Breakdown of lignins, lignin model compounds, and hydroxy-aromatics, to C1 and C2 chemicals via metal-free oxidation with peroxide or persulfate under mild conditions†
Adam Wu,
Jean Michel Lauzon,
Indah Andriani and
Brian R. James*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: brj@chem.ubc.ca
First published on 7th April 2014
Abstract
The aromatic components of lignin model compounds and lignins are degraded in basic, aqueous solutions using H2O2 or K2S2O8, even at ambient temperatures, to mainly MeOH, formate, carbonate, and oxalate. The MeOH derives from aromatic OMe substituents, and the other products are formed from other substituents and/or C–H bonds in the aromatic rings.
Lignocellulosic biomass is an attractive, potential renewable source of biofuels and other useful aromatic compounds, and research interest this area has intensified enormously in the last decade;1–3 it is evident that progress in the lignin area2 is slower than in the cellulose area,3 presumably because of the more complex nature of the lignin structure. The interest in lignin is particularly significant within the pulp and paper industry, which generates large amounts of this currently largely ‘waste’ material.4 Lignin is a cross-linked, racemic and phenolic macromolecule comprised of three basic phenyl propane monomers (p-hydroxycinnamyl, coniferyl and sinapyl alcohols) connected by a network of C–C and C–O bonds.2d,5 Phenol, guaiacol (1), and syringyl derivatives (2–4), may be considered basic structural units of lignin and, along with vanillic (5) and veratric (6) acids (Chart 1) are loosely termed lignin model compounds (LMCs) in this current paper; the Chart also shows examples of so-called ‘dimeric’ LMCs (i.e. containing two aromatic rings) that contain, like lignins, a benzylic OH group (7 and 8).
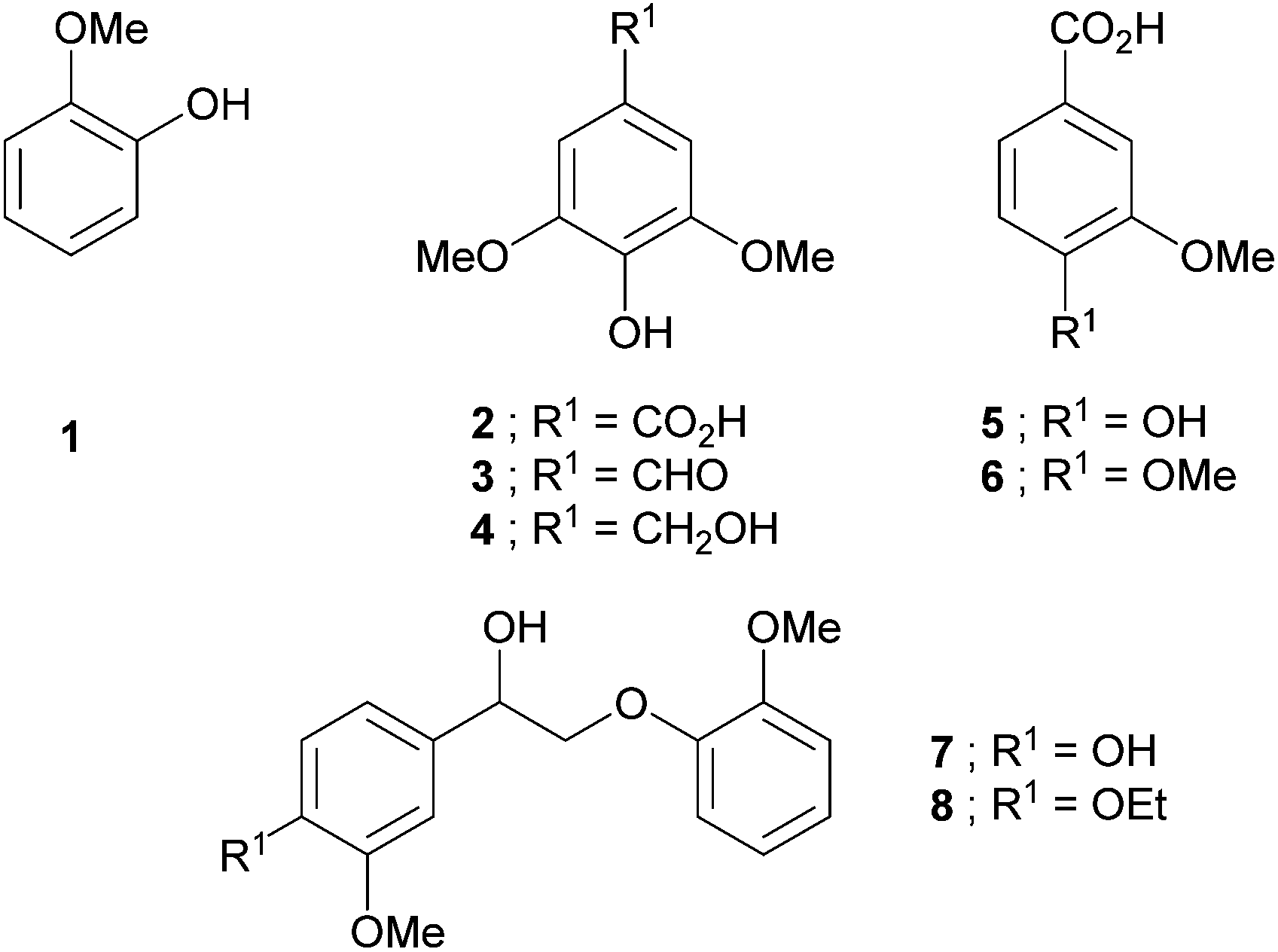 | ||
| Chart 1 Lignin model compounds (LMCs). | ||
