2 Among these alternatives, hydrogen stands out as a highly promising energy carrier due to its exceptional energy density.3,4 However, traditional methods for hydrogen production, which rely on fossil fuels, inherently lead to carbon dioxide (CO2) emissions.5,6 To mitigate these issues, the production of green hydrogen through water electrolysis powered by renewable energy sources—such as solar and wind—emerges as an ideal solution.7,8 Existing water electrolysis technologies9,10 primarily include alkaline water electrolysis (AWE) and polymer electrolyte membrane (PEM) electrolysis, both of which necessitate high-purity water. This requirement limits their scalability due to the unequal availability and scarcity of freshwater resources.11 Consequently, seawater, as a virtually unlimited resource, presents a compelling option for seawater electrolysis (SWE).12 The electrolysis process in seawater involves two half-reactions: the hydrogen evolution reaction (HER) at the cathode13 and the oxygen evolution reaction (OER) at the anode.14 While seawater holds considerable promise, its intricate composition presents notable challenges for efficient and sustainable SWE.11,15SWE is generally categorized into two types: indirect seawater electrolysis (ISE) and direct seawater electrolysis (DSE).16,17 DSE and ISE differ primarily in their pre-treatment requirements. ISE necessitates desalination methods, such as reverse osmosis or electrodialysis, to convert seawater into highly pure water before electrolysis, leading to additional costs and energy losses (up to 7% of the total system cost).18,19 In contrast, DSE directly utilizes untreated seawater, adding alkaline electrolyte (e.g., 1 M KOH) to suppress the chlorine evolution reaction (CER) and enhance OER.20 This approach results in alkaline effluent with residual ions (e.g., Cl−, Mg2+, Ca2+) and unreacted bases, complicating discharge management. Strategies for managing DSE effluent often involve diluting it with seawater or employing acid and base neutralization. However, these approaches can be detrimental to the marine environment and result in the chemical energy from the neutralization process being released as heat, offering little economic advantage.21,22 Alternatively, the recirculation systems can filter precipitates (e.g., Mg(OH)2, Ca(OH)2) to minimize waste but require efficient filtration and periodic alkaline replenishment.23 Resource recovery options exist, such as extracting Mg(OH)2 and Ca(OH)2 for industrial uses (e.g. construction materials, CO2 sequestration)24 and hypochlorite for disinfection,25 enhancing process economics. DSE is increasingly favored over ISE due to advancements in electrocatalysis and electrolysis cell design,26 demonstrating competitive hydrogen production from offshore wind farms at distances over 40–50 km.27 Moreover, DSE's compatibility with fuel cells enables integrated energy conversion and storage, offering a pathway to harness surplus renewable electricity while providing clean drinking water and renewable energy to arid regions, supporting future energy transitions.28 However, the industrialization of DSE still faces challenges, particularly concerning catalyst efficiency and system stability, which are critical for large-scale hydrogen production.29 There exists a considerable body of research focused on three main categories of seawater electrolysis: natural seawater electrolysis (NSE), characterized by a pH of approximately 8.1 and without the addition of strong bases like NaOH or KOH;11 alkaline seawater electrolysis (ASE), wherein NaOH or KOH is added to adjust the alkalinity of the seawater;30 and simulated seawater electrolysis (SSE), which typically involves a 0.5 M NaCl solution, potentially modified with NaOH or KOH.31 Given that above techniques use either untreated seawater or only simple physical pre-treatment (e.g., filtration, disinfection), NSE and ASE can be categorized collectively as DSE. Both DSE and SSE-related research aims to provide valuable technical guidance for DSE.
In recent years, advancements in novel electrode materials,12,32 electrolysis cells,33,34 and system design35,36 have propelled the development of DSE (Fig. 1). The interplay between catalyst activity, stability, and selectivity must be carefully balanced, and various types of electrolysis cells require optimization based on specific electrode and catalyst design criteria.28 Additionally, optimizing the coupling configurations of renewable energy sources with electrolysis cells and auxiliary machines is crucial for enhancing overall system stability.
 | ||
| Fig. 1 Possible challenges about long-term stability in DSE. Reproduced with permission.37 Copyright 2015, The Royal Society of Chemistry. Reproduced with permission.38 Copyright 2024, American Chemical Society. Reproduced with permission.39 Copyright 2024, Springer Nature. Reproduced with permission.40 Copyright 2024, Elsevier. Reproduced with permission.41 Copyright 2019, American Chemical Society. Reproduced with permission.42 Copyright 2023, ASME. | ||
In this review, we aim to elucidate the mechanisms underlying the poor stability associated with DSE technologies. We will first provide a detailed overview of the challenges that influence the long-term stability of DSE technologies. Subsequently, we will discuss effective approaches for enhancing the long-term stability of both OER and HER electrodes, encompassing material development, surface modifications, and structural optimization. Furthermore, we will conduct a thorough evaluation of the diverse electrolysis cell designs employed in DSE, including membrane-based and membrane-less configurations, as well as low- and high-temperature electrolysis systems. Additionally, we will analyze stability improvement strategies tailored for SWE systems coupled with intermittent renewable energy sources. Finally, we will summarize the critical stability challenges that require attention in the future development of DSE systems.
2 General description and stability challenges of direct seawater electrolysis
DSE is the same as conventional water electrolysis in terms of the main reactions, including OER and HER. However, since seawater is different from acid and alkali solutions as well as pure water, various dissolved ions are present.43 For example, seawater contains 55 wt% Cl− and 31 wt% Na+ by mass,44 which is the reason why a 0.5 M NaCl solution is usually used as a simulated seawater.45 Among them, Cl− will undergo CER and compete with OER at the anode, which may form toxic chloride or hypochlorite ions (Fig. 2(a) and (b)).46,47 Main anions and cations such as SO42−, Mg2+ and Ca2+ and microbial bacteria/microorganisms are also present. Mg2+ and Ca2+ will form Mg(OH)2 and Ca(OH)2 precipitates (Fig. 2(c)) in environments with locally elevated pH, which occurs at pH![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) ∼9.5, resulting in blockage of the active site, resulting in physical degradation.48,49 Furthermore, microbial bacteria and microorganisms can contribute to biofouling during seawater electrolysis, also blocking the active sites on the electrodes and the pores of the membrane.50 It is difficult to avoid issues such chemical and physical/mechanical degradation to cell components of seawater electrolysis,16 which deteriorates their ability to stabilize for long-term operation. For very small amounts of ions such as Br−, F−, etc.,29 they may adsorb onto the electrode surface. In this review, we are not concerned with them because of their low amount.
∼9.5, resulting in blockage of the active site, resulting in physical degradation.48,49 Furthermore, microbial bacteria and microorganisms can contribute to biofouling during seawater electrolysis, also blocking the active sites on the electrodes and the pores of the membrane.50 It is difficult to avoid issues such chemical and physical/mechanical degradation to cell components of seawater electrolysis,16 which deteriorates their ability to stabilize for long-term operation. For very small amounts of ions such as Br−, F−, etc.,29 they may adsorb onto the electrode surface. In this review, we are not concerned with them because of their low amount.
 | ||
| Fig. 2 (a) The Pourbaix diagram of the DSE. (b) Maximum permitted overpotentials for OER electrocatalysts. Reproduced with permission.44 Copyright 2024, The Royal Society of Chemistry. (c) Pourbaix diagram of HER and the competitive reduction of impurity cations in seawater. Reproduced with permission.51 Copyright 2021, The Royal Society of Chemistry. (d) Main long-term stability challenges for DSE: chemical degradation and physical/mechanical degradation. | ||
2.1 Working principle
The reaction mechanism of DSE is analogous to electrolysis technologies using pure water, the main reaction is 2H2O → 2H2+ O2 with the production of oxygen and hydrogen from water, and the cell voltage requires a standard thermodynamic potential of 1.23 V.44,52 Actually, a higher external voltage is required to drive the reaction.32 It specifically consists of two half-reactions, anode oxidation to produce oxygen and cathode reduction to produce hydrogen. Depending on the differences in acidic (H+) and alkaline (OH−) electrolyte, the cathodic/anodic half-reactions are as follows:50,53Acidic:
| Anode: 2H2O → O2 + 4H+ + 4e− (E° = 1.23 V) |
| Cathode: 4H+ + 4e− → 2H2 (E° = 0.00 V) |
Alkaline:
| Anode: 4OH− → O2 + 2H2O + 4e− (E° = 0.40 V) |
| Cathode: 4H2O + 4e− → 2H2 + 4OH− (E° = −0.83 V) |
However, due to the complex composition of seawater, challenges associated with DSE including potential side reactions, Cl− (∼0.54 M) plays a major hindering role in the electrolysis process.54 In the context of the reaction dynamics, the substantial presence of Cl−, paired with the limited availability of OH− and the analogous thermodynamic overpotentials associated with the CER and the OER, positions CER as a formidable side reaction at the anode. Under acidic or alkaline conditions at room temperature, competitive CER are described respectively:55
Acidic:
| 2Cl− → Cl2 + 2e− (E° = 1.36 V) |
Alkaline:
| Cl− + 2OH− → ClO− + H2O + 2e− (E° = 0.89 V) |
It is important to highlight that, according to thermodynamics, the OER is somewhat superior to the CER across a generalized pH range.28 Dionigi et al.46 conducted an in-depth analysis of Pourbaix plots that incorporated chloride and oxygen chemistry (Fig. 2(a)). They discovered that the potential difference between the CER and the OER is influenced by the pH of the electrolyte. Specifically, at pH < 3, the potential difference ranges from 180 to 350 mV, whereas in alkaline conditions (pH > 7.5), this potential difference can reach as high as 490 mV (Fig. 2(b)). This increased potential difference favors the selective occurrence of the OER.56 However, the kinetics of the four-electron OER is considerably slower than that of the two-electron CER. This disparity leads to significant overpotentials and reduces the thermodynamic advantage of OER, particularly in acidic conditions where the potential gap is at its narrowest.12 The result indicates that the CER is more advantageous compared to the OER, especially at high current densities. Therefore, based on the alkaline conditions inhibiting CER, alkaline water electrolysis (AWE) cell and anion exchange membrane water electrolysis (AEMWE) cell appear to hold greater promise for DSE compared to proton exchange membrane water electrolysis (PEMWE) cell.45 In addition, this may be the reason why some of the studies have taken ASE. The primary difference between NSE and ASE is that ASE employs natural seawater as the electrolyte and is supplemented with an alkaline additive (typically 1 M NaOH/KOH), precisely controlling the operating potential of ASE to shift the electrolysis conditions toward those of alkaline water electrolysis, which enhances the competitiveness of OH−.56 This effectively suppresses the competitive reactions and corrosive effects, thereby significantly improving the durability of electrolyzers. Unlike conventional water electrolysis where only H2O is consumed, enabling acids and bases to persist within the electrolyzer over extended operation to maintain required pH conditions (with only periodic replenishment of high-purity water needed), ASE systems face inherent limitations. The continuous replenishment of natural seawater in ASE leads to progressive ion enrichment in the electrolyte. Primarily, Ca2+ and Mg2+ ions persistently deplete OH− through precipitation formation, gradually lowering environmental pH and generating difficult-to-remove precipitates.48,49 Consequently, ASE operation necessitates a flowing electrolyte system with continuous alkaline seawater renewal, posing substantial commercialization challenges—particularly considering that DSE is economically viable mainly when deployed on offshore wind platforms where routine maintenance proves impractical.35,36 Additionally, the high price of strong alkalis (e.g., $800 per ton for KOH)1 is also a barrier to the widespread practical application of ASE. NSE has an undeniable cost advantage in practical applications. We consider that ASE is an improvement of NSE in DSE, and although the cost aspect needs to be improved, all related research aims to provide an effective indirect solution for the lasting development of DSE.
2.2 Mechanisms of chemical degradation
DSE is a technology that directly utilizes seawater as the electrolyte, fundamentally falling within the domain of water electrolysis. Consequently, the mechanisms of long-term performance degradation observed in conventional water electrolysis systems also apply to DSE. For instance, the dissolution of anode catalyst materials due to over-oxidation,57 passivation of bipolar plates and porous transport layers (PTLs) under high-potential conditions,3 and degradation of membranes caused by radical attacks and metal ion poisoning,3etc. However, it is critical to emphasize that the operational environment of DSE is significantly exacerbated by the complex ionic composition of seawater. Compared to alkaline or acidic electrolytes, the lower conductivity of seawater generally results in lower electrolysis efficiency.58 Moreover, due to the similar overpotentials of OER and CER, seawater electrolysis faces a significant challenge of simultaneously releasing toxic chlorine species at the anode. As we mentioned earlier, Cl2 (acidic condition)/ClO− (alkaline condition) produced during the side-reaction CER oxidizes and corrodes the catalyst as well as other key components (Fig. 2(d)) (bipolar plates, porous transport layers, etc. are usually made of metal (e.g. nickel, titanium or steel) or carbon).59 The oxide layer of the metal components causes an increase in ohmic loss as the current increases,60 and the anode potential further increases and exacerbates electrochemical and chemical degradation as described above. For example, the most commonly used catalyst for HER in pure water electrolysis is Pt,61 but in DSE, the stability of Pt is poor due to the strong interactions between Cl− and Pt preventing the adsorption of the reactants, and ultimately,62 Pt atoms absorbing large amount of Cl− begin to dissolve (Fig. 2(d)).632.3 Mechanisms of physical/mechanical degradation
Seawater, functioning as a non-buffering agent, causes the pH levels to rise locally at the cathode and fall at the anode during the electrolysis process. The pH near the anode can decrease by roughly 9 units, while at the cathode, it can decrease by roughly 5 units at a current density of 10 mA cm−2,64 the local pH increase due to the production of OH− by the HER during seawater electrolysis causes the precipitation of metal oxides (MgO and CaO) or hydroxides (Mg(OH)2 and Ca(OH)2),51 which occurs at a pH 9.5.65 These insoluble objects not only cover the electrodes and inhibit the mass transfer of H2O,66 resulting in a poor HER stability, but also block other components (e.g., diaphragms or membranes) and cause the membrane dehydration at the cathode, accelerating the chemical degradation of ionomer and membrane (Fig. 2(d)). In addition, for membranes such as AEM, the presence of Cl− can compete with the OH− conductivity of the AEM and increase additional ohmic losses. Increasing the OH− concentration can partially offset the competitive effect,67 but precipitation formation is also exacerbated. Therefore, this method is not informative and reducing precipitation formation is the more viable option. In addition, there are many organic molecules and microorganisms in seawater. They are first adsorbed on the surface of electrodes and membrane and then begin to form a conditioning film, providing a basis for microbial attachment. Minutes or hours later, microorganisms such as sulphate-reducing bacteria and algal spores colonize these surfaces by secreting extracellular polymeric substances (EPS) to form biofilms. After that, mature biofilms form multilayered microbial communities.68 In the coming days or weeks, macroalgae, barnacles, shellfish, and other visible fouling organisms will attach to the biofilm, creating fouling communities that further promote corrosion and biofouling.69,70 Biofouling can also block the active sites on the electrodes and the pores of the membrane, showing the obvious physical/mechanical degradation for DSE under long-term operation (Fig. 2(d)).50 Anti-biofouling measures for membranes and electrodes are not explicitly mentioned in DSE. We can borrow from seawater desalination. Some surface modification techniques are employed, such as hydrophilic coatings like polyethylene glycol (PEG) and polydopamine (PDA) reduce foulant adhesion. PDA-poly(sodium 4-styrene sulfonate) (PSS)/TiO2 modified AEMs and PSS&PDA-M membranes exhibit strong fouling resistance.71 Moreover, the formation and accumulation of gas bubbles generated in response to OER and HER involving gas evolution in water electrolysis usually bring about undesired covering of the active sites causing additional overpotentials.10 The impact of a large number of bubbles at high currents can also disrupt the interaction between the catalyst and the electrolyte (Fig. 2(d)).29,72 In addition, due to the complexity of seawater, other degradation phenomena in DSE may introduce unique synergistic mechanisms with bubbles behaviors, which are different from general water electrolysis. For example, compared with fresh water, high salinity reduces the surface tension of seawater and increases the adhesion of bubbles to the electrode, which prolongs the bubble residence time.73,74 The attachment of bubbles on the electrode surface will hinder the flow of electrolyte and cause a local microenvironment.75 The anode oxygen bubbles-covered area consumes OH− due to OER, the local pH value of the surrounding area is reduced, and the Cl− concentration increases relatively, which may trigger the CER. When HER occurs at the cathode, OH− is produced and the local pH increases, but the hydrogen bubbles hinder the diffusion of OH−, resulting in the rapid precipitation (Mg(OH)2 and Ca(OH)2) at the edge of the bubble. Therefore, the above degradation constitutes a major challenge and bottleneck for the long-term stability of DSE and is the reason why the long-term durability of DSE (NSE and ASE) is poorer than that of SSE.
