
Palladium-catalysed ortho-acylation of 6-anilinopurines/purine nucleosides via C–H activation†
Srinivasarao Allu and
K. C. Kumara Swamy*
School of Chemistry, University of Hyderabad, Hyderabad 500 046, Telangana, India. E-mail: kckssc@yahoo.com; kckssc@uohyd.ac.in
First published on 22nd October 2015
Abstract
Purinyl N1 directed ortho-acylation of 6-anilinopurines was achieved via C(sp2)–H bond activation in the presence of [Pd]-catalyst using aldehydes or α-oxocarboxylic acids as the acylating source. A wide variety of purine appended 2′-aminoacetophenones/benzophenones are isolated in good to excellent yields. These catalytic transformations are also successfully applied to 6-anilinopurine nucleosides.
Introduction
Aryl ketones are important building blocks in several biologically active natural products, pharmaceuticals and agrochemicals.1 Thus, construction of these motifs always attracts considerable attention from synthetic chemists. Among the numerous methods developed for the synthesis of aryl ketones, Friedel–Crafts acylation is the most accepted synthetic procedure.2 However, poor regioselectivity, limited functional group tolerance and using an over-stoichiometric amount of Lewis acid catalyst limit the scope of this reaction. Therefore, it is highly desirable to develop a mild and efficient method for the synthesis of aryl ketones.Recently transition metal catalysed direct C–H bond acylation of arenes has been reported by several pioneering groups using aldehydes,3 α-oxocarboxylic acids,4 alcohols,5 toluene derivatives,6 benzylamines,7 benzyl chlorides/bromides,8 carboxylic acids,9 diketones10 or benzylic ethers11 as acylating source with the aid of directing groups. This direct acylation reaction is more atom economic and environmentally friendly alternative to the Friedel–Crafts acylation, which is commonly used for the synthesis of aryl ketones. Purine could also be used as a directing group for the ortho-C–H functionalization of aryl moieties.12 As our ongoing work on C–H activation13 and modification of nucleoside derivatives,14 we herein report the palladium-catalysed ortho-acylation of 6-anilinopurines with aldehydes/α-oxocarboxylic acids via purinyl N1 directed C–H bond activation. More importantly, this study provides the purine appended 2′-aminoacetophenones/benzophenones, which may be used as active pharmaceutical ingredients.15
Results and discussion
To achieve ortho-acylation, we have initiated our studies by performing the reaction of 6-anilinopurine 1a with 1-heptanal in the presence of Pd(OAc)2 (5 mol%) with TBHP (3 equiv.) as an oxidant under neat conditions at 110 °C for 24 h. We were happy to find that ortho-acylated purine derivative 2a was obtained in 38% isolated yield (Table 1, entry 1). Inspired by this result, we proceeded to maximise the yield of the product 2a by varying the reaction parameters as depicted in Table 1. Among the solvents DMF, NMP, CH3CN, DCE, dioxane, toluene, xylene and AcOH, only dioxane gave moderate yield (44%). In the remaining cases, product 2a yield was poor or negligible (Table 1, entries 2–9). Among the palladium catalysts, only palladium acetate gave better conversion to the acylated product 2a (Table 1, entries 10–12). Proper oxidant is also crucial for this reaction. Compared to TBHP, benzoyl peroxide or H2O2 or benzoquinone gave lower yields of the product 2a (Table 1, entries 13–15). Gratifyingly, further optimisation revealed that reaction proceeds better in dioxane/AcOH/DMSO (7/2/1, v/v/v) solvent mixture to afford the ortho-acylated product in good yield of 62%. Increasing the amount of catalyst Pd(OAc)2 from 5 mol% to 10 mol% improved the yield of the product 2a to 74% after isolation (Table 1, entry 17). Thus, the optimised reaction conditions for the present reaction were: Pd(OAc)2 (10 mol%), TBHP (3 equiv.) and dioxane/AcOH/DMSO (7/2/1, v/v/v, 3 mL) at 110 °C (oil bath temperature) for 24 h.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (5 mol%) | Oxidant | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), 1-heptanal (0.6 mmol), oxidant (3 equiv.), solvent (3 mL), 110 °C (oil bath temperature).b Isolated yields. ND = not detected.c This solvent system was earlier used by Ge and coworkers.4b | ||||
| 1 | Pd(OAc)2 | TBHP | — | 38 |
| 2 | Pd(OAc)2 | TBHP | DMF | Trace |
| 3 | Pd(OAc)2 | TBHP | NMP | 21 |
| 4 | Pd(OAc)2 | TBHP | CH3CN (90 °C) | Trace |
| 5 | Pd(OAc)2 | TBHP | DCE (90 °C) | 23 |
| 6 | Pd(OAc)2 | TBHP | Dioxane | 44 |
| 7 | Pd(OAc)2 | TBHP | Toluene | 10 |
| 8 | Pd(OAc)2 | TBHP | Xylene | 32 |
| 9 | Pd(OAc)2 | TBHP | AcOH | Trace |
| 10 | PdCl2 | TBHP | Dioxane | 18 |
| 11 | PdCl2(CH3CN)2 | TBHP | Dioxane | 24 |
| 12 | PdCl2(PPh3)2 | TBHP | Dioxane | Trace |
| 13 | Pd(OAc)2 | Benzoyl peroxide | Dioxane | ND |
| 14 | Pd(OAc)2 | H2O2 | Dioxane | 31 |
| 15 | Pd(OAc)2 | Benzoquinone | Dioxane | 16 |
| 16 | Pd(OAc)2 | TBHP | Dioxane/AcOH/DMSO (7/2/1, v/v/v) | 62 |
| 17 | Pd(OAc)2 (10 mol%) | TBHP | Dioxane/AcOH/DMSO (7/2/1, v/v/v)c | 74 |

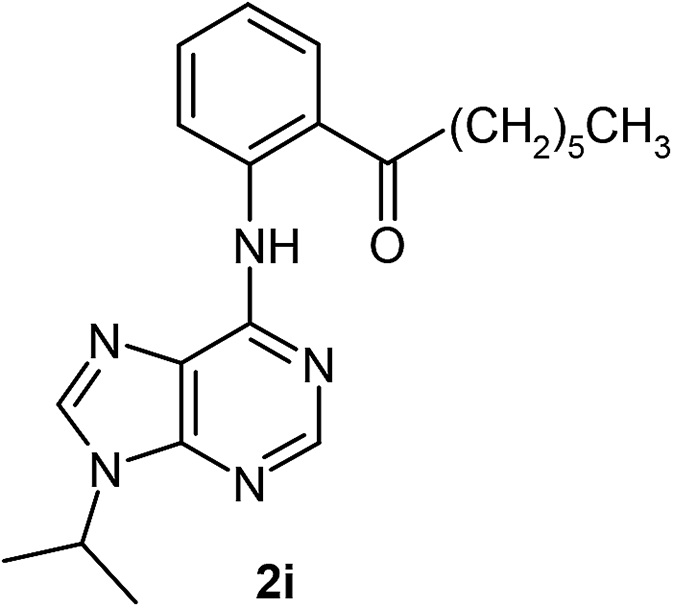
After having the optimised reaction conditions in hand, we examined the substrate scope by varying the 6-anilinopurine derivatives and aldehydes (Table 2). Anilines bearing electron-donating or withdrawing substituents underwent cross-dehydrogenative coupling (CDC) with heptanal smoothly and produced the corresponding acylated derivatives 2a–2h in good to excellent yields (61–74%). Bromo and chloro functional groups were well tolerated and afforded ortho-acyl derivatives 2d and 2e. These products can pave way for further manipulation via cross-coupling reactions utilising the –Br/–Cl functionalities. The reaction is highly regioselective when performed with meta-substituted amines. In these cases, only one regioisomer was observed and the sterically less hindered C–H position was acylated. Under these catalytic conditions ortho-substituted 6-anilinopurines did not furnish the corresponding acylated derivatives.4a,5b We have also examined the effect of purine N9-substituent on the course of the reaction. Thus, the reaction of (9-isopropyl-9H-purin-6-yl)-phenyl-amine 1i with heptanal afforded the ortho-acylated derivative 2i in good yield (70%). The generality of the methodology was then extended to 6-anilinopurine nucleoside 1j with 1-heptanal and 1-hexanal; in both the cases we have isolated the corresponding ortho-acylated nucleosides 2j and 2k respectively, in decent yields (54–56%). The reaction worked well in the case of simple 2-anilinopyrimidine, though.
|
|||
|---|---|---|---|
| Entry no. | Substrate | Product | Yieldb (%) |
| a Reaction conditions: amine 1 (0.3 mmol), aldehyde (0.6 mmol), Pd(OAc)2 (10 mol%), TBHP (3 equiv.) and dioxane/AcOH/DMSO (7/2/1, v/v/v, 3 mL) at 110 °C (oil bath temperature) for 24 h.b Isolated yield. | |||
| 1 |  |
 |
74 |
| 2 |  |
 |
71 |
| 3 | 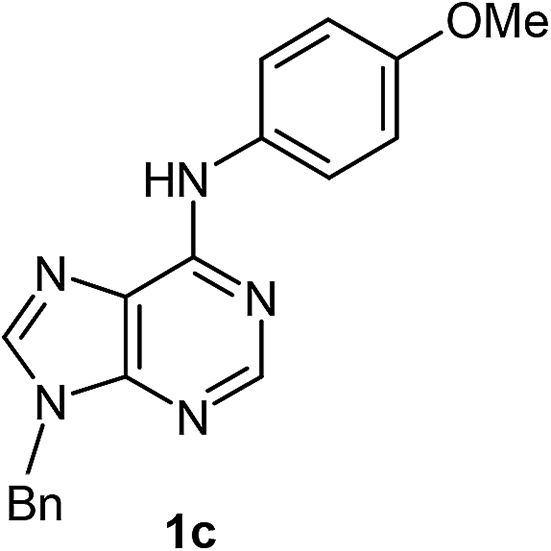 |
 |
72 |
| 4 |  |
 |
66 |
| 5 |  |
 |
62 |
| 6 |  |
 |
68 |
| 7 |  |
 |
61 |
| 8 |  |
 |
63 |
| 9 | 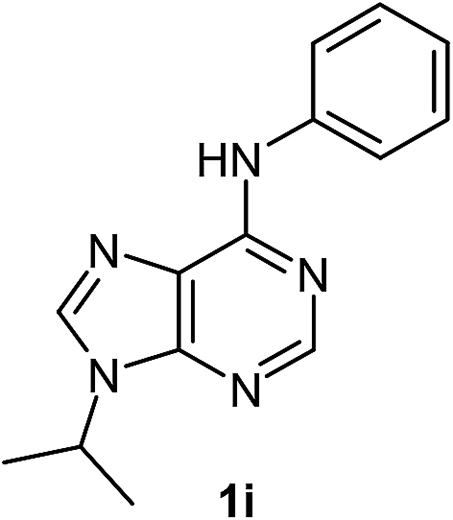 |
 |
70 |
| 10 |  |
 |
56 |
| 11 | 1j |  |
54 |
| 12 |  |
 |
68 |
| 13 | 1a |  |
71 |
| 14 | 1a | 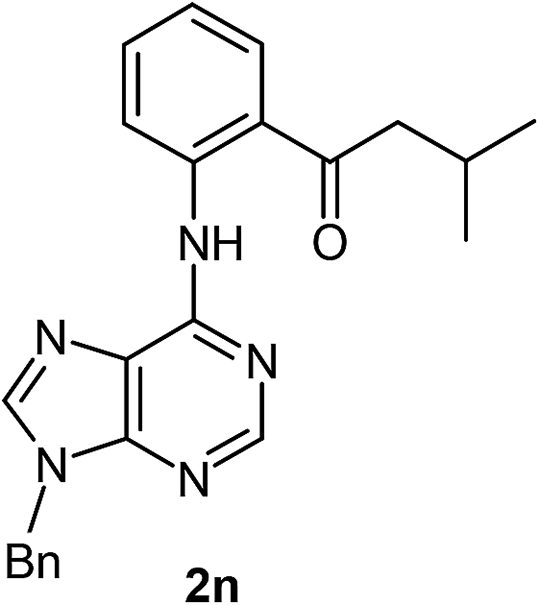 |
68 |
| 15 | 1a |  |
60 |
| 16 | 1a |  |
54 |

We then investigated the effect of a wide range of alkyl aldehydes. The reaction worked well with isovaleraldehyde (a branched aldehyde) and produced the acylated derivative 2n in good yield (68%). Cyclohexane carboxaldehyde also participated in this coupling and gave the corresponding ketone 2o in 60% yield. It is noteworthy that citronellal, a monoterpenoid could also provide the acylated derivative 2p in moderate yield (54%). Rather surprisingly, aryl aldehydes underwent oxidation to the corresponding carboxylic acids under these conditions. Thus, we observed the reactivity difference between the other directing group assisted cross dehydrogenative coupling (CDC) reaction of aryl aldehydes with the purine directed CDC reactions.3 The structure of one of the acylated derivatives (2m) was further confirmed by using X-ray crystallography (Fig. 1).
 | ||
| Fig. 1 Molecular structure of compound 2m. Selected bond lengths [Å] with esd's in parentheses: C(24)–O(1) 1.2263(17), C(24)–C(23) 1.490(2), N(10)–C(18) 1.3964(19), N(10)–C(6) 1.3621(18), N(9)–C(11) 1.4604(18). | ||
Plausible mechanistic pathway for the ortho-acylation using aldehydes
Based on previous reports,3 a plausible pathway is outlined for Pd-catalysed ortho-acylation in Scheme 1. Initially, through the chelate-directed C–H activation of purine N1 atom, the six-membered cyclopalladated intermediate I is formed. The reaction of aldehyde with TBHP generates reactive acyl and OH radicals which react with intermediate I to produce the Pd(IV) intermediate II.3 Finally, species II undergoes reductive elimination leading to the formation of acylated derivative 2a (or 2b–2p). The active Pd(II) is regenerated for next catalytic cycle.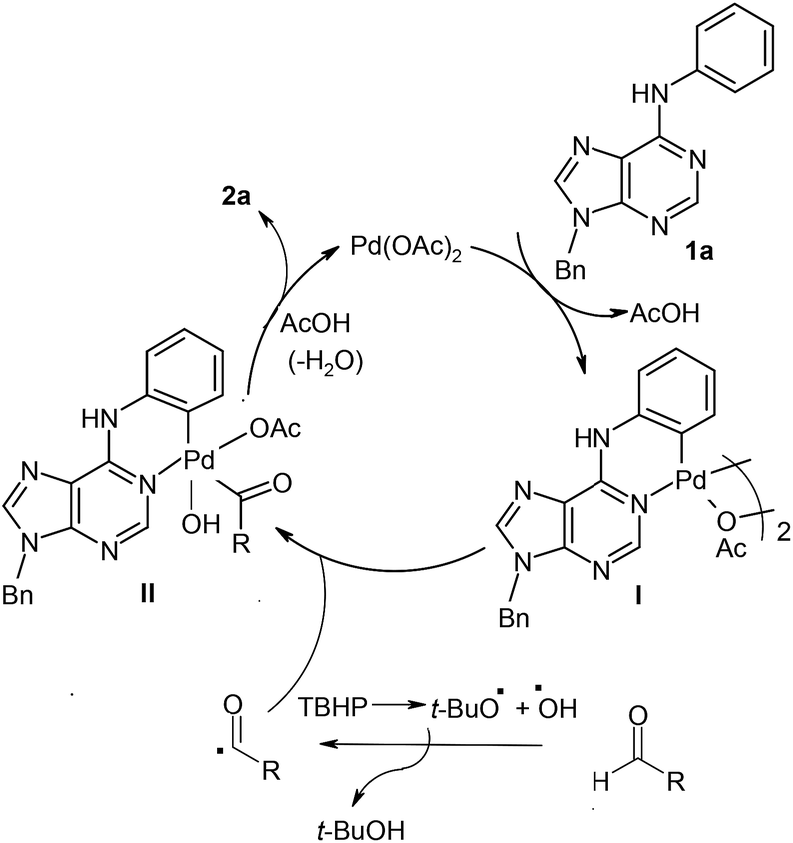 | ||
| Scheme 1 Plausible reaction pathway for the formation of ortho-acylated derivatives from 6-anilinopurines with aldehydes. | ||
Palladium-catalysed C(sp2)–H bond acylation of 6-anilinopurines with α-oxocarboxylic acids
The above Pd(OAc)2/TBHP catalytic system was applicable to alkyl aldehydes only; aryl aldehydes under those catalytic conditions were oxidized to the corresponding carboxylic acids. We overcame this drawback by choosing α-oxocarboxylic acid as the acylating source. Ag2O and K2S2O8 were used as oxidant and co-oxidant respectively (Scheme 2), under conditions similar to that available in the literature.4b Thus, the reaction of 6-anilinopurine 1a with phenylglyoxylic acid in the presence of PdCl2 (10 mol%) afforded the corresponding acylated derivative 3a in good yield (64%). Although Pd(OAc)2 also worked, the yield was lower (50%). Under these catalytic conditions, we have examined the substrate scope with respect to 4-Cl and 3-OMe substituted anilinopurines (1e and 1j) also. In both the cases, the corresponding ortho-aroyl derivative was isolated in good yield. Arylglyoxylic acids containing Me, F or Br functional groups are well tolerated under these conditions and afford the ortho-aroyl derivatives 3d–3g in good yields. It is noteworthy that the reaction also proceeded smoothly with 2-ketobutyric acid (an alkyl α-oxocarboxylic acid) by furnishing the ortho-acyl derivative 3h in 63% yield. Thus, our protocol is applicable for both aryl and alkyl α-oxocarboxylic acids. | ||
| Scheme 2 Synthesis of ortho-aroyl 6-anilinopurines using α-oxocarboxylic acid as acylating source. | ||
Conclusions
In summary, we have developed an efficient method for the Pd-catalysed oxidative ortho-acylation of 6-anilinopurines with alkyl aldehydes via C–H bond activation. A broad range of functional groups and a variety of alkyl aldehydes were well tolerated under these catalytic conditions. This protocol was also successfully applied on 6-anilinopurine nucleoside. 2′-Aminobenzophenones are synthesized by using α-oxocarboxylic acids as the acylating source. Further studies on the mechanistic pathway are currently going on in our laboratory.Experimental section
General comments
Solvents were dried according to known methods as appropriate.16 1H, 13C spectra (1H, 400 MHz; 13C, 100 MHz) were recorded using a 400 MHz spectrometer in CDCl3 with shifts referenced to SiMe4 (δ = 0). IR spectra were recorded on an FTIR spectrophotometer. Melting points were determined by using a local hot-stage melting point apparatus and are uncorrected. Elemental analyses were carried out on a CHN analyser. Mass spectra were recorded using LC-MS and HRMS (ESI-TOF analyser) equipment.General procedure for the ortho-acylation of 6-anilinopurine derivatives with aldehydes: synthesis of compounds 2a–2p
A mixture of 6-anilinopurine (0.3 mmol) and Pd(OAc)2 (10 mol%) was taken in a Schlenk tube under N2. To this, dioxane/AcOH/DMSO (7/2/1, v/v/v, 3 mL) solvent mixture was added and stirring was continued at rt for 10 min. To this mixture, aldehyde (0.6 mmol) and TBHP (0.9 mmol) were added. The contents were heated with stirring at 110 °C (oil bath temperature) for 24 h. After cooling to rt, the reaction mixture was extracted with EtOAc (3 × 30 mL) and water. The combined organic phase was washed with brine solution (30 mL), dried over anh. Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using n-hexane–EtOAc (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as the eluent.
:1) mixture as the eluent.
General procedure for the ortho-acylation of 6-anilinopurine derivatives with α-oxocarboxylic acids: synthesis of compounds 3a–3h
A mixture of 6-anilinopurine (0.3 mmol), PdCl2 (10 mol%), Ag2O (0.3 mmol), K2S2O8 (0.3 mmol) and α-oxocarboxylic acid (0.6 mmol) was taken in a Schlenk tube under N2 atmosphere. To this, dioxane/AcOH/DMSO (7/2/1, v/v/v, 3 mL) mixture was added and the contents stirred at 110 °C (oil bath temperature) for 24 h. After cooling to rt, the reaction mixture was extracted with EtOAc (3 × 30 mL) and washed with water (30 mL). The combined organic phase was washed with brine solution (30 mL), dried over anh. Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography on silica gel using n-hexane–EtOAc (3:2) mixture as the eluent.
X-ray data
X-ray data for compound 2m was collected using Mo Kα (λ = 0.71073 Å) radiation. The structure was solved and refined by standard methods.17![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.249(3), b = 9.685(3), c = 14.060(5) Å, α = 94.073(6), β = 91.653(6), γ = 107.489(6), V = 1067.2(6) Å3, Z = 2, μ = 0.079 mm−1, data/restraints/parameters: 3797/0/276, R indices (I > 2σ(I)): R1 = 0.0443, wR2 (all data) = 0.1258. CCDC no. 1423000.†
, a = 8.249(3), b = 9.685(3), c = 14.060(5) Å, α = 94.073(6), β = 91.653(6), γ = 107.489(6), V = 1067.2(6) Å3, Z = 2, μ = 0.079 mm−1, data/restraints/parameters: 3797/0/276, R indices (I > 2σ(I)): R1 = 0.0443, wR2 (all data) = 0.1258. CCDC no. 1423000.†Acknowledgements
We thank Department of Science & Technology (DST, New Delhi) for financial support, single crystal X-ray diffractometer and HRMS facility, KCK thanks DST for a J. C. Bose fellowship and UGC for a one-time grant. ASR thanks CSIR (New Delhi) for a fellowship.References
- (a) Y. Deng, Y.-W. Chin, H. Chai, W. J. Keller and A. D. Kinghorn, J. Nat. Prod., 2007, 70, 2049 CrossRef CAS PubMed; (b) P. J. Harrington and E. Lodewijk, Org. Process Res. Dev., 1997, 1, 72 CrossRef CAS; (c) K. R. Romines, G. A. Freeman, L. T. Schaller, J. R. Cowan, S. S. Gonzales, J. H. Tidwell, C. W. Andrews, D. K. Stammers, R. J. Hazen, R. G. Ferris, S. A. Short, J. H. Chan and L. R. Boone, J. Med. Chem., 2006, 49, 727 CrossRef CAS PubMed; (d) H. Ogita, Y. Isobe, H. Takaku, R. Sekine, Y. Goto, S. Misawa and H. Hayashi, Bioorg. Med. Chem., 2002, 10, 3473 CrossRef CAS.
- G. A. Olah, Friedel–Crafts Chemistry, Wiley, New York, 1973 Search PubMed.
- (a) X. Jia, S. Zhang, W. Wang, F. Luo and J. Cheng, Org. Lett., 2009, 11, 3120 CrossRef CAS PubMed; (b) O. Baslé, J. Bidange, Q. Shuai and C.-J. Li, Adv. Synth. Catal., 2010, 352, 1145 CrossRef PubMed; (c) C.-W. Chan, Z. Zhou and W.-Y. Yu, Adv. Synth. Catal., 2011, 353, 2999 CrossRef CAS PubMed; (d) Y. Wu, B. Li, F. Mao, X. Li and F. Y. Kwong, Org. Lett., 2011, 13, 3258 CrossRef CAS PubMed; (e) C. Li, L. Wang, P. Li and W. Zhou, Chem.–Eur. J., 2011, 17, 10208 CrossRef CAS PubMed; (f) F. Szabó, J. Daru, D. Simkó, T. Z. Nagy, A. Stirling and Z. Novák, Adv. Synth. Catal., 2013, 355, 685 CrossRef PubMed; (g) C.-W. Chan, Z. Zhou, A. S. C. Chan and W.-Y. Yu, Org. Lett., 2010, 12, 3926 CrossRef CAS PubMed; (h) H. Li, P. Li and L. Wang, Org. Lett., 2013, 15, 620 CrossRef CAS PubMed; (i) S. Sharma, J. Park, E. Park, A. Kim, M. Kim, J. H. Kwak, Y. H. Jung and I. S. Kim, Adv. Synth. Catal., 2013, 355, 332 CAS; (j) X.-B. Yan, Y.-W. Shen, D.-Q. Chen, P. Gao, Y.-X. Li, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Tetrahedron, 2014, 70, 7490 CrossRef CAS PubMed; (k) M. Sun, L.-K. Hou, X.-X. Chen, X.-J. Yang, W. Sun and Y.-S. Zang, Adv. Synth. Catal., 2014, 356, 3789 CrossRef CAS PubMed; (l) M. Yi, X. Cui, C. Zhu, C. Pi, W. Zhu and Y. Wu, Asian J. Org. Chem., 2015, 4, 38 CrossRef CAS PubMed; (m) Q. Zhang, C. Li, F. Yang, J. Li and Y. Wu, Tetrahedron, 2013, 69, 320 CrossRef CAS PubMed; (n) A. Banerjee, S. K. Santra, S. Guin, S. K. Rout and B. K. Patel, Eur. J. Org. Chem., 2013, 1367 CrossRef CAS PubMed; (o) S. K. Santra, A. Banerjee and B. K. Patel, Tetrahedron, 2014, 70, 2422 CrossRef CAS PubMed; (p) A. Banerjee, A. Bera, S. K. Santra, S. Guin and B. K. Patel, RSC Adv., 2014, 4, 8558 RSC; (q) Q. Tian, P. He and C. Kuang, Org. Biomol. Chem., 2014, 12, 7474 RSC; (r) G. Kumar and G. Sekar, RSC Adv., 2015, 5, 28292 RSC; (s) J. Zhao, H. Fang, C. Xie, J. Han, G. Li and Y. Pan, Asian J. Org. Chem., 2013, 2, 1044 CrossRef CAS PubMed; (t) J.-H. Chu, S.-T. Chen, M.-F. Chiang and M.-J. Wu, Organometallics, 2015, 34, 953 CrossRef CAS; (u) Z.-J. Cai, C. Yang, S.-Y. Wang and S.-J. Ji, J. Org. Chem., 2015, 80, 7928 CrossRef CAS PubMed; (v) X.-F. Wu, Chem.–Eur. J., 2015, 21, 12252 CrossRef CAS PubMed.
- (a) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS PubMed; (b) M. Li and H. Ge, Org. Lett., 2010, 12, 3464 CrossRef CAS PubMed; (c) H. Li, P. Li, H. Tan and L. Wang, Chem.–Eur. J., 2013, 19, 14432 CrossRef CAS PubMed; (d) Z.-Y. Li, D.-D. Li and G.-W. Wang, J. Org. Chem., 2013, 78, 10414 CrossRef CAS PubMed; (e) J. Yao, R. Feng, Z. Wu, Z. Liu and Y. Zhang, Adv. Synth. Catal., 2013, 355, 1517 CrossRef CAS PubMed; (f) C. Pan, H. Jin, X. Liu, Y. Cheng and C. Zhu, Chem. Commun., 2013, 49, 2933 RSC; (g) H. Li, P. Li, Q. Zhao and L. Wang, Chem. Commun., 2013, 49, 9170 RSC; (h) B. Xu, W. Liu and C. Kuang, Eur. J. Org. Chem., 2014, 2576 CrossRef CAS PubMed.
- (a) F. Xiao, Q. Shuai, F. Zhao, O. Baslé, G. Deng and C.-J. Li, Org. Lett., 2011, 13, 1614 CrossRef CAS PubMed; (b) Y. Yuan, D. Chen and X. Wang, Adv. Synth. Catal., 2011, 353, 3373 CrossRef CAS PubMed; (c) H. Tang, C. Qian, D. Lin, H. Jiang and W. Zeng, Adv. Synth. Catal., 2014, 356, 519 CrossRef CAS PubMed; (d) J. Park, A. Kim, S. Sharma, M. Kim, E. Park, Y. Jeon, Y. Lee, J. H. Kwak, Y. H. Jung and I. S. Kim, Org. Biomol. Chem., 2013, 11, 2766 RSC; (e) M. Kim, S. Sharma, J. Park, M. Kim, Y. Choi, Y. Jeon, J. H. Kwak and I. S. Kim, Tetrahedron, 2013, 69, 6552 CrossRef CAS PubMed; (f) L. Hou, X. Chen, S. Li, S. Cai, Y. Zhao, M. Sun and X.-J. Yang, Org. Biomol. Chem., 2015, 13, 4160 RSC; (g) Q. Zhang, F. Yang and Y. Wu, Tetrahedron, 2013, 69, 4908 CrossRef CAS PubMed.
- (a) S. Guin, S. K. Rout, A. Banerjee, S. Nandi and B. K. Patel, Org. Lett., 2012, 14, 5294 CrossRef CAS PubMed; (b) Z. Xu, B. Xiang and P. Sun, RSC Adv., 2013, 3, 1679 RSC; (c) Z. Yin and P. Sun, J. Org. Chem., 2012, 77, 11339 CrossRef CAS PubMed; (d) Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC; (e) J. Weng, Z. Yu, X. Liu and G. Zhang, Tetrahedron Lett., 2013, 54, 1205 CrossRef CAS PubMed; (f) F. Xiong, C. Qian, D. Lin, W. Zeng and X. Lu, Org. Lett., 2013, 15, 5444 CrossRef CAS PubMed; (g) H. Song, D. Chen, C. Pi, X. Cui and Y. Wu, J. Org. Chem., 2014, 79, 2955 CrossRef CAS PubMed; (h) Y. Zheng, W.-B. Song, S.-W. Zhang and L.-J. Xuan, Tetrahedron, 2015, 71, 1574 CrossRef CAS PubMed.
- Q. Zhang, F. Yang and Y. Wu, Chem. Commun., 2013, 49, 6837 RSC.
- (a) G. Zhang, S. Sun, F. Yang, Q. Zhang, J. Kang, Y. Wu and Y. Wu, Adv. Synth. Catal., 2015, 357, 443 CrossRef CAS PubMed; (b) A. Behera, W. Ali, S. Guin, N. Khatun, P. R. Mohanta and B. K. Patel, RSC Adv., 2015, 5, 33334 RSC.
- J. Lu, H. Zhang, X. Chen, H. Liu, Y. Jiang and H. Fu, Adv. Synth. Catal., 2013, 355, 529 CrossRef CAS PubMed.
- W. Zhou, H. Li and L. Wang, Org. Lett., 2012, 14, 4594 CrossRef CAS PubMed.
- S. Han, S. Sharma, J. Park, M. Kim, Y. Shin, N. K. Mishra, J. J. Bae, J. H. Kwak, Y. H. Jung and I. S. Kim, J. Org. Chem., 2014, 79, 275 CrossRef CAS PubMed.
- (a) M. K. Lakshman, A. C. Deb, R. R. Chamala, P. Pradhan and R. Pratap, Angew. Chem., Int. Ed., 2011, 50, 11400 CrossRef CAS PubMed; (b) H.-M. Guo, L.-L. Jiang, H.-Y. Niu, W.-H. Rao, L. Liang, R.-Z. Mao, D.-Y. Li and G.-R. Qu, Org. Lett., 2011, 13, 2008 CrossRef CAS PubMed; (c) H.-M. Guo, W.-H. Rao, H.-Y. Niu, L.-L. Jiang, G. Meng, J.-J. Jin, X.-N. Yang and G.-R. Qu, Chem. Commun., 2011, 47, 5608 RSC; (d) R. R. Chamala, D. Parrish, P. Pradhan and M. K. Lakshman, J. Org. Chem., 2013, 78, 7423 CrossRef CAS PubMed; (e) H. J. Kim, M. J. Ajitha, Y. Lee, J. Ryu, J. Kim, Y. Lee, Y. Jung and S. Chang, J. Am. Chem. Soc., 2014, 136, 1132 CrossRef CAS PubMed; (f) A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS PubMed; (g) M. A. Ali, X. Yao, H. Sun and H. Lu, Org. Lett., 2015, 17, 1513 CrossRef CAS PubMed.
- (a) R. Rama Suresh and K. C. Kumara Swamy, J. Org. Chem., 2012, 77, 6959 CrossRef PubMed; (b) S. Allu and K. C. Kumara Swamy, J. Org. Chem., 2014, 79, 3963 CrossRef CAS PubMed; (c) R. N. P. Tulichala and K. C. Kumara Swamy, Chem. Commun., 2015, 51, 12008 RSC; (d) S. Allu and K. C. Kumara Swamy, Adv. Synth. Catal., 2015, 357, 2665 CrossRef CAS PubMed.
- (a) K. C. Kumara Swamy, S. Allu, V. Srinivas, E. Balaraman and K. V. P. Pavan Kumar, Cryst. Growth Des., 2011, 11, 2302 CrossRef CAS; (b) K. C. Kumara Swamy, E. Balaraman and N. Satish Kumar, Tetrahedron, 2006, 62, 10152 CrossRef PubMed.
- (a) V. Gayakhe, Y. S. Sanghvi, I. J. S. Fairlamb and A. R. Kapdi, Chem. Commun., 2015, 51, 11944 RSC; (b) A. Nayak, G. Chandra, I. Hwang, K. Kim, X. Hou, H. O. Kim, P. K. Sahu, K. K. Roy, J. Yoo, Y. Lee, M. Cui, S. Choi, S. M. Moss, K. Phan, Z.-G. Gao, H. Ha, K. A. Jacobson and L. S. Jeong, J. Med. Chem., 2014, 57, 1344 CrossRef CAS PubMed; (c) M. Legraverend and D. S. Grierson, Bioorg. Med. Chem., 2006, 14, 3987 CrossRef CAS PubMed; (d) V. E. Marquez and M.-I. Lim, Med. Res. Rev., 1986, 6, 1 CrossRef CAS PubMed.
- D. D. Perrin, W. L. F. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon, Oxford, 1986 Search PubMed.
- (a) G. M. Sheldrick, SADABS, Siemens Area Detector Absorption Correction, University of Göttingen, Germany, 1996 Search PubMed; (b) G. M. Sheldrick, SHELX-97 – A program for crystal structure solution and refinement, University of Göttingen, 1997 Search PubMed; (c) G. M. Sheldrick, SHELXTL NT Crystal Structure Analysis Package, Analytical X-ray System, Version 5.10, Bruker AXS, WI, USA, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra. CCDC 1423000. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra18447a |
| This journal is © The Royal Society of Chemistry 2015 |