DOI:
10.1039/C5RA14928B
(Paper)
RSC Adv., 2015,
5, 92096-92106
Improved oxygen reduction activity on silver-modified LaMnO3–graphene via shortens the conduction path of adsorbed oxygen
Received
27th July 2015
, Accepted 22nd October 2015
First published on 22nd October 2015
Abstract
Silver-modified LaMnO3–reduced graphene oxide (RGO) composites are synthesized via a sol–gel method with citric acid as a chelating agent. The as-prepared nanocomposites are characterized via X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy, transmission electron microscopy, and X-ray photoelectron spectroscopy. The XRD results show that decorating with Ag does not change the perovskite structure and it is metal form. The electrocatalytic activities of the composites for the oxygen reduction reaction are evaluated. The 2 wt% Ag/LaMnO3–RGO as the air cathode catalyst shows a high voltage plateau. The corresponding oxygen reduction reaction mainly favors a four-electron transfer process, exhibits a maximum cathodic current density of 5.45 mA cm−2 at 1600 rpm, and displays good stability (i/i0 = 92.9% at −0.3 V after 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s with a rotation rate of 1600 rpm), which is better than that of the commercial Pt/C (20 wt% Pt on carbon) electrocatalyst at the same testing conditions. Such excellent catalytic activity is attributed to the synergistic effect of Ag, LaMnO3, and graphene in the composite, which provide numerous reactive sites and the modification of Ag shortens the conduction path of adsorbed oxygen, thus reducing charge transfer resistance and improving cathode performance.
000 s with a rotation rate of 1600 rpm), which is better than that of the commercial Pt/C (20 wt% Pt on carbon) electrocatalyst at the same testing conditions. Such excellent catalytic activity is attributed to the synergistic effect of Ag, LaMnO3, and graphene in the composite, which provide numerous reactive sites and the modification of Ag shortens the conduction path of adsorbed oxygen, thus reducing charge transfer resistance and improving cathode performance.
1. Introduction
New energy storage and conversion devices have aroused attention because of the increasing energy demand of our society. In theory, metal–air battery with high theoretical specific energy density and low environmental effect provides one of the most promising solutions to this demand.1 As the main cathode reaction for metal–air battery, oxygen reduction reaction (ORR) is of great importance and requires highly efficient electro-catalysts to speed up the reaction rate because of the sluggish kinetics. At present, Pt or Pt alloy2–5 are known to be the most efficient catalysts for ORR because of their relatively high activity and stability. However, the high price and the scarcity of Pt are key issues that hamper the large-scale applications of clean energy technology. Therefore, alternative catalysts with high durability have been actively pursued, such as MnO2,6,7 IrO2,8 N-doped carbon material,9–12 and several transition metal oxides,13,14 which is an important way to improve the commercial applicability of metal–air battery.
Among these candidates, perovskite materials with the formula ABO3 have been extensively studied because of their relatively high activity and stability for the ORR at alkaline conditions, other than their cost advantage and being environmentally benign.15–19 A study has reported that LaMnO3 exhibited promising catalytic activity for ORR because of its defective cation deficient lattice and excellent oxygen mobility.20 However, a major disadvantage of LaMnO3 catalysts is related to its ease in aggregating and poor conductivity. To overcome the aggregation, one approach was compounding with high surface area and chemically stable support matrix to provide numerous reactive sites for LaMnO3. Compared with active carbon, carbon nanotubes and other porous carbon materials,21–26 graphene exhibits excellent performance because of its high surface area, good chemical and environmental stability, excellent electrical conductivity and strong adhesion to catalyst particles.27,28 Recently, there are some attempts to combine noble metal or oxides catalyst with graphene as electrocatalysts for ORR in alkaline electrolytes. For instance, Ng et al.29 observed that the combination of graphene and La0.5Ce0.5Fe0.5Ni0.5O3 as a cathode material had a high cycle life for Li–air batteries. Sung et al.30 fabricated graphene-oxide-intercalated layered manganese oxides as an efficient ORR catalyst in alkaline media. Jin et al.31 electrochemically deposited silver nanoclusters on nitrogen-doped graphene as efficient electrocatalyst for oxygen reduction reaction. Therefore, LaMnO3 was supported by graphene may be an effective approach to decreased the agglomeration.
The as-prepared graphene using a modified Hummer preparation have a lot of oxygenic functional groups, such as carboxyl, carbonyl group and other resultant defects, which lead to low conductivity. Therefore, an effective strategy to improve the conductivity of LaMnO3–graphene composites is by introducing noble metals. Among these,32–34 Ag is considered as the most attractive contender because it has good catalytic activity, high electrical conductivity, and relatively low cost. Ag is also proven as an excellent electrocatalyst for the ORR with the ability to catalyze the direct four electron (4e−) reaction and the HO2− anion disproportionation. Recently, Su et al.35 deposited Ag nanoparticles into the porous (Ba, Sr)(Co, Fe)O3 cathodes via a modified electroless deposition process. Liu et al.7 reported Ag nanoparticle-modified MnO2 nanorods catalyst as an air electrode in Zn–air battery. These studies indicated that the electron charge-transfer process of the oxygen reduction over the air electrode was significantly improved using a proper amount of silver-modified material.33,36
In this paper, silver-modified LaMnO3–reduced graphene oxide (RGO) composites were successfully synthesized, and the Ag content in the composites was optimized. The composites show an interconnected porous structure with uniform deposition of LaMnO3 and Ag nanoparticles (NPs). In studying the electrocatalytic activities of composites, we found that silver-modified LaMnO3–RGO exhibit a more positive onset potential, higher cathodic density, lower H2O2 yield, and higher electron transfer number for the ORR in alkaline media than LaMnO3. Additionally, silver-modified LaMnO3–RGO showed better durability than the commercial Pt/C catalyst, which proves to be an efficient catalyst for Zn–air battery.
2. Experimental
2.1 Synthesis of Ag-modified LaMnO3–RGO composites
In this study, thin layer graphene materials were synthesized from graphite powder (Qingdao Graphite Company) via an improved Hummer preparation method according to ref. 29. The Ag-modified LaMnO3–graphene composites were synthesized via the sol–gel technique. In the first step, 100 mL of graphene water suspension (the mass ratio of graphene to perovskite was 10 wt%) was subjected to ultrasonic vibration for 1 h. The second, stoichiometric amounts of 0.1 M La(NO3)3 and Mn(NO3)2 solution were mixed with varied amounts of 0.1 M AgNO3 solution. Citric acid (CA) was added to the mixture as a chelating agent, and the mole ratio of the former nitrates to CA was controlled around 1:1. Subsequently, the graphene suspension was interfused to the four suspension solutions. Moreover, the surface-active agent alkylphenol polyoxyethylene (OP-10) was also added to prevent the gel from cracking in the drying process. NH3·H2O was added slowly to adjust the pH value to approximately 9.0. The mixed solution was heated at 60 °C on a hot plate to initiate the polymerization reaction and to evaporate the excrescent water until the sample transformed into a gel, which was initially heated at 350 °C for 5 h in air to obtain a precursor powder and then at 600 °C for 4 h in vacuum to produce the final samples. A series of Ag-modified LaMnO3–graphene composites with varied Ag mass contents (1, 2, and 4 wt%) to perovskite were prepared. The synthesis of LaMnO3–graphene composites is similar to that of Ag/LaMnO3–graphene composites.
2.2 Characterization
The phase identification of the composites was characterized by X-ray diffraction (XRD, D/max-2500/pc) with Cu Kα radiation λ = 1.5405 Å. Raman spectra were recorded on a microscopic confocal Raman spectrometer (Labram HR 800) with an excitation of 514 nm laser light. The morphology of the composites was investigated by field-emission scanning electron microscopy (FE-SEM, Hitachi S-4800) with energy dispersive spectrometer (EDS) and transmission electron microscopy (TEM, JEOL-2010) with an accelerating voltage of 200 kV. The binding energy of the elements was measured at room temperature by X-ray photoelectron spectroscopy (XPS, ESCALAB250) with a monochromated Al–Mg X-ray source (Al hν = 1486.6 eV; Mg hν = 1253.6 eV).
2.3 Preparation of the thin-film electrode
Briefly, 10 mg mixed powder (the ratio of catalyst:acetylene black was 1:1) was mixed with 4 mL isopropanol and 25 μL Nafion solution (5 wt%, DuPont, USA), then stirred ultrasonically for 30 min to form a homogeneous suspension. A total of 25 μL ink was dropped onto the rotate disk electrode (RDE, 5 mm in diameter) or the disk of the rotating ring-disk electrode (RRDE), and the solvent was evaporated at room temperature. The mixed powder loading on the glassy carbon (GC) disk is 317 μg cm−2. The loading of the commercial Pt/C and Ag powder (20 nm, Beijing Xing Rong Source Technology Co. Ltd) are the same. The RRDE electrode consisted of a catalyst-coated GC disk (5 mm diameter, 0.196 cm2 of geometric surface area) surrounded by a Pt ring (0.125 cm2 of geometric surface area).
2.4 Electrochemical measurement
The electrochemical measurements were performed using a three electrode system in 0.1 M KOH electrolyte solution: the GC disk electrode coated with catalyst thin film is served as the working electrode, a platinum foil and Hg/HgO electrode serving as counter and reference electrodes, respectively. The electrochemical characteristics and the activity of the composites toward the ORR were evaluated mainly by RDE and RRDE techniques in O2 saturated 0.1 M KOH solution by using a Pine electrochemical system (AFMSRX rotator and AFCBP1 bipotentiostat). For all the RRDE measurements, the ring potential was held at 0.5 V vs. Hg/HgO in order to oxidize any HO2− generated in alkaline solution.37
2.5 Fabrication and characterization of the zinc–air battery
A homemade zinc–air single battery with a polished zinc plate and the air electrode as the anode and the cathode, respectively, was fabricated. The cathode was made as follows: a certain amount of catalyst, acetylene black, 1 wt% poly(tetrafluoroethylene) (PTFE) and ethanol were mixed to form homogeneous slurry, which was coated onto the nickel foam current collector (1 × 1 cm) using a blade. Subsequently, the air electrode was calcined at 60 °C in air for 24 h to remove residual moisture. The final loading of active composition was 10 mg cm−2, which was calculated based on the mass of composites and acetylene black in the cathode electrodes. The back side of Ni foam was covered by the gas diffusion layer (a proofed breathable film) with 4 MPa forming pressure, in order to prevent the electrolyte from leaking out, supply air flow channels and reaction sites in the cathode of zinc–air batteries.38 The discharging curve was determined on a Neware Battery test instrument (CT-3008W-5V5Ma-S4, ShenZhen Neware Instrument Company) at 20 °C with the electrolyte being 6 M KOH. The AC impedance spectra were also measured on an electrochemical workstation (832C; Shanghai CH Instrument Company) at 20 °C with a frequency range between 106 to 10−1 Hz, while the signal amplitude was 10 mV.
3. Results and discussions
3.1 XRD and Raman spectra analysis
The crystal structures of the samples were characterized via XRD, as shown in Fig. 1a. The prepared RGO displays a broad (002) peak at approximately 24.5° and a weak (100) peak at approximately 44°, which suggests that the interplanar carbon bonds of the pristine graphite was broken and that RGO nanosheets were formed.10,29 The diffraction data of LaMnO3 are in agreement with the characteristic peaks of perovskite phase (PDF reference code 50-0297). Compared with those of pure LaMnO3, the main diffraction peaks of LaMnO3–RGO composites remain similar to the composites, which indicate that low-content RGO cannot change the perovskite type structure. For the 2 wt% Ag/LaMnO3–RGO composite, the XRD pattern presents the diffraction index of LaMnO3 as well as the characteristic peaks of Ag. Several peaks located at 2θ values of 38.3°, 44.2°, 64.4°, 77.4°, and 81.5° can be assigned to the pure Ag metal phase (111), (200), (220), (311), and (222) crystalline planes, respectively, based on the standard Ag (PDF 04-0783) shown in Fig. 1a. Furthermore, no additional peaks for Ag2O, AgO, or other silver oxides were detected, which suggests that Ag is highly chemically compatible with perovskite-type materials in its metal form. The average crystallite sizes of LaMnO3 and Ag microcrystal in 2 wt% Ag/LaMnO3–RGO composites were approximately 23 and 20 nm, respectively, which was calculated in terms of the Scherrer's formula based on the diffraction peak of LaMnO3 (200) and Ag (111).
 |
| | Fig. 1 (a) XRD patterns of RGO, LaMnO3, LaMnO3–RGO, 2 wt% Ag/LaMnO3–RGO (the ratio of RGO to perovskite oxides was 10 wt%) over the 2θ range of 5–90° and standard patterns of LaMnO3 and Ag; (b) Raman spectroscopy of LaMnO3, RGO, 2 wt% Ag/LaMnO3–RGO. | |
Fig. 1b is the Raman spectroscopy of LaMnO3, RGO, 2 wt% Ag/LaMnO3–RGO sample. The typical Raman spectrum of LaMnO3 is about in 650, 1558 and 2690 cm−1. The absorption peak nearby 1558 cm−1 is accord to the high frequency vibration mode of MnO6 octahedral structure. The peak at 650 and 2690 cm−1 are due to the lattice vibration and stretching vibration of Mn–O bond, respectively. The existing of G band at 1582 cm−1 and D band at 1351 cm−1 in the spectrum of RGO indicates the disordered sp2 structure. The Raman spectrum of 2 wt% Ag/LaMnO3–RGO sample is very similar to that observed in the RGO, and with an increased ID/IG intensity ratio compared to that in RGO. This change suggests a surface-enhanced Raman effect of Ag.
3.2 SEM-TEM analysis
Fig. 2a shows the FE-SEM image of the LaMnO3 samples. It can be seen that the LaMnO3 particles are well crystallized, but the phenomenon of agglomeration is serious, and the average particle size is 30–40 nm. The as-prepared RGO consists of the characteristic wrinkle-like thin nanosheets (Fig. 2b) with an average thickness of 1.2 nm, which indicates that a thin RGO nanosheet was obtained. A typical FE-SEM image of 2 wt% Ag/LaMnO3–RGO composite is shown in (Fig. 2c and d). The synthesized composite exhibits a porous structure. Because the content of RGO in the composite is little (10%), LaMnO3 and metallic Ag nanoparticles homogenously grew along the surface of RGO sheets without agglomeration, so that the RGO was completely covered. As catalyst of air electrode, its porous structure resulted in an increase in the three-phase region and the improvement of the mass transfer process. The EDS spectrum of 2 wt% Ag/LaMnO3–RGO composite shown in Fig. 2e indicates the presence of C, O, Mn, La, and Ag elements. The C signal should mainly originate from RGO nanosheets. The Ag signal seems to be weak, which may be a result of the low Ag content. The high resolution TEM image of the 2 wt% Ag/LaMnO3–RGO is shown in Fig. 2f, in which the interplanar distances of LaMnO3 is approximately 0.274 nm, which corresponds to the (200) crystalline planes as that of perovskite. In addition, the lattice spacing of the nanoparticle given in Fig. 2f is 0.238 nm, which corresponds to the Ag (111) crystalline planes. This result is consistent with the result calculated from the XRD analysis. Accordingly, the formation to anchor perovskite nanoparticles onto the RGO with the modification of silver may be proposed.
 |
| | Fig. 2 (a) FE-SEM image of the LaMnO3, (b) TEM image of RGO, (c and d) FE-SEM, (e) EDS, (f) HRTEM images of the 2 wt% Ag/LaMnO3–RGO composite. | |
3.3 XPS analysis
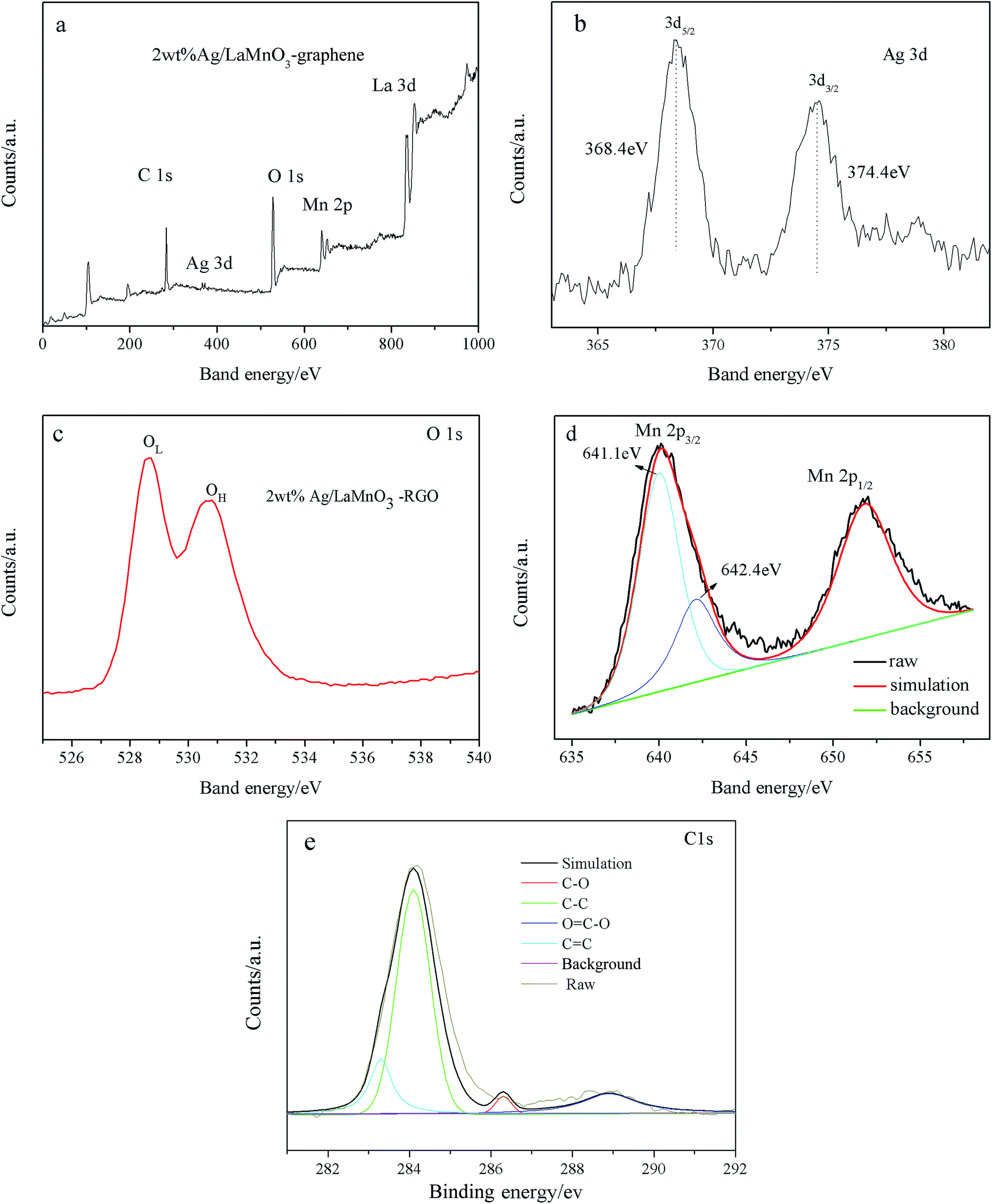
The information about the chemical element valence of 2 wt% Ag/LaMnO3–RGO was investigated via XPS. As shown in Fig. 3a, the presented peaks of C1s, Ag3d, O1s, Mn2p, and La3d demonstrate the accurate elemental composition of the composite. In the Ag3d spectrum (Fig. 3b), the obtained Ag3d peaks (368.4 eV for Ag3d5/2 and 374.4 eV for Ag3d3/2) should be attributed to the overlapped peaks of the 3d binding energy of metallic silver.39 Fig. 3c displays the O1s spectra for the composites with the 2 wt% Ag/LaMnO3–RGO. The O1s spectra had two separate peaks at 528.6 (designated as OL) and 530.7 eV (designated as OH). Where 528.6 eV represents the crystal lattice oxygen (β oxygen), which corresponds to the Mn–O–Mn bond of MnO6 and 530.7 eV represents the activated chemisorbed oxygen (α oxygen).40 The α oxygen can be incorporated into oxygen vacancies (Vo), which redounds to the ORR. The deconvolution of corresponding Mn2p spectrum (Fig. 3d) indicates the two peaks, namely, Mn2p3/2 at 641 and Mn2p1/2 at 653 eV. The range of Mn2p3/2 could be divided into two peaks by fitting the location at approximately 642.4 and 641.1 eV, which corresponds to Mn(III) and Mn(IV), respectively. Fig. 3e shows the C1s spectra of composite. The peak located at 284.6 eV is related to sp2 hybridized carbon in the RGO, which corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The binding energies of 283.4 eV, 287.3 eV and 289.2 eV were assigned to the C–C bond, C–O bond and O–CO (–COO–) bond, respectively. The existence of the O–CO (–COO–) band is result from residual oxygenous groups in RGO.
C bond. The binding energies of 283.4 eV, 287.3 eV and 289.2 eV were assigned to the C–C bond, C–O bond and O–CO (–COO–) bond, respectively. The existence of the O–CO (–COO–) band is result from residual oxygenous groups in RGO.
 |
| | Fig. 3 XPS spectra surveys scan (a), Ag3d (b), high resolution O1s (c), Mn2p (d) and C1s (e) of the obtained 2 wt% Ag/LaMnO3–RGO. | |
3.4 ORR activity
Linear sweep voltammetry (LSV) on RDE was performed to further investigate the ORR activity. The LaMnO3, LaMnO3–RGO and Ag/LaMnO3–RGO (the mass ratio of Ag to perovskite was varied 1–4%)-modified GC disk electrodes at rotation rates from 400 rpm to 1600 rpm are shown in Fig. 4a–e, respectively, which show a typical increasing current density with a higher rotation speed, as expected. For comparison, the polarization curves of LaMnO3, LaMnO3–RGO and Ag/LaMnO3–RGO with different Ag loadings are displayed in Fig. 4f at a rotation rate of 1600 rpm. The ORR properties of different catalysts are given in Table 1. The activity of the LaMnO3 was poorer than those of the LaMnO3–RGO, which may be attributed to their synergistic effect. Graphene is not only as supports for nanoparticles, but also as electronic conductive channels. The porous structure of composites increased the three-phase interface area, where ORR occurred. Thus, doping Ag enhanced the ORR activity to a particular degree with a higher disk current density and a more positive onset potential than those of LaMnO3. Apparently, the onset potential of Ag/LaMnO3–RGO was −0.09 V, whereas the value was −0.21 V for LaMnO3 at 1600 rpm. The half wave potential of Ag/LaMnO3–RGO positively shifted to a value. When the content was 2 wt%, resulted in a highest disk current density (5.45 mA cm−2). Further increases in the Ag content (4 wt%) did not improve the cathode performance. Ag as a precious metal is expensive; thus, the optimum Ag content is 2 wt%.
 |
| | Fig. 4 Typical ORR polarization curves on GC supported thin film LaMnO3 (a), LaMnO3–RGO (b), 1 wt% Ag/LaMnO3–RGO (c), 2 wt% Ag/LaMnO3–RGO (d) and 4 wt% Ag/LaMnO3–RGO (e) in O2 saturated 0.1 M KOH solution with a scan rate of 5 mV s−1 and different rotation rates; LaMnO3, LaMnO3–RGO and Ag/LaMnO3–RGO with an RDE rotation speed of 1600 rpm (f), Koutecky–Levich plots for the ORR at −0.70 V (vs. Hg/HgO) (g). | |
Table 1 ORR comparison of different catalysts in O2 saturated 0.1 M KOH at 1600 rpm
| Sample |
Onset potential (V) |
Half-wave potential (V) |
Limiting current density (mA cm−2) |
Electron transfer number |
| LaMnO3 |
−0.21 |
−0.48 |
3.08 |
3.20 |
| LaMnO3–RGO |
−0.13 |
−0.34 |
3.59 |
3.52 |
| 1 wt% Ag/LaMnO3–RGO |
−0.09 |
−0.32 |
4.30 |
3.91 |
| 2 wt% Ag/LaMnO3–RGO |
−0.09 |
−0.30 |
5.45 |
3.96 |
| 4 wt% Ag/LaMnO3–RGO |
−0.09 |
−0.32 |
4.51 |
3.93 |
A detailed study about the relationship between the current density and the rotation rate was performed to further investigate the electrocatalytic ORR mechanism and the dominant process. ORR kinetics analysis can be conducted using Koutecky–Levich (K–L) equations.
| | |
Id−1 = idl−1 + ik−1 = (Bω½)−1 + ik−1
| (1) |
where
Id is the measured current density,
idl and
ik are the kinetic and film diffusion-limiting current densities, respectively,
B is the reciprocal of the slope,
ω is the angular velocity of disk,
n is the number of electrons in the oxygen reduction,
F is the Faraday constant (96
500 C mol
−1),
Co is O
2 volume concentration (1.14 × 10
−6 mol cm
−3),
ν is the kinematic viscosity of the electrolyte (0.01 cm
2 s
−1), and
Do is the diffusion coefficient of O
2 in 0.1 M KOH (1.73 × 10
−5 cm
2 s
−1).
41
The corresponding K–L plots of these five catalysts at −0.70 V (vs. Hg/HgO) are shown in Fig. 4g. Id−1 and ω−½ have a linear relationship, and the slopes of Ag/LaMnO3–RGO are closer to the 4e− transfer pathway than those of LaMnO3 and LaMnO3–RGO. Based on the slopes of K–L plots, the electron transfer number of Ag/LaMnO3–RGO with different Ag loadings during the reaction are calculated to be approximately 3.91, 3.96, and 3.93, respectively. On the other hand, the number of electrons transferred from LaMnO3 and LaMnO3–RGO is 3.2 and 3.5, which indicates that the ORR on LaMnO3 and LaMnO3–RGO may proceed via both two-electron and four-electron transfer pathways.
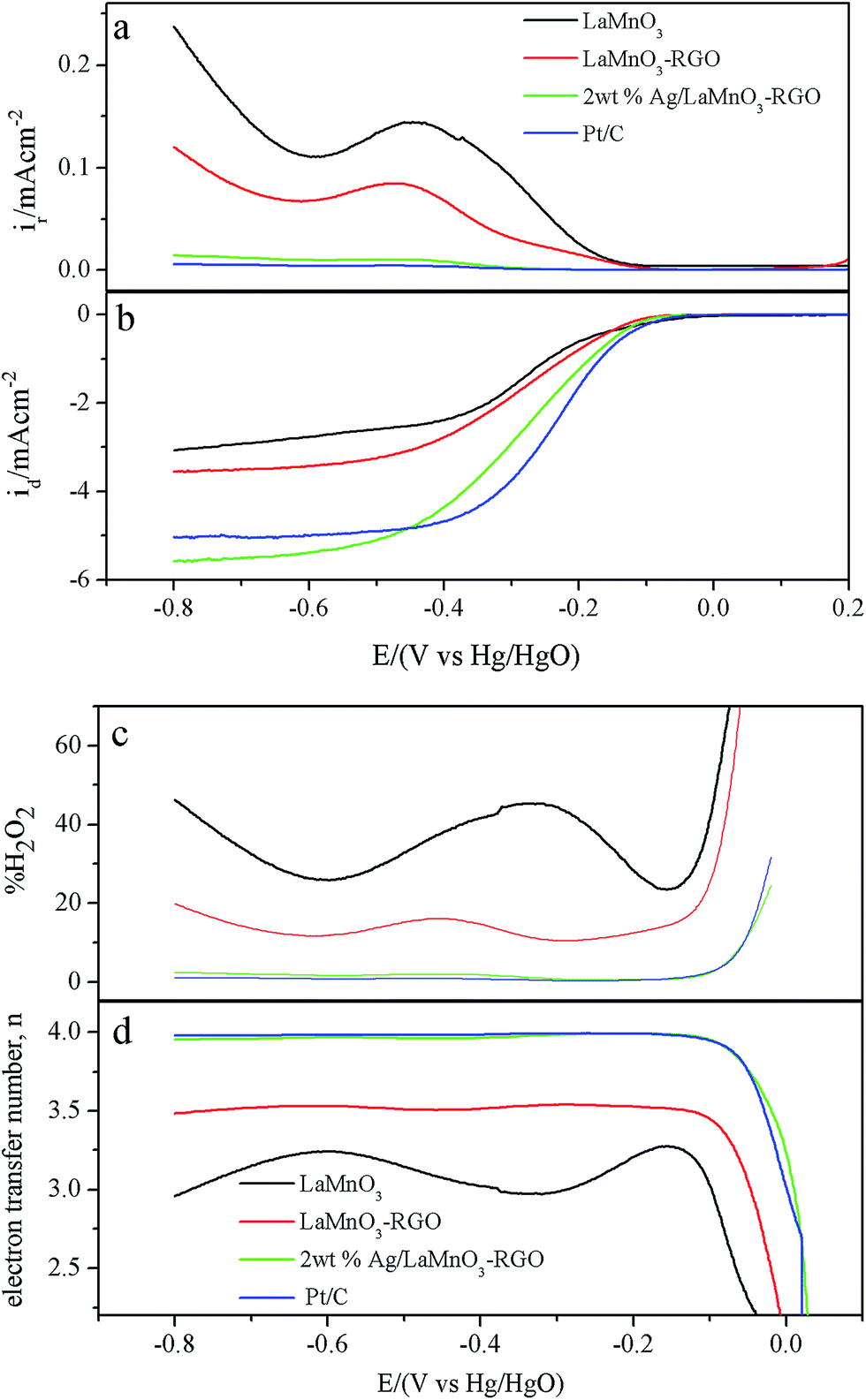
The stability of the electrocatalysts is clearly a critical issue for metal–air battery applications. The durability of 2 wt% Ag/LaMnO3–RGO for the ORR was evaluated via a chronoamperometric method. It can be seen from Fig. 5a, the stability of Ag in alkaline media is well, but the onset potential, half-wave potential and limiting current density are all lower than that 2 wt% Ag/LaMnO3–RGO composite. The 2 wt% Ag/LaMnO3–RGO as a cathode catalyst for ORR shows good performance in terms of its higher diffusion limiting current density (5.45 mA cm−2) though its half wave potential is lower than that of commercial Pt/C. For comparison, the durability of commercial Pt/C catalyst was also evaluated using an identical measurement system, and the obtained results are shown in Fig. 5b. The current densities of both catalysts initially decrease with time. The ORR current density of 2 wt% Ag/LaMnO3–RGO presents a relatively slow downtrend and maintained 92.9% of their initial currents even after 30000 s of continuous operation. On the other hand, a rapid 16% loss of the current density was observed for the commercial Pt/C catalyst. Such excellent stability was probably attributed to the intimate interaction between Ag, LaMnO3, and RGO. From the structural characterization results, the high specific surface area and the polyporous nature of Ag/LaMnO3–RGO not only facilitate the diffusion of oxygen but also provide a substantial reaction interface for the ORR.
 |
| | Fig. 5 (a) A comparison among the LSV curves of Ag, 2 wt% Ag/LaMnO3–RGO and Pt/C at 1600 rpm, (b) chronoamperometric responses for the ORR of 2 wt% Ag/LaMnO3–RGO and Pt/C at −0.30 V vs. Hg/HgO in O2 saturated 0.1 M KOH solution at a rotation rate of 1600 rpm. | |
RRDE technique was employed to further study the ORR mechanism via the analysis of the Pt ring current (Fig. 6a and b). To verify the ORR catalytic pathways of the catalysts, the hydroperoxide generated from the ring-disk current density at the working electrode and the overall numbers of electrons transferred were calculated based on the following equations.42
| |
 | (3) |
| |
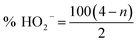 | (4) |
where
id and
ir are the disk and ring currents on the electrodes, respectively, and
N = 0.22 is the disk electrode collection efficiency. Overall, an increase in the ring current density implies that the production of HO
2− results in a reduction in the value of
n. As shown in
Fig. 6c, the measured HO
2− yield is approximately 40% for LaMnO
3 over the potential range of −0.8 V to −0.15 V and significantly decreases to a low value of 25% for LaMnO
3–RGO. The measured HO
2− yield is further decreased to below 2.5% after the modification of silver, which indicated Ag catalyzed the direct 4e
− reaction pathway as well as the HO
2− decomposition. The corresponding electron number transferred during the ORR process varies in the reverse order, as shown in
Fig. 6d, and these values are 3.2, 3.5 and 3.96 for LaMnO
3, LaMnO
3–RGO and 2 wt% Ag/LaMnO
3–RGO, respectively. This result is in agreement with those obtained from the K–L plots based on RDE measurements. The measured HO
2− yield (below 2.5%) and the electron transfer number (3.96) for 2 wt% Ag/LaMnO
3–RGO are close to those for commercial Pt/C, whose HO
2− yield, is approximately 2% and electron transfer number is 3.98 (
Fig. 6c and d). This result indicates that a 4e
− transfer pathway dominates the reaction. These results suggest that the synthesized 2 wt% Ag/LaMnO
3–RGO is a promising ORR catalyst.
 |
| | Fig. 6 (a) Ring current and (b) disk current density obtained with LSVs on RRDE for LaMnO3, LaMnO3–RGO, 2 wt% Ag/LaMnO3–RGO and Pt/C in O2 saturated 0.1 M KOH. The disk potential was scanned at 5 mV s−1 with the electrode rotated at 1600 rpm and the ring potential was fixed at 0.5 V; (c) determined peroxide percentage and (d) calculated electron transfer number n at various potentials based on the corresponding RRDE data. | |
3.5 Mechanism for the enhanced ORR activity
ORR over pure perovskite oxides proceeds via an oxygen ion reaction mechanism,43 whereas that of the silver-modified LaMnO3–RGO hybrid catalyst involves a synergistic effect mechanism. Based on the SEM and HRTEM images, Ag and LaMnO3 particles are homogeneously dispersed on RGO nanosheets and exhibit a special porous structure. Therefore, nanoparticles dispersed on RGO can provide numerous reactive sites. In addition, silver is an excellent catalyst because of its conductivity, superior oxygen solubility and mobility between oxygen and silver. Fig. 7b shows the processes of ORR on Ag/LaMnO3–RGO cathode, which may further explain the reason for the improvement of LaMnO3–RGO catalytic performance by Ag. An appropriate model proposed by van Heuveln44 is shown in Fig. 8.
 |
| | Fig. 7 ORR mechanism of the silver-modified LaMnO3–RGO. | |
 |
| | Fig. 8 Processes of ORR on 2 wt% Ag/LaMnO3–RGO cathode. | |
When ORR occurred on the surface of LaMnO3, oxygen is adsorbed and dissociates on the surface of LaMnO3 to adsorbed oxygen (Oad) (step 1). This Oad obtains an electron, which results in the formation of a negatively charged oxygen ion (Oad−) (step 2). Subsequently, such oxygen species diffuses to the three phase boundary (TPB) reaction zone (step 3) where another electron is obtained and is quickly incorporated into an oxygen vacancy Vo via a charge transfer process because of the high concentration of active sites for ORR over the RGO surface. Finally, a lattice oxygen Oo× is generated (step 4). When the LaMnO3–RGO is decorated with a proper amount of silver, the Ag nanoparticles not only create the additional effective surface sites for the adsorption and dissociation of oxygen species but also provide a short path for oxygen diffusion to the TPB reaction zone (steps I–III).45,46 The oxygen molecule from the gas phase is adsorbed on the surface of Ag, thereby resulting in the formation of adsorbed oxygen (Oad) (I). This process is more rapid than only LaMnO3 is used because the reaction barrier of O2 dissociation on silver surfaces is significantly reduced and Ag has favorable conductivity for oxygen (O). Such oxygen species then directly diffuses through Ag to the three-phase boundary (II), and the OTPB finally obtains 2e− prior to integration with an oxygen vacancy Vo to generate a lattice oxygen Oo× (III),44 which accelerates the ORR, thereby resulting in an improvement of the cathode electrochemical performance.
3.6 Zn–air battery activity
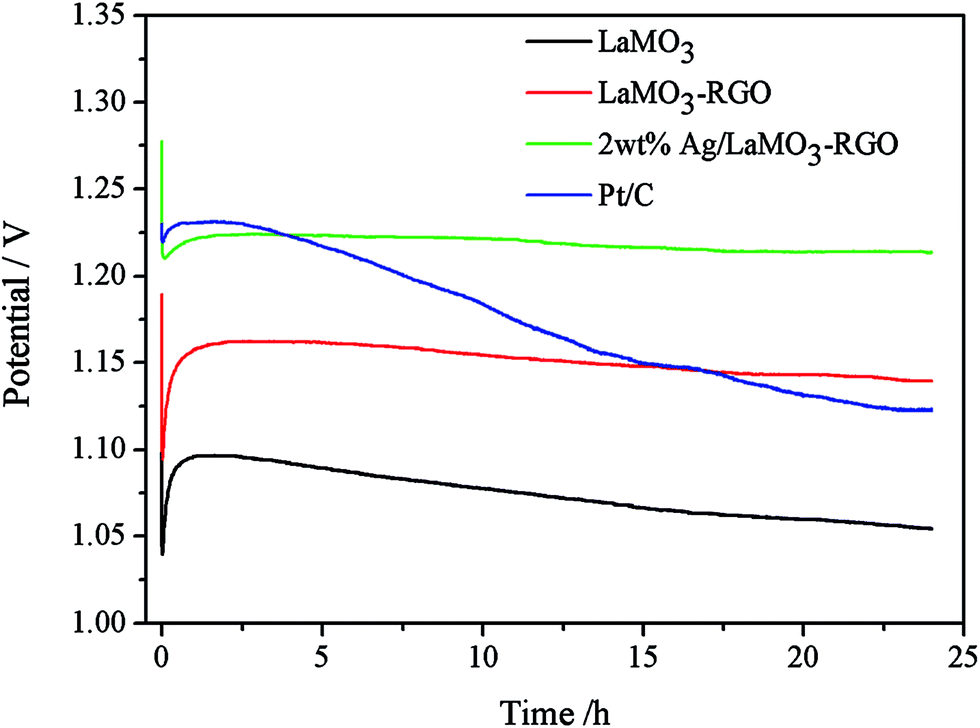
Galvanostatic discharge studies of cathodes with varied samples in a single cell were performed in the 6 M KOH electrolyte at a discharge current density of 30 mA cm−2 (3 A g−1), and the corresponding curves are shown in Fig. 9. The discharge curves of all catalysts initially increase because of the short immersion period in the electrolyte, which resulted in the insufficient contact of the active material with the electrolyte. Aside from Pt/C, LaMnO3–RGO and 2 wt%Ag/LaMnO3–RGO both reached a steady state and presented a relatively slow decay rate because of the changes in electrolyte concentration or in electrode microstructure. The voltage plateau response for the commercial Pt/C catalyst decreases nearly 10% after 24 h. These results indicate that either LaMnO3–RGO or 2 wt% Ag/LaMnO3–RGO has superior durability than Pt/C catalyst, which can be ascribed to the following reasons. RGO appears to be a chemically stable and a high-surface area support material for catalyst. Ag and perovskite particles that supported RGO homogeneously with a porous structure can provide large surface area and numerous reactive sites, therefore, increase the three-phase interface area, provide sufficient transmission paths for O2, which is in favor of ORR. This phenomenon results in the production of the steady discharge voltage plateau. In addition, the impregnation of silver in the LaMnO3–RGO electrode result in a higher discharge voltage plateau than Pt/C and an excellent discharge performance because Ag has a high electrical conductivity.
 |
| | Fig. 9 Discharge curves of air electrodes for different sample. | |
Although ORR over an electrode is complicated, the impedance spectra could be separated into two zones for all the cathodes with varied samples, which suggest that at least two different electrode processes limited the ORR. The resistance at high frequency is probably associated with the charge transfer processes, which may include the electron and ion transfer processes. The low-frequency straight line is ascribed to diffusion processes, including the adsorption–desorption of oxygen, oxygen diffusion at the gas cathode interface, and the surface diffusion of intermediate oxygen species.47 Indeed, the impedance spectra are evaluated by fitting the spectra to an equivalent circuit, as shown in the inset. L is the equivalent inductance, C1 is the limit capacitance, and R1 is the ohmic resistance between the reference electrode and the electrode current collector. Fig. 10 shows the EIS characteristic of different air electrodes, and the solid line is a fitted curve. The corresponding fitting parameters are listed in Table 2. Both the charge transfer resistance and surface diffusion resistance are significantly reduced after LaMnO3 was compounded with RGO. The former resistance is attributed to R2 caused by the faradaic reactions and the double-layer capacitance C2 on the grain surface. On the other hand, the latter resistance is ascribed to the Warburg resistance W and R3. Silver possess good conductivity and it is a good catalyst for oxygen surface adsorption, dissociation of molecular oxygen into atomic oxygen, and oxygen surface diffusion. Therefore, the charge transfer resistance and surface diffusion resistance are further reduced for 2 wt% Ag/LaMnO3–RGO. Moreover, Ag and perovskite particles are homogeneously distributed on RGO nanosheets with a porous structure, which not only shortens the conduction path of absorbed oxygen but also expands the active zone for O2 adsorption of both Ag particles surface and Ag/LaMnO3–RGO interfacial regions.
 |
| | Fig. 10 Nyquist plots of the assembled Zn–air battery with LaMnO3, LaMnO3–RGO and 2 wt% Ag/LaMnO3–RGO cathodes. | |
Table 2 Results of fitting EIS for the assembled Zn–air battery with varied samples
| Sample |
L |
R1 |
C1 |
R2 |
C2 |
R3 |
W |
| LaMnO3 |
4.24 × 10−8 |
0.8082 |
0.03404 |
3.106 |
7.24 × 10−7 |
39.47 |
0.01346 |
| LaMnO3–RGO |
4.4 × 10−8 |
0.764 |
0.051 |
2.17 |
1.2 × 10−6 |
18.63 |
0.028 |
| 2 wt% Ag/LaMnO3–RGO |
4.61 × 10−8 |
0.552 |
0.085 |
1.44 |
1.774 × 10−6 |
4.75 |
0.045 |
4. Conclusions
In this study, silver-modified LaMnO3–reduced graphene oxide (RGO) composites are synthesized via a sol–gel method with citric acid as a chelating agent. The composites exhibited a porous structure, and modification of Ag shortened the conduction path of absorbed oxygen and expanded the active zone for O2 adsorption, thereby presenting superior electrocatalytic properties for ORR. Although the half wave potential for 2 wt% Ag/LaMnO3–RGO is not as good as that of the Pt/C, the long-term stability and the voltage plateau under same discharge current density are more excellent than that of the Pt/C catalyst. All these features indicate that silver-modified LaMnO3–RGO is a promising material for the next generation of highly efficient ORR electrocatalysts in metal–air batteries.
Acknowledgements
The authors gratefully acknowledge the support of the National Natural Science Foundation of China (No. 51402253) and the Independent Research Program for the Young Teachers of Yanshan University (No. 14LGA007).
Notes and references
- Y. M. Liu, S. Chen, X. Quan, H. T. Yu, H. M. Zhao, Y. B. Zhang and G. H. Chen, J. Phys. Chem. C, 2013, 117, 14992 CrossRef CAS.
- Y. C. Xing, Y. Cai, M. B. Vukmirovic, W. P. Zhou, H. Karan, J. X. Wang and R. A. Radoslav, J. Phys. Chem. Lett., 2011, 1, 3238 CrossRef.
- R. S. Vojislav, F. Ben, S. M. Bongjin, G. F. Wang, N. R. Philip, A. L. Christopher and M. M. Nenad, Science, 2007, 315, 493 CrossRef PubMed.
- T. H. Yeh, C. W. Liu, H. S. Chen and K. W. Wang, Electrochem. Commun., 2013, 31, 125 CrossRef CAS.
- R. F. Wang, H. Li, S. Ji, H. Wang and Z. Q. Lei, Electrochim. Acta, 2010, 55, 1519 CrossRef CAS.
- C. Shi, G. L. Zang, Z. Zhang, G. P. Sheng, Y. X. Huang, G. X. Zhao, X. K. Wang and H. Q. Yu, Electrochim. Acta, 2014, 132, 239 CrossRef CAS.
- F. W. Goh Thomas, Z. L. Liu, X. M. Ge, Y. Zong, G. J. Du and T. S. Andy Hor, Electrochim. Acta, 2013, 114, 598 CrossRef.
- N. Sasikala, K. Ramya and K. S. Dhathathreyan, Energy Convers. Manage., 2014, 77, 545 CrossRef CAS.
- W. Y. Wong, W. R. W. Daud, A. B. Mohamad, A. A. H. Kadhum, K. S. Loh, E. H. Majlan and K. L. Lim, Electrochim. Acta, 2014, 129, 47 CrossRef CAS.
- L. Wang, F. X. Yin and C. X. Yao, Int. J. Hydrogen Energy, 2014, 39, 15913 CrossRef CAS.
- S. Y. Gao and K. R. Geng, Nano Energy, 2014, 6, 44 CrossRef CAS.
- H. F. Dong, Y. Zhao, Y. F. Tang, S. C. Burkert and A. Star, ACS Appl. Mater. Interfaces, 2015, 7, 10734 Search PubMed.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082 CrossRef CAS PubMed.
- W. Yang, J. Salim, C. Ma, Z. H. Ma, C. W. Sun, J. Q. Li, L. Q. Chen and Y. Kim, Electrochem. Commun., 2013, 28, 13 CrossRef CAS.
- S. Velraj and J. H. Zhu, J. Power Sources, 2013, 227, 48 CrossRef CAS.
- K. N. Jung, J. H. Jung, W. B. S. Im, K. Yoon, K. H. Shin and J. W. Lee, ACS Appl. Mater. Interfaces, 2013, 20, 9902 Search PubMed.
- J. J. Xu, D. Xu, Z. L. Wang, H. G. Wang, L. L. Zhang and X. B. Zhang, Angew. Chem., 2013, 125, 3979 CrossRef.
- F. Emiliana, M. Rhiyaad, L. Pieter, C. Olaf, K. Rudiger and J. S. Thomas, ChemElectroChem, 2013, 2, 338 Search PubMed.
- C. Jin, X. C. Cao, F. L. Lu, Z. R. Yang and R. Z. Yang, Int. J. Hydrogen Energy, 2013, 38, 10389 CrossRef CAS.
- F. L. Lu, J. Sui, J. M. Su, C. Jin, M. Shen and R. Z. Yang, J. Power Sources, 2014, 271, 55 CrossRef CAS.
- Y. L. Li, X. F. Li, D. S. Geng, Y. J. Tang, R. Y. Li, J. P. Dodelet, L. Michel and X. L. Sun, Carbon, 2013, 64, 170 CrossRef CAS.
- Y. N. Zhang, H. M. Zhang, J. Li, M. R. Wang, H. J. Nie and F. X. Zhang, J. Power Sources, 2013, 240, 390 CrossRef CAS.
- N. Vijayadurga, W. L. Jong, P. K. Swaminatha, G. Wu and N. P. Branko, J. Power Sources, 2008, 183, 34 CrossRef.
- H. Wang, R. F. Wang, H. Li, Q. F. Wang, J. Kang and Z. Q. Lei, Int. J. Hydrogen Energy, 2011, 36, 839 CrossRef CAS.
- Y. Shen, D. Sun, L. Yu, W. Zhang, Y. Y. Shang, H. R. Tang, J. F. Wu, A. Y. Cao and Y. H. Huang, Carbon, 2013, 62, 288 CrossRef CAS.
- X. J. Song and D. B. Zhang, Energy, 2014, 70, 223 CrossRef CAS.
- Y. L. Li, J. J. Wang, X. F. Li, D. S. Geng, R. Y. Li and X. L. Sun, Chem. Commun., 2011, 47, 9438 RSC.
- Y. Eunjoo and H. S. Zhou, ACS Nano, 2011, 5, 3020 CrossRef PubMed.
- L. X. Wang, A. Mahbuba, W. Kapila, S. Steven and K. Y. S. Ng, J. Power Sources, 2013, 234, 8 CrossRef CAS.
- Y. P. Hee, J. S. Tae, I. Han, H. J. Jong, A. Docheon and J. Y. Sung, Electrochem. Commun., 2014, 41, 35 CrossRef.
- S. Jin, M. Chen, H. F. Dong, B. Y. He, H. T. Lu, L. Su, W. H. Dai, Q. C. Zhang and X. J. Zhang, J. Power Sources, 2015, 274, 1173 CrossRef CAS.
- J. J. Lv, S. S. Li, A. J. Wang, L. P. Mei, J. R. Chen and J. J. Feng, Electrochim. Acta, 2014, 136, 521 CrossRef CAS.
- Y. Lin, R. Ran and Z. P. Shao, Int. J. Hydrogen Energy, 2010, 35, 8281 CrossRef CAS.
- H. Huang, Y. Y. Meng, L. Alec, D. Arthur and L. S. Steven, J. Phys. Chem. C, 2013, 117, 25352 CrossRef CAS.
- R. Su, Z. Lv, S. P. Jiang, Y. B. Shen, W. H. Su and K. F. Chen, Int. J. Hydrogen Energy, 2013, 38, 2413 CrossRef CAS.
- Y. M. Guo, Y. Liu, R. Cai, D. J. Chen, R. Ran and Z. P. Shao, Int. J. Hydrogen Energy, 2012, 37, 14492 CrossRef CAS.
- J. Wu, C. Jin, Z. R. Yang, J. H. Tian and R. Z. Yang, Carbon, 2015, 82, 562 CrossRef CAS.
- M. Kohei, I. K. Ken, A. Takeshi, F. Tomokazu, K. Kazuo and O. Zempachi, J. Mater. Chem., 2011, 21, 1913 RSC.
- J. X. Shu, Z. H. Wang, G. Q. Xia, Y. Y. Zheng, L. H. Yang and W. Zhang, Chem. Eng. J., 2014, 252, 374 CrossRef CAS.
- H. Yang, J. X. Zhang, G. J. Lin, T. Xian and J. L. Jiang, Adv. Powder Technol., 2013, 24, 242 CrossRef CAS.
- C. Jin, X. C. Cao, L. Y. Zhang and R. Z. Yang, J. Power Sources, 2013, 241, 225 CrossRef CAS.
- F. Emiliana, M. Rhiyaad, L. Pieter, C. Olaf, K. Rudiger and J. S. Thomas, ACS Catal., 2014, 4, 1061 CrossRef.
- J. J. Liu, X. M. Jin, W. W. Song, F. Wang, N. Wang and Y. Song, Chin. J. Catal., 2014, 35, 1173 CrossRef CAS.
- Z. H. Wang, C. Xu, Z. L. Lou, J. S. Qiao, B. Y. Ren and K. N. Sun, Int. J. Hydrogen Energy, 2013, 38, 1074 CrossRef CAS.
- T. Z. Sholklapper, V. Radmilovic, C. P. Jacobson, S. J. Visco and D. L. C. Jonghe, J. Power Sources, 2008, 175, 206 CrossRef CAS.
- J. H. Wang, M. L. Liu and M. C. Lin, Solid State Ionics, 2006, 177, 939 CrossRef CAS.
- W. Zhou, R. Ran, R. Cai, Z. P. Shao, W. Q. Jin and N. P. Xu, J. Power Sources, 2009, 186, 244 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.