
On the constitution and thermodynamic modelling of the system Ti–Ni–Sn†
M. Gürthabc,
A. Grytsivabc,
J. Vrestald,
V. V. Romakae,
G. Giesterf,
E. Bauerbc and
P. Rogl*ab
aInstitute of Material Chemistry and Research, University of Vienna, Währingerstrasse 42, A-1090 Wien, Austria
bChristian Doppler Laboratory for Thermoelectricity, Wien, Austria. E-mail: peter.franz.rogl@univie.ac.at
cInstitute of Solid State Physics, TU-Wien, Wiedner Hauptstrasse, 8-10, A-1040 Wien, Austria
dMasaryk University, CEITEC, Kamenice 753/5, Brno, Czech Republic
eDepartment of Materials Science and Engineering, Lviv Polytechnic National University, Ustiyanovycha Str. 5, 79013 Lviv, Ukraine
fInstitute of Mineralogy and Crystallography, University of Vienna, Althanstrasse 14, A-1090 Wien, Austria
First published on 14th October 2015
Abstract
Phase equilibria of the system Ti–Ni–Sn have been determined for the isothermal section at 950 °C based on X-ray powder diffraction (XPD) and electron probe microanalysis (EPMA) of about 60 ternary alloys in as cast and annealed state. The section is characterized by the formation of four ternary compounds labelled τ1 to τ4. Whereas two of the ternary compounds are found without significant homogeneity regions: τ1-TiNiSn (half Heusler phase, MgAgAs-type), τ3-Ti2Ni2Sn (U2Pt2Sn-type), τ4-(Ti1−x−ySnxNiy)Ni3 with AuCu3-type exhibits a solution range (0.22 ≤ x ≤ 0.66 and 0.22 ≥ y ≥ 0.02) and a particularly large homogeneity region is recorded for τ2-Ti1+yNi2−xSn1−y (Heusler phase, MnCu2Al-type). Extended solid solutions starting from binary phases at 950 °C have been evaluated for Ti5Ni1−xSn3 (filled Mn5Si3 = Ti5Ga4-type; 0 ≤ x ≤ 1), Ti1−xSnxNi3 (TiNi3-type; 0 ≤ x ≤ 0.27) and (Ti1−xNix)1−ySny (CsCl-type) reaching a maximum solubility at x = 0.53, y = 0.06). From differential thermal analysis (DTA) in alumina crucibles under argon a complete liquidus surface has been elucidated revealing congruent melting for τ2-TiNi2Sn at 1447 °C, but incongruent melting for τ1-TiNiSn (pseudobinary peritectic formation: ![[small script l]](https://www.rsc.org/images/entities/char_e146.gif) + τ2 ↔ τ1 at 1180 °C), τ3-Ti2Ni2Sn (peritectic formation: L + τ2 + Ti5NiSn3 ↔ τ3 at 1151 °C) and τ4-Ti1−xSnxNi3 (peritectic formation: L + TiNi3 + (Ni) ↔ τ4 at 1157 °C). A Schultz–Scheil diagram for the solidification behavior was constructed for the entire diagram involving 20 isothermal four-phase reactions in the ternary. For a thermodynamic CALPHAD assessment of the ternary diagram we relied on the binary boundary systems as modelled in the literature. As thermodynamic data in the ternary system were only available in the literature for the compounds TiNi2Sn and TiNiSn, heat of formation data were supplied by our density functional theory (DFT) calculations for Ti2Ni2Sn, as well as for the solid solutions, which were modelled for Ti1−xSnxNi3, Ti5Ni1−xSn3 and (Ti1−xNix)1−ySny. Thermodynamic calculation was performed with the Pandat software and finally showed a reasonably good agreement for all the 20 invariant reaction isotherms involving the liquid.
+ τ2 ↔ τ1 at 1180 °C), τ3-Ti2Ni2Sn (peritectic formation: L + τ2 + Ti5NiSn3 ↔ τ3 at 1151 °C) and τ4-Ti1−xSnxNi3 (peritectic formation: L + TiNi3 + (Ni) ↔ τ4 at 1157 °C). A Schultz–Scheil diagram for the solidification behavior was constructed for the entire diagram involving 20 isothermal four-phase reactions in the ternary. For a thermodynamic CALPHAD assessment of the ternary diagram we relied on the binary boundary systems as modelled in the literature. As thermodynamic data in the ternary system were only available in the literature for the compounds TiNi2Sn and TiNiSn, heat of formation data were supplied by our density functional theory (DFT) calculations for Ti2Ni2Sn, as well as for the solid solutions, which were modelled for Ti1−xSnxNi3, Ti5Ni1−xSn3 and (Ti1−xNix)1−ySny. Thermodynamic calculation was performed with the Pandat software and finally showed a reasonably good agreement for all the 20 invariant reaction isotherms involving the liquid.
1 Introduction
The so-called “Half Heusler” (HH) compound TiNiSn, known as an n-type semiconductor since 1986,1,2 has hitherto displayed a high potential for exceptional efficiency in thermoelectric (TE) generators converting (waste) heat into electricity. Thermoelectric research has, therefore, focused mainly on improvement of ternary intermetallic compounds based on TiNiSn, which crystallize with the non-centrosymmetric cubic MgAgAs-type structure (space group F4![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Beside skutterudites and clathrates, HH compounds are promising candidates for high temperature thermoelectric applications because they inherit a tuneable electronic structure, which can be modified through (i) doping/substitution on its three metal sublattices, (ii) engineering of a generally narrow band gap, and (iii) nanostructuring via ball-milling and precipitation of secondary system inherent phases as the most prominent among many other techniques. An overview on the thermoelectric properties of HH alloys, reaching up to 700 °C a ZTmax ∼ 1.0 for p-type Ti{FexCo1−x}{SnySb1−y} and a ZTmax ∼ 1.2 for n-type {Ti1−u−vZruHfv}Ni{Sn1−wSbw} can be found from a recent review article.3 However, the biggest disadvantage of HH alloys is their relatively high thermal conductivity, which has to be decreased in order to increase the thermoelectric performance of these materials. Large scale production and particularly nanostructuring of TE-materials by precipitation of preferably system inherent phases, however, needs a profound knowledge not only of isothermal phase relations, temperature dependent solubilities and vacancy concentrations but also of the solidification behaviour in each ternary subsystem of any multi-component TiNiSn-based alloy system. Although several papers in the literature provide (a) phase relations in the isothermal section of Ti–Ni–Sn at 800 °C (ref. 4) (see also refs. therein), (b) melting temperatures of TiNiSn (Tm = 1182 °C;5 Tm = 1182 °C (ref. 6)), TiNi2Sn (Tm = 1447 °C (ref. 5)), (c) DFT heat of formation data for various binary and ternary compounds,5,7 (d) calorimetric heat of formation data8 and (e) coefficient of thermal expansion αave = 11.3 × 10−6 K−1 (40–690 °C),6 to our best knowledge hitherto no liquidus projection exists. For most reports in the literature the homogeneity regions of the Heusler and the half Heusler phase play no important role, but several authors gave proof to the non-stoichiometry of TiNi2−ySn (0 < y < 0.04, 800 °C;4 0 < y < 0.22, as cast5) and of TiNi1+ySn (0 < y < 0.10, as cast;5 0 < y < 0.06, 900 °C;9 −0.06 < y < 0.08, at 1100 °C (ref. 10)).
m). Beside skutterudites and clathrates, HH compounds are promising candidates for high temperature thermoelectric applications because they inherit a tuneable electronic structure, which can be modified through (i) doping/substitution on its three metal sublattices, (ii) engineering of a generally narrow band gap, and (iii) nanostructuring via ball-milling and precipitation of secondary system inherent phases as the most prominent among many other techniques. An overview on the thermoelectric properties of HH alloys, reaching up to 700 °C a ZTmax ∼ 1.0 for p-type Ti{FexCo1−x}{SnySb1−y} and a ZTmax ∼ 1.2 for n-type {Ti1−u−vZruHfv}Ni{Sn1−wSbw} can be found from a recent review article.3 However, the biggest disadvantage of HH alloys is their relatively high thermal conductivity, which has to be decreased in order to increase the thermoelectric performance of these materials. Large scale production and particularly nanostructuring of TE-materials by precipitation of preferably system inherent phases, however, needs a profound knowledge not only of isothermal phase relations, temperature dependent solubilities and vacancy concentrations but also of the solidification behaviour in each ternary subsystem of any multi-component TiNiSn-based alloy system. Although several papers in the literature provide (a) phase relations in the isothermal section of Ti–Ni–Sn at 800 °C (ref. 4) (see also refs. therein), (b) melting temperatures of TiNiSn (Tm = 1182 °C;5 Tm = 1182 °C (ref. 6)), TiNi2Sn (Tm = 1447 °C (ref. 5)), (c) DFT heat of formation data for various binary and ternary compounds,5,7 (d) calorimetric heat of formation data8 and (e) coefficient of thermal expansion αave = 11.3 × 10−6 K−1 (40–690 °C),6 to our best knowledge hitherto no liquidus projection exists. For most reports in the literature the homogeneity regions of the Heusler and the half Heusler phase play no important role, but several authors gave proof to the non-stoichiometry of TiNi2−ySn (0 < y < 0.04, 800 °C;4 0 < y < 0.22, as cast5) and of TiNi1+ySn (0 < y < 0.10, as cast;5 0 < y < 0.06, 900 °C;9 −0.06 < y < 0.08, at 1100 °C (ref. 10)).
In order to shed light on the complicated synthesis procedures for single-phase TiNiSn-based alloys, the present paper is intended to provide (1) a liquidus surface for the entire Ti–Ni–Sn phase diagram i.e. precise information on the solidification paths, and (2) phase relations in an isothermal section at 950 °C. As the TiNiSn system is a subsystem of a multi component thermoelectric alloy system, for which a thermodynamic (pre-) calculation can save extensive experimental work, the present paper will furthermore provide a thermodynamic assessment of the Ti–Ni–Sn ternary. This CALPHAD-type modelling is based on existing assessments for the binary boundary systems as well as relies on experimental thermodynamic data for the ternary compounds and will be backed by DFT energies of formation wherever needed in the modelling.
2 Experimental details
2.1. Sample preparation and characterization
Pure elements in form of Ti-, Ni-rods, Sn-shot or bars with a minimum purity of 99.95 mass% from Alfa Aesar were used for the preparation of about 60 alloys with various compositions for the system Ti–Ni–Sn. First stoichiometric amounts of Ti and Ni were arc melted together under 6 N argon and then the proper amount of Sn was added. The reguli were flipped 3 times for a better homogenization. Afterwards the samples were vacuum sealed in quartz ampullae (which were backfilled at RT with 200 mbar Ar), annealed at different temperatures for 7 days (200 °C → 950 °C with a heating rate of 10°C min−1) and water quenched. Some samples were further annealed in evacuated quartz ampullae (which were backfilled at RT with 200 mbar Ar) at different temperatures in Al2O3 crucibles for better equilibration.For sample characterization we used Scanning Electron Microscopy (SEM), Electron Probe Microanalysis (EPMA) and X-ray Powder Diffraction (XPD). The microstructure and chemical composition of the alloys were analyzed by SEM on a Zeiss Supra 55 VP equipped with an energy dispersive X-ray (EDX) detector operated at 20 kV. Samples for EPMA were prepared by standard metallographic methods. In some cases polishing was performed under glycerine instead of water to avoid oxidation and/or hydrolysis of samples. X-ray powder diffraction profiles for all alloys were collected from a HUBER-Guinier image plate with monochromated CuKα1-radiation. For Rietveld refinements we used the FULLPROF program,11 whilst precise lattice parameters were obtained by least square methods with program STRUKTUR12 employing pure Ge (99.9999%) as internal standard (aGe = 0.5657906 nm).
Single crystals of Ti5NiSn3 were isolated from alloy Ti53Ni11Sn34 annealed at 1100 °C for 5 days. The crystals were inspected on an AXS-GADDS texture goniometer for quality and crystal symmetry prior to X-ray single crystal (XSC) intensity data collection on a four-circle Nonius Kappa diffractometer (CCD area detector and graphite monochromated MoKα radiation, λ = 0.071069 nm). Orientation matrix and unit cell parameters were derived using the program DENZO (Nonius Kappa CCD, Program Package, Nonius Delft, The Netherlands). Besides psi-scans no additional absorption correction was necessary because of the rather regular crystal shape and small dimensions of the investigated specimens. The structure was solved by direct methods and refined with the SHELXS-97 and SHELXL-97 programs,13 respectively.
The differential thermal analyses (DTA) measurements were performed on annealed samples in a Netzsch 404 Pegasus DSC (differential scanning calorimetry) equipment in Al2O3-crucibles under a stream of 6 N argon and a heating rate of 5 K min−1. The equipment was calibrated in the temperature range from 300 to 1400 °C against pure metal standards supplied by Netzsch to be within ±1 °C.
2.2. Thermodynamic modelling
| ΔH = 103[Etot(TiaNibSnc) − a(Etot(Ti)/j) − b(Etot(Ni)/k) − c(Etot(Sn)/l)]/(a + b + c), |
For the thermodynamic description of the respective phase structures, commonly used thermodynamic models were applied with respect to reference Gibbs energies of components in given phase ϕ, which have a polynomial form:18
 | (1) |
The Gibbs energy of a given phase like liquid and solid solution or a compound is expressed as a sum of several contributions:
 | (2) |
The particular terms in case of solution phases like liquid or solid solutions are described as follows:
 | (3a) |
 | (3b) |
 | (3c) |
 | (3d) |
| Lɸi,j,k = 0Lɸi,j,kxi + 1Lɸi,j,kxj + 2Lɸi,j,kxk | (3e) |
nLɸi,j = an + bnT + cnT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnT lnT
| (3f) |
Thermodynamic modelling of intermetallic phases is based on the compound energy formalism in which the particular terms are described as follows:
 | (4a) |
 | (4b) |
 | (4c) |
The difference in the reference Gibbs energy for a given real or hypothetical compound i:j (0Gi:j) of an intermetallic phase and the Gibbs energies of the elements i, j in their Standard Element Reference states (SER) (0Gi, 0Gj) is given by the equation:
| Δ0Gi:j = 0Gi:j − ν0iGi − ν0jGj = ΔH − TΔS | (5) |
At T = 0 K, one may write ΔH(T = 0) = ΔE(T = 0), i.e. the difference in enthalpies is equal to the difference of total energies. These total energy differences have been calculated ab initio at the equilibrium volume in the present paper. The difference in enthalpies ΔH, at finite temperature is then obtained as (Kirchhoff's law):
| ΔH = ΔE + ∫ΔCpdT | (6) |
In the region without phase transformation, entropy can be expressed as:
| ΔS = ∫(ΔCp/T)dT | (7) |
In general, the heat capacity difference ΔCp is temperature dependent, and in the simplest case it can be described by a linear function:
| ΔCp = a + bT | (8) |
Substitution of the enthalpic and entropic term with the relations (6)–(8) in eqn (5) yields after integration:
|
Δ0Gi:j = ΔE + a(1 − lnT)T − (b/2)T2
| (9) |
This equation has been employed in the phase diagram calculations. ΔE is calculated ab initio and ΔCp is optimized as a curve fitting parameter to the experimental phase equilibrium data.
3 Results
3.1. The binary boundary systems
Information on the three binary phase diagrams is based on the compilation by Massalski.21 Assessments of experimental phase diagram data and thermodynamic modelling are available from various research groups: Ni–Sn,22,23 Ti–Sn,24 and Ti–Ni.25–27 Detailed crystallographic data on unary and binary boundary phases reported in the literature (ref. 21–43) and obtained in the current work are summarized in Table S1 (ESI†); data for ternary compounds are listed in Table 1.| Phase, temperature range (°C) | Space group, prototype | Lattice parameters (nm) | Comments/references | |
|---|---|---|---|---|
| a | c | |||
| a This work. | ||||
| τ1-TiNiSn | F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m 3m |
0.5927 | — | Ref. 44 |
| MgAgAs | 0.59332(6) | — | Ref. 4 | |
| 0.59349(1) | — | a | ||
| Ti1+yNi2−xSn1−y | 0.59655(4) | — | x = 0.779, y = −0.008 at 1100 °C; in equilibrium with τ2a | |
| τ2-Ti1+yNi2−xSn1−y | Fmm |
0.6097(3) | — | x = 0, y = 0, z = 0 (ref. 4) |
| MnCu2Al | 0.60973(4) | — | x = 0, y = 0, z = 0,a see also Fig. 9 | |
| 0.60710(5) | — | x = 0.344, y = 0.038 at 950 °C; in equilibrium with τ1a | ||
| 0.60503(8) | — | x = 0.008, y = 0.344 at 1100 °C; in equilibrium with TiNia | ||
| 0.6049(2) | — | x = −0.651, y = −0.121 at 1100 °C; in equilibrium with TiNi3 and Ni3Sn2a | ||
| τ3-Ti2+xNi2Sn1−x | P42/mnm | 0.68168(4) | 0.64379(6) | x = 0 (ref. 4) |
| U2Pt2Sn | 0.68273(6) | 0.63850(4) | x = 0.195 in equilibrium with TiNi and Ti5NiSn3 at 950 °C | |
| τ4-(Ti1−x−ySnxNiy)Ni3 | Pmm |
0.36316(5) | Ti14.0Ni80.5Sn5.5; x = 0.22 and y = 0.22; see also Fig. 10 | |
| AuCu3 | 0.36904(7) | Ti6.2Ni77.4Sn16.4; x = 0.66 and y = 0.1 | ||
| τ4′ | Unknown | At ∼ Ti16–14Ni75Sn9–11 | ||
| τ5 | Unknown | At ∼ Ti52Ni23Sn25 | ||
3.2. Phase equilibria in the ternary system Ti–Ni–Sn
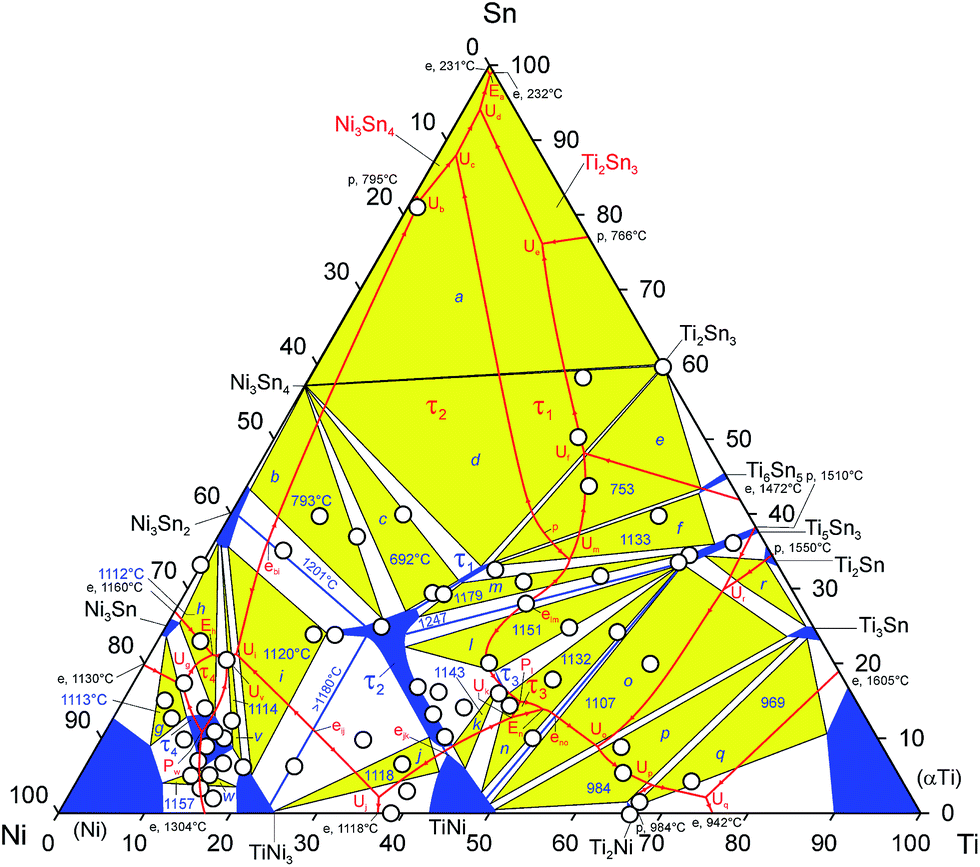
In order to determine the phase equilibria in the ternary system, about 60 alloys were investigated in as-cast state and after annealing at 800 and 950 °C (7 days). In some cases these temperatures were not sufficient for equilibration of the samples and therefore they were additionally annealed for 7 days in the temperature range from 450 to 770 °C (for Sn contents > 40 at%) and at 1050, 1080 and 1100 °C (for alloys containing high melting compounds such as TiNi3, TiNi, Ti5Sn3 and τ2).Combined evaluation of EDX and XPD data in equilibrated samples defined the number of three-phase fields, which are labelled alphabetically in the figures from “a” to “w”. The temperatures of the invariant four-phase reactions have been determined with DTA on equilibrated samples. Microstructures of the as-cast samples were used to define the primary crystallization fields and reactions during solidification. The phase equilibria established in the system are presented as projections of liquidus- and solidus surfaces (Fig. 1 and 2), as a melting-crystallization diagram (Fig. 3) and in form of a Schultz–Scheil diagram (Fig. 4) for the solidification behavior in the entire system involving 20 isothermal four-phase reactions in the ternary. Phase equilibria at 950 °C are presented as an isothermal section in Fig. 5. A summary of the phases involved in invariant reactions is available from Table 2. Phase compositions of the selected alloys, which have been investigated in different states, are presented in Table S2 (ESI†) which contains links to the SEM images (Fig. 6–8) that document respective statements.
 | ||
| Fig. 1 Liquidus projection of Ti–Ni–Sn. The labels inside the circles denote indices for the microstructures of as-cast samples presented in Fig. 6 (open circles) and Fig. 8 (gray circles). The small triangles show compositions of eutectics observed in as-cast samples. Composition of the phases measured by EPMA is listed in Table S2 (ESI†). | ||
 | ||
| Fig. 2 Solidus projection with corresponding temperatures, determined by DSC measurements. The labels inside the circles denote indexes for the microstructures of the samples annealed at 950 °C (Fig. 7, open circles) and other temperatures Fig. 8 (gray and semi-filled circles). | ||
 | ||
| Fig. 3 Sub-solidus surface with superimposed mono-variant lines from the liquidus. The composition of phases involved in the invariant reactions is summarized in Table 2. | ||
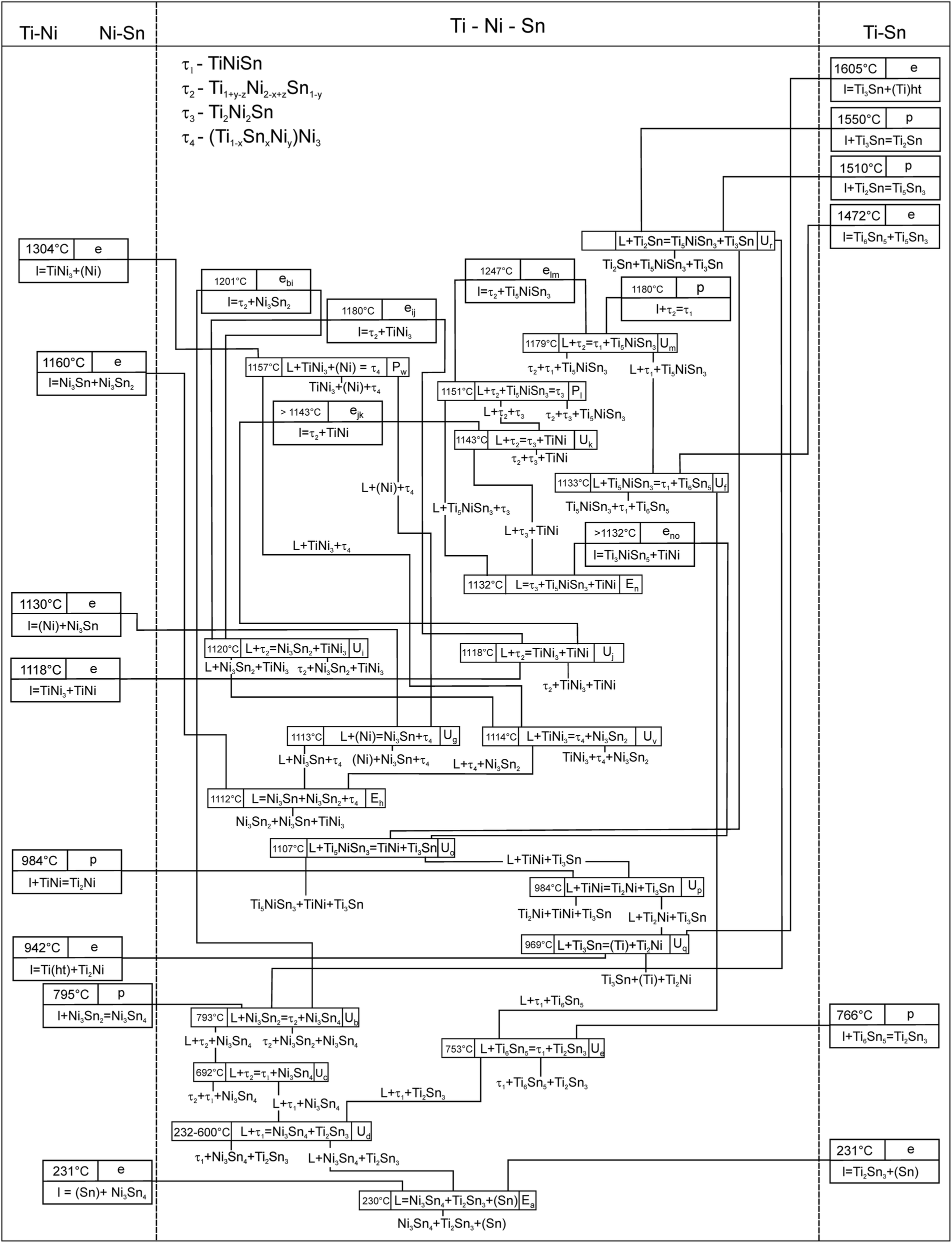 | ||
| Fig. 4 Schultz–Scheil diagram for the ternary Ti–Ni–Sn system. The homogeneity regions: Ti1−xSnxNi3, Ti5NixSn3, (Ti1−xNix)1−ySny and Ti1+yNi2−xSn1−y are noted as TiNi3, Ti5NiSn3, (Ti1−xNix)1−ySny and τ2. | ||
 | ||
| Fig. 5 Isothermal section at 950 °C. The labels inside the circles denote indices for the microstructures of the annealed samples presented in Fig. 7. | ||
| Reaction (exp.) | Phase | Ti | Ni | Sn | Type t °C | Reaction (calc.) | Phase | Ti | Ni | Sn | Type t °C |
|---|---|---|---|---|---|---|---|---|---|---|---|
| L + Ti2Sn ↔ Ti5Sn3(Ni) + Ti3Sn | L | 62 | 8 | 30 | Ur | L + Ti2Sn ↔ Ti5Sn3(Ni) + Ti3Sn | L | 63.6 | 4.0 | 32.4 | U |
| Ti2Sn | 65.0 | 1.0 | 34.0 | n.d. | Ti2Sn | 67.2 | 0.3 | 32.5 | 1510.4 | ||
| Ti5Sn3(Ni) | 57.5 | 8 | 34.5 | Ti5Sn3(Ni) | 60.5 | 3.2 | 36.3 | ||||
| Ti3Sn | 74 | 1 | 25 | Ti3Sn | 75.1 | 0.0 | 24.9 | ||||
| L + τ2 ↔ Ti5Sn3(Ni) + τ1 | L | 42 | 24 | 34 | Um | L + τ2 + Ti5Sn3(Ni) ↔ τ1 | L | 34.5 | 29.5 | 36.0 | P |
| τ2 | 28.4 | 45.3 | 26.3 | 1179 | τ2 | 24.4 | 51.2 | 24.4 | 1168.5 | ||
| τ1 | 31.2 | 37.4 | 31.5 | τ1 | 33.3 | 33.3 | 33.4 | ||||
| Ti5Sn3(Ni) | 56.4 | 8.6 | 35.0 | Ti5Sn3(Ni) | 56.3 | 9.9 | 33.8 | ||||
| L + Ti5Sn3(Ni) ↔ τ1 + Ti6Sn5 | L | 37 | 15 | 48 | Uf | L + Ti5Sn3(Ni) ↔ τ1 + Ti6Sn5 | L | 34.0 | 26.1 | 39.9 | U |
| Ti5Sn3(Ni) | 57.9 | 5.9 | 36.2 | 1133 | Ti5Sn3(Ni) | 56.4 | 9.7 | 33.9 | 1154.8 | ||
| τ1 | 33.2 | 33.6 | 33.2 | τ1 | 33.3 | 33.3 | 33.3 | ||||
| Ti6Sn5 | 52.7 | 4.5 | 42.8 | Ti6Sn5 | 54.6 | 0.8 | 44.6 | ||||
| L + τ2 + Ti5Sn3(Ni) ↔ τ3 | L | 46 | 39 | 15 | P1 | L + τ2 ↔ Ti5Sn3(Ni) + τ3 | L | 40.9 | 39.3 | 19.8 | U |
| τ2 | 41.2 | 40 | 18.8 | 1151 | τ2 | 24.9 | 51.7 | 23.4 | 1156.4 | ||
| Ti5Sn3(Ni) | 55.6 | 11.1 | 33.3 | Ti5Sn3(Ni) | 56.1 | 10.2 | 33.7 | ||||
| τ3 | 28.9 | 48.9 | 22.2 | τ3 | 40.0 | 40.0 | 20.0 | ||||
| L + τ2 ↔ τ3 + (TiNi) | L | 46 | 41 | 13 | Uk | L + τ2 ↔ τ3 + (TiNi) | L | 40.7 | 42.0 | 17.3 | U |
| τ2 | 41.1 | 49.8 | 9.1 | 1143 | τ2 | 25.0 | 51.9 | 23.1 | 1151.0 | ||
| τ3 | 42.3 | 40.4 | 17.3 | τ3 | 40.0 | 40.0 | 20.0 | ||||
| (TiNi) | 44.0 | 50.0 | 6.0 | (TiNi) | 31.3 | 48.7 | 20.0 | ||||
| L + τ2 ↔ (TiNi3) + (TiNi) | L | 36 | 62 | 2 | Uj | L ↔ (TiNi3) + τ2 + (TiNi) | L | 31.7 | 60.9 | 7.4 | E |
| τ2 | 37.8 | 52 | 10.2 | 1118 | τ2 | 25.1 | 52.5 | 22.4 | 1130.8 | ||
| (TiNi3) | 25 | 74.9 | 0.1 | (TiNi3) | 25.0 | 75.0 | 0.0 | ||||
| (TiNi) | 41.3 | 53.2 | 5.5 | (TiNi) | 37.0 | 54.5 | 8.5 | ||||
| L ↔ τ3 + (TiNi) + Ti5Sn3(Ni) | L | 49 | 37 | 14 | En | L ↔ τ3 + (TiNi) + Ti5Sn3(Ni) | L | 43.5 | 39.8 | 16.7 | E |
| τ3 | 43.9 | 40.2 | 15.9 | 1132 | τ3 | 40.0 | 40.0 | 20.0 | 1139.9 | ||
| (TiNi) | 47.5 | 49.8 | 2.7 | (TiNi) | 32.6 | 49.3 | 18.1 | ||||
| Ti5Sn3(Ni) | 55.6 | 11.1 | 33.3 | Ti5Sn3(Ni) | 56.1 | 10.2 | 33.7 | ||||
| L + Ti5Sn3(Ni) ↔ (TiNi) + Ti3Sn | L | 58 | 33 | 9 | Uo | L ↔ Ti5Sn3(Ni) + (TiNi) + Ti3Sn | L | 53.2 | 35.1 | 11.7 | E |
| Ti5Sn3(Ni) | 55.6 | 11.1 | 33.4 | 1107 | Ti5Sn3(Ni) | 56.5 | 9.6 | 33.9 | 1108.1 | ||
| (TiNi) | 49.3 | 49.6 | 1.1 | (TiNi) | 38.2 | 51.8 | 10.0 | ||||
| Ti3Sn | 72.5 | 3.8 | 23.7 | Ti3Sn | 75.0 | 0.1 | 24.9 | ||||
| L + (TiNi3) + (Ni) ↔ τ4 | L | 12.0 | 77.0 | 11.0 | Pw | L + (TiNi3) ↔ (Ni) + τ4 | L | 11.8 | 74.6 | 13.6 | U |
| (TiNi3) | 18.7 | 77.9 | 3.4 | 1157 | (TiNi3) | 13.5 | 77.6 | 8.9 | 1061.8 | ||
| (Ni) | 10.2 | 86.2 | 3.6 | (Ni) | 10.6 | 79.3 | 10.1 | ||||
| τ4 | 13.7 | 80.8 | 5.5 | τ4 | 13.4 | 72.4 | 14.2 | ||||
| L + (Ni) ↔ Ni3Sn + τ4 | L | 6.1 | 77.0 | 16.9 | Ug | L + (Ti Ni3) ↔ (Ni) + Ni3Sn | L | 8.0 | 74.2 | 17.8 | E |
| (Ni) | 6.2 | 84.9 | 8.9 | 1113 | (Ni) | 7.5 | 78.9 | 13.6 | 1045.8 | ||
| Ni3Sn | 0.8 | 75.6 | 23.6 | Ni3Sn | 1.5 | 73.7 | 24.8 | ||||
| τ4 | 8.6 | 78.5 | 12.9 | τ4 | 11.3 | 72.3 | 16.4 | ||||
| L + (TiNi3) ↔ Ni3Sn2 + τ4 | L | 9.0 | 70.0 | 21.0 | Uv | L + (TiNi3) ↔ τ2 + τ4 | L | 14.8 | 71.1 | 14.1 | U |
| (TiNi3) | 17.9 | 75.4 | 6.7 | 1114 | TiNi3 | 18.6 | 76.2 | 5.2 | 1066.7 | ||
| Ni3Sn2 | 0.9 | 63.1 | 36.0 | τ2 | 24.0 | 52.4 | 23.6 | ||||
| τ4 | 15.9 | 75.7 | 8.4 | τ4 | 14.5 | 72.1 | 13.4 | ||||
| L ↔Ni3Sn + τ4 + Ni3Sn2 | L | 7.0 | 72.0 | 21.0 | Eh | L ↔ Ni3Sn + τ4 + Ni3Sn2 | L | 8.5 | 70.7 | 20.8 | E |
| Ni3Sn | 1.2 | 73.3 | 25.5 | 1112 | Ni3Sn | 1.9 | 72.7 | 25.4 | 1045.5 | ||
| τ4 | 11.8 | 75.5 | 12.7 | τ4 | 11.2 | 72.0 | 16.8 | ||||
| Ni3Sn2 | 0.4 | 63.4 | 36.2 | Ni3Sn2 | 2.9 | 62.6 | 34.5 | ||||
| L + τ2 ↔ (Ti Ni3) + Ni3Sn2 | L | 10.0 | 69.0 | 21.0 | Ui | L ↔ τ2 + τ4 + Ni3Sn2 | L | 10.6 | 67.5 | 21.9 | E |
| τ2 | 18.9 | 57 | 24.1 | 1120 | τ2 | 23.5 | 52.1 | 24.4 | 1044.6 | ||
| (TiNi3) | 19.6 | 75.3 | 5.1 | τ4 | 12.0 | 71.7 | 16.3 | ||||
| Ni3Sn2 | 1.8 | 62.6 | 35.6 | Ni3Sn2 | 4.1 | 62.3 | 33.6 | ||||
| L + (Ti Ni) ↔ Ti3Sn + Ti2Ni | L | 67.0 | 29.5 | 3.5 | Up | L ↔ (Ti Ni) + Ti3Sn + Ti2Ni | L | 66.8 | 30.9 | 2.3 | E |
| (TiNi) | 50.3 | 49.2 | 0.5 | 984 | (TiNi) | 45.3 | 54.4 | 0.3 | 957.5 | ||
| Ti3Sn | 73.9 | 2.5 | 23.6 | Ti3Sn | 75.9 | 1.1 | 23.0 | ||||
| Ti2Ni | 65.1 | 33.4 | 1.5 | Ti2Ni | 66.7 | 33.3 | 0.0 | ||||
| L + Ti3Sn ↔ Ti2Ni + (Ti) | L | 74 | 24 | 2 | Uq | L ↔ Ti3Sn + Ti2Ni + (Ti) | L | 68.6 | 29.3 | 2.1 | E |
| Ti3Sn | 74.9 | 1.5 | 23.6 | 969 | Ti3Sn | 76.2 | 1.2 | 22.6 | 957.2 | ||
| Ti2Ni | 66 | 33.1 | 0.9 | Ti2Ni | 66.7 | 33.3 | 0.0 | ||||
| (Ti) | 83.6 | 5.8 | 10.6 | (Ti) | 76.1 | 0.7 | 23.2 | ||||
| L + Ni3Sn2 ↔ τ2 + Ni3Sn4 | L | 1 | 17 | 82 | Ub | L + Ni3Sn2 ↔ τ2 + Ni3Sn4 | L | 8.3 | 27.4 | 64.3 | U |
| Ni3Sn2 | 0 | 56.2 | 43.8 | 793 | Ni3Sn2 | 0.1 | 56.2 | 43.7 | 748.0 | ||
| τ2 | 24.6 | 49.6 | 25.8 | τ2 | 23.6 | 51.0 | 25.4 | ||||
| Ni3Sn4 | 0.5 | 42.4 | 57.1 | Ni3Sn4 | 0.0 | 44.0 | 56.0 | ||||
| L + Ti6Sn5 ↔ τ1 + Ti2Sn3 | L | 18 | 6 | 76 | Ue | L + Ti6Sn5 ↔ τ1 + Ti2Sn3 | L | 14.7 | 3.5 | 81.8 | U |
| Ti6Sn5 | 52.8 | 4 | 43.2 | 753 | Ti6Sn5 | 54.5 | 0.0 | 45.5 | 738.7 | ||
| τ1 | 33.4 | 33.2 | 33.4 | τ1 | 33.3 | 33.3 | 33.4 | ||||
| Ti2Sn3 | 40.3 | 0 | 59.7 | Ti2Sn3 | 40.0 | 0.0 | 60.0 | ||||
| L + τ2↔ τ1 + Ni3Sn4 | L | 2 | 10 | 88 | Uc | L + τ2↔ τ1 + Ni3Sn4 | L | 8.4 | 24.8 | 66.8 | U |
| τ2 | 26.8 | 46.3 | 26.9 | 692 | τ2 | 23.6 | 51.0 | 25.4 | 716.7 | ||
| τ1 | 30.8 | 38.1 | 31.1 | τ1 | 33.3 | 33.3 | 33.4 | ||||
| Ni3Sn4 | 0 | 42.9 | 57.1 | Ni3Sn4 | 0.0 | 43.8 | 56.2 | ||||
| L ↔ Ni3Sn4 + Ti2Sn3 + (Sn) | L | 0.2 | 0.6 | 99.2 | Ea | ||||||
| Ni3Sn4 | 0 | 42.9 | 57.1 | ∼232 | |||||||
| Ti2Sn3 | 40 | 0 | 60 | ||||||||
| (Sn) | 0.8 | 0.8 | 98.4 | ||||||||
| L + τ1 ↔ Ni3Sn4 + Ti2Sn3 | L | 2 | 4 | 94 | Ud | ||||||
| τ1 | 32.6 | 34.6 | 32.8 | 232–600 | |||||||
| Ni3Sn4 | 0 | 42.9 | 57.1 | ||||||||
| Ti2Sn3 | 40 | 0 | 60 | ||||||||
| + τ2 ↔ τ1 |
|
37.5 | 25.0 | 37.5 | pcm | ||||||
| τ2 | 27.5 | 45.0 | 27.5 | >1179 | |||||||
| τ1 | 31.0 | 38.0 | 31.0 | ||||||||
| + τ2 ↔ τ3 |
|
40.8 | 39.3 | 19.8 | p | ||||||
| τ2 | 1156.4 | ||||||||||
| τ3 | |||||||||||
| ↔ τ2 + Ni3Sn2 |
|
6.3 | 57.6 | 36.1 | ebi | ↔ τ2 + Ni3Sn2 |
|
9.7 | 56.8 | 33.5 | e |
| τ2 | 23 | 52 | 25 | 1201 | τ2 | 1129.4 | |||||
| Ni3Sn2 | 1 | 59.4 | 39.6 | Ni3Sn2 | |||||||
| ↔ τ2 + (TiNi3) |
|
24.2 | 64.9 | 10.9 | eij | ↔ τ2 + (TiNi3) |
|
24.8 | 66.0 | 9.2 | e |
| τ2 | 23.6 | 52.7 | 23.7 | >1180 | τ2 | 1197.2 | |||||
| TiNi3 | 23.8 | 75.4 | 0.8 | TiNi3 | |||||||
| ↔ τ2 + (TiNi) |
|
40.5 | 51 | 8.5 | ejk | ↔ τ2 + (TiNi) |
|
35.9 | 51.3 | 12.7 | e |
| τ2 | 40 | 51 | 9 | >1143 | τ2 | 1220.8 | |||||
| (TiNi) | 41 | 51 | 8 | (TiNi) | |||||||
| ↔ τ2 + Ti5Sn3(Ni) |
|
40 | 32 | 28 | elm | ↔ τ2 + Ti5Sn3(Ni) |
|
37.8 | 34.3 | 27.8 | e |
| τ2 | 28 | 48 | 24 | 1247 | τ2 | 1209.5 | |||||
| Ti5Sn3(Ni) | 55.6 | 11.1 | 33.3 | Ti5Sn3(Ni) | |||||||
| ↔ (TiNi) + Ti3Sn |
|
54.8 | 35.0 | 10.3 | e | ||||||
| (TiNi) | 1111.5 | ||||||||||
| Ti3Sn | |||||||||||
| ↔ (TiNi) + Ti5Sn3(Ni) |
|
51 | 36 | 13 | eno | ↔ (TiNi) + Ti5Sn3(Ni) |
|
45.9 | 38.8 | 15.3 | e |
| (TiNi) | 48.6 | 49.6 | 1.8 | >1132 | (TiNi) | 1144.6 | |||||
| Ti5Sn3(Ni) | 55.6 | 11.1 | 33.3 | Ti5Sn3(Ni) | |||||||
| ↔ Ti2Ni + Ti3Sn |
|
67.7 | 30.1 | 2.2 | e | ||||||
| Ti2Ni | 957.8 | ||||||||||
| Ti3Sn | |||||||||||
 | ||
| Fig. 6 Microstructure of selected as-cast samples. The composition of the samples is marked with respective indices inside of the open circles on Fig. 1. | ||
 | ||
| Fig. 7 Microstructures of the selected samples annealed at 950 °C. Labels inside of the triangles denote the respective three phase fields (Fig. 1–5). Compositions of the phases are listed in Table S2 (ESI†). The compositions of the samples are marked with respective indices inside of the open circles on Fig. 5. | ||
 | ||
| Fig. 8 Microstructures of selected as-cast and annealed samples. Labels inside of the triangles denote the respective three-phase fields (Fig. 1–5). Composition of the phases after EPMA is listed in Table S2 (ESI†). The composition of the as-cast samples is marked with respective indices inside of the gray circles on Fig. 1. The annealed samples are denoted with indices inside gray and semi-filled circles on Fig. 2. | ||
Investigation of the alloys in as-cast state (Fig. 6 and 8) shows, that the Heusler phase (HP, τ2-TiNi2Sn, MnCu2Al type) has the largest field of primary crystallization. This phase melts congruently at 1447 °C (ref. 5) and exhibits a wide homogeneity region at sub-solidus temperatures. Thus in the sample with nominal composition TiNi2Sn, the HP crystallizes primarily (Ti25.2Ni49.7Sn25.1, at.%), followed by small grains of τ2 with composition Ti15Ni61Sn24 (at.%) and the crystallization ends by solidification of Ni3Sn2 (Fig. 6a). After annealing at 950 °C (Fig. 7a), an almost single-phase sample of τ2 was obtained with small residual amounts of Ni3Sn2. In order to determine details in constitution, a sample with composition Ti18Ni58Sn24 was prepared and investigated in three states. The as-cast specimen shows primary τ2 with a composition close to stoichiometric TiNi2Sn (Ti23.3Ni52.2Sn24.4), Ni3Sn2 and a eutectic structure with the composition Ti7.5Ni71.7Sn20.8 (Fig. 6f). The samples annealed at 950 °C and 1100 °C (Fig. 7f and 8f) reveal the three-phase field “i”: τ2 + Ni2Sn3 + TiNi3 (Ti1−xSnxNi3). However, the composition of τ2 in the annealed specimens differs significantly: Ti23.0Ni52.3Sn24.7 at 950 °C but Ti18.9Ni57.0Sn24.1 at 1100 °C, respectively. The Rietveld refinement of the sample annealed at 1100 °C confirms the crystal structure of the Heusler phase with Ti/Ni substitution in the 4b site (½, ½, ½, , ). At high temperatures the homogeneity region of this phase extends further towards the binary phase TiNi. Thus, the sample Ti37Ni50Sn13 in as-cast state defines τ2 and TiNi with compositions Ti29.5Ni50.0Sn20.6 and Ti41.3Ni50.7Sn7.9, respectively. However, after annealing at 1100 °C this sample exhibits almost single-phase τ2 with a composition in the range from Ti35.9Ni50.3Sn13.8 to Ti38.2Ni50.6Sn11.2. A two-phase gap between TiNi and τ2 is defined to exist in the composition range between 6 and 9 at% Sn considering EPMA data obtained from samples Ti36Ni58Sn6 and Ti43Ni49Sn8 annealed at 1100 °C. Temperatures of invariant reaction that were measured on these samples by DTA were defined to be 1118 °C (Uj) and 1146 °C (Uk). Taking into account the extended homogeneity region of τ2, a structural chemical formula for the Heusler phase may be expressed as Ti1+yNi2−xSn1−y. The homogeneity region of this phase at 1100 °C extends towards binary Ni–Sn, τ1 and TiNi reaching compositions Ti19Ni57Sn24, Ti27Ni46Sn27 and Ti41Ni50Sn9, respectively. Details on atom site preference in the crystal structure of τ2 will be discussed in Section 3.3.
In contrast to the full Heusler phase (τ2), the half Heusler phase (HH, τ1, MgAgAs-type) has a much lower thermodynamic stability, resulting in a smaller primary crystallization field. τ1 forms incongruently (Fig. 8k) by the invariant peritectic reaction l + τ2 ↔ τ1 at 1180 °C. τ1 shows a homogeneity region that tends toward τ2. Thus, the sample Ti29Sn42Ni29 after annealing at 1100 °C (Fig. 8n) reveals two compositions for the two Heusler phases (τ1-Ti30.8Sn38.1Ni31.1 and τ2-Ti26.8Sn46.2Ni26.9 in at%) and Ni3Sn4, which forms from liquid during quenching. The crystallization of as-cast samples with tin contents outside the section Ni3Sn2–τ2–τ1–Ti5Sn3 finishes with the formation of a tin rich liquid (Fig. 6b, c, o, 8a, b, d, g and k). Consequently, all samples from this region, that were annealed at 800 and 950 °C, were found to be in equilibrium with the Sn-rich liquid (see fields labelled “s”, “t”, “u” in Fig. 5). Such a behavior suggests a cascade of transition type reactions (U) with temperatures decreasing from this section to the Sn-rich corner of the diagram. In order to produce equilibrium samples for the determination of these reaction temperatures, the samples were annealed in the temperature range from 450 to 770 °C. Comparing the microstructures of the as-cast alloys with those annealed at different temperatures (Fig. 7a–c, 8c, e and h) and considering the temperatures determined by DTA, four invariant reactions were established: Ub, Uc, Ud, Ue.
Besides of the Heusler phase (τ2), the solid solution based on binary Ti5Sn3 (Ti5NixSn3) also plays a dominant role in the formation of phase equilibria in the ternary system. Although this phase forms incongruently in the binary system and crystallizes in a narrow field from the liquid, the primary field of crystallization in the ternary extends up to 40 at% Ni (Fig. 1 and 3). The solubility of nickel in Ti5Sn3 reaches a maximum extent at Ti5NiSn3, significantly increasing the thermodynamic stability of this phase (see DFT calculations in Section 3.4.2.1).
As-cast samples located within the composition triangle Ti5NiSn3–Ti2Ni2Sn–TiNi and in the primary crystallization field of Ti5NiSn3 show similar but rather complicated microstructures. As an example Fig. 8j represents the crystallization of four phases in the as cast sample Ti48Ni34Sn18: primary Ti5NixSn3 (Ti55.5Ni11.4Sn33.0) followed by the crystallization of τ2 (Ti37.8Ni49.5Sn12.7), τ3-Ti2Ni2Sn (Ti43.8Ni40.1Sn16.1), TiNi (Ti44.8Ni49.2Sn6.0) and a two-phase eutectic TiNi + Ti5NixSn3 (Ti49.1Ni38.0Sn12.9).
Eutectics with similar compositions are observed in several other samples (see small triangles in Fig. 3) and they are mainly located inside the three-phase region: τ3 + TiNi + Ti5NiSn3. For example Fig. 6k shows the microstructure of the as-cast sample Ti53Ni23Sn24 with a primary crystallization of Ti5NixSn3 (Ti56.1Ni10.7Sn33.2) and a eutectic with the composition: Ti48.8Ni38.2Sn13.0. As the composition of the liquid of the afore mentioned samples never crosses the tie-line TiNi − Ti5NiSn3 during crystallization, we can define the eutectic type reaction, En: L ↔ TiNi + Ti2Ni2Sn + Ti5NixSn3, and the quasi-binary reaction, emax,no: l ↔ TiNi + Ti5NiSn3. At higher titanium content, we already observe the crystallization of Ti3Sn. In the sample Ti60Ni20Sn20, Ti3Sn with the composition Ti72.9Ni2.4Sn24.7 solidifies after Ti5NiSn3 (Ti56.5Ni11.0Sn32.5) (Fig. 6l). Besides Ti3Sn, the microstructure shows two eutectics with very close compositions ∼ Ti56Ni35Sn9 (TiNi + Ti5NiSn3, bright eutectic) and ∼Ti57Ni33Sn10 (TiNi + Ti3Sn, dark eutectic). From the location of these eutectics (see Fig. 3) the transition type reaction Uo was assigned: L + Ti5NiSn3 ↔ TiNi + Ti3Sn. The crystallization of as-cast samples from this region (Fig. 7l–n), the phase composition and temperatures measured on the samples annealed at 950 °C (Fig. 6l–n), allow us to define another cascade of transition type reactions: Uo (1107 °C), Up (984 °C) and Uq (969 °C). Phase equilibria involving Ti2Sn (phase region “r”) were not investigated, because the equilibration occurs at temperatures exceeding the technical limits of our furnaces and DTA.
The sample with stoichiometry τ3-Ti2Ni2Sn, in as-cast state (Fig. 7i), shows primary crystallization of Ti5NiSn3 (Ti55.4Ni11.7Sn32.9), than solidifies as τ2 with two compositions (Ti27.7Ni48.1Sn24.1 and Ti32.5Ni48.8Sn18.8) and subsequently we observe the crystallization of τ3 with composition Ti43.6Ni40.1Sn16.2. The crystallization is finished by solidification of TiNi (Ti43.1Ni49.6Sn7.3) and the formation of a two-phase eutectic TiNi + Ti5NixSn3 with the composition Ti49.3Ni37.0Sn13.8. Such a crystallization behavior indicates a peritectic formation of this phase via the reaction Pl: L + τ2 + Ti5NixSn3 ↔ τ3. The sample annealed at 800 and 950 °C was not completely equilibrated and contains four phases (τ2, τ3, Ti5NixSn3 and TiNi), however, after annealing at 1050 °C (Fig. 8i), TiNi disappears and only three equilibrium phases remain: τ2 (Ti28.9Ni48.8Sn22.3), τ3 (Ti41.2Ni40.0Sn18.8) and Ti5NixSn3 (Ti55.7Ni11.0Sn33.3). The temperature of the invariant equilibrium P1 at 1151 °C is derived from DTA on the sample annealed at 1050 °C. A similar morphology that confirms the peritectic crystallization of τ3 is observed for the as-cast sample Ti40Ni46Sn14 (Fig. 7h) indicating a peritectic solidification of TiNi (Ti41.6Ni49.9Sn8.5) around τ2 with compositions varying from Ti30.7Ni49.5Sn19.9 to Ti29.4Ni49.4Sn21.2. The solidification in this sample also ends in the two-phase eutectic TiNi + Ti5NixSn3 with composition Ti50.6Ni34.5Sn14.9. All these observations establish a transition type reaction Uk: L + τ2 ↔ τ3 + TiNi at 1143 °C.
Another four-phase equilibrium, that involves the Heusler phase (τ2) and binary TiNi is documented for the sample Ti37Ni57Sn6. In as-cast state (Fig. 6g), we observe primary crystallization of τ2 with compositions Ti33Ni51Sn16 and Ti36Ni53Sn11 for big and small grains, and a fine eutectic with overall composition Ti36Ni61Sn13: the eutectic consists of TiNi (Ti40Ni55Sn5) and TiNi3, and significantly coagulates after annealing at 950 °C for 10 days (Fig. 7g). As the composition of the eutectic lies outside of the three-phase triangle τ2 + TiNi3 + (Ti1−xNix)1−ySny (field “j” on Fig. 3), a transition type reaction Uj is defined: L + τ2 ↔ TiNi3 + (Ti1−xNix)1−ySny. However, the temperature of this reaction, determined by DTA is 2 °C lower than the respective reaction l ↔ TiNi3 + TiNi (1118 °C) in the binary system.
Binary TiNi3 has the highest melting point (1380 °C) within the Ti–Ni system and also exhibits rather extended Ti/Sn substitution (Ti1−xSnx)Ni3 up to xmax = 0.27 at 1080 °C. However, at a higher tin content, the hexagonal structure of (Ti1−xSnx)Ni3 undergoes a structural transformation into a cubic structure with AuCu3-type: τ4-(Ti1−x−ySnxNiy)Ni3. The homogeneity region of this phase extends at 1080 °C from x = 0.22 and y = 0.22 (in equilibrium with (Ti1−xSnx)Ni3 and (Ni)) to x = 0.54 and y = 0.06 (in equilibrium with Ni3Sn and (Ni)). However, the maximal solubility of tin in τ4 at temperatures below solidus increases to 16.4 at% (x = 0.66 and y = 0.1) at 950 °C) as determined from an annealed alloy Ti5Ni80Sn15 (Fig. 7d) in equilibrium with Ni3Sn and (Ni).
The sample in as cast state shows primary crystallization of (Ni) with grains of τ4-(Ti1−x−ySnxNiy)Ni3 solidifying around them, and both phases are embedded in a Ni3Sn matrix (Fig. 6d). In alloy Ti10Ni76Sn14, τ4 crystallizes as a primary phase with composition Ti13.0Ni77.2Sn9.8 and solidification finishes in a two-phase eutectic (τ4 + Ni3Sn) with composition Ti5.8Ni74.6Sn19.6. This type of solidification indicates incongruent formation of this phase during a peritectic reaction. But our guess on the formation of this phase in a three-phase peritectic, L + (Ti1−xSnx)Ni3 ↔ τ4, was ruled out by the observation that for the as-cast alloys with nickel contents from 75 to 81 at% Ni the last portion of the liquid becomes depleted by nickel. Such a solidification behavior agrees well with a cascade of reactions, peritectic Pw: L + (Ni) + (Ti1−xSnx)Ni3 ↔ τ4 followed by a transition type reaction Ug: L + (Ni) ↔ Ti1−xSnx)Ni3 + Ni3Sn.
The temperature of the invariant reaction Ug (1113 °C) is only one degree higher than that determined for Eh: L ↔ Ni3Sn + Ni3Sn2 + τ4 (1112 °C), which is evident from the microstructure of the as-cast alloy with nominal composition Ti7Ni70Sn23 (Fig. 6e): we observe primary crystallization of Ni3Sn2, continued by a solidification of NiSn3 and the final liquid crystallizes in form of a eutectic with composition Ti7.5Ni71.7Sn20.8. The composition of this eutectic lies outside of the tie-triangle Ni3Sn + Ni3Sn2 + τ4 determined on a sample annealed at 950 °C (Fig. 7e, Table S2 (ESI†)), but it is located inside of this three-phase field as it was established for the sample annealed at 1080 °C. It should be noted, that this invariant eutectic is much finer than the two-phase mono-variant eutectic l → (Ti1−xSnx)Ni3 + Ni3Sn2 that is observed in the as-cast sample Ti18Ni58Sn24 (Fig. 7f). The microstructure of the latter sample also documents a secondary crystallization of Ni3Sn2 after primary τ2. Considering this and the fact that the composition of this mono-variant eutectic (Ti10Ni69Sn21) is located slightly outside of the three-phase field (Ti1−xSnx)Ni3 + Ni3Sn2 + τ2 (“i” in Fig. 3), the invariant reaction Ui: L + τ2 ↔ (Ti1−xSnx)Ni3 + Ni3Sn2 was established. Another transition-type invariant three-phase reaction Uv: L + (Ti1−xSnx)Ni3 ↔ τ4 + Ni3Sn2 located between the afore mentioned reactions was established at 1114 °C. The respective three-phase field on the solidus is rather narrow and we were unable to detect the eutectic structure that would document this invariant reaction.
An additional uncertainty in the determination of phase equilibria in this region arises from the characterization of the samples by XPD that show the existence of a further novel phase with composition Ti1−xSnxNi3 (0.36 ≤ x ≤ 0.48), which exists in the Ni-poor part of the solid solution of τ4-(Ti1−xSnxNiy)Ni3. In order to determine details on the crystal structure and phase equilibria involving this phase, (labelled as τ4′-Ti1−xSnxNi3) additional investigations need to be performed.
The invariant four-phase reactions established in the system are summarized in a Schulz–Scheil diagram (Fig. 4), and in order to complete this crystallization scheme, some additional invariant three-phase reactions (emax) are requested. Besides the above discussed quasi-binary eutectic emax,no: l ↔ TiNi + Ti5NiSn3, the high melting τ2 phase forms a set of invariant eutectics with the solid solutions of binary compounds Ti5Sn3 (elm), TiNi (ejk), TiNi3(ejk) and Ni3Sn2 (ebi). The microstructure of the as-cast alloy Ti8Ni57Sn35 (Fig. 8m) represents a fine eutectic structure (with composition Ti6.3Ni57.6Sn36.1) that forms via the invariant reaction ebi: l ↔ τ2 + Ni3Sn2. The temperature of this reaction is 1201 °C, being above the neighboring invariant reactions Ub (793 °C) and Ui (1120 °C). This provides an additional proof for this quasi-binary eutectic. The isothermal section at 950 °C (see Fig. 6) represents phase equilibria similar to those reported earlier at 800 °C (ref. 4) for exception that two additional three-phase fields involving τ4 are present in the up-dated section. The difference in extension of the homogeneity regions for solid solutions may be interpreted as the typical shrinkage of single phase fields with decrease of temperature as well as by the general difficulties of equilibration of samples at low temperatures. In this context we need to mention difficulties in the homogenization of alloys containing the high melting compounds τ2, Ti5NixSn3, TiNi and (Ti1−xSnx)Ni3 at 950 °C and even at sub-solidus temperatures (1050–1100 °C). Therefore thermodynamic modelling of the phase diagram is more suitable to describe the equilibrium compositions at temperatures below 1000 °C.
3.3. Crystal structures and homogeneity regions of ternary phases
With respect to the importance of crystallographic data for defining models for the thermodynamic parameters we performed a number of Rietveld refinements for τ1-TiNiSn, τ2-Ti1+yNi2−xSn1−y, (Ti1−xSnx)Ni3 and Ti5NixSn3 (see Table S3 (ESI†)). The half Heusler phase (τ1) shows a narrow homogeneity region that is slightly off-stoichiometric on the tin-rich side Ti32.6Ni34.6Sn32.8 at 1100 °C, and Ti32.6Ni34.3Sn33.1 at 950 °C) and extends up to Ti30.9Ni37.8Sn31.3 in equilibrium with τ2 at 1100 °C. Rietveld refinement for TiNiSn confirms full atom order in the structure (Table S3 (ESI†)). The Heusler phase (τ2-Ti1+yNi2−xSn1−y) exhibits the biggest homogeneity region that extends at 1100 °C to Ti27Ni46Sn27 (x = 0.30 and y = 0.00 in equilibrium with τ1), to Ti41Ni50Sn9 (x = 0 and y = 0.64 in equilibrium with TiNi) and to Ti19Ni57Sn24 (x = −0.65 and y = −0.12 in equilibrium with TiNi3 and Ni3Sn2).Rietveld refinements (Table S3 (ESI†)) confirm a full order for stoichiometric TiNi2Sn, whereas Ti/Sn substitution occurs in sites 4a (0, 0, 0) and 4b (½, ½, ½), respectively for tin-poor and nickel-rich extensions of the homogeneity region. The refinement did not reveal any substantial deviation from a full occupancy of Ni in site 8c (¼, ¼, ¼). It seems that this site is only affected for compositions that extend from stoichiometric τ2-TiNi2Sn towards τ1-TiNiSn. Considering the close crystallographic relation between both Heusler phases and TiNi (all are derivatives of the W-type structure, we plotted the compositional dependence of the lattice parameters for the section TiNi–TiNi2Sn (Fig. 9). One can see that lattice parameters increase with increasing tin content in the 4a site (0, 0, 0) and the dependence shows a positive deviation from Vegard's law. Maximal lattice parameters (a ≈ 0.610 nm) are observed for compositions near stoichiometric τ2-TiNi2Sn and the unit cell shrinks when Sn-atoms substitute for Ti in the 4b site (½, ½, ½). Due to the small difference in lattice parameters for τ2 and the TiNi-based solution (Ti1−xNix)1−ySny the XPD reflexes for these phases strongly overlap and we were unable to perform an unambiguous deconvolution of the diffraction maxima and to determine precise lattice parameters for the equilibrium compositions. However, these compounds appear as well separated individual phases on SEM images in as-cast (Fig. 6h and 8o) and annealed (Fig. 7g) alloys, allowing a reliable determination of the compositions by EPMA. A much more significant decrease of the lattice parameters from a ≈ 0.610 nm to a ≈ 0.593 nm is observed for the structural change from τ2-TiNi2Sn to τ1-TiNiSn and because of this fact these two phases are well distinguishable in XPD profiles.
 | ||
| Fig. 9 Compositional dependence of lattice parameters for homogeneity regions for the phases: TiNi, τ1-TiNiSn and τ2-TiNi2Sn. The composition of the solid solution is expressed as Ti1+yNi2−xSn1−y. Extensions of the homogeneity regions at 1100 °C are marked with dashed lines. Literature (open symbols), squares;32,36,44–51 circles;1,5,9,52–56 triangles-up.57 | ||
Compositional dependence of lattice parameters for the new compound τ4-(Ti1−x−ySnxNiy)Ni3 with AuCu3-type structure is shown in Fig. 10. Lattice parameters increase with increase of tin content and this dependence extrapolates to the value of a = 0.3738 nm reported for the high pressure modification of Ni3Sn which also adopts the AuCu3 type structure.
 | ||
| Fig. 10 Compositional dependence of lattice parameters for the homogeneity region of τ4-(Ti1−x−ySnxNiy)Ni3 (0.02 ≤ x ≤ 0.22) for samples annealed at 950 °C (circles) and 1080 °C (squares). Diamonds: calculated values (this work). Triangle: isotypic high-pressure modification of Ni3Sn.28 | ||
The close structural relation between AuCu3 and the crystal structure of Ni (Cu-type) allows a second order transformation between these structures. However, the phase separation between these structures was clearly documented by SEM images for sample Ti5Ni80Sn15 annealed at 950 °C (Fig. 7d) and was confirmed by XPD showing two sets of diffraction spectra of cubic structures with primitive (τ4) and face-centered (Ni) lattices. We note that primitive X-ray diffraction peaks from the τ4 lattice almost vanish at low Sn contents (zero intensity for Ti0.786Sn0.214Ni3) due to the fact that the electron density in both crystallographic sites becomes similar. This may lead to possible misinterpretation of XPD patterns of this phase with the fcc nickel-based solid solution.
Furthermore, we subjected the ternary solid solution Ti5NixSn3 (Hf5CuSn3-type structure; i.e. a filled variant of binary Ti5Sn3 with the Mn5Si3-type) to a detailed crystallographic investigation. Due to non-equilibrium solidification of as-cast samples Ti5NixSn3 with x = 0.9 and 1.0, EPMA line scans showed that the nickel content in this phase increases from 6.3 at% Ni at the grain centre to 10.2 at% Ni at the rim of the grains. This heterogeneity results in some broadening of the XPD reflections. Annealing at 1100 °C for 6 days provides a complete homogenization of the composition, but a comparison of XPD profiles for as-cast and annealed samples shows that the half-width of only some reflections is reduced whilst others show a clear split suggesting a structural transformation in this phase. Several single crystals were selected from the annealed sample but inspection on an AXS-GADDS texture goniometer did not reveal any significant distortion of the unit cell. One crystal specimen was characterized by means of four-circle Nonius Kappa diffractometer intensity data. Although the measured data were processed in triclinic symmetry, single crystal refinement fully complies with the symmetry and atom site distribution of the Hf5CuSn3-type structure (ordered Ti5Ga4-type; for crystallographic details see Table 3).
| Parameter | Ti5Ni0.96Sn3 |
| EPMA, at% | Ti56.0Ni10.0Sn34.0 |
| Refinement; at% | Ti55.8Ni10.7Sn33.5 |
| a; c [nm] | 0.81440(1); 0.555922(9) |
| Reflections in refinement | 304Fo > 4sig(Fo) of 309 |
| Number of variables | 16 |
| RF = ∑|F0 − Fc|/∑F0 | 0.0298 |
| RInt | 0.0303 |
| wR2 | 0.0595 |
| GOF | 1.143 |
| Extinction (Zachariasen) | 0.0021 |
| Ti1 in 6g (x, 0, ¼), x; occ. | 0.2550(4); 1.00(1) |
| U11; U22 | 0.052(1); 0.0227(9) |
| U33; U23 = U13 = 0; U12 [nm2] | 0.0252(8); 0.0114(4) |
| Sn1 in 6g (x, 0, ¼), x; occ. | 0.61161(8); 1.002(7) |
| U11; U22 | 0.0253(3); 0.0077(2) |
| U33; U23 = U13 = 0; U12 [nm2] | 0.0117(2); 0.0038(1) |
| Ti2 in 4d (1/3, 2/3, 0), occ. | 0.996(8) |
| U11 = U22; U33 | 0.0098(1); 0.0063(5) |
| U23 = U13 = 0; U12 [nm2] | 0.0045(2) |
| Ni in 2b (0, 0, 0), occ. | 0.96(2) |
| U11 = U22; U33 | 0.094(3); 0.066(3) |
| U23 = U13 = 0; U12 [nm2] | 0.047(2) |
| Residual density: max; min | 2.31; −3.42 |
| Principal mean square atomic | Ti1 0.0620, 0.0252, 0.0227 |
| Displacements (U) | Sn1 0.0312, 0.0117, 0.0077 |
| Ti2 0.0091, 0.0091, 0.0063 | |
| Ni 0.0945, 0.0945, 0.0660 |
3.4. Thermodynamic assessment of the Ti–Ni–Sn system
3.4.2.1. DFT calculations of ternary compounds TiNiSn, TiNi2Sn, Ti2Ni2Sn and of the solid solutions Ti5Ni1−xSn3, Ti1−xSnxNi3 and (Ti1−xNix)1−ySny.
Ti5Ni1−xSn3. The modelling of the Ti5Ni1−xSn3 solid solution consisted of two steps. At the first step the ground state energy of Ti5Sn3 with optimised crystal structure was calculated (Table 4). The negative enthalpy of formation (Table 5) proves the possibility of its formation. At the second step the chemical composition of the compound was modified by introducing additional Ni atoms into the 2b site to fit the Ti5NiSn3 composition. After that the ground state energy of Ti5NiSn3 with optimised crystal structure (Table 4) was calculated. Comparing the enthalpy of formation of Ti5Sn3 and Ti5NiSn3 (Table 5) one can see that it is lower for Ti5NiSn3, which means that the filling of the 2b site with Ni atoms is energetically favourable and predicts the formation of the interstitial solid solution Ti5Ni1−xSn3. For further thermodynamic calculations the ground state energy and the enthalpy of formation were calculated for the hypothetical compound Ni5NiNi3 (Table 5) with the same structure as Ti5NiSn3 and optimised crystal structure geometry (Table 4).
| Atom | Wyckoff | x/a | y/b | z/c |
|---|---|---|---|---|
| Ti5Sn3 (a = 0.8102119, c = 0.5453824 nm) | ||||
| Ti1 | 6g | 0.242235 | 0 | 0.25 |
| Ti2 | 4d | 1/3 | 2/3 | 0 |
| Sn1 | 6g | 0.608723 | 0 | 0.25 |
|
||||
| Ti5NiSn3 (a = 0.8250848, c = 0.5511669 nm) | ||||
| Ti1 | 6g | 0.256722 | 0 | 0.25 |
| Ti2 | 4d | 1/3 | 2/3 | 0 |
| Sn1 | 6g | 0.610405 | 0 | 0.25 |
| Ni1 | 2b | 0 | 0 | 0 |
|
||||
| Ni5NiNi3 (a = 0.6970264, c = 0.4840398 nm) | ||||
| Ni1 | 6g | 0.333297 | 0 | 0.25 |
| Ni2 | 4d | 1/3 | 2/3 | 0 |
| Ni3 | 6g | 0.666532 | 0 | 0.25 |
| Ni4 | 2b | 0 | 0 | 0 |
|
||||
| SnNi3 (a = 0.535448, c = 0.8647618 nm) | ||||
| Sn1 | 2a | 0 | 0 | 0 |
| Sn2 | 2d | 1/3 | 2/3 | 3/4 |
| Ni1 | 6g | 1/2 | 0 | 0 |
| Ni2 | 6h | 0.177094 | 0.354189 | 1/4 |
|
||||
| TiNi3 (a = 0.5137315, c = 0.8382629 nm) | ||||
| Ti1 | 2a | 0 | 0 | 0 |
| Ti2 | 2d | 1/3 | 2/3 | 3/4 |
| Ni1 | 6g | 1/2 | 0 | 0 |
| Ni2 | 6h | 0.171415 | 0.34283 | 1/4 |
|
||||
| Ti0.5Sn0.5Ni3 (a = 0.5237464, c = 0.8544106 nm) | ||||
| Ti1 | 2a | 0 | 0 | 0 |
| Sn2 | 2d | 1/3 | 2/3 | 3/4 |
| Ni1 | 6g | 1/2 | 0 | 0 |
| Ni2 | 6h | 0.171782 | 0.343565 | 1/4 |
|
||||
| Sn0.5Ti0.5Ni3 (a = 0.5248831, c = 0.8498294 nm) | ||||
| Sn1 | 2a | 0 | 0 | 0 |
| Ti2 | 2d | 1/3 | 2/3 | 3/4 |
| Ni1 | 6g | 1/2 | 0 | 0 |
| Ni2 | 6h | 0.176477 | 0.352954 | 1/4 |
|
||||
| Ti2Sn3 (a = 0.6013709, b = 2.0176674, c = 0.7073997 nm) | ||||
| Ti1 | 8f | 0 | 0.080577 | 0.047598 |
| Ti2 | 8e | 1/4 | 0.33657 | 1/4 |
| Sn1 | 8f | 0 | 0.120959 | 0.426385 |
| Sn2 | 8f | 0 | 0.224113 | 0.073991 |
| Sn3 | 8e | 1/4 | 0.479599 | 1/4 |
| Compound | ΔH (meV per atom) | References |
|---|---|---|
| TiNiSn | −547.083 | This work |
| −715 | Ref. 5 | |
| −549 | Ref. 7 | |
| TiNi1.25Sn | −493.933 | This work |
| TiNi1.5Sn | −466.162 | This work |
| TiNi1.75Sn | −461.613 | This work |
| TiNi2Sn | −472.872 | This work |
| −622 | Ref. 5 | |
| Ti2Ni2Sn | −468.209 | This work |
| −485 | Ref. 5 | |
| Ti5NiSn3 | −404.251 | This work |
| −388 | Ref. 5 | |
| Ti5Sn3 | −385.435 | Ref. 5 |
| −348 | This work | |
| Ni5NiNi3 | +242.338 | This work |
| Ti2Sn3 | −354.214 | This work |
| TiNi3 | −524.463 | This work |
| −478 | Ref. 5 | |
| TiNi3 (Cu3Au-type) | −532.018 | This work |
| −750 | Ref. 59 | |
| −484 (LMTO-ASA) | Ref. 60 | |
| −488 (FP-LMTO) | Ref. 60 | |
| SnNi3 | −219.215 | This work |
| SnNi3 (Cu3Au-type) | −240.291 | This work |
| Ti0.5Sn0.5Ni3 | −341.340 | This work |
| Ti0.5Sn0.5Ni3 (Cu3Au-type) | −341.886 | This work |
| Sn0.5Ti0.5Ni3 | −318.140 | This work |
| TiNi | −390.862 | This work |
| −411 | Ref. 5 | |
| −610 | Ref. 59 | |
| −395 (LMTO-ASA) | Ref. 60 | |
| −373 (FP-LMTO) | Ref. 60 | |
| Ti0.875Sn0.125Ni | −364.133 | This work |
| Ti0.75Sn0.25Ni | −378.446 | This work |
| Ti0.625Sn0.375Ni | −413.794 | This work |
| Ti0.25Sn0.75Ni | −185.791 | This work |
| NiSn | −15.954 | This work |
| Ni0.75Ti0.25Sn | −106.858 | This work |
| Ni0.5Ti0.5Sn | +24.483 | This work |
| Ni0.25Ti0.75Sn | −141.487 | This work |
| TiSn | −101.017 | This work |
| TiSn0.75Ni0.25 | −126.421 | This work |
| TiSn0.5Ni0.5 | −219.074 | This work |
| TiSn0.25Ni0.75 | −232.767 | This work |
Ti1−xSnxNi3. The substitutional (Ti by Sn) solid solution based on TiNi3 was modelled in two ways. In the first case the Ti2 atoms in the 2d position and in the second case the Ti1 atoms in the 2a site were substituted by Sn atoms yielding the compositions Ti0.5Sn0.5Ni3 and Sn0.5Ti0.5Ni3, respectively. The ground state energy and the enthalpy of formation (Table 5) were calculated for SnNi3, TiNi3, Ti0.5Sn0.5Ni3, and Sn0.5Ti0.5Ni3 with a completely relaxed geometry (Table 4). The comparison of Ti0.5Sn0.5Ni3 and Sn0.5Ti0.5Ni3 at the same composition (x = 0.5) shows that the configuration of Ti0.5Sn0.5Ni3 is more preferable due to the lower enthalpy of formation value. The analysis of the ΔH values of TiNi3, SnNi3, and Ti0.5Sn0.5Ni3 predicts that the formation of the solid solution at T = 0 K is not possible. However, at higher temperature the formation of Ti1−xSnxNi3 is still possible due to the entropy contribution.
The calculations were also carried out for the high-pressure modifications (Cu3Au-type) of TiNi3 and Ni3Sn binaries and the intermediate composition Ti0.5Sn0.5Ni3. For the intermediate composition the unit cell was doubled in all three directions of the basis vectors (2a × 2b × 2c) giving in total 32 atoms in the supercell and the symmetry was reduced to P1. The optimised values of the lattice parameter a for TiNi3, Ni3Sn, and Ti0.5Sn0.5Ni3 are 0.36077234, 0.37354910, and 0.367873 nm, respectively. The slope of the a(x) dependence in the range 0 ≤ x ≤ 0.5 is higher than for the higher x values. Most likely this is due to the impact of s- and p-states of Sn atoms that could increase the covalent contribution into the system of chemical bonds.
(Ti1−xNix)1−ySny. For the solid solution based on binary TiNi three isotypic structures were modelled: TiNi, TiSn, and NiSn with optimized lattice parameter a = 0.302378757, 0.3430739, and 0.32247303 nm, respectively. The calculated ΔH values of the hypothetical solid solutions between TiNi, TiSn, and NiSn are collected in Table 5. For the intermediate compositions the unit cell was doubled in all three directions of the basis vectors (2a × 2b × 2c) giving in total 16 atoms in the supercell and the symmetry was reduced to P1. The composition Ti0.5Sn0.5Ni is omitted in Table 5 as it corresponds to the TiNi2Sn compound.
TiNi1+xSn. The interstitial solid solution based on the half-Heusler TiNiSn phase (Ti in 4a, Ni in 4c, Sn in 4b) was modelled by reducing the symmetry of the crystal to P1 and successively filling of four available voids (4d site) with Ni atoms. According to the calculated ΔH values (Table 5) the formation of limited solid solutions between TiNiSn and TiNi2Sn at T = 0 K is not possible (Table 5). For Ti2Sn3 and Ti2Ni2Sn the calculated ΔH values that were used in thermodynamic calculations are collected in Table 5 and the optimised crystallographic data of the Ti2Sn3 compound are listed in Table 4. The calculated ΔH values for the selected compounds in Ti–Ni–Sn system using full-potential elk code (GGA-PBE) are mostly in a good agreement with those obtained with pseudopotential VASP code (GGA-PBE),5 except for TiNiSn and TiNi2Sn (Table 5). This is strange, as for TiNiSn the ΔH values obtained using the same VASP code and GGA-PBE approximation by Colinet et al.7 are comparable with our results (Table 5). The heat of formation values for binary TiNi (Table 5) obtained with FLAPW, pseudopotential,5 LMTO-ASA,60 and FP-LMTO60 methods are comparable with experimental data collected in ref. 59, while the value calculated using the semi-empirical tight-binding method59 significantly differs.
The calculated ΔH value for the high-pressure modification of TiNi3 (Table 5) using the FLAPW method is comparable with those obtained by LMTO-ASA and FP-LMTO methods,60 while for the semi-empirical tight-binding method59 the ΔH value is significantly higher. It is interesting to note, that calculations showed higher absolute values of ΔH for the high-pressure modifications of TiNi3, Ni3Sn and intermediate Ti0.5Sn0.5Ni3 in comparison with the normal-pressure modification.
Douglas et al. observed an increase of lattice parameter a for the TiNi1+xSn solid solution in the range 0 ≤ x ≤ 0.1 according to the XRD data.5 The comparison of optimized (DFT) and experimental values of lattice parameter with a set from ref. 5 (Fig. 11) shows that the theoretical and interpolated experimental dependencies are almost parallel, while the slope of the reference dependence is more horizontal. Such deviation in slope means that the x(Ni) values corresponding to the lattice parameters in ref. 5 might be significantly lower. To explain the increase of lattice parameter we have modelled a structure that consists of four TiNiSn unit cells where one Ni atom from each of them diffused to form one full-Heusler TiNi2Sn unit cell. This is a model of the coherent grain boundary between TiNiSn and TiNi2Sn phases. The crystal structure geometry optimisation for this model shows that the lattice parameters of TiNiSn and TiNi2Sn unit cells vary in the ranges 0.58693 ÷ 0.58686 and 0.60766 ÷ 0.60762 nm, respectively (Fig. 12).
 | ||
| Fig. 11 Concentration dependences of optimized (DFT), experimental and reference5 values of lattice parameter a for the TiNi1+xSn solid solution. | ||
 | ||
| Fig. 12 The initial and relaxed geometry of the hypothetical TiNiSn–TiNi2Sn structure. | ||
The projection of the electron localization function (Fig. 13) shows localized maxima between Ni and Sn atoms within the half-Heusler (HH) cell and somewhat lower in the full-Heusler (FH) cell. The calculations predict that the presence of the TiNi2Sn phase strongly affects the TiNiSn lattice parameter value due to the stress on the grain boundary between Heusler and half-Heusler phases. The distribution of the total density of states (Fig. 14) for TiNiSn, TiNi1.25Sn, TiNi2Sn, and TiNiSn–TiNi2Sn grain boundary confirmed that TiNiSn is a semiconductor and TiNi2Sn is characterized by a metallic type of conductivity. In the case of TiNi1.25Sn (MgAgAs-type) the additional Ni atoms in position 4d generate a new band inside the energy gap and eliminate it. Thus the material predicted has metallic type of conductivity similar to TiNi2Sn. The DOS distribution at the grain boundary of TiNiSn–TiNi2Sn significantly differs from those in pure TiNiSn and TiNi2Sn. The main difference between TiNiSn and TiNi2Sn is that the energy bands are strongly delocalised. Like in the case of TiNi1.25Sn and TiNi2Sn the electrical conductivity at the grain boundary is predicted to be metallic. The deformation of the crystal structure at the TiNiSn–TiNi2Sn grain boundary leads to additional phonon scattering and decrease of the thermal conductivity of the material. The increased electrical conductivity and decreased thermal conductivity should have a positive effect on the value of the thermoelectric figure of merit (Z).
 | ||
| Fig. 13 Distribution of the electron localization function along the lattice plane in the TiNiSn–TiNi2Sn structure. | ||
 | ||
| Fig. 14 Distribution of the total density of states (per formula unit) for TiNiSn, TiNi1.25Sn, TiNi2Sn, and for the grain boundary TiNiSn–TiNi2Sn. The Fermi level (EF) is at E = 0 eV. | ||
3.4.2.2 Results of thermodynamic modelling. For thermodynamic modelling of the stability of ternary compounds TiNiSn, TiNi2Sn and Ti2Ni2Sn, the DFT calculated values of the formation energy (ΔH) (Table 5) were used and the entropic term was optimized. Similarly, DFT data were used for Ti2Sn3. Data for Ti5Sn3 were taken from ref. 24 and the line solubility to Ti5NiSn3 was optimized. The mutual solubility of TiNi3 and SnNi3 was modelled on the basis of binary data from ref. 23 and 26 and optimized. Stability of TiNi was modelled as B2_BCC and BCC_A2 binary phases25,26 with optimized solubility of Sn. Note, both phases TiSn and NiSn are in the respective binary systems. The parameters used for the calculations are presented in Table 6. The results of the thermodynamic optimisation are summarized in a liquidus surface (Fig. 15), in a series of isopleths: Ni–TiSn, Sn–TiNi, NiSn–TiSn (Fig. 16), as well as in a series of isothermal sections: 800 °C, 900 °C, 950 °C (Fig. 17), 1050 °C and at 1200 °C (Fig. 18).
| Phase | Ref. |
|---|---|
| a This work.b Optimization.c DFT. | |
| LIQUID/(Ni,Sn,Ti) | |
| (0)L(LIQUID,NI,Ti) = −153707.39 + 34.859449 × T |
26 |
| (1)L(LIQUID,NI,TI) = −81824.755 + 25.809901 × T |
26 |
| (2)L(LIQUID,NI,TI) = −10.077897 × T | 26 |
| (0)L(LIQUID,SN,TI) = −91598.90 − 0.9416 × T |
24 |
| (1)L(LIQUID,SN,TI) = 45682.64 − 12.1045 × T |
24 |
| (0)L(LIQUID,NI,SN) = −104602.87 + 197.8089 × T − 21.6959 × T × LN |
23 |
| (1)L(LIQUID,NI,SN) = −30772.17 + 52.5528 × T − 7.56094 × T × LN(T) |
23 |
| (2)L(LIQUID,NI,SN) = 6582.31 | 23 |
| Ternary parameter | |
| (0)L(LIQUID,NI,SN,TI) = −10000 |
a,b |
|
|
| Ni3Sn2/(Ni,Va)1 (Ni,Va)1 (Ni,Sn,Ti)1 | |
| (0)G(NI3SN2,NI:NI:NI) = 31637.299 + 355.176064 × T − 66.288 × T × LN(T) − 0.0145221 × T × 2 |
23 |
| (0)G(NI3SN2,VA:NI:NI) = 21091.532 + 236.784043 × T − 44.192 × T × LN(T) − 0.0098814 × T × 2 |
23 |
| (0)G(NI3SN2,NI:VA:NI) = 21091.532 + 236.784043 × T − 44.192 × T × LN(T) − 0.0098814 × T × 2 |
23 |
| (0)G(NI3SN2,VA:VA:NI) = 10545.766 + 118.58688 × T − 22.096 × T × LN(T) − 0.0048407 × T × 2 |
23 |
| (0)G(NI3SN2,NI:NI:SN; 0) = 2 × GHSERNI + GHSERSN − 83734.818 + 14.6888165 × T |
23 |
| (0)G(NI3SN2,VA:NI:SN) = 5000 + GHSERNI + GHSERSN | 23 |
| (0)G(NI3SN2,NI:VA:SN) = GHSERNI + GHSERSN − 52177.98 + 10.774 × T |
23 |
| (0)G(NI3SN2,VA:VA:SN) = GHSERSN + 20000.0 |
23 |
| (0)L(NI3SN2,NI:NI,VA:SN) = −9784.4 − 12.385 × T | 23 |
| (1)L(NI3SN2,NI:NI,VA:SN) = +12000.0 |
23 |
| Ternary parameters | |
| (0)G(NI3SN2,NI:NI:TI) = GHSERTI + 2 × GHSERNI − 76451.5 + 3.08647 × T |
a |
| (0)G(NI3SN2,VA:NI:TI) = GHSERTI + GHSERNI + 123451.5 + 3.08647 × T |
a |
| (0)G(NI3SN2,NI:VA:TI) = GHSERTI + GHSERNI − 67451.5 + 3.08647 × T |
a |
| (0)G(NI3SN2,VA:VA:TI) = GHSERTI + 5451.5 + 3.08647 × T | a |
| (0)L(NI3SN2,NI:NI:TI,SN) = −50000 |
a |
|
|
| Ni3Sn4/(Ni)0.25(Ni,Sn)0.25(Sn)0.5 | |
| (0)G(NI3SN4,NI:NI:SN) = −25078.56 + 4.291 × T + 0.5 × GHSERNI + 0.5 × GHSERSN |
23 |
| (0)G(NI3SN4,NI:SN:SN) = +7613.24 + 8.749 × T + 0.25 × GHSERNI + 0.75 × GHSERSN | 23 |
| (0)L(NI3SN4,NI:NI,SN:SN) = −52928.16 |
23 |
|
|
| Ni3Sn_LT_TYPE/(Ni,Sn)0.75(Ni,Sn)0.25 | |
| (0)G(NI3SN_LT_TYPE,NI:NI) = +6300 + GHSERNI | 23 |
| (0)G(NI3SN_LT_TYPE,SN:NI) = +5000 + 0.25 × GHSERNI + 0.75 × GHSERSN | 23 |
| (0)G(NI3SN_LT_TYPE,NI:SN) = −28408 + 7.0009 × T + 0.75 × GHSERNI + 0.25 × GHSERSN |
23 |
| (0)G(NI3SN_LT_TYPE,SN:SN) = +5000 + GHSERSN | 23 |
|
|
| FCC_A1/(Ni,Sn,Ti)1 (Va)1 | |
| (0)L(FCC_A1,NI,TI:VA) = −98143 + 6.706 × T |
26 |
| (1)L(FCC_A1,NI,TI:VA; 1) = −62430 |
26 |
| (0)TC(FCC_A1,NI:VA) = +633 | 18 |
| (0)BMAGN(FCC_A1,NI:VA) = +0.52 | 18 |
| (0)TC(FCC_A1,NI,TI:VA) − 2500s | 26 |
| (1)TC(FCC_A1,NI,TI:VA) = −3000 | 26 |
| (2)TC(FCC_A1,NI,TI:VA) = +1300 | 26 |
| (0)BM(FCC_A1,NI,TI:VA) = 50000 |
26 |
| (0)L(FCC_A1,NI,SN:VA) = −69507.35 + 74.5697727 × T − 8.0319551 × T × LN(T) |
23 |
| (1)L(FCC_A1,NI,SN:VA) = −12395.19 |
23 |
| (0)TC(FCC_A1,NI,SN:VA) = −6000 | 22 |
| (1)TC(FCC_A1,NI,SN:VA) = 3000 | 22 |
| (0)BMAGN(FCC_A1,NI,SN:VA) − 6.8002 | 23 |
| (1)BMAGN(FCC_A1,NI,SN:VA) = 4.3689 | 23 |
| Ternary parameter | |
| (0)L(FCC_A1,NI,SN,TI:VA) = −170000 |
a |
|
|
| BCC_A2/(Ni,Ti,Sn,Va)1(Va)3 | |
| (0)L(BCC_A2,NI,TI:VA) = −97427 + 12.112 × T |
26 |
| (1)L(BCC_A2,NI,TI:VA1) = −32315 |
26 |
| (0)TC(BCC_A2,NI,TI:VA) = −575 | 26 |
| (0)G(BCC_A2,VA:VA) = 80 × T | 26 |
| (0)BMAGN(BCC_A2,NI,TI:VA) = −0.85 | 26 |
| (0)L(BCC_A2,NI,SN:VA) = +2369 + 43.736 × T | 23 |
| (1)L(BCC_A2,NI,SN:VA) = −654762.56 + 63.272352 × T |
23 |
| (2)L(BCC_A2,NI,SN:VA) = +689895.17 − 94.74087 × T |
23 |
| (0)L(BCC_A2,SN,TI:VA) = −142089.52 + 28.14226 × T |
24 |
| (1)L(BCC_A2,SN,TI:VA) = 42811.467 |
24 |
| Ternary parameter | |
| (0)L(BCC_A2,NI,SN,TI:VA) = +30000 |
a |
|
|
| Disordered part of BCC_B2, identical with BCC_A2 | |
|
|
| A2_BCC/(Ni,Ti,Sn)1(Va)3 | |
| (0)L(A2_BCC,NI,TI:VA; 0) = −97427 + 12.112 × T |
26 |
| (1)L(A2_BCC,NI,TI:VA) = −32315 |
26 |
| (0)TC(A2_BCC,NI,TI:VA) = −575 | 26 |
| (0)BMAGN(A2_BCC,NI,TI:VA; 0) = −0.85 | 26 |
|
|
| HCP_A3/(Ni,Ti,Sn)1(Va)0.5 | |
| (0)L(HCP_A3,NI,TI:VA) = −20000 |
26 |
| (0)L(HCP_A3,NI,SN:VA) = 2000 | a |
| (0)G(HCP_A3,SN,TI:VA) = −127549.582 + 23.2048828 × T |
24 |
| (1)G(HCP_A3,SN,TI:VA) = 64500.46 + 7.7566 × T |
24 |
| (2)G(HCP_A3,SN,TI:VA) = 31287.55 |
24 |
|
|
| BCC_B2/(Ni,Ti,Sn)1(Ni,Ti,Sn)1(Va)3 | |
| (0)G(BCC_B2,NI:TI:VA) = −33193 + 10.284 × T |
26 |
| (0)G(BCC_B2,TI:NI:VA) = −33193 + 10.284 × T |
26 |
| (0)G(BCC_B2,NI:SN:VA) = −40000 + 11 × T |
a |
| (0)G(BCC_B2,SN:NI:VA) = −40000 + 11 × T |
a |
| (0)G(BCC_B2,TI:SN:VA) = −40000 + 11 × T |
a |
| (0)G(BCC_B2,SN:TI:VA) = +40000 + 11 × T |
a |
| (0)L(BCC_B2,TI:NI,TI:VA) = +60723.7 − 15.4024 × T |
26 |
| (0)L(BCC_B2,NI,TI:TI:VA) = +60723.7 − 15.4024 × T |
26 |
| (0)L(BCC_B2,NI:NI,TI:VA) = −55288.8 + 25.4416 × T |
26 |
| (0)L(BCC_B2,NI,TI:NI:VA) = −55288.8 + 25.4416 × T |
26 |
| (2)L(BCC_B2,NI:NI,TI:VA) = +6010.11 + 3.95974 × T | 26 |
| (2)L(BCC_B2,NI,TI:NI:VA) = +6010.11 + 3.95974 × T | 26 |
| Ternary parameters | |
| (0)L(BCC_B2,TI,SN:NI:VA) = −69000 + 2 × T |
23 |
| (0)L(BCC_B2,NI,SN:NI:VA) = −69000 + 2 × T |
23 |
|
|
| BCT_A5/(Ni,Sn,Ti)1 | |
| (0)L(BCT_A5,SN,TI) = 50000 |
24 |
| (0)L(BCT_A5,NI,SN) = −21500 |
23 |
|
|
| Ti3Sn/(Ti)3(Ni,Sn,Va)1 | |
| (0)G(TI3SN,TI:SN) = 3 × GHSERTI + GHSERSN − 141133.07 + 1.1272 × T |
24 |
| (0)G(TI3SN,TI:VA) = 3 × GHSERTI + 15000 |
24 |
| Ternary parameter | |
| (0)G(TI3SN,TI:NI; 0) = 3 × GHSERTI + GHSERNI − 45000 |
a |
|
|
| Ti2Sn/(Ti,Va)2(Sn,Ni,Va)1 | |
| (0)G(TI2SN,TI:SN) = 2 × GHSERTI + GHSERSN − 122344.77 + 6.0034 × T |
24 |
| (0)G(TI2SN,TI:VA) = 2 × GHSERTI + 10000 |
24 |
| (0)G(TI2SN,VA:SN) = GHSERSN + 5000 | 24 |
| (0)G(TI2SN,VA:VA) 298.15, 300000 |
24 |
| (0)L(TI2SN,TI:SN,VA) = −34085.17 |
24 |
| (0)L(TI2SN,TI,VA:SN) = −49803.91 + 24.4710 × T |
24 |
| Ternary parameters | |
| (0)G(TI2SN,VA:NI) = GHSERNI + 15000 |
a |
| (0)G(TI2SN,TI:NI) = 2 × GHSERTI + GHSERNI − 70000 + 6.0034 × T |
a |
|
|
| Ti5Sn3/(Ti)5(Sn)3(Ni,Va)1 | |
| (0)G(TI5SN3,TI:SN:VA) = 5 × GHSERTI + 3 × GHSERSN − 330180 + 5.3 × T |
24 |
| Ternary parameter | |
| (0)G(TI5SN3,TI:SN:NI) = 5 × GHSERTI + 3 × GHSERSN + GHSERNI − 395000 + 2.3 × T |
a |
|
|
| Ti6Sn5/(Ti)6(Ti,Sn,Ni)5 | |
| (0)G(TI6SN5,TI:SN) = 6 × GHSERTI + 5 × GHSERSN − 468938.25 + 5.3729 × T |
24 |
| Ternary parameters | |
| (0)G(TI6SN5,TI:NI) = 6 × GHSERTI + 5 × GHSERNI − 300000.25 + 5.3729 × T |
a |
| (0)L(TI6SN5,TI:TI,SN,NI) = 40000 |
a |
|
|
| TiNi3/(Ni,Ti)1(Ni,Sn,Ti)3 | |
| (0)G(TINI3,NI:NI) = +4 × GNIHCP | 25 |
| (0)G(TINI3,NI:TI) = −157744 + 18.6544 × T + 3 × GNIHCP + GHSERTI |
26 |
| (0)G(TINI3,TI:NI) = +157744 − 18.6544 × T + GNIHCP + 3 × GHSERTI |
26 |
| (0)G(TINI3,TI:TI) = +4 × GHSERTI | 25 |
| (0)L(TINI3,NI:NI,TI) = +143216 − 101.776 × T |
26 |
| (1)L(TINI3,NI:NI,TI) = +109156 − 66.448 × T |
26 |
| (0)L(TINI3,TI:NI,TI) = +20000 |
26 |
| (0)L(TINI3,NI,TI:TI) = +60000 |
26 |
| Ternary parameters | |
| (0)G(TINI3,TI:SN) = −100000 + GHSERSN + 3 × GDHCTI |
a |
| (0)G(TINI3,NI:SN) = −80000 + GHSERSN + 3 × GDHCNI |
a |
| (0)L(TINI3,NI:NI,SN,TI) = −110000 |
a |
| (0)L(TINI3,TI:NI,SN,TI) = −150000 |
a |
|
|
| Ti2Ni/(Ni,Ti)2(Ni,Ti)1 | |
| (0)G(TI2NI,TI:NI) = +3 × GTI2NI | 26 |
| (0)G(TI2NI,NI:TI) = +2 × GLAVNI + GLAVTI + 30000 − 3 × GTI2NI |
26 |
| (0)L(TI2NI,NI,TI:NI) = +60000 |
26 |
| (0)L(TI2NI,NI:NI,TI) = +60000 |
26 |
| (0)L(TI2NI,TI:NI,TI) = +60000 |
26 |
| (0)L(TI2NI,NI,TI:TI) = +60000 |
26 |
| GTI2NI = 0.333333 × GHSERNI + 0.666667 × GHSERTI – 27514.218 + 2.85345219 × T |
26 |
|
|
| Ti2Sn3/(TI)2(SN)3 | |
| (0)G(TI2SN3,TI:SN) = 2 × GHSERTI + 3 × GHSERSN − 170900 + 4.886 × T |
a,c |
|
|
| Ternary compounds | |
|
|
| NiSnTi/(Ni)1(Sn)1(Ti)1 | |
| (0)G(NISNTI,NI:SN:TI) = +GHSERNI + GHSERTI + GHSERSN − 150455 + 14.2 × T |
a,c |
|
|
| Ni2SnTi2/(Ni)2(Sn)1(Ti)2 | |
| (0)G(NI2SNTI2,NI:SN:TI) = +2 × GHSERNI + 2 × GHSERTI + GHSERSN − 225876 + 10.51 × T |
a,c |
|
|
| Ni2SnTi/(Ni,Sn,Ti)2(Sn,Va)1(Ti,Va)1 | |
| (0)G(NI2SNTI,NI:SN:TI) = +2 × GHSERNI + GHSERTI + GHSERSN − 176384 + 2.83 × T |
a,c |
| (0)G(NI2SNTI,NI:SN:VA) = +2 × GHSERNI + GHSERSN + 31200 |
a |
| (0)G(NI2SNTI,NI:VA:TI) = +2 × GHSERNI + GHSERTI + 31200 |
a |
| (0)G(NI2SNTI,NI:VA:VA) = +2 × GHSERNI + 31200 |
a |
| (0)L(NI2SNTI,NI:SN:TI,VA) = −49000 |
a |
| (2)L(NI2SNTI,NI:SN:TI,VA) = −139000 |
a |
| (0)L(NI2SNTI,NI:SN,VA:TI) = −149000 |
a |
|
|
| Ti5Sn5Ni30/(Sn,Ti)1(Ni,Va)3 | |
| (0)G(TI5SN5NI30,SN:NI) = 3 × GHSERNI + GHSERSN − 131928 + 68.0 × T |
a |
| (0)G(TI5SN5NI30,TI:NI) = 3 × GHSERNI + GHSERTI − 131928 + 34.0 × T |
a |
| (0)L(TI5SN5NI30,SN:NI,VA) = −140000 |
a |
| (0)L(TI5SN5NI30,TI:NI,VA) = −100000 |
a |
| (0)L(TI5SN5NI30,SN,TI:NI) = −130000 |
a |
 | ||
| Fig. 15 System Ti–Ni–Sn; calculated liquidus projection (top) and 3D view (bottom). | ||
 | ||
| Fig. 16 System Ti–Ni–Sn; calculated isopleths Ni–TiSn, Sn–TiNi and NiSn–Ti–Sn. | ||
 | ||
| Fig. 17 System Ti–Ni–Sn; calculated isothermal sections at 800, 900 and 950 °C. | ||
 | ||
| Fig. 18 System Ti–Ni–Sn; calculated isothermal sections at 1050 and 1200 °C. | ||
The calculation respects well the experimental melting points Tm of the three ternary compounds: τ1 (1169 °C calculated/versus 1179 °C experimental), τ2 (1141 °C/1447 °C), τ3 (1156 °C/1151 °C). Although the overall shape of the liquidus surface is close to the experimental data, several calculated reaction types differ from those experimentally described (for details see the comparison of calculated with experimental reactions given in Table 2). Thus more parameters for liquid and solids may be needed for better consistency between calculation and experiment.
4 Conclusions
From a combined activity of experimental investigation (XPD, EMPA, DTA) and CALPHAD calculation we have established the phase relations in the system Ti–Ni–Sn. The system is characterized by the formation of four ternary compounds labelled τ1 to τ4. A particularly large homogeneity region is recorded for τ2-Ti1+yNi2−xSn1−y (Heusler phase, MnCu2Al-type).Extended solid solutions starting from binary phases at 950 °C have been evaluated for Ti5Ni1−xSn3 (filled Mn5Si3 = Ti5Ga4-type; 0 ≤ x ≤ 1), Ti1−xSnxNi3 (TiNi3-type; 0 ≤ x ≤ 0.27) and (Ti1−xNix)1−ySny (CsCl-type) reaching a maximum solubility at x = 0.53, y = 0.06). From DTA measurements in alumina crucibles under argon a complete liquidus surface has been elucidated revealing congruent melting for τ2-TiNi2Sn at 1447 °C, but incongruent melting for τ1-TiNiSn (pseudobinary peritectic formation: + τ2 ↔ τ1 at 1180 °C), τ3-Ti2Ni2Sn (peritectic formation: L + τ2 + Ti5NiSn3 ↔ τ3 at 1151 °C) and τ4-(Ti1−x−ySnxNiy)Ni3 (peritectic formation: L + TiNi3 + (Ni) ↔ τ4 at 1157 °C). A Schultz–Scheil diagram for the solidification behavior was constructed for the entire diagram involving 26 isothermal reactions in the ternary.
As for a CALPHAD assessment of the ternary diagram thermodynamic data in the ternary system were only available in the literature for the compounds TiNi2Sn and TiNiSn, heat of formation data were supplied by our DFT calculations for Ti2Ni2Sn, as well as for the solid solutions, which were modelled for Ti1−xSnxNi3, Ti5Ni1−xSn3 and (Ti1−xNix)1−ySny. Thermodynamic calculation was performed with the Pandat software and finally showed a reasonably good agreement for all the 20 invariant reaction isotherms involving the liquid.
Acknowledgements
This research was supported by the Grant Agency of the Czech Republic (Project No. GA14-15576S), by the Project CEITEC-Central European Institute of Technology (CZ.1.05/1.1.00/02.0068) from the European Regional Development Fund and by the Ministry of Education of the Czech Republic (7AMB15AT002). Part of the research was funded by the Austrian FWF under project P24380. The authors are furthermore grateful to some lattice parameter evaluations made by Philipp Sauerschnig.References
- R. V. Skolozdra, Y. V. Stadnyk and E. E. Starodynova, Ukr. Fiz. Zh., 1986, 31(8), 1258 CAS.
- Y. M. Goryachev, S. V. Kal'chenko, R. V. Skolozdra and Yu. V. Stadnyk, Elektron. Stroenie i Svoistva Tugoplavk. Soed., I Met. AN USSR. In-t Probl. Materialoved., Kiev, 1991, p. 81.
- G. Schierning, R. Chavez, R. Schmechel, B. Balke, G. Rogl and P. Rogl, Transl. Mater. Res., 2015, 2, 025001 CrossRef.
- V. V. Romaka, P. Rogl, L. Romaka, Y. Stadnyk, N. Melnychenko, A. Grytsiv, M. Falmbigl and N. Skryabina, J. Solid State Chem., 2013, 197, 103 CrossRef CAS.
- J. E. Douglas, C. S. Birkel, N. Verma, V. M. Miller, M.-S. Miao, G. D. Stucky, T. M. Pollock and R. Seshadri, J. Appl. Phys., 2014, 115, 043720 CrossRef.
- D.-Y. Jung, K. Kurosaki, C.-E. Kim, H. Muta and S. Yamanaka, J. Alloys Compd., 2010, 489, 328 CrossRef CAS.
- C. Colinet, P. Jund and J.-C. Tédenac, Intermetallics, 2014, 46, 103 CrossRef CAS.
- M. Yin and P. Nash, Data presented at CALPHAD XLIII, Changsha. Hunan, China, June 1st to June 6th, 2014 Search PubMed.
- R. A. Downie, D. A. MacLaren, R. I. Smith and J. W. G. Bos, Chem. Commun., 2013, 49, 4184 RSC.
- H. Hazama, M. Matsubara, R. Asahi and T. Takeuchi, J. Appl. Phys., 2011, 110, 063710 CrossRef.
- J. Rodriguez-Carvajal, FULLPROF, a program for Rietveld refinement and pattern matching analysis, Abstract of the satellite meeting on powder diffraction of the XV congress, p. 127, Int. Union of Crystallography, Talence, France, 1990, see also Physica B., 1993, 55, 192 Search PubMed.
- W. Wacha, Program STRUKTUR, Master thesis, University of Technology Vienna, 1989.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- ELK, Program package, http://elk.sourceforge.net/.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- Pandat software, version 2014 2.0, CompuTherm LLC, Madison, Wisconsin, USA, 2014, http://www.computherm.com/ Search PubMed.
- H. L. Lukas, S. G. Fries and B. Sundman, Computational Thermodynamics, Cambridge Univ. Press, Cambridge, 2007 Search PubMed.
- A. T. Dinsdale, Calphad, 1991, 15, 317 CrossRef CAS.
- O. Redlich and A. Kister, Ind. Eng. Chem. Res., 1948, 40, 345 CrossRef.
- Y. M. Muggianu, M. Gambino and J. P. Bros, J. Chim. Phys. Phys.-Chim. Biol., 1975, 72, 83 CAS.
- Binary Alloy Phase Diagrams, ed. T. B. Massalski, H. Okamoto, P. R. Subramanian and L. Kacprzak, ASM International, Materials Park, Ohio, USA, 2nd edn, December 1990, xxii, pp. 3589 Search PubMed.
- H. S. Liu, J. Wang and Z. P. Jin, Calphad, 2004, 28, 363 CrossRef CAS.
- A. Zemanova, A. Kroupa and A. Dinsdale, Monatsh. Chem., 2012, 143, 1255 CrossRef CAS.
- F. Yin, J.-C. Tedenac and F. Gascoin, Calphad, 2007, 31(3), 370 CrossRef CAS.
- P. Bellen, K. C. Hari Kumar and P. Wollants, Z. Metallkd., 1996, 87, 12 Search PubMed.
- J. de Keyzer, G. Cacciamani, N. Dupin and P. Wollants, Calphad, 2009, 33, 109 CrossRef CAS.
- E. Povoden-Karadeniz, D. C. Cirstea, P. Lang, T. Wojcik and E. Kozeschnik, Calphad, 2013, 41, 128 CrossRef CAS.
- F. C. Cannon, Mater. Res. Soc. Symp. Proc., 1984, 22, 113 Search PubMed.
- P. Pietrokowsky and E. P. Frink, Trans. Am. Soc. Met., 1957, 49, 339 Search PubMed.
- H. Nowotny, H. Auer Welsbach, J. Bruss and A. Kohl, Monatsh. Chem., 1959, 90, 15 CrossRef CAS.
- J. C. Schuster, M. Naka and T. Shibayanagi, J. Alloys Compd., 2000, 305, L1 CrossRef CAS.
- K. Schubert, K. Frank and R. Gohle, Naturwissenschaften, 1963, 50, 41 CrossRef CAS.
- J. H. N. van Vucht, H. A. C. M. Bruning, H. C. Donkerloot and A. H. Gomes de Mesquita, Philips Res. Rep., 1964, 19, 407 CAS.
- H. Kleinke, M. Waldeck and P. Gütlich, Chem. Mater., 2000, 12, 2219 CrossRef CAS.
- S. Sridharan, H. Nowotny and S. F. Wayne, Monatsh. Chem., 1983, 114, 127 CrossRef CAS.
- E. L. Semenova and Y. V. Kudryavtsev, J. Alloys Compd., 1994, 203, 165 CrossRef CAS.
- W. Buhrer, R. Gotthardt, A. Kulik, O. Mercier and F. Staub, J. Phys. F: Met. Phys., 1983, 13, L77 CrossRef.
- J. L. Glimois, P. Forey, R. Guillen and J. L. Feron, J. Less-Common Met., 1987, 134, 221 CrossRef CAS.
- M. Wilkens, J. Wegst, E. Gunzel, W. Burkhardt, H. G. Meissner, W. Schutt, K. Schubert and P. Esslinger, Naturwissenschaften, 1956, 43, 248 Search PubMed.
- S. K. Shadangi, M. Sing, S. C. Panda and S. Bhan, Cryst. Res. Technol., 1986, 21, 867 CrossRef CAS.
- H. Fjellvag and A. Kjekshus, Acta Chem. Scand., Ser. A, 1986, 40, 23 CrossRef.
- W. Jeitschko and B. Jaberg, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1982, 38, 598 CrossRef.
- M. K. Bhargava and K. Schubert, J. Less-Common Met., 1973, 33, 181 CrossRef CAS.
- V. A. Romaka, Y. V. Stadnyk, D. Fruchart, T. I. Dominuk, L. P. Romaka, P. Rogl and A. M. Goryn, Semiconductors, 2009, 43, 1124 CrossRef CAS.
- H. P. Stüwe and Y. Shimomura, Z. Metallkd., 1960, 51, 180 Search PubMed.
- G. R. Purdy and J. G. Parr, Trans. Am. Inst. Min., Metall. Pet. Eng., 1961, 221, 636 CAS.
- P. Duwez and J. L. Taylor, Trans. Am. Inst. Min., Metall. Pet. Eng., 1950, 188, 1173 CAS.
- D. M. Poole and W. Hume Rothery, J. Inst. Met., 1954/55, 83, 473 Search PubMed.
- J. L. Taylor and R. W. Floyd, J. Inst. Met., 1951/52, 80, 577–587 Search PubMed.
- T. V. Philip and P. A. Beck, Trans. Am. Inst. Min., Metall. Pet. Eng., 1957, 209, 1269 Search PubMed.
- F. J. J. van Loo, G. F. Bastin and A. J. H. Leenen, J. Less-Common Met., 1978, 57, 111 CrossRef CAS.
- A. Shinogi, M. Tanaka and K. Endo, J. Phys. Soc. Jpn., 1978, 44, 774 CrossRef CAS.
- E. A. Görlich, K. Latka, A. Szytula, D. Wagner, R. Kmiec and K. Ruebenbauer, Solid State Commun., 1978, 25, 661 CrossRef.
- W. Jeitschko, Metall. Trans., 1970, 1(11), 3159 CAS.
- J. Pierre, R. V. Skolozdra and Y. V. Stadnyk, J. Magn. Magn. Mater., 1993, 128, 93 CrossRef CAS.
- P. G. van Engen, K. H. J. Buschow and M. Erman, J. Magn. Magn. Mater., 1983, 30, 374 CrossRef CAS.
- H. Hazama, M. Matsubara, R. Asahi and T. Takeuchi, J. Appl. Phys., 2011, 110, 063710 CrossRef.
- E. Parthé, L. Gelato, B. Chabot, M. Penzo, K. Cenzual and R. Gladyshevskii, TYPIX Standardized Data and Crystal Chemical Characterization of Inorganic Structure Types, Springer, Berlin, Germany, 1994 Search PubMed.
- P. Mikušik and Š. Pick, Solid State Commun., 1993, 86(7), 467 CrossRef.
- A. Pasturel, C. Colinet, D. Nguyen Manh, A. T. Paxton and M. van Schilfgaarde, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52(21), 15176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S3. See DOI: 10.1039/c5ra16074j |
| This journal is © The Royal Society of Chemistry 2015 |