DOI:
10.1039/C5RA16137A
(Paper)
RSC Adv., 2015,
5, 92292-92302
cRGD-functionalized redox-sensitive micelles as potential doxorubicin delivery carriers for αvβ3 integrin over expressing tumors
Received
11th August 2015
, Accepted 20th October 2015
First published on 21st October 2015
Abstract
Polymeric micelles as nanocarrier-based drug delivery systems provide an innovative platform for selectively delivering active molecules and offer better antitumour activity. However, circulation stability in vivo, controllable drug intracellular release and high targeting efficiency are several practical challenges for micelles. Therefore, we developed cRGD-modified and shell crosslinked micelles (RSCMs) based on amphiphilic poly(styrene-co-maleic anhydride) (SMA). RSCMs exhibited a spherical shape and were homogeneous with an average diameter of 106.1 nm and a low polydispersity of 0.087. In comparison, after crosslinking the micelles possessed better stability against dilution and stronger redox-response towards dithiothreitol (DTT). The SMA micelles displayed high drug loading capacity for hydrophobic DOX with a drug loading of approximately 14.1–19.2% (w/w) and an encapsulation efficiency of 72.1–82.7% (w/w). The in vitro release studies of shell crosslinked micelles showed that DOX release was minimal (<25%, 24 h) under physiological conditions. However, in the presence of 10 mM DTT, accelerated release of DOX was achieved (60%, 24 h). MTT assays in B16F10 cells indicated that RSCMs displayed low cytotoxicity up to a concentration of 500 μg ml−1. Moreover, the IC50 value showed that cRGD-DOX-ss-M could be more effective than other groups. In vitro, cellular uptake was further studied with confocal laser scanning microscopy and flow cytometry, both the qualitative and quantitative results demonstrated that cRGD-DOX-ss-M possessed much better specificity to cancer cells and superior stimulated release property in cytoplasm. Notably, cRGD-DOX-ss-M could more efficiently be delivered to and release into the nuclei of αvβ3 integrin over expressing tumor cell line (B16F10) than counterparts of integrin-deficient tumor cells (Hela). Thus, these cRGD-modified redox-sensitive micelles have appeared as a technology platform with great potential for targeted anticancer drug delivery and release for integrin over expressing tumor cells.
1. Introduction
Chemotherapy is still the main treatment strategy for a broad range of solid tumors.1 However, the clinical efficacy of therapies and survival benefit of the patient are limited by disadvantages of chemotherapeutic drugs such as low bioavailability, dose-limiting toxicities or drug delivery barriers.2 Nanocarrier-based drug delivery systems (DDS) provide an innovative platform for selectively delivering active molecules and offer better antitumor activity,3 among which polymeric micelles have emerged as a promising system, and have been widely studied in preclinical4,5 and clinical trials.6 Clinical studies have demonstrated that polymeric micelles overcome some limitations and delivered various drugs such as paclitaxel, doxorubicin or cisplatin6–8 with remarkable antitumor efficacy. However, stability in vivo,9,10 controllable drug-release profiles11 and high targeting efficiency of the tumor site12–14 present several practical challenges for micelles as drug delivery systems.
To improve circulation stability of polymeric micelles and prevent drug leakage in blood circulation, the micelles could be crosslinked through a reversible bond under tumor microenvironment stimuli,15 such as pH,16,17 redox signals18–21 and multiple stimuli,18,22 which remain circulation stability and control activated drugs release in response to specific stimuli. Among the stimuli, redox potential is often used to trigger the intracellular drug delivery. Especially, disulfide crosslinking provides an opportunity for triggered drug to release due to the existence of abundant reducing substances including glutathione (GSH) in cancer cell,23 which has recently been explored for reversible stabilization of polymeric micelles.24 On the other hand, strategy to achieve cancer-targeted drug delivery is the utilization of unique cell-surface molecules markers that are specifically overexpressed in the cancerous tissues.25 One such molecule such as the ανβ3 integrin, which can specifically recognize the peptide containing RGD (Arg-Gly-Asp) sequence, is a promising strategy for tumor-targeting treatment.26 RGD receptors are overexpressed by tumor vessels and by a wide variety of human carcinoma cells including melanoma, breast, prostate, pancreas and colon. For example, RGD peptide analogues functionalized therapeutic systems such as micelles,27,28 liposome29 and nanoparticle carriers30,31 have improved therapeutic efficiency and tumor selectivity.
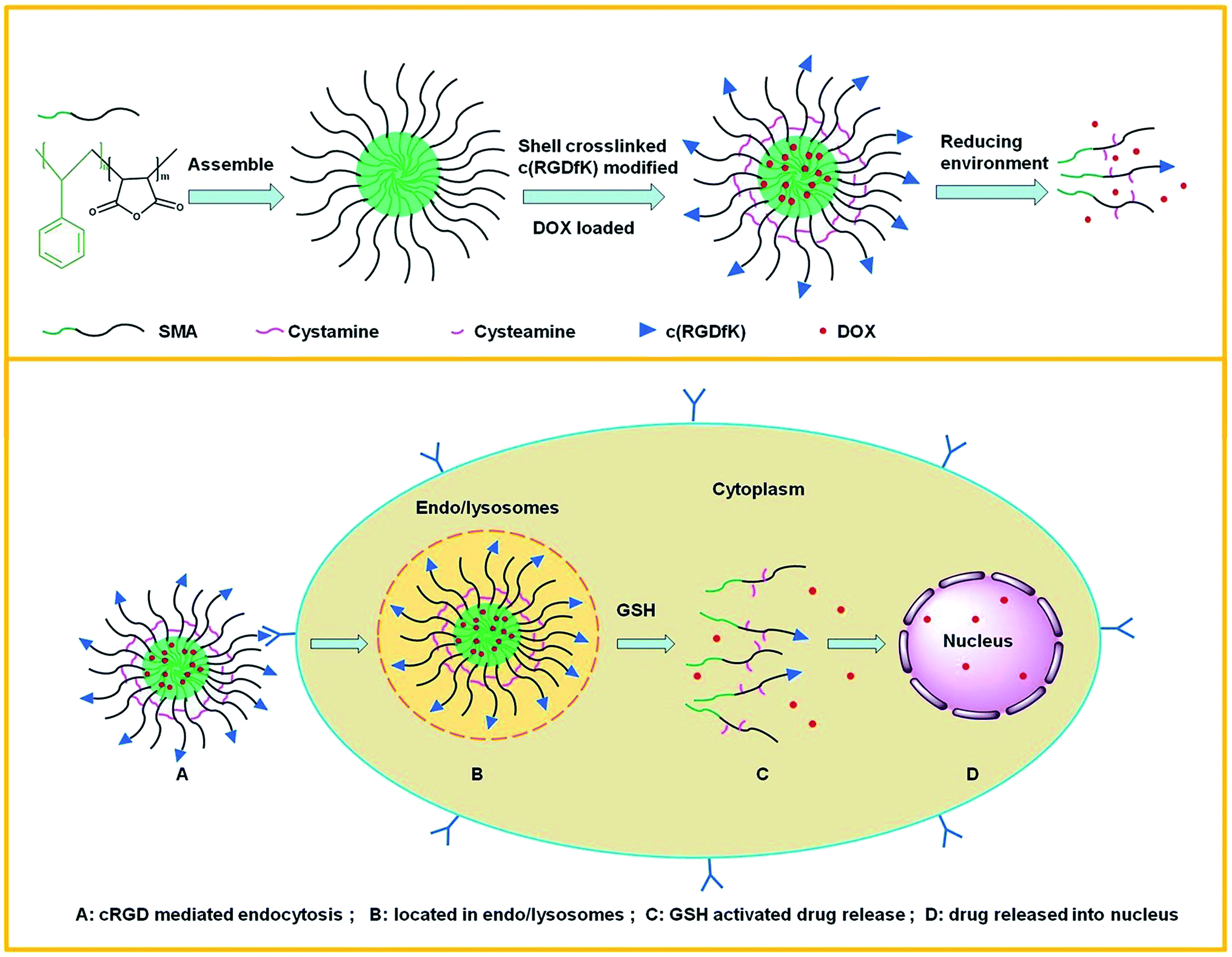
In order to fully exert targeted antitumor effect and controllable drug-release of the drug delivery, we build RGD peptide analogues functionalized redox crosslinking polymeric micelles. Herein, we selected a cyclic pentapeptide c(Arg-Gly-Asp-d-Phe-Lys) (cRGDfK or named cRGD), which can selectively bind to ανβ3 integrin with high affinity,27,28 liganding to the surface of the micelles polymer. Hydrophobic doxorubicin (DOX) as a model drug was encapsulated into poly(styrene-co-maleic anhydride) micelles which were further crosslinked by cystamine to improve circulation stability and achieve redox-sensitive intracellular delivery (Fig. 1). Although the coupling of RGD peptides modified nanocarriers were found to be able to target tumor blood vessels,26 several strategies including circulation stability, controllable drug intracellular release and high targeting efficiency used in one polymeric micelles delivery system has not been reported. In this work, we evaluated the physicochemical properties of the cRGD-functionalized polymer micelles, and investigated stability against dilution, drug-release profile in response to the reducing environment of tumor cells. The effects of the micelles on cellular uptake were investigated in murine malignant melanoma cell line (B16F10) (ανβ3 integrin-positive) and human cervix adenocarcinoma cell line (Hela) (ανβ3 integrin-negative). It was expected that the cRGD-modified and redox-sensitive shell crosslinked micelles could increase the stability of the micelles in circulation, control drug intracellular release and enhance the targeted delivery of DOX into αvβ3 integrin over expressing tumor cells.
 |
| | Fig. 1 Schematic illustration of cRGD-functionalized redox-sensitive core-crosslinked SMA micelles for active loading and intracellular microenvironment triggered release of DOX. | |
2. Materials and methods
2.1 Materials
Poly(styrene-co-maleic anhydride) (SMA, molecular weight 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 760 Da) with a molar styrene to maleic anhydride ratio of 5:1 in the backbone was purchased from Polyscope Polymers Co, Ltd, (Geleen. Netherlands). c(RGDfK) peptide was synthesized by Shanghai ABBiocheem Co, Ltd, (Shanghai, China). Pyrene and 1,4-dithiothreitol (DTT) were purchased from Shanghai Aladdin Chemistry Co, Ltd, (Shanghai, China). Doxorubicin hydrochloride (DOX-HCl) was obtained from Xianghe Shunda fine chemical Co, Ltd, (Wuhan, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI) and penicillin streptomycin were purchased from Biosharp (Hefei, China). RPMI-1640 and DMEM medium was purchased from Hyclone (Utah, USA). Trypsin was purchased from Gibco-BRL Life Technologies (Carlsbad, CA). Fetal bovine serum was purchased from Sijiqing Biology Engineering Materials Co, Ltd, (Zhejiang, China). All other chemicals used were analytical grade and used without further treatment.
760 Da) with a molar styrene to maleic anhydride ratio of 5:1 in the backbone was purchased from Polyscope Polymers Co, Ltd, (Geleen. Netherlands). c(RGDfK) peptide was synthesized by Shanghai ABBiocheem Co, Ltd, (Shanghai, China). Pyrene and 1,4-dithiothreitol (DTT) were purchased from Shanghai Aladdin Chemistry Co, Ltd, (Shanghai, China). Doxorubicin hydrochloride (DOX-HCl) was obtained from Xianghe Shunda fine chemical Co, Ltd, (Wuhan, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI) and penicillin streptomycin were purchased from Biosharp (Hefei, China). RPMI-1640 and DMEM medium was purchased from Hyclone (Utah, USA). Trypsin was purchased from Gibco-BRL Life Technologies (Carlsbad, CA). Fetal bovine serum was purchased from Sijiqing Biology Engineering Materials Co, Ltd, (Zhejiang, China). All other chemicals used were analytical grade and used without further treatment.
2.2 Cell cultures
The mouse malignant melanoma cell line (B16F10) and human cervix adenocarcinoma cell line (Hela) were purchased from American Type Culture Collection (ATCC). The cells were cultured in RPMI-1640 and DMEM medium respectively containing 10% (v/v) fetal bovine serum (FBS) and 1% antibiotics (penicillin G, 100 unit per ml; streptomycin, 100 unit per ml) at 37 °C in a 5% CO2 atmosphere. At intervals, cells were passaged using 0.25% trypsin–EDTA solution before confluence.
2.3 Micellar preparations
2.3.1 Preparation of SMA micelles and redox-sensitive shell crosslinked micelles. The non-crosslinked micelles (NCMs) were prepared by the conventional dialysis method.32 Briefly, SMA (40 mg) was dissolved in 2 ml N,N′-dimethylformamide (DMF) and the solution (20 mg ml−1) was added dropwise to 20 ml ultra-pure water under stirring conditions. After the mixture was stirred for 1 h at room temperature (r.t.), DMF was isolated by dialysis (MWCO 14000) against 1 L deionized water for 24 h. The dialysis medium was refreshed at least 3 times. The shell crosslinked micelles (SCMs) were fabricated by the crosslinking of cystamine dihydrochloride. The molar ratio of maleic anhydride, cystamine dihydrochloride and sodium bicarbonate was 1:0.25:1. Firstly, 20 ml NCMs were catalytic hydrolysed by sodium bicarbonate (2.52 mg, 0.03 mM). After the suspension was stirred for 30 min, cystamine dihydrochloride (1.69 mg, 0.0075 mM) was added and further reacted for 12 h. Finally, the micelles were dialyzed against deionized water for 24 h to remove redundant crosslinker.
2.3.2 Preparation of cRGD-modified crosslinked micelles. The cRGD-modified crosslinked micelles (RSCMs) were prepared by one-step self-assembly method. Briefly, the NCMs was crosslinked by cystamine dihydrochloride as previously described. The molar ratio of maleic anhydride and cRGD peptide was 8:1, cRGD peptide (2.32 mg, 0.004 mM, 0.125 equiv. versus maleic anhydride) was added to the crosslinked micelles and incubated at 4 °C for 12 h. After incubation, the cRGD-modified crosslinked micelles were purified by dialysis.
2.3.3 Preparation of DOX-loaded micelles. DOX was chosen as a model drug to assess the DOX loaded NCMs (DOX-M), SCMs (DOX-ss-M) and RSCMs (cRGD-DOX-ss-M). The DOX-loaded polymeric micelles were prepared by dialysis method, while the whole procedure was performed in the dark. Typically, 2 ml of DOX-HCl solution in DMF (5.0 mg ml−1) was pretreated by 20 μl of triethylamine for 6 hours. Then SMA (20 mg ml−1) and DOX (5 mg ml−1) dissolved in DMF were added dropwise to 20 ml ultra-pure water, stirring for 5 h at r.t. to allow for drug encapsulation. In the end, the mixture was dialyzed to eliminate unload DOX and the organic solvent. Then DOX-loaded micelles were crosslinked and modified as described in 2.3.1 and 2.3.2.
2.4 Characteristics of micelles
2.4.1 Determination of critical micellar concentration. The critical micellar concentration (CMC) of RSCMs was determined using pyrene as a fluorescent probe. The concentration of the block polymer was varied from 1 × 10−5 to 1 mg ml−1 and the concentration of pyrene was fixed at 0.6 μM. Fluorescence spectra were recorded using a fluorescence spectrometer (PerkinElmer LS55, USA) with an excitation wavelength of 334 nm. The emission fluorescence at 373 and 384 nm was monitored. The CMC was estimated at the crosspoint when extrapolating the intensity ratio I384/I373 in low and high concentration regions.
2.4.2 Measurement of zeta potential, size distribution and TEM. The size and zeta potential of micelles were determined using dynamic light scattering (DLS). Measurements were carried out at 25 °C by a Zetasizer Nano-ZS90 (Malven, Worcestershire, UK) with a 632.8 nm He–Ne laser using back-scattering detection. The shape of micelles was observed using transmission electron microscopy (TEM, JEOL JEM-2100F, Japan) at an accelerating voltage of 200 kV. The samples were prepared by casting one drop of micelle suspension on carbon-coated copper grids, followed by staining with 1% phosphotungstic acid.
2.4.3 Stability of non-crosslinked micelles and shell crosslinked micelles. Physical stability of the NCMs and SCMs were investigated by DLS. The samples were diluted by organic solvent (DMF, 1:10) and extensive dilution (water, 1:5000), their size were measured by dynamic light scattering (DLS). The redox-responsiveness evaluation of SCMs in responsive to a reductive environment in vitro was monitored by DLS measurement. The sample solution at a concentration below the CMC was bubbled with N2 for 15 min. Then, DTT was added to yield the final DTT concentration of 10 mM, the operation was in a shaking bed at 200 rpm and 37 °C. The change of micelle size was determined by DLS at predetermined time intervals.
2.4.4 Detection of drug loading content and drug loading efficiency. To determine drug loading content (DLC) and drug loading efficiency (DLE), various DOX formulations were centrifuged at 10000 rpm for 30 minutes to collect sediments and washed off the surface residual DOX. Then the sediments were freeze-dried to obtain the weights of DOX loaded micelles. The freeze-dried powders were dissolved in DMSO and determined by measuring the fluorescent intensity (excitation: 482 nm, emission: 556 nm) using a fluorescence spectrophotometer-(PerkinElmer LS55, USA). Wherein, calibration curve was obtained with DOX/DMSO solutions with different DOX concentrations. The DLC and DLE were calculated according to the following equations:
| DLC (%) = (weight of loaded DOX/weight of DOX loaded micelles) × 100% |
| DLE (%) = (weight of loaded DOX/weight of added DOX) × 100% |
2.5 In vitro drug-release behaviors of DOX-loaded micelles
The in vitro release of DOX from DOX-ss-M was investigated at 37 °C in 0.1 M phosphate buffer solution (PBS, pH 7.4) with or without 10 mM DTT. Typically, 1 ml DOX-ss-M solution was transferred to a dialysis tube with a MWCO of 8000–14000 and then immersed into either 25 ml PBS with no DTT or 10 mM DTT, continuously shaken (200 rpm) at 37 °C. At desired time intervals, 3 ml of the solution was obtained from the reservoir for fluorescence measurement (excitation: 480 nm, emission: 570 nm) and 3 ml of fresh media were replenished. The release experiments were conducted in triplicate, and the results presented are the average data with standard deviations. In a similar way, the release of DOX from DOX-M in PBS was also determined.
2.6 In vitro cytotoxicity assay
The cytotoxicity of blank micelles (NCMs and RSCMs), DOX-loaded micelles (DOX-M and cRGD-DOX-ss-M) and free DOX solution were performed by MTT assay using B16F10 cells. Cells were seeded onto 96-well plates at a density of 8000 cell per well in 100 μL of RPMI-1640 containing 10% FBS and incubated for 24 h to reach 80% confluency. The medium was removed with a fresh medium containing the empty micelles, DOX-loaded micelles and free DOX with varying concentrations. After 24 h or 48 h, the medium was replaced by 100 μL fresh RPMI-1640 and 20 μL of MTT solution (5 mg ml−1) was added. After incubation for another 4 h, 150 μL of DMSO was added and shaken for 10 min. The absorbance at 570 nm of each well was measured using a microplate reader (BioTek Epoch, USA). The cell viability (%) was determined by comparing the absorbance with control wells containing only cell culture medium. Data are presented as average ± SD (n = 4). The IC50 value (the concentration that inhibited cell growth by 50%) of different formulation was calculated using GraphPad Prism software.
2.7 In vitro cellular uptake
The cellular uptake and intracellular release behaviors of the DOX-loaded micelles were investigated by confocal laser scanning microscopy (CLSM) and flow cytometry (FCM) against B16F10 cells (ανβ3 integrin-positive cells) and Hela cells (ανβ3 integrin negative cells). When examined by CLSM, B16F10 cells or Hela cells, which have been cultured in a confocal dish and incubated with different DOX-loaded micelles and free DOX. After different incubation time intervals (2 h, 6 h), the culture medium was removed and the cells were washed three times with PBS. Then the cells were fixed with 4% paraformaldehyde for 15 min and cell nuclei were stained with DAPI for 10 min. Confocal microscopy images were taken using CLSM (Perkin Elmer, UltraVIEW VoX, USA).
For FCM analysis, B16F10 cells or Hela cells were seeded in 6-well plates (105 cells per well). After presetting incubation, the cells were collected by trypsin and centrifuged at 1000 rpm for 5 min, washed with cold PBS twice, and resuspended in 300 μL of cold PBS. Finally, the cell suspensions were filtered through 300-mesh nylon mesh and analyzed for fluorescent intensity with FCM (BD AccuriC6, USA) through the fluorescence channel 2 (FL2).
2.8 Statistical analysis
Data was expressed as means ± the standard deviation (SD) obtained from three separate experiments. The significances of the differences were determined using Student's t-test two-tailed for each paired experiment. A P-value < 0.05 was considered statistically significant in all cases.
3. Results and discussion
3.1 Micellization and characterization
A series of measurements were employed to verify the formations of the micelles self-assembled from amphiphilic SMA copolymers. Firstly, the NCMs, SCMs and RSCMs had a narrow size distribution in water with polydispersity index (PDI, <0.10) and an average diameter about 100 nm determined from the DLS measurement, no obvious change in size and PDI after crosslinking by cystamine or cRGD modification were observed (Table 1). Zeta potentials of the NCMs, SCMs and RSCMs determined by DLS were −26.1 mV, −33.4 mV and −31.5 mV, respectively, indicating favorable stability (Table 1). The slight decrease in zeta potential after crosslinking might be due to the existence of a small number of free carboxyl groups on the surface. Moreover, the negative surface charges of the micelles could reduce clearance by reticulo-endothelial system (RES) and prolong circulation time due to the low absorption of plasma proteins.33 The TEM images showed that the RSCMs exhibited a spherical shape and homogeneous distribution (Fig. 2a and b), which was consistent with the results obtained from DLS. Using pyrene as a fluorescence probe, the CMC of the SMA micelles was determined to be 2.5 μg ml−1 (Fig. 2d), which guarantee the micelle to keep a favorable stability in bodily fluids before reaching tumor sites.
Table 1 Size and zeta potential of blank micelles determined by DLS
| Sample |
Size (nm) |
PDI |
Zeta potential (mV) |
| NCMs |
104.50 ± 2.13 |
0.093 ± 0.025 |
−26.1 ± 1.8 |
| SCMs |
95.43 ± 4.58 |
0.073 ± 0.041 |
−33.4 ± 1.9 |
| RSCMs |
106.12 ± 3.75 |
0.087 ± 0.041 |
−31.5 ± 3.2 |
 |
| | Fig. 2 (a) and (b) TEM image of RSCMs at different magnification. (c) Size distribution of RSCMs by DLS. (d) CMC of RSCMs derived from the plot of I384/I373 ratio vs. micelle concentration using pyrene as a probe. | |
The stability of NCMs and SCMs against extensive dilution and organic solvent were investigated using DLS. As shown in Fig. 3a, the apparent increase in size for NCMs could be observed, wherein the size of the NCMs increased from 100 nm to 160 nm after 5000 fold dilution with water and swelled to 460 nm after 10 fold dilution with DMF. In contrast, no obvious size change was observed for SCMs upon the same volume dilution (Fig. 3b). The results suggested that SCMs could retain the micellar structure even at concentrations below the CMC or oily reagent, which is highly favored for bioapplications of micelles. In the process of intravenous administration, polymer micelles were usually susceptible to dilution below CMC, which may lead to the dissociation of micelles.34 Therefore, the stability of the system is very important during infusion. In addition, after long-term storage at r.t. for over 6 month, there were no significant changes of SCMs in the average particle size, which demonstrated long-term stability of the SCMs.
 |
| | Fig. 3 Comparison of stability of NCMs (a) and SCMs (b) against dilution by water (5000 fold) and DMF (10 fold). (c) Redox-induced size variation of SCMs in response to 10 mM DTT with time followed by DLS. | |
To verify the redox-responsive cleavage of the disulfide bond in SCMs, the change of SCMs size in response to 10 mM DTT was monitored by DLS at various time intervals. As shown in Fig. 3c, the size change was observed after 15 min, in which large aggregates with a diameter about 550 nm, and the originally unimodal peak changed into multimodal peaks in the size distribution. As time went on, the portion that represented aggregation (550 nm) increased over time. Such variation in the size was likely due to the reductive cleavage to disassemble the micelles. These results indicated that the SMA micelles crosslinked with the disulfide was able to decrosslink under a reducing environment.
The basic requirements for polymeric micelles as drug-delivery systems include high drug-loading capacity, biodegradability and controllable drug-release profiles. DOX is one of the most potent antitumor agents to treat a wide variety of solid malignant tumors by interacting with topoisomerase II, which was used as a model anti-cancer drug to evaluate the capability of the SMA micelles as a drug carrier. The SMA micelles displayed high drug loading capacity of 19.2 ± 2.1% (w/w) for hydrophobic DOX and encapsulation efficiency of 82.7 ± 1.7% (w/w). By comparison, the drug loading capacity of SMA micelles was higher than that of previously reported polymer micelles such as PEG-PCL based micelles35 and chitosan-based polymeric micelles.36 It might be due to that high proportion styrene of SMA could form larger hydrophilic core to load DOX through physically trapping. After crosslinking by cystamine or cRGD modification, the loading efficiency and the entrapment efficiency of DOX-ss-M and cRGD-DOX-ss-M had only slightly decreased to 14.1 ± 1.5%, 15.3 ± 2.8% and 72.1 ± 2.2%, 75.6 ± 3.1% (Table 2). However, the DOX-loaded micelles exhibited smaller size than blank micelles (76.11–80.34 nm & 95.43–106.12 nm), which may owe to hydrophobic interaction between insolubility DOX and the hydrophobic cores of polymeric micelles (Tables 1 and 2). It should be noted that micelles with a small size of less than 100 nm is more likely to avoid the elimination from RES and promote the EPR effect.37,38
Table 2 Characteristics of DOX-loaded micelles
| Sample |
Size (nm) |
PDI |
Zeta potential (mV) |
DLC (%) |
DLE (%) |
| DOX-M |
80.34 ± 4.32 |
0.169 ± 0.032 |
−29.1 ± 2.3 |
19.2 ± 2.1 |
82.7 ± 1.7 |
| DOX-ss-M |
69.64 ± 5.56 |
0.178 ± 0.029 |
−37.6 ± 3.5 |
14.1 ± 1.5 |
72.1 ± 2.2 |
| cRGD-DOX-ss-M |
76.11 ± 3.77 |
0.176 ± 0.052 |
−35.5 ± 2.2 |
15.3 ± 2.8 |
75.6 ± 3.1 |
3.2 In vitro drug-release behaviors of DOX-loaded micelles
The DOX release curves from the formulations (DOX-M, DOX-ss-M) were investigated in PBS (0.1 M, pH 7.4) with or without 10 mM DTT at 37 °C. In the absence of DTT (Fig. 4), DOX-M quickly released with a 50.0% cumulative release of DOX within 8 h, followed by a slow release up to 24 h reaching about 60.0% cumulative release. The rapid release at the physiological environment (pH 7.4, PBS) due to unstability of micelles would lead to drug leakage before reaching the tumor site, therefore induce side effects and reduce therapeutic effects, which is a major challenge for micelles as drug delivery in vivo.22 By comparison, the release of DOX from the DOX-ss-M was less than 25% cumulative release under the same condition over a period of 24 h. This remarkable decrease implied that the shell crosslinking of the micelles could maintain the micellar structure to entrap the drug in the hydrophobic region to prevent DOX leakage. However, in the presence of 10 mM DTT, the release rate of DOX-ss-M obviously increased, in which approximately 60% of DOX was released within 8 h. It thereby corroborated our hypothesis that DOX-ss-M could be selectively released at the tumor site under a reducing environment, which resulted from reductive cleavage of disulfide bonds.39
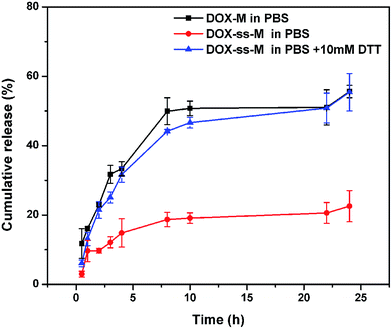 |
| | Fig. 4 In vitro release of DOX in the presence or absence of 10 mM DTT from DOX-ss-M and DOX-M. Data are presented as mean ± SD (n = 3). | |
3.3 In vitro cytotoxicity assay
Murine melanoma B16F10 cells which overexpress integrin αvβ3,26 was used as the models of tumor neovascular endothelial cells. The biocompatibility of the blank micelles and the cytotoxicity of DOX-loaded micelles against B16F10 cells were determined by MTT assays. In Fig. 5a and b, both NCMs and RSCMs exhibited no obvious cytotoxicity (cell viabilities ≥ 80%) up to a tested concentration of 500 μg ml−1 when incubation for 24 h and 48 h, indicating that the micelles as drug delivery vehicles have excellent biocompatibility and low-toxicity.
 |
| | Fig. 5 Cytotoxicity of blank micelles (NCMs and RSCMs), DOX-loaded micelles (DOX-M and cRGD-DOX-ss-M) and free DOX using B16F10 cells. (a) Blank micelles, 24 h incubation, (b) blank micelles, 48 h incubation, (c) drug loaded micelles and free DOX, 24 h incubation, (d) drug loaded micelles and free DOX, 48 h incubation. Data are presented as mean ± SD (n = 4). | |
To evaluate whether the SMA micelles formulations influenced the cytotoxicity of DOX, the viability of B16F10 cells incubated with DOX-loaded micelles (DOX-M, cRGD-DOX-ss-M) and free DOX for 24 h and 48 h were determined by MTT. As shown in Fig. 5c and d, both free DOX and DOX-loaded micelles caused a dose-dependent and time-dependent increase in cytotoxicity. When the concentration of DOX increased from 0.01 to 10 μg ml−1, the viability of B16F10 cells incubated after 24 and 48 h with free DOX decreased from 96% to 35% and 89% to 20%, while those treated with cRGD-DOX-ss-M decreased from 95% to 11% and 82% to 4%, respectively. As expected, the viability of B16F10 cells incubated with cRGD-DOX-ss-M exhibited significantly higher toxicity than free DOX either after 24 or 48 h. The different cytotoxicity might result from the difference in their cellular uptake. Furthermore, after treatment for 48 h, the IC50 values of cRGD-DOX-ss-M (0.1807 μg ml−1), which was 1.6 and 0.5 fold lower than the values of free DOX (0.4696 μg ml−1) and DOX-M (0.2763 μg ml−1) respectively. The enhanced cytotoxicity of cRGD-DOX-ss-M was attributed to cRGD peptide-mediated endocytosis and more drug release via the stimuli of reducing environment of tumor cell.
3.4 In vitro cellular uptake
CLSM was firstly employed to qualitatively observe the internalization process as a function of time in the ανβ3 integrin-positive cells (B16F10) and ανβ3 integrin negative cells (Hela) cultured with various DOX formulations (Fig. 6). At the same incubation time, the DOX fluorescence intensity in B16F10 and Hela cells was enhanced when DOX was loaded into micelles (Fig. 6). This difference was likely due to easier access of small particles (70–80 nm) to the cells.38 From all the overlapped color in the merged image, the red fluorescence from various DOX-loaded micelles was visibly observed mainly in cell cytoplasm at first (Fig. 6a and c), but eventually the fluorescence spreading to the nuclei and distinctly increased (Fig. 6b and d). This may be due to that DOX-loaded micelles needed to release in the cytosol, followed by diffusion into the nuclei. Both in B16F10 and Hela cells, the redox-responsive DOX-ss-M showed higher fluorescence than DOX-M, owed to the facilitated release of DOX responding to intracellular reducing environment. Moreover, the ανβ3 integrin-positive B16F10 cells treated with cRGD-DOX-ss-M exhibit significantly higher cellular uptake than DOX-ss-M, while not in ανβ3 integrin negative Hela cells. It was a powerful proof which indicated the enhanced cellular uptake via high affinity of cRGD peptide to over-expressed ανβ3 integrin.
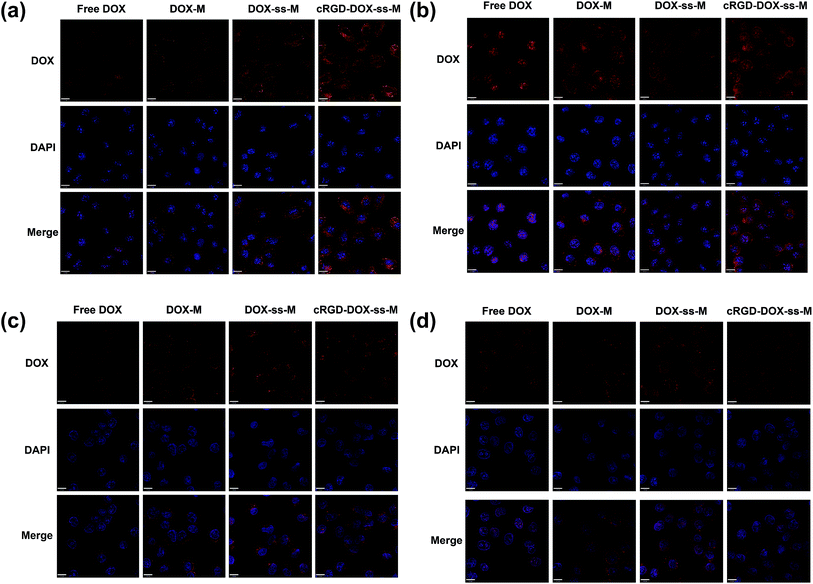 |
| | Fig. 6 Confocal laser scanning microscope images of B16F10 cells (ανβ3 integrin-positive) ((a) 2 h, (b) 6 h) and Hela cells (ανβ3 integrin-negative) ((c) 2 h, (d) 6 h) that were treated with free DOX and DOX-loaded micelles. Nuclei were stained with DAPI. DAPI staining images and DOX images were merged. | |
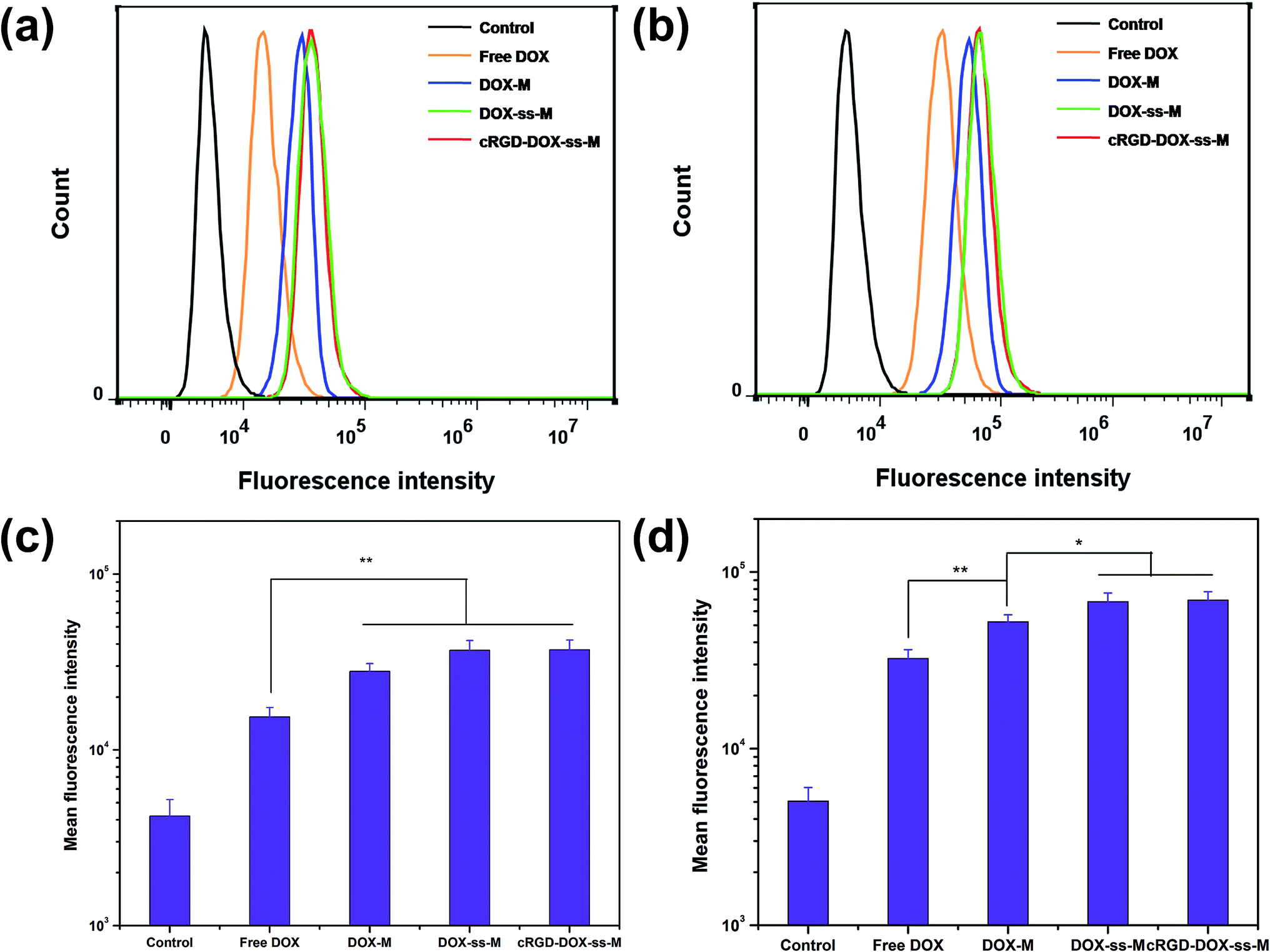
As expected, the quantitative flow cytometry analysis (FACS) results were also in agreement with the CLSM result. Cells without any DOX treatment only showed the auto fluorescence, so they were presented as the negative control. Both in B16F10 cells (Fig. 7) and Hela cells (Fig. 8), compared to that without redox-responsive micelles, redox-responsive micelles revealed higher fluorescence (DOX-ss-M & DOX-M, cRGD-DOX-ss-M & DOX-M). Furthermore, from Fig. 7a and b, cRGD-DOX-ss-M presented the strongest DOX fluorescence in B16F10 cells after 2 h and 6 h incubation, confirming again that the roles of integrin-mediated process in the cellular uptake and cellular redox response in the drug release. Fig. 7c and d demonstrated the quantitative determination of the fluorescence, we could find that the cellular fluorescence intensity in cRGD-DOX-ss-M treated cells was 1.28 and 1.53 times that of DOX-ss-M and DOX-M treated cells after 2 h, 1.60 and 2.42 times that of DOX-ss-M and DOX-M treated cells after 6 h, respectively. While Hela cells incubated with the cRGD-DOX-ss-M and DOX-ss-M showed equivalent fluorescence intensity after same time interval (Fig. 8), which is well consistent with the CLSM assay. From all cellular uptake results, we concluded that the cRGD peptide-decorated redox-responsive polymeric micelles, cRGD-DOX-ss-M, had the ability to convey anticancer drug to target cancer cells and enhance the intracellular release.
 |
| | Fig. 7 Flow cytometry analyses and mean fluorescence intensity contrasting the level of cellular uptake between B16F10 cells incubated with free DOX and various DOX loaded micelles for 2 h ((a) and (c)) and 6 h ((b) and (d)). Statistical significance:*P < 0.05, **P < 0.01. | |
 |
| | Fig. 8 Flow cytometry analyses and mean fluorescence intensity contrasting the level of cellular uptake between Hela cells incubated with free DOX and various DOX loaded micelles for 2 h ((a) and (c)) and 6 h ((b) and (d)). Statistical significance:*P < 0.05, **P < 0.01. | |
4. Conclusions
In summary, we have demonstrated that cRGD-modified and shell-crosslinked micelles (RSCMs) based on amphiphilic polymer SMA could efficiently deliver and redox-responsed release DOX into cancer cells with αvβ3 integrin over expressing, achieving preferable antitumor effect. The terminal groups of hydrophobic chains in this copolymer were modified with c(RGDfK) peptide, resulting in a high selectivity to tumor cells that αvβ3 integrin over expressing; some hydrophobic segments were crosslinked by cystamine dihydrochloride, endowing an optimal redox-sensitivity to stimulate the intracellular drug release. A series of measurements demonstrated that the intelligent micelles possessed desirable features, including favourable stability, high drug-loading capacity, low cytotoxicity, and controlled release property. Notably, both the cytotoxicity assay and cellular uptake analysis revealed these active targeting and redox-responsive micelles had excellent killing effect on B16F10 cells, due to the rapid deliver to the target cells and abundant release in intracellular microenvironment. Therefore, the cRGD-decorated redox-sensitive micelles have the potential to be developed as an effective targeted drug delivery system for cancer chemotherapy.
Acknowledgements
The authors would be supported by the National Natural Science Foundation of China (81201197), Hubei Province Natural Science Fund Project (2015CFB588 and 2014CFA080), and Talents Program from Hubei University of Technology (BSQD12049). The authors thank Yanghao (Ph.D.) at Huazhong University of Science and Technology, China for the helpful discussion.
Notes and references
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- H. Cabral, J. Makino, Y. Matsumoto, P. Mi, H. L. Wu, T. Nomoto, K. Toh, N. Yamada, Y. Higuchi, S. Konishi, M. R. Kano, H. Nishihara, Y. Miura, N. Nishiyama and K. Kataoka, ACS Nano, 2015, 9, 4957–4967 CrossRef CAS PubMed.
- T. Wei, J. Liu, H. L. Ma, Q. Cheng, Y. Y. Huang, J. Zhao, S. D. Huo, X. D. Xue, Z. C. Liang and X. J. Liang, Nano Lett., 2013, 13, 2528–2534 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- R. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104, 593–598 CrossRef CAS PubMed.
- T. Hamaguchi, T. Doi, T. Eguchi-Nakajima, K. Kato, Y. Yamada, Y. Shimada, N. Fuse, A. Ohtsu, S. Matsumoto, M. Takanashi and Y. Matsumura, Clin. Cancer Res., 2010, 16, 5058–5066 CrossRef CAS PubMed.
- Y. H. Bae and H. Yin, J. Controlled Release, 2008, 131, 2–4 CrossRef CAS PubMed.
- S. C. Owen, D. P. Y. Chan and M. S. Shoichet, Nano Today, 2012, 7, 53–65 CrossRef CAS.
- F. H. Meng, R. Cheng, C. Deng and Z. Y. Zhong, Mater. Today, 2012, 15, 436–442 CrossRef CAS.
- S. Bhatnagar and V. V. K. Venuganti, J. Nanosci. Nanotechnol., 2015, 15, 1925–1945 CrossRef CAS.
- L. Zhao, N. Li, K. Wang, C. Shi, L. Zhang and Y. Luan, Biomaterials, 2014, 35, 1284–1301 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021–3035, 10.1039/b701217a.
- X. Guo, C. Shi, J. Wang, S. Di and S. Zhou, Biomaterials, 2013, 34, 4544–4554 CrossRef CAS PubMed.
- D. H. Kim, Y. K. Seo, T. Thambi, G. J. Moon, J. P. Son, G. Li, J. H. Park, J. H. Lee, H. H. Kim, D. S. Lee and O. Y. Bang, Biomaterials, 2015, 61, 115–125 CrossRef CAS PubMed.
- Y. J. Wu, D. F. Zhou, Y. X. Qi, Z. G. Xie, X. S. Chen, X. B. Jing and Y. B. Huang, RSC Adv., 2015, 5, 31972–31983 RSC.
- Y. G. Su, Y. W. Hu, Y. Z. Du, X. Huang, J. B. He, J. You, H. Yuan and F. Q. Hu, Mol. Pharmaceutics, 2015, 12, 1193–1202 CrossRef CAS PubMed.
- J. Li, T. Yin, L. Wang, L. Yin, J. Zhou and M. Huo, Int. J. Pharm., 2015, 483, 38–48 CrossRef CAS PubMed.
- Y. W. Hu, Y. Z. Du, N. Liu, X. Liu, T. T. Meng, B. L. Cheng, J. B. He, J. You, H. Yuan and F. Q. Hu, J. Controlled Release, 2015, 206, 91–100 CrossRef CAS PubMed.
- Y. P. Li, K. Xiao, W. Zhu, W. B. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS PubMed.
- P. Kuppusamy, H. Li, G. Ilangovan, A. J. Cardounel, J. L. Zweier, K. Yamada, M. C. Krishna and J. B. Mitchell, Cancer Res., 2002, 62, 307–312 CAS.
- L. L. Wu, Y. Zou, C. Deng, R. Cheng, F. H. Meng and Z. Y. Zhong, Biomaterials, 2013, 34, 5262–5272 CrossRef CAS PubMed.
- G. J. Chen, L. W. Wang, T. Cordie, C. Vokoun, K. W. Eliceiri and S. Q. Gong, Biomaterials, 2015, 47, 41–50 CrossRef CAS PubMed.
- F. Danhier, A. Le Breton and V. Preat, Mol. Pharmaceutics, 2012, 9, 2961–2973 CrossRef CAS PubMed.
- C. Y. Zhan, B. Gu, C. Xie, J. Li, Y. Liu and W. Y. Lu, J. Controlled Release, 2010, 143, 136–142 CrossRef CAS PubMed.
- M. Cai, M. Y. Ye, X. X. Shang, H. H. Sun, M. X. Liu, H. M. Sun, Z. Ma and H. D. Zhu, RSC Adv., 2015 10.1039/c5ra16137a.
- C. Rangger, A. Helbok, E. von Guggenberg, J. Sosabowski, T. Radolf, R. Prassl, F. Andreae, G. C. Thurner, R. Haubner and C. Decristoforo, Int. J. Nanomed., 2012, 7, 5889–5900 CrossRef CAS PubMed.
- E. A. Murphy, B. K. Majeti, L. A. Barnes, M. Makale, S. M. Weis, K. Lutu-Fuga, W. Wrasidlo and D. A. Cheresh, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9343–9348 CrossRef CAS PubMed.
- F. Danhier, B. Vroman, N. Lecouturier, N. Crokart, V. Pourcelle, H. Freichels, C. Jerome, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 140, 166–173 CrossRef CAS PubMed.
- X. P. Duan, J. S. Xiao, Q. Yin, Z. W. Zhang, H. J. Yu, S. R. Mao and Y. P. Li, Nanotechnology, 2014, 25, 125102 CrossRef PubMed.
- X. P. Duan and Y. P. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- X. X. Chuan, Q. Song, J. L. Lin, X. H. Chen, H. Zhang, W. B. Dai, B. He, X. Q. Wang and Q. Zhang, Mol. Pharmaceutics, 2014, 11, 3656–3670 CrossRef CAS PubMed.
- N. Nasongkla, X. Shuai, H. Ai, B. D. Weinberg, J. Pink, D. A. Boothman and J. M. Gao, Angew. Chem., Int. Ed., 2004, 43, 6323–6327 CrossRef CAS PubMed.
- L. L. Cai, P. Liu, X. Li, X. Huang, Y. Q. Ye, F. Y. Chen, H. Yuan, F. Q. Hu and Y. Z. Du, Int. J. Nanomed., 2011, 6, 3499–3508 CAS.
- C. L. Shi, X. Guo, Q. Q. Qu, Z. M. Tang, Y. Wang and S. B. Zhou, Biomaterials, 2014, 35, 8711–8722 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. H. Meng, C. Deng, J. Feijen and Z. Y. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.