Penrose nanotiles: design of the thin and thick rhomb molecules to self-assemble into a quasicrystal†
Dimitri N. Laikov*
Chemistry Department, Moscow State University, 119991 Moscow, Russia. E-mail: laikov@rad.chem.msu.ru
First published on 7th April 2014
Abstract
New organic molecules are designed, with synthesis in mind, to fit the shape of the thin and thick rhombs of the Penrose tiling and self-assemble by hydrogen bonding into nearly flat sheets (that may stack face-to-face) following the matching rules.
The aesthetics of pure mathematics is sometimes only one step away from an empirical discovery of new physical phenomena. So is the case with the Penrose tiling1,2 – it was only ten years later that the metallic quasicrystals were discovered3 whose structure can be explained4 by the aperiodic filling of space. The analogy between the tiling problems of mathematics and the packing of atoms in solids is not full, however, as the interatomic potentials, unlike the rigid-body contacts, allow for much flexibility and the thermodynamics comes into play in the material world. Numerical simulations of quasicrystals by simple lattice models5 may draw a qualitative picture, but the oversimplified interaction potentials have not much to do with the metallic bonding. A molecule, unlike an atom, can have a tile-like shape, so a logical question arises whether molecular quasicrystals can be found in Nature. The so-called liquid quasicrystals6,7 are micellar aggregates where each micelle is a supramolecular assembly of many tree-like molecules and is believed to behave more like a spherical particle. To the best of our knowledge, no experimental observation of truly molecular quasicrystals has been reported yet. We find only one line of theoretical research8,9 on a molecular Penrose tiling in which the molecules are put into each of its nodes and make hydrogen bonds along the rhomb edges – to get the bonds at angles multiple of π/5 the authors took a tenfold-symmetric 10,5-coronene core and attached ethynylcarboxylic groups to it. Unfortunately, no synthesis of 10,5-coronene is known and this antiaromatic molecule seems likely to be nearly as unstable as its analog pentalene.10
One may wonder if two molecules can have the shape of the Penrose rhomb tiles and bind to each other following the matching rules, and if yes, what are the smallest molecules of this kind. This can be made a well-defined combinatorial problem, but the answer is unlikely to be found by a direct computer search, as there are far too many combinations to test. It is the chemical intuition of the human mind coupled with computer modelling that can solve this problem – we are pleased to have designed the meaningful new molecules that are likely to be the sought ones, we report our theoretical findings here and hope that the art and science of organic synthesis would one day materialize them.
Our design of the molecular tiles goes through the imaginative generation of ideas of their structure and the computational tests of their fitness, the underlying design principles are: (a) to be tile-like, the molecules should be flat and rigid – fused aromatic six- and five-membered rings and other conjugated groups are the natural building blocks; (b) the bends with angles ≈nπ/5 are made with the five-membered rings; (c) to make the matching rules followed, at least three hydrogen bonds with the right pattern on each rhomb edge are needed. Full geometry optimization of molecular clusters with up to 70 molecules (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 010 atoms) was done by our parametrizable electronic structure model11 with a modification of the linear-scaling density-matrix algorithms12,13 implemented in our parallel computer code. This computational model is fast enough for testing the binding properties of tens of molecules a working day, a few hundreds of atoms each, and accurate enough for at least a semi-quantitative analysis of intermolecular interactions including hydrogen bonding and π-stacking.
010 atoms) was done by our parametrizable electronic structure model11 with a modification of the linear-scaling density-matrix algorithms12,13 implemented in our parallel computer code. This computational model is fast enough for testing the binding properties of tens of molecules a working day, a few hundreds of atoms each, and accurate enough for at least a semi-quantitative analysis of intermolecular interactions including hydrogen bonding and π-stacking.
It took us about a hundred days of such work within two years to find the first matching molecules (Fig. 1), the thick 1 and the thin 2. We see that these tiles fit together when we look at the energy-minimized structures of molecular clusters that come from the optimization starting from a hand-drawn guess, one such tiling shown in Fig. 2, although not ideally flat, is only slightly wavy (as seen from the side) and the energy cost for the flattening is low – its energy would rise by as little as 3.7 kcal mol−1 if it were constrained to C5h symmetry. The binding energies between molecules in such clusters are close to additive and show an almost linear scaling with the number of edge links – all the structures drawn schematically in Fig. 3 have been optimized and the atomic coordinates are given in the ESI.† All eight kinds of tiling nodes are there, with thick:thin count from 2:1 to 3:4, as well as the two unique series of five-fold symmetric clusters of growing size – 5:0, 20:5, 45:25, and 5:0, 20:10, 30:10. We believe that if we would go on computing the bigger clusters of our molecules in these series, we would always get only the slightly wavy nearly flat structures – we are unaware of any exact mathematical theorem underlying this problem of imperfectly flat aperiodic tilings, and we, as chemists, would like to ask a bright mathematical mind for any help.
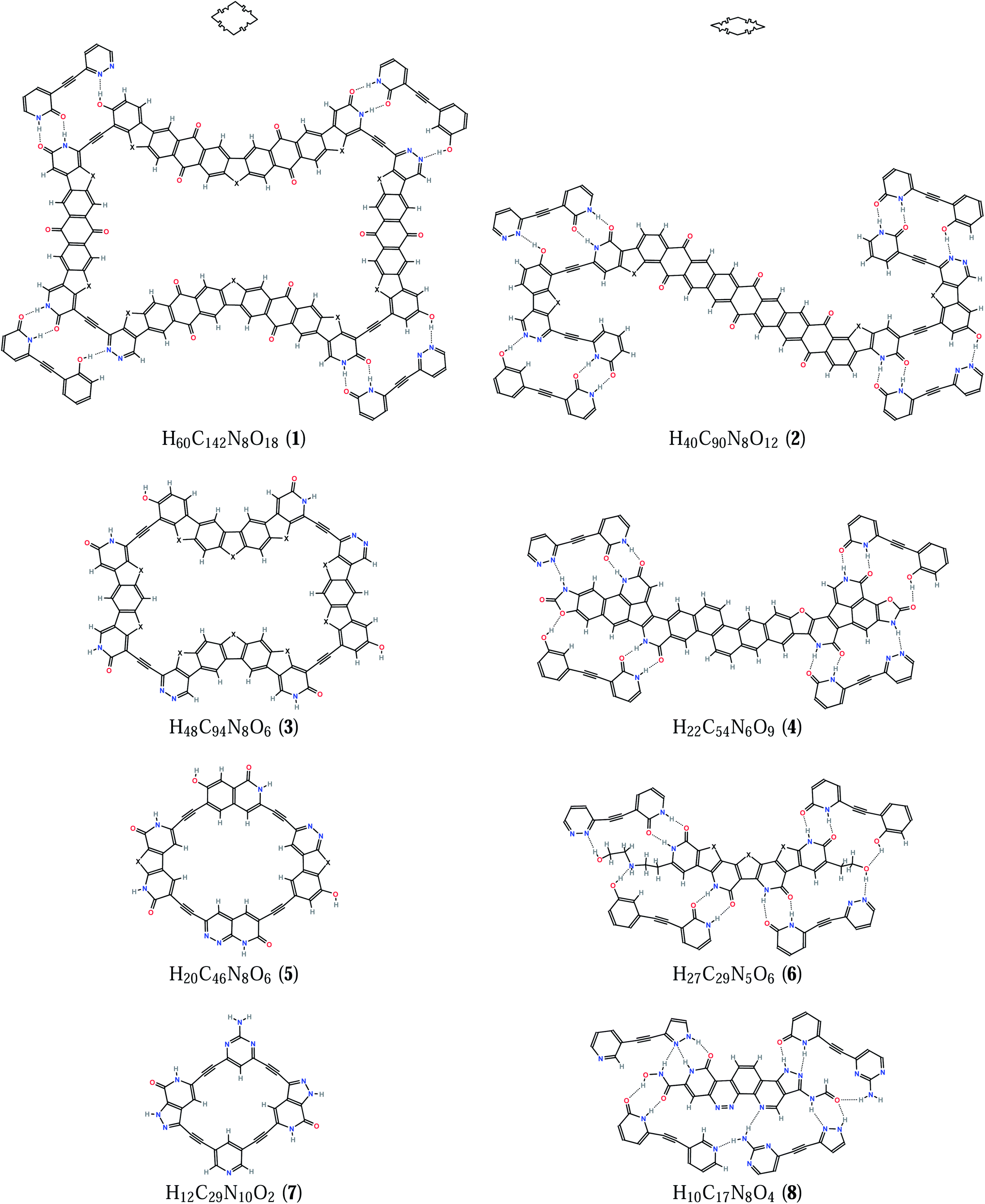 | ||
| Fig. 1 Matching rhomb tile molecules and fragments of their neighbors (X = CH2). | ||
 | ||
| Fig. 2 A tiling cluster of 20 thick and 10 thin rhomb molecules (1)20(2)10, top and side view. | ||
 | ||
| Fig. 3 Binding energies (kcal mol−1) of molecular tiling clusters: (1)n(2)m, (3)n(4)m, (5)n(6)m, (7)n(8)m, the values in brackets are for C5h-symmetry constrained geometries. | ||
Besides the edge-to-edge binding within the two-dimensional tiling layers, the stacking of the flat molecules to form columns should also be studied. We have optimized the structures of the face-to-face stacked clusters (5:0)2, (5:0)3, and (20:5)2 and get a nearly additive binding energies of ∼248 kcal mol−1 (thick–thick) and ∼208 kcal mol−1 (thin–thin) for the stacked pairs, these are quite large compared to ∼40 kcal mol−1 for the edge links but still meaningful for such big flat molecules with 228 and 150 atoms. (Our computational model11 seems to overestimate both kinds of interactions by ∼25% as judged by some limited calculations on smaller fragments by more rigorous methods, but this semiquantitative level of accuracy is fairly enough for this work.) As best seen in the (20:5)2 case, putting the atoms slightly off-top by ∼1 Å in the two layers is enough to relieve the electrostatic repulsion and allows the two-dimensional aperiodic tiling to be repeated periodically along the third dimension – we expect this kind of quasicrystal to be the most stable phase at room temperature.
As for the chemical practicality, we believe that molecules 1 and 2 are not too challenging and may even allow a convergent synthesis – the ethynyl linkers between the (aromatic) six-membered rings can be attached using Pd-catalyzed cross-coupling14 of (aryl) halogenides and ethynyltrimethylsilane as a reagent. The five-membered rings with X = CH2, CO, NH, O, and S can be chosen, furthermore, some X = C(Si(CH3)3)2 and alike would help to make the molecules bulkier and thus weaken the stacking. If we need to put many six-membered rings in a row, we keep in mind that the polyacenes up to heptacene are known15 but are getting too unstable at that end, so we take polyacenequinones of some kind instead, as they are more stable and easier to make,16 the synthesis of a nonacenetriquinone is known17 and it gives us courage to put such a fragment in the middle of our thin rhomb molecule. Some bulky side groups instead of hydrogens can be added to improve the solubility and to somehow fill the holes between the molecules as seen in Fig. 2, on the other hand, layered crystalline organic materials with pores of this size have been newly discovered,18 and our design may become the first nanoporous molecular quasicrystal.
We have also sought smaller molecular tiles that might be less synthetically challenging, and a natural way to shrink our big thick rhomb is to throw out some of its six-membered rings – we replace every three fused ones with just one and get a rather nice molecule 3. One may wonder why we have not started from this very one, and the truth is we did, but we then gave up looking for the matching thin rhomb molecule, as the space left for it seemed too narrow to fit all the needed hydrogen bonds, it was only much later that we did another brainstorming and came up with a slim stiff molecular tile 4. These two smaller molecules fit each other well enough to build free-standing one-layer tilings, the computed binding energies (Fig. 3) of ∼39 kcal mol−1 for each edge link are very close to what we get for the bigger ones. The 20:10 cluster shown in Fig. 4 may seem too unsmooth, but it would cost an energy of 48.3 kcal mol−1 to bring it from C1 to C5h symmetry – still not so much divided over 30 molecules or 4030 atoms. The stacking energies are now lower: ∼150 kcal mol−1 (thick–thick) and ∼100 kcal mol−1 (thin–thin). As before, there is some space for chemical variation and substitution – various X groups in 3 can be used, the five aromatic six-membered rings in the middle of 4 can be rearranged and even more rings can be fused in many ways, bulky groups can be attached to aromatic carbons instead of H atoms. Needless to say, the chemical synthesis of such molecules would be still too far from straightforward.
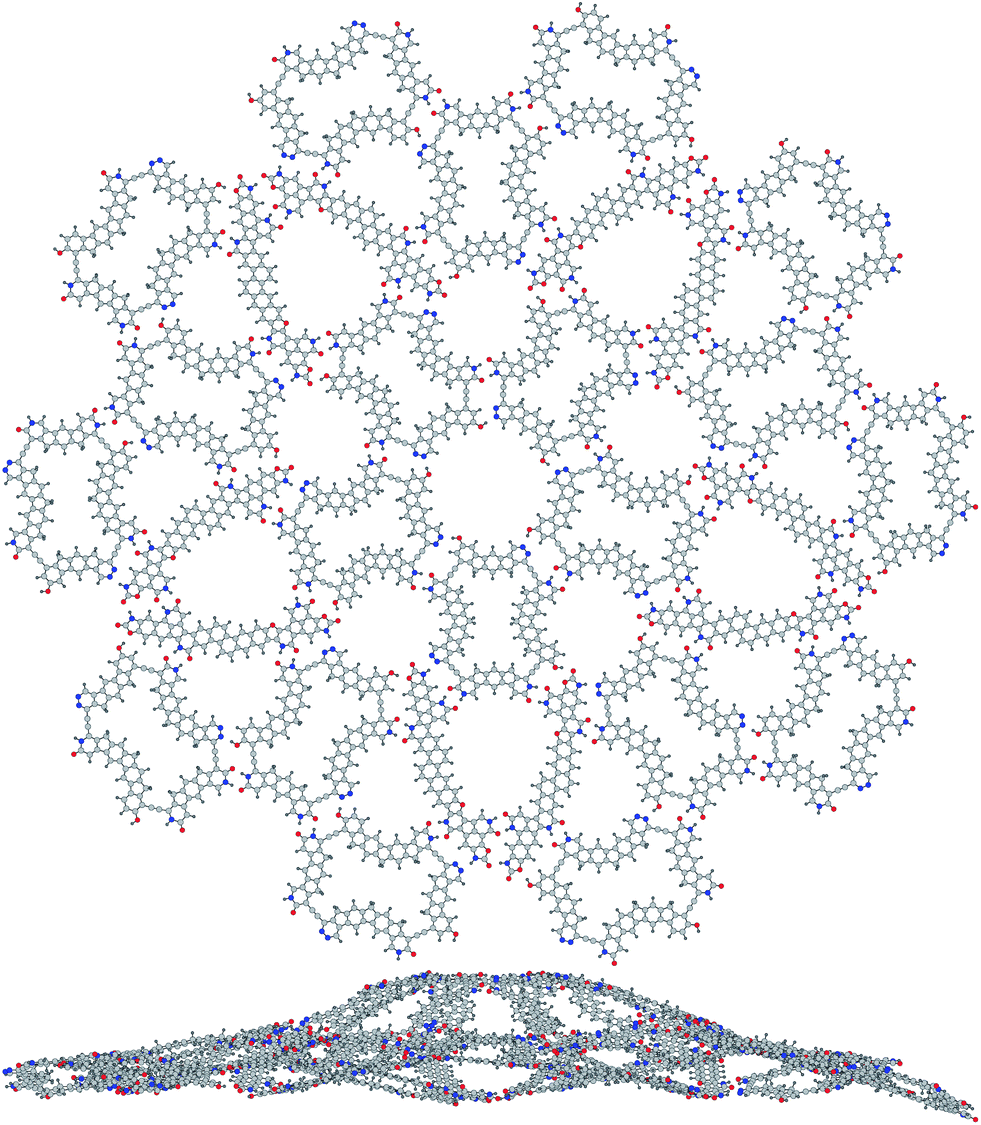 | ||
| Fig. 4 A molecular tiling cluster (3)20(4)10, top and side view. | ||
The finding of the smaller matching pair of molecules has cheered us up to try harder to seek out even smaller ones, and we have found the way to shrink 3 even further still keeping the same hydrogen-bonding receptors at nearly the same angles to get 5. The space left for the thin rhomb molecule is now so narrow that we could not fit any monolithic molecule there that would have made all the hydrogen bonds needed, but we have come up with 6 that has two flexible chains reaching out to the very corners and making two hydrogen bonds each. Without these two side chains there would be such an unwanted cross-talk between the unbound dangling groups of the neighboring molecules that the matching rules would be no more working and the tiling would break down. As we put the molecules 5 and 6 together to build our one-layer tilings (Fig. 3), only the biggest 30:10 and 45:25 clusters hold together, but the smaller ones lose the flatness and fold wildly into heaps of stacks. This does not mean, however, that we must fail here to get the three-dimensional quasicrystal, as already the two-layer tilings, such as the C5-symmetric one in Fig. 5, become stable and well-built, there seems to be some hydrogen bonding between the layers (also thanks to the hydroxyl groups of the flexible chains), the stacking energies are less additive, roughly ∼110 kcal mol−1 (thick–thick) and ∼70 kcal mol−1 (thin–thin), coming close to those of the four edge links.
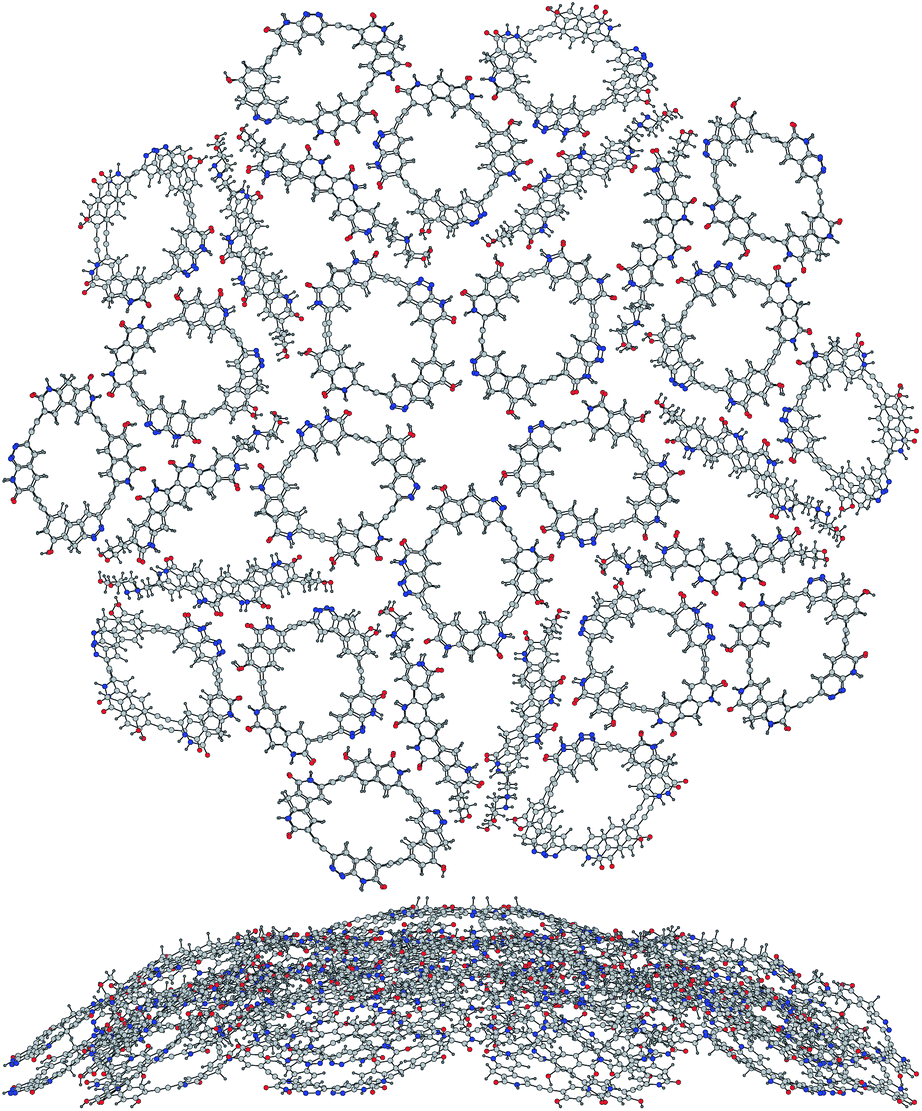 | ||
| Fig. 5 Top and side view of a two-layer molecular tiling cluster [(5)20(6)10]2. | ||
To get the smallest molecular tiles, we have to take some other hydrogen-bonding groups, as there seems to be no way to further shrink the backbone. Of all known two-ring heterocycles, we find a pyrazolopyridone (1H-pyrazolo[3,4-c]pyridin-7(6H)-one) to be unique in forming a cyclic hydrogen-bonded pentamer – we believe it to be the smallest building block to be put on the two sharp-angle sides of the thick rhomb. Thus we have designed 7 and 8 that might be the smallest and simplest molecules of the kind. As before, most one-layer tilings (Fig. 3) do not hold together now (although the 20:5 one did), but the two-layer tilings such as the one in Fig. 6 look very well and the computed stacking energies are ∼80 kcal mol−1 for both thick–thick and thin–thin pairs. Given the known synthesis of the pyrazolopyridone,19 the commercial availability of 2-aminopyrimidine and 3,5-dibromopyridine, and the broad applicability of Pd-catalyzed cross-coupling14 of aryl halides with (trimethylsilyl)acetylene followed by desilylation and a second cross-coupling, the molecule 7 is crying for synthesis and calls the molecule 8 to follow.
 | ||
| Fig. 6 Top and side view of a two-layer molecular tiling cluster [(7)30(8)10]2. | ||
Looking forward to that happy day when the first quasicrystal of this kind would be made, we have computed the X-ray diffraction patterns (Fig. 7 and 8) of our molecular clusters, the incoming wave of length λ = 1.5418 Å (Cu Kα) falling normal to the tiling's plane and the outgoing wavevector projection components in the range ±2π/(5λ). As the cluster size grows, we see the 10-fold symmetric pattern to sharpen, and we believe that those first to synthesize and crystallize these substances would be rewarded with this striking witness of the new material's aperiodic structure.
 | ||
| Fig. 7 Computed diffraction patterns (negative) of (1)20(2)5 (top) and (1)45(2)25 (bottom). | ||
 | ||
| Fig. 8 Computed diffraction patterns (negative) of (3)20(4)5 (top) and (3)45(4)25 (bottom). | ||
Our theoretical design of molecular Penrose “nanotiles” inspires further work towards the first practical observation of molecular quasicrystals – it gives the go-ahead to the chemical synthesis of such molecules and the study of their self-assembly. A deeper theoretical insight into the thermodynamics and kinetics of these binary mixtures from large-scale molecular dynamics simulations with no periodic boundary conditions would also be of great help to avoid the dead ends in the experimental work by showing under what conditions the quasicrystalline phase may exist. The design of the smaller (or smallest) molecules of this kind allowing for the easier (or easiest) chemical synthesis would be another way to go. We believe that one day the two substances made up of the thin and thick rhomb-shaped molecules will be mixed in the golden ratio and do a chemical computation to solve the aperiodic puzzle.
The inspiration to this work came in part from the two lectures given in Moscow by Dan Shechtman in 2012 and by Sir Roger Penrose in 2013. The author thanks Roald Hoffmann for comments and encouragement.
References
- R. Penrose, Bull. Inst. Math. Appl., 1974, 10, 266 Search PubMed.
- R. Penrose, Math. Intel., 1979, 2, 32 CrossRef.
- D. Shechtman, I. Blech, D. Gratias and J. W. Cahn, Phys. Rev. Lett., 1984, 53, 1951 CrossRef CAS.
- D. Levine and P. J. Steinhardt, Phys. Rev. Lett., 1984, 53, 2477 CrossRef CAS.
- L.-H. Tang and M. V. Jarić, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 4524 CrossRef.
- X. Zeng, G. Ungar, Y. Liu, V. Percec, A. E. Dulcey and J. K. Hobbs, Nature, 2004, 428, 157 CrossRef CAS PubMed.
- G. Ungar, V. Percec, X. Zeng and P. Leowanawat, Isr. J. Chem., 2011, 51, 1206 CrossRef CAS.
- Z. Zhou and K. D. M. Harris, ChemPhysChem, 2006, 7, 1649 CrossRef CAS PubMed.
- Z. Zhou and K. D. M. Harris, J. Phys. Chem. C, 2008, 112, 16186 CAS.
- T. Bally, S. Chai, M. Neuenschwander and Z. Zhu, J. Am. Chem. Soc., 1997, 119, 1869 CrossRef CAS.
- D. N. Laikov, J. Chem. Phys., 2011, 135, 134120 CrossRef PubMed.
- X.-P. Li, R. W. Nunes and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 10891 CrossRef CAS.
- A. H. R. Palser and D. E. Manolopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 12704 CrossRef CAS.
- K. Sonogashira, J. Organomet. Chem., 2002, 653, 46 CrossRef CAS.
- M. M. Payne, S. R. Parkin and J. E. Anthony, J. Am. Chem. Soc., 2005, 127, 8028 CrossRef CAS PubMed.
- T. Chiba, P. W. Kenny and L. L. Miller, J. Org. Chem., 1987, 52, 4327 CrossRef CAS.
- W. C. Christopfel and L. L. Miller, J. Org. Chem., 1986, 51, 4169 CrossRef CAS.
- S. Dalapati, S. Jin, J. Gao, Y. Xu, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 17310 CrossRef CAS PubMed.
- D. Chapman and J. Hurst, J. Chem. Soc., Perkin Trans. 1, 1980, 2398 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic coordinates and energies of the molecular systems. See DOI: 10.1039/c4ra01354a |
| This journal is © The Royal Society of Chemistry 2014 |