Catalytic oxidative or hydrogenolysis breakdown of LMCs of the type shown in Chart 1 has been reported for decades, the most recent studies using V-, Cu-, Ni-, and Ru-based systems;2c,6 however, reports on potentially useful breakdown of lignins (as opposed to fuel value by non-selective, complete combustion to CO2)7 are, as expected, more limited,1b,2b,8 although discussion on catalytic model systems has sometimes been embellished to imply effectiveness for lignins!9 The first report on the use of a Ru-catalyst did not appear until 2010,10 which is surprising considering that Ru species appear to be the most widely used in catalytic oxidations; indeed, every type of catalyzed organic oxidation process (with or without bond-cleavage) can be exemplified by a Ru system, particularly using ruthenium-oxo species.11 Griffith's group had reported in 1993 an oxidation of 3,4-dimethoxybenzyl alcohol (veratryl alcohol) to the acid (6) using RuCl3·3H2O in the presence of KBrO3 (bromate) or K2S2O8 (persulfate) as co-oxidants in basic aqueous solution at room temperature (r.t., ∼20 °C), these generating, respectively, perruthenate (RuO4−) or ruthenate (RuO42−);12 other benzyl alcohols and aldehydes, and cinnamyl alcohol and aldehyde, were all similarly oxidized to the corresponding carboxylic acid. The findings were presented as new, efficient methods for selective oxidation of organic alcohols and aldehydes, and no mention was made of LMCs. The bromate systems were catalytic in RuO4−, whereas the persulfate systems were stoichiometric and not catalytic in RuO42−.12 Based on this report, we initiated a project in 2010 using such RuO4− or RuO42− systems for oxidation of LMCs and lignins.
This current communication describes the surprising and remarkable discovery that, even in the absence of a Ru-oxo species, the aromatic components of LMCs and lignins can be oxidized by S2O82− and by H2O2 with up to complete loss of aromaticity in basic aqueous solutions even at ambient temperatures; control experiments gave ≤15% consumptions, showing that it is the oxidants that effect the degradation to products, mainly MeOH, formate, carbonate, and oxalate. Non-catalysed, complete combustion of aromatics typically requires temperatures of 500 °C.7
Each of the commercially available model substrates (1–6), the synthesized dimers 7 and 8,6a and several lignin samples13 were reacted in an NMR tube under the general conditions shown in Scheme 1. The use of H2O2 (inexpensive and green)14 seemed particularly attractive, and was the primary oxidant used for this study.
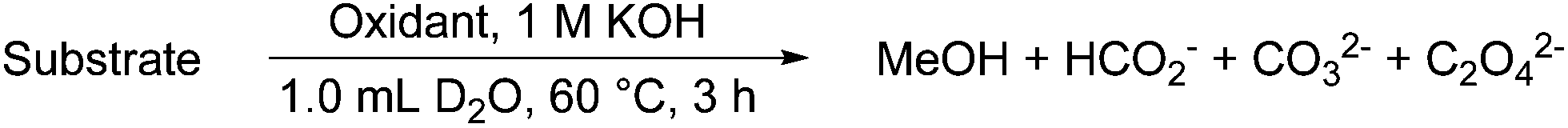 | ||
| Scheme 1 Generalized reaction scheme for oxidation of LMCs and lignins. | ||
The initial 1H NMR spectra were recorded at r.t. prior to addition of the oxidant, and the tube was then heated to 60 °C with substrate consumption monitored over time by the decreasing intensities of aromatic 1H NMR signals in the δH 7.5–6.5 region. A 3 h reaction time was generally used to define a standard set of conditions because 100% consumption of the monomer LMCs were often seen at this time; remaining aromatic content in the lignins ranged from 1 to 22%. The reaction mixture was allowed to cool to room temperature before pivalic acid was added as a standard to determine the yields of MeOH and formate (Table 1), which both possess an 1H NMR signal. The other major products formed were carbonate and oxalate, which we tried to quantify for substrates 2, 3, and the highest yielding lignin (11) using quantitative 13C{1H} NMR spectroscopy (Table 2).15
| Substratea | Oxidantb | Consumptn (%) | MeOH (mmol) | HCO2− (mmol) |
|---|---|---|---|---|
| a 0.050 mmol in 1 mL D2O with 1.0 M KOH, except for 7 (0.025 mmol).b 0.50 mmol H2O2, 0.25 mmol K2S2O8.c Not fully dissolved; dimer 8 was insoluble.d 15 mg of lignin sample used. | ||||
| Guaiacol (1) | H2O2 | 61 | 0.025 | 0.021 |
| K2S2O8 | 94 | 0.028 | 0.008 | |
| Syringic acid (2) | H2O2 | 89 | 0.073 | 0.022 |
| K2S2O8 | 100 | 0.069 | 0.002 | |
| Syringyl aldehyde (3) | H2O2 | 100 | 0.096 | 0.085 |
| K2S2O8 | 100 | 0.086 | 0.016 | |
| Syringyl alcohol (4) | H2O2 | 100 | 0.100 | 0.093 |
| K2S2O8 | 100 | 0.096 | 0.013 | |
| Veratric acid (5) | H2O2 | 40 | 0.004 | 0.003 |
| K2S2O8 | 96 | 0.020 | 0.002 | |
| Vanillic acid (6) | H2O2 | 4 | 0.000 | 0.000 |
| K2S2O8 | 51 | 0.025 | 0.000 | |
| LMC dimer (7)c | H2O2 | n/a | 0.022 | 0.039 |
| K2S2O8 | n/a | 0.019 | 0.002 | |
| Ligninsd | ||||
| 9 | H2O2 | 80 | 0.040 | 0.046 |
| K2S2O8 | 84 | 0.018 | 0.006 | |
| 10 | H2O2 | 97 | 0.022 | 0.031 |
| K2S2O8 | 95 | 0.033 | 0.007 | |
| 11 | H2O2 | 99 | 0.093 | 0.050 |
| K2S2O8 | 89 | 0.090 | 0.004 | |
| 12 | H2O2 | 82 | 0.047 | 0.054 |
| K2S2O8 | 82 | 0.019 | 0.003 | |
| 13 | H2O2 | 91 | 0.055 | 0.033 |
| K2S2O8 | 78 | 0.037 | 0.003 | |
Each experiment was carried out at least twice, with data showing variation of up to ±15% in the substrate consumptions and yields;16 the average values are presented in Table 1. Slow degradation of the LMCs is observed at room temperature; for example, consumption of syringyl alcohol (4) using H2O2 is ∼20% is after 2 h, ∼35% after 4 h, and 100% after 48 h and NMR data confirm production of the same four major products. Preliminary room temperature studies with lignin 11 are promising with >90% consumption observed after 24 h; signals for MeOH, formate, carbonate, and oxalate are all present in the 13C{1H} NMR spectrum.
The quantitative data for MeOH and formate derived from 1H NMR data for substrates 2, 3, and 11 with H2O2 are considered in reasonable agreement with those determined by 13C{1H} NMR data (Table 2). Interestingly, the 13C{1H} NMR spectra reveal other C-containing products beside carbonate and oxalate.17 For example, for 3, the total carbon detected by 13C{1H} NMR is in the 87–97% range of the theoretical value of 0.450 mmol [0.050 mmol substrate × 9 (the number of C-atoms)];18 the amounts of MeOH, HCO2−, CO32−, C2O42−, and unidentified C-atoms (integration of seven smaller resonances), are 0.080, 0.079, 0.080, 0.046, and 0.11 mmol, respectively (Table S5†). Similar data for 2 (Table S4†) show 74–86% of the theoretical value; corresponding data for the lignin (Table S6†) show the same four major products and only 14% unidentified C-atoms. Of note, the same unidentified 13C{1H} NMR resonances are seen for the lignin and the syringyl substrates.
The highest substrate consumptions (>90%) are realized with 1–5, particularly using S2O82− as the oxidant, with the syringyl substrates 2–4 giving the most MeOH. The MeOH is not formed via hydrolysis in the basic conditions, consistent with a study showing that OMe substituents within aromatic compounds are stable toward hydrolysis in 20% NaOH at 100 °C.19 Nevertheless the MeOH likely derives from the OMe groups since the data for 2–4, which contain two OMe substituents, compared with those for 1 and 5 with one OMe, suggest a correlation between the number of OMe groups and MeOH production (and substrate consumption). Support for this is found in corresponding data for o- and p-ethoxyphenols which, with the H2O2/KOH treatment, generate EtOH and formate, analogous to the reactivity of 1. The data for 6, which contains two OMe groups, does not fit the correlation, and implies that the presence of an OH group (as in 5) in some way ‘activates’ the system.
NMR studies show that MeOH, formate, and oxalate are not oxidised under the standard conditions using H2O2, and similarly the syringyl reactants 2–4 are not oxidised stepwise (i.e. alcohol to aldehyde to acid) prior to de-aromatization. That the formate is not formed from the MeOH is supported further by data for 3 and 4, where the amount of MeOH produced corresponds to (or approaches) that expected for formation from the OMe groups. The dimer model 7 (with two OMe groups) did not completely dissolve under the reaction conditions even when 0.025 mmol of substrate was used, but both MeOH and formate are formed. Qualitatively the data suggest that the MeOH likely comes from just one OMe, again likely the one in the phenoxide ring. Dimer 8 was essentially insoluble, and thus gave no MeOH or formate. Of interest, the data for lignin 11 correspond closely to complete conversion of the OMe substituents to MeOH (∼17%), since an independent 1H NMR method gives a more precise value of 18.7%. Such a breakdown of lignins to MeOH could provide a simple method for determining their OMe content, typically in the 11.4–22.9% range (by weight),20 as determined traditionally via conversion to MeI by treatment with HI.21
A lower pH solution was also tested for all the substrates using K2CO3 because carbonate had been used in the studies of Griffith et al. for in situ formation of RuO4− (KOH had been used for generating RuO42−).11,12 However, lower consumptions (≤20%) were generally seen for the LMCs, and none of the lignins was fully soluble in the K2CO3 solution. Substrate 2 on treatment with RuO42− (formed by addition of RuCl3·3H2O to the S2O82−/KOH system) resulted in less MeOH (0.034 mmol) and more formate (0.012 mmol) than in the absence of Ru;17 the same outcome was seen with lignin 11. A separate reaction showed that RuO42− did indeed promote the oxidation of MeOH and formate in the S2O82−/KOH solutions; at 60 °C over 3 h, ∼84% of MeOH was oxidized to a mixture of formate and carbonate.
A common feature of H2O2 and K2S2O8 in oxidations is that the mechanisms typically involve generation of radicals (˙OH22 and ˙SO4−,23 respectively), and a radical process at the aromatic methoxy centre to form MeOH seems plausible. The possibility of the presence of trace transition metals promoting Fenton-type free-radical oxidations with the H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 was ruled out by the non-effect of added diethylenetriaminepentaacetic acid (DTPA), commonly used for sequestering metal ions in pulp and paper industry.24 Surprisingly, the exclusion of O2 has no effect on the degradation reactions, as shown by studying the syringic acid (2) system under Ar; similarly, exclusion of ambient light (by wrapping the NMR tube in aluminium foil) had no effect on the consumption and product formation. Photochemical induced degradation of LMCs and lignins in alkaline H2O2 solutions has been widely studied,25 but there are no reports on formation of the C1 and C2 compounds found herein. Further, Stahl's group has reported the use of conditions similar to ours (H2O2 in 2 M NaOH at 50 °C for 10 h) in a 1:1:1 H2O–THF–MeOH solvent mixture for C–C bond cleavage of a dimer LMC to give mainly 1 and 6 with no evidence for breakdown of the aromatic rings.2b Solvent effects clearly play a dominant role in peroxide oxidation processes, and further work is necessary to gain insight into the mechanism.
22 was ruled out by the non-effect of added diethylenetriaminepentaacetic acid (DTPA), commonly used for sequestering metal ions in pulp and paper industry.24 Surprisingly, the exclusion of O2 has no effect on the degradation reactions, as shown by studying the syringic acid (2) system under Ar; similarly, exclusion of ambient light (by wrapping the NMR tube in aluminium foil) had no effect on the consumption and product formation. Photochemical induced degradation of LMCs and lignins in alkaline H2O2 solutions has been widely studied,25 but there are no reports on formation of the C1 and C2 compounds found herein. Further, Stahl's group has reported the use of conditions similar to ours (H2O2 in 2 M NaOH at 50 °C for 10 h) in a 1:1:1 H2O–THF–MeOH solvent mixture for C–C bond cleavage of a dimer LMC to give mainly 1 and 6 with no evidence for breakdown of the aromatic rings.2b Solvent effects clearly play a dominant role in peroxide oxidation processes, and further work is necessary to gain insight into the mechanism.
Irrespective of the mechanism, formation of MeOH from the methoxy group of 1 might generate catechol, and so this (and phenol) were also subjected to the oxidation conditions using H2O2 or K2S2O8 (at 1 M KOH). Complete consumption of the substrates was observed, though, consistent with the absence of OMe groups, no MeOH is formed. Notably, the major products present were formate, carbonate, and oxalate. In contrast, oxidation of catechol and phenol in aqueous solution by O2 (which requires high temperature and pressure) is complex and can plausibly generate many products, including formate.26
De-polymerization of lignin to, for example, phenolic-type chemicals in the hope of establishing its use as a feedstock for the sustainable production of bulk chemicals remains, of course, a key goal1,2 and hopefully, by fine-tuning our demonstrated mild oxidation conditions especially using a variety of solvent mixtures (with or without the involvement of transition metal catalysts), such a useful type of degradation can be accomplished.
We thank NSERC Lignoworks for financial support, and Dr. Andrew Lewis at Simon Fraser University for assistance with, and use of, the Bruker AVANCE 600 MHz NMR spectrometer.
Notes and references
- (a) E. Heracleous and A. Lemonidou, Platinum Met. Rev., 2013, 57, 101–109 CrossRef; (b) P. Azadi, R. Carrasquillo-Flores, Y. J. Pagan-Torres, E. I. Gurbuz, R. Farnood and J. A. Dumesic, Green Chem., 2012, 14, 1573–1576 RSC; (c) S. R. Collinson and W. Thielemans, Coord. Chem. Rev., 2010, 254, 1854–1870 CrossRef CAS PubMed.
- (a) R. Thilakaratne, T. Brown, Y. Li, G. Hu and R. Brown, Green Chem., 2014, 16, 627–636 RSC; (b) A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed; (c) S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed; (d) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- (a) B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC; (b) A. Osatiashtiani, A. F. Lee, D. R. Brown, J. A. Melero, G. Morales and K. Wilson, Catal. Sci. Technol., 2014, 4, 333–342 RSC.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Green Chem., 2012, 14, 3295–3303 RSC.
- (a) Q. Yang, J. Shi, L. Lin, L. Peng and J. Zhuang, Bioresour. Technol., 2012, 123, 49–54 CrossRef CAS PubMed; (b) S. Shimizu, T. Yokoyama, T. Akiyama and Y. Matsumoto, J. Agric. Food Chem., 2012, 60, 6471–6476 CrossRef CAS PubMed; (c) W. K. El-Zawawy, M. M. Ibrahim, M. N. Belgacem and A. Dufresne, Mater. Chem. Phys., 2011, 131, 348–357 CrossRef CAS PubMed; (d) Y. S. Kim, H.-M. Chang and J. F. Kadla, Holzforschung, 2008, 62, 38–49 CAS.
- (a) A. Wu, B. O. Patrick, E. Chung and B. R. James, Dalton Trans., 2012, 41, 11093–11106 RSC; (b) B. Sedai, C. Díaz-Urrutia, R. T. Baker, R. Wu, L. A. P. Silks and S. K. Hanson, ACS Catal., 2011, 1, 794–804 CrossRef CAS; (c) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- (a) S. Kang, X. Li, J. Fan and J. Chang, Renewable Sustainable Energy Rev., 2013, 27, 546–558 CrossRef CAS PubMed; (b) D. Shen, J. Hu, R. Xiao, H. Zhang, S. Li and S. Gu, Bioresour. Technol., 2013, 130, 449–456 CrossRef CAS PubMed.
- W. Partenheimer, Adv. Synth. Catal., 2009, 351, 456–466 CrossRef CAS.
- J. Urquhart, in Chemistry World, June 2011, p. 23 Search PubMed.
- J. M. Nichols, L. M. Bishop, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2010, 132, 12554–12555 CrossRef CAS PubMed.
- W. P. Griffith, Ruthenium Oxidation Complexes: Their Uses as Homogenous Organic Catalysts, Springer, London, 2010 Search PubMed.
- A. J. Bailey, W. P. Griffith, S. I. Mostafa and P. A. Sherwood, Inorg. Chem., 1993, 32, 268–271 CrossRef CAS.
- Alkali lignin (from Aldrich) (9), pyrolytic lignin (10), a CH2Cl2-soluble fraction Kraft lignin (from Prof. J. Kadla, Faculty of Forestry, University of British Columbia) (11), indulin AT Kraft lignin (from MeadWestvaco Corp.) (12), and a lignin from Lignol Energy Corp. (13).
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- See ESI (pages S2–S3) for details on spectroscopic equipment and techniques used.†.
- The experimental error was relatively large when H2O2 was used due to decomposition in aqueous alkaline media. See ESI for details.†.
- See ESI for detailed tabulated data (Tables S2–S6) and spectra for reactions involving substrates 2 (Fig. S1–S4), 3 (Fig. S5), and 11 (Fig. S6).†.
- This “suboptimal mass balance” has also been observed by Stahl's group for a dimer LMC substrate where discrepancies between the conversion of substrate and yield of product were seen. See ref. 2b.
- A. V. Wacek, W. Limontschew and C. Aas, Ind. Eng. Chem., 1957, 49, 1389–1390 CrossRef.
- D. Fengel and G. Wegener, Wood: Chemistry, Ultrastructure, Reactions, De Gruyter, Berlin, 1989, ch. 6, p. 152 Search PubMed.
- (a) H. Li, X.-S. Chai, M. Liu and Y. Deng, J. Agric. Food Chem., 2012, 60, 5307–5310 CrossRef CAS PubMed; (b) H. Goto, K. Koda, G. Tong, Y. Matsumoto and G. Meshitsuka, J. Wood Chem. Technol., 2006, 26, 81–93 CrossRef CAS.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds: Mechanistic Principles and Synthetic Methodology Including Biochemical Processes, Academic Press, New York, 1981, ch. 3, pp. 33–71 Search PubMed.
- (a) H.-R. Lin, Eur. Polym. J., 2001, 37, 1507–1510 CrossRef CAS; (b) K.-C. Huang, R. A. Couttenye and G. E. Hoag, Chemosphere, 2002, 49, 413–420 CrossRef CAS.
- J. R. Hart, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011, vol. 13, pp. 573–578 Search PubMed.
- O. Lanzalunga and M. Bietti, J. Photochem. Photobiol., B, 2000, 56, 85–108 CrossRef CAS.
- H. R. Devlin and I. J. Harris, Ind. Eng. Chem. Fundam., 1984, 23, 387–392 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tabulated experimental results and associated NMR spectra. See DOI: 10.1039/c4ra00742e |
| This journal is © The Royal Society of Chemistry 2014 |