
Regulation of the cathode inner Helmholtz plane in dilute ether electrolytes using an electric-field-responsive solvent for high-voltage lithium metal batteries†
Junru
Wu‡
ab,
Ziyao
Gao‡
ab,
Zhiqiang
Fu
ab,
Lili
Lin
ab,
Xianshu
Wang
 *c,
Yun
Zhao
*a,
Lele
Peng
a,
Feiyu
Kang
ab,
Zhenyu
Guo
d,
Maria-Magdalena
Titirici
d,
Yunlong
Zhao
e,
Xiulin
Fan
f and
Baohua
Li
*a
*c,
Yun
Zhao
*a,
Lele
Peng
a,
Feiyu
Kang
ab,
Zhenyu
Guo
d,
Maria-Magdalena
Titirici
d,
Yunlong
Zhao
e,
Xiulin
Fan
f and
Baohua
Li
*a
aShenzhen Key Laboratory on Power Battery Safety Research and Institute of Materials Research, Tsinghua Shenzhen International Graduate School, Tsinghua University, Shenzhen 518055, China. E-mail: libh@sz.tsinghua.edu.cn; yzhao.zjut@hotmail.com
bSchool of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
cNational and Local Joint Engineering Research Center of Lithium-ion Batteries and Materials Preparation Technology, Key Laboratory of Advanced Battery Materials of Yunnan Province, Faculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: xswang2016@m.scnu.edu.cn
dDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK
eDyson School of Design Engineering, Imperial College London, London SW7 2AZ, UK
fState Key Laboratory of Silicon and Advanced Semiconductor Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China
First published on 18th March 2025
Abstract
Raising the cut-off voltage to 4.6 V can boost the energy density of lithium metal batteries (LMBs) with the LiCoO2 cathode. However, the unfavorable cathode electrolyte interphase (CEI) formed on the high-voltage cathode when using Li metal-compatible ether electrolytes leads to a short lifespan of batteries. Here, we harness the battery intrinsic electric field to manipulate the cathode inner Helmholtz plane (IHP) during the battery's charge by simply introducing an electric-field-responsive solvent into an ether electrolyte. Specifically, 2,2-difluoroethyl trifluoromethanesulfonate (DTS) as a weak solvated co-solvent preferentially adsorbs on the cathode surface under electric field activation. Owing to the majority of “high-voltage fragile” 1,2-dimethoxyethane ether molecules squeezed out from the IHP by DTS, a LiF-rich and organics-less stable CEI film is formed, which minimizes cathode cracking and structure degradation under high-voltage. The DTS-containing electrolyte shows superior cycling stability of LMBs under harsh conditions of 4.6 V, 4.2 mA h cm−2 cathode loading and 15 μL less electrolyte. Furthermore, the feasibility of this concept is validated in practical A h-level pouch full cells. This work highlights the significance of the IHP modulation and interfacial chemistry, providing an alternative pathway towards ether-based electrolyte engineering for high-voltage LMBs.
Broader contextHigh-voltage lithium metal batteries represent the practical solution for achieving a battery energy density of 500 W h kg−1. However, the traditional carbonate and ether electrolytes used in this type of battery are either unstable with lithium metal or unstable with the high-voltage cathode or both. For example, lithium metal compatible ether electrolytes undergo continuous oxidative decomposition on the cathode surface at high voltage, leading to unstable cathode electrolyte interphase (CEI) formation and poor cycling performance. Previous research in the literature mainly focused on the solvation structure rather than the adsorption characteristics and coordination environment of ether molecules within an electric double layer (EDL) during battery operation. In this study, we introduce a solvent that is responsive to an electric field into the ether electrolyte. The role of this additional electric field responsive solvent is that it effectively regulates the distributed species in the cathode inner Helmholtz plane (IHP). Specifically, the 2,2-difluoroethyl trifluoromethanesulfonate (DTS) molecule demonstrates enhanced absorption on the cathode during the battery's charging, squeezing most 1,2-dimethoxyethane (DME) molecules away from the IHP. The DTS-enriched IHP promotes the generation of a LiF-rich and stable CEI film with less organic components, enabling Li‖LiCoO2 cells to operate stably under harsh conditions of 4.6 V, ultrahigh cathode loading and less electrolyte. These advances bring great promise for the high voltage ether electrolyte design toward next generation high-energy LMBs. |
Introduction
In the post-lithium (Li) -ion battery era, a variety of battery devices for energy storage are booming.1,2 Among them, Li metal batteries (LMBs) are attractive in terms of high energy density when coupled with layered transition metal oxide (LTMO) cathodes.3,4 Represented by the LiCoO2 cathode, raising the cut-off voltage from 4.2 V to 4.6 V achieves a remarkable 50% increase in specific capacity (from 140 mA h g−1 to 210 mA h g−1).5 However, such high energy density battery systems impose stringent requirements on electrolyte chemistry, particularly the good compatibility with the Li metal anode and high-voltage LiCoO2. Unfortunately, applying commercial carbonate electrolytes to LMBs yields inferior cycle life arising from the vulnerable solid electrolyte interphase (SEI) on the Li anode, which leads to dendritic Li growth and dead Li formation.6–10 The ether electrolytes possess superior stability to Li anodes, yet their poor anti-oxidant ability cannot stabilize high-voltage cathodes, leading to limitations for the applications of LMBs.11,12The stability of the electrolytes towards high voltage cathodes depends on the properties of the cathode–electrolyte interphases (CEIs), whose interfacial chemistry is highly associated with adsorbed species in the electric double layer (EDL) near the cathode surface and Li+ solvation.13 Particularly, the internal structure of the EDL (i.e., the layer nearest to the electrode surface), defined as the inner Helmholtz plane (IHP), is dominated by anions and molecules that intensely interact with the electrode surface.14 The solvated Li+ configurations with a large-size are excluded from the IHP and reside in the outer Helmholtz plane (OHP).15 It can be expected that in dilute ether electrolytes, free ether molecules, e.g., 1,2-dimethoxyethane (DME), prevail in the IHP. These species are prone to decompose and then form an unstable CEI rich in organics, which impairs the ion transport kinetics and deteriorates the electrochemical performance and the structural integrity of cathodes.16 Early-developed ether electrolytes for high-voltage LMBs were formulated by nearly equivalent molar ratios of solvents/Li salts without or with “inert” fluorinated diluents, known as highly concentrated electrolytes (HCEs)17,18 and localized HCEs (LHCEs).19,20 It was found that the oxidative stability window of ether electrolytes could be widened by reducing the number of unbound “high-voltage vulnerable” ether molecules.21 Nevertheless, the increased dosage of Li salt and the usage of hydrofluoroether diluents inevitably pose high costs and environmental challenges.22 Recently, several molecular design strategies on the ether backbone have been proposed to improve the intrinsic oxidative potential of ether solvents, including selective methylation,23,24 management of –O–CH2–O– sections,25 and substitution with the functional segment.26 However, due to the reduced ionic conductivity and the complexity of the solvent preparation process, their development remains in infancy. Moreover, previous studies predominantly focused on the solvation structure, while the in-depth exploration on adsorption characteristics and coordination environment of ether molecules within the EDL during battery operation has been neglected. This is crucial for robust CEI construction on high-voltage cathodes using ether electrolytes, although it remains challenging.
Herein, we demonstrate that the battery intrinsic electric field drives the specific adsorption of a weakly solvated co-solvent, 2,2-difluoroethyl trifluoromethanesulfonate (DTS), to replace conventional DME in the IHP, enabling the desirable CEI chemistry on the 4.6 V LiCoO2 cathode. The introduction of DTS into the diluted ether electrolyte (1.0 mol L−1 lithium difluoro(oxalato)borate dissolved in DME), namely, the LiDFOB–DME:DTS electrolyte, achieves an electrochemical stability window of up to 5.5 V. Calculation and characterization evidence that the DTS molecules, with weak binding to Li+, exhibit enhanced interactions with cathodes during the battery's charging, which maximises their competitive adsorption on the cathode surface, thus “squeezing” the most DME molecules away from the IHP. The DTS-abundant IHP facilitates the formation of thin and stable LiF-rich/organics-less CEI films, which protects aggressive cathodes from harmful electrolyte erosion and crystal structure degradation. Paired with high-voltage LiCoO2 cathodes (area loading ∼2 mA h cm−2), the electrolytes enable LMBs to maintain over 90% capacity retention after 400 cycles at 4.55 V. Furthermore, under highly challenging conditions of less electrolyte of 10 μL, cathode loading over 4 mA h cm−2, or a cut-off voltage of 4.6 V, the cell still shows superior electrochemical performances. Meanwhile, the practical applicability is underscored through successful operation in the A h-level pouch cells. Our work provides a new insight into the advanced electrolyte design and aggressive CEI chemistry, especially for ether-based systems, which is of significant importance for stable and high-energy LMB technologies.
Results and discussion
The absorption behavior of electric-field-responsive solvent within the IHP
Density functional theory (DFT) calculations were conducted to obtain the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of salt and solvent molecules, correlating to the oxidative and reductive decomposition potential.27 As shown in Fig. S1 (ESI†), the DTS molecule possesses the lowest HOMO energy of −9.954 eV among these components, which demonstrates the superior oxidation stability of DTS. Furthermore, the adsorption properties of solvent molecules on the surface of cathodes were analyzed. As shown in Fig. 1a, both DME and DTS molecules are connected to the cathode through the coordination of O atoms in the ether and sulfonyl groups with the Co atoms in LiCoO2. The violin plots in Fig. 1b present that the average adsorption energy of DME (−0.8 eV) is higher than that of DTS (−0.5 eV) in the absence of the electric field. The introduction of an electric field leads to a significant reduction in both adsorption energies. Particularly, the DTS molecule exhibits the lowest average adsorption energy (−1.5 eV), which indicates its strong preference for adhering on the LiCoO2 surface upon the activation from the electric field. Furthermore, the dipole moment (μ) and dielectric constant (ε) were evaluated to confirm the different responsiveness on the electric field between DME and DTS. The DTS molecule displays higher μ and ε values (1.53 Debye and 32.25) than those of the DME molecule (1.48 Debye and 6.9,24 Fig. S2, ESI†), indicating the discrepancy in dipole, thereby leading to the difference in adsorption behavior onto the electrified electrode surface.28,29 To verify the effects of the DTS molecule in the electrolyte, 1 M LiDFOB dissolved in DME (marked as the “LiDFOB–DME” electrolyte) was used as a reference. The optimal amount of DTS was added to the LiDFOB–DME electrolyte as an experimental sample and the details for DTS optimization were discussed, as shown in Fig. S3 (ESI†). In the following discussion, the optimizing LiDFOB–DME:DTS electrolyte refers to 1.0 M LiDFOB dissolved in a mixture of DME and DTS with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. Further insight into the aggregation state of each component in the electrolyte system at the electrified interface is acquired by molecular dynamics (MD) simulations, as depicted in Fig. S4a and b (ESI†). For the pristine DME electrolyte, a large number of DME molecules in the range of 1.5 nm gather on the cathode surface under positive polarization (Fig. 1c and d), while a dramatic decrease in DME at the interface is observed when DTS is added, as displayed in Fig. 1e. Instead, a significant accumulation of DTS molecules is found on the cathode surface (Fig. 1f), dominating the species adhered to the cathodes.
:3. Further insight into the aggregation state of each component in the electrolyte system at the electrified interface is acquired by molecular dynamics (MD) simulations, as depicted in Fig. S4a and b (ESI†). For the pristine DME electrolyte, a large number of DME molecules in the range of 1.5 nm gather on the cathode surface under positive polarization (Fig. 1c and d), while a dramatic decrease in DME at the interface is observed when DTS is added, as displayed in Fig. 1e. Instead, a significant accumulation of DTS molecules is found on the cathode surface (Fig. 1f), dominating the species adhered to the cathodes.
 | ||
| Fig. 1 Mechanism analysis of the adsorption distributions of solvent molecules on the cathode. (a) Scheme of adsorption for different solvents on the LiCoO2 surface. (b) Violin plot depicting the adsorption energy of different solvents. The electric field intensity is set as 0.5 eV Å−1. White points represent the median values of adsorption energy for solvents. (c) and (e) Molecular number density along with the distance from the electrified LiCoO2 cathode in (c) LiDFOB–DME and (e) LiDFOB–DME:DTS electrolytes. The MD simulation snapshot of DME molecules distribution is shown in the inset. (d) and (f) MD simulation snapshots of DME and DTS molecules distributions in (d) LiDFOB–DME and (f) LiDFOB–DME:DTS electrolytes. (g) and (h) Schematic illustration of distributed species in the IHP on the LiCoO2 cathode in (g) LiDFOB–DME and (h) LiDFOB–DME:DTS electrolytes. | ||
The introduction of DTS improves the high voltage stability of ether-based electrolytes because of the aggregation of free DTS molecules on the cathode driven by the electric field (Fig. 1b). The underlying reason might be that the introduction of the DTS molecules with electric field responsiveness alters the Li+ solvation configurations in the OHP and diffuse layer, synergistically influencing the adsorbed species in the IHP on the cathode surface. The electrostatic potential (ESP) distribution of solvent molecules shows that the DME molecules present concentrated negative charge on oxygen atoms, while electron clouds are uniformly distributed within the whole DTS molecule, leading to the significantly weakened binding of Li ions with DTS (Fig. S5, ESI†). Meanwhile, MD simulations combined with FTIR and Raman experiments verify that in the LiDFOB–DME electrolyte, several DME molecules are coordinated with Li ions and the remaining DME molecules are in a free state (Fig. S6a and S7, ESI†). The peak at 725 cm−1 in the Raman spectrum of LiDFOB salt corresponds to the O–B–O deformation vibration of the anion, which shifts with various forms of ionic association between anions and cations.30,31 As shown in Fig. S7b and Table S1 (ESI†), the LiDFOB–DME electrolyte contains 82.5% uncoordinated DFOB− anions (i.e., solvent-separated ion pairs (SSIPs, 702.5 cm−1)) and 17.5% contact ion pairs (CIPs, 710.0 cm−1, DFOB− bound to one Li+) in the solvation shell. These results suggest that many free DME molecules enter the IHP in the LiDFOB–DME electrolyte, which will suffer from continuous oxidative decomposition on the cathode surface at high voltage (Fig. 1g). By contrast, for the LiDFOB–DME:DTS electrolyte, the DTS molecules not only hardly participate in the inner Li+ solvation sheath but instead promote the complexation of Li ions with DME molecules, thus reducing the quantity of free DME molecules (Fig. S6b, ESI†). The coordination environment of the DFOB− anion comprises 55.8% SSIPs, 40.0% CIPs and 4.2% aggregated ions (AGGs, 718 cm−1, corresponding to DFOB− bound to more than Li+, Fig. S7b and Table S2, ESI†). FTIR spectra of electrolytes and their components show similar trends (Fig. S8, ESI†). The peak corresponding to the anion in the LiDFOB–DME:DTS electrolyte is blue-shifted as compared to that of the LiDFOB–DME electrolyte, indicating a more incomplete dissociation of the anion and more cation–anion clusters. These are consistent with the difference in the distribution of DFOB− anions on the electrified cathode surface. It seems that within 0.7 nm from the surface, the number density of DFOB− in the DTS-containing electrolyte is lower than that in the LiDFOB–DME electrolyte (Fig. S4b, d and e, ESI†), probably attributed to the more anions entering the solvation sheath and forming larger nano-sized ion clusters that reside in the OHP.15,32 In this way, a considerable amount of anti-oxidation DTS molecules preferentially enter the IHP, which contributes to the formation of a stable CEI and inhibits the decomposition of DME on the electrode surface (Fig. 1h). To summarize, the weak binding of DTS to Li ions and the strong interaction with the electrified electrode synergistically achieve the enrichment of free DTS molecules on the cathode surface, thus maximally preventing DME molecules into the IHP. These above characteristics of the LiDFOB–DME:DTS electrolyte result in slightly lower ionic conductivity (Fig. S9, ESI†) but significantly improved electrochemical oxidative stability and (Fig. S10, ESI†) increased Li ion transfer number (tLi+) (Fig. S11 and Table S3, ESI†).
To clarify the underlying mechanism of the high voltage stability of the LiDFOB–DME:DTS electrolyte, the evolution of the absorbed species on the LiCoO2 surface during the charging process was investigated by in situ Raman spectroscopy. As shown in Fig. 2a, for the cathode using the LiDFOB–DME electrolyte, the peaks at 845 cm−1, 818 cm−1, and 361 cm−1 are assigned to the C–O–C bend, C–O stretch, and C–O stretch/CH2 rock/C–C stretch of the DME molecule,33 respectively. These peaks appear at the beginning of the battery charging and almost disappear after a while, indicating that DME molecules are first adsorbed on the cathode surface and subsequently depleted due to massive oxidative decomposition (Fig. 2b and c). By contrast, after introducing the DTS solvent, the peaks corresponding to DME cease to appear and are replaced by those peaks of DTS, at positions of 763 cm−1, 330 cm−1, and 306 cm−1 belonging to the C–S stretch/SO3 symmetric bend, SO3 rock and C–S torsion,34 respectively (Fig. 2a, d and e). Notably, throughout the charging process, the peaks of DTS are weakened rather than disappearing completely, which suggests that some of the DTS molecules undergo electrochemical reactions and then form a protective film, and the rest are consistently located in the EDL, effectively isolating the DME from the electrode (Fig. 2d and e).
 | ||
| Fig. 2 Electric double layer (EDL) structure evolution during the charging process. (a) Raman spectra of different electrolytes and the corresponding pure solvent. Voltage versus time profiles and in situ Raman spectra near the interface of the LiCoO2 electrode during the charging process in (b) and (c) LiDFOB–DME and (d) and (e) LiDFOB–DME:DTS electrolytes. | ||
Electrochemical performance evaluation
The cyclic voltammetry (CV) and galvanostatic cycling tests of Li‖LiCoO2 cells were conducted to prove the superiority of the LiDFOB–DME:DTS electrolyte. As shown in Fig. S12 (ESI†), the LiDFOB–DME:DTS electrolyte exhibits highly overlapping peaks and reversible electrochemical behavior during the CV scan compared to the other two electrolytes, implying negligible side reactions and stable interface formation. Fig. 3a displays the long-term cycling performance of Li‖LiCoO2 cells using various electrolytes within the voltage range of 3–4.55 V. The capacity of cells with carbonate and LiDFOB–DME electrolytes shows fast decay after 80 cycles and 160 cycles, respectively. An increased charging and discharging voltage polarization was also observed for these two electrolytes (∼0.50 V at 170 cycles for the former and ∼0.33 V at the 200th cycle for the latter) (Fig. S13, S14a, ESI† and Fig. 3b). This indicates the structural deterioration of cathode materials after cycling at high voltage and byproduct accumulation on the surface from the side reactions of the electrolyte. Meanwhile, the low average coulombic efficiency (CE) of cells in the LiDFOB–DME electrolyte, only 98.3% within 160 cycles, also demonstrates the continuous oxidative decomposition of this electrolyte and the constant loss of active Li during the cycling. In sharp contrast, the LiDFOB–DME:DTS electrolyte enables the cell to stably operate for 400 cycles with a high capacity retention of 90.3% and an average CE of over 99.0% (Fig. 3a), along with a smaller overpotential of 0.1 V even at the 300th cycle (Fig. 3b and Fig. S14b, ESI†). Additionally, the improvement in rate performance with the LiDFOB–DME:DTS electrolyte as compared to carbonate and ether electrolytes is shown in Fig. 3c, delivering a capacity of 172 mA h g−1 at 1C, 150 mA h g−1 at 2C and 135 mA h g−1 at 3C for Li‖LiCoO2 full cells. The corresponding charge/discharge voltage profiles of the cells at different rates also emphasize this enhancement (Fig. S15, ESI†).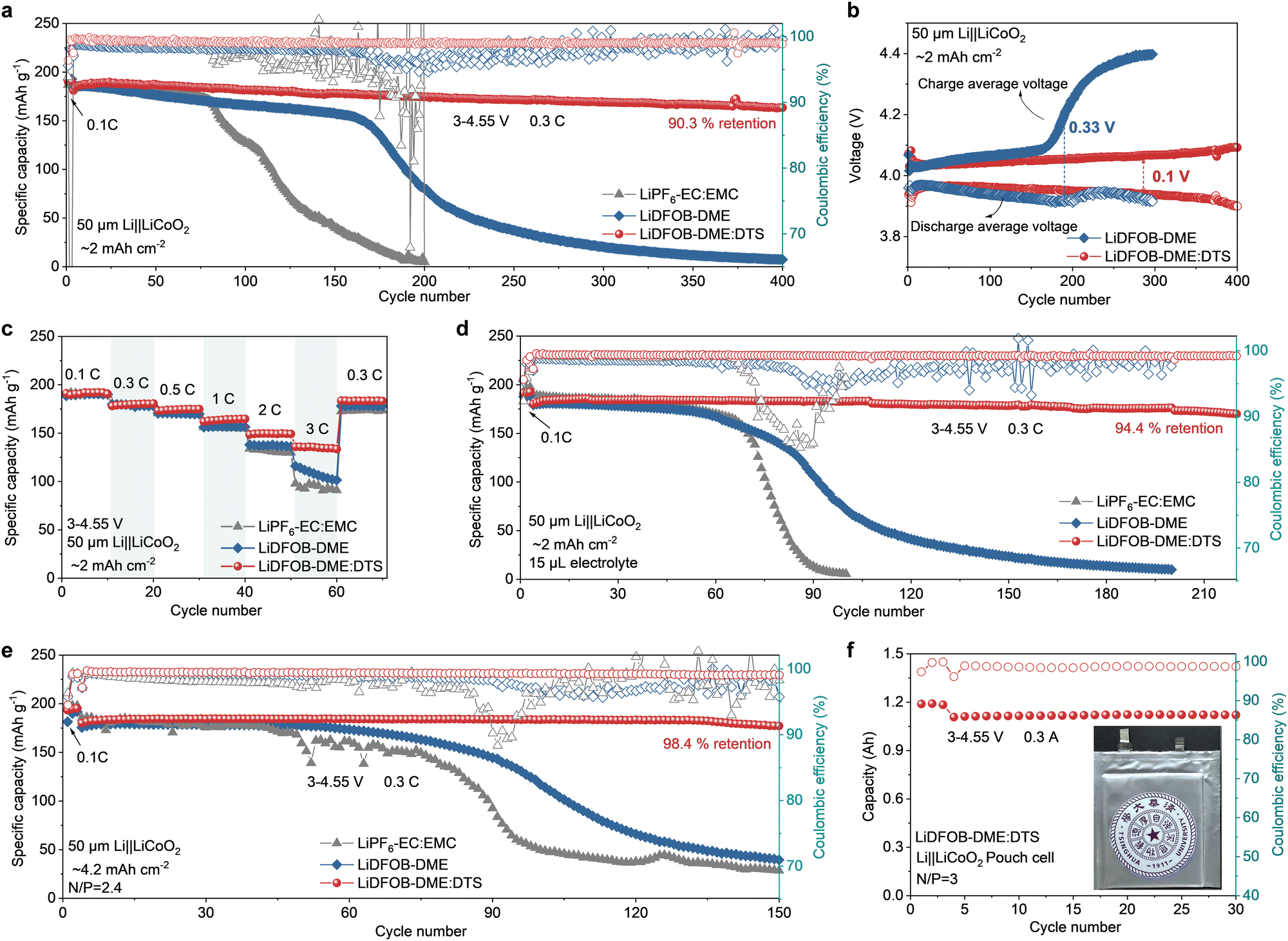 | ||
| Fig. 3 Electrochemical performance of Li‖LiCoO2 cells. (a) Cycling performance of Li‖LiCoO2 cells with different electrolytes in the voltage range of 3–4.55 V at 25 °C. (b) The charge and discharge average voltage of Li‖LiCoO2 batteries with different electrolytes during cycling. (c) Rate performance of Li‖LiCoO2 cells under different C rates. (d) Cycling performance of Li‖LiCoO2 cells with the 15 μL electrolyte. (e) Cycling performance of Li‖LiCoO2 cells with an N/P ratio of 2.4 in the LiDFOB–DME:DTS electrolyte. (f) Cycling performance of Li‖LiCoO2 pouch cells with the LiDFOB–DME:DTS electrolyte. The inset shows the optical image of the pouch cell. | ||
To confirm the prolonged cycling test of practical LMBs, harsh testing conditions are employed. As shown in Fig. 3d, when the electrolyte dosage is reduced to 15 μL, the capacity of Li‖LiCoO2 cells using carbonate and LiDFOB–DME electrolytes drops rapidly probably due to excessive electrolyte depletion and undesirable ion diffusion inside the electrode. By sharp contrast, the LiDFOB–DME:DTS electrolyte allows the battery to retain a high capacity retention of 94.4% after 220 cycles, with a high average CE of 99.2%. Paired with cathodes with a capacity of 4.2 mA h cm−2 (i.e., an area capacity ratio of negative (10 mA h cm−2) to positive electrodes (N/P) ∼2.4), the LMB with LiDFOB–DME:DTS delivers a high specific capacity of 177.2 mA h g−1 at the 150th cycle while those with other two electrolytes suffer from capacity decline after 50 cyles (Fig. 3e). In addition, when the higher cathode capacity of 4.2 mA h cm−2 and an extremely limited amount of 10 μL (E/C ∼ 2.9 g A h−1) are employed, the LiDFOB–DME:DTS electrolyte still enables the cell maintain improved cycling stability (Fig. S16, ESI†). In a scaled-up cell format, A h-class Li‖LiCoO2 pouch cells (area capacity ratio of negative/positive: 10 mA h cm−2/3.23 mA h cm−2, N/P ratio = 3) were further evaluated as shown in Fig. 3f. Specifically, the pouch cell with the DTS-based electrolyte can deliver a reversible capacity of about 1.1 A h, yielding high reversibility without any capacity decay within 30 cycles.
The interfacial chemistry on the cathode
To elucidate the underlying mechanism of the improved stability of LiCoO2 cathodes in the LiDFOB–DME:DTS electrolyte, the morphology and microstructure of the cycled LiCoO2 cathode were characterized by field emission scanning electron microscopy (FE-SEM) and transmission electron microscopy (TEM). The LiCoO2 electrodes disassembled from the cell cycling in the carbonate and DME electrolytes show severe particle cracking, some of which lose electrical contact with the current collector (Fig. 4a and Fig. S17a, ESI†). Meanwhile, the particle surface appears to be rough, probably due to the accumulation of by-products from the continuous oxidative decomposition of the electrolyte (the inset in Fig. 4a and Fig. S17b, ESI†). These results are simultaneously confirmed by TEM characterization. The CEI film formed in the carbonate electrolyte is uneven and thick, together with an 11 nm-thick disordered spinel phase (Fig. S18, ESI†). In contrast to the layered structure, such rather compact structures of rock salts are detrimental to Li ions’ migration, thus resulting in severe polarization and capacity loss of the battery.35 Likewise, for the LiDFOB–DME electrolyte, a non-uniform CEI film with a thickness of 9 nm and a spinel phase of ∼11 nm accumulated was observed on the cathode surface (Fig. 4b and c). It is noteworthy that the LiCoO2 electrode cycling in the LiDFOB–DME:DTS electrolyte exhibits a more intact and smooth surface without obvious cracks (Fig. 4d). Additionally, a thinner and more homogenous CEI film (about 1.8 nm) is formed on the LiCoO2 surface, which maintains a typical layered structure in the bulk (Fig. 4e and f). | ||
| Fig. 4 Characterization of the cathode particle and CEI morphology. (a) and (d) The cross-sectional FE-SEM images of the LiCoO2 cathode cycled in (a) LiDFOB–DME and (d) LiDFOB–DME:DTS electrolytes. The FE-SEM morphologies of the cycled cathode are shown in the inset. (b)–(f) HRTEM images of the LiCoO2 electrode disassembled from the full discharged Li‖LiCoO2 cell after 100 cycles within the voltage range of 3.0–4.55 V in (b) and (c) LiDFOB–DME and (e) and (f) LiDFOB–DME:DTS electrolytes. (g) and (h) DRT profiles of LiCoO2‖LiCoO2 cells at different temperatures in (g) LiDFOB–DME and (h) LiDFOB–DME:DTS electrolytes. | ||
To further evaluate the Li+ transfer process at the interface on the LiCoO2 cathode, electrochemical impedance spectroscopy (EIS) evolution upon the temperature of the symmetrical cells with LiCoO2 electrodes at a discharged state of 50% was measured (Fig. S19, ESI†). The two intertwined electrochemical processes of Li ion transport at interface and charge transfer are decoupled by the distribution of relaxation times (DRT) technique to enable accurate identification and quantification of specific kinetics on similar time scales.36,37Fig. 4g and h present DRT profiles converted from Nyquist plots of symmetric LiCoO2‖LiCoO2 cells at various temperatures in LiDFOB–DME and LiDFOB–DME:DTS electrolytes. It can be observed that the peak in the relaxation time range of 10−5 to 10−4 s (corresponding to the Li+ transport across the CEI) for the samples in the DTS-containing electrolyte is significantly lower than those without DTS. Meanwhile, in terms of the resistance in the time range of 10−4 to 10−3 s representing the charge transfer process, the LiDFOB–DME-DTS sample also shows a lower value at different temperatures. However, when the temperature increases to 333 K, an abnormal increase in the ion transport impedance of CEI is seen for the cell using the LiDFOB–DME electrolyte (the inset of Fig. 4g). This indicates the CEI proliferation on the electrode surface, which probably associates with aggravated electrolyte decomposition at high temperature.
The composition and structure of the CEI on the LiCoO2 cathode were systematically investigated to identify the interface chemistry associated with different EDL structures and absorption behaviors. From X-ray photoelectron spectroscopy (XPS) with Ar+ depth profiling, a large amount of organic compounds (C 1s spectra and O 1s spectra) and trace LiF/P-containing species appear on the surface of the cycled cathode in the carbonate electrolyte (Fig. S20a–d, ESI†), originating from the decomposition of LiPF6 salt.38 As the sputtering time reaches 1 min, a sharp decrease in the percentage of C-containing species suggests that the organics are predominantly concentrated in the outer layers of the CEI (Fig. S20e, ESI†). For the LiDFOB–DME electrolyte, the as-formed CEI is mainly composed of organic species and a small amount of B/F-species originating from LiDFOB salt decomposition (Fig. 5a–c).39 Although the proportion of C species decreases slightly with increasing sputtering time, it remains at a relatively high level (Fig. 5d). In comparison, for the LiCoO2 cathode surface from the cell cycling in the LiDFOB–DME:DTS electrolyte, less organic components (e.g., C–C, C–O) are detected in the CEI, together with more inorganic species (e.g., LiF), indicating that the degradation of the ether solvent is effectively suppressed (Fig. 5a–c and the right part of Fig. 5d). Meanwhile, as the etching depth increases, the signals of B–F/B–O are weakened (Fig. 5b), whereas the LiF content increases. (Fig. 5c). The peak at ∼159 eV in S 2p spectra related to the Li2S component (Fig. S21, ESI†)37 implies that the adsorbed DTS preferentially decomposes to form a stable CEI with the inorganics-rich inner layer (Fig. 5c).
 | ||
| Fig. 5 Interfacial chemistry at LiCoO2 cathodes. (a)–(c) XPS etching results of the CEI on the LiCoO2 cathode after 100 cycles in LiDFOB–DME and LiDFOB–DME:DTS electrolytes with (a) C 1s, (b) B 1s, and (c) F 1s spectra. (d) Quantified atomic concentrations of the detected elements on the CEI films on LiCoO2 cathodes after 100 cycles in different electrolytes. (e)–(g) Intensity sputter profiles of (e) C2H−, (f) BO2−, and (g) LiF2− measured by TOF-SIMS. The insets show the 3D reconstruction distribution map of C2H−, BO2−, and LiF2− showing the CEI structure and chemistry of cathodes retrieved from Li‖LiCoO2 cells after 100 cycles in different electrolytes. (h) and (i) Kelvin probe force microscopy interfacial potential images of LiCoO2 cathodes after 100 cycles in (h) LiDFOB–DME and (i) LiDFOB–DME:DTS electrolytes. (j) and (k) Schematic of CEI formation and cycled LiCoO2 cathode particles in (j) LiDFOB–DME and (k) LiDFOB–DME:DTS electrolytes. | ||
The interfacial chemistry of the CEI was further validated by time-of-flight secondary ion mass spectrometry (TOF-SIMS). From the 3D distribution and profile depth (Fig. S22, ESI†), it can be seen that the signal of C2H− (mass charge ratio (m/z) = 25) in the carbonate electrolyte is distributed in the outer layer, and its content is much more than that of the species (LiF2−, m/z = 45), PO2− (m/z = 63) from salt decomposition. This demonstrates that the CEI is dominated by organic species from serious carbonate decomposition. In the LiDFOB–DME electrolyte, although the C2H− signal shows a similar trend (Fig. 5e), BO2− (m/z = 43) and LiF2− signals are visible throughout the whole etching process (Fig. 5f and g). The signal of BO2− decreases rapidly in the first 10 nm of the etching depth and then stabilizes at 40 nm, which implies that both the DME solvent and LiDFOB salt are involved in the formation of the CEI layer. By comparison, for the cycled LiCoO2 in the LiDFOB–DME:DTS electrolyte, it is apparent that the signal of C2H− and BO2− is prominently weaker than that of the other two samples. Notably, the abundance of LiF2− illustrates that a significant amount of DTS molecules absorbs on the cathode surface and participates in the formation of the cathode|electrolyte interface. Meanwhile, Kelvin probe force microscopy (KPFM) images of cycled LiCoO2 exhibit the average roughness of surface potential decrease from 31.8 mV for the LiDFOB–DME electrolyte to 29.3 mV for the LiDFOB–DME:DTS electrolyte, suggesting that DTS promotes the formation of the CEI layer with a more uniform surface potential distribution (Fig. 5h and i). This facilitates the generation of a homogeneous electric field and the transportation of Li+ at the interface.40
In brief, in the LiDFOB–DME electrolyte, the predominant DME molecules in the EDL mainly contributes to the formation of an organic-rich CEI, together with the decomposition of some LiDFOB salt to form borides and fluorides on the cathode surface. However, these compounds are difficult to stabilize the LiCoO2 electrode, leading to an extensive structural degradation of the LiCoO2 cathode after cycling, manifested by the generation of spinel phases and obvious cracks. These results further exacerbate the electrolyte corrosion towards cathode particles and cause poor cycling stability. After adding DTS, benefiting from the electric field-driven preferential adsorption of DTS molecules, the aggregation of DME molecules on the surface and their continuous oxidation are effectively restrained, which significantly reduces the accumulation of organic by-products at the interface. Such a thin and LiF-rich CEI with superior stability can protect cathode materials from structural deterioration and electrolyte attack, thus prolonging their cycle life at high voltages.41
The morphology of disassembled Li anodes (Li+ stripping from the anode) from Li‖LiCoO2 batteries cycled in various electrolytes and the composition of the resulting SEI were also characterized. For carbonate and DME electrolytes, SEM images show some Li dendrites and a large accumulation of electrolyte by-products on the surface (Fig. S23a and b, ESI†). By contrast, the Li anode in the LiDFOB–DME:DTS electrolyte presents a smooth and dendrites-free morphology (Fig. S23c, ESI†). XPS depth profiling and elemental analyses reveal that the SEI formed in the carbonate and LiDFOB–DME electrolytes is bilayered, with the outer layer consisting mainly of organic compounds (surface C elements up to ∼36% and 39%, respectively), and a small amount of inorganic species in the inner layer (F elemental percentage of 5–15%, Fig. S24 and S25, ESI†). By contrast, in the LiDFOB–DME:DTS electrolyte, the as-formed SEI structure tends to be mosaic, with a large amount of LiF distributed in the organic matrix, and the proportion of C elements is not higher than 20% in the whole profiling range (Fig. S26, ESI†). The LiF-rich SEI with higher mechanical strength and interfacial energy can effectively minimize the generation of Li dendrites, achieving high-reversibility Li deposition/stripping.42,43 This improvement is further confirmed by the voltage evolution of symmetric Li‖Li batteries at a constant current of 0.5 mA cm−2 (Fig. S27, ESI†). The overpotential of the Li‖Li symmetric batteries using the carbonate electrolyte increases gradually after 250-hour cycling, with a large fluctuation after 420 hours. In the LiDFOB–DME electrolyte, the symmetric cell experiences a sudden drop in overpotential after 180 hours of cycling, probably due to the penetration of Li dendrites inducing a soft short-circuit inside battery. In comparison, the LiDFOB–DME:DTS electrolyte allows the symmetric cell to sustain a stable voltage hysteresis within 50 mV with no oscillation in the whole cycling.
The stability and structure evolution of cathodes operating at higher voltage
To further demonstrate and evaluate the positive effects of the as-designed DTS-containing electrolyte, the constant voltage float test was performed in Li‖LiCoO2 cells under a higher cut-off voltage of 4.6 V versus Li/Li+ (the inset of Fig. 6a). Compared to the LiDFOB–DME electrolyte with a quasi-steady-state leakage current of ∼30 μA cm−2 after the first charge, the introduction of DTS displays a smaller leakage current of 16 μA cm−2 during a 48 hour full-process. This represents a diminished side reaction rate and the passivated surface towards the cathode, consistent with the self-discharge test. As shown in Fig. 6a, after 48 hours of rest, the fully charged battery using the LiDFOB–DME:DTS electrolyte possesses an open-circuit voltage of 4.51 V, higher than that of the LiDFOB–DME electrolyte (4.42 V). Followingly, the long-term cycling performance of the Li‖LiCoO2 battery under the high voltage of 4.6 V was examined to verify the availability of the LiDFOB–DME:DTS electrolyte (Fig. 6b). The capacity of high-voltage cells with the carbonate-based electrolyte suffers from a dramatic drop after 75 cycles, together with a decline in CEs. In the LiDFOB–DME electrolyte, the cell delivers a discharge capacity of 147 mA h g−1 at the 150th cycle with an average CE value as low as 97.7%. In contrast, the LiDFOB–DME:DTS electrolyte can maintain a capacity retention of 87.5% after 300 cycles at 0.5C and an ultrahigh average CE of >99.2%, which is inextricably associated with the formed superior cathode|electrolyte interface. | ||
| Fig. 6 The stability and structural evolution of LiCoO2 charged to 4.6 V. (a) Voltage versus time curves display the self-discharge of Li‖LiCoO2 cells with different electrolytes. The inset shows the chronoamperometry test at 4.6 V at 1st cycle and a constant voltage charge process in Li‖LiCoO2 cells with different electrolytes. (b) Cycling performance of Li‖LiCoO2 cells with different electrolytes in the voltage range 3–4.6 V at 25 °C. (c) and (d) Voltage–capacity curve and the in situ XRD pattern of the LiCoO2 electrode using (c) LiDFOB–DME and (d) LiDFOB–DME:DTS electrolytes during the initial cycle within a voltage range of 3–4.6 V at 0.1C. | ||
In situ XRD measurements were used to explore the influence of interfacial chemistry on the Li+ insertion/extraction behavior within the voltage range of 3.0–4.6 V, as displayed in Fig. 6c and d. During the charging process, the Li+ extraction from the layered structure triggers the contraction and expansion between the Li slabs in the c-axis direction, which can be demonstrated by the variation in characteristic peaks of (003).44 It is worth noting that the lattice transformation in the c-axis direction makes the most contribution to the overall volume change during cycling.45 According to the shift of the (003) peak, it is calculated that the maximum c-axis contraction of LiCoO2 in the LiDFOB–DME electrolyte is 3.0%, while that value for LiDFOB–DME:DTS is 2.8% upon charging (Fig. S28 and S29 and Table S4, ESI†) during the repeated cycles, the slightly smaller shrinkage in the DTS-containing sample can alleviate the internal stress during repeated cycles and suppress crack formation.
In addition, compared with the sample in the LiDFOB–DME electrolyte, the appearance of characteristic peaks located at 19.5° and the increased intensity at 37.6° corresponding to (101) confirm the enhanced O3/H1-3 phase transition for LiCoO2 in the LiDFOB–DME:DTS electrolyte at the end of the charging46 (Fig. S28 and S29, ESI†). The formation of the H1-3 phase attributed to deeper delithiation demonstrates that more Li ions can be extracted from LiCoO2 in the LiDFOB–DME:DTS electrolyte, resulting in higher reversible capacity at the same cut-off voltage.47,48 This is consistent with the higher initial CE of the Li‖LiCoO2 cell using the LiDFOB–DME:DTS electrolyte (95.5%) compared to that of the LiDFOB–DME electrolyte (90.4%, Fig. 6b). When discharged to 3.0 V, the (003) peak of LiCoO2 in the LiDFOB–DME electrolyte suffers from an obvious broadening and cleavage. This indicates that the uncontrollable generation of byproducts on the LiCoO2 surface leads to irreversible phase transition and severe phase heterogeneity, thus impeding the transportation of Li+ and increasing the polarization of the cell. By contrast, the sample in the LiDFOB–DME:DTS electrolyte shows a subtle peak shift and attenuation, suggesting the high reversibility of phase transition during the whole cycling. This is credited to the stable interfacial chemistry derived from the DTS solvent, which effectively inhibits side reactions at the interface and renders desirable Li+ transport kinetics upon high voltage operation.
Conclusions
In summary, we demonstrate the external electric field-driven specific absorption of weakly solvated co-solvent DTS introduced to the LiDFOB–DME electrolyte can alter the species in the IHP on the high-voltage cathode. Benefiting from weak binding between Li+ and DTS combined with its strong absorption to the cathode under the external electric field, DTS preferentially dominates the IHP excluding most of the DME molecules, as demonstrated through in situ Raman spectroscopy. The as-obtained LiDFOB–DME:DTS electrolyte exhibits high oxidation stability and promotes inorganic LiF dominated interphase formation, which prevents the cathode materials from structure deterioration and crack generation. Consequently, the cells using the 50 μm-Li anode and LiCoO2 cathode of 2 mA h cm−2 loading maintain stability over 400 cycles between 3 and 4.55 V, with a high capacity retention of 92% and a small overpotential. Even by increasing the cut-off voltage to 4.6 V, the cell can deliver 87.5% of the initial capacity at the 300th cycle, achieving high Li+ extraction/intercalation reversibility. The industrial A h-class pouch batteries are assembled to demonstrate their great potential for commercial applications. Our work sheds new perspectives for advanced high-voltage electrolyte design and CEI interfacial configuration, prompting the development of practical high-voltage LMBs.Experimental section
Materials
Lithium hexafluorophosphate salt (LiPF6, 99.99%) and lithium difluoro(oxalato)borate salt (LiDFOB, 99.9%) were purchased from Capchem, China. Ethylene carbonate (EC, DoDoChem, 99.98%), ethyl methyl carbonate (EMC, DoDoChem, 99.9%), dimethyl ether (DME, DoDoChem, 99.95%), and 2,2-difluoroethyl trifluoromethanesulfonate (DTS, Alfa Aesar, 98%) were dried with 4 Å molecular sieves (Sigma-Aldrich) before use. Molarities (M, mol L−1), used to denote the salt concentration in the electrolytes, were calculated based on the number of moles of salt and the volume of solvents. The chemical storage and electrolyte preparation were carried out in an Ar-filled glove box (MBraun) with moisture and oxygen concentrations <0.01 ppm. The commercial carbonate electrolyte (1.0 M LiPF6 dissolved in EC/EMC (1:1, by volume), marked as “LiPF6–EC:EMC”) was purchased from DoDoChem. The LiDFOB–DME electrolyte was prepared by dissolving 1.0 M LiDFOB into the pure DME solvent. The LiDFOB–DME:DTS electrolyte was prepared by dissolving 1.0 M LiDFOB into the mixture of DME and DTS with different volume ratios of 1:1, 1:2, 1:3 or 1:4.
The LiCoO2 cathode with a LiCoO2:super-P:PVDF weight ratio of 96.4:1.8:1.8 was purchased from Guangdong Canrd New Energy Technology Co., Ltd. The mass loading of the active materials was about 11.0–11.5 or 22.0–22.5 mg cm−2. Celgard 2500 separators (25 μm, polypropylene, PP) were purchased from Celgard, USA.
Materials’ characterization
1H NMR spectra were recorded on a Bruker AVANCE III 400 with chloroform-d6 as the solvent. Raman spectra were acquired using a miniature laser confocal Raman spectrometer (Horiba LabRAM HR800, France) with a 532 nm laser at room temperature. Each electrolyte was sealed in a capillary tube. FTIR spectra of pure solvents and electrolytes were recorded with a Bruker Vertex 70 instrument. The oxidative stability of the electrolyte was investigated by linear sweep voltammetry (LSV) tests on Li‖stainless steel cells. The oxidation potential value of the electrolyte was determined as the voltage at which the current was increased to 20 μA. The ionic conductivity of electrolyte samples was obtained from electrochemical impedance spectroscopy (EIS) by immersing two platinum black blocking electrodes into an electrolyte sample. EIS tests were performed over a frequency range of 105 Hz to 100 Hz with an alternating potential amplitude of 5 mV on a VMP3 multichannel electrochemical workstation (Bio Logic Scientific Instruments, France). Li‖LiCoO2 coin cells underwent 3 formation cycles followed by delithiation to 50% depth of discharge (DOD) at 0.1C and 25 °C, and then disassembly and reassembly of the LiCoO2‖LiCoO2 symmetric cells. Finally, the cycled cells were kept at 293, 303, 313, 323, and 333 K to record the temperature-dependent EISs. The distribution of relaxation times (DRT) of these impedance data is generated using DRT tools, a MATLAB graphical user interface (GUI) for computing DRT based on Tikhonov regularization with continuous function discretization. The Li ion transfer number (tLi+) of the electrolyte sample was measured using the method reported by Abraham et al.49 The procedure was as follows: a symmetric Li‖Li cell was assembled and polarization currents, including the initial (I°) and steady-state (Iss) current values, were recorded at a small polarization potential (ΔV) of 10 mV. Simultaneously, the initial and steady-state values of the bulk resistance ( and Rssb) and electrode–electrolyte interface resistance (
and Rssb) and electrode–electrolyte interface resistance ( and Rssi) were measured by EIS before and after constant potential polarization. The value of tLi+ was calculated from the following equation:
and Rssi) were measured by EIS before and after constant potential polarization. The value of tLi+ was calculated from the following equation: | (1) |
Electrochemical measurements
CR2032 coin cells were assembled in a high-purity Ar-filled glovebox (O2 < 0.01 ppm, H2O < 0.01 ppm). For the evaluation of the compatibility of electrolytes with Li metal, galvanostatic cycling measurements were performed at 0.5 mA cm−2 in symmetrical Li‖Li cells with repeated 1 h charge–1 h discharge cycles.Li‖LiCoO2 cells were assembled and subjected to galvanostatic cycling at room temperature. The cells comprised 50 μm Li foil with the LiCoO2 (2 mA h cm−2) electrode and an electrolyte amount of 50 μL, 15 μL or 10 μL. Coin cells were cycled in a voltage range of 3.0 to 4.55 V or 4.6 V for three formation cycles at 0.1C or 0.2C (1C = 190 mA g−1), followed by long-term cycling at either 0.3C or 0.5C. The cyclic voltammetry (CV) curves of the Li‖LiCoO2 full cells were measured on a VMP3 multichannel electrochemical workstation (Bio Logic Science Instruments, France) in the voltage range of 3–4.55 V for 5 cycles at a scanning rate of 0.1 mV s−1. Li‖LiCoO2 pouch cells with 1.2 A h, consisting of eight layers of 50 μm Li foil, six layers of double-sided LiCoO2 cathodes (3.23 mA h cm−2) and 6 mL electrolyte (N/P ∼ 3), were subjected to a constant-current (CC) protocol. After the first two formation cycles at 0.1 A, the battery was cycled at 0.3 A. The volume of each pouch cell is about 16.6 cm3 (6.8 cm × 5.8 cm × 0.42 cm).
DFT calculation
The calculation of adsorption energy on the LiCoO2 surface was performed with the Vienna ab initio simulation package (VASP),51–54 using the Perdew–Burke–Ernzerhof functional55 of DFT with a Hubbard U correction and the projector augmented wave method. The effective U value with 5.3 eV of Co was adopted. The valence electrons were described by plane-wave basis sets with a plane-wave cut-off energy of 500 eV. Four layers of LiCoO2 (104) were used, with a vacuum layer of 20 Å. Only the two top layers were fully relaxed, whereas the two bottom layers were fixed during the geometry optimizations. The convergence criteria were set to 1 × 10−5 eV energy differences to solve the electronic wavefunctions. All the geometries (atomic coordinates) were converged to within 1 × 10−2 eV Å−1 for the maximal components of forces.MD simulation
All MD simulations were conducted using the GROMACS 2019.356 to investigate the electrode–electrolyte interface structure. Parameters for Li+, DFOB−, DME, DMC and DTS were generated with the antechamber module of Amber1857 using the general Amber force field (GAFF),58 with partial charges set to fit the electrostatic potential generated with B3LYP/6-31G(d) by RESP.59 The electrolyte system was placed in a periodic cubic box, in which the NPT ensemble (constant number of particles, temperature and pressure) was applied. The temperature and pressure were controlled via a V-rescale thermostat60 (298.15 K) and a Parrinello–Rahman barostat61 (the reference pressure 1 bar). The cutoff radius for neighbor searching and nonbonded interactions was taken to be 12 Å, and all bonds were constrained using the LINCS algorithm.62 The system was fully equilibrated after 100 ns simulation and another 100 ns was run to collect the date for statistical analysis. For the electrode–electrolyte simulation, a planar LiCoO2(104) surface was used and the other simulation settings were the same as above. The size of the planar LiCoO2(104) surface is 5.17 nm × 5.40 nm. In the LiDFOB–DME system, the number of Li+, DFOB− and DME is 100, 100, and 962, respectively. In the LiDFOB–DME:DTS system, the number of Li+, DFOB−, DME, and DTS is 100, 100, 241, and 568, respectively. The ratio of each type of molecule is based on the molar ratio of each molecule of the system. All computed structures in MD simulations were illustrated using VMD.63 The radial pair distribution function g(r) was also calculated through VMD.Author contributions
X. S. Wang, Y. Zhao, and B. H. Li conceived and designed this work. J. R. Wu and Z. Y. Gao conducted the experiments and carried out the characterization. Z. Q. Fu performed and interpreted the DFT calculations and MD simulations. L. L. Lin assisted TEM characterization. J. R. Wu and Baohua Li prepared the manuscript with the assistance of L. L. Peng, F. Y. Kang, Z. Y. Guo, M. M. Titirici, Y. L. Zhao, and X. L. Fan. All authors discussed the results and contributed to the final version of the paper.Data availability
The data supporting this article are available within the article and the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52261160384 and 52302278), Shenzhen Basic Research Special Fund (Natural Science Foundation) Key Projects (JCYJ20220818101004009), the Shenzhen Engineering Research Center on Key Technology of Next-Generation Power and Energy-Storage Battery (XMHT20230108012), and the Shenzhen Key Laboratory on Power Battery Safety Research (ZDSYS201707271615073). The authors also acknowledge the Materials and Devices Testing Center at Tsinghua Shenzhen International Graduate School, Shenzhen 518055, China.References
- L. Yu, J. Huang, S. Wang, L. Qi, S. Wang and C. Chen, Adv. Mater., 2023, 35, 2210789 CrossRef CAS PubMed.
- S.-J. Yang, J.-K. Hu, F.-N. Jiang, H. Yuan, H. S. Park and J.-Q. Huang, InfoMat, 2024, 6, e12512 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- J. Xiang, L. Yang, L. Yuan, K. Yuan, Y. Zhang, Y. Huang, J. Lin, F. Pan and Y. Huang, Joule, 2019, 3, 2334–2363 CrossRef CAS.
- W. Zheng, G. Liang, H. Guo, J. Li, J. Zou, J. A. Yuwono, H. Shu, S. Zhang, V. K. Peterson, B. Johannessen, L. Thomsen, W. Hu and Z. Guo, Energy Environ. Sci., 2024, 17, 4147–4156 RSC.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 7 Search PubMed.
- J.-G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS PubMed.
- J.-F. Ding, Y.-T. Zhang, R. Xu, R. Zhang, Y. Xiao, S. Zhang, C.-X. Bi, C. Tang, R. Xiang, H. S. Park, Q. Zhang and J.-Q. Huang, Green Energy Environ., 2023, 8, 1509–1530 CrossRef CAS.
- J. Xiang, Y. Zhao, L. Yuan, C. Chen, Y. Shen, F. Hu, Z. Hao, J. Liu, B. Xu and Y. Huang, Nano Energy, 2017, 42, 262–268 CrossRef CAS.
- X.-R. Chen, C. Yan, J.-F. Ding, H.-J. Peng and Q. Zhang, J. Energy Chem., 2021, 62, 289–294 CrossRef CAS.
- S. Di Tommaso, P. Rotureau and C. Adamo, J. Phys. Chem. A, 2012, 116, 9010–9019 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- C. Yan, R. Xu, Y. Xiao, J.-F. Ding, L. Xu, B.-Q. Li and J.-Q. Huang, Adv. Funct. Mater., 2020, 30, 1909887 CrossRef CAS.
- C. Yan, H. Yuan, H. S. Park and J.-Q. Huang, J. Energy Chem., 2020, 47, 217–220 CrossRef.
- C. Yan, H.-R. Li, X. Chen, X.-Q. Zhang, X.-B. Cheng, R. Xu, J.-Q. Huang and Q. Zhang, J. Am. Chem. Soc., 2019, 141, 9422–9429 CrossRef CAS PubMed.
- H. Wang, J. Zhang, H. Zhang, W. Li, M. Chen, Q. Guo, K. C. Lau, L. Zeng, G. Feng, D. Zhai and F. Kang, Cell Rep. Phys. Sci., 2022, 3, 100919 CrossRef CAS.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef PubMed.
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- A.-M. Li, O. Borodin, T. P. Pollard, W. Zhang, N. Zhang, S. Tan, F. Chen, C. Jayawardana, B. L. Lucht, E. Hu, X.-Q. Yang and C. Wang, Nat. Chem., 2024, 16, 922–929 CrossRef CAS PubMed.
- E. Park, J. Park, K. Lee, Y. Zhao, T. Zhou, G. Park, M.-G. Jeong, M. Choi, D.-J. Yoo, H.-G. Jung, A. Coskun and J. W. Choi, ACS Energy Lett., 2023, 8, 179–188 CrossRef CAS.
- Z. Li, H. Rao, R. Atwi, B. M. Sivakumar, B. Gwalani, S. Gray, K. S. Han, T. A. Everett, T. A. Ajantiwalay, V. Murugesan, N. N. Rajput and V. G. Pol, Nat. Commun., 2023, 14, 868 CrossRef CAS PubMed.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- D. D. Méndez-Hernández, P. Tarakeshwar, D. Gust, T. A. Moore, A. L. Moore and V. Mujica, J. Mol. Model., 2013, 19, 2845–2848 CrossRef PubMed.
- S.-Y. Jun, K. Shin, Y. Lim, S. Kim, H. Kim, C. Y. Son and W.-H. Ryu, Small Struct., 2024, 5, 2300578 CrossRef CAS.
- P.-C. Li, Z.-Q. Zhang, Z.-W. Zhao, J.-Q. Li, Z.-X. Xu, H. Zhang and G. Li, Adv. Mater., 2024, 36, 2406359 CrossRef CAS PubMed.
- J. L. Allen, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Power Sources, 2011, 196, 9737–9742 CrossRef CAS.
- S.-D. Han, J. L. Allen, E. Jónsson, P. Johansson, D. W. McOwen, P. D. Boyle and W. A. Henderson, J. Phys. Chem. C, 2013, 117, 5521–5531 CrossRef CAS.
- S. Mao, J. Mao, Z. Shen, Q. Wu, S. Zhang, J. Zhang and Y. Lu, Nano Lett., 2023, 23, 7014–7022 CrossRef CAS PubMed.
- H. Yoshida and H. Matsuura, J. Phys. Chem. A, 1998, 102, 2691–2699 CrossRef CAS.
- S. F. Parker and L. Zhong, R. Soc. Open Sci., 2018, 5, 171574 CrossRef PubMed.
- W. Zhao, K. Wang, X. Fan, F. Ren, X. Xu, Y. Liu, S. Xiong, X. Liu, Z. Zhang, M. Si, R. Zhang, W. van den Bergh, P. Yan, C. Battaglia, T. Brezesinski and Y. Yang, Angew. Chem., Int. Ed., 2023, 62, e202305281 CrossRef CAS PubMed.
- Y. Lu, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Joule, 2022, 6, 1172–1198 CrossRef CAS.
- Z. Li, Y.-X. Yao, M. Zheng, S. Sun, Y. Yang, Y. Xiao, L. Xu, C.-B. Jin, X.-Y. Yue, T. Song, P. Wu, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2024, e202409409 Search PubMed.
- W. Zhang, L. Hong, Y. Zhang, P. Mei, B. Ai, Y. Zhang, C. Zhou and X. Bao, Angew. Chem., Int. Ed., 2024, e202409069 Search PubMed.
- J. Wu, Z. Gao, Y. Wang, X. Yang, Q. Liu, D. Zhou, X. Wang, F. Kang and B. Li, Nano-Micro Lett., 2022, 14, 147 CrossRef CAS PubMed.
- X. Liu, B. Wen, G. Zhong, X. Cheng, C. Jian, Y. Guo, Y. Huang, J. Ma, P. Shi, L. Chen, D. Zhang, S. Wu, M. Liu, W. Lv, Y.-B. He and F. Kang, Sci. China Mater., 2024, 67, 1947–1955 CrossRef CAS.
- H. Wan, J. Xu and C. Wang, Nat. Rev. Chem., 2024, 8, 30–44 CrossRef CAS PubMed.
- X. Fan, X. Ji, F. Han, J. Yue, J. Chen, L. Chen, T. Deng, J. Jiang and C. Wang, Sci. Adv., 2018, 4, eaau9245 CrossRef CAS PubMed.
- J. Wu, Z. Gao, Y. Tian, Y. Zhao, Y. Lin, K. Wang, H. Guo, Y. Pan, X. Wang, F. Kang, N. Tavajohi, X. Fan and B. Li, Adv. Mater., 2023, 35, 2303347 CrossRef CAS PubMed.
- L. Wang, J. Ma, C. Wang, X. Yu, R. Liu, F. Jiang, X. Sun, A. Du, X. Zhou and G. Cui, Adv. Sci., 2019, 6, 1900355 CrossRef PubMed.
- G. G. Amatucci, J. M. Tarascon and L. C. Klein, J. Electrochem. Soc., 1996, 143, 1114 CrossRef CAS.
- L. Liu, L. Chen, X. Huang, X.-Q. Yang, W.-S. Yoon, H. S. Lee and J. McBreen, J. Electrochem. Soc., 2004, 151, A1344 CrossRef CAS.
- X. Tan, Y. Zhang, S. Xu, P. Yang, T. Liu, D. Mao, J. Qiu, Z. Chen, Z. Lu, F. Pan and W. Chu, Adv. Energy Mater., 2023, 13, 2300147 CrossRef CAS.
- H. Ren, J. Hu, H. Ji, Y. Huang, W. Zhao, W. Huang, X. Wang, H. Yi, Y. Song, J. Liu, T. Liu, M. Liu, Q. Zhao and F. Pan, Adv. Mater., 2024, 2408875 CrossRef CAS PubMed.
- K. M. Abraham, Z. Jiang and B. Carroll, Chem. Mater., 1997, 9, 1978–1988 CrossRef CAS.
- C. G. Pope, J. Chem. Educ., 1997, 74, 129 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- D. Case, I. Ben-Shalom, S. Brozell, D. Cerutti, T. Cheatham III, V. Cruzeiro, T. Darden, R. Duke, D. Ghoreishi and M. Gilson, AMBER 18, University of California, San Francisco, 2018 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 2002, 97, 10269–10280 CrossRef.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, Phys. Rev. Lett., 1980, 45, 1196–1199 CrossRef CAS.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee05818f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |