Efficient catalytic-free method to produce α-aryl cycloalkanones through highly chemoselective coupling of aryl compounds with oxyallyl cations†
Juan Luoab,
Hui Zhoua,
Jiwei Hua,
Rui Wangc and
Qiang Tang*ab
aChongqing Key Laboratory of Biochemistry and Molecular Pharmacology, College of Pharmacy, Chongqing Medical University, Chongqing 400016, P. R. China. E-mail: tangqiang@cqmu.edu.cn
bThe Faculty of Laboratory Medicine, Chongqing Medical University, Chongqing 401331, P. R. China
cSchool of Pharmacy & Bioengineering, Chongqing University of Technology, Chongqing 400054, P. R. China
First published on 25th March 2014
Abstract
Catalytic-free coupling of aryl compounds and α-halo cycloketones via in situ generated oxyallyl cation intermediates is reported here. The reactions efficiently afford α-naphthol cycloalkanones with moderate to excellent yields. Electron-rich aromatic compounds are also used to produce the corresponding α-aryl cycloalkanones, and in some cases, analytically pure products are obtained after simple filtration followed by evaporation.
Introduction
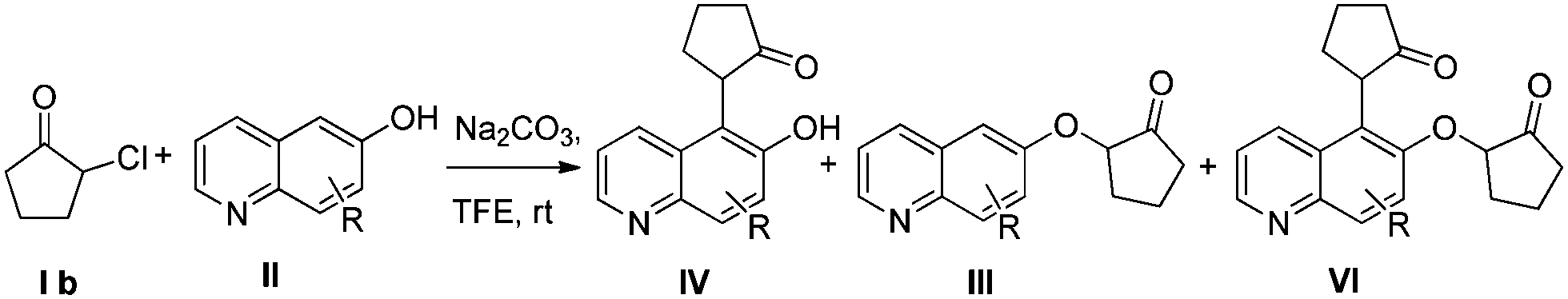
α-Aryl cycloalkanones are important building blocks for concise preparation of medicines or organic functional materials, such as naphthonone,1 brazan,2 and electroluminescence devices.3 The synthesis of α-aryl cycloalkanones, particularly α-naphthol cycloalkanones, can be realized by nucleophilic substitution of α-hydroxylketones with oxygen protected naphthols via Grignard reagents and final deprotection,4 or by direct electrophilic substitution of α-hydroxylketones with phenols under acidic conditions.5 α-Arylation of β-dicarbonyl compounds, as a powerful technology, needs to be catalyzed by transition metals,6 organocatalysts7 or enzymes.8 Pappo and colleagues have recently reported an efficient cross-dehydrogenative coupling reaction between phenols and α-substituted β-ketoesters catalyzed by iron trichloride.9It has been reported that reaction of β-naphthol with α-cyclohexanone in xylene under refluxing temperature produces compound V directly.10 However, a high temperature is necessary, the yield is moderate (22–57%), and a large amount of by-product III is evolved. It is worth noting that direct substitution of α-haloketones with naphthols under basic conditions produces phenol ethers III (Scheme 1) as the main product.11
 | ||
| Scheme 1 Different reaction pathways of α-haloketones with naphthols. | ||
Here we report an efficient catalyst-free coupling of unprotected naphthols and oxyallyl cation intermediates generated from α-halocycloketones at room temperature. Oxyallyl cations have been extensively explored for more than half a century.12 The literature is replete with examples of cycloaddition reactions of such species, especially (4 + 3) cycloaddition reactions.13 Moreover, aromatic groups have been extensively used to trap oxyallyl cations in the context of Nazarov cyclization.14 However, there are only a few reports on the interrupted cycloaddition reactions of aromatic compounds with oxyallyl cations derived from α-halo ketones.15 Recently, MacMillan and colleagues and ourselves both reported an efficient synthesis of α-indole carbonyl compounds via electrophilic aromatic substitution of unprotected indoles to in situ generated oxyallyl cations.16 To further expand the reaction scope, we started to use naphthols as nucleophiles to react with oxyallyl cations generated from α-halo cycloalkanones. To our delight, the reaction proceeded smoothly and produced α-naphthol cycloalkanones IV (Scheme 1) which are eventually transformed to product V (Scheme 1) with high yield.
Results and discussion
We first explored the reaction between 2-chlorocyclohexanone (Ia) and 2-naphthol (IIb). At room temperature, most of the starting materials remain unreacted in common organic solvents such as DMF, DMSO, THF, toluene, Et2O, CH2Cl2, EtOAc, CH3CN, toluene and xylene (Table 1, entries 1 and 2). Although cough medicine, product 1, is evolved when xylene is used as a solvent under high temperature, 33% yield of ether by-product 2, which is not transformed to product 1 within the prolonged reaction time, is also obtained (Table 1, entry 3). In the case where water was used as a solvent, hydrolysis of 2-chlorocyclohexanone becomes the preferred reaction pathway to produce compound 3 serving as the main product,17 while very little of compound 1 could be isolated (Table 1, entry 4). Owing to their high ionizing power and low nucleophilicity, fluorinated alcohols, such as trifluoroethanol (TFE) and hexafluoro isopropanol (HFIP), are the best solvents for generation of oxyallyl cations.18 So the fluorinated alcohols were then evaluated (Table 1, entries 5–11). Under the reaction conditions of Na2CO3 in TFE at room temperature, a clean reaction with high yield is realized (Table 1, entry 5). Higher temperature (Table 1, entry 6) or higher ionizing power (Table 1, entry 7) is beneficial for the reaction rate rather than the reaction efficiency. With TFE as the solvent, the choice of base has a significant effect on the reaction purity and yield. Organic bases, e.g. pyrrolidine and Et3N, effectively initiate the reaction (Table 1, entries 10 and 11). However, when a relatively weak base, e.g. sodium bicarbonate, was chosen, virtually no reaction takes place (Table 1, entry 8). In contrast, when a strong base, e.g. NaOH, was used, the reaction becomes complex with formation of Favorskii rearrangement adduct 5 among the side products (Table 1, entry 9).19
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Temperature (°C) | Time (h) | Productb (yield%) |
| a Reaction condition: I (0.5 mmol), II (0.5 mmol), base (0.6 mmol) in solvent (1 mL).b Isolated yields.c DMF, DMSO, THF, Et2O, toluene, CH2Cl2, EtOAc, CH3CN.d Yield of minor product was determined by crude NMR integration.e No other product was isolated. TFE = 2,2,2-trifluoroethanol; HFIP = hexafluoro-2-propanol. | |||||
| 1 | Na2CO3 | Solventsc | 25 | 48 | 1 (<5) |
| 2 | Na2CO3 | Xylene | 25 | 48 | 1 (<5) |
| 3 | Na2CO3 | Xylene | 140 | 24 | 1/2 (42/33)d |
| 4 | Na2CO3 | H2O | 25 | 12 | 1/3 (8/85)d |
| 5 | Na2CO3 | TFE | 25 | 12 | 1 (91)e |
| 6 | Na2CO3 | TFE | 80 | 10 | 1/4 (88/5)d |
| 7 | Na2CO3 | HFIP | 25 | 10 | 1 (86)e |
| 8 | NaHCO3 | TFE | 25 | 48 | 1 (<5) |
| 9 | NaOH | TFE | 25 | 4 | 1/5 (48/5)d |
| 10 | Pyrrolidine | TFE | 25 | 48 | 1/4 (71/6)d |
| 11 | Et3N | TFE | 25 | 48 | 1/4 (80/6)d |

Then we examined various naphthols in reaction with 2-chlorocyclopentanone. Both protected and unprotected β-naphthols are alkylated at C-1 to produce compounds IV or V in moderate to excellent yields (Table 2, entries 1–6). Bromo-substituted naphthols show excellent outcomes no matter what the substitution position of bromo is (Table 2, entries 3–5). For α-naphthols, the reaction shows excellent chemoselectivity, naphthols are regioselectively alkylated at C-4, and no C-2 alkylated product is obtained (Table 2, entries 7 and 8). To further examine the regioselectivity, 4-chloro-1-naphthol was selected as substrate (Table 2, entry 8). No C-2 alkylated product was detected under our standard reaction conditions. Most of the starting material is recovered after reacting for 12 h.
|
||||
|---|---|---|---|---|
| Entry | Haloketone | Naphthol | Product | Yield |
| a I (0.5 mmol), II (0.5 mol), Na2CO3 (0.6 mmol) in TFE (1 mL).b Isolated yields.c Ratio of diastereomers was determined by integration of benzylic proton signals in 1H NMR spectrum.d No another isomer was detected.e The structure is confirmed by 2D NMR spectra (COSY, HMQC and HMBC).f No desired product is obtained.g The 1H and 13C NMR spectra are the same as that reported in the literature.4ah Ic (1.0 mmol), IIi (0.5 mol), Na2CO3 (1.1 mmol) in HFIP (1 mL).i The ether product (15), 2-(naphthalen-2-yloxy)pentan-3-one, is also isolated. | ||||
| 1 | 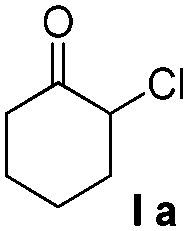 |
 |
 |
91% (dr > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)c :1)c |
| 2 |  |
 |
 |
92% (dr = 10:1)c |
| 3 | 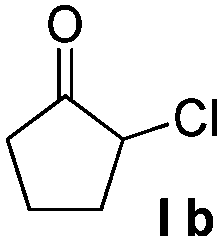 |
 |
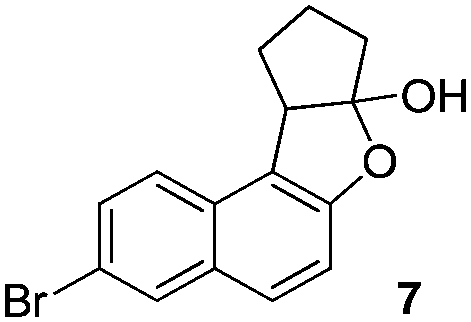 |
93% (dr = 10:1)c |
| 4 |  |
 |
 |
93% (dr = 8:1)c |
| 5 |  |
 |
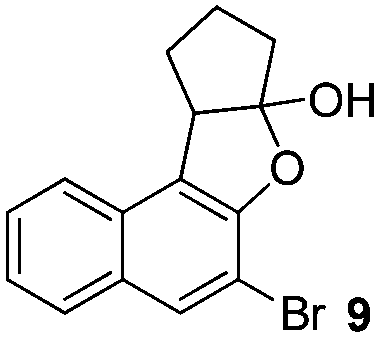 |
94% (dr = 8:1)c |
| 6 | 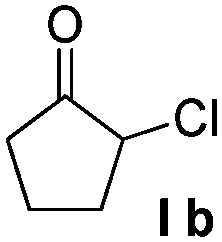 |
 |
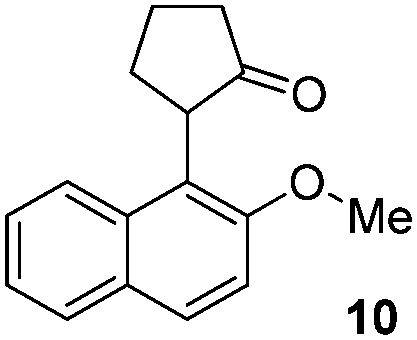 |
88% |
| 7 |  |
 |
 |
89%d,e |
| 8 |  |
 |
 |
—f |
| 9 | 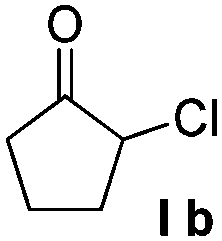 |
 |
 |
87%d,g |
| 10 |  |
 |
 |
11%h,i |

Noncyclic haloketones were also examined. Unfortunately, when 1,1-dichloropropan-2-one or 1,3-dichloropropan-2-one was used, we found that most of the 2-naphthol remained unreacted. Similarly, for substrates 1,3-dibromo-3-methylbutan-2-one and 1-chloro-1,3-diphenylpropan-2-one, a small amount of 2-naphthol is consumed without isolating any desired products. Only 2-bromo-3-pentanone produces a small amount of condensed product 14 in the presence of excessive haloketone (Table 2, entry 10).
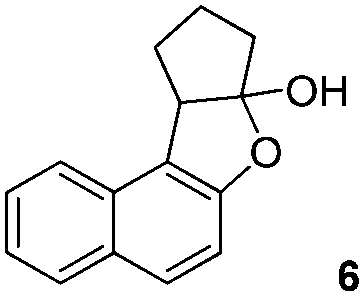
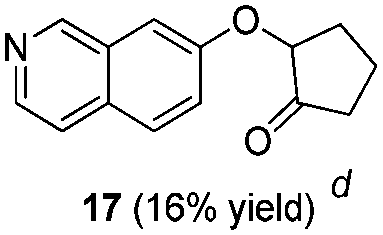
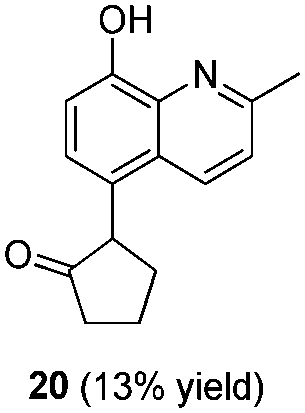
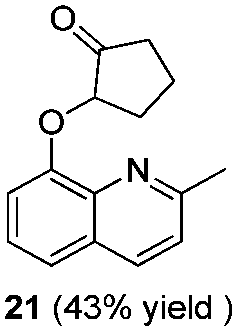
To explore the reaction scope further, we screened different hydroxyquinolines, wherein 5-hydroxy and 7-hydroxy isoquinoline gave acceptable yields of α-aryl cycloalkanones along with their corresponding ether products III (Table 3, entries 1 and 2). It should be noted that product 16 is the ketone isomer, not the hemiketal form. When excessive haloketone Ib was used, disubstituted product 22 was obtained as a mixture of stereoisomers (Table 3, entry 3).
|
||||
|---|---|---|---|---|
| Entry | Quinine | Product IV | Product III | Product VI |
| a Isolated yield.b I (0.5 mmol), II (0.5 mol), Na2CO3 (0.6 mmol) in TFE (1 mL).c I (1.5 mmol), II (0.5 mol), Na2CO3 (1.6 mmol) in TFE (1 mL).d Yield of product 15 was determined by integration of proton signals in 1H NMR spectrum.e Not detected.f Ratio of diastereomers was determined by integration of proton signals in 1H NMR spectrum.g The structure is confirmed by 2D NMR spectra (COSY, HMQC and HMBC). | ||||
| 1b |  |
 |
 |
—e |
| 2b |  |
 |
 |
—e |
| 3c |  |
 |
 |
 |
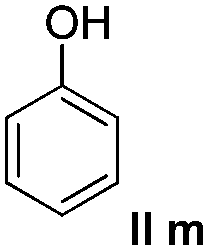
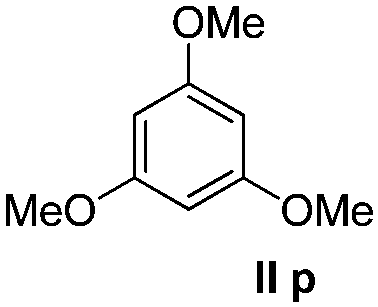
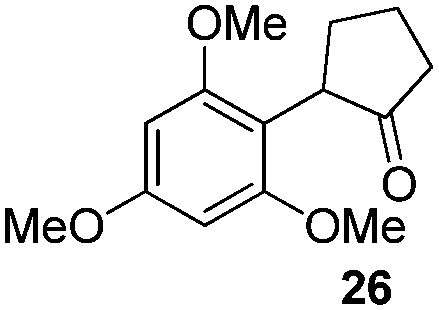
We think that the reaction mechanism is just like that of Friedel–Crafts alkylation, so we used different types of aromatic compound to react with 2-chlorocyclopentanone. We found that an electron-rich system of aromatic compounds plays an important role in the reaction efficiency. When phenol (IIm) was used as a substrate, only ether product 23 is obtained (Table 4, entry 1). For the reaction of substrates IIn and IIo, the main products are, respectively, ether 24 and amine 25, both of which are accompanied by several other complex compounds. However, when the more electron-rich compound IIp was used, an almost quantitative reaction takes place. After simple filtration followed by evaporation, analytically pure product 26 is obtained (Table 4, entry 4). The reaction efficiency is dramatically lowered if there is an electron-withdrawing group on the arene ring. Moderate yield is obtained in the presence of substrate IIq, while no reaction occurs for substrate IIr (Table 4, entries 5 and 6).









Conclusions
We developed a highly efficient and practical method for chemoselective coupling of α-halo cycloalkanones with electron-rich aromatic compounds, especially naphthols, at room temperature. No protection of naphthol substrates and no catalysts are required in our protocol. α-Aryl cycloalkanone 26 is obtained in quantitative yield, and analytically pure products are obtained after simple filtration followed by evaporation. An electron-rich system of aryl compounds plays an important role in the reaction efficiency. Further research on the interrupted cycloaddition reaction of oxyallyl cations is being pursued in our lab.Experimental section
General information
Nuclear magnetic resonance spectra (1H and 13C) were recorded on JEOL ECA400 (400 MHz), Bruker AV300 (300 MHz), AV400 (400 MHz), AV500 (500 MHz) or BBFO400 (400 MHz) spectrometers. 1H NMR chemical shifts were recorded in parts per million (ppm, δ) relative to tetramethylsilane (δ 0.00) or DMSO (δ = 2.50, singlet), and the splitting patterns were designated as singlet (s), doublet (d), triplet (t), quartet (q), dd (doublet of doublets); m (multiplets), etc. All first-order splitting patterns were assigned on the basis of the appearance of the multiplet. Splitting patterns that could not be easily interpreted were designated as multiplet (m) or broad (br). High resolution mass spectral analysis (HRMS) was performed on a Finnigan MAT 95 XP mass spectrometer (Thermo Electron Corporation). Analytical thin-layer chromatography (TLC) was carried out on Merck 60 F254 pre-coated silica gel plates (0.2 mm thickness). Visualization was performed using a UV lamp or chemical stains such as KMnO4 and 2,4-dinitrophenyl hydrazine solutions.Commercially available materials purchased from Alfa Aesar or Aldrich were used as received, apart from α-haloketones that were further purified via distillation or column chromatography over silica gel prior to use. Some of the α-chloroketones (2-bromo-3-pentanone and 1,3-dibromo-3-methylbutan-2-one) were prepared using a literature method.20
Typical procedure for metal-free coupling of aromatic compounds with α-haloketones
A 4 mL vial equipped with a magnetic stir bar was charged with fresh distilled α-halo ketone I (0.5–1.5 mmol), aromatic compound II (0.5 mmol) and TFE or HFIP (1.0 mL). Anhydrous Na2CO3 (0.6–1.6 mmol) was added to the reaction mixture and stirred at room temperature. After completion of the reaction (about 12–24 h, monitored by TLC or crude 1H NMR analysis), the reaction mixture was filtered through a celite pad using Et2O or CH2Cl2 and the filtrate was concentrated under reduced pressure. The crude residue was further purified by silica gel flash chromatography using EtOAc/hexanes as eluent to give pure products. In some cases, analytically pure products could be obtained merely by simple filtration and evaporation under reduced pressure.:1), Mp: 76–79 °C, 11% yield. 1H NMR (400 MHz, CDCl3) δ = 8.08 (dd, J = 11.5, 8.8 Hz, 1H), 7.31 (dd, J = 8.7, 3.2 Hz, 1H), 7.20–7.08 (m, 2H), 5.07–4.82 (m, 1H), 3.92 (dd, J = 18.8, 8.8 Hz, 1H), 2.76 (d, J = 2.1 Hz, 3H), 2.67–2.49 (m, 3H), 2.49–2.33 (m, 3H), 2.33–2.13 (m, 4H), 2.13–1.98 (m, 1H), 1.98–1.80 (m, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ = 218.07, 217.80, 214.13, 213.94, 157.91, 157.86, 152.57, 152.51, 140.74, 140.52, 132.69, 132.52, 128.47, 128.26, 126.70, 126.58, 124.15, 123.85, 122.30, 122.21, 113.27, 112.37, 81.38, 81.16, 51.47, 51.07, 38.71, 38.61, 35.44, 35.39, 31.80, 31.69, 29.40, 25.40, 21.04, 21.02, 17.22 ppm. HRMS (ESI) calcd for C20H22NO3 (M + 1)+: 324.1600, found: 324.1603.Acknowledgements
The project was sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, and the Scientific and Technological Research Program of Chongqing Municipal Education Commission.Notes and references
- (a) M. J. Mousseron, US2882201A, 1959; (b) E. Campaigne and R. F. Weddleton, J. Heterocycl. Chem., 1986, 23, 1625–1628 CrossRef CAS PubMed.
- (a) A. Martínez, M. Fernández, J. C. Estévez, R. J. Estévez and L. Castedo, Tetrahedron, 2005, 61, 1353–1362 CrossRef PubMed; (b) J. N. Chatterjea and K. D. Banerji, Chem. Ber., 1965, 98, 2738–2741 CrossRef CAS PubMed; (c) R. H. Callighan and M. S. Morgan, J. Org. Chem., 1964, 29, 983–985 CrossRef CAS; (d) J. N. Chatterjea, Experientia, 1956, 12, 371–372 CrossRef CAS; (e) R. A. Robinson and E. Mosettig, J. Am. Chem. Soc., 1939, 61, 1148–1151 CrossRef CAS.
- (a) M. Kawamura and S.-s. Chiba, EP2436679A1, 2009; (b) U. Mitschke and P. Bauerle, J. Mater. Chem., 2000, 10, 1471–1507 RSC; (c) J. Salbeck, Ber. Bunsenges. Phys. Chem., 1996, 100, 1667–1677 CrossRef CAS PubMed; (d) E. Campaigne and S. W. Osborn, J. Heterocycl. Chem., 1968, 5, 655–661 CrossRef CAS PubMed; (e) T. K. Chaitanya and R. Nagarajan, Org. Biomol. Chem., 2011, 9, 4662–4670 RSC.
- (a) M. Góra, M. K. Łuczyński and J. J. Sepioł, Synthesis, 2005, 2005, 1625–1630 CrossRef PubMed; (b) M. Rettig, A. Sigrist and J. Rétey, Helv. Chim. Acta, 2000, 83, 2246–2265 CrossRef CAS; (c) C. M. Nachtsheim and A. W. Frahm, Arch. Pharm., 1989, 322, 187–197 CrossRef CAS PubMed.
- (a) S. A. Belapure, Z. G. Beamer, J. E. Bartmess and S. R. Campagna, Tetrahedron, 2011, 67, 9265–9272 CrossRef CAS PubMed; (b) K. R. Prabhakar, V. P. Veerapur, P. Bansal, K. P. Vipan, K. M. Reddy, A. Barik, B. K. D. Reddy, P. Reddanna, K. I. Priyadarsini and M. K. Unnikrishnan, Bioorg. Med. Chem., 2006, 14, 7113–7120 CrossRef CAS PubMed; (c) S. K. Kundu and A. Pramanik, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2004, 43, 595–603 Search PubMed; (d) S. K. Kundu, A. Patra and A. Pramanik, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2004, 43, 604–611 Search PubMed; (e) S. Das, A. Pramanik, R. Fröhlich and A. Patra, Tetrahedron, 2004, 60, 10197–10205 CrossRef CAS PubMed.
- (a) E. Motti, N. Della Ca, D. Xu, S. Armani, B. M. Aresta and M. Catellani, Tetrahedron, 2013, 69, 4421–4428 CrossRef CAS PubMed; (b) V. Bhat, J. A. Mackay and V. H. Rawal, Org. Lett., 2011, 13, 3214–3217 CrossRef CAS PubMed; (c) N. V. Sastry Mudiganti, S. Claessens and N. De Kimpe, Tetrahedron, 2009, 65, 1716–1723 CrossRef PubMed; (d) X. Guo, R. Yu, H. Li and Z. Li, J. Am. Chem. Soc., 2009, 131, 17387–17393 CrossRef CAS PubMed; (e) M. P. DeMartino, K. Chen and P. S. Baran, J. Am. Chem. Soc., 2008, 130, 11546–11560 CrossRef CAS PubMed; (f) J. Christoffers, T. Werner, W. Frey and A. Baro, Eur. J. Org. Chem., 2003, 2003, 4879–4886 CrossRef PubMed.
- (a) J. Alemán, B. Richter and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 5515–5519 CrossRef PubMed; (b) S. Kobbelgaard, M. Bella and K. A. Jørgensen, J. Org. Chem., 2006, 71, 4980–4987 CrossRef CAS PubMed.
- J. Pietruszka and C. Wang, Green Chem., 2012, 14, 2402–2409 RSC.
- R. Parnes, U. A. Kshirsagar, A. Werbeloff, C. Regev and D. Pappo, Org. Lett., 2012, 14, 3324–3327 CrossRef CAS PubMed.
- (a) T. S. Osdene and P. B. Russell, J. Org. Chem., 1966, 31, 2646–2648 CrossRef CAS; (b) F. Ebel, Helv. Chim. Acta, 1929, 12, 3–16 CrossRef CAS PubMed.
- (a) W.-J. Bai, J.-H. Xie, Y.-L. Li, S. Liu and Q.-L. Zhou, Adv. Synth. Catal., 2010, 352, 81–84 CrossRef CAS PubMed; (b) J. A. Murphy, A. G. J. Commeureuc, T. N. Snaddon, T. M. McGuire, T. A. Khan, K. Hisler, M. L. Dewis and R. Carling, Org. Lett., 2005, 7, 1427–1429 CrossRef CAS PubMed; (c) O. Orovecz, P. Kovács, P. Kolonits, Z. Kaleta, L. Párkányi, É. Szabó and L. Novák, Synthesis, 2003, 2003, 1043–1048 CrossRef; (d) G. Lauktien, F.-J. Volk and A. W. Frahm, Tetrahedron: Asymmetry, 1997, 8, 3457–3466 CrossRef CAS; (e) H. W. Dürbeck, C. G. B. Frischkorn and K. Hilpert, Tetrahedron, 1971, 27, 2927–2937 CrossRef; (f) M. K. M. Dirania and J. Hill, J. Chem. Soc. C, 1969, 2144–2147 RSC; (g) K. L. Williamson, R. T. Keller, G. S. Fonken, J. Szmuszkovicz and W. S. Johnson, J. Org. Chem., 1962, 27, 1612–1615 CrossRef CAS; (h) F. G. Bordwell and M. W. Carlson, J. Am. Chem. Soc., 1970, 92, 3370–3377 CrossRef CAS.
- (a) A. G. Lohse and R. P. Hsung, Chem.–Eur. J., 2011, 17, 3812–3822 CrossRef CAS PubMed; (b) M. Harmata, Chem. Commun., 2010, 46, 8886–8903 RSC; (c) M. Harmata, Chem. Commun., 2010, 46, 8904–8922 RSC; (d) D. A. Foley and A. R. Maguire, Tetrahedron, 2010, 66, 1131–1175 CrossRef CAS PubMed; (e) M. Harmata, Adv. Synth. Catal., 2006, 348, 2297–2306 CrossRef CAS PubMed; (f) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem.–Eur. J., 2006, 12, 3438–3447 CrossRef CAS PubMed; (g) J. Huang and R. P. Hsung, Chemtracts, 2005, 18, 207–214 CAS; (h) Y. Tamaru, Eur. J. Org. Chem., 2005, 2005, 2647–2656 CrossRef PubMed; (i) B. Niess and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2005, 44, 26–29 CrossRef CAS PubMed; (j) I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934–1949 CrossRef CAS PubMed; (k) M. Harmata and P. Rashatasakhon, Tetrahedron, 2003, 59, 2371–2395 CrossRef CAS; (l) M. Harmata, Acc. Chem. Res., 2001, 34, 595–605 CrossRef CAS PubMed; (m) J. H. Rigby and F. C. Pigge, Org. React., 1997, 51, 351–478 CAS; (n) M. Harmata, Tetrahedron, 1997, 53, 6235–6280 CrossRef CAS; (o) J. Mann, Tetrahedron, 1986, 42, 4611–4659 CrossRef CAS; (p) T. T. Tidwell, Angew. Chem., Int. Ed., 1984, 23, 20–32 CrossRef PubMed; (q) H. M. R. Hoffmann, Angew. Chem., 1984, 96, 29–48 CrossRef CAS PubMed; (r) H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1984, 23, 1–19 CrossRef PubMed; (s) R. Noyori and Y. Hayakawa, Org. React., 1983, 29, 163–344 CAS; (t) R. Noyori, Acc. Chem. Res., 1979, 12, 61–66 CrossRef CAS; (u) H. Takaya, S. Makino, Y. Hayakawa and R. Noyori, J. Am. Chem. Soc., 1978, 100, 1765–1777 CrossRef CAS; (v) H. M. R. Hoffmann, Angew. Chem., Int. Ed., 1973, 12, 819–835 CrossRef PubMed.
- (a) B. Föhlisch, R. Flogaus, G. H. Henle, S. Sendelbach and S. Henkel, Eur. J. Org. Chem., 2006, 2006, 2160–2173 CrossRef PubMed; (b) D. I. MaGee, E. Godineau, P. D. Thornton, M. A. Walters and D. J. Sponholtz, Eur. J. Org. Chem., 2006, 2006, 3667–3680 CrossRef PubMed; (c) B. Föhlisch, H. Korfant, H. Meining and W. Frey, Eur. J. Org. Chem., 2000, 2000, 1335–1344 CrossRef; (d) B. Lo, S. Lam, W.-T. Wong and P. Chiu, Angew. Chem., Int. Ed., 2012, 51, 12120–12123 CrossRef CAS PubMed.
- (a) Y. Kwon, R. McDonald and F. G. West, Angew. Chem., Int. Ed., 2013, 52, 8616–8619 CrossRef CAS PubMed; (b) S. A. Bonderoff, T. N. Grant, F. G. West and M. Tremblay, Org. Lett., 2013, 15, 2888–2891 CrossRef CAS PubMed; (c) J. Boudreau, M.-A. Courtemanche, V. M. Marx, D. Jean Burnell and F.-G. Fontaine, Chem. Commun., 2012, 48, 11250–11252 RSC; (d) D. Lebœuf, V. Gandon, J. Ciesielski and A. J. Frontier, J. Am. Chem. Soc., 2012, 134, 6296–6308 CrossRef PubMed; (e) Z.-X. Ma, S. He, W. Song and R. P. Hsung, Org. Lett., 2012, 14, 5736–5739 CrossRef CAS PubMed; (f) W. T. Spencer, M. D. Levin and A. J. Frontier, Org. Lett., 2010, 13, 414–417 CrossRef PubMed; (g) J. Huang, D. Lebœuf and A. J. Frontier, J. Am. Chem. Soc., 2011, 133, 6307–6317 CrossRef CAS PubMed; (h) T. N. Grant, C. J. Rieder and F. G. West, Chem. Commun., 2009, 5676–5688 RSC; (i) T. Jin and Y. Yamamoto, Org. Lett., 2008, 10, 3137–3139 CrossRef CAS PubMed; (j) C. J. Rieder, R. J. Fradette and F. G. West, Heterocycles, 2010, 80, 1413–1427 CrossRef CAS PubMed; (k) V. M. Marx and D. J. Burnell, J. Am. Chem. Soc., 2010, 132, 1685–1689 CrossRef CAS PubMed; (l) P. Cao, X.-L. Sun, B.-H. Zhu, Q. Shen, Z. Xie and Y. Tang, Org. Lett., 2009, 11, 3048–3051 CrossRef CAS PubMed; (m) C. J. Rieder, R. J. Fradette and F. G. West, Chem. Commun., 2008, 1572–1574 RSC; (n) T. N. Grant and F. G. West, J. Am. Chem. Soc., 2006, 128, 9348–9349 CrossRef CAS PubMed; (o) T. D. White and F. G. West, Tetrahedron Lett., 2005, 46, 5629–5632 CrossRef CAS PubMed; (p) A. Yungai and F. G. West, Tetrahedron Lett., 2004, 45, 5445–5448 CrossRef CAS PubMed; (q) J. A. Bender, A. E. Blize, C. C. Browder, S. Giese and F. G. West, J. Org. Chem., 1998, 63, 2430–2431 CrossRef CAS.
- (a) M. Harmata, C. Huang, P. Rooshenas and P. R. Schreiner, Angew. Chem., Int. Ed., 2008, 47, 8696–8699 CrossRef CAS PubMed; (b) J. Leitich and I. Heise, Eur. J. Org. Chem., 2001, 2001, 2707–2718 CrossRef.
- (a) Q. Tang, X. Chen, B. Tiwari and Y. R. Chi, Org. Lett., 2012, 14, 1922–1925 CrossRef CAS PubMed; (b) M. N. Vander Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075–3079 RSC.
- (a) T. Utsukihara, H. Nakamura, M. Watanabe and C. Akira Horiuchi, Tetrahedron Lett., 2006, 47, 9359–9364 CrossRef CAS PubMed; (b) D. J. Aitken, C. Gauzy and E. Pereira, Tetrahedron Lett., 2004, 45, 2359–2361 CrossRef CAS PubMed; (c) M. Schmittel and M. Röck, Chem. Ber., 1992, 125, 1611–1620 CrossRef CAS PubMed; (d) P. D. Bartlett and G. F. Woods, J. Am. Chem. Soc., 1940, 62, 2933–2938 CrossRef CAS.
- (a) A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421–8426 CrossRef CAS PubMed; (b) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 2004(18), 29 Search PubMed; (c) H. Schaal, T. Haber and M. A. Suhm, J. Phys. Chem. A, 1999, 104, 265–274 CrossRef; (d) W. J. Middleton and R. V. Lindsey, J. Am. Chem. Soc., 1964, 86, 4948–4952 CrossRef CAS.
- (a) B. Föhlisch, T. Franz and G. Kreiselmeier, Eur. J. Org. Chem., 2005, 2005, 4687–4698 CrossRef PubMed; (b) B. Föhlisch, A. Radl, R. Schwetzler-Raschke and S. Henkel, Eur. J. Org. Chem., 2001, 2001, 4357–4365 CrossRef; (c) A. W. Fort, J. Am. Chem. Soc., 1962, 84, 4979–4981 CrossRef CAS.
- (a) K. Tanemura, T. Suzuki, Y. Nishida, K. Satsumabayashi and T. Horaguchi, Chem. Commun., 2004, 470–471 RSC; (b) K. M. Brummond and K. D. Gesenberg, Tetrahedron Lett., 1999, 40, 2231–2234 CrossRef CAS; (c) R. R. Fraser and F. Kong, Synth. Commun., 1988, 18, 1071–1077 CrossRef CAS.
- (a) K. Chung, S. M. Banik, A. G. De Crisci, D. M. Pearson, T. R. Blake, J. V. Olsson, A. J. Ingram, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 7593–7602 CrossRef CAS PubMed; (b) D. B. Ramachary and C. F. Barbas, Org. Lett., 2005, 7, 1577–1580 CrossRef CAS PubMed.
- B. Foehlisch, R. Joachimi and S. Reiner, J. Chem. Res., Miniprint, 1993, 7, 1701–1730 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical shifts assignments, and spectral data for compounds. See DOI: 10.1039/c4ra01043d |
| This journal is © The Royal Society of Chemistry 2014 |