3 Electrode materials and designs for securing long-term stability
Most DSE electrode designs focus on activity and selectivity, but the low stability due to degradation (seawater-induced degradation such as corrosion and blockage, etc. and degradation common to pure water electrolysis such as catalyst detachment, agglomeration and bubble blockage, etc.) also needs to be overcome for DSE. However, highly stable materials sometimes limit their activity, so catalytic activity and stability should be balanced during the design of DSE catalysts.28 The stability of the electrodes can be improved by optimizing the electrode materials to enhance their chemical degradation resistance, by surface modification to improve the selectivity of the OER and HER electrodes in the DSE, and by structural design to rapidly discharge bubbles and improve the formation and distribution of precipitates (Table 1).| Catalysts | Electrolyte | j [mA cm−2] | η [mV] | Stability | Ref. |
|---|---|---|---|---|---|
| OER | |||||
| NSE | |||||
| Cr2O3–CoOx | Seawater | 400 | 760 | 230 h@160 mA cm−2 | 76 |
| Mo3Se4–NiSe | Seawater | 10 | 166 | 50 h@500 mA cm−2 | 77 |
| PtPd–Ti | Seawater | 130 | 600 | 12 h@2.0 V (vs. RHE) | 78 |
| Zr–Co3O4/CP | Seawater | 100 | 570 | 160 h@100 mA cm−2 | 79 |
| CuS@CoOOH | Seawater | 10 | 290 | 72 h@10 mA cm−2 | 80 |
| HPS-NiMo | Seawater | 500 | 595 | 120 h@1.8 V (vs. RHE) | 81 |
| P/RP-SNCF | Seawater | 10 | 340 | 100 h@10 mA cm−2 | 82 |
| CoFeOF/nickel foam (NF) | Seawater | 100 | 280 | 145 h@400 mA cm−2 | 83 |
| ASE | |||||
| NiFe/NiSx-NF | 1.0 M KOH + seawater | 400 | 560 | 1000 h@400 mA cm−2 | 84 |
| MnCo/NiSe/NF | 1.0 M KOH + seawater | 1000 | 460.2 | 200 h@500 mA cm−2 | 85 |
| CoFe–Ni2P/NF | 6.0 M KOH + seawater | 500 | 304 | 600 h@500 mA cm−2 | 86 |
| RuMoNi/NF | 1.0 M KOH + seawater | 1000 | 470 | 3000 h@500 mA cm−2 | 87 |
| Ru/NiFeOOH | 1.0 M KOH + seawater | 500 | 330 | 400 h@100 mA cm−2 | 88 |
| NiFeO–CeO2/NF | 1.0 M KOH + seawater | 1000 | 408 | 200 h@1000 mA cm−2 | 89 |
| Ni3S2@NiFe LDH/NF | 1.0 M KOH + seawater | 1000 | 340 | 100 h@200 mA cm−2 | 90 |
| NiMoN@NiFeN/NF | 1.0 M KOH + seawater | 1000 | 398 | 100 h@500 mA cm−2 | 91 |
| Fe–Ni2Pv−x | 1.0 M KOH + seawater | 100 | 180 | 100 h@100 mA cm−2 | 92 |
| Co/P–Fe3O4@IF | 1.0 M KOH + seawater | 100 | 290 | 100 h@500 mA cm−2 | 93 |
| (NiFe)C2O4/NF | 1.0 M KOH + seawater | 100 | 280 | 600 h@1000 mA cm−2 | 94 |
| NiFe–CuCo LDH | 6.0 M KOH + seawater | 100 | 259 | 500 h@500 mA cm−2 | 95 |
| Fe2P/Ni3N/NF | 1.0 M KOH + seawater | 1000 | 340 | 40 h@500 mA cm−2 | 96 |
| Ru–Ni(Fe)P2/NF | 1.0 M KOH + seawater | 1000 | 361 | 50 h@600 mA cm−2 | 97 |
| Fe2P/Ni1.5Co1.5N/N2P | 1.0 M KOH + seawater | 500 | 307 | 40 h@100 mA cm−2 | 98 |
| Ni-doped FeOOH | 1.0 M KOH + seawater | 200 | 350 | 80 h@100 mA cm−2 | 99 |
| Fe–Ni–O–N/NFF | 1.0 M KOH + seawater | 1000 | 289 | 40 h@500 mA cm−2 | 100 |
| S-(Ni,Fe)OOH | 1.0 M KOH + seawater | 1000 | 462 | 100 h@100 mA cm−2 | 101 |
| Ni2P–Fe2P/NF | 1.0 M KOH + seawater | 1000 | 431 | 48 h@100 mA cm−2 | 102 |
| SSE | |||||
| NiFe-LDH/NF | 1.0 M NaOH + 0.5 M NaCl | 100 | 270 | 180 h@400 mA cm−2 | 103 |
| Ir/CoFe-LDH | 6.0 M NaOH + 2.8 M NaCl | 10 | 202 | 1000 h@800 mA cm−2 | 104 |
| Co3−xPdxO4 | 1 M PBS + 0.5 M NaCl | 10 | 370 | 250 h@100 mA cm−2 | 105 |
| Se_NiFe LDH | 1.0 M NaOH + 1.0 M NaCl | 400 | 670 | 600 h@100 mA cm−2 | 106 |
| NiCoHPi@Ni3N/NF | 1.0 M KOH + 0.5 M NaCl | 100 | 365 | 120 h@200 mA cm−2 | 107 |
| NiMoFe/NM | 1.0 M NaOH + 0.5 M NaCl | 100 | 241 | 1500 h@100 mA cm−2 | 108 |
| FeCr–Ni3S2 | 1.0 M KOH + 0.5 M NaCl | 500 | 286 | 500 h@500 mA cm−2 | 109 |
| F-NiFe-LDH | 1.0 M KOH + 0.5 M NaCl | 500 | 306 | 1000 h@1000 mA cm−2 | 110 |
| HER | |||||
| NSE | |||||
| Mo5N6 | Seawater | 10 | 260 | 100 h@20 mA cm−2 | 111 |
| CoxMo2−xC/MXene/NC | Seawater | 40 | 440 | 225 h@4 mA cm−2 | 63 |
| Pt@mh-3D MXene | Seawater | 30 | 360 | 250 h@10 mA cm−2 | 112 |
| Ti/NiCo | Seawater | 120 | 1000 | 10 h@-1.0 V (vs. RHE) | 113 |
| PF–NiCoP/NF | Seawater | 10 | 287 | 20 h@-0.29 V (vs. RHE) | 114 |
| Ptat–CoP MNSs/CFC | Seawater | 10 | 300 | 24 h@10 mA cm−2 | 115 |
| Ni5P4 | Seawater | 10 | 144 | 10 h@100 mA cm−2 | 116 |
| PtRuMo | Seawater | 100 | 800 | 20 h@-0.8 V (vs. RHE) | 117 |
| CoMoC/Mxene/NC | Seawater | 15 | 500 | 225 h@-0.5 V (vs. RHE) | 63 |
| Pt@mh-3D MXene | Seawater | 10 | 280 | 250 h@10 mA cm−2 | 112 |
| PtNb–Nb2O5 | Seawater | 500 | 440 | 360 h@-0.44 V (vs.RHE) | 118 |
| Pt/WO2 | Seawater | 10 | 290 | 500 h@100 mA cm−2 | 119 |
| ASE | |||||
| NiCoP–Cr2O3 | 1.0 M NaOH + seawater | 4000 | 275 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h@500 mA cm−2 000 h@500 mA cm−2 |
120 |
| F-FeCoPv@IF | 1.0 M KOH + seawater | 1000 | 210 | 20 h@100 mA cm−2 | 121 |
| Cu2S@NiS@Ni/NiMo | 1.0 M KOH + seawater | 1000 | 250 | 2000 h@500 mA cm−2 | 122 |
| Ru/MoO2@NiMoO4 | 1.0 M KOH + seawater | 1000 | 184 | 50 h@1000 mA cm−2 | 20 |
| Ru/Cd0.02Se4 | 1.0 M KOH + seawater | 10 | 6.3 | 50 h@10 mA cm−2 | 123 |
| Pt–Ni@NiMoN | 1.0 M KOH + seawater | 10 | 11 | 80 h@200 mA cm−2 | 124 |
| Pt–Ni3S2/Co9S8-Sv | 1.0 M KOH + seawater | 18 | 10 | 300 h@100 mA cm−2 | 125 |
| HW-NiMoN-2h | 1.0 M KOH + seawater | 1000 | 130 | 70 h@1000 mA cm−2 | 126 |
| Pt–Ni3N@V2O3/NF | 1.0 M KOH + seawater | 10 | 21 | 500 h@500 mA cm−2 | 127 |
| HPS–NiMo | 1.0 M KOH + seawater | 10 | 34 | 240 h@500 mA cm−2 | 128 |
| Co/P–Fe3O4@IF | 1.0 M KOH + seawater | 500 | 261 | 100 h@500 mA cm−2 | 76 |
| SA-MoO2/Ni3(PO4)2/NF | 1.0 M KOH + seawater | 10 | 46 | 10 h@100 mA cm−2 | 129 |
| Ni2P–Fe2P | 1.0 M KOH + seawater | 1000 | 389 | 40 h@500 mA cm−2 | 102 |
| NiMoN@NiFeN | 1.0 M KOH + seawater | 1000 | 218 | 100 h@500 mA cm−2 | 130 |
| SSE | |||||
| Ni–Co@Fe–Co PBA | 1 M KOH + 0.5 M NaCl | 183 | 10 | 24 h@50 mA cm−2 | 131 |
| NiCoPv@NF | 1 M KOH + 0.5 M NaCl | 1000 | 237 | 50 h@100 mA cm−2 | 132 |
| B,V-Ni2P | 1 M KOH + 0.5 M NaCl | 100 | 162 | 100 h@500 mA cm−2 | 133 |
| NiS–FeS@IF | 1 M KOH + 0.5 M NaCl | 500 | 322 | 500 h@1000 mA cm−2 | 134 |
| Ni-SN@C | 1 M KOH + 0.5 M NaCl | 10 | 23 | 24 h@10 mA cm−2 | 135 |
| a-NiCoS/c-CeOx | 1 M KOH + 0.5 M NaCl | 10 | 79 | 50 h@50 mA cm−2 | 136 |
3.1 Electrocatalytic materials and mechanisms for degradation resistance
Rational design of the catalyst composition is an effective way to enhance its corrosion resistance. Mn-based catalysts have been rigorously studied, demonstrating an OER selectivity that exceeds 90% in a 0.5 M NaCl aqueous solution across a range of pH levels.137 Jiang et al.138 developed a range of MnO2-based catalysts incorporating various dual dopants to enhance their performance as OER catalysts. They found that the inclusion of Fe and V as dopants in γ-MnO2 effectively strengthened the Mn–O bond and increased corrosion resistance (Fig. 3(a) and (b)). This resulted in the Mn–Fe–V composition exhibiting remarkable stability coupled with a high OER efficiency of 87.96%, maintained over a 500-hour operation period at a current density of 1 A cm−2 in alkaline NaCl solutions (Fig. 3(c)). Similarly, Ji et al.139 fabricated NiFe layered double hydroxide (LDH)/FeOOH heterostructures deposited on NiFe foam (Fig. 3(d)), achieving strong interfacial interactions. These heterostructures used for overall alkaline simulated seawater splitting with stable operation for 105 hours at a current density of 100 mA cm−2 (Fig. 3(e)). The enhanced performance was attributed to the heterojunctions, which facilitated the formation of NiOOH species that provided active sites, while the presence of FeOOH contributed significantly to corrosion resistance. | ||
| Fig. 3 The element content changes in the surface oxide compositions of the different elements doped manganese dioxide-coated electrodes before and after electrolysis: (a) Fe and (b) V. (c) The durability of the different elements doped manganese dioxide-coated electrodes. (a)–(c) Reproduced with permission.138 Copyright 2012, Elsevier. (d) TEM image of the NiFe LDH/FeOOH sample. (e) The durability test of the NiFe LDH/FeOOH in alkaline NaCl solutions. (d) and (e) Reproduced with permission.139 Copyright 2021, American Chemical Society. (f) The durability test and the SEM images of Pt–Ru–Mo-decorated Ti mesh. Reproduced with permission.117 Copyright 2021, The Royal Society of Chemistry. (g) The durability test of Mo5N6. (h) The N K-edge XANES of Mo5N6 and MoN before and after HER. (i) Mo LIII edge of Mo5N6 before and after HER. Reproduced with permission.111 (g)–(i) Copyright 2018, American Chemical Society. | ||
In the field of HER catalyst, alloying platinum-based materials with transition metals such as Fe, Ni, or Mo has been shown to potentially improve their corrosion resistance. Li et al.117 showcased a Pt–Ru–M (where M denotes Cr, Fe, Co, Ni, or Mo) alloy on a titanium mesh, achieving commendable stability exceeding 172 hours for seawater HER (Fig. 3(f)). They elucidated that this enhanced stability was due to the preferential dissolution of the M species over that of Pt and Ru, given that M species were more thermodynamically inclined to dissolve in the presence of corrosive Cl−. Jin et al.111 reported that nitrogen-rich Mo5N6 exhibits impressive durability, lasting 100 hours at an overpotential of 300 mV in seawater (Fig. 3(g)). The material's structural stability is attributed to its abundant strong metal–nitrogen bonds and the high valence state of molybdenum, which enhances its resistance to detrimental Cl− (Fig. 3(h) and (i)).
3.2 Electrode surface modifications
Improving the selectivity of the catalyst is an effective strategy to enhance main reactions and avoid CER side reaction,140,141 so electrode surface modification is introduced as a more promising and widely adopted approach to increase the OER and HER selectivity.There are two main ways to carry out the electrode surface modification, including the introduction of other anion ions84 by electrolyte additives/in situ generation and formation of a built-in electric field layer at the electrode–electrolyte interface to repel Cl− and the construction of Cl− a protective layer or blocking layer to decrease the local concentration of Cl− near the electrode. Firstly, Ma et al.103 offered a fresh perspective on surface sulfate additives in enhancing the stability of Ni-based electrodes in DSE. The presence of SO42− as an additive in the electrolyte tends to be preferentially adsorbed onto the anode surface. This adsorption creates an electrostatic repulsive force that effectively pushes Cl− away from the bulk solution. This repulsive influence of SO42− is also observed with NiFe LDH nanoarrays on nickel foam anodes, leading to stable performance at a current density of 400 mA cm−2 over 500 h in real seawater conditions. This stability is reported to be 3 to 5 times greater compared to operations conducted in an electrolyte lacking Na2SO4. Fan et al.142 developed a stable earth-rich LDH electrocatalyst for seawater electrolysis, achieving 2800 hours of operation at 2.0V versus RHE (Fig. 4(a)). Intercalating carbonate ions and anchoring graphene quantum dots improved corrosion resistance by reducing chloride ion adsorption (Fig. 4(b)). In addition, Liu et al.143 described a cobalt ferricyanide/cobalt phosphide (CoFePBA/Co2P) anode that can electrolyze alkaline seawater for more than 1000 hours at 1000 mA cm−2 (Fig. 4(c)) without corroding due to its self-generated PO43− and Fe(CN)63− layers repelling Cl− through electrostatic repulsion during operation (Fig. 4(d)). Tsao et al.106 electrodeposited NiFe LDH onto Se_NiFe foam. During the OER process in alkaline seawater, Se species were in situ oxidized to SeO3− or SeO4−, which could diffuse into the NiFe LDH layer, effectively repelling Cl− through electrostatic repulsion (Fig. 4(e)). Gong et al.144 developed a NiFe@DG core–shell nanocatalyst with a defective graphene coating that creates a built-in electric field at the electrode–electrolyte interface, repelling corrosive Cl− and protecting active metal sites. The electrolysis cell with NiFe@DG||Pt/C showed excellent performance in seawater with 1 M KOH, achieving current densities of 10 mA cm−2 and 100 mA cm−2 at low voltages of 1.496 V and 1.602 V, respectively, while operating stably for 1000 hours (Fig. 4(f)). Zhang et al.145 also created a strong built-in electric field at the (Ni, Fe) OOH/Ni12P5 interface, improving oxygen-evolution kinetics while reducing Cl− adsorption.
 | ||
| Fig. 4 (a) Stability test of CoFe–Ci@GQDs/NF at 2.0V versus RHE in alkaline simulated seawater for 2800h. (b) Schematic diagram of CoFe–Ci@GQDs protection mechanism against Cl− corrosion in seawater. (a) and (b) Reproduced with permission.142 Copyright 2024, Springer Nature. (c) Stability test at 1000 mA cm−2 in electrolytes: 6 M NaOH + saturated NaCl vs. 6 M NaOH + seawater. (d) Adsorbed Cl− quantification on various electrodes in 6 mM NaOH + 2.8 mM NaCl via ion chromatography. (c) and (d) Reproduced with permission.143 Copyright 2023, Wiley. (e) The mechanism of the improvement of stability of the Se_NiFe_LDH electrode. Reproduced with permission.106 Copyright 2021, Elsevier. (f) The stability tests and mechanism of enhanced durability for NiFe@DG compared to NiFe/G. Reproduced with permission.144 Copyright 2023, American Chemical Society. (g) Stability tests of Co3–xPdxO4 catalysts on MnO2 protected Ni foam substrates at current densities of 200 mA cm−2. Reproduced with permission.105 Copyright 2023, Wiley. (h) The electrocatalytic stability test at 20 mA cm−2 and representation of the enhanced stability achieved through the deposition of CeOx on the OER catalyst. Reproduced with permission.146 Copyright 2018, Wiley. | ||
For the construction of a protective layer or blocking layer, Nocera's research group reports that coatings such as Fe2O3, MnO2 and PbO2 will help to inhibit Cl−.147 Wang et al.105 added a MnO2 protective layer to nickel foam to prevent Cl− corrosion, then deposited Co3−xPdxO4 on the modified substrate. After 450 hours of continuous operation at 200 mA cm−2 in natural seawater, the potential remained stable (Fig. 4(g)). Keisuke Obata et al.146 found that a CeOx layer deposited at the anode protects the NiFeOx electrocatalyst from iron loss, ensuring stable performance. This layer permits OH− and O2 to pass while blocking Cl−, without impairing the underlying activity of OER catalyst (Fig. 4(h)).
The primary challenges associated with HER electrocatalysts include corrosion resulting from the strong interactions between Cl− and Pt, as well as the precipitation of Mg2+/Ca2+ at the cathode, both of which significantly compromise performance and long-term stability. Surface modifications can be employed to mitigate the effects of Cl− corrosion and prevent the precipitation. Xu et al.122 developed a Cu2S@NiS@Ni/NiMo electrode demonstrating high HER activity with a 250 mV overpotential at 1000 mA cm−2 and exceptional durability over 2000 h at 500 mA cm−2 in 1 M NaOH and seawater (Fig. 5(a)). The NiS layer provides sulfur for protective polyanion-rich coatings, effectively resisting Cl− corrosion (Fig. 5(b)). Additionally, Ni3N@C/NF, featuring Ni3N nanosheets coated with a carbon shell, protects Ni3N from poisoning and corrosion by Cl−, achieving the sustained performance over 100 h in 1 M KOH and seawater (Fig. 5(c)).148 For the improvement of precipitation, OH− can be captured or limited, or Mg2+/Ca2+ can be repelled from reaching the electrode surface. Guo et al.11 showed that OH− can be captured by an introduced Lewis acid layer (creating a dynamic localized acidic environment) (Fig. 5(d)), such as Cr2O3, TiO2 and V2O3 layer proposed by Hu et al.127 Bao et al.119 exploited the in situ formation of hydrotungsten bronze (HxWOy) due to the reversible hydrogen insertion/excretion behavior of WO2 to act as a proton ‘sponge’ to store H+ and create an acid-like local environment (Fig. 5(e)), culminating in the proposal of a Pt/WO2 catalyst and realizing the long-term stability of >500 hours at 100 mA cm−2 in natural seawater electrolysis (Fig. 5(f)).
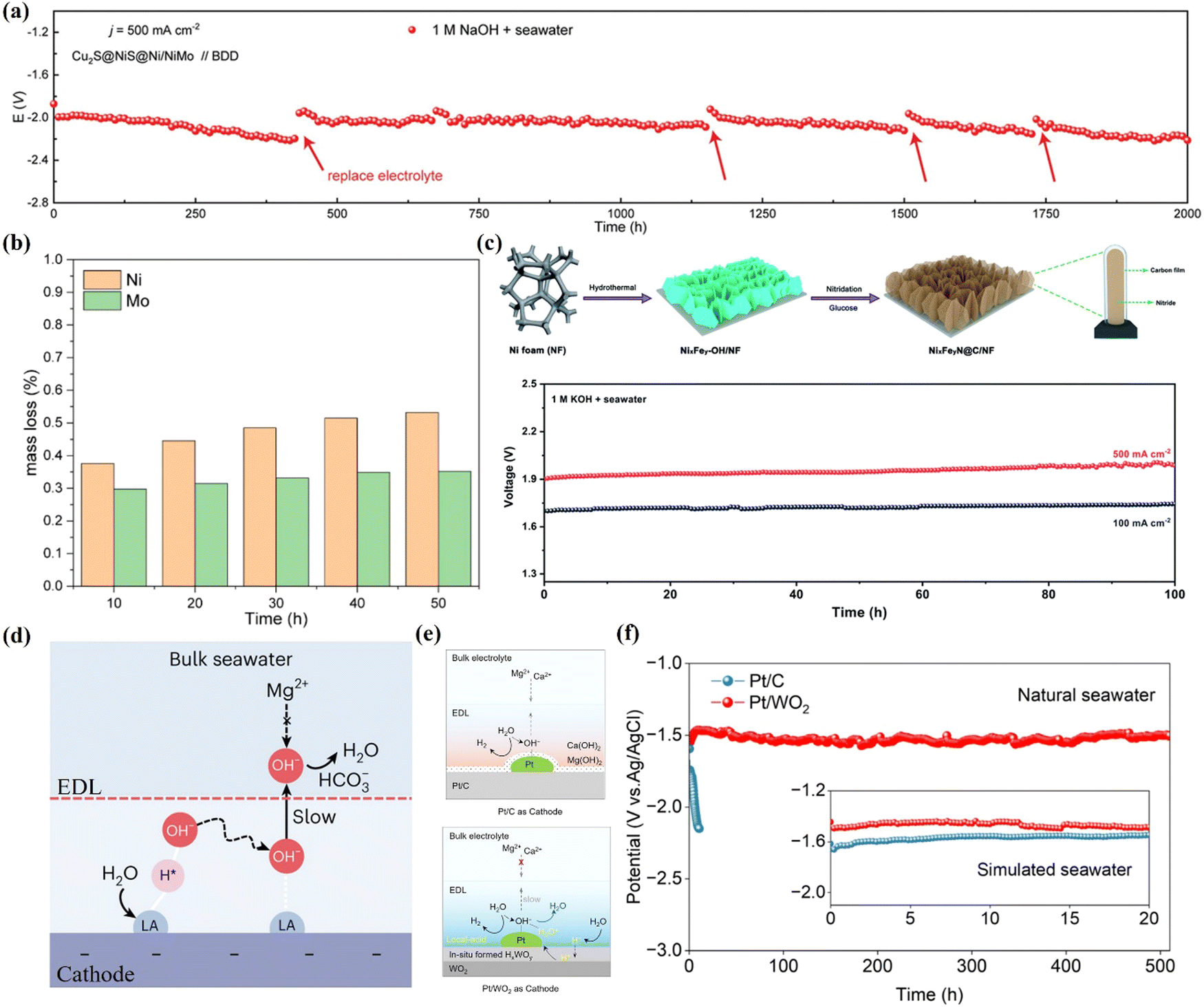 | ||
| Fig. 5 (a) Stability test of BDD//Cu2S@NiS@Ni/NiMo at 500 mA cm−2 in 1 M NaOH + seawater. (b) Time-dependent mass loss measurement of Cu2S@NiS@Ni/NiMo under 400 mA cm−2. (a) and (b) Reproduced with permission.122 Copyright 2023, Wiley. (c) The preparation process of NixFeyN@C/NF and stability tests at 100 and 500 mA cm−2. Reproduced with permission.148 Copyright 2021, The Royal Society of Chemistry. (d) Schematic illustrating the generation of a local alkaline microenvironment at the Lewis acid-modified cathode, enhancing HER while inhibiting precipitate formation. Reproduced with permission.11 Copyright 2023, Springer Nature. (e) Schematic comparison of the HER mechanisms on Pt/C and Pt/WO2. (f) Stability testing at 100 mA cm−2 over Pt/WO2 and Pt/C in natural seawater under Ar. Reproduced with permission.119 Copyright 2024, American Chemical Society. | ||
3.3 Electrode structures and configurations
Firstly, optimizing the electrode structure and configuration can significantly improve its own electrochemical active surface area (ECSA), or activity. Nonetheless, the intrinsic electrochemical activity of electrode is not completely harnessed.149 In particular, the control of the electrode structure encompasses aspects such as the thickness of the porous structure, the arrangement of catalytic sites, micro- and nano-pore structures and hierarchical pore structures. These factors play a crucial role in modulating the interfacial engineering of mass transport and electron and ion conduction throughout the reactions at the electrodes150,151 and in optimizing the effective use of the catalytic sites.10 Most importantly, it can also improve the unstable operation caused by chemical and physical/mechanical degradation in seawater electrolysis (accumulation of bubbles during OER and HER reactions, precipitation of metal oxides or hydroxides covering the cathode electrodes). This can affect ion transport and corrosion resistance at the interface, as well as interfacial delamination over long periods of operation all further affecting long-term stability.152It was found that a heterogeneous bimetallic phosphide, Ni2P–Fe2P, on Ni foam is an effective bifunctional catalyst for seawater electrolysis through an in situ growth-ion exchange phosphidation method. The resulting porous, ultrathin nanosheets (7.4 nm) exhibited hydrophilic properties (Fig. 6(a)), enhancing specific surface area (Fig. 6(b)), electrolyte diffusion, and bubble release, which improved the electrocatalyst's stability at high current densities.102 In addition, a similar function was achieved by the multilayer crystalline-amorphous heterostructured electrode material NF/(CoMo)0.85Se@FeOOH proposed by Li et al.153 (Fig. 6(c)), featuring a porous architecture for enhanced active site exposure and mass transfer. This also emphasizes the importance of constructing porous structures and achieving ideal surface superhydrophobicity and superhydrophilicity to ensure adequate exposure to active sites and efficient mass transfer.154 The multilayered architecture, featuring a (CoMo)0.85Se core with a FeOOH shell and an in situ formed transition metal (oxy)hydroxide outer layer enriched with polyatomic anions (MoOxn− and SeOxn−), demonstrates excellent mechanical stability and resistance to chloride corrosion in harsh seawater conditions. Liu et al.155 proposed a design, Os–Ni4Mo/MoO2 micropillar arrays (Fig. 6(d)) with strong metal-support interaction (MSI), for seawater electrolysis. Strong MSI between Os and Ni4Mo/MoO2 enhances the catalyst's surface electronic structure, lowering the reaction barrier and boosting catalytic activity. In addition, this design not only enhances electron and mass transfer, but also creates a dual Cl− repelling layer using electrostatic force to protect active sites from Cl− attack in seawater oxidation, consisting of strong Os–Cl adsorption and an in situ-formed MoO42− layer. Consequently, it shows excellent stability, with a degradation of just 0.37 μV h−1 after 2500 hours of seawater oxidation. For the precipitation, Liang et al.156 developed a novel honeycomb-like 3D structure with longitudinal micro-channels for a microscopic bubble/precipitate traffic system, allowing for robust anti-precipitation in seawater (Fig. 6(e)). This ordered cathode generates uniform small-sized bubbles that aid in self-cleansing (Fig. 6(e)). Liu et al.157 designed a seawater HER electrode with a Ni(OH)2 nanofiltration membrane grown on nickel foam at room temperature (Fig. 6(f)). The positively charged Ni(OH)2 membrane, featuring nanometer-scale cracks, selectively hinders the transfer of Mg2+/Ca2+, reducing precipitation by 98.3% (Fig. 6(g)). This design ensures rapid of transfer OH− and H2O for enhanced HER activity and stability than the Nikel foam electrode. In terms of electrode–membrane interface, Frisch et al.152 adopted a combined catalyst coated membrane (CCM) and catalyst coated substrate (CCS) approach (a lower (2 mgLDH cm−2) and a higher (8 mgLDH cm−2) amount on the membrane (CCM) and on the PTL (CCS), respectively.), enhancing catalyst adhesion and reducing delamination at the interface. Since there are fewer studies on the improvement of membrane–electrode interface for DSE, we can draw on the relevant strategies in AEMWE. Wan et al.158 proposed a swelling-assisted transfer strategy to build ordered anode catalyst layers (ACL) on AEM (Fig. 6(h)). Specifically, a three-dimensional interlocked ACL/AEM interface, formed by direct membrane deposition method, can be perfectly transferred to the ordered ACL to AEM, thus permitting vertically orientated through-hole ACL structures and aligned ionic layer layers for OH− transfer. Stable operation was achieved for 700 h in pure water-feeding mode at ∼1.7 V with a current density of 1000 mA cm−2 due to the strong adhesion between the membrane and CL. Hu et al.159 presented an easy method to prepare patterned membranes by casting a polymer solution to the surface of commercially available monocrystalline silicon plates that have a pyramidal pattern on their surface (Fig. 6(i)). The prepared membranes show 39% permeability and 23% enhancement in electrochemical surface area as compared to flat membranes with the same catalyst loadings.
 | ||
| Fig. 6 (a) Schematic illustration of the formation of Ni2P–Fe2P/NF and chronopotentiometric curves at 100 and 500 mA cm−2 for overall water/sweater splitting in 1 M KOH seawater. (b) SEM images of Ni2P–Fe2P/NF. (a) and (b) Reproduced with permission.102 Copyright 2020, Wiley. (c) Synthetic procedure for the NF/(CoMo)0.85Se@FeOOH nanosheet array and wetting characteristics of bare NF, NF/(CoMo)0.85Se, and NF/(CoMo)0.85Se@FeOOH. Reproduced with permission.153 Copyright 2023, American Chemical Society. (d) Schematic illustration of dual Cl− repelling layer and chronopotentiometry curves of Os–Ni4Mo/MoO2 for OER. Reproduced with permission.155 Copyright 2024, Wiley. (e) SEM images of the honeycomb-like 3D electrode and its mechanism of repelling precipitation. Reproduced with permission.156 Copyright 2024, Elsevier. (f) The number of Mg2+, Cl−, and H2O passing through the charged and uncharged Ni(OH)2 nanosheet. (g) The mass of precipitation on the Ni(OH)2–Pt–NF and Pt–NF electrodes. Reproduced with permission.157 Copyright 2023, The Royal Society of Chemistry. (h) Details of the novel swell-assisted transfer method. and schematic illustration of the general fabrication method of the 3D-ordered ACL in an MEA. Reproduced with permission.158 Copyright 2024, The Royal Society of Chemistry. (i) Preparation of patterned membrane and schematic diagram of membrane electrode with patterned membrane. Reproduced with permission.159 Copyright 2024, American Chemical Society. | ||
4 Electrolysis cell designs for continuous and large-scale production
The electrode referenced earlier plays a crucial role in DSE and has been the subject of numerous studies aimed at enhancing its long-term stability. Challenging seawater environments and severe operational conditions, such as elevated current densities and high temperatures, impose significant demands on catalyst performance. Regrettably, many existing catalyst engineering approaches fall short of fulfilling the rigorous requirements for practical applications. The design of the reactors and their components also significantly influences overall stability. Additionally, various strategies for improving stability are tied to different electrolyte and reactor configurations. In this section, we will provide a comprehensive description of these aspects.4.1 Membrane-less electrolysis cells
The most common type of membrane-less electrolysis cell is the single-compartment electrolysis cell, which utilizes a three-electrode system. This design is frequently employed in laboratory-scale catalyst screening experiments.17 However, there remains a disconnect between the testing conducted with these electrolysis cells and actual production processes, a topic we will not delve into here. Membrane-less electrolysis cells hold significant promise for DSE due to their streamlined design, which features fewer components compared to membrane electrolysis cells.30 However, they demand higher catalyst activity and selectivity, making it still in need of further research. For example, macro membrane-less electrolysis cells face challenges like gas crossover,160 but various methods have been developed to mitigate this issue. One traditional approach utilizes the Segré–Silberberg effect (relies on shear-induced lift to maintain neutrally buoyant particles at a fixed distance from the center in laminar flow), where controlling bubbles around electrodes via a flow rate gradient helps separate H2 and O2 into distinct effluent channels (Fig. 7(a)).37 For instance, using two flow-through electrodes made of porous metal mesh positioned face-to-face allows the electrolyte to carry H2 and O2 into separate channels (Fig. 7(b)), reducing gas crossover.161 O’Neil et al.162 tried angling these electrodes and adding insulating plates (Fig. 7(c)), resulting in an increase in ohmic impedance but minimized gas separation (Fig. 7(d)). Alternatively, Esposito et al.163 implemented an asymmetric mesh electrode (Pt deposited onto only outer surfaces of Ti mesh) that leveraged buoyancy for gas separation (Fig. 7(e)), achieving a crossover rate of about 1%, though with subpar electrolytic efficiency (Fig. 7(f)). It operates stably for 75 minutes under continuous illumination with 0.5 M H2SO4, coupled with a floating photovoltaic module. Consequently, microfluidic membrane-less electrolysis cells have been developed to improve both gas crossover and high ohmic resistance issues and they have no need for pumps. De et al.164 integrated bifunctional nickel nitride (Ni3N/Ni) catalysts into electrolysis cells (Fig. 7(g)), achieving 6-hours stable operation. However, at higher current densities, additional strategies are needed. Although this approach is still in its early stages, there is significant potential for enhancing its activity, stability, and electrolysis efficiency. However, the key issue is that its size is too small for industrial DSE with high currents. In addition, a new type of membrane-less electrolysis cells using pure water conditions has been designed. Hodges et al.165 proposed a capillary-fed electrolysis (CFE) cell concept, enabling bubble-free operation for hydrogen and oxygen evolution electrodes. Their alkaline capillary feed electrolysis cells outperform commercial cells, achieving a voltage of only 1.51 V at 500 mA cm−2 and 85 °C. It showed stable performance over a long period of time lasting from one day to 30 days at 80 °C (Fig. 7(h)) and room temperature and is considered by the authors of this paper to be of interest in future DSE. | ||
| Fig. 7 (a) The membrane-less electrolysis cell separates the produced gas utilizing Segré–Silberberg effect. Reproduced with permission.37 Copyright 2015, The Royal Society of Chemistry. (b) Mechanism of membrane-free divergent electrode-flow-through electrolysis. Reproduced with permission.161 Copyright 2015, Elsevier. (c) Membrane-less electrolysis cell utilizing angled mesh flow-through electrodes and a single-component device structure. (d) Solution resistance as a function of angle between electrodes and buildup of bubbles on the electrodes in stagnant solution and in a flowing electrolyte with fluid velocity of 13.2 cm s−1 during electrolysis at 2.5 V. (c) and (d) Reproduced with permission.162 Copyright 2016, The Electrochemical Society. (e) Detailed schematic of a passive membrane-less electrolysis cell utilizing buoyancy-driven product separation. (f) The comparison of hydrogen cross-over percentage is presented for both symmetric and asymmetric electrodes under different angles, with operations conducted at a current density of 20 mA cm−2. Reproduced with permission.163 Copyright 2023, The Royal Society of Chemistry. (g) Separation of gas products at an intermediate position and at the exit when the flow rate varies. Reproduced with permission.164 Copyright 2021, American Chemical Society. (h) Design concept for capillary-fed electrolysis cell and its 30 days stability test. Reproduced with permission.165 Copyright 2022, Springer Nature. | ||
4.2 Membrane-based electrolysis cells
In DSE, modifications to the electrode structure have been described in detail in Section 3.2. Next, we focus on the impact of operating modes and other key components on long-term stability. The typical mode of operation is to use a symmetrical feed of alkalized seawater, however, asymmetrical feeding may avoid the competitive CER at the anode. Strasser et al.166 describe an electrolysis cell that circulates neutral seawater at the cathode and pure KOH at the anode, demonstrating excellent corrosion resistance and OER selectivity. Starting at 1.7 V with a current density of 200 mA cm−2, the voltage rose by 8–10 mV every 12 hours, accumulating to about 100 mV over 100 hours (Fig. 8(a)). While the NiFe-LDH electrodes remained stable, the degradation was linked to the anode catalyst, collector, or membrane components. Although few studies have been done on the key components such as flow fields in DSE, there have also been some studies into structural designs to facilitate bubble removal in AMEWE using KOH or water.167 Duan et al.168 introduced a novel concave–convex mastoid polar plate that enhances gas–liquid distribution uniformity in the flow channel. Using a three-dimensional model for numerical simulations, they optimized the structure with a multi-objective genetic algorithm. The optimized design achieved an 8.40% improvement in electrolyte flow rate uniformity while maintaining a lower pressure drop compared to the original structure (Fig. 8(b)), which can be beneficial for the long-term stable operation.3 In addition, Shi et al.169 designed a pH asymmetric electrolysis cells (Fig. 8(c)) incorporating a Na+ exchange membrane for direct seawater decomposition to prevent Cl− corrosion by blocking Cl− transport to the anode, and mitigated Mg2+ and Ca2+ precipitation in near-neutral seawater (pH < 9.5). The output cathode electrolyte is maintained at pH 8.5 at 100 mA cm−2, showing a stable voltage response. After 120 hours of water-splitting, only a slight voltage increase is needed to reach 100 mA cm−2 after refreshing the solution, confirming the system's durability (Fig. 8(d)). Alternatives to the anode include electronic mediators or small organic molecules that can replace slow OERs, enabling reactions at low voltages to avoid OERs and potentially eliminate chlorine emissions,44,170 which is not what the review is exploring.
 | ||
| Fig. 8 (a) Symmetric alkalinized 0.5 M NaCl feed and asymmetric 0.5 M NaCl feed at the cathode and 0.5 M KOH feed at the anode. Reproduced with permission.166 Copyright 2020, The Royal Society of Chemistry. (b) Machine learning aids the optimization process of flow field in AWE.168 Copyright 2024, Elsevier. (c) Asymmetric electrolysis cell with Na+ exchange membrane. (d) Long-term stability test of the asymmetric electrolysis cell with Na+ exchange membrane at 100 mA cm−2 and Cl− concentration in anode electrolytes. (c) and (d) Reproduced with permission.169 Copyright 2023, Springer Nature. (e) Schematic of an electrolysis cell with anode: seawater vapor (80% relative humidity) and cathode: dry N2 and current density versus time at an applied voltage of 1.6 V for different feedstock at the electrolysis tank anode. Reproduced with permission.171 Copyright 2016, The Royal Society of Chemistry. (f) Schematic of an electrolysis cell using a vapor feed at the anode and saltwater at the cathode and the effect of Na+ in solution on the overpotential caused by pH gradients. Reproduced with permission.172 Copyright 2021, The Royal Society of Chemistry. (g) Schematic of an electrolysis cell with BPM. Reproduced with permission.33 Copyright 2023, Elsevier. (h) Mechanism of seawater acidification via synergistic effects between inorganic precipitation on the cathode surface and proton flux from BPM. (i) pH variation of bulk seawater during 1000 hours of BPMWE operation at 20 mA cm−2, along with changes in concentrations of Mg2+ and Ca2+. (h) and (i) Reproduced with permission.173 Copyright 2023, The Electrochemical Society. | ||
178 and SO42−179 permeation across the membrane is also inhibited, the main reason is that, for example, H+ flux inhibits K+ crossover to the cathode, which also highlights an interesting approach to mitigating contaminant ion crossover during the electrolysis process. Kim et al.179 created a locally alkaline environment in the anode chamber with KOH solution and a locally acidic environment in the cathode chamber with H2SO4 solution in BPMWE. This setup optimized pH conditions for each half-reaction, reducing anodic competition for CER and Ca2+/Mg2+ precipitation at the cathode. Additionally, an optimized porous cathode with an open region and BPM facilitated proton generation through water dissociation (Fig. 8(h)).180 The reduction of protons near the cation exchange layer, along with proton flow through the open region, minimized inorganic scaling and acidified the seawater, resulting in BPMWE stability for 1000 hours (Fig. 8(i)).173 The advantages and challenges of various electrolysis cells (e.g. AWE, AEMWE, PEMWE, BPMWE) under seawater conditions are summarized in Table 2.
| Type | Advantages | Challenges |
|---|---|---|
| AWE3,9 | 1. Mature, commercially available | 1. Low efficiency and current density |
| 2. Lower material costs | 2. Gas crossover | |
| 3. Robust design | 3. Slow response to variable conditions | |
| AEMWE3,9,45 | 1. High efficiency and current density | 1. Limited membrane durability |
| 2. Compact, modular design | 2. Low OH− conductivity | |
| 3. High-purity hydrogen production | 3. Precipitation blocking membrane and electrodes | |
| 4. Lower material costs | ||
| PEMWE4,174,181 | 1. High efficiency and current density | 1. Expensive catalysts and components |
| 2. Compact, modular design | 2. More prone to CER | |
| 3. High-purity hydrogen production | 3. Ca2+/Mg2+, etc. displace H+ in membrane175 | |
| BPMWE177,179 | 1. Separate anode/cathode environments | 1. Less mature |
| 2. Flexible anode/cathode pH control | 2. Complex membrane fabrication | |
| 3. Reduce Cl2 and precipitation | ||
 | ||
| Fig. 9 (a) Examples of some typical degradation pathways of membrane materials. Reproduced with permission.28 Copyright 2020, Elsevier. (b) The schematic structure of high-molecular-weight hexyltrimethylammonium-tethered polycarbazoles (HQPC-TMAs) is presented, along with the electrolysis performance of HQPC-TMA-2.4 in 1.0 M KOH and 1.0 M KOH + seawater. Additionally, a 1000-hour durability test of the 1.0 M KOH electrolyzer, operated at 1.0 A cm−2 and 60 °C.184 Copyright 2024, The Royal Society of Chemistry. (c) Molecular structures of AEM: branched poly(terphenyl piperidinium) (b-PTP), AEI: Sustainion XB-7, and CEI: Nafion, along with a schematic representation of the various catalyst-PTL interfaces and stability tests at 10 A cm−2 (Pt/C as the cathode. The operational conditions included a cell temperature of 80 °C and 1 M KOH as the electrolyte). Reproduced with permission.185 Copyright 2024, Wiley. (d) Chemical structures of ionomers (PTPA, PBPA, and PFTA-20) and an illustration of CCS || CCM MEA configuration is presented. Additionally, a long-term durability test of AEMWE was conducted at a constant current density of 1.0 A cm−2 at 80 °C. Reproduced with permission.186 Copyright 2025, Elsevier. | ||
| Ion | Concentration (mol kg−1 (H2O)) | Effect |
|---|---|---|
| Cl− | 0.56576 | CER (corrosion),45 reducing the conductivity166 |
| Na+ | 0.48616 | No significant adverse effects reported |
| Mg2+ | 0.05475 | Precipitate covering the membrane28 |
| SO42− | 0.02927 | Affecting the OH− transport and reducing the conductivity45 |
| Ca2+ | 0.01065 | Precipitate covering the membrane28 |
| K+ | 0.01058 | No significant adverse effects reported |
| HCO3− | 0.00183 | Concentration polarization,183 reducing the conductivity187 |
| Br− | 0.00087 | Corrosion and poison29 |
| CO32− | 0.00027 | Concentration polarization,183 reducing the conductivity187 |
| F− | 0.00007 | Corrosion and poison29 |
| Biofouling | NA | Poison and covering membrane188 |
| OH− | 0.00001 | No adverse effects reported |
Improved electrode design strategies for degradation (bubble/precipitation coverage, CER corrosion, etc.) are also beneficial to the performance of the membrane in long-term operation, as described in detail in Section 3. In addition, some specific strategies have been proposed to address the above membrane degradation. Firstly, Kim et al.184 developed high-molecular-weight hexyltrimethylammonium-tethered polycarbazoles (HQPC-TMA-x) membrane that exhibited high ionic conductivity, mechanical robustness, and alkaline stability. HQPC-TMA-x also helps mitigate ionomer adsorption issues on electrodes due to the polycarbazole backbone. It demonstrated outstanding performance in both pure water and direct seawater electrolysis. HQPC-TMA-2.4 also showed impressive durability, maintaining a high current density of 1.0 A cm−2 for 1000 hours, with minimal irreversible degradation rates of 52 and 6 μV h−1 for platinum group metal (PGM)-free cells, respectively (Fig. 9(b)). In addition, from the point of view of enhanced membrane conductivity (strong OH− transport) and strong adhesion of the ionomer, Zheng et al.185 used a branched poly(terphenyl piperidinium) (b-PTP) AEM189 that proved to be robust, and a blended ionomer combining strong adhesive Nafion ionomer and strong OH− transport capacity of the Sustainion ionomer, which allowed the AEMWE to operate for more than 800 hours at 10 A cm−2 (Fig. 9(c)). Li et al.186 designed an anion exchange ionomer (AEI) featuring a non-rotatable fluorenyl backbone that minimizes phenyl adsorption and enhances oxidation stability, resulting in 1800 h with a low voltage of 1.6 V at 1.0 A cm−2. Notably, they proposed a hybrid MEA design, combining a coated-catalyst substrate anode and a coated-catalyst membrane cathode, addressing membrane swelling and enhancing MEA stability through strong bonding between the anode catalyst layer and the porous substrate (Fig. 9(d)). Although the above studies did not directly conduct long-term stability tests under seawater electrolysis conditions, they still provide guidance for the design of durable membranes and ionomers for DSE.
4.3 Solid oxide-based electrolysis cells
In addition to the above-mentioned electrolysis cells operating at low temperatures, high-temperature solid oxide electrolysis cells (SOECs) using seawater operating at thermally driven high temperatures (800–1000 °C) have also been reported. In a high-temperature SOEC, steam is evaporated from the water and transported to the cathode to produce H2, and the resulting O2 passes through the solid oxide membrane to the anode to form O2.190 Operating at thermally driven high temperatures results in lower costs relative to electrical energy. In addition, high operating temperatures increase reaction kinetics and improve overall electrolysis efficiency. Theoretically, the energy conversion efficiency of a SOEC can exceed 100% if the absorbed heat is considered to be free of charge. The absence of any impurities in the water vapor also greatly benefits the lifetime of the membrane and catalyst. However, seawater electrolysis in SOEC can be problematic, including the long-term durability requirements of the electrodes and damage to the electrolysis cells from trace impurities in seawater vapor.191 Liu et al.192 used SOEC to split untreated seawater, achieving a hydrogen production rate of 183 mL min−1 and a degradation rate of 4.0% over 420 hours at a constant current density of 200 mA cm−2. The energy conversion efficiency was 72.47%, without reusing high-temperature exhaust gas. However, they also found that ppm salts were still present in the vapor and that the tubes or cells could clog at certain points after 420 hours of operation. Not only that, high temperatures may require catalysts with excellent thermal stability,193 for example, Hauch et al.194 reported that continuous steam electrolysis at high current densities (1000 mA cm−2) caused significant microstructural degradation at the oxygen electrode–electrolyte interface and a marked increase in ohmic resistance. Most of the catalyst engineering strategies proposed in the current reviews have focused on low-temperature water electrolysis, with little involvement in high-temperature water electrolysis. Whether all strategies can be applied to high-temperature water electrolysis is not clear, and thus they are still in the early stages of development.4.4 Electrolysis cell integration and scaling-up
Integrating different technologies into a single system offers the potential to improve efficiency, reduce costs, and address complex challenges more effectively. Nocera et al.195,196 recently developed the forward osmosis water electrolysis (FOWS) cells, streamlining seawater electrolysis by combining reverse osmosis desalination with freshwater electrolysis. This system utilizes brine directly, mitigating issues with impurities and energy losses. The FOWS consists of a single electrochemical chamber separated from seawater by a semi-permeable membrane, with a 0.8 M NaPi buffer solution on one side and 0.6 M NaCl on the other. The use of cellulose acetate membranes to filter out anions and cations such as Ca2+, Mg2+, CO32− and Cl− from seawater from the hydrolysis region (Fig. 10(a)). Continuous operation requires balancing freshwater consumption and introduction by adjusting the concentration gradient and electrolysis current density (Fig. 10(b)). With 0.8 M NaPi and a platinum catalyst, the system maintained a stable electrolysis at 250 mA cm−2 for 48 hours (Fig. 10(c)), achieving nearly 100% Faraday efficiency. However, challenges like gas crossover due to the lack of a barrier between hydrogen and oxygen, and Cl− diffusion from seawater remain. Logan et al.197 suggested using an osmosis assembly between electrodes to separate gases (Fig. 10(d)), but Cl− diffusion and HER catalyst selectivity and stability issues in seawater need further addressing. | ||
| Fig. 10 (a) Key components of the FOWS cell for use in contaminated water sources and their operation process. (b) The rates of water influx (red) and outflux (blue). (c) Stability test at 250 mA with a 0.8 M NaPi inner electrolyte solution and 0.6 M NaCl outer electrolyte solution. (a)–(c) Reproduced with permission.196 Copyright 2020, National Academy of Sciences. (d) The membrane divides the electrodes into two chambers, with desalinated water collected from the catholyte. Reproduced with permission.197 Copyright 2021, Elsevier. (e) Actual picture of the integrated stack of the AWE with porous PTFE membrane. Reproduced with permission.34 Copyright 2022, Springer Nature. (f) Electrolysis of seawater using a water phase-transition migration mechanism through porous PTFE membrane. (g) The average ion concentrations in the innovative DSE cell and Xinghua Bay seawater. (h) Water migration behavior of the porous PTFE membrane before and after 10 days of use. Reproduced with permission.39 Copyright 2024, Springer Nature. | ||
Based on the above research about the integration of in situ seawater purification technologies and water electrolysis technologies, Xie et al.34 developed an innovative DSE system for hydrogen production, operating at 250 mA cm−2 for over 3200 hours. This system integrates an in situ water purification mechanism that uses a self-driven phase transition process and seawater electrolysis. It features a hydrophobic, porous polytetrafluoroethylene (PTFE) membrane as a gas pathway, alongside a concentrated KOH solution acting as a self-dampening electrolyte (Fig. 10(e)). The difference in vapor pressure between seawater and the KOH allows for the evaporation of seawater, which then diffuses through the membrane and is re-liquefied, effectively filtering out contaminants like Cl−, Mg2+, and SO42−. This results in a continuous supply of pure water for hydrogen production from seawater. Due to the efficient isolation of impurities in the electrolysis cells, the integrated cell stack utilizes commercial MoNi/Ni foam for the anode and PtNi mesh for the cathode (Fig. 10(f)). Additionally, in collaboration with China Dongfang Electric Corporation, the team developed the “Dongfu-1,” the first offshore wind-driven hydrogen production platform without desalination.39 During sea trials in May 2023, it produced 1.3 N m3 h−1 of hydrogen, exceeding the target energy consumption of 5 kW h N m−3, and operated for over 240 hours with over 99.99% ionic barrier efficiency against seawater impurities (Fig. 10(g)) and there is no change in water migration in PTFE membranes (Fig. 10(h)), ensuring hydrogen purity levels of 99.9% to 99.99%.
For large-scale DSE, there is still a trade-off between economic factors and performance stability for different electrode materials during the transition from laboratory scale to industrial scale. Specifically, commercial Pt-based electrocatalysts can only operate in natural seawater for less than 1 hour due to toxic substances,198 while commercial RANEY® Ni suffers severe Cl− corrosion.199 Commercial IrO2200 and RuO2201 also face selectivity and durability challenges. Therefore, directly applying existing commercial catalysts to DSE for large-scale applications is impractical. Most ampere-level DSE electrodes currently employ self-supported electrodes developed on metal foam substrates. Metal foams act not only as conductors and carriers but also enable their own metal ions to replace metal salt precursors, directly participating in electrocatalyst assembly. This facilitates the growth of diverse nanostructures (e.g., nanowires, nanosheets, nanoarrays), effectively enhancing catalytic activity while reducing costs and stability uncertainties associated with ionomer binders.202 Taking nickel foam (NF) as an example, although its intrinsic DSE catalytic activity is modest (η = 573 at 10 mA cm−2),203 its catalytic performance and durability can be dramatically improved through catalyst loading and modification (see Table 1). Combined with low cost, NF emerges as a strong candidate for DSE electrodes. Additionally, metal meshes/felts offer the advantages of lower cost, higher mechanical strength, and superior chemical stability.204 Relative studies have explored stainless steel mesh (SSM) substrates as electrodes for various electrochemical reactions. For example, Lyu et al.205 investigated commercial 316 SS and 304 SS as OER electrodes in natural seawater electrolysis. They found that 304 SS exhibited inferior corrosion resistance compared to 316 SS in neutral and mildly alkaline seawater electrolytes due to direct metal dissolution and CER. Increasing Mo content in 304/316 SS can enhance their stability and performance in seawater electrolysis. Notably, SSM has demonstrated performance surpassing commercial RuO2 and IrO2.202 These cost-effective, durable substrates also enable the development of 3D electrodes with controlled microstructures, enhancing local mass transfer to accommodate industrial-level high current densities while maintaining catalytic activity. We strongly recommend utilizing these advantageous catalytic substrates in DSE electrode R&D to further improve performance, selectivity, and long-term durability.
Compared to catalyst design, DSE device engineering presents greater challenges. Integrated DSE systems require balancing catalyst cost with prolonged durability. While academia has extensively studied seawater electrolysis catalysts and proposed numerous strategies to address harsh operating conditions, two critical gaps remain: (1) most studies overlook practical challenges of real seawater in application scenarios; (2) practical implementation demands innovative catalyst engineering and optimization of catalyst layer characteristics (uniformity, thickness) alongside other components like porous transport layers (PTL material properties, hydrophilicity, thickness). Furthermore, catalyst synthesis methods profoundly impact scalable production, essentially requiring a paradigm shift from materials chemistry to chemical engineering. Despite catalysis being a well-established field, knowledge about catalyst mass production remains scarce, though it ultimately determines process feasibility. Industrially, catalyst mass-production formulas remain closely guarded secrets, still dominated by empirical approaches. Taking widely studied NiFe-LDH in DSE as an example, electrochemical deposition and in situ growth (e.g., hydrothermal methods) offer simple and scalable fabrication. However, these methods suffer from time/energy intensiveness, typically requiring hours (even the fastest electrodeposition takes 3–15 min206). Notably, Tian et al. developed an ultrafast method by immersing Prussian blue analogs (PBA) chemically deposited on NF into 1 M KOH solution, achieving NiFe-LDH nanoparticle immobilization on NF within 10–90 s without post-treatment. The NiFe-LDH-20s/NF electrode demonstrates exceptional activity with low overpotential (∼0.240 V at 10 mA cm−2), small Tafel slope (38 mV dec−1), and stable performance during 15-h multi-step chronopotentiometric testing (50–500 mA cm−2).207 With advancing catalytic technologies, academia urgently needs to deepen understanding of catalyst scale-up to comprehensively bridge synthesis–mechanism–process relationships. This requires interdisciplinary expertise spanning chemistry (synthesis, physics, analysis), materials science, and chemical/mechanical engineering (reactor design). We therefore advocate for cross-disciplinary collaborative development in the transition of DSE technologies from laboratory to industrial scale. A comparison table of key performance indicators for different types of electrolysis cells (AWE, PEM, AEMWE and BPMWE) is given for reference (Table 4).
| Anode | Cathode | Electrolyte | T (°C) | Performance | Ref. |
|---|---|---|---|---|---|
| AWE | |||||
| MHCM-z-BCC | NiMoS | Neutral phosphate-buffered seawater | 25 | 2.1 V@10 mA cm−2, current density dropped to 100% after 10 h | 208 |
| NiNS | NiNS | Neutral phosphate-buffered seawater | 25 | 1.8 V@48.3 mA cm−2 | 209 |
| Co–Fe2P | Co–Fe2P | 0.5 M NaCl + 1.0 M KOH | 25 | 1.69 V@100 mA cm−2, 22 h@100 mA cm−2 | 210 |
| SSM | Ni–MoN | 1 M KOH + seawater | 25 | 1.783 V@500 mA cm−2, 100 h@500 mA cm−2 | 211 |
| NCMS/NiO | NCMS/NiO | 5 M KOH + seawater | 80 | 1.98 V@1000 mA cm−2, 30 d@100 mA cm−2 | 212 |
| S-(Ni,Fe)OOH | NiMoN | 1 M KOH + seawater | 25 | 1.837 V@500 mA cm−2, 100 h@500 mA cm−2 | 101 |
| Mo–Ni3S2/NF | PtNi mesh | 30 wt% KOH (inner) + seawater (outer) | 25 | 3200 h@250 mA cm−2 | 34 |
| NiFeP@Ag | Cu2S@Ni | 1 M KOH + seawater | 25 | 1.95 V@400 mA cm−2, 1200 h@400 mA cm−2 | 213 |
| PEMWE | |||||
| FeOx | FeOx | 0.6 M NaCl + 0.1 M KOH | 25 | 30 min | 214 |
| Ir black | Pt/C | Anode: water vapor | 80 | 2.37 V@500 mA cm−2, 1.5 h@500 mA cm−2 | 172 |
| Cathode: 0.5 M NaCl | |||||
| (NiFe)C2O4/NF | Pt/C | 1 M KOH + seawater | 25 | 2.4 V@500 mA cm−2, 150 h@500 mA cm−2 | 94 |
| CoOx–Cr2O3 | CoOx–Cr2O3 | Seawater | 60 | 1.87 V@1000 mA cm−2, 100 h@500 mA cm−2 | 11 |
| AEMWE | |||||
| Pt/C | Co3−xPdxO4 on MnO2-NF | Anode: 0.5 M NaCl + 1.0 M PBS | 25 | 65 h@100 mA cm−2 | 105 |
| Cathode: humidified N2 | |||||
| Pb2Ru2O7−x | Pt/C | 0.6 M NaCl + 0.1 M NaOH | 25 | 1.8 V@275 mA cm−2, 5 h@200 mA cm−2 | 140 |
| Fe, P-NiSe2 NFs | Fe, P-NiSe2 NFs | Anode: N2 | 25 | 1.7 V@305 mA cm−2, 1.8 V@200 h | 215 |
| Cathode: seawater | |||||
| NiFe-LDH | Pt/C | Anode: 0.5 M NaCl | 25 | 12 h@1.7 V | 166 |
| Cathode: 0.5 M KOH | |||||
| RuMoNi | RuMoNi | 1 M KOH + seawater | 80 | 1.72 V@1000 mA cm−2, 240 h@500 mA cm−2 | 87 |
| Ni–FeOOH | Pt/C | 1 M KOH + seawater | 25 | 1.7 V@469 mA cm−2, 15 h@500 mA cm−2 | 99 |
| NiFe-LDH/Ni@NixSy | CoP/C/Ni@NIxPy | 1 M KOH + seawater | 60 | 2 V@1000 mA cm−2, 100 h | 152 |
| BPMWE | |||||
| Ir@Ti fiber | Pt@Ti fiber | Anode: 0.5 M KOH | 25 | 1000 h@100 mA cm−2 | 173 |
| Cathode: seawater | |||||
| Pt@Ti mesh | Pt@Ti mesh | Anode: 0.1 M NaOH | — | Cell voltage was changed from −3.0 to –3.5 V (100 h@10 mA cm−2) | 216 |
| Cathode: sea salt solution | |||||
| IrOx nanoparticles | Pt black | Seawater for both electrolytes | RT | Cell voltage increased by 0.90 V (>6 h operation; 250 mA cm−2) | 33 |
| Porous Pt | Porous Pt | Anode: 0.5 M NaOH | 25 | Cell voltage was stable at −2.3 V (100 h; 20 mA cm−2) | 158 |
| Cathode: seawater | |||||
5 System-level integration and considerations for long-term stability
The coupling of renewable energy sources such as wind turbines with electrolysis cells is an important system integration for future DSE technology for green hydrogen production. Specifically, the system-level integration is mainly composed of electrolysis cells and some auxiliary machines such as wind power generation system, seawater pumps, pipes, gas–water separation, hydrogen purification and storage systems.40 In offshore wind power generation, wind turbines initially produce direct current (DC) energy, which is necessary for the electrolysis cells. However, a transformer is required to convert this to the appropriate voltage level. Pumps are used to supply seawater to the electrolysis cells continuously. Then, electrolysis cells produce hydrogen and oxygen at the cathode and anode respectively. To purify hydrogen and oxygen, it is essential to incorporate a cooling system and a drying phase to manage issues linked to water vaporization and the potential transfer of water across the membrane. The separation of the generated gases from the electrolyte is facilitated by a gravity-operated separator drum, which is typically installed at the top of the electrolysis cells. Actually, the lifespan of these auxiliary equipment is also a key factor in the long-term and stable operation of the entire DSE. For the corrosion of some ions in seawater such as Cl− and biofouling, inlet and outlet pipes and other auxiliary machines require corrosion-resistant treatment to avoid leaks that could lead to unstable operation of the system, such as using corrosion-resistant materials (titanium alloys, nickel-based coatings, polymer-lined pipework, etc.)217–219 or applying anti-fouling coatings. In addition, local chlorine generation has been shown to delay biofouling.220 Maritime workers employ technologies such as cold spray221 and plasma spray222 to apply specialized coatings—like copper-doped polymers, metals, and silver-doped antifouling solutions—onto marine engineering equipment.223 These targeted antifouling strategies aim to enhance their lifespan while minimizing environmental impact. Although copper and silver are effective for their antibacterial and antifouling properties, their potential ecological risks necessitate stringent regulation and ongoing improvements.224 Here, we present typical effective antifouling references for consideration (Fig. 11(a) and (b)).225,226 For the byproducts, such as precipitates (e.g., Mg(OH)2, Ca(OH)2),227,228 hypochlorite or hypochlorous acid,229,230etc. Some effective practical engineering guidance for the treatment of byproducts is provided. For example, sodium hypochlorite (NaOCl)231 is formed by mixing chlorine gas from the anode with NaOH from the cathode in seawater electrolysis, and the resulting concentration of sodium hypochlorite is similar to that found in household and industrial bleach.232 In addition, Badjatya et al.228 adopted seawater electrolysis to produce Mg(OH)2 as a precursor for Mg-based cement. After converting Mg(OH)2 into MgCO3 by reacting with CO2 in a low-temperature carbonation curing step, the resulting Mg-based cement can exhibit compressive strength comparable to that achieved by Portland cement after only 2 days of curing. Furthermore, compressors may be required to store the produced hydrogen at pressures ranging from 1 to 30 bar. For safe storage, the generated hydrogen can also be contained in metal hydride tanks. Depending on the targeted purity level, additional drying of hydrogen can be carried out through cooling, dehydrogenation, and variable pressure adsorption to remove any residual water before it is stored or introduced into the required distribution network. The location of the electrolysis cells in relation to the offshore wind farm not only affects the installation, but also the long-term stability of the whole system and ultimately its operating costs. Three different configurations233 for electrolysis cells deployment have been considered (Fig. 11(c)): | ||
| Fig. 11 (a) Illustration of the antifouling mechanism for Cu–Ti composite and corrosion processes in Cu–Fe laser-cladded coatings. Reproduced with permission.225 Copyright 2021, Elsevier. (b) Schematic representation of corrosion processes in Cu–Fe laser-cladded coatings. Reproduced with permission.226 Copyright 2023, Elsevier. (c) Integrated electrolysis system configurations: onshore, offshore, and distributed. Reproduced with permission.233 Copyright 2024, Elsevier. (d) Illustration of the seawater chamber connected to the ocean. (e) Illustration of the overall system consisting of power supply, electrolysis module, and auxiliary module. (f) The diagram shows the wind turbine network, fluctuation in turbine power with wind speed, and pressure and stress distribution of the floating platform in a fluctuating environment. (d)–(f) Reproduced with permission.39 Copyright 2024, Springer Nature. | ||
(a) Onshore:35 in this scenario, electricity generated offshore is transmitted through undersea cables to offshore substations, where the voltage is increased. The electricity is then further conveyed via high-voltage cables to an onshore electrolysis cell for large-scale hydrogen production. While this configuration may facilitate the construction and upkeep of electrolysis cells, it could lead to greater energy losses due to the lengthy transmission distances and the challenges associated with managing high-voltage power offshore. In addition to maintaining plant components and systems, ensuring the stable operation of electrolysis cells is a significant challenge. Renewable energy sources, like wind and solar, experience variability due to seasonal changes in wind patterns and solar intensity. This fluctuation in power input can lead to frequent starts and stops of the entire system, potentially causing harm to the electrodes of the electrolysis cells and diminishing their longevity.234 Therefore, there is an analysis indicating that a single 7 MW cell stack undergoes 87 cycles per day. However, by utilizing three 2.33 MW cell stacks, this cycling can be notably reduced to just 35 cycles per day. This reduction could potentially enhance the longevity of the electrolysis cells, as frequent cycling contributes to the degradation of cell performance. On the downside, using multiple stacks may lead to higher capital costs, although the cost per kW for electrolysis cell stacks tends to decrease as their power capacity increases.235
(b) Offshore:236 in this arrangement, electricity produced by offshore wind turbines is sent to a proximate offshore electrolysis cell. This system converts electricity into pressurized hydrogen, which is subsequently transported to the mainland through pipelines. This approach minimizes energy losses and may reduce the costs associated with long-distance electrical transmission. However, oxygen and hydrogen bubble dynamics, as well as the performance of liquid–gas separators, can be affected by the orientation of the equipment due to the motion of the offshore platform. Be aware that bubbles can cause physical/mechanical degradation in the electrolysis cells, Liu et al.39 have proposed an innovative floating electrolysis system that harnesses wind and wave energy for direct electrolysis using dynamic seawater. The heart of this system is an electrolysis module developed by the Xie's group, which leverages the principle of aqueous phase transfer and integrates various modules, including energy storage, current conversion, hydrogen detection, and transport, for enhanced synergy. The electrolysis cell is designed to float within a seawater tank, addressing seawater fluctuations to improve system stability. To ensure the platform remains stable against displacement from cables or adverse weather conditions, it features counterweight modules and four anchoring points. Additionally, seawater tanks connected to the ocean via controlled valves help mitigate wave impact while facilitating direct interaction with the electrolysis cells. Weighing about 48.5 tons, the platform is engineered to withstand wave forces. A sealed connection between the seawater tank and the electrolysis cells prevents sloshing and overflowing into the floating structure. The collaborative functionality of the offshore wind energy storage module, hydrogen detection module, and other auxiliary components ensures reliable and efficient operation of the system.
(c) Distributed:237 this configuration entails the installation of small-scale electrolysis cells directly on each wind turbine, enabling immediate conversion of electricity into hydrogen at the generation site. The produced hydrogen is then compressed and conveyed to the mainland. Individual (hydrogen) produced by each unit is transported through risers and can be collected in subsea manifolds for output. By localizing production, this setup decreases transmission energy losses and enhances the overall efficiency of hydrogen generation. The primary benefit of this configuration is that if one electrolysis cell malfunctions, the remaining wind turbines can still effectively generate hydrogen.238 Relative strategies are employed to enhance the stability of systems like floating offshore wind (FOW) platforms (Fig. 11(d) and 11(e)). These strategies must account for adequate space for the electrolysis cell system and ensure robust mechanical stability to maintain safety and performance once the electrolysis facility is integrated into the foundation (Fig. 11(f)).239
For the three configurations mentioned above, in the case of centralized onshore electrolysis, energy is transmitted to shore via a submarine high-voltage direct current (HVDC) cable. This approach is practical for nearby offshore locations, but it faces challenges such as energy losses and limited flexibility for future expansion. Conversely, both decentralized and centralized offshore systems utilize submarine hydrogen pipelines for energy transfer, which offer advantages over high-voltage cables. Hydrogen pipelines provide greater capacity for expansion and are generally more economical, as they are not constrained by the transmission limits of a single cable. Decentralized offshore systems are particularly advantageous due to their modular nature, allowing for continuous hydrogen production even if one electrolysis cell or turbine fails. However, they face challenges related to operational complexity and maintenance, necessitating further validation in offshore conditions.238 On the other hand, centralized offshore systems can compete effectively, particularly in terms of simplified maintenance for individual turbines. While centralized systems are generally less complex, a significant drawback is that hydrogen production ceases if a failure occurs in the system. Furthermore, economic analyses240 reveal that distributed hydrogen production is the most cost-effective option, with a levelized cost of hydrogen (LCoH2) at $13.34 per kg, followed closely by centralized production at $13.66 per kg, and land-based production at $14.10 per kg. Rogeau et al.241 also indicate that offshore electrolysis is significantly cheaper in terms of LCoH2, particularly in deep-water scenarios (over 100 m), where the decentralized option is expected to gain favor over centralized systems in the coming years. However, these findings require further investigation and discussion to be fully validated while considering long-term stability and economy of system.
Finally, we analyzed and projected the future of three prominent seawater hydrogen production demonstration projects in China. The 100 kW-level seawater electrolysis system (20 N m3 h−1) developed by the Dalian Institute of Chemical Physics (DICP) achieves a cell voltage of 1.59 V and DC power consumption of 3.80 kW h N−1 m−3 h−1 H2−1 at a current density of 3000 A m−2 through high-performance electrodes and wide-power-adaptation processes. The hydrogen purity exceeds 99.999%, with successful adaptation to offshore wind power fluctuations.242 Xie Heping's team pioneered the phase transition migration-driven in situ direct seawater electrolysis technology. Utilizing PTFE membrane gas–liquid isolation and vapor pressure differential mechanisms, this desalination-free hydrogen production method was validated by a 10 N m3 h−1 prototype operating continuously for 240 hours in Xinghua Bay, Fujian. A 1.2 N m3 h−1 floating platform for seawater hydrogen production via renewable energy achieved the first direct integration with offshore wind power under challenging conditions (wind scale 3–8, wave height 0.3–0.9 m) in Xinghua Bay, maintaining stable operation for 10 days.243 The “Integrated Offshore Green Hydrogen-Methanol-Ammonia Production System” jointly developed by National Energy Group Hydrogen Technology Co., Ltd, Yantai CIMC Raffles Offshore Engineering Co., Ltd, and others has received China Classification Society (CCS) certification. Combining off-grid PEM/alkaline electrolysis with green ammonia/methanol storage technologies, it represents China's first marine hydrogen demonstration project featuring offshore mobile platform-based green hydrogen production coupled with hydrogen-based chemical processes.244
6 Summary and outlook
Direct seawater electrolysis (DSE) presents a promising avenue for sustainable hydrogen production, leveraging abundant ocean resources to generate clean fuel. However, several stability challenges arise due to the complex composition of seawater, which includes various ions and microorganisms that can lead to chemical and physical degradation of the electrolysis system. Key issues include the degradation of traditional pure water electrolysis, competitive chlorine evolution reactions (CER), ion-induced precipitation, and biofouling, which damage electrodes and other components materials. These degradation processes can lead to reduced overall efficiency, limiting the longevity of the systems used for DSE.In pursuit of longer-term stability, many catalysts have been tested in alkaline natural seawater and demonstrated significant performance, but there has been no further discussion on how these catalysts function in natural seawater. Compared to alkaline seawater solutions, natural seawater has lower conductivity, and its pH is closer to neutral or weakly alkaline. As a result, under the same conditions, NSE of DSE encounters a higher overpotential than ASE of DSE, making it difficult to achieve sufficient industrial-grade current density within the OER potential range at this pH. Additionally, due to the high overpotentials and the complex and harsh electrolyte environment of natural seawater, NSE electrodes are subjected to more intense chemical and electrochemical corrosion. It is important to note that the design of long-durability NSE cathodes also requires careful modulation around the key pH-related ion (OH−) and the development of NSE anode electrode catalytic materials requires greater emphasis on enhancing the water dissociation capability of electrocatalysts under neutral or weak alkaline conditions, improving OER selectivity and increasing the corrosion resistance of electrode materials. Generally, NSE exhibits poorer performance and durability compared to ASE and SSE. However, some high-performing catalysts provide effective guidance, such as the Cr2O3–CoOx, introducing a Lewis acid layer (Cr2O3) was introduced onto the catalyst surface (CoOx) to split H2O and generate localized surface alkalinity, thereby suppressing the release of chlorine gas captured at the anode. Additionally, the strong binding between OH− and the Lewis acid layer reduced the capture of OH− by Mg2+ and Ca2+ cations present in the seawater electrolyte, enabling anti-precipitation at the cathode.
Moreover, the design of electrolysis cells, whether membrane-less systems or membrane-based systems—plays a crucial role in enhancing operational stability. Membrane-less electrolyzers, leveraging hydrodynamic control (e.g., Segré–Silberberg effect, asymmetric electrodes) or capillary-fed designs, minimize gas crossover but face scalability limitations and require highly selective catalysts. Membrane-based systems, such as mature AMEWE, the stability of AEMs significantly impacts the long-term operation of DSE. Membrane degradation in DSE can be attributed to basic and seawater effects. Strategies to enhance membrane performance include developing robust high-molecular-weight polycarbazole membranes and blending ionomers that improve adhesion and ionic conductivity. Notable advances include hybrid MEA designs that strengthen bonding between catalyst layers and membranes. In addition, BPMWE have shown particular promise due to their ability to restrict chloride ion transport (achieving <0.005% Cl− crossover), thereby minimizing corrosive side reactions. However, further optimization is still needed to adapt to asymmetric seawater feed to cross the laboratory to commercial level. For integrated systems, such as phase-transition PTFE membrane designs, demonstrate promise by combining in situ purification with electrolysis, achieving >3200 hours of stable operation and >99.99% impurity rejection. Morever, it can be believed that the synergizing hydrogen production with byproduct valorization (e.g., Mg(OH)2 for cement) could further enhance economic viability in the future.
Looking ahead, the future of DSE also aligns with the broader transition towards offshore renewable energy generation. The successful integration of advanced DSE technologies within maritime environments is pivotal. This will necessitate not only continued innovations in electrolysis cell designs and materials but also the establishment of robust support systems for hydrogen purification and transport. System-level integration—including consideration of auxiliary components like separators, compressors, and energy management systems—will be essential for maximizing efficiencies and ensuring reliable hydrogen production. With ongoing developments in offshore wind projects incentivizing hydrogen production directly at sea, three configurations (onshore, offshore, distributed) for hydrogen production using offshore electrolysis are compared. Although they have their pros and cons, there's significant potential for decentralized production capabilities to emerge, minimizing logistical barriers and improving economic viability. Several stability challenges arise in electrolysis systems due to the complex composition of seawater, intermittent fluctuations in renewable energy, and dynamic marine environmental disturbances, such as waves, temperature variations, and mechanical stresses. These factors can adversely affect the long-term stability of these systems, highlighting the need for innovations in dynamic control and buffering strategies: (1) to stabilize operating temperatures in offshore environments, it is essential to design integrated phase-change materials (PCMs) or incorporate active cooling and heating loops. (2) Additionally, real-time platform stabilization technologies, such as dynamic counterweight systems or thrusters, can mitigate wave-induced oscillations, reducing mechanical stresses on electrolysis modules and gas–liquid separators. (3) Implementing hybrid energy storage solutions can buffer intermittent power inputs and minimize cycling degradation. (4) Real-time monitoring through embedded sensor networks will enable tracking of temperature variations and environmental changes, allowing for AI-driven predictive maintenance. (5) At the laboratory scale, we can pre-simulate waves, temperature variations, and conduct accelerated aging studies alongside intermittent power conditions. Characterizing electrode and membrane electrode assembly and linking them to electrochemical signals will contribute to building a database that enables AI-driven alarm signals to identify maintenance needs under actual operating conditions.
Overall, while the path to efficient and stable seawater electrolysis remains fraught with challenges, ongoing research and innovation position this technology as a central player in the quest for clean hydrogen production. By addressing the multifaceted aspects of stability and integration, DSE could potentially transform the renewable energy landscape and contribute substantially to global decarbonization efforts. Continued collaborative efforts among researchers, engineers, and industry stakeholders will be crucial to realizing the full potential of DSE in the near future.
Data availability
This review paper does not contain any primary data generated or analyzed by the authors. All data discussed in this review are derived from previously published studies, which are appropriately cited in the manuscript. Readers are encouraged to refer to the original publications for detailed datasets and methodologies. If further information is required, the corresponding author is available for consultation and can provide guidance on accessing the referenced studies.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The work described in this paper was supported by a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (project no. 15308024), a grant from the Research Institute for Smart Energy at The Hong Kong Polytechnic University (CDB2) and a grant from Chongqing Talents (CSTB2024YCJH-KYXM0082).References
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS.
- D. Guan, B. Wang, J. Zhang, R. Shi, K. Jiao, L. Li, Y. Wang, B. Xie, Q. Zhang, J. Yu, Y. Zhu, Z. Shao and M. Ni, Energy Environ. Sci., 2023, 16, 4926–4943 RSC.
- L. Wu, Z. Pan, S. Yuan, X. Shi, Y. Liu, F. Liu, X. Yan and L. An, Chem. Eng. J., 2024, 488, 151000 CrossRef CAS.
- L. Wu, G. Zhang, B. Xie, C. Tongsh and K. Jiao, Int. J. Green Energy, 2021, 18, 541–555 CrossRef CAS.
- B. Zhang, C. Zhu, Z. Wu, E. Stavitski, Y. H. Lui, T.-H. Kim, H. Liu, L. Huang, X. Luan, L. Zhou, K. Jiang, W. Huang, S. Hu, H. Wang and J. S. Francisco, Nano Lett., 2020, 20, 136–144 CrossRef CAS PubMed.
- M. A. Achomo, A. Kumar, N. R. Peela and P. Muthukumar, Int. J. Hydrogen Energy, 2024, 58, 1640–1672 CrossRef CAS.
- M. Aghaie, M. Mehrpooya and F. Pourfayaz, Energy Convers. Manag., 2016, 124, 141–154 CrossRef CAS.
- B. S. Zainal, P. J. Ker, H. Mohamed, H. C. Ong, I. M. R. Fattah, S. M. A. Rahman, L. D. Nghiem and T. M. I. Mahlia, Renewable Sustainable Energy Rev., 2024, 189, 113941 CrossRef CAS.
- L. Wu, Z. Pan, S. Yuan, X. Huo, Q. Zheng, X. Yan and L. An, Energy AI, 2024, 18, 100411 CrossRef.
- S. Yuan, C. Zhao, X. Cai, L. An, S. Shen, X. Yan and J. Zhang, Prog. Energy Combust. Sci., 2023, 96, 101075 CrossRef.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS.
- Z. Zhu, L. Luo, Y. He, M. Mushtaq, J. Li, H. Yang, Z. Khanam, J. Qu, Z. Wang and M.-S. Balogun, Adv. Funct. Mater., 2024, 34, 2306061 CrossRef CAS.
- H. Yu, J. Wan, M. Goodsite and H. Jin, One Earth, 2023, 6, 267–277 CrossRef.
- S. Zhang, Y. Wang, S. Li, Z. Wang, H. Chen, L. Yi, X. Chen, Q. Yang, W. Xu, A. Wang and Z. Lu, Nat. Commun., 2023, 14, 4822 CrossRef CAS PubMed.
- W. He, X. Li, C. Tang, S. Zhou, X. Lu, W. Li, X. Li, X. Zeng, P. Dong, Y. Zhang and Q. Zhang, ACS Nano, 2023, 17, 22227–22239 CrossRef CAS PubMed.
- J. Liang, Z. Li, X. He, Y. Luo, D. Zheng, Y. Wang, T. Li, B. Ying, S. Sun, Z. Cai, Q. Liu, B. Tang and X. Sun, Mater. Today, 2023, 69, 193–235 CrossRef CAS.
- M. A. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC.
- J. Liu, S. Duan, H. Shi, T. Wang, X. Yang, Y. Huang, G. Wu and Q. Li, Angew. Chem., Int. Ed., 2022, 134, e202210753 CrossRef.
- D. Liu, Y. Cai, X. Wang, Y. Zhuo, X. Sui, H. Pan and Z. Wang, Energy Environ. Sci., 2024, 17, 6897–6942 RSC.
- C. Deng and G.-M. Weng, Nano Energy, 2024, 126, 109674 CrossRef CAS.
- E. C. La Plante, X. Chen, S. Bustillos, A. Bouissonnie, T. Traynor, D. Jassby, L. Corsini, D. A. Simonetti and G. N. Sant, ACS EST Eng., 2023, 3, 955–968 CrossRef CAS PubMed.
- M. Qi, M. Qin, H. Wang, B. Lin, J. Chen, X. Shi, X. Du, S. Mao, J. Zhao, H. Zhang, L. Xi and Y. Wang, Appl. Catal., B, 2024, 356, 124259 CrossRef CAS.
- Q. Chen, Y. Choo, N. Akther, H. K. Shon and G. Naidu, Chem. Eng. J., 2024, 494, 153007 CrossRef CAS.
- E. Jwa, W. Lee, S. Choi, Y.-C. Jeung, K. S. Hwang, J.-H. Han and N. Jeong, Desalination, 2024, 580, 117580 CrossRef CAS.
- P. Farràs, P. Strasser and A. J. Cowan, Joule, 2021, 5, 1921–1923 CrossRef.
- B. A. Franco, P. Baptista, R. C. Neto and S. Ganilha, Appl. Energy, 2021, 286, 116553 CrossRef CAS.
- H. Fei, R. Liu, T. Liu, M. Ju, J. Lei, Z. Wang, S. Wang, Y. Zhang, W. Chen, Z. Wu, M. Ni and J. Wang, Adv. Mater., 2024, 36, 2309211 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, P. Fornasiero, G. Tian, P. Strasser and X.-Y. Yang, Angew. Chem., Int. Ed., 2024, 136, e202412087 CrossRef.
- A. Badreldin, A. El Ghenymy, A.-R. Al-Zubi, A. Ashour, N. Hassan, A. Prakash, M. Kozusznik, D. V. Esposito, S. Ui Solim and A. Abdel-Wahab, J. Power Sources, 2024, 593, 233991 CrossRef CAS.
- X. Zhou, G. Wei, C. Liu, Q. Zhao, H. Gao, W. Wang, X. Zhao, X. Zhao and H. Chen, J. Colloid Interface Sci., 2024, 670, 709–718 CrossRef CAS PubMed.
- C. Feng, M. Chen, Z. Yang, Z. Xie, X. Li, S. Li, A. Abudula and G. Guan, J. Mater. Sci. Technol., 2023, 162, 203–226 CrossRef CAS.
- D. H. Marin, J. T. Perryman, M. A. Hubert, G. A. Lindquist, L. Chen, A. M. Aleman, G. A. Kamat, V. A. Niemann, M. B. Stevens, Y. N. Regmi, S. W. Boettcher, A. C. Nielander and T. F. Jaramillo, Joule, 2023, 7, 765–781 CrossRef CAS.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- S. Ramakrishnan, M. Delpisheh, C. Convery, D. Niblett, M. Vinothkannan and M. Mamlouk, Renewable Sustainable Energy Rev., 2024, 195, 114320 CrossRef CAS.
- H. Meskher, A. R. Woldu, P. K. Chu, F. Lu and L. Hu, EcoEnergy, 2024, 2, 630–651 CrossRef CAS.
- S. M. H. Hashemi, M. A. Modestino and D. Psaltis, Energy Environ. Sci., 2015, 8, 2003–2009 Search PubMed.
- H. Liu, M. Yu, X. Tong, Q. Wang and M. Chen, Chem. Rev., 2024, 124, 10509–10576 Search PubMed.
- T. Liu, Z. Zhao, W. Tang, Y. Chen, C. Lan, L. Zhu, W. Jiang, Y. Wu, Y. Wang, Z. Yang, D. Yang, Q. Wang, L. Luo, T. Liu and H. Xie, Nat. Commun., 2024, 15, 5305 Search PubMed.
- D. Niblett, M. Delpisheh, S. Ramakrishnan and M. Mamlouk, J. Power Sources, 2024, 592, 233904 CrossRef CAS.
- R. d’Amore-Domenech and T. J. Leo, ACS Sustainable Chem. Eng., 2019, 7, 8006–8022 CrossRef.
- J. Shi, R. Xia, X. Zheng, R. Yao and J. Zheng, A Review on Hydrogen Production From Ocean Renewable Energy and the Application Status, 2023 Search PubMed.
- L. Ai, Y. Tian, T. Xiao, J. Zhang, C. Zhang and J. Jiang, J. Colloid Interface Sci., 2024, 673, 607–615 CrossRef CAS.
- L. Chen, C. Yu, J. Dong, Y. Han, H. Huang, W. Li, Y. Zhang, X. Tan and J. Qiu, Chem. Soc. Rev., 2024, 53, 7455–7488 RSC.
- Y. Wang, M. Wang, Y. Yang, D. Kong, C. Meng, D. Zhang, H. Hu and M. Wu, Chem. Catal., 2023, 3, 100643 CrossRef CAS.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- J. S. Ko, J. K. Johnson, P. I. Johnson and Z. Xia, ChemCatChem, 2020, 12, 4526–4532 CrossRef CAS.
- J. Chang and Y. Yang, Renewables, 2023, 1, 415–454 CrossRef.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- J. Corbin, M. Jones, C. Lyu, A. Loh, Z. Zhang, Y. Zhu and X. Li, RSC Adv., 2024, 14, 6416–6442 RSC.
- W. Zheng, L. Y. S. Lee and K.-Y. Wong, Nanoscale, 2021, 13, 15177–15187 Search PubMed.
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC.
- X.-J. Zhai, Q.-X. Lv, J.-Y. Xie, Y.-X. Zhang, Y.-M. Chai and B. Dong, Chem. Eng. J., 2024, 153187 CrossRef CAS.
- J. Yu, B.-Q. Li, C.-X. Zhao and Q. Zhang, Energy Environ. Sci., 2020, 13, 3253–3268 RSC.
- B. Xu, J. Liang, X. Sun and X. Xiong, Green Chem., 2023, 25, 3767–3790 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- F. Meharban, C. Lin, X. Wu, L. Tan, H. Wang, W. Hu, D. Zhou, X. Li and W. Luo, Adv. Energy Mater., 2024, 14, 2402886 CrossRef CAS.
- J. Niklas Hausmann, R. Schlögl, P. W. Menezes and M. Driess, Energy Environ. Sci., 2021, 14, 3679–3685 RSC.
- C. Yang, G. Rousse, K. Louise Svane, P. E. Pearce, A. M. Abakumov, M. Deschamps, G. Cibin, A. V. Chadwick, D. A. Dalla Corte, H. Anton Hansen, T. Vegge, J.-M. Tarascon and A. Grimaud, Nat. Commun., 2020, 11, 1378 CrossRef CAS PubMed.
- S. M. Hoseinieh, F. Ashrafizadeh and M. H. Maddahi, J. Electrochem. Soc., 2010, 157, E50 CrossRef CAS.
- Y. Hu, M. Tu, T. Xiong, Y. He, M. Mushtaq, H. Yang, Z. Khanam, Y. Huang, J. Deng and M.-S. Balogun, J. Energy Chem., 2025, 103, 282–293 CrossRef CAS.
- Y.-Y. Ma, C.-X. Wu, X.-J. Feng, H.-Q. Tan, L.-K. Yan, Y. Liu, Z.-H. Kang, E.-B. Wang and Y.-G. Li, Energy Environ. Sci., 2017, 10, 788–798 RSC.
- X. Wu, S. Zhou, Z. Wang, J. Liu, W. Pei, P. Yang, J. Zhao and J. Qiu, Adv. Energy Mater., 2019, 9, 1901333 CrossRef.
- M. Auinger, I. Katsounaros, J. C. Meier, S. O. Klemm, P. U. Biedermann, A. A. Topalov, M. Rohwerder and K. J. J. Mayrhofer, Phys. Chem. Chem. Phys., 2011, 13, 16384–16394 RSC.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- X. Lu, J. Pan, E. Lovell, T. H. Tan, Y. H. Ng and R. Amal, Energy Environ. Sci., 2018, 11, 1898–1910 Search PubMed.
- S. Dresp, F. Dionigi, S. Loos, J. Ferreira de Araujo, C. Spöri, M. Gliech, H. Dau and P. Strasser, Adv. Energy Mater., 2018, 8, 1800338 Search PubMed.
- H. Jin, J. Wang, L. Tian, M. Gao, J. Zhao and L. Ren, Mater. Des., 2022, 213, 110307 CrossRef CAS.
- D. M. Yebra, S. Kiil and K. Dam-Johansen, Prog. Org. Coat., 2004, 50, 75–104 CrossRef CAS.
- Y. Li and C. Ning, Bioact. Mater., 2019, 4, 189–195 Search PubMed.
- I. G. Wenten, M. Z. Bazant and K. Khoiruddin, Sep. Purif. Technol., 2024, 345, 127228 CrossRef CAS.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Catal., 2020, 10, 81–92 CrossRef CAS.
- N. Rajapakse, M. Zargar, T. Sen and M. Khiadani, Sep. Purif. Technol., 2022, 289, 120772 CrossRef CAS.
- J. Das, S. Mandal, A. Borbora, S. Rani, M. Tenjimbayashi and U. Manna, Adv. Funct. Mater., 2024, 34, 2311648 CrossRef CAS.
- P. A. Kempler, R. H. Coridan and L. Luo, Chem. Rev., 2024, 124, 10964–11007 CrossRef CAS PubMed.
- A. W. Tricker, J. K. Lee, J. R. Shin, N. Danilovic, A. Z. Weber and X. Peng, J. Power Sources, 2023, 567, 232967 CrossRef CAS.
- M. B. Poudel, N. Logeshwaran, S. Prabhakaran, A. R. Kim, D. H. Kim and D. J. Yoo, Adv. Mater., 2024, 36, 2305813 CrossRef CAS PubMed.
- J. Zheng, Appl. Surf. Sci., 2017, 413, 72–82 CrossRef CAS.
- X. Ju, H. Guo, Z. Tao, A. Niu, H. Wei, J. S. Chen, X. He, H. Xian, X. Sun, Q. Kong and T. Li, J. Colloid Interface Sci., 2025, 683, 189–196 CrossRef CAS PubMed.
- P. Gu, Y. Song, Y. Fan, X. Meng, J. Liu, G. Wang, Z. Li, H. Sun, Z. Zhao and J. Zou, Adv. Energy Mater., 2024, 2403657 Search PubMed.
- Y. Zhang, S. Jeong, E. Son, Y. Choi, S. Lee, J. M. Baik and H. Park, ACS Nano, 2024, 18, 16312–16323 CrossRef CAS PubMed.
- F. Liu, R. Hu, H. Qiu, H. Miao, Q. Wang and J. Yuan, J. Alloys Compd., 2022, 913, 165342 CrossRef CAS.
- S. A. Patil, A. C. Khot, V. D. Chavan, I. Rabani, D. Kim, J. Jung, H. Im and N. K. Shrestha, Chem. Eng. J., 2024, 480, 146545 CrossRef CAS.
- Y. Kuang, M. J. Kenney, Y. Meng, W.-H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M.-C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- R. Andaveh, A. Sabour Rouhaghdam, J. Ai, M. Maleki, K. Wang, A. Seif, G. Barati Darband and J. Li, Appl. Catal., B, 2023, 325, 122355 CrossRef CAS.
- C. Huang, Q. Zhou, L. Yu, D. Duan, T. Cao, S. Qiu, Z. Wang, J. Guo, Y. Xie, L. Li and Y. Yu, Adv. Energy Mater., 2023, 13, 2301475 CrossRef CAS.
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H.-M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed.
- L. Zhou, Z.-H. Huang, F. Kang and R. Lv, Chem. Eng. J., 2023, 458, 141457 CrossRef CAS.
- H. Zhang, Z. Bi, P. Sun, A. Chen, T. Wågberg, X. Hu, X. Liu, L. Jiang and G. Hu, ACS Nano, 2023, 17, 16008–16019 CrossRef CAS PubMed.
- J.-T. Ren, L. Chen, W.-W. Tian, X.-L. Song, Q.-H. Kong, H.-Y. Wang and Z.-Y. Yuan, Small, 2023, 19, 2300194 Search PubMed.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 Search PubMed.
- X. Liu, Q. Yu, X. Qu, X. Wang, J. Chi and L. Wang, Adv. Mater., 2024, 36, 2307395 Search PubMed.
- T. Cui, J. Chi, K. Liu, J. Zhu, L. Guo, H. Mao, X. Liu, J. Lai, H. Guo and L. Wang, Appl. Catal., B, 2024, 357, 124269 CrossRef CAS.
- Z. Li, Y. Yao, S. Sun, J. Liang, S. Hong, H. Zhang, C. Yang, X. Zhang, Z. Cai, J. Li, Y. Ren, Y. Luo, D. Zheng, X. He, Q. Liu, Y. Wang, F. Gong, X. Sun and B. Tang, Angew. Chem., Int. Ed., 2024, 63, e202316522 CrossRef CAS PubMed.
- L. Yu, J. Xiao, C. Huang, J. Zhou, M. Qiu, Y. Yu, Z. Ren, C.-W. Chu and J. C. Yu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202382119 Search PubMed.
- W. Ma, D. Li, L. Liao, H. Zhou, F. Zhang, X. Zhou, Y. Mo and F. Yu, Small, 2023, 19, 2207082 CrossRef CAS PubMed.
- D. Wu, B. Liu, R. Li, D. Chen, W. Zeng, H. Zhao, Y. Yao, R. Qin, J. Yu, L. Chen, J. Zhang, B. Li and S. Mu, Small, 2023, 19, 2300030 Search PubMed.
- F. Zhang, Y. Liu, F. Yu, H. Pang, X. Zhou, D. Li, W. Ma, Q. Zhou, Y. Mo and H. Zhou, ACS Nano, 2023, 17, 1681–1692 CrossRef CAS PubMed.
- Y. S. Park, J. Lee, M. J. Jang, J. Yang, J. Jeong, J. Park, Y. Kim, M. H. Seo, Z. Chen and S. M. Choi, J. Mater. Chem. A, 2021, 9, 9586–9592 RSC.
- Y. Wang, W. Yu, B. Zhou, W. Xiao, J. Wang, X. Wang, G. Xu, B. Li, Z. Li, Z. Wu and L. Wang, J. Mater. Chem. A, 2023, 11, 1886–1893 RSC.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS.
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang, Z. Tian, Z. Lu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 22740–22744 Search PubMed.
- X. Duan, Q. Sha, P. Li, T. Li, G. Yang, W. Liu, E. Yu, D. Zhou, J. Fang, W. Chen, Y. Chen, L. Zheng, J. Liao, Z. Wang, Y. Li, H. Yang, G. Zhang, Z. Zhuang, S.-F. Hung, C. Jing, J. Luo, L. Bai, J. Dong, H. Xiao, W. Liu, Y. Kuang, B. Liu and X. Sun, Nat. Commun., 2024, 15, 1973 CrossRef CAS PubMed.
- N. Wang, P. Ou, S.-F. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao, Y. Yan, J. Zhang, Z. Wang, R. Dorakhan, A. Badreldin, A. Abdel-Wahab, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Adv. Mater., 2023, 35, 2210057 CrossRef CAS PubMed.
- W.-H. Hung, B.-Y. Xue, T.-M. Lin, S.-Y. Lu and I.-Y. Tsao, Mater, Today Energy, 2021, 19, 100575 CAS.
- H. Sun, J. Sun, Y. Song, Y. Zhang, Y. Qiu, M. Sun, X. Tian, C. Li, Z. Lv and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 22061–22070 CrossRef CAS PubMed.
- L. Shao, X. Han, L. Shi, T. Wang, Y. Zhang, Z. Jiang, Z. Yin, X. Zheng, J. Li, X. Han and Y. Deng, Adv. Energy Mater., 2024, 14, 2303261 CrossRef CAS.
- K. Huang, H. Qi, W. Zeng, L. Guo, C. Lian, H. Liu and J. Hu, Sep. Purif. Technol., 2025, 364, 132364 Search PubMed.
- J. Mu, C. Yu, X. Song, L. Chen, J. Zhao and J. Qiu, Adv. Funct. Mater., 2025, 2423965 CrossRef.
- H. Jin, X. Liu, A. Vasileff, Y. Jiao, Y. Zhao, Y. Zheng and S.-Z. Qiao, ACS Nano, 2018, 12, 12761–12769 CrossRef CAS PubMed.
- L. Xiu, W. Pei, S. Zhou, Z. Wang, P. Yang, J. Zhao and J. Qiu, Adv. Funct. Mater., 2020, 30, 1910028 CrossRef CAS.
- Y. Zhang, P. Li, X. Yang, W. Fa and S. Ge, J. Alloys Compd., 2018, 732, 248–256 CrossRef CAS.
- Q. Lv, J. Han, X. Tan, W. Wang, L. Cao and B. Dong, ACS Appl. Energy Mater., 2019, 2, 3910–3917 CrossRef CAS.
- S. Ye, W. Xiong, P. Liao, L. Zheng, X. Ren, C. He, Q. Zhang and J. Liu, J. Mater. Chem. A, 2020, 8, 11246–11254 Search PubMed.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M.-S. Jie, T. Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 Search PubMed.
- H. Li, Q. Tang, B. He and P. Yang, J. Mater. Chem. A, 2016, 4, 6513–6520 RSC.
- N. Nie, D. Zhang, Z. Wang, W. Yu, S. Ge, J. Xiong, Y. Gu, B. Yang, J. Lai and L. Wang, Appl. Catal., B, 2022, 318, 121808 CrossRef CAS.
- D. Bao, L. Huang, Y. Gao, K. Davey, Y. Zheng and S.-Z. Qiao, J. Am. Chem. Soc., 2024, 146, 34711–34719 CrossRef CAS PubMed.
- Q. Sha, S. Wang, L. Yan, Y. Feng, Z. Zhang, S. Li, X. Guo, T. Li, H. Li, Z. Zhuang, D. Zhou, B. Liu and X. Sun, Nature, 2025, 639, 360–367 Search PubMed.
- J. Zhu, J. Chi, T. Cui, L. Guo, S. Wu, B. Li, J. Lai and L. Wang, Appl. Catal., B, 2023, 328, 122487 CrossRef CAS.
- W. Xu, T. Ma, H. Chen, D. Pan, Z. Wang, S. Zhang, P. Zhang, S. Bao, Q. Yang, L. Zhou, Z. Tian, S. Dai and Z. Lu, Adv. Funct. Mater., 2023, 33, 2302263 Search PubMed.
- X. Gu, M. Yu, S. Chen, X. Mu, Z. Xu, W. Shao, J. Zhu, C. Chen, S. Liu and S. Mu, Nano Energy, 2022, 102, 107656 Search PubMed.
- H. Hu, Z. Zhang, Y. Zhang, T. Thomas, H. Du, K. Huang, J. P. Attfield and M. Yang, Energy Environ. Sci., 2023, 16, 4584–4592 RSC.
- S. Li, Y. Zhuo, D. Liu, H. Pan and Z. Wang, Appl. Catal., B, 2024, 355, 124188 CrossRef CAS.
- M. Ning, Y. Wang, L. Wu, L. Yang, Z. Chen, S. Song, Y. Yao, J. Bao, S. Chen and Z. Ren, Nano-Micro Lett., 2023, 15, 157 CrossRef CAS PubMed.
- H. Hu, Z. Zhang, L. Liu, X. Che, J. Wang, Y. Zhu, J. P. Attfield and M. Yang, Sci. Adv., 2024, 10, eadn7012 CrossRef CAS PubMed.
- S. Jeong, U. Kim, S. Lee, Y. Zhang, E. Son, K.-J. Choi, Y.-K. Han, J. M. Baik and H. Park, ACS Nano, 2024, 18, 7558–7569 CrossRef CAS PubMed.
- J. Lu, S. Chen, Y. Zhuo, X. Mao, D. Liu and Z. Wang, Adv. Funct. Mater., 2023, 33, 2308191 Search PubMed.
- M. Wen, Y. Cao, B. Wu, T. Xiao, R. Cao, Q. Wang, X. Liu, H. Xue, Y. Yu, J. Lin, C. Xu, J. Xu and B. OuYang, Nat. Commun., 2021, 12, 5106 CrossRef CAS PubMed.
- H. Zhang, J. Diao, M. Ouyang, H. Yadegari, M. Mao, M. Wang, G. Henkelman, F. Xie and D. J. Riley, ACS Catal., 2023, 13, 1349–1358 Search PubMed.
- Y. Yu, W. Zhou, X. Zhou, J. Yuan, X. Zhang, L. Wang, J. Li, X. Meng, F. Sun, J. Gao and G. Zhao, ACS Catal., 2024, 14, 18322–18332 CrossRef CAS.
- T. Zhao, S. Wang, C. Jia, C. Rong, Z. Su, K. Dastafkan, Q. Zhang and C. Zhao, Small, 2023, 19, 2208076 CrossRef CAS PubMed.
- J. Fan, X. Ma, J. Xia, L. Zhang, Q. Bi and W. Hao, J. Colloid Interface Sci., 2024, 657, 393–401 Search PubMed.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S.-Z. Qiao, Adv. Mater., 2021, 33, 2007508 CrossRef CAS PubMed.
- H.-M. Zhang, Y. Gao, J. Li, J. Sun, D. Wang, L. Wang and Y. Meng, Fuel, 2024, 375, 132652 CrossRef CAS.
- Z. Kato, M. Sato, Y. Sasaki, K. Izumiya, N. Kumagai and K. Hashimoto, Electrochim. Acta, 2014, 116, 152–157 CrossRef CAS.
- N. Jiang and H. Meng, Surf. Coat. Technol., 2012, 206, 4362–4367 CrossRef CAS.
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60, 17371–17378 Search PubMed.
- P. Gayen, S. Saha and V. Ramani, ACS Appl. Energy Mater., 2020, 3, 3978–3983 CrossRef CAS.
- X. Xu, Y. Pan, Y. Zhong, C. Shi, D. Guan, L. Ge, Z. Hu, Y.-Y. Chin, H.-J. Lin, C.-T. Chen, H. Wang, S. P. Jiang and Z. Shao, Adv. Sci., 2022, 9, 2200530 CrossRef CAS PubMed.
- R. Fan, C. Liu, Z. Li, H. Huang, J. Feng, Z. Li and Z. Zou, Nat. Sustain., 2024, 7, 158–167 CrossRef.
- W. Liu, J. Yu, M. G. Sendeku, T. Li, W. Gao, G. Yang, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309882 CrossRef CAS PubMed.
- Z. Gong, J. Liu, M. Yan, H. Gong, G. Ye and H. Fei, ACS Nano, 2023, 17, 18372–18381 CrossRef CAS PubMed.
- S. Zhang, Y. Wang, X. Wei, L. Chu, W. Tian, H. Wang and M. Huang, Appl. Catal., B, 2023, 336, 122926 CrossRef CAS.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 130, 1632–1636 CrossRef.
- T. P. Keane, S. S. Veroneau, A. C. Hartnett and D. G. Nocera, J. Am. Chem. Soc., 2023, 145, 4989–4993 CrossRef CAS PubMed.
- B. Wang, M. Lu, D. Chen, Q. Zhang, W. Wang, Y. Kang, Z. Fang, G. Pang and S. Feng, J. Mater. Chem. A, 2021, 9, 13562–13569 RSC.
- J. Tang, C. Su and Z. Shao, Exploration, 2024, 4, 20220112 CrossRef CAS PubMed.
- L. Shao, X. Han, L. Shi, T. Wang, Y. Zhang, Z. Jiang, Z. Yin, X. Zheng, J. Li, X. Han and Y. Deng, Adv. Energy Mater., 2024, 14, 2303261 CrossRef CAS.
- J. Na, H. Yu, S. Jia, J. Chi, K. Lv, T. Li, Y. Zhao, Y. Zhao, H. Zhang and Z. Shao, J. Energy Chem., 2024, 91, 370–382 CrossRef CAS.
- M. L. Frisch, T. N. Thanh, A. Arinchtein, L. Hager, J. Schmidt, S. Brückner, J. Kerres and P. Strasser, ACS Energy Lett., 2023, 8, 2387–2394 CrossRef CAS.
- P. Li, S. Zhao, Y. Huang, Q. Huang, Y. Yang and H. Yang, ACS Catal., 2023, 13, 15360–15374 CrossRef CAS.
- T. Kou, S. Wang, R. Shi, T. Zhang, S. Chiovoloni, J. Q. Lu, W. Chen, M. A. Worsley, B. C. Wood, S. E. Baker, E. B. Duoss, R. Wu, C. Zhu and Y. Li, Adv. Energy Mater., 2020, 10, 2002955 CrossRef CAS.
- D. Liu, X. Wei, J. Lu, X. Wang, K. Liu, Y. Cai, Y. Qi, L. Wang, H. Ai and Z. Wang, Adv. Mater., 2024, 36, 2408982 CrossRef CAS PubMed.
- J. Liang, Z. Cai, X. He, Y. Luo, D. Zheng, S. Sun, Q. Liu, L. Li, W. Chu, S. Alfaifi, F. Luo, Y. Yao, B. Tang and X. Sun, Chem, 2024, 10, 3067–3087 CAS.
- Q. Liu, Z. Yan, J. Gao, H. Fan, M. Li and E. Wang, Chem. Sci., 2023, 14, 11830–11839 RSC.
- L. Wan, J. Liu, D. Lin, Z. Xu, Y. Zhen, M. Pang, Q. Xu and B. Wang, Energy Environ. Sci., 2024, 17, 3396–3408 RSC.
- C. Hu, Y. J. Lee, Y. Ma, X. Zhang, S. W. Jung, H. Hwang, H. K. Cho, M.-G. Kim, S. J. Yoo, Q. Zhang and Y. M. Lee, ACS Energy Lett., 2024, 9, 1219–1227 CrossRef CAS.
- D. V. Esposito, Joule, 2017, 1, 651–658 CrossRef CAS.
- M. I. Gillespie, F. van der Merwe and R. J. Kriek, J. Power Sources, 2015, 293, 228–235 CrossRef CAS.
- G. D. O’Neil, C. D. Christian, D. E. Brown and D. V. Esposito, J. Electrochem. Soc., 2016, 163, F3012–F3019 CrossRef.
- J. T. Davis, J. Qi, X. Fan, J. C. Bui and D. V. Esposito, Int. J. Hydrogen Energy, 2018, 43, 1224–1238 CrossRef CAS.
- B. S. De, P. Kumar, N. Khare, J.-L. Luo, A. Elias and S. Basu, ACS Appl. Energy Mater., 2021, 4, 9639–9652 CrossRef CAS.
- A. Hodges, A. L. Hoang, G. Tsekouras, K. Wagner, C.-Y. Lee, G. F. Swiegers and G. G. Wallace, Nat. Commun., 2022, 13, 1304 CrossRef CAS PubMed.
- S. Dresp, T. N. Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 Search PubMed.
- Q. Xu, L. Zhang, J. Zhang, J. Wang, Y. Hu, H. Jiang and C. Li, EnergyChem, 2022, 4, 100087 CrossRef CAS.
- X. Duan, X. Xiang, J. Chen, A. Zhou, J. Xiao, J. Wen and S. Wang, Int. J. Hydrogen Energy, 2024, 55, 1505–1513 CrossRef CAS.
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14, 3934 CrossRef CAS PubMed.
- T. Liu, C. Lan, M. Tang, M. Li, Y. Xu, H. Yang, Q. Deng, W. Jiang, Z. Zhao, Y. Wu and H. Xie, Nat. Commun., 2024, 15, 8874 CrossRef CAS PubMed.
- S. Kumari, R. Turner White, B. Kumar and J. M. Spurgeon, Energy Environ. Sci., 2016, 9, 1725–1733 RSC.
- R. Rossi, D. M. Hall, L. Shi, N. R. Cross, C. A. Gorski, M. A. Hickner and B. E. Logan, Energy Environ. Sci., 2021, 14, 6041–6049 RSC.
- J.-H. Han, J. Electrochem. Soc., 2023, 170, 093506 CrossRef CAS.
- L. Wu, L. An, D. Jiao, Y. Xu, G. Zhang and K. Jiao, Appl. Energy, 2022, 323, 119651 CrossRef CAS.
- R. Yoshimura, S. Wai, Y. Ota, K. Nishioka and Y. Suzuki, Catalysts, 2022, 12, 934 CrossRef CAS.
- J. Li, J. Sun, Z. Li and X. Meng, Int. J. Hydrogen Energy, 2022, 47, 29685–29697 CrossRef CAS.
- S. Z. Oener, M. J. Foster and S. W. Boettcher, Science, 2020, 369, 1099–1103 CrossRef CAS PubMed.
- M. A. Blommaert, J. A. H. Verdonk, H. C. B. Blommaert, W. A. Smith and D. A. Vermaas, ACS Appl. Energy Mater., 2020, 3, 5804–5812 CrossRef CAS.
- B. Kim, H. K. Shon, D. S. Han and H. Park, Desalination, 2023, 551, 116431 CrossRef CAS.
- J.-H. Han, ChemSusChem, 2022, 15, e202200372 CrossRef CAS PubMed.
- G. Moon, J. Lim, B. Kim, D. S. Han and H. Park, Green Chem., 2025, 27, 982–1005 RSC.
- A. Zhegur, N. Gjineci, S. Willdorf-Cohen, A. N. Mondal, C. E. Diesendruck, N. Gavish and D. R. Dekel, ACS Appl. Polym. Mater., 2020, 2, 360–367 CrossRef CAS.
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 4, 2549–2561 CrossRef CAS.
- S. Kim, S. H. Yang, S.-H. Shin, H. J. Cho, J. K. Jang, T. H. Kim, S.-G. Oh, T.-H. Kim, H. Han and J. Y. Lee, Energy Environ. Sci., 2024, 17, 5399–5409 RSC.
- Y. Zheng, W. Ma, A. Serban, A. Allushi and X. Hu, Angew. Chem., Int. Ed., 2025, 64, e202413698 CrossRef CAS PubMed.
- L. Li, C. Lin, X. Ma, Y. Ma, A. Zhu, Z. Xie and Q. Zhang, Chem. Eng. J., 2025, 508, 160916 CrossRef CAS.
- H. Becker, J. Murawski, D. V. Shinde, I. E. L. Stephens, G. Hinds and G. Smith, Sustainable Energy Fuels, 2023, 7, 1565–1603 RSC.
- H. Saada, B. Fabre, G. Loget and G. Benoit, ACS Energy Lett., 2024, 9, 3351–3368 CrossRef CAS.
- Y. Zheng, A. Serban, H. Zhang, N. Chen, F. Song and X. Hu, ACS Energy Lett., 2023, 8, 5018–5024 CrossRef CAS.
- W. Zhang, M. Liu, X. Gu, Y. Shi, Z. Deng and N. Cai, Chem. Rev., 2023, 123, 7119–7192 CrossRef CAS PubMed.
- C. K. Lim, Q. Liu, J. Zhou, Q. Sun and S. H. Chan, J. Power Sources, 2017, 342, 79–87 CrossRef CAS.
- Z. Liu, B. Han, Z. Lu, W. Guan, Y. Li, C. Song, L. Chen and S. C. Singhal, Appl. Energy, 2021, 300, 117439 CrossRef CAS.
- J. P. Stempien, Q. Liu, M. Ni, Q. Sun and S. H. Chan, Electrochim. Acta, 2014, 147, 490–497 Search PubMed.
- A. Hauch, R. Küngas, P. Blennow, A. B. Hansen, J. B. Hansen, B. V. Mathiesen and M. B. Mogensen, Science, 2020, 370, eaba6118 CrossRef CAS PubMed.
- S. S. Veroneau, A. C. Hartnett, A. E. Thorarinsdottir and D. G. Nocera, ACS Appl. Energy Mater., 2022, 5, 1403–1408 CrossRef CAS.
- S. S. Veroneau and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2024855118 CrossRef CAS PubMed.
- B. E. Logan, L. Shi and R. Rossi, Joule, 2021, 5, 760–762 CrossRef.
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2023, 9, eadi7755 CrossRef CAS PubMed.
- Q.-Y. Fang, J.-J. Gu, R.-X. He, W.-W. Ni, J. Ran, N. Deng and J.-B. He, J. Solid State Electrochem., 2025, 1–9 Search PubMed.
- O. Horner, D. P. Wilkinson and E. L. Gyenge, J. Electrochem. Soc., 2024, 171, 094502 CrossRef CAS.
- K. Kishor, S. Saha, A. Parashtekar and R. G. S. Pala, J. Electrochem. Soc., 2018, 165, J3276–J3280 CrossRef CAS.
- A. Mayyas, M. Ruth, B. Pivovar, G. Bender and K. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, 2019 Search PubMed.
- X. Lyu, J. Li, C. J. Jafta, Y. Bai, C. P. Canales, F. Magnus, Á. S. Ingason and A. Serov, J. Environ. Chem. Eng., 2022, 10, 108486 CrossRef CAS.
- H. Hu, X. Wang, J. P. Attfield and M. Yang, Chem. Soc. Rev., 2024, 53, 163–203 Search PubMed.
- X. Lyu, Y. Bai, J. Li, R. Tao, J. Yang and A. Serov, J. Environ. Chem. Eng., 2023, 11, 109667 CrossRef CAS.
- J. Wang, L. Ji and Z. Chen, ACS Catal., 2016, 6, 6987–6992 Search PubMed.
- S. Mitchell, N.-L. Michels and J. Pérez-Ramírez, Chem. Soc. Rev., 2013, 42, 6094 RSC.
- S. Hsu, J. Miao, L. Zhang, J. Gao, H. Wang, H. Tao, S. Hung, A. Vasileff, S. Z. Qiao and B. Liu, Adv. Mater., 2018, 30, 1707261 CrossRef PubMed.
- Y. Zhao, B. Jin, A. Vasileff, Y. Jiao and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 8117–8121 RSC.
- S. Wang, P. Yang, X. Sun, H. Xing, J. Hu, P. Chen, Z. Cui, W. Zhu and Z. Ma, Appl. Catal., B, 2021, 297, 120386 CrossRef CAS.
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou, X. Xing, T. Tong, Y. Yao, J. Bao, L. Yu, S. Chen and Z. Ren, Adv. Mater., 2022, 34, 2201774 Search PubMed.
- S. Seenivasan and D.-H. Kim, J. Mater. Chem. A, 2022, 10, 9547–9564 RSC.
- W. Xu, Z. Wang, P. Liu, X. Tang, S. Zhang, H. Chen, Q. Yang, X. Chen, Z. Tian, S. Dai, L. Chen and Z. Lu, Adv. Mater., 2024, 36, 2306062 CrossRef CAS PubMed.
- B. C. M. Martindale and E. Reisner, Adv. Energy Mater., 2016, 6, 1502095 CrossRef.
- J. Chang, G. Wang, Z. Yang, B. Li, Q. Wang, R. Kuliiev, N. Orlovskaya, M. Gu, Y. Du, G. Wang and Y. Yang, Adv. Mater., 2021, 33, 2101425 CrossRef CAS PubMed.
- J.-H. Han, E. Jwa, H. Lee, E. J. Kim, J.-Y. Nam, K. S. Hwang, N. Jeong, J. Choi, H. Kim, Y.-C. Jeung and T. D. Chung, Chem. Eng. J., 2022, 429, 132383 CrossRef CAS.
- A. Surbled, N. Larché and D. Thierry, Corrosion Management of Seawater Cooling Systems, Elsevier, 2024, pp. 67–128 Search PubMed.
- D. A. Shifler, LaQue's Handbook of Marine Corrosion, John Wiley & Sons, Ltd, 2022, pp. 667–689 Search PubMed.
- A. Banerjee and B. K. Banerjee, in 2015 International Conference on Sustainable Mobility Applications, Renewables and Technology (SMART), 2015, pp. 1–12.
- T. Matos, V. C. Pinto, P. J. Sousa, M. S. Martins, E. Fernández and L. M. Goncalves, Chem. Eng. J., 2024, 500, 156836 CrossRef CAS.
- H. Guosheng, X. Lukuo, L. Xiangbo and W. Hongren, Int. J. Electrochem. Sci., 2016, 11, 8738–8748 Search PubMed.
- K. Xu, J. Tian, S. Xie and P. Wu, J. Therm. Spray Technol., 2023, 32, 2170–2186 CrossRef CAS.
- X. Shi, H. Liang and Y. Li, Coatings, 2024, 14, 1454 CrossRef CAS.
- S. Pourhashem, A. Seif, F. Saba, E. G. Nezhad, X. Ji, Z. Zhou, X. Zhai, M. Mirzaee, J. Duan, A. Rashidi and B. Hou, J. Mater. Sci. Technol., 2022, 118, 73–113 Search PubMed.
- J. Tian, K. Xu, J. Hu, S. Zhang, G. Cao and G. Shao, J. Mater. Sci. Technol., 2021, 79, 62–74 CrossRef CAS.
- H. Ma, Z. Liu, J. Li, Q. Liu, J. Zhang and T. Wei, J. Alloys Compd., 2023, 940, 168820 CrossRef CAS.
- L. D. Ellis, A. F. Badel, M. L. Chiang, R. J.-Y. Park and Y.-M. Chiang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12584–12591 CrossRef CAS PubMed.
- P. Badjatya, A. H. Akca, D. V. Fraga Alvarez, B. Chang, S. Ma, X. Pang, E. Wang, Q. Van Hinsberg, D. V. Esposito and S. Kawashima, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2114680119 CrossRef CAS PubMed.
- B. Singh, A. Goyal, S. Verma, L. Singh and A. Draksharapu, Sustainable Energy Fuels, 2024, 8, 5131–5146 RSC.
- J. N. Bamba, A. T. Dumlao, R. M. Lazaro, D. D. Matienzo and J. Ocon, Curr. Opin. Electrochem., 2024, 48, 101592 CrossRef CAS.
- L. Huang, X. Huang, J. Yan, Y. Liu, H. Jiang, H. Zhang, J. Tang and Q. Liu, J. Hazard. Mater., 2023, 442, 130024 CrossRef CAS PubMed.
- M. I. Alvarado-Ávila, E. Toledo-Carrillo and J. Dutta, ACS Omega, 2022, 7, 37465–37475 CrossRef PubMed.
- J.-W. Ding, Y.-S. Fu and I.-Y. Lisa Hsieh, Energy Convers. Manage.: X, 2024, 24, 100770 CAS.
- S. Wang, A. Lu and C.-J. Zhong, Nano Converg., 2021, 8, 4 CrossRef CAS PubMed.
- Wind Speed & Power Generated Dataset For Floating Offshore 7 MW and 15 MW Turbine, https://data.ncl.ac.uk/articles/dataset/Wind_Speed_Power_Generated_Dataset_For_Floating_Offshore_7_MW_and_15_MW_Turbine/24516718/1, (accessed December 19, 2024).
- Z. Wang, H. Wang, T. Sant, Z. Zhao, R. Carriveau, D. S.-K. Ting, P. Li, T. Zhang and W. Xiong, Int. J. Hydrogen Energy, 2024, 71, 1266–1282 CrossRef CAS.
- C. A. Rodríguez Castillo, B. Yeter, S. Li, F. Brennan and M. Collu, Wind Energy Sci., 2024, 9, 533–554 CrossRef.
- O. S. Ibrahim, A. Singlitico, R. Proskovics, S. McDonagh, C. Desmond and J. D. Murphy, Renewable Sustainable Energy Rev., 2022, 160, 112310 CrossRef CAS.
- Floating Offshore Wind Farms, ed. L. Castro-Santos and V. Diaz-Casas, Springer International Publishing, Cham, 2016 Search PubMed.
- D. Jang, K. Kim, K.-H. Kim and S. Kang, Energy Convers. Manag., 2022, 263, 115695 CrossRef CAS.
- A. Rogeau, J. Vieubled, M. De Coatpont, P. Affonso Nobrega, G. Erbs and R. Girard, Renewable Sustainable Energy Rev., 2023, 187, 113699 CrossRef CAS.
- China Science Daily Demonstration operation of a 100-kilowatt-level high-efficiency seawater electrolysis hydrogen production system, https://www.dicp.cas.cn/xwdt/mtcf/202502/t20250217_7528001.html, (accessed March 25, 2025).
- Shenzhen University Academician Xie Heping's team: Solving the century-old problem of “direct electrolysis of seawater to produce hydrogen” – China Energy Network, https://www.cnenergynews.cn/guonei/2023/03/28/detail_20230328131624.html, (accessed March 25, 2025).
- China Classification Society issues AIP certificate for China's first “one-stop offshore green hydrogen, alcohol and ammonia production operation system”, https://www.ccs.org.cn/ccswz/articleDetail?id=202404070877984720, (accessed March 25, 2025).
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |