
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linker-cluster cooperativity in confinement of proline-functionalized Zr-based metal–organic frameworks and its effect on the organocatalytic aldol reaction†
Zarfishan Dilruba‡
a,
Ardeshir D. Yeganeh‡ b,
Sofia Kolinc,
Sadia Noora,
Hassan Shatlab,
Carl Wielanda,
Bo-Hung Yua,
Katrin Gugelerd,
Anna Zensa,
Johannes Kästnerd,
Deven P. Estese,
Kristyna Pluhackova*c,
Simon Krause*b and
Sabine Laschat*a
b,
Sofia Kolinc,
Sadia Noora,
Hassan Shatlab,
Carl Wielanda,
Bo-Hung Yua,
Katrin Gugelerd,
Anna Zensa,
Johannes Kästnerd,
Deven P. Estese,
Kristyna Pluhackova*c,
Simon Krause*b and
Sabine Laschat*a
aInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: sabine.laschat@oc.uni-stuttgart.de
bNanochemistry Department, Max Planck Institute for Solid State Research, Heisenbergstr. 1, D-70569 Stuttgart, Germany. E-mail: s.krause@fkf.mpg.de
cStuttgart Center for Simulation Science (SC SimTech), Cluster of Excellence EXC 2075 SimTech, Universität Stuttgart, Universitätsstr. 32, D-70569 Stuttgart, Germany. E-mail: kristyna.pluhackova@simtech.uni-stuttgart.de
dInstitut für Theoretische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
eInstitut für Technische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany
First published on 29th April 2025
Abstract
Metal organic frameworks (MOFs) provide unique opportunities for molecular heterogeneous catalysis by mimicking the active sites of enzymes. However, understanding and controlling the interaction between the metal node and the organic linker carrying the catalytic unit and the resulting confinement effects remain challenging. Here, in a combined theoretical and experimental approach, Zr-UiO-67-MOFs with ortho-N-acylproline-functionalized biphenyl-dicarboxylate linkers were prepared and compared with the corresponding MOFs with regioisomeric meta-linkers. As benchmark catalysis, the organocatalytic aldol reaction of p-nitrobenzaldehyde and cyclohexanone was studied. Experimental results revealed that the ortho-linker accelerated the aldol reactions, whereas the regioisomeric meta-linker decreased the reaction rate, which was rationalized by pore blocking of the meta-linker via molecular dynamics simulations. Moreover, the acid modulator used in the MOF preparation also played a critical role in the formation of acetal byproducts through competing acid catalysis. Our study provides novel insights into the cooperative catalysis between the linker-attached organocatalyst and the MOF metal center.
Introduction
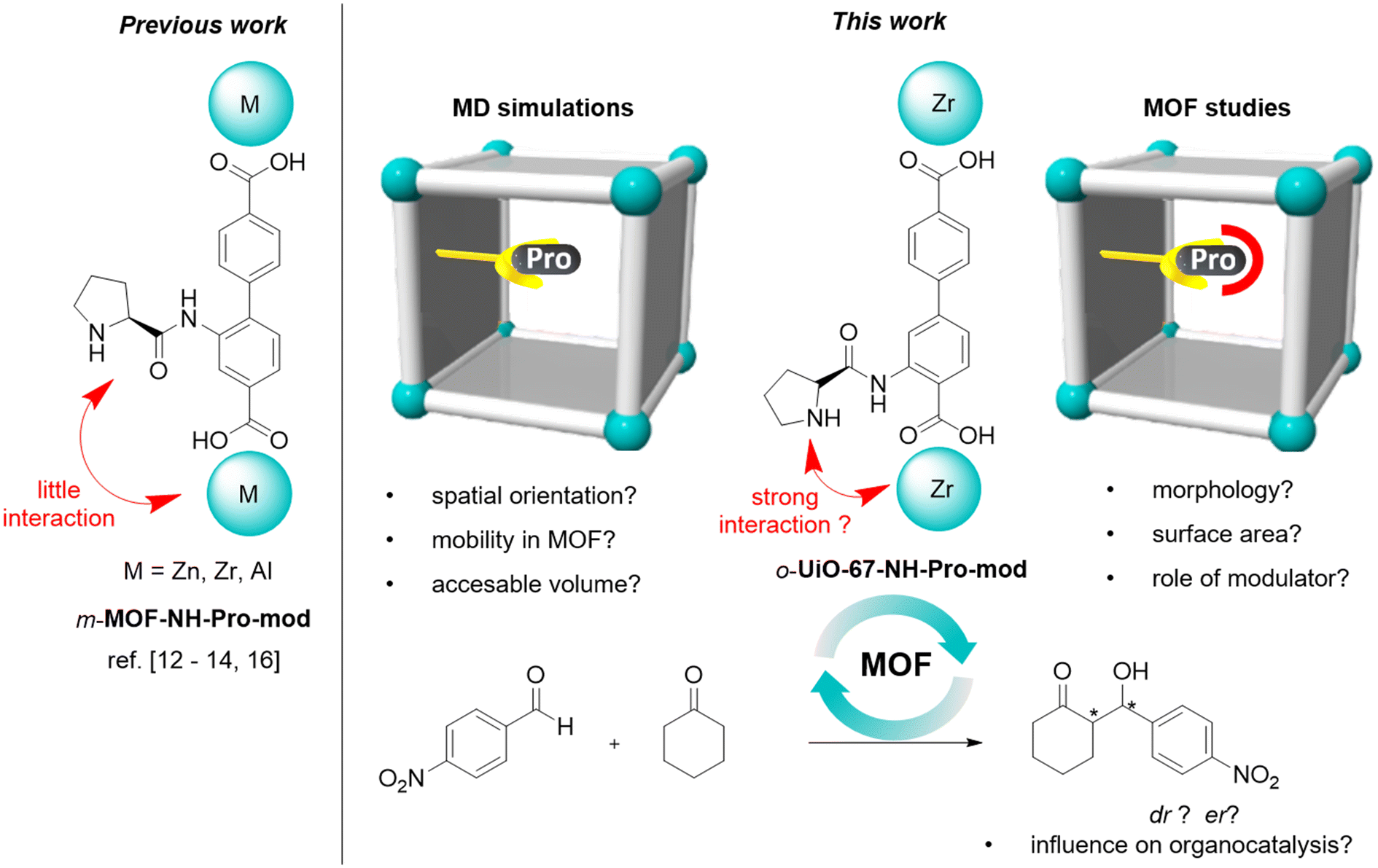
Defined spatial co-arrangement of active sites inside defined confined space is a critical feature of the catalytic activity and selectivity in enzymes. Metal–organic frameworks (MOFs) are porous crystalline solids consisting of organic linkers and inorganic nodes of various geometries, which determine the structure, pore size, and topology of the crystal lattice.1–3 By changing the size of the linkers while keeping the nodes constant, a series of isoreticular MOFs can be prepared.1,4,5 The high porosity, structural tailorability and controllable properties render MOFs as particularly attractive materials for molecular heterogeneous catalysis which may allow to mimic sophisticated microenvironments compared to enzymes.6,7 The catalytically active sites can be introduced into the nanoporous framework either as part of the building blocks, by post-synthetic covalent attachment to the linker, or by coordination onto unsaturated coordination site of the inorganic node.8,9 Due to the long-range order and nanoporosity, the chemical environment of individual active sites is, in theory, well defined and comparable throughout the whole crystal. Keeping in mind that the size compatibility of the substrates and the framework enabling the reactants to access the MOF-encapsulated catalysts and products to escape from them, MOFs enable heterogeneous molecular catalysis under confinement carrying a great potential for enhanced selectivity and activity. Given its hybrid nature, the incorporation of organocatalysts in MOFs has been targeted in the past.3,10,11In these first studies, the linker lengths, solvent, presence of defects and bulky functional groups, as well as the presence of coordination sites of the inorganic node in close proximity of the organocatalyst have been shown to influence the conversion, diastereo- and enantioselectivity.12 For example, seminal work has been published on organocatalytic aldol additions13 with proline-functionalized MOFs,12,14,15 in which L-proline or derivatives were either directly attached to the linker pre- or postsynthetically (e.g. IRMOF-10,12a UiO-67,16a,b MUF-77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12e) or coordinated to the metal node (e.g. DUT-67, H2N-MIL-101,12d MIL-101(Cr), CUP-1).17 In particular the latter is interesting for Zr-based MOFs in which modulators (monocarboxylic acids) are used to control the crystallization process, but are also incorporated as capping agents to produce low-connective metal nodes and structural defects (missing linker, missing cluster). The study on organocatalytic activity of DUT-67 utilized this aspect by incorporating L-proline as a capping agent on an 8-connective Zr-cluster.12f Organocatalytic studies showed a cooperative effect between the Lewis acid sites of the Zr-node and the active organocatalytic site of L-proline. However, in this approach the presence of other modulators (TFA, acetic acid) as well as the ill-defined distribution and dynamic attachment/detachment of molecular proline via reversible coordination chemistry results in a complex active site environment and difficult interpretation of the actual catalytic mechanism and mixed effects on selectivity. This is in stark contrast to MOFs in which proline is covalently attached (e.g. via amide formation) to the linker backbone and spatially fixed within the whole crystal lattice.
12e) or coordinated to the metal node (e.g. DUT-67, H2N-MIL-101,12d MIL-101(Cr), CUP-1).17 In particular the latter is interesting for Zr-based MOFs in which modulators (monocarboxylic acids) are used to control the crystallization process, but are also incorporated as capping agents to produce low-connective metal nodes and structural defects (missing linker, missing cluster). The study on organocatalytic activity of DUT-67 utilized this aspect by incorporating L-proline as a capping agent on an 8-connective Zr-cluster.12f Organocatalytic studies showed a cooperative effect between the Lewis acid sites of the Zr-node and the active organocatalytic site of L-proline. However, in this approach the presence of other modulators (TFA, acetic acid) as well as the ill-defined distribution and dynamic attachment/detachment of molecular proline via reversible coordination chemistry results in a complex active site environment and difficult interpretation of the actual catalytic mechanism and mixed effects on selectivity. This is in stark contrast to MOFs in which proline is covalently attached (e.g. via amide formation) to the linker backbone and spatially fixed within the whole crystal lattice.
Interestingly, the majority of proline-functionalized MOFs are based on biphenyldicarboxylate linkers for which so far only the meta-proline amide-functionalized linkers were studied (Schemes 1 and 2). Using this linker geometry, the proline points inwards the respective pores and is spatially isolated from the inorganic nodes.12 We anticipate that MOFs with a proline function in ortho-position to the framework forming carboxylate and consequently in close proximity to the inorganic node might influence the organocatalysis by cooperative interaction with the metal node while still covalently fixating the proline within the framework for a defined confined microenvironment. Such cooperativity effects between catalytically active sites and support surfaces are very poorly understood, but have the potential to both hinder or enhance catalysis.12g,18–21 Nießing et al. previously demonstrated such cooperativity between the metal node and the proline active site, proposing that electrophile activation can influence enantioselectivity. Their findings indicate that aldehyde coordination occurs at a distant metal center rather than the one adjacent to the proline substituent due to ring strain12d. Being able to control the distance between the metal node and the organocatalyst via substitution changes to the organic linker in MOFs has the potential to yield unprecedented insights into the role of the interaction between catalysts and supports.
 | ||
| Scheme 1 Comparison of o-UiO-67-NH-Pro-mod with the known MOFs with meta functionalized linker. | ||
 | ||
| Scheme 2 Synthesis of new proline-functionalized linker ortho-5 and Zr-MOF o-UiO-67-NH-Pro-mod. | ||
In this work, we detail the synthesis and characterization of UiO-67 derivatives based on ortho- and meta-proline amide biphenyldibenzoate linkers and investigate their performance in heterogeneous asymmetric organocatalytic aldol reactions. We identify that next to the proline position on the linker also the nature and concentration of the modulator used in the synthesis as well as the solvent used in the organocatalytic reactions have strong effects on structural features, selectivity, and activity of the catalyst. Moreover, we detail how the morphology, surface area and modulator used in the MOF synthesis takes on an active role in acetal formation as competing side reaction. Additionally, molecular dynamics (MD) simulations are performed to study the spatial orientation of the proline catalysts and the mobility of guest molecules and accessible volume in the MOFs. These properties play a critical role for catalytic efficiency, as they can determine the accessibility of reactants to catalytic sites and ensure that products can be removed promptly (Scheme 1).
Results and discussion
In order to characterize the impact of proline position relative to the metal center in UiO-67 on the catalysis of the aldol reaction we first synthesized the respective proline amide functionalized benzoate linkers.Synthesis of organic proline-functionalized linker
As outlined in Scheme 2 the synthesis of ortho-proline-functionalized linker and soluble catalyst commenced with the Suzuki coupling of methyl 2-amino-4-bromo-benzoate 1 and boronic acid 2 in the presence of 4 mol% of Pd(OAc)2, followed by condensation of N-Boc-proline 3 with diisopropylcarbodiimide (DIC) and N,N-Dimethylpyridin-4-amin (DMAP) to give the N-Boc-protected dimethylester 4 in 61% overall yield over 2 steps. Deprotection of the N-Boc group with TFA yielded the diester ortho-5 suitable for homogeneous reference catalysis. Saponification of 4 with KOH in THF gave the N-Boc-protected linker ortho-6.Synthesis and characterization of proline-functionalized UiO-67 MOFs
The synthesis of proline-functionalized UiO-67-type MOFs (visualized in Fig. 1) follows the procedures previously published by using the Boc-protected linkers ortho-6 and meta-6, respectively, to prevent side reactions of non-protected proline.16a In previous reports, it was found that upon solvothermal synthesis of meta-6 with benzoic acid in DMF at 120 °C for 4 days, the Boc group was thermally cleaved during the MOF synthesis, liberating the catalytically active secondary amine.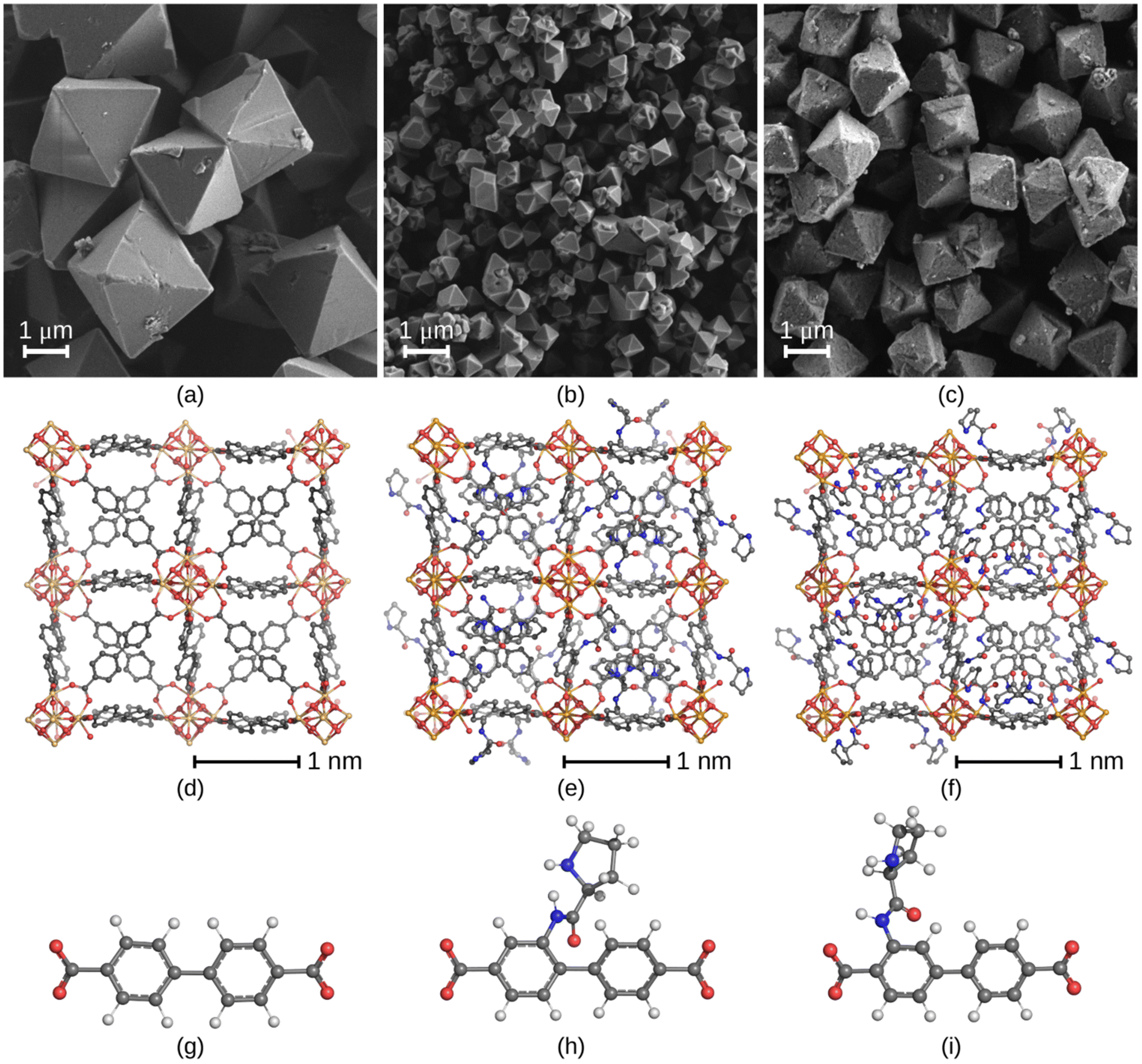 | ||
| Fig. 1 UiO-67 and UiO-67-NH-Pro structures synthesized and studied in this work. SEM pictures of UiO-67 (a), m-UiO-67-NH-Pro (b) and o-UiO-67-NH-Pro (c) generated with the acetic acid modulator. The respective molecular representations of one defect-free unit cell (d–f), and the individual linkers (g–i). In the experimental structure of UiO-67 all linkers were manually substituted for proline-modified linkers with proline in position meta or ortho to obtain m-UiO-67-NH-Pro and o-UiO-67-NH-Pro, respectively.16c For visualization of UiO-67 type MOFs with defects, see ESI Fig. S22.† Oxygen atoms are shown in red, nitrogen atoms in blue, zirconium atoms in yellow and carbon atoms in grey. Hydrogen atoms are white in the bottom row but omitted for clarity in the unit cell representation. | ||
To study the influence of the modulator on the deprotection, framework crystallization, and ultimately catalytic activity we synthesized o-UiO-67-Pro by solvothermal synthesis in DMF with TFA (120 °C, 24 h, further denoted to as o-UiO-67-NH-Pro-TFA), BA (120 °C, 96 h, further denoted to as o-UiO-67-NH-Pro-BA),16a AcOH (120 °C, 96 h, further denoted to as o-UiO-67-NH-Pro-AcOH). The same reaction procedures were carried out using meta-6 as the organic linker and resulted in the MOFs further denoted to as m-UiO-67-NH-Pro-TFA, m-UiO-67-NH-Pro-BA, m-UiO-67-NH-Pro-AcOH. In addition, we synthesized UiO-67 with the three modulators as a non-functionalized reference, further denoted as UiO-67-TFA, UiO-67-BA, and UiO-67-AcOH. For all reactions, a white microcrystalline powder was obtained that was washed with fresh DMF (3 times over 2 days), suspended and washed in acetone (5 times over 2 days), and dried in dynamic vacuum for at least 16 h (<3 × 10–3 kPa, 80 °C).
The structure and phase purity of the samples was evaluated by PXRD on as made, and dried samples. As shown in Fig. 2, the PXRD patterns for both functionalized and non-functionalized MOF samples, synthesized using all three modulators, align with the predicted powder pattern for UiO-67.12c,h
 | ||
| Fig. 2 (a) PXRD patterns of the as-made MOF samples with TFA as modulator; (b) nitrogen adsorption of as-made MOF samples with TFA as modulator (filled circles) and desorption (empty circles) isotherms at 77 K. | ||
Following the specified washing and drying procedures, the reflections observed in some ortho and meta proline-functionalized samples became broader, suggesting a potential reduction in crystallinity and/or long-range order. Nonetheless, the samples showed improved crystallinity upon re-immersion in the reaction solution, indicating that these materials maintain their long-range order under the reaction conditions (details can also be seen in Fig. S1–S3†).
The Boc-deprotection during synthesis was examined by analyzing the 1H NMR spectra of the digested samples in a NaOH/D2O solution.22 Both ortho- and meta-proline functionalized UiO-67 samples, synthesized with TFA as a modulator, underwent in situ Boc deprotection within 24 h, as indicated by the disappearance of the methyl peaks associated with the Boc at the chemical shift of around 1 and 1.2 ppm for o-UiO-67-NH-Pro-TFA and 1.2 ppm for m-UiO-67-NH-Pro-TFA (Fig. S7–S9†). In contrast, the ortho- and meta-proline functionalized MOFs synthesized with BA and AcOH as modulators showed partial Boc deprotection (80 to 96% deprotection estimated from 1H NMR), after an extended reaction time of 4 days (Fig. S10 and S11†).
Previous research by Kutzscher et al.16a highlighted the key role of the strong Lewis acidity of Zr4+ ions in facilitating the in situ Boc deprotection of proline, typically requiring a reaction time of 96 h. However, our observations showed a significant acceleration of Boc deprotection when TFA as a much stronger Brønsted acid was used as a modulator, reducing the time for complete deprotection to only 24 h.
Given these findings, we proceeded with a thermal post-synthetic Boc deprotection treatment,12a which involved placing the MOF samples in a microwave vial containing DMF and heating them at 180 °C for 3 h. The effectiveness of this treatment was confirmed by 1H NMR spectroscopy, as indicated by the absence of peaks corresponding to the Boc group post-treatment, as illustrated in Fig. S7–S9.† The 1H NMR analysis of digested samples also allows us to evaluate the presence of modulators in the MOF samples. Indeed, we observe the presence of all three types of modulators in all three MOF systems in different concentrations (see Table S1†). Given that the concentrations of the modulators in the MOFs are in a comparable range, we expect a comparable impact on the catalysis. In the solid framework, these modulators act as capping ligands at the clusters, resulting in missing linker defects,23 that can give additional access to Lewis acid sites as well as additional nanoporosity.
We have performed thermogravimetric analysis (TGA) to obtain the chemical composition of the MOFs. Since the MOFs were dried under mild conditions and containing residual solvent and solvent decomposition products (as indicated by the H NMR spectra of the digested samples), along with the possibility of having missing cluster defects, the defect quantification using a combination of TGA and 1H NMR may not have yielded a fully accurate chemical composition (Table S2†). Nevertheless, detailing the chemical composition and quantifying defects is crucial, as these factors can influence the MOF's pore structure, chemical properties, and ultimately, its catalytic performance.
We have taken SEM micrographs to analyze the morphology of the MOFs. The bare and functionalized UiO MOFs in each modulator system, have comparable crystal size and morphology within the distinct modulator system (Fig. S13–S15†). This consistency within each modulator group allows us to state that crystal size and morphology effects will play a critical factor influencing the observed differences in catalytic properties.
To evaluate the impact of the functionalization on the porosity of the samples, we recorded nitrogen adsorption isotherms at 77 K. In Fig. 2 (bottom), the MOF samples synthesized with TFA as a modulator exhibit a type-Ib isotherm. A decrease in nitrogen uptake is evident when proline functional groups are introduced, which can be attributed to the occupancy of the porosity with proline and a partial collapse of the framework upon solvent removal. The isotherms of the MOFs synthesized with AcOH and BA can be found in the ESI (Fig. S4 and S5†).
Homogeneous and heterogeneous catalysis
In order to compare the catalytic activity of o-UiO-67-NH-Pro with the known m-UiO-67-NH-Pro,16a the organocatalytic aldol reaction of 4-nitrobenzaldehyde 7 with cyclohexanone 8 to the syn- and anti-aldol products syn-9 and anti-9 was studied as benchmark reaction (Table 1).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | mol% | Time [d] | Solvent | Yielda 9 [%] | Yielda 10 [%] | 9 dra syn/anti |
syn-9 erb,c minor/major | anti-9 erb (S,R)/(R,S) |
| a Determined from the 1H NMR spectrum of the crude product using mesitylene as the external standard.b Determined by HPLC on a CHIRALPAK® AD-H column, 250 × 4.6 mm, 5 μm, hexane : i-PrOH (90:10), 0.8 mL min−1, 254 nm, 22 °C.c syn-9: minor (R,R) or (S,S); major (R,R) or (S,S). |
|||||||||
| (1) | ortho-5 | 5 | 7 | CDCl3 | 15 | 0 | 27:73 |
43:57 |
41:59 |
| (2) | ortho-5 | 5 | 7 | DMSO | 26 | 0 | 33:67 |
56:44 |
29:71 |
| (3) | meta-5 | 5 | 9 | CDCl3 | 7 | 0 | 38:62 |
35:65 |
09:91 |
| (4) | ortho-5 | 5 | 7 | MeOH | 22 | 40 | 40:60 |
59:41 |
27:73 |
| (5) | meta-5 | 5 | 7 | MeOH | 6 | 15 | 13:87 |
43:57 |
17:83 |
| (6) | ortho-5 | 20 | 1 | CDCl3 | >99 | 0 | 20:80 |
44:56 |
36:64 |
| (7) | meta-5 | 20 | 3 | CDCl3 | >99 | 0 | 25:75 |
59:41 |
20:80 |
| (8) | o-UiO-67-NH-Pro-TFA | 5 | 7 | MeOH | 9 | 9 | 91:9 |
42:58 |
35:65 |
| (9) | m-UiO-67-NH-Pro-TFA | 5 | 7 | MeOH | 5 | 75 | 61:39 |
56:44 |
05:95 |
| (10) | o-UiO-67-NH-Pro-TFA | 20 | 7 | DMSO | 59 | 0 | 79:21 |
50:50 |
50:50 |
| (11) | o-UiO-67-NH-Pro-TFA | 20 | 7 | CDCl3 | 95 | 0 | 81:19 |
50:50 |
49:51 |
| (12) | m-UiO-67-NH-Pro-TFA | 20 | 7 | DMSO | 17 | 0 | 81:19 |
50:50 |
43:57 |
| (13) | m-UiO-67-NH-Pro-TFA | 20 | 7 | CDCl3 | 25 | 0 | 78:22 |
53:47 |
44:56 |
| (14) | UiO-67-TFA (0% Pro) | 20 | 7 | CDCl3 | 60 | 0 | 79:21 |
50:50 |
48:52 |

First, the soluble proline-functionalized linkers ortho-5, meta-5 were examined under homogeneous conditions. When employing a catalyst loading of 5 mol%, the aldol reaction did not go to completion even after 7 days, regardless of the tested solvent (CDCl3, MeOH, DMSO). For example, ortho-5 gave after 7 days in CDCl3 15% of aldol 9 with anti-9 as the major diastereomer [dr (syn-9/anti-9) = 27:73, er syn-9 = 43:57, anti-9 = 41:59] and in DMSO 26% of 9 [dr (syn-9/anti-9) = 33:67, er syn-9 = 56:44, anti-9 = 29:71] (Table 1, entries 1 and 2).
In case of meta-5 yields were even lower: 7% 9 [dr (syn-9/anti-9) = 38:62, er syn-9 = 35:65, anti-9 9:91] (entry 3). However, in MeOH the acetal 10 was isolated as the major product in 40% for ortho-5 (15% for meta-5), whereas the aldol products 9 were obtained only as minor byproducts (22% for ortho-5, 6% for meta-5) with similar diastereomeric and enantiomeric ratios as discussed above (entries 4 and 5). Monitoring the homogeneous catalysis in CDCl3 by 1H NMR over 7 days revealed a higher reaction rate for ortho-5 as compared to meta-5 (Fig. S9 and S10†). The reactivity difference between the regioisomers was clearly visible, when higher catalyst loadings were employed. For 20 mol% ortho-5 the reaction was complete after 1 day [dr (syn-9/anti-9) = 20:80, er syn-9 = 44:56, anti-9 = 36:64], whereas 20 mol% meta-5 required 3 days for completion [dr (syn-9/anti-9) = 25:75, er syn-9 = 59:41, anti-9 = 20:80] (entries 6 and 7). In general, for the homogeneous catalysis diastereoselectivities of 9 differed only little between ortho-5 and meta-5. In all cases the anti-aldol anti-9 was the major diastereomer. In contrast, with respect to the enantioselectivity anti-aldols anti-9 possessed higher er values as compared to syn-aldols syn-9 and er values for ortho-5 were somewhat smaller than for meta-5 (For further details, see Tables S4, S5 and Schemes S4–S7† for proposed mechanism).
Preliminary heterogeneous catalysis experiments with 5 mol% of o-UiO-67-NH-Pro-mod gave low yields (<12%) even after 7 days in CDCl3 with both AcOH, BA or TFA modulators which did not reveal any trends (for all optimization experiments on heterogeneous catalysis, see Table S6†). Use of MeOH as the solvent resulted in preferred formation of the acetal 10, e.g. 5 mol% of o-UiO-67-NH-Pro-TFA gave 9% of acetal 10 and 9% aldol 9, while m-UiO-67-NH-Pro-TFA gave 75% of 10 and only 5% 9 (entries 8 and 9). A long-term experiment utilizing 5 mol% of o-UiO-67-NH-Pro-TFA further supported this observation (Fig. S11†). When a hot filtration test was carried out after 2 days, the NMR yield of the acetal 10 increased with increasing time, while the concentration of the aldol 9 remained constant (Fig. S11b†). These results indicate that the heterogeneous catalysis takes place as anticipated only in the presence of the solid catalyst, whereas the acetal formation proceeded after removal of the solid catalyst, presumably via leached TFA modulator. Fortunately, at 20 mol% loading with the proline-modified Zr-MOFs containing TFA modulator the heterogeneous catalysis of the aldol addition was significantly accelerated. As shown in Table 1, entry 10, in DMSO o-UiO-67-NH-Pro-TFA gave 59% aldol 9, preferably as the syn-aldol syn-9 (dr 79:21), but racemic for both syn- and anti-diastereomers [er syn-9 = 50:50, anti-9 = 48:52]. Under similar conditions m-UiO-67-NH-Pro-TFA gave only 17% 9 with similar diastereoselectivity [dr (syn-9/anti-9) = 81:19, er syn-9 = 50:50, anti-9 = 43:57] (entry 12). Gratifyingly, the reactivity of the Zr-MOFs could be further improved by using CDCl3 as the solvent. Thus, o-UiO-67-NH-Pro-TFA gave 95% aldol 9 [dr (syn-9/anti-9) = 81:19, er syn-9 = 50:50, anti-9 = 49:51] after 7 days (entry 11), whereas m-UiO-67-NH-Pro-TFA produced 25% aldol 9 [dr (syn-9/anti-9) = 78:22, er syn-9 = 53:47, anti-9 = 44:56] (entry 13). It should be noted, that UiO-67-TFA (0% Pro) (20 mol% in CDCl3) was already catalytically active, yielding 60% of 9 [dr (syn-9/anti-9) = 79:21, er syn-9 = 50:50, anti-9 = 48:52] with the syn-9 as major diastereomer (entry 14).
Thus, both the regioisomer of the proline-functionalized linker as well as the solvent had a major impact on the reaction rate and yields. Presumably, the reversal of the diastereoselectivity from the anti-preference under homogeneous conditions to the syn-preference under heterogeneous conditions is due to the Lewis acid catalysis taking place at the Zr nodes of the MOF in agreement with Kaskel's earlier report.16a
This rationale was further supported by catalytic control experiments with ZrOCl2·8 H2O (15 mol%) in CDCl3, which gave 5% aldol 9 albeit as an almost equimolar mixture of diastereomers [dr (syn-9/anti-9) = 55:45] (entry 13, Table S7†). An additional control experiment employing ZrOCl2·8 H2O (15 mol%) in MeOH yielded already after 20 h 96% acetal 10 (entry 14), indicating that in MeOH the Zr-catalyzed acetalization is strongly favoured over the aldol reaction.12a,b,e,21,24
While MeOH accelerates the acetal formation at the expense of the aldol reaction, DMSO or CDCl3 gave higher yields of the aldol product as compared to MeOH. The beneficial effect of polar solvents on the aldol reaction has been recently examined in theoretical studies by Gavali,25 Lustosa26 and Świderek.27 The increased yields of CDCl3 as compared to DMSO might be due to Brønsted acid catalysis accelerating both condensation step towards the iminium ion, iminium ion – enamine equilibrium, as well as final hydrolysis to release the aldol product. On the other hand, diastereoselectivities were little affected by the solvent, presumably due to the cyclic transition states. The proposed mechanistic details of homogenous and heterogenous catalysis are depicted in Schemes S3–S5 in the ESI.†
It should be noted that the involvement of Lewis acid catalysis by the Zr nodes has also been discussed by Kaskel for UiO-66 with L-proline defect sites.12f The Lewis acid catalysis by the Zr node would also explain the experimental observation, that the unmodified MOF UiO-67-TFA (0%Pro) (Table 1, entry 14) gave a similar diastereoselectivity as compared to the proline-modified MOFs (entries 8–13). According to Kaskel the reversed diastereoselectivity might also be explained by the close proximity of the proline units from the other linker units in the MOF pore.16a Moreover, Telfer has emphasized that differences in the diastereomeric transition states are very small and these energetic differences are influenced by weak interactions of the catalytic intermediates with modulators and solvents.12e
Finally, hot filtration tests for the catalytic reactions with 20 mol% of catalysts were carried out in CDCl3, which revealed that for both UiO-67-TFA as well as proline-modified MOFs o-UiO-67-NH-Pro-TFA, m-UiO-67-NH-Pro-TFA heterogeneous catalysis took place, i.e. the yield of the aldol 9 remained constant after removal of the catalyst indicating no leaching of active linkers into the reaction mixture as shown in Fig. 3.
 | ||
| Fig. 3 Comparison of hot filtration UiO-67-TFA, o-UiO-67-NH-Pro-TFA and m-UiO-67-NH-Pro-TFA in the presence of 20 mol% catalyst in CDCl3. | ||
Moreover, the following qualitative trend regarding the reaction rate was observed: o-UiO-67-NH-Pro-TFA > UiO-67-TFA ≥ m-UiO-67-NH-Pro-TFA (Fig. 3).
These observations indicate that the spatial orientation of the proline group within the framework plays a pivotal role in the catalytic activity and potentially other factors that may influence the heterogeneous reaction such as mass transport in the nanoporous frameworks.
Molecular dynamics simulations of proline-functionalized MOF
Consequently, the spatial orientation and arrangement of the proline groups in the UiO-67 frameworks and its influence on the mobility of the individual molecules in the porous frameworks was studied by all-atom molecular dynamics (MD) simulations.Our averaged MD simulations revealed markedly different spacing of the NH group on the proline catalyst and the metal centers for o-UiO-67-NH-Pro and m-UiO-67-NH-Pro (Fig. 4, top row). In detail, the NH group is most of the time 0.7–0.9 nm away from the nearest Zr in m-UiO-67-NH-Pro. In o-UiO-67-NH-Pro, on the other hand, the NH group positions itself significantly closer to the nearest Zr, namely 0.3–0.7 nm. Thus, for more than 50% of the time the proline's NH group interacts either directly with the oxygens/hydroxyl ions of the metal center or indirectly via methanol OH group. Similar trend is observed for the distance of the enamine “spacer” NH group and the metal center (shown in ESI Fig. S23†). Whereas the distance of 0.7–0.8 nm in the m-UiO-67-NH-Pro system is too large to simultaneously stabilize the aldehyde interacting with the TS3′/4′ (see Scheme S6†) by both the metal center and the spacer NH, the spacing of 0.3–0.5 nm in the o-UiO-67-NH-Pro is suitable for simultaneous stabilization, thus most likely improving the syn/anti selectivity of the reaction.
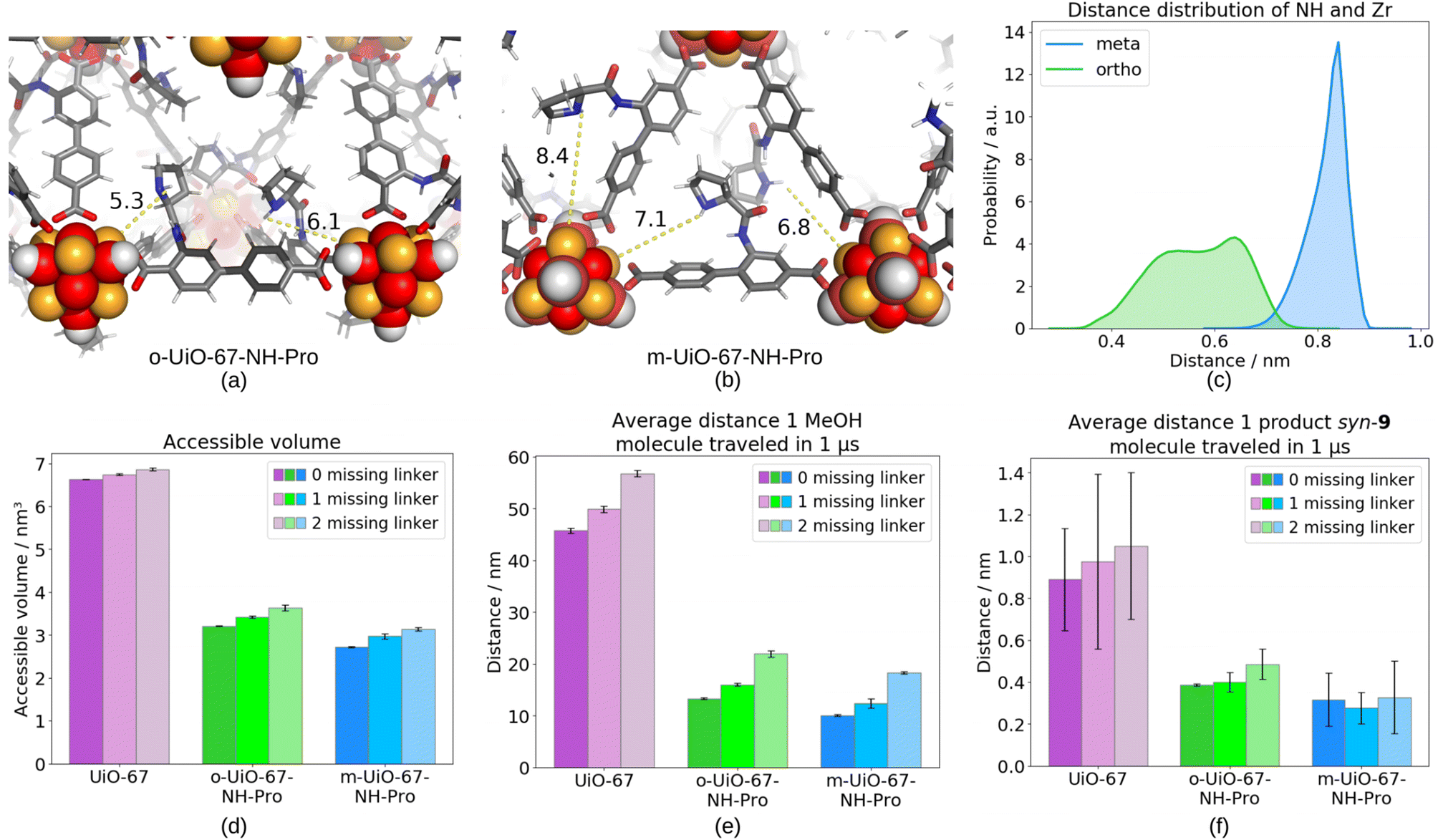 | ||
| Fig. 4 Exemplary positions of the proline catalysts described by the distance of the NH-group to the nearest Zr atom in o-UiO-67-NH-Pro (a) and m-UiO-67-NH-Pro (b). Oxygen atoms are shown in red, nitrogen atoms in blue, zirconium atoms in orange, carbon atoms in gray, hydrogen atoms in white. (c) Minimal distance distributions of the NH-group of the catalyst to the Zr atom of the metal cluster in respect of catalyst position in defect-free MOFs. (d) Accessible volumes in each system. (e) Average distance that MeOH traveled in 1 μs in each system. (f) Average distance that the product syn-9 covered in 1 μs in each system. The errors denote standard deviations. | ||
The drastic reduction (by 51.6% in case of o-UiO-67-NH-Pro and 58.9% in m-UiO-67-NH-Pro compared to UiO-67) of the accessible volume by the attachment of the catalyst on the linker is depicted in Fig. 4, bottom left. Notably, o-UiO-67-NH-Pro retains a higher accessible volume compared to m-UiO-67-NH-Pro, which suggests that the catalyst next to the metal cluster occupies less space. Fig. 4, bottom middle shows that both the presence of the catalyst inside the MOF and the catalyst's position influence the movement of methanol as a guest molecule in the pore. In agreement with the higher accessible volume of o-UiO-67-NH-Pro, methanol diffuses better in the o-UiO-67-NH-Pro compared to m-UiO-67-NH-Pro, yet the mobility is even more reduced than the accessible volume, in detail methanol travels only 29% and 22% of the average distance in o-UiO-67-NH-Pro and m-UiO-67-NH-Pro compared to UiO-67, respectively. The reduced mobility of methanol in m-UiO-67-NH-Pro results from the catalyst blocking the pore windows and from higher entangling of methanol with the catalyst. In o-UiO-67-NH-Pro, on the other hand, the catalyst is positioned at the outer edge of the linker, resulting in more open pore windows and thus increased methanol mobility.
Similarly, the movement of the substrates is much easier in catalyst-free UiO-67. Transport properties are more drastically impacted for the larger substrate and product molecules of the aldol reaction. Even though the total distance the substrates travel within 1 μs in UiO-67 is more than ten times shorter than that of methanol, the substrates readily pass from one cage to another passing within 1 μs almost 2 unit cells (the unit cell has dimensions of x = y = z = 2.69 nm). The presence of the catalyst strongly restricts also the movement of the reactants. Substrate 7 reaches only 9% and 16% of the traveled distance in o-UiO-67-NH-Pro and m-UiO-67-NH-Pro compared to the catalyst-free UiO-67, respectively (ESI Fig. S25 and S26†). Substrate 8 is also drastically slowed down, in detail to 8% in o-UiO-67-NH-Pro and to 8% in m-UiO-67-NH-Pro. The comparably high error bars in the distances the substrates travel does not allow for distinction of the effect of the catalyst position on the mobility of the substrates in the timescale of our simulations. The situation becomes even more severe for the bicyclic syn-9 product as can be seen in Fig. 4, bottom right. While the product can move slowly but steadily through the catalyst-free UiO-67 (the average traveled distance of 0.89 nm per 1 μs corresponds to 1.4 transitions from pore to pore per 1 μs), it gets stuck in the smaller, tetrahedral pore in presence of the catalyst (0 pore to pore transitions in o-UiO-67-NH-Pro and 0.1 per 1 μs in m-UiO-67-NH-Pro). Obviously, more extensive sampling is required to ultimately analyze the product diffusion. In order to test the ability of the product to diffuse through UiO-67 with the catalyst attached on the linker either in the meta or ortho position, additional simulations were performed at a higher temperature of 813 K. The great number of product transitions from pore to pore has enabled us to calculate average travelled distance per 1 μs, amounting to 6.89 ± 1.56 nm and 11.24 ± 2.61 nm for o-UiO-67-NH-Pro and m-UiO-67-NH-Pro, respectively. These values, being even larger that the distances the substrates travel at 313 K through catalyst-free UiO-67, prove the general ability of the product to diffuse through o-UiO-67-NH-Pro and m-UiO-67-NH-Pro. Yet, the slow diffusion at experimental temperature of 313 K can clearly limit the reaction efficiency by insufficient mass transport of reagents and inhibition by immobilized products. Thus, improving the mobility of the product through the MOF, e.g., by utilizing a mixed linker approach or by extending the linker size or utilizing dynamic aspects of the framework, is holding a great promise for improving the UiO-67-NH-Pro-catalyzed aldol condensation of 4-nitrobenzaldehyde with cyclohexanone.
To estimate the role of missing linker defects, one or two linkers per simulation cell were substituted by two or four TFAs, respectively, and additional simulations have been conducted. The defect-rich MOFs show only a slight increase in the overall accessible pore volume (see Fig. 4 bottom left) but the defects systematically increase the mobility of methanol (shown as average traveled distance per 1 μs in Fig. 4 bottom middle). The removal of linkers creates additional pathways and larger pores, thus increasing methanol diffusion. However, the structural alterations introduced by missing-linker defects with TFA modulator, fail to significantly alter the mobility of substrates 7 and 8 (see ESI Fig. S25 and S26†), as well as of the product syn-9 (shown in Fig. 4 bottom right). Given that the traveled distance of the substrates and the product exhibit only minimal variation in the defective systems compared to the defect-free UiO-67, the steric hindrance imposed by the proline catalyst persists in restricting the overall mobility of larger molecules.
Conclusion
This study highlights how confinement and positioning of active site groups on the linker in UiO-67 MOFs influence catalysis, distinguishing it from MOF systems with lack of positional variability and further addressing the importance of understanding structure–function relationships in nanoporous MOF catalysts. As demonstrated by computational methods, by placing the proline group and its “spacer” enamine group in closer proximity to the metal clusters we achieve higher diffusivity of the molecules in the ortho-functionalized MOF while also providing spatial proximity of the proline and enamine “spacer” groups to the metal cluster, enabling better coordination of the transition states. We also identify the critical role of the acidic modulator in the in situ deprotection of the Boc group from the proline-based linker as well as their influence on the catalytic formation of acetals when reactions are carried out in methanol. We envision that our approach is viable to the construction of multifunctional active catalysts in other chemical transformations in which both Lewis acid and proline sites will act in a cooperative manner. Our extensive computational analysis indicates that linker functionalization may come at the price of reduced substrate/product mobility in the nanoporous network of MOFs and that individual missing linker defects do not alleviate this effect if substituted by TFA. The limited access of active sites in particular in the crystal interior by blocking of pore windows by the proline groups is an aspect we intend to study further also by experimental methods. To the best of our knowledge, our study is the first to consider these aspects with respect to catalytic activity in nanoporous confinement of MOFs12d,28 and we believe this to be a critical area of research for the improved design of active molecular MOF catalysts.Author contributions
Z.D. and S.N. synthesized the linkers and performed catalytic experiments, C.W. and B.H.Y. optimized the linker synthesis. A.D.Y. and H.Sh. synthesized and characterized the MOFs. K.G. performed DFT calculations. S.Ko. performed and analyzed MD simulations. D.P.E. provided his expertise on molecular heterogeneous catalysis. A.Z. checked the data. A.D.Y., A.Z., S.L., K.P. and S.Kr. wrote the manuscript. S.L. supervised the linker synthesis and catalytic experiments and coordinated the research. All authors proofread the manuscript and agreed to the final version.Data availability
Data supporting this study are contained in the manuscript and ESI.† Raw data is openly available from (DaRUS) at (@sadia+ Ardi URL will be generated after publication).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Generous financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 358283783 – SFB 1333/2 2022, subprojects A08, B05, B08, C04, C07 and HBFG, shared instrumentation (Grant No. INST 41/897-1 FUGG for 700 MHz-NMR), the Ministerium für Wissenschaft, Forschung und Kunst des Landes Baden-Württemberg, and the Fonds der Chemischen Industrie is gratefully acknowledged. K. P. and S. Ko. acknowledge the support by the Deutsche Forschungsgemeinschaft under Germany's Excellence Strategy – EXC 2075-390740016 and by the Stuttgart Center for Simulation Science (SC SimTech). S. K. and A. D. Y. acknowledge financial support by the Carl-Zeiss-Stiftung within the NEXUS program. The authors also gratefully acknowledge the scientific support and HPC resources provided by the Erlangen National High Performance Computing Center (NHR@FAU) of the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) under the NHR project MoTrNanoMat. NHR funding is provided by federal and Bavarian state authorities. NHR@FAU hardware is partially funded by the German Research Foundation (DFG) – 440719683. Open Access funding provided by the Max Planck Society.References
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Reticular synthesis and the design of new materials, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Introduction to metal–organic frameworks, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- B. I. Ahmad, K. T. Keasler, E. E. Stacy, S. Meng, T. J. Hicks and P. J. Milner, MOFganic Chemistry: Challenges and Opportunities for Metal–Organic Frameworks in Synthetic Organic Chemistry, Chem. Mater., 2023, 35, 4883–4896 CrossRef CAS PubMed.
- S. Pandey, B. Demaske, O. A. Ejegbavwo, A. A. Berseneva, W. Setyawan, N. Shustova and S. R. Phillpot, Electronic structures and magnetism of Zr-, Th-, and U-based metal-organic frameworks (MOFs) by density functional theory, Comput. Mater. Sci., 2020, 184, 109903 CrossRef CAS.
- N. Stock and S. Biswas, Synthesis of metal-organic frameworks (MOFs): routes to various MOF topologies, morphologies, and composites, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- E. B. Anderson and M. R. Buchmeiser, Catalysts immobilized on organic polymeric monolithic supports: from molecular heterogeneous catalysis to biocatalysis, ChemCatChem, 2012, 4, 30–44 CrossRef CAS.
- R. Greifenstein, T. Ballweg, T. Hashem, E. Gottwald, D. Achauer, F. Kirschhöfer, M. Nusser, G. Brenner-Weiß, E. Sedghamiz and W. Wenzel, MOF–Hosted Enzymes for Continuous Flow Catalysis in Aqueous and Organic Solvents, Angew. Chem., Int. Ed., 2022, 61, e202117144 CrossRef CAS PubMed.
- J. Dai and H. Zhang, Recent advances in catalytic confinement effect within micro/meso–porous crystalline materials, Small, 2021, 17, 2005334 CrossRef CAS PubMed.
- C. H. Sharp, B. C. Bukowski, H. Li, E. M. Johnson, S. Ilic, A. J. Morris, D. Gersappe, R. Q. Snurr and J. R. Morris, Nanoconfinement and mass transport in metal–organic frameworks, Chem. Soc. Rev., 2021, 50, 11530–11558 RSC.
- X. Li, J. Wu, C. He, Q. Meng and C. Duan, Asymmetric catalysis within the chiral confined space of metal–organic architectures, Small, 2019, 15, 1804770 CrossRef PubMed.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. De Vos and N. Stock, Cerium-based metal organic frameworks with UiO-66 architecture: synthesis, properties and redox catalytic activity, Chem. Commun., 2015, 51, 12578–12581 RSC.
- (a) D. J. Lun, G. I. Waterhouse and S. G. Telfer, A general thermolabile protecting group strategy for organocatalytic metal− organic frameworks, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS PubMed; (b) C. Kutzscher, H. C. Hoffmann, S. Krause, U. Stoeck, I. Senkovska, E. Brunner and S. Kaskel, Proline functionalization of the mesoporous metal− organic framework DUT-32, Inorg. Chem., 2015, 54, 1003–1009 CrossRef CAS PubMed; (c) G. Nickerl, M. Leistner, S. Helten, V. Bon, I. Senkovska and S. Kaskel, Integration of accessible secondary metal sites into MOFs for H2S removal, Inorg. Chem. Front., 2014, 1, 325–330 RSC; (d) S. Nießing, C. Czekelius and C. Janiak, Immobilisation of catalytically active proline on H2N-MIL-101 (Al) accompanied with reversal in enantioselectivity, Catal. Commun., 2017, 95, 12–15 CrossRef; (e) L. Liu, T.-Y. Zhou and S. G. Telfer, Modulating the performance of an asymmetric organocatalyst by tuning its spatial environment in a metal–organic framework, J. Am. Chem. Soc., 2017, 139, 13936–13943 CrossRef CAS PubMed; (f) K. D. Nguyen, C. Kutzscher, S. Ehrling, I. Senkovska, V. Bon, M. de Oliveira Jr, T. Gutmann, G. Buntkowsky and S. Kaskel, Insights into the role of zirconium in proline functionalized metal-organic frameworks attaining high enantio-and diastereoselectivity, J. Catal., 2019, 377, 41–50 CrossRef CAS; (g) G. Zhang, R. Chong, X. Zhou, J. Yang, Y. Bai, Z. H. Zhang and J. Lin, Positional Isomerism: A Novel Paradigm for Enhancing Iodine Adsorption in Functionalized Metal–Organic Frameworks, Inorg. Chem., 2024, 63, 22288–22296 CrossRef CAS PubMed; (h) D. K. Sannes, S. Øien-Ødegaard, E. Aunan, A. Nova and U. Olsbye, Quantification of linker defects in UiO-type metal–organic frameworks, Chem. Mater., 2023, 35, 3793–3800 CrossRef CAS.
- Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Catalytic enantioselective aldol reactions, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC.
- Y. Zhang, J. Guo, J. Zhang, X. Qiu, X. Zhang, J. Han, B. Zhang, C. Long, Y. Shi and Z. Yang, Metal-organic frameworks enable regio-and stereo-selective functionalization of aldehydes and ketones, Chem, 2022, 8, 1688–1704 CAS.
- M. Zhou, E.-S. M. El-Sayed, Z. Ju, W. Wang and D. Yuan, The synthesis and applications of chiral pyrrolidine functionalized metal–organic frameworks and covalent-organic frameworks, Inorg. Chem. Front., 2020, 7, 1319–1333 RSC.
- (a) C. Kutzscher, G. Nickerl, I. Senkovska, V. Bon and S. Kaskel, Proline functionalized UiO-67 and UiO-68 type metal–organic frameworks showing reversed diastereoselectivity in aldol addition reactions, Chem. Mater., 2016, 28, 2573–2580 CrossRef CAS; (b) G. Zhang, R. Chong, X. Zhou, J. Yang, Y. Bai, Z. H. Zhang and J. Lin, Positional Isomerism: A Novel Paradigm for Enhancing Iodine Adsorption in Functionalized Metal–Organic Frameworks, Inorg. Chem., 2024, 63(46), 22288–22296 CrossRef CAS PubMed; (c) N. Ko, J. Hong, S. Sung, K. E. Cordova, H. J. Park, J. K. Yang and J. Kim, A significant enhancement of water vapour uptake at low pressure by amine-functionalization of UiO-67, Dalton Trans., 2015, 44, 2047–2051 RSC.
- L. Lili, Z. Xin, R. Shumin, Y. Ying, D. Xiaoping, G. Jinsen, X. Chunming and H. Jing, Catalysis by metal–organic frameworks: proline and gold functionalized MOFs for the aldol and three-component coupling reactions, RSC Adv., 2014, 4, 13093–13107 RSC.
- S. E. Maier, O. Bunjaku, E. Kaya, M. Dyballa, W. Frey and D. P. Estes, Surface immobilized Cu-1,10-phenanthroline complexes with α-aminophosphonate groups in the 5-position as heterogenous catalysts for efficient atom-transfer radical cyclizations, Dalton Trans., 2023, 52, 8442–8448 RSC.
- S. E. Maier, T. Nagel, M. Turan, E. Kaya, W. Frey, M. Dyballa and D. P. Estes, Comparison of the Catalytic Activity of Surface-Immobilized Copper Complexes with Phosphonate Anchoring Groups for Atom Transfer Radical Cyclizations and Additions, Organometallics, 2024, 43, 233–241 CrossRef CAS.
- S. Maier, S. P. Cronin, M.-A. Vu Dinh, Z. Li, M. Dyballa, M. Nowakowski, M. Bauer and D. P. Estes, Immobilized Platinum Hydride Species as Catalysts for Olefin Isomerizations and Enyne Cycloisomerizations, Organometallics, 2021, 40, 1751–1757 CrossRef CAS.
- H.-H. Nguyen, Z. Li, T. Enenkel, J. Hildebrand, M. Bauer, M. Dyballa and D. P. Estes, Probing the Interactions of Immobilized Ruthenium Dihydride Complexes with Metal Oxide Surfaces by MAS NMR: Effects on CO2 Hydrogenation, J. Phys. Chem. C, 2021, 125, 14627–14635 CrossRef CAS.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Defect engineering: tuning the porosity and composition of the metal–organic framework UiO-66 via modulated synthesis, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- L. Liu, Z. Chen, J. Wang, D. Zhang, Y. Zhu, S. Ling, K.-W. Huang, Y. Belmabkhout, K. Adil and Y. Zhang, Imaging defects and their evolution in a metal-organic framework at sub-unit-cell resolution, Nat. Chem., 2019, 11, 622–628 CrossRef CAS PubMed.
- U. S. Arrozi, H. W. Wijaya, A. Patah and Y. Permana, Efficient acetalization of benzaldehydes using UiO-66 and UiO-67: Substrates accessibility or Lewis acidity of zirconium, Appl. Catal., A, 2015, 506, 77–84 CrossRef CAS.
- A. S. Gavali, P. J. Maliekal, V. A. Naik and P. M. Badani, Decoding the impact of solvents in altering the conversion rates and stereoselectivity in proline-catalyzed asymmetric aldol reaction, Theor. Chem. Acc., 2024, 143, 14, DOI:10.1007/s00214-023-0.
- D. M. Lustosa, S. Barkai, I. Domb and A. Milo, Effect of Solvents on Proline Modified at the Secondary Sphere: A Multivariate Exploration, J. Org. Chem., 2022, 87, 1850–1857 CrossRef CAS PubMed.
- K. Świderek, A. R. Nödling, Y.-H. Tsai, L. Y. P. Luk and V. Moliner, Reaction mechanism of organocatalytic Michael addition of nitromethane to cinnamaldehyde: a case study on catalyst regeneration and solvent effects, J. Phys. Chem. A, 2018, 122, 451–459 CrossRef PubMed.
- K. Hemmer, M. Cokoja and R. A. Fischer, Exploitation of Intrinsic Confinement Effects of MOFs in Catalysis, ChemCatChem, 2021, 13, 1683–1691 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi02724h |
| ‡ These co-authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |