DOI:
10.1039/C5RA16741H
(Communication)
RSC Adv., 2015,
5, 92596-92601
Targeted delivery of a novel peptide–docetaxel conjugate to MCF-7 cells through neuropilin-1 receptor: reduced toxicity and enhanced efficacy of docetaxel†
Received
19th August 2015
, Accepted 22nd October 2015
First published on 22nd October 2015
Abstract
We have designed a novel peptide–docetaxel conjugate, which delivers docetaxel specifically to cancer cell targeting neuropilin-1 (NRP-1) receptor, thus enhancing its efficacy and acts more aggressively in breast cancer cells.
Delivery of therapeutic molecules in a targeted fashion to the tumor site is extremely important for the treatment of cancer, because it helps in increasing the drug concentration to the tumor site and reduces the side effects. For this purpose, understanding the molecular signature of the tumor site and development of specific ligands to target these molecular signatures are crucial in the development of potential therapeutics.1 Docetaxel (DX), approved by the FDA as a therapeutic molecule, shows anticancer activity against various cancers including breast and prostate.2 DX affects normal microtubule dynamic instability by targeting β-tubulin to promote microtubule polymerization,3 and bipolar spindle structure to induce mitotic block in proliferative cancer cells.4 Although DX is used as chemotherapeutic agent for various cancers, the efficacy of this drug is poor and shows non-selective toxicity towards to normal cell.5 Further, when DX was used in combination with prednisone in chemotherapy to improve survival efficacy, that resulted in insignificant improvement with increasing toxicities.2e,6 To overcome these issues, many groups are trying to improve the efficacy and selectivity of DX to tumor site through development of new delivery systems.7 However, all these attempts did not show significant success in terms of target specific DX delivery to minimize dose concentration and nonspecific toxicity. For this purpose, further extensive research is necessary for the development of a high potential novel DX-conjugate, which can deliver DX in a targeted manner to specific cancer cell. In this work, for the first time, we designed a novel DX–peptide conjugate targeting neuropilin-1 (NRP-1) receptor. NRP-1, a transmembrane protein, known as a receptor for vascular endothelial growth factor (VEGF) and member of class 3 semaphorin protein family, plays a crucial role in cell internalization pathway.8 Moreover, NRP-1 is over-expressed in various advanced stage tumor cell and plays a significant role in tumor progression, but it is absent in normal cell.9 Here, we report that a short peptide, CGNKRTR (tLyP-1), which is known to penetrate tumor cell through an NRP-1 dependent CendR internalization pathway,10 conjugated with DX exhibiting reduced toxicity and enhanced efficacy. LyP-1 (CGNKRTRGC) is a cyclic tumor homing peptide bound with cell surface receptor protein p32 in tumor, undergoes proteolytic cleavage into the truncated form of CGNKRTR, known as tLyP-1. However, tLyP-1 has less binding affinity to the p32 receptor, but can directly bind to the NRP-1 receptor on the cancer cell surface and for subsequent internalization.10 Accordingly, we used truncated form of Lyp1, CGNKRTR (tLyP-1) for conjugation with DX. We synthesized CGNKRTR peptide using Wang resin in a microwave peptide synthesizer. DX was conjugated with maleimide in presence of catalytic amount of dimethyl amino pyridine (DMAP). DX and DMAP were taken in a round bottle flask containing dichloromethane and stirred at 0 °C for 30 minutes under nitrogen atmosphere. Subsequently, 3-(maleimido) propionic acid N-hydroxysuccinimide ester was added to the mixture and stirred for overnight at 4 °C under nitrogen atmosphere, which resulted in maleimido modified docetaxel (MDX). MDX was further conjugated with CGNKRTR peptide through a covalent conjugation of thiol group (–SH) of the N-terminal Cys with the maleimido group to produce peptide modified DX (PMDX) (Fig. 1a, Scheme 1, ESI†). Similarly, FITC-PMDX was prepared by a conjugation of FITC-CGNKRTR peptide with MDX through a covalent interaction between thiol (–SH) of the peptide and the maleimido group of MDX (ESI†). All compounds were purified through HPLC followed by characterization through NMR and MALDI-TOF-MS (ESI, Fig. S1–S5†).
 |
| | Fig. 1 (a) Synthetic scheme of MDX and PMDX from DX. Cartoon representation of tLyP-1 peptide conjugated with DX binding with over-expressed NRP-1 receptor in cancer cell. (b) FACS data showing high expression of NRP-1 receptor in cell surface of MCF-7 cell. (c) FACS data showing absence of NRP-1 receptor in cell surface of HEK293T cell. | |
First, we have confirmed the over-expression of NRP-1 in MCF-7 cells (human breast cancer) compare to HEK293T (human embryonic kidney cells) cells by FACS (Fig. 1b and c and S6†). Thus, in this manuscript we have used MCF-7 and HEK293T to compare between the cancer and normal cell line. It is described before that CGNKRTR peptide binds with the NRP-1 receptor and helps in cellular internalization process.10 Therefore, we have found the binding of FITC-PMDX on the cell membrane of MCF7 cells (Fig. S7†). Next, we have also showed the binding of PMDX with NRP-1 by docking study (Fig. 2a and b and S8†). Here, we have used Lys-Pro-Arg (KPR) peptide, derived from tuftsin peptide known to bind with NRP-1, to compare the binding site of our PMDX.11 The analysis showed that the carboxyl end group of PMDX interacting with Thr349, Ser346, Tyr353, Glu348 and Trp301 of NRP-1 through H-bonding (Fig. 2b and S8†). The docking study further suggested that the PMDX bound to the NRP-1 receptor in a similar fashion of KPR peptide and also followed the C-end rule (CendR). Efficacy of PMDX and DX were evaluated through a cell viability assay in MCF-7 cells, where NRP-1 receptor was over-expressed. MCF-7 cells treated with DX and PMDX for 4 h, at 125, 250, 500 and 1000 nM concentrations, were found to have differences in the cell viability of 3.7%, 6.1%, 9.2% and 18.7%, respectively, indicating that the percent viability of MCF-7 cells was higher in DX treated cells compared to the PMDX treated cells in different concentrations. Although differences in cell viability at lower concentration are less, at higher concentration differences are significant. This is due to the fact that PMDX is a targeted drug conjugate and thus uptake was enhanced at higher concentration. This interesting result clearly indicates that PMDX is more potent than DX (Fig. 2c). As an initial control experiment, we also examined the toxicity of CGNKRTR peptide and found that this peptide is non-cytotoxic to the MCF-7 cells (Fig. 2d). Next, we examined whether NRP-I dependent cellular internalization of PMDX is the only path for entering the PMDX into MCF-7 cells or not, using blocking experiments. For this purpose, MCF-7 cells were incubated with CGNKRTR peptide for 2 h and then treated with different concentrations of PMDX for 24 h and found that killing of MCF-7 cells was not significant and percent cell viability also not changed with increasing concentration of PMDX. This interesting result indicates that incubation of MCF-7 cells with CGNKRTR peptide resulted in a saturation of binding of CGNKRTR peptide with NRP-1 receptor on the MCF-7 cell surface, which inhibits further binding of CGNKRTR peptide attached with MDX. This experiment further suggested that internalization of PMDX occurred exclusively through NRP-1 dependent pathway (Fig. 2d). Thus, PMDX appears to be more selective and a targeted pro-drug of DX. To check the toxicity of PMDX compared to the DX, we performed cell viability assay in normal HEK293T cell line. We observed that the differences in percent viability of HEK293T cells after treatment with DX and PMDX for 24 h at 12.5, 25, 50 and 100 nM concentrations were 65.5%, 67.6%, 65.8% and 69.1%, respectively (Fig. 2e). This result indicate poor cellular uptake of PMDX compared to the DX in HEK293T cells. This is due to the low abundance of NRP-1 receptor on the HEK293T cells, indicating that PMDX is less toxic to normal cells compared to DX (Fig. 2e). We also performed cellular uptake study in MCF-7 and HEK293T cells after treatment with 10 μM solution of FITC-PMDX for 1 h. We found 2.5 times higher uptake of PMDX in MCF-7 cells compared to the HEK293T cells (Fig. 2f and S9†), suggesting that CGNKRTR peptide helps in cellular internalization of PMDX through NRP-1 receptors in MCF-7 cells. We have also analyzed the targeted ability of PMDX in other cell lines such as A549 (NRP1+), MDA-MB-231 (NRP1+, ER−, HER2−) and MDA-MB-453 (NRP1−, ER−, HER2+) cell lines. Interestingly, we have found that PMDX shows more cytotoxicity compared to the DX in case of NRP-1 positive cell lines whereas it shows less cytotoxicity compared to the DX in case of NRP-1 negative cell line. These results are similar as observed in case of MCF-7 (NRP1+, ER+, HER2−) and HEK293T (NRP1−) cell (Fig. S10†). The above experimental results fully addressed the limitation of DX in terms of non-specificity towards targeted tumor cell through peptide conjugation with DX at in vitro cellular level. Excellent selectivity of PMDX in cancer cell death compared to normal cell motivated us to investigate that whether PMDX affects intracellular biological activity similar like DX or not. For this purpose, we decided to investigate the effect of DX and PMDX on microtubule network, cell cycle, pathways of cell death, p53 and p21 protein regulation in MCF-7 cells, after treatment with DX and PMDX. It is known that DX binds with β-tubulin, which perturbs normal intracellular microtubule dynamics,3 and induces mitotic arrest of cancer cells.4 Now, the question is whether PMDX also affects microtubule networks in similar manner or not. We observed a bundle like structure of microtubule network of MCF-7 cells after treatment with DX and PMDX (Fig. 3 and 4). This indicates that PMDX retains its activity as mitotic inhibitor similar like DX, since we know that DX inhibits mitosis through stabilization of microtubule and forms intracellular microtubule bundle like structures (Fig. 3, S11 and S12†). Next, we incubated MCF-7 cells with FITC-PMDX for 12 h and observed a similar result as earlier. Interestingly, we observed that there was no co-localization of green and red signal indicating that DX became free from the conjugate after entering into the MCF-7 cell (Fig. 4).
 |
| | Fig. 2 (a) Image showing PMDX (green and blue stick structure) bound on the surface of NRP-1 (brown colour) receptor. (b) Image showing binding partners between PMDX and NRP-1, dotted line indicating H-bonding between amino acids of both PMDX and NRP-1. (c) Comparison of cell viability of DX and PMDX in MCF-7 cells (p < 0.05; N = 6; N = number of experiments and p = statistical significance). MCF-7 cells were treated with DX and PMDX for 4 h, followed by the media was changed and incubated for another 44 h in fresh media without the drug. (d) Cell viability of CGNKRTR, PMDX in MCF-7 cells after blocking with peptide and without blocking with peptide (p < 0.05; N = 6). MCF-7 cells were treated for 24 h. (e) Comparison of cell viability of DX and PMDX in HEK293T cells (p < 0.05; N = 6). HEK293T cells were treated with DX and PMDX for 24 h, (f) comparison of cellular uptake of 10 μM solution of FITC-PMDX in MCF-7 and HEK293T cells after 1 h incubation. | |
 |
| | Fig. 3 Microtubule network of MCF-7 cells after treatment with 50 nM solution of DX and PMDX for 24 h. MCF-7 cells were treated with monoclonal anti-alpha tubulin (EP1332Y) as a primary antibody which specifically can bind to the alpha tubulin of cellular microtubules. Images were taken in 40× objective mode of a fluorescence microscope. Threads of intracellular microtubules were not prominent after treatment with both DX and PMDX due to the formation of bundles of intracellular microtubules. Scale bar corresponds to 20 μm. | |
 |
| | Fig. 4 Microtubule network of MCF-7 cells after treatment with 10 μM solution of FITC-PMDX for 12 h. (a) Image at 488 nm channel indicating uptake of green coloured FITC-PMDX. (b) Red colored bundle like structure of microtubule at 561 nm channel indicating uptake of PMDX and its interaction. (c) Merged image indicating that DX was released from PMDX after internalization, as we did not observe co-localization of green colored segment with red colored microtubule bundles. MCF-7 cells were treated with monoclonal anti-alpha tubulin (EP1332Y) as a primary antibody which specifically binds to the alpha tubulin of cellular microtubules. Images are captured under a confocal microscope. Scale bar corresponds to 20 μm. | |
Next, we compared the effect of DX and PMDX on inhibition of cell growth by studying the cell cycle of MCF-7 cells. We found that cells were arrested in G2/M phase after treatment with DX and PMDX. In case of PMDX, percentage of cells in G0/G1 phase was decreased by 3.2 unit compared to DX and 3.8 unit increase in percentage of cells in G2/M + S phase. This data further confirmed that PMDX retained its activity as mitotic inhibitor like DX, but PMDX was more potent than DX, as we observed that 3.8 units higher population of MCF-7 cells in the G2/M + S phase compared to DX (Fig. 5). As we know that DX induces the activation of p53 protein,12 we were interested to compare the activation level of p53 of MCF-7 cells after treatment with DX and PMDX. From immunoblotting experiment, we have observed higher expression of p53 in PMDX treated MCF-7 cells in compared to DX treated cells. In contrast, lesser expression of p21 was observed in case of PMDX treated cells in compared to that of DX treated cells. Further, the protein expression has been quantified and normalized against loading control (α-tubulin). This result shows a two fold rise in p53 protein expression in PMDX in compared to DX treated MCF7 cell. Whereas, p21 protein relatively less expressed in case of PMDX treated cells. These results indicate higher accumulation of oncogenic repressor protein p53 in MCF-7 cell after treatment with same concentration of PMDX in compared to DX (Fig. 6). Further, we have evaluated its cellular localization using immunocytochemistry experiment. From the microscopic images, we found that the red fluorescence was localized on nucleus of both DX and PMDX treated cells, indicating the activation of p53 after treatment with both compounds. Interestingly, higher red fluorescence was observed in case of PMDX treated cells compared to DX indicating high activation of p53 in case of PMDX treated MCF-7 cells (Fig. 7a, b and S13–S15†). This result also suggests that CGNKRTR peptide conjugation with DX increased the efficacy in MCF-7 cells. Further, we studied the activation of p21 protein in MCF-7 cells after treatment with DX and PMDX following similar method described above. We observed that p21 activation similar like p53 (Fig. 7a and b, S13, S14 and S16†).
 |
| | Fig. 5 FACS analysis of cell-cycle of MCF-7 cells after treatment with 50 nM DX and PMDX for 24 h. Treated and control MCF-7 cells were fixed using 70% ice cold ethanol. The cells were washed by PBS and incubated with a solution of PI and RNase A at room temperature for 45 min, before analysis. The working concentration of PI and RNase A was 100 μg mL−1 and 10 μg mL−1 respectively. | |
 |
| | Fig. 6 (a) Immunoblotting experiment showing activation of p53 and p21 proteins in MCF-7 cell after treatment with PMDX and DX. (b) Bar diagram shows higher expression of p53 protein and lesser expression of p21 protein in MCF-7 cell after treatment with PMDX compared to the DX (p < 0.05). | |
 |
| | Fig. 7 (a) Activation of p53 and p21 in MCF-7 cells after treatment with 50 nM DX for 24 h. (b) Activation of p53 and p21 in MCF-7 cells after treatment with 50 nM PMDX for 24 h. Scale bar corresponds to 20 μm. | |
We studied the cellular death pathway of MCF-7 cell after treatment with DX and PMDX. For that purpose, initially we examined through microscope about the binding of annexin V-FITC and internalization of propidium iodide (PI) into the MCF-7 cells after treatment with DX and PMDX. We observed green and red signal in both DX and PMDX (Fig. S17†) treated cells. Subsequently, we performed apoptosis assay using annexin V-FITC and propidium iodide (PI) by flow cytometry. We treated MCF-7 cells with DX and PMDX followed by treatment with annexin V-FITC and PI for FACS analysis. FACS data showed that 2.25 times of high population of apoptotic cell death in case of PMDX compared to DX (Fig. 8a–d). The combined data concluded that PMDX induced apoptosis of MCF-7 cells in more potent fashion than DX.
 |
| | Fig. 8 FACS analysis of MCF-7 cells after treatment with annexin V-FITC and propidium iodide. (a) Control cells, (b) cells treated with DX, (c) cells treated with PMDX. (d) Bar diagram represents comparison of healthy (Q3) and apoptotic cells (Q4 + Q2) population (p < 0.05). MCF-7 cells were treated with 1000 nM solution of DX and PMDX for 4 h, after that media was changed and incubated for another 44 h in fresh media without the drug. At the time of analysis cells were incubated with annexin V and PI for 15 min at 37 °C. | |
We observed that novel PMDX is highly specific and potential in 2D cancer cells those have higher NRP1 receptor on cell surface. However, successful drug in monolayer cell culture often fail to show activity in in vivo system.13 To address this issue, we have used 3D spheroid model, which has been recently being emphasized for screening the effect of the therapeutic molecules after evaluation in 2D cell culture system.13 Further this model is found to be relevant for therapeutic evaluation, as it mimics the in vivo model both in structural and molecular aspects.14 Thus, we have interested to check whether our novel conjugate of DX will affect the growth of in vivo like MCF-7 spheroid model which mimic tumor complex microenvironment. In brief, we have developed the spheroid culture system for MCF-7 using liquid overlay technique and growth kinetics of this spheroid has been monitored in each day interval up to 10 days.15 Effect of the PMDX has been monitored accordingly against only DX and untreated control. The spheroids were treated with drugs from second day. Volume of the spheroids was taken as parameter to analyze the spheroids growth kinetics. In case of DX treated group, we observed, significant reduction in spheroids growth after 6th day and gradually shrink up to 10th day. This is due to the lower penetration or availability of bare DX at different layer of the spheroid system. Interestingly, we observed inhibition of the growth after 2nd day (or 24 h) of incubation with PMDX. This result indicates that, the peptide modification of the DX, not only results in increased cellular uptake of DX, but also enhances its distribution all around the spheroid 3D complex environment, resulting both higher and earlier growth delay in comparison to the bare DX (Fig. 9a and b). Thus we observed that the PMDX has more anti-cancer potential in both monolayer cells and in vivo tumor mimicking 3D spheroid cells.
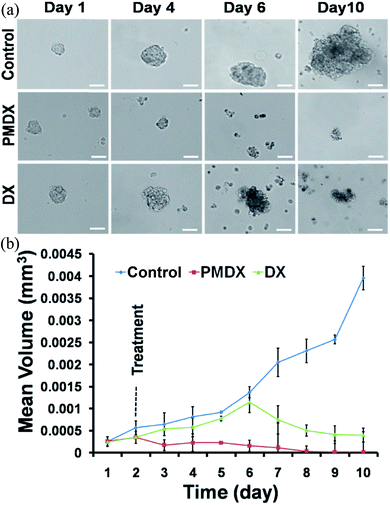 |
| | Fig. 9 (a) Bright field images of MCF-7 tumor spheroid. (b) Graphical representation shows the size reduction of tumor spheroid after treatment with PMDX and DX with respect to control (p < 0.05). | |
Conclusions
In summary, we have synthesized a novel PMDX conjugate for targeted and selective delivery of DX specific to cancer cells through NRP-1 receptor. This conjugate exhibited superior potential than DX in cell cytotoxicity, cell cycle arrest at G2/M phase, activation of p53 and p21 proteins. Further, we also showed that enhanced apoptotic death of MCF-7 cells by PMDX compared to DX. Finally, we have shown that PMDX acts better than DX in inhibiting the growth of tumor mimicking 3D spheroid model. We envision that our novel PMDX conjugate will function as a better and selective anticancer drug for in vivo experiments.
Acknowledgements
Authors wish to thank CSIR-IICB for FACS facility, NCCS-Pune, Dr D. Biswas and Mr M. K. Ghosh, IICB Kolkata for cell lines, Dr P. Chakrabarti for accessing the lab facility and Dr R. Natarajan for reading the manuscript. AS, PK, BJ and PM thank CSIR, SM thanks UGC, and SG thanks DST-Ramanujan for their fellowships, respectively. SG kindly acknowledges DST, India (SR/SO/BB-0102/2012) for financial assistance.
Notes and references
-
(a) E. Ruoslahti, Nat. Rev. Cancer, 2002, 2, 83–90 CrossRef PubMed;
(b) E. Ruoslahti, Drug Discovery Today, 2002, 7, 1138–1143 CrossRef CAS PubMed;
(c) E. Ruoslahti, S. N. Bhatia and M. J. Sailor, J. Cell Biol., 2010, 188, 759–768 CrossRef CAS PubMed.
-
(a) G. I. Georg, T. C. Boge, Z. S. Cheruvallath, J. S. Clowers, G. C. B. Harriman, M. Hepperle and H. Park, TAXOL®: Science and Applications, CRC Press, New York, 1995, pp. 317–378 Search PubMed;
(b) G. I. Georg, G. C. B. Harriman, D. G. Vander Velde, T. C. Boge, Z. S. Cheruvallath, A. Datta, M. Hepperle, H. Park, R. H. Himes and L. Jayasinghe, Taxane Anticancer Agents: Basic Science and Current Status, American Chemical Society, Washington DC, 1995, pp. 217–231 Search PubMed;
(c) D. Guénard, F. Guéritte-Vogelein and P. Potier, Acc. Chem. Res., 1993, 26, 160–167 CrossRef;
(d) M. Suffness, Taxol: Science and Applications, CRC Press, New York, 1995 Search PubMed;
(e) D. P. Petrylak, C. M. Tangen, M. H. Hussain, P. N. Lara, J. A. Jones, M. E. Taplin, P. A. Burch, D. Berry, C. Moinpour, M. Kohli, M. C. Benson, E. J. Small, D. Raghavan and E. D. Crawford, N. Engl. J. Med., 2004, 351, 1513–1520 CrossRef CAS PubMed;
(f) I. F. Tannock, R. de Wit, W. R. Berry, J. Horti, A. Pluzanska, K. N. Chi, S. Oudard, C. Théodore, N. D. James, I. Turesson, M. A. Rosenthal and M. A. Eisenberger, N. Engl. J. Med., 2004, 351, 1502–1512 CrossRef PubMed.
- M. J. Piccart, Bull. Mem. Acad. R. Med. Belg., 1998, 153, 285–292 CAS , discussion 92.
- K. S. Cunha, M. L. Reguly, U. Graf and H. H. de Andrade, Mutagenesis, 2001, 16, 79–84 CrossRef CAS PubMed.
- U. Vaishampayan and M. Hussain, Expert Rev. Anticancer Ther., 2008, 8, 269–281 CrossRef CAS PubMed.
- W. W. Tan, Cancer Control: Journal of the Moffitt Cancer Center, 2006, 13, 194–198 Search PubMed.
-
(a) S. Sundaram, C. Durairaj, R. Kadam and U. B. Kompella, Mol. Cancer Ther., 2009, 8, 1655–1665 CrossRef CAS PubMed;
(b) F. Esmaeili, M. H. Ghahremani, S. N. Ostad, F. Atyabi, M. Seyedabadi, M. R. Malekshahi, M. Amini and R. Dinarvand, J. Drug Targeting, 2008, 16, 415–423 CrossRef CAS PubMed;
(c) H. Kim, Y. Lee, I. H. Lee, S. Kim, D. Kim, P. E. Saw, J. Lee, M. Choi, Y. C. Kim and S. Jon, J. Controlled Release, 2014, 178, 118–124 CrossRef CAS PubMed;
(d) N. Goodarzi, M. H. Ghahremani, M. Amini, F. Atyabi, S. N. Ostad, N. ShabaniRavari, N. Nateghian and R. Dinarvand, Chem. Biol. Drug Des., 2014, 83, 741–752 CrossRef CAS PubMed.
-
(a) S. Takagi, T. Tsuji, T. Amagai, T. Takamatsu and H. Fujisawa, Dev. Biol., 1987, 122, 90–100 CrossRef CAS PubMed;
(b) S. Soker, S. Takashima, H. Q. Miao, G. Neufeld and M. Klagsbrun, Cell, 1998, 92, 735–745 CrossRef CAS PubMed;
(c) A. L. Kolodkin, D. V. Levengood, E. G. Rowe, Y. T. Tai, R. J. Giger and D. D. Ginty, Cell, 1997, 90, 753–762 CrossRef CAS PubMed;
(d) T. Teesalu, K. N. Sugahara, V. R. Kotamraju and E. Ruoslahti, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16157–16162 CrossRef CAS PubMed.
-
(a) L. M. Ellis, Mol. Cancer Ther., 2006, 5, 1099–1107 CrossRef CAS PubMed;
(b) D. R. Bielenberg, C. A. Pettaway, S. Takashima and M. Klagsbrun, Exp. Cell Res., 2006, 312, 584–593 CrossRef CAS PubMed;
(c) Y. Cao, L. Wang, D. Nandy, Y. Zhang, A. Basu, D. Radisky and D. Mukhopadhyay, Cancer Res., 2008, 68, 8667–8672 CrossRef CAS PubMed.
- L. Roth, L. Agemy, V. R. Kotamraju, G. Braun, T. Teesalu, K. N. Sugahara, J. Hamzah and E. Ruoslahti, Oncogene, 2012, 31, 3754–3763 CrossRef CAS PubMed.
- C. W. Vander Kooi, M. A. Jusino, B. Perman, D. B. Neau, H. D. Bellamy and D. J. Leahy, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6152–6157 CrossRef CAS PubMed.
- H. H. ndez-Vargas, J. Palacios and G. Moreno-Bueno, Oncogene, 2007, 26, 2902–2913 CrossRef PubMed.
- F. Hirschhaeuser, H. Menne, C. Dittfeld, J. West, W. Mueller-Klieser and L. A. Kunz-Schughart, J. Biotechnol., 2010, 148, 3–15 CrossRef CAS PubMed.
- J. Friedrich, C. Seidel, R. Ebner and L. A. Kunz-Schughart, Nat. Protoc., 2009, 4, 309–324 CrossRef CAS PubMed.
- J. B. do Amaral, P. Rezende-Teixeira, V. M. Freitas and G. M. Machado-Santelli, Tissue Eng., Part C, 2011, 17, 1097–1107 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra16741h |
| ‡ These authors contribute equally. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 








