
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The energy storage application of core-/yolk–shell structures in sodium batteries
Anurupa
Maiti
 *,
Rasmita
Biswal
,
Soumalya
Debnath
and
Anup
Bhunia
*
*,
Rasmita
Biswal
,
Soumalya
Debnath
and
Anup
Bhunia
*
Department of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, India. E-mail: anu.inorganic@gmail.com; bhunia1988@gmail.com; rs.biswal14@gmail.com; soumalyabethua@gmail.com
First published on 13th May 2024
Abstract
Materials with a core–shell and yolk–shell structure have attracted considerable attention owing to their attractive properties for application in Na batteries and other electrochemical energy storage systems. Specifically, their large surface area, optimum void space, porosity, cavities, and diffusion length facilitate faster ion diffusion, thus promoting energy storage applications. This review presents the systematic design of core–shell and yolk–shell materials and their Na storage capacity. The design of different metal structures with different shapes and their corresponding synthesis methods are also highlighted. Moreover, changes in capacity with a variation in the carbon moiety and porosity in terms of applications are highlighted. Furthermore, to compete with the dominant Li batteries in the market, materials with low-cost large-scale production and high active mass loading need to be developed. Thus, both yolk- and core–shell structures have been designed considering their significant structural advantages for application in Na batteries and their impacts on the rate capacity and reversible capacity. This review also demonstrates the advantages of yolk–shell and core–shell structures in faster Na ion transportation and excellent cycling stability. Furthermore, the synergistic effect of the core–shell structure enhances reversible capacity. Additionally, the conducting coating of the encapsulated structure increases electron transport, and a higher exposure of the electrode to the electrolyte is further beneficial for the growth of a stable SEI layer. Considering these advantages and disadvantages, this review may help guide the future advancement of sodium batteries (SIBs) in upcoming research based on the advantages of the core–shell and yolk–shell morphology. Finally, future perspectives regarding machine learning (ML) to access better performances in Na batteries are discussed.
 Anurupa Maiti | Dr Anurupa Maiti obtained her bachelor's degree from Vidyasagar University, India, and PhD from the Indian Institute of Technology Kharagpur, in 2022. Following her doctoral studies, she conducted postdoctoral research at the Indian Institute of Science Education and Research, Kolkata. In addition to her research endeavors, Dr Maiti has pursued a PG-Diploma in Machine Learning, showcasing her multidisciplinary interests. Her research focuses on electrochemistry, particularly in the realm of organic electrocatalysis, coupled with applications of machine learning. This combination of expertise suggests a forward-looking approach to tackling challenges at the intersection of chemistry and data science. |
1. Introduction
The storage of sustainable energy has become an important topic owing to the increasing demand for renewable clean energy in daily life. In this case, high-temperature (HT) Na–S battery technology was first developed for application in electric vehicles (EV) by Ford Motor Company in 1966.1 Faradion (UK), Novasis (USA), HiNa (China), and Tiamat (France) are some of the renowned companies who have evolved their expertise towards sodium battery technology in the last few years.2 The leading Chinese company CATL (market value of almost $200 billion) is focused on developing batteries with high energy density, reaching 160 W h kg−1 to date, and is targeted to reach a value of 300 W h kg−1 for sodium batteries.3 Moreover, the Chinese automaker JAC Group and tech company ‘HiNa Battery’ developed Na-battery technology named ‘Hua Xianzi’, a compact electric vehicle that can travel 155 miles on a single charge.3 Most sodium battery startup companies were established after 2010, and by 2030, the Na e-vehicle will be a new market competitor to Li-ion batteries. The Na-ion battery company manufacturers have started their business with the total market value of 6–60 billion RMB (Renminbi, official currency of China), except for CATL (1329.643 billion RMB).3Recently, numerous advancements have been made in battery technology and its different integral systems. Multifold modifications are employed on different battery components such as the anode, cathode, electrolyte, membrane, and thermal management system. Researchers have employed different technologies in several battery systems to enhance their performance. In the last decade, numerous reviews and publications have focused on the components of sodium batteries such as their separator, cathode, anode and electrolyte.1,4 Alternatively, herein, we highlight the various aspects of the sodium battery briefly. Importantly, the focus of this review is to help guide the selection of appropriate cathodic and anodic materials based on a thorough survey of the previous literature.
Sodium (Na) is a much cheaper metal compared to lithium (Li), potassium (K), and magnesium (Mg) and its redox potential is comparatively more positive [E°(Na+/Na0) = −2.71 V vs. the standard hydrogen electrode] than that of Li metal.1 Hence its operating voltage window (greater than 2 V can be considered), theoretical capacity (1672 mA h g−1) and operating temperature (operating at 20–300 °C) will facilitate a higher capacity compared to lithium.1 However, the ionic radius of sodium is larger (1.02 Å) than that of lithium (0.76 Å) and it is heavier (Na: 23 g mol−1vs. Li: 7 g mol−1), consequently producing a lower volumetric capacity.5 However, despite these shortcomings, it is interesting to note that metallic Na is abundant and cheap compared to Li and due to its low cost and toxicity, it is an ideal alternative for the future production of electrical vehicles (EV).6 Moreover, lithium extraction from mines is very costly, laborious and produces huge CO2 emissions.7 Hence, lithium batteries are not ecofriendly and their main cathodic components such as cobalt and nickel are also carcinogenic elements. Importantly, the melting point of sodium is higher than that of lithium, and thus it can operate at very high temperatures (above 300 °C). Sodium batteries are also less explosive in a higher temperature range and can operate in a wide temperature range of 20 °C to 300 °C. Therefore, for long-distance travel, the decrease in capacity is less associated with temperature. Hence, sodium batteries are considered market competitors in the future.5 However, high operational temperatures can cause severe damage to their liquid system and electrolyte, limiting their industrial application. Furthermore, sodium batteries demonstrate a promising performance for the storage of renewable energy from solar cells, power grids and electric vehicles given that they safely work at a higher temperature and have lower air sensitivity compared to lithium.8 Gratifyingly, the research on achieving an operational battery temperature in the intermediate position is ongoing with various modifications and several changes already applied for battery system improvement.9 The main disadvantages associated with sodium batteries are their low energy density, low cyclic stability and operational temperature, which are also a major area of research and further improvements are needed. Moreover, considering the advancement of battery technology to achieve ultrafast charging (80% charge within 5 min), the recyclability of cathode materials has been proposed recently. Hence, we need to focus on the recyclability and higher cyclability of the components of sodium batteries for their industrialization.10
In Na-batteries, Na-metal is used as the anode material, while the cathode is generally a higher surface carbon, liquid sulphur or a layered oxide material.11 Besides, the medium, electrolyte, electrolyte additive, highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy gap of the electrolyte and additive, cathode and anode materials, electrolyte viscosity, and temperature are the key factors affecting the performance of sodium batteries.11 Presently, although the aqueous sodium battery is a low-cost market option, its narrow operational window limits its usefulness. Hence, organic electrolytes are comparatively better suited to achieve a high specific capacity.12 Recent studies focused on the improvement of doping in the inner side of the core, increasing the porosity of carbon materials, and using a high-conductive electrolyte to in situ engineer the material.1 Doping can also increase the electron density at the metal sites, which will enhance the storage of ions in both the cathode and anode. Moreover, the construction of the cathode and anode with different hard carbons enhances the Na storage performance. Initially, Prussian-based materials were used as the cathode material for sodium storage; however, the negative ion material does not provide stable structure.13 Hence, further design is needed for the application of sodium batteries. In this case, the rational design of the yolk–shell structure enables the controlled growth of the shell and core structures, where the void between shell and core can facilitate the Na+ insertion/extraction process, which are systematically synthesized by controlling the ratio of the precursor reagents.14
Sodium-ion batteries involve mainly three mechanistic pathways, i.e., charge insertion mechanisms for intercalation, alloying, and conversion.15 The term “insertion” has also been employed to describe the introduction of foreign species (atoms, ions, and molecules) into the host lattice.15 Furthermore, the term intercalation is used in batteries, which is also one type of topographical insertion of a guest molecule in batteries. The term intercalation is associated with major structural changes during the alloying possess, i.e. a sodium ion is intercalated in the host lattice by insertion as a guest molecule. The significant difference between insertion and intercalation involves volume changes.15 In the case of the intercalation process, large-size sodium ions cause volume changes, which create a higher activation energy as a result of loss of capacity. Besides the intercalation method, alloying is another widely investigated process, where Na+ ions are inserted into the host matrix and form an alloy-based compound.
The importance of the alloying reaction is a lower cell voltage operation with higher gravimetric capacity compared to the intercalation process.15 However, higher mechanical stress and loss of electrical contact are the critical drawbacks of this process. Hence, several improvements in the alloying process are extensively demanded.16 The main issues are related to large volume expansions, typical voltage hysteresis during discharging cycles and sluggish energy storage process.17 The LUMO and the HOMO energy gap of the electrolyte in SIBs determines their thermodynamic stability window.18 The critical issue of Na-ion batteries is the degradation of their performance at low temperatures below 0 °C, where the Na+ ion loses its diffusion ability and at higher temperature, which is not applicable for EV application due to safety concerns.19 Hence, a wide operation temperature range should be the focus of future research for the commercialization of SIBs. In this case, several chemical modifications such as heteroatom doping20 and formation of composites and covalent networks have been applied, but there is a long way to go to achieve the target values for industrialization processes like lithium batteries.15 Furthermore, to compete in the commercial market with Li-ion batteries, some challenging drawbacks arising from SIBs have to be addressed. The construction of Na-ion batteries with a low-temperature range, Na+ migration, and higher operational voltage should be controlled to achieve the maximum performance.19 In general, the SIB electrolytes are a mixture of organic electrolytes with dissolved sodium salts. However, the organic electrolytes utilized for SIBs are linear/cyclic carbonates and ethers of non-degradable sodium salts. The electrolyte is prepared by mixing 1.0 M of NaClO4 or NaPF6 salt in ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), diethyl carbonate (DEC), and ethyl methyl carbonate (EMC) solvents.21
Yolk–shell structures have been rationally designed due to their capability to alleviate volume expansion and accumulate numerous ions in their cavity.22 In general, a yolk–shell structure has a suitable void space to facilitate electron transfer and its co-tuneable phases can enhance the rate capability of sodium storage. The scalable and easily accessible methods for the preparation of yolk–shell structures provide diffusion channels, which also influence their sodium storage capacity.23 The synthesized yolk–shell structures provide a shorter Li+/Na+ diffusion distance and enhance the penetration of ions in the anode or cathode.24 Also, the integration of a carbon shell on yolk–shell structures prevents corrosion from the electrolyte and improves the conductivity of the materials. Furthermore, any undesirable reactions are completely disrupted from the electrolyte by the protective shell structures. The controlled synthesis of yolk–shell structures can tune their thickness, size, and shell volume, which is capable of regulating their sodium storage performances.25 In addition, the integration of yolk–shell structures affects the dielectric constant, conductivity and penetration of the electrolyte. The design of scalable yolk–shell structures requires low-cost equipment with a setup to achieve a wide range of temperatures.26 Yolk–shell structures integrated with conducting carbon shells increase the rate capability and cycle stability. Further, although microspheres, flakes, flowers, and 2D layered structures are also attractive for application in SIBs, the recent literature suggests that yolk–shell/core shell structures with different morphologies can achieve the maximum practical significance.27 Recent developments in the low-cost synthesis of core–shell structures also demonstrate the facile integration of electrode materials. Although a comparative overview provides insight into the mechanism, it depends on the material design, conductive platform, mesoporous channel, etc. Core–shell and yolk–shell materials enhance the reversible capacity, battery cyclability, rate capacity and lifetime in comparison to other nanomaterials.28–30 The void of the yolk aids the growth of a stable SEI layer on the carbon shell, and hence increases the cycle stability.31 The carbon shell also stops the pulverization of the core and stops the volume expansion during cycling.31 Moreover, the multi-shell core or yolk shell structure enhances the synergistic effect and maintains long-term interaction.32 Hence, by changing the reaction conditions, solvent ratio and surface integration, the performances can be maximized. The notable cycle stability of yolk–shell and hollow core–shell structures is slightly higher compared to core–shell structures. Thus, this review comprehensively discusses the design of yolk–shell and core–shell structures, their controllable parameters, and surface integration. Importantly, the related commercialization of lithium-ion batteries and sodium-ion batteries and recent topics of machine learning on batteries are also discussed. The synthetic procedures and role of yolk–shell/core–shell structures across wide electrochemical applications are proposed in the critical discussion. Finally, a summary of our critical views on sodium batteries are presented in the last section.
2. Effects controlling the sodium storage performances
According to recent research, various cathode materials in SIBs such as Na-based layered oxides,33 sodium superionic conductor materials34 and fluorophosphates,35 have attracted considerable attraction. However, the major challenge associated with these cathode materials is their close-packed oxygen ion array, which hinders the diffusion of sodium ions.29 However, the future modification of cathode materials can enable high sodium ion storage, long life, and high rate capability.30 Although layered rock salt structures have been shown to be promising materials for sodium storage due to their intercalation property, they show poor cycle stability due to their volume expansion during the alloying reaction.36Again, doping can maximize the reversible capacity by improving the reactivity at the surface/interface region by reducing the solid electrolyte interfacial resistance.37 Doping causes a reduction in the kinetic barrier of Na+ ion movement in the intrinsic structure. Moreover, a higher quantity of dopant can control the cyclability and rate capacity performance.38 Doping of nitrogen atoms in FeS2/Fe3O4@C,39 yolk-like TiO2,38 MoSe2-C,40 and Bi2S3@mesoporous carbon41 nanocomposites resulted in a higher reversible capacity. Furthermore, introducing a higher degree of pyrrolic nitrogen,42 multi-metal interaction43 promotes the reversible capacity and rate capacity.44 Covalent assembly with inorganic materials also facilitates the ion diffusion kinetics, outstanding mechanical stability and interlayer electron transfer.45 Moreover, a hierarchical porous structure facilitates the charge transfer kinetics and acceleration of electrolyte transport, which improve the reaction kinetics.46 Covalent triazine frameworks as facile and low-cost synthesized networks deliver a high reversible capacity due to their two-dimensional exfoliated few-layered structure.47 A coating of carbon layer also regulates the superior sodium storage performances.48 A recent investigation also showed that a carbon coating on the cathode and anode increases the conductivity of their materials and increases their performances.49 The optimum void space also accumulates charge, exhibiting the best cyclic performances.50 It was found that a nano-size (∼15 nm) pore diameter could sustain over 80% capacity retention during 1300 cycles at 5 A g−1.51 However, it was observed that 10–20 nm carbon layer and pore volume of around 10 nm resulted in a capacity of 198 mA h g−1 at 20 A g−1 over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.50 It was also noticed that a yolk size of ∼100–200 nm and carbon thickness of 20 nm with the average pore radius of 2 nm resulted in 90.8% capacity retention after 1000 cycles at 1 A g−1.52 Hence, it is preferable to maintain the pore size of 10 nm to obtain a higher electrochemical performance. Moreover, the optimum hollow structure can prevent large volume expansion and enhance the electronic conductivity, which also result in an enhancement in stability and rate capability.53,54
000 cycles.50 It was also noticed that a yolk size of ∼100–200 nm and carbon thickness of 20 nm with the average pore radius of 2 nm resulted in 90.8% capacity retention after 1000 cycles at 1 A g−1.52 Hence, it is preferable to maintain the pore size of 10 nm to obtain a higher electrochemical performance. Moreover, the optimum hollow structure can prevent large volume expansion and enhance the electronic conductivity, which also result in an enhancement in stability and rate capability.53,54
Researchers developed a new electrospinning technique for SIB application, where a large surface area and high porosity could be obtained.55 Additionally, the defective carbon material prepared through the electrospinning method also resulted in an increase in the reversible capacity.56 It has also been noted that crystalline materials in regular form show higher intercalation of sodium ions through their regular plane. The preferential orientation of the (102) and (104) facets of layered oxides could improve the capacity and rate performance.57 Furthermore, the ball-milling process increased the ionic conductivity of the material compared to the materials synthesized via other methods, where the tetragonal form provides a greater capacity than the cubic form.58 Recent advances showed that the anode material in sodium batteries is very important in controlling their reversible capacity and cycle stability. The electrode using the standard poly(vinylidene difluoride) binder exhibits the weakest electrochemical properties, while that employing sodium alginate binder supplies the best reversible capacity and higher cycle stability with higher capacity retention due to its high ionic conductivity.59 The SIB electrolyte also enhances the capacity by regulating the solvent and salt concentrations.60 For industrialisation, a bulk amount of materials is needed for their proper application. Among the synthesis methods, the spray drying method is the one of the facile, simple methods for large-scale production.61 The proper choice of the substrate for the current collector also has a great impact on the performance of SIBs. Among the foils, aluminium foil is a low-cost material, simple, and easily synthesized as the current collector.62 The standardization of anode materials can also potentially impact the storage performances of SIBs. Among the anode materials, sulphides, phosphides, selenides, and nitrides are used due to their high storage capacity in SIBs.63–65 A recent investigation showed that phosphide materials offer several advantages such as higher energy density and superior kinetics.66 Among the phosphorus materials, black phosphorus shows the highest reversible capacity and lowest ion diffusion coefficient.67 Further, ultra-stable cycle performances can be controlled by tuning the thickness of the material.68 Morphology construction is another vital parameter for regulating the storage capacity. In this case, compared to hierarchal micropore structures, hollow urchin-type structures showed a higher reversible capacitance due to their adjustable porous channels and void space.69 Moreover, 2D graphene-based compounds with abundant oxygen-containing functional moieties facilitate the storage of sodium ions. The honeycomb-like structure is one of the structures that promotes the sodium ion storage capacity, which is favourable for a high sodium storage capacity.70 The voltage window is another factor for improving the capacity. A literature study showed that an anode material containing redox pairs exhibiting a voltage plateau at 3.4 V (vs. Na+/Na) in the range of 1.0–4.0 V delivered a higher reversible capacity but inferior cycling stability.71 Also, to overcome safety issues such as membrane damage and flammable liquids, well-designed and modified membranes have been engineered recently.72 A polymeric electrolyte membrane such as PFSA-Na showed high ionic conductivity, outstanding thermal stability, and excellent mechanical flexibility in a wide temperature range.73 The charge–discharge capacity and coulombic efficiency also depend on the electrolyte. It was noted that a polymeric gel electrolyte containing polyvinylidene fluoride-hexafluoropropylene (PVDF-HFP) resulted in a higher coulombic efficiency.74
3. Yolk shell and core shell
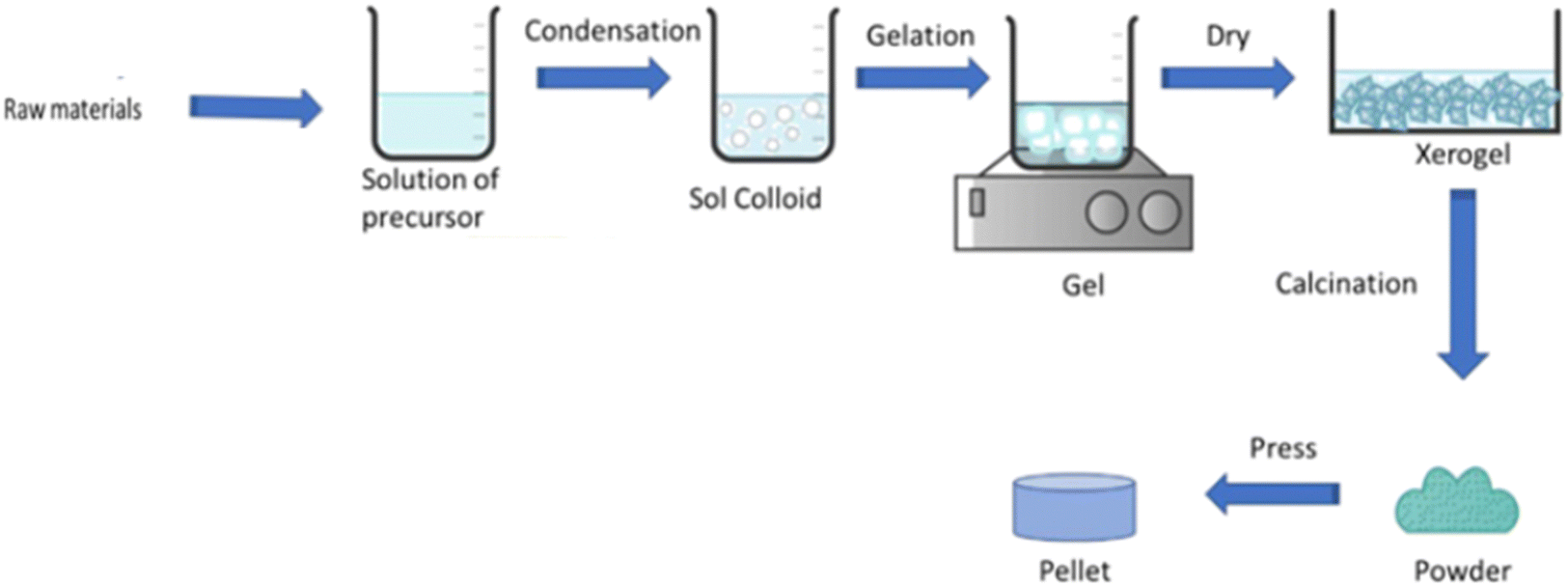
Yolk–shell NPs consist of a movable core/shell with optimum void or outer shell with the same or different materials. The void space is generated through the removal of the sacrificial layer or partial etching of the outer shell. The inner core or yolk material generally consists of different metals or metal oxides, non-metal oxides, metal sulfides, phosphides, selenides, bimetallic compounds, etc.27,38,43,75 Furthermore, porosity can be introduced in the material by different template etching methods. Core–shell structures consist of a core/sacrificial shell/outer shell structure. Recently, many studies have been reported on yolk shell and core shell structures with different applications.76,77 Generally, the core consists of an inorganic metal or polymetallic small materials, which is further covered by hard templates such as different carbon and hard templates.78 The core shell or yolk shell can be synthesized through different template-assisted or template-free methods such as galvanic replacement, Ostwald ripening, and self-assembly methods. These methods are systematically described in the following sections. Fig. 1 depicts the formation of the core and yolk shell structure.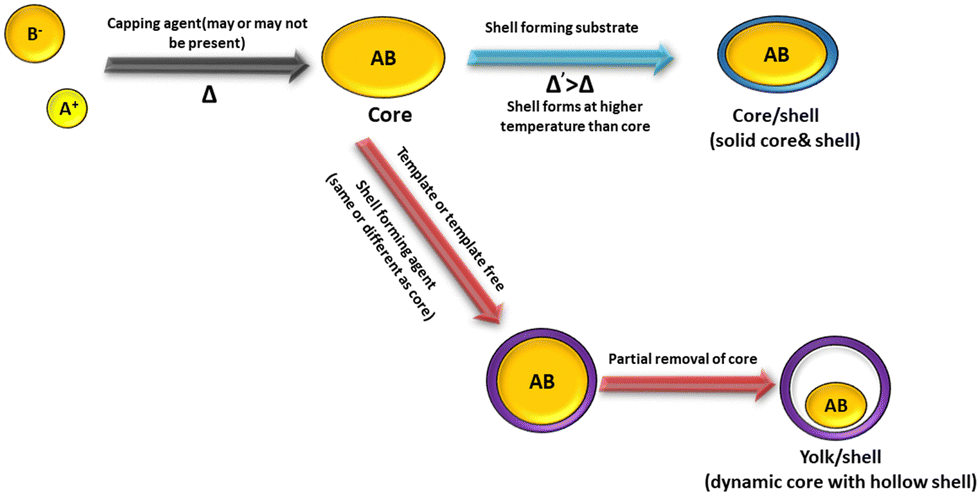 | ||
| Fig. 1 Schematic representation of the core–shell and yolk–shell structure. | ||
3.1. Synthesis of yolk shell
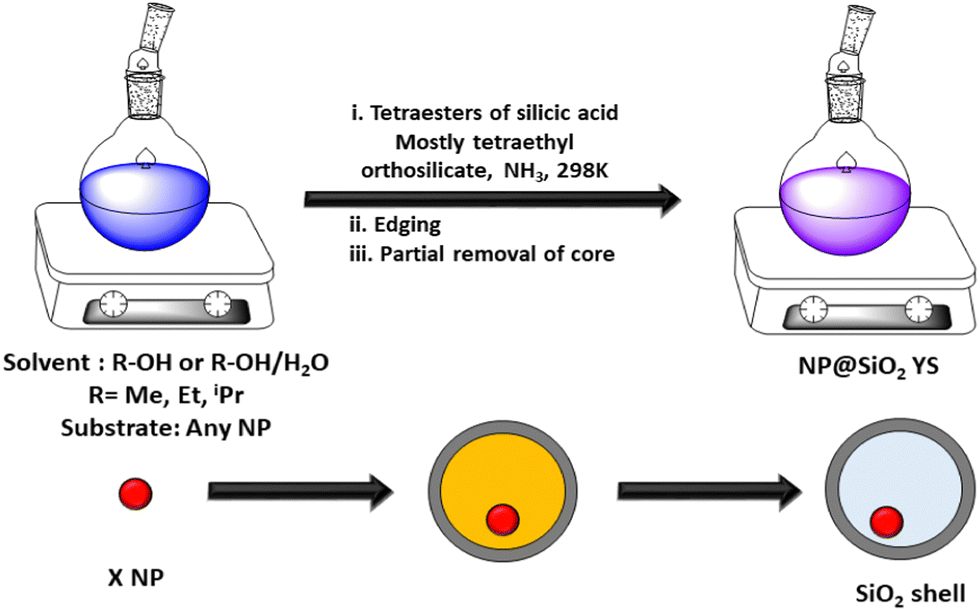
Sacrificial templates can be classified as hard and soft templates. Templates such as silica, Fe3O4, V2O5, Cu2O, carbon, FeOOH, gold, and AgBr are generally used as hard templates.80 Silica is a well-known hard template, which is usually removed by hydrofluoric acid or in hot concentrated alkaline/ammonia solution.81 The etching of silica though HF creates water-soluble H2SiF6 due to the high electronegativity of fluorine. The alkali etching process is slightly slower than acid etching and alkali treatment generates soluble sodium silicate. However, the acid etching process requires more safety measures and precaution compared to alkali medium. Notably, the ammonia etching of silica provides a void space of 50 nm in the material. Besides the silica template, several familiar hard templates such as Au/TiO2,79 Pd/C,82 Pd/CeO2,83 γ-Fe2O3/Y2O3,84 Pd/SiO2,85 and Fe3O4/SiO286 have been used for the construction of yolk–shell structures. Besides them, many other inorganic sacrificial templates have emerged for synthesis of yolk–shell structures such as V2O5,87 Ni,88 Cu2O,89 and AgBr.90 Metal salts and metal oxides produce an acid-soluble metal core, which is removed by the dissolution process. In addition, metal hydroxides synthesized by different methods followed by calcination, produce metal oxide-like yolk–shell structures.91 In general, the removal of any volatile or any chemical product can also produce yolk–shell structures. Further, multi-shell yolk shell structures could be obtained through electrostatic interaction among different structure directing agents with silica. Multi-shell yolk–shell structures were obtained via the successive addition of bis(trimethoxysilyl)ethane and TEOS.92,93 The co-structure-directing agent APTES was attached to the SiO2 core–shell structure through electrostatic attraction. The thickness of the silica-shell could be controlled by varying the concentration of TEOS as the silica source. Importantly, the partial etching of the core structure produced a yolk–shell structure.94,95 The Dou group96 demonstrated the preparation of yolk–shell FeS@carbon nanospheres, which showed a high capacity of approximately 545 mA h g−1 over 100 cycles. These nanostructures enabled the Na/FeS@C battery to achieve an ultrahigh energy density of approximately 438 W h kg−1, making them suitable for commercial applications. Their method involved coating Fe3O4 nanoparticles with a sacrificial layer of condensed silica using the Stöber method, followed by the addition of resorcinol formaldehyde (RF) via the sol–gel process. The resulting Fe3O4@C nanospheres underwent a carbonization process through calcination, and the sacrificial silica layer was removed using concentrated NaOH to produce the yolk–shell structure.
In the realm of soft template-based approaches, the synthesis of yolk–shell nanostructures has attracted significant attention. The synthesis of yolk–shell nanostructures involves the complete removal of the soft template through thermal annealing or calcination. However, the void size in the core–shell structure is the most important factor influencing the sodium storage capacity, which allows the active materials to expand easily without damaging the outer protective shell. Further, a too large void space reduces the volumetric energy density.97 Hence, the desirable thickness of the void should be tailored to obtain a higher reversible capacity and cycling performance.97 Researchers also used surfactants such as an aqueous mixture of lauryl sulfonate betaine (LSB, a zwitterionic surfactant) and sodium dodecyl benzenesulfonate (SDBS, an anionic surfactant), and Triton X-100 surfactant98–100 to increase the electrostatic interaction among the precursors. The soft template CTAB in ethylene glycol medium produced microsphere structures and yolk–shell structures were obtained after the acid etching process.101 F127 is another important fluorocarbon surfactant, which electrostatically interacts with the core particles to generate mesostructures after the subjecting them to calcination.102 In addition, sulfur serves as a sacrificial template for yolk–shell nanostructures. The sulfur core can be dissolved in various organic solvents, including toluene, carbon disulfide, xylene, carbon tetrachloride, and benzene.103 Furthermore, the sulfur core template can also be eliminated by increasing the calcination temperature. In the case of the sacrificial template, silver can be removed by employing a dissolved NH3 solution, resulting in the formation of a silver amine complex.104 Additionally, β-FeOOH nanosheets served as a sacrificial template, which could be fully reduced to Fe3O4.105 A mixture of two oxide or nitrate salts in concentrated sodium hydroxide solution also produced yolk–shell structures.106 In summary, yolk–shell synthesis involves intricate control of the sacrificial templates, etching processes, and precursor addition to achieve the desired void spaces, which are crucial for high-performance applications such as energy storage systems. However, sacrificial templates are associated with high cost and achieving integral yolk–shell structures with the desired morphology can be challenging. Additionally, the removal of the template can present difficulties, adding to the complexity of the process. In the case of hard templates such as silica, their removal involves chemical processes such as etching. Alternatively, soft templates are typically removed through thermal annealing or calcination. Further, the void size is important in yolk–shell structures for achieving the optimal performance in applications such as energy storage. Various surfactants and reagents are used to enhance the electrostatic interactions among the precursors and facilitate the removal of the template. Achieving precise control of the sacrificial templates and etching processes is crucial for obtaining the desired void spaces in yolk–shell structures, which are essential for high-performance applications. However, the challenges related to cost, morphology control, and template removal complexity need to be addressed.
| Materials | Current density (A g−1) | Capacity | Cycle | Template | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| FeS/C | 0.1 | 300.4 mA h g−1 | 10000 |
Iron naphthenate and mercaptoethanol | 81.1% after 10000 cycles |
27 |
| NC TiO2-Y | 20C | 115.9 mA h g−1 | — | Diethylenetriamine | 38 | |
| SnSe2/NiSe2@NC | 3 | 322.7 mA h g−1 | 7500 | Dopamine | — | 43 |
| Sb@Void@GDY | 1 | 325 mA h g−1 | 8000 | Hexaethynylbenzene | 74% after 8000 cycles | 28 |
| Co3O4-C | 0.5 | 311 mA h g−1 | 200 | Polyvinylpyrilidone | 31 | |
| FeS2@C | 0.1 | 511 mA h g−1 | 100 | Polydopamine | — | 29 |
| CoSe/C | 16 | 361.9 mA h g−1 | — | ZIF-67 | — | 131 |
| Fe3N@C | 2 | 248 mA h g−1 | 300 | Polyacrylonitrile | — | 132 |
| CoS2/VS4@NC | 5 | 417.28 mA h g−1 | 700 | ZIF-67 | — | 32 |
| ZnS@C | 1 | 211.1 mA h g−1 | 5700 | CTAB | — | 133 |
3.2. Galvanic replacement
Galvanic displacement is a simple, low-cost, facile technique, where room temperature stirring of the reactants produces high exposed edges and facets in yolk shell structures.134 Galvanic replacement reactions lead to a controllable size, shape, hollow interior, and different morphologies using a sacrificial template, where two different electrochemical potentials provide yolk–shell nanostructures.135 Generally, the lower standard reduction potential material is treated as the sacrificial layer and the higher standard electrode potential metal is used as the shell. For example, when a copper strip was immersed into a higher reduction potential silver nitrate salt solution (Ag+/Ag), the copper strip is gradually corroded in the solution as Cu2+. Hence, some of the exposed sites of the copper are deposited on the Ag crystal and replace some Ag atoms.135 In the galvanic replacement reaction, a seed growth reagent such as CTAB and PVP is needed for metal growth and a higher reduction potential metal salt solution is prepared for seed growth.135 The hollow core is chosen as a presynthesized template such as any lower reduction potential microsphere, nanocage, and nanobox.136 As the reaction gradually proceeds, the different presynthesized template morphologies gradually dissolved and co-deposited with the higher reduction potential material. The galvanic replacement reaction also occurs with non-metallic solutions such as sulphur and selenium. Here, excess non-metallic solution is used as the template upon which metal is nucleated and excess nonmetal is further removed by choosing the appropriate solvent.137 Several studies reported that an anti-galvanic reaction may also occur if the size of the higher reduction material such as Au is used less than 3 nm, and then it can reduce the lower reduction potential Ag+.138 Further, the alkali medium of the salt solution may favour the multi-shell core–shell structure compared to aqueous medium.138 The shell thickness and voids are also regulated by galvanic replacement. This strategy can be utilized to prepare different types of multi-shell-alloy. The Xia group prepared yolk–shell structure Pd@AuxCu1−x yolk–shell nanocages where Pd@Cu core–shell nano-cubes were placed in solutions of HAuCl4.139 Tao and co-workers synthesized Au@AgPd nanoparticles, where the pre-synthesized Au@Ag core shell was treated with Pd2+ ions. Subsequently, galvanic replacement of silver by Pd produced open-mouth yolk–shell-type structures (Fig. 2).139,140 This galvanic replacement was further modified by the Kirkendall effect. This Kirkendall effect has been designated as a classical phenomenon where diffusion occurs at the interface between two metals with different mobility. The Kirkendall effect is a similar phenomenon to galvanic replacement, where the higher diffusion coefficient metal provides a higher vacancy flux, while the lower diffusion coefficient material provides less vacancy. The different vacancy flux creates a yolk–shell structure. The excess vacancies near the higher diffusion coefficient are called ‘Kirkendall voids’. The Kirkendall effect occurs if atomic diffusion occurs via vacancies and each component possesses different mobility.141 A hollow-shell is created by oxidation, phosphidation, or chalcogenation through the Kirkendall process. Various composites such as Sn@void@SnO/SnO2, FePt@Fe2O3, and Au/Fe2O3 have been prepared through the Kirkendall effect.142–144 In 2013, the Oh group145 developed a unique galvanic replacement process, where pre-synthesized square prism-shaped Mn3O4 nanocrystals (∼20 nm) were kept in an iron(II) perchlorate solution. It was observed that the ionic Fe2+ gradually underwent oxidative precipitation to form Fe2O3, while Mn(III) was reduced to Mn(II) and dissolved in the reaction mixture. Hence, the Fe2O3 slowly replaced some of the Mn(III) and deposited on the Mn3O4 nanocrystals. Upon increasing the concentration of iron(II) perchlorate, the size of the hollow interior increased. They showed that the presence of clear facet-like structures in the FFT spot pattern of the exposed nanocage-like γ-Fe2O3 structures. This exposed nanocage-like structure exhibited superior lithium storage compared to other materials.145 Galvanic replacement is very simple and cost effective but its complexity, long reaction time, scalability, purity and diffusion control of ions make its application in batteries. Table 2 presents a summary of the different materials prepared through galvanic replacement reaction towards SIB application. In summary, galvanic displacement is a straightforward and cost-effective method for producing yolk–shell structures with high exposed edges and facets. It involves a controlled reaction between two different electrochemical potentials, typically using a lower standard reduction potential material as the sacrificial layer and a higher standard electrode potential metal as the shell. Seed growth reagents aid in metal growth, and presynthesized templates dictate the hollow core morphology. This process can also occur with non-metallic solutions such as sulfur or selenium. Galvanic replacement, often modified by the Kirkendall effect, allows the regulation of the shell thickness and voids, and the creation of multi-shell alloys. However, although it is simple and cost-effective, challenges in terms of scalability, purity, and ion diffusion control hinder its application in battery technology. Table 2 shows some electrochemical performances using galvanic replacement for the preparation of yolk shell structures. | ||
| Fig. 2 (a) Low-magnification TEM image (a) of OM-YS Au@AgPd NPs and schematic representation of the synthetic procedure for open-mouthed, yolk–shell Au@AgPd NPs via the galvanic reaction of Au@Ag NPs with Pd2+ ions at room temperature. Reprinted with permission from ref. 140. ©2015, the Royal Society of Chemistry. | ||
| Materials | Current density | Capacity | Cycle | Capacity retention | Energy density (W h kg−1) | Method | Ref. |
|---|---|---|---|---|---|---|---|
| Sb NTs | 1 A g−1 | 342 mA h g−1 | 6000 | 74% @6000 | 252 | Galvanic replacement | 146 |
| Sb@C@TiO2 | 1 A g−1 | 193 mA h g−1 | 4000 | 67% after 400 cycles | 179 | Galvanic replacement | 147 |
| Sb/TiO2 | 2 A g−1 | 245 mA h g−1 | — | 87% after 100 cycles | — | Galvanic replacement | 148 |
| Sb⊂CTHNs | 0.1 A g−1 | 607.2 mA h g−1 | 100 | — | — | Galvanic replacement | 149 |
3.3. Ostwald ripening
Ostwald ripening refers to the creation of yolk–shell structures using the solubility property of crystal aggregations. This process is based on the Gibbs–Thomson influence, which facilitates the growth of larger particles, and the dissolution of unstable smaller particles with a critical size (Fig. 3).150 In this process, larger nanoparticles break into smaller ones and aggregate to form a larger core molecule, which possesses shell molecules. The small molecules aggregate to the centre to create a spherical aggregate. Yolk–shell nanostructures have been synthesized via symmetric and asymmetric aggregation, where small nanoparticles aggregate around the solid spheres. These small crystallites are dissolved during the recrystallization to make the void space move towards the outer shell, while their movable core part consists of solid spheres.151,152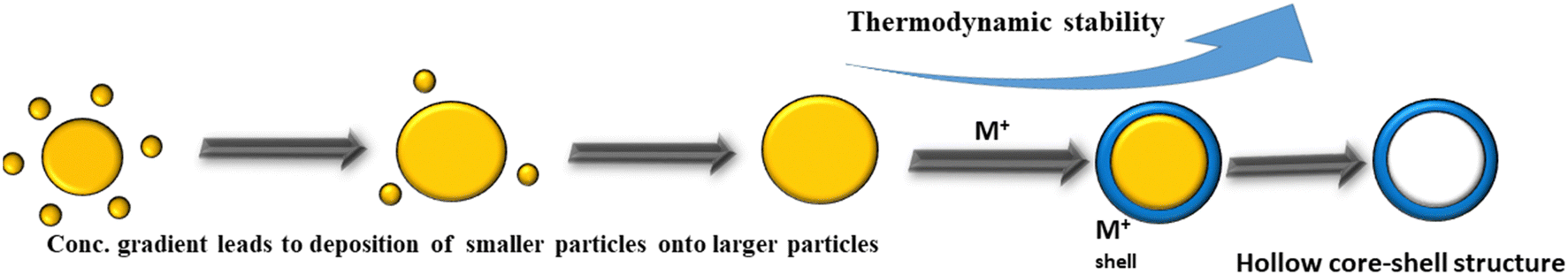 | ||
| Fig. 3 Schematic design of Ostwald ripening. | ||
Generally, this method is the most common strategy to generate hollow materials. In this process, generally, a solvent mixture upon long-term hydrothermal treatment without the addition of any template generates mesoporous-like structures. The surface area varies from 110–150 m2 g−1 and the extra void space is capable of accommodating the lithium atom in its structure.153 The Ostwald ripening method is the best-suited method for generating hollow mesoporous materials compared to other methods given that it does not involve the use of hazardous chemicals. The α-Fe2O3/SnO2 yolk–shell structure was synthesized by the Ostwald ripening mechanism, where the SnO2 nanoparticles were prepared from K2SnO3·3H2O and deposited around the α-Fe2O3 NPs. Here, the hydrothermal treatment of SnO2 nanoparticles resulted in their dissolution and created a void space.154 Researchers also employed template-free Ostwald ripening processes for the synthesis of Fe3O4/Co3O4,155 Pt/CeO2,91 and Au/Cu2O156 nanostructures. The Bao group summarized the Ostwald ripening process for the preparation of materials for the fabrication of lithium-ion batteries, where solid TiO2 spheres, VO2 hollow microspheres, MnO2 hollow structures, and Fe3O4 hollow nanospheres were prepared by treating their metal salt by the hydrolysis method or heating in the hydrothermal method.157 Often, the polarity of the solvent also increases the yield of the materials. The Zhaou group demonstrated the synthesis of Na3V2(PO4)2O2F (NVPF) by Ostwald ripening, where larger nanoparticles produced the shell with several inner nanospaces and pores under solvothermal conditions. This material demonstrated exceptional cycling stability (0.023% capacity decay per cycle after 2000 cycles at 20C).158 Park and co-workers synthesized a goat pupil-like configuration of S-doped SnSe yolks and hollow carbon shells through the Ostwald ripening method, which also showed excellent cycle stability.159 Later, the same group showed the generation of Co3O4-C hollow yolk–shell through the Ostwald ripening method for super performances towards lithium and sodium batteries.31 Zhang et al. also prepared yolk-like TiO2 through the Ostwald ripening method, which showed a high rate capability.38 The template-free approach using Ostwald ripening can potentially offer advantages such as simplicity, reduced cost, and scalability compared to methods involving the use of external templates. However, optimizing the process parameters and understanding the underlying mechanisms of Ostwald ripening are crucial for achieving the desired nanostructure characteristics and properties. In summary, Ostwald ripening is a method for creating yolk–shell structures by leveraging the solubility properties of crystal aggregations, driven by the Gibbs–Thomson influence. This method can be used for SIB application given that it is a very straightforward reaction for yolk shell production. Table 3 shows a summary of the electrochemical performances of yolk shell structures using Ostwald ripening.
| Materials | Current density | Capacity (mA h g−1) | Cycle | Method | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| NC TiO2-Y | 1C | 200.7 | 550 | Ostwald ripening | 95.5% after 3000 cycles | 38 |
| Co/(NiCo)Se2 | 0.2 A g−1 | — | 80 | Ostwald ripening | 96% after 10th cycles | 160 |
| Na3V2(PO4)2O2F | 50C | 85.4 | — | Ostwald ripening | 84% after 200 cycles | 158 |
| Co3O4–C | 2 A g−1 | 619 | 400 | Ostwald ripening | — | 31 |
| S-doped SnSe | 0.5 A g−1 | 186 | 1000 | Ostwald ripening | 74% after 2nd cycle | 159 |
3.4. Ultrasonic spray pyrolysis (USP)
Ultrasonic spray pyrolysis (USP) is a scalable continuous one-step approach for the synthesis of hollow- and yolk–shell nanostructures via the decomposition and evaporation method. This process involves the creation of an aerosol (liquid–gas interface) from a liquid precursor solution, which is subsequently transformed into micro-droplets. The microdroplets are transported into a furnace via a gas flow. Inside the furnace, decomposition of the precursor and solvent evaporation occur, leading to the formation of yolk–shell nanostructures. The volatile organic compounds evaporate during the heating process, leaving behind void spaces and resulting in the formation of a yolk–shell structure. Firstly, micro-droplets are generated using USP, followed by evaporation of the solvent. The Suslick group reported the synthesis of a composite using (NH4)2MoS4 and silica as the precursors in a liquid, which was then ultrasonically nebulized into microdroplets, carried by a gas flow into a furnace, where the precursor underwent decomposition to create the SiO2/MoS2 composite. After treatment with HF, a porous MoS2 network was obtained, where the surface area and porosity were controlled by the precursor solution.161 The same group162 published a study using a Sunbeam model 696, 1.7 MHz, where a household ultrasonic humidifier (<$30) was utilized to conduct the ultrasonic spray pyrolysis method. Silica colloid, styrene, ethylene glycol methacrylate, AIBN and Co2(CO)8 were used as the template and metal precursors. In this process, SDS was used as the surfactant. The solution was first nebulized into an inert gas stream, which carried the resulting droplets into the first furnace (200 °C), where the solvent evaporated, and the organic monomer polymerized. Subsequently, the resulting solution was fed to the hotter furnace (700 °C), where the fully pyrolyzed organic polymer produced porosity. However, the conversion of this process required 6–8 h and the obtained yield was 40%. The surface area was also likely to be around 200 m2 g−1.162 Further, without the use of a template, ultrasonic pyrolysis results in a very low surface. Mai et al.163 reported the preparation of yolk–shell-structured anatase TiO2 microspheres, where titanium(IV) bis(ammonium lactato)dihydroxide (TiBALDH) and sucrose were used as the precursor solution, which was fed into an ultrasonic atomizer. The black product after treatment in a tube furnace at 600 °C for 5 h in air produced yolk–shell TiO2 microspheres.163 Herein, Fig. 4 shows the ultrasonic pyrolysis technique, where titanium(IV) bis(ammonium lactato)dihydroxide and sucrose in water was used as the precursor solution. Here, the TiO2−x microspheres prepared via the facile spray-pyrolysis showed remarkable long-term cycling stability.163 In general, for SIB application, titanium/nickel or iron prepared via the spray pyrolysis method have been found to be promising materials.164 The ultrasonic pyrolysis method is indeed scalable and versatile. However, it also has certain limitations, such as the high cost and complexity of the required instrumentation, as well as challenges related to the preparation and compatibility of the precursors. These factors make it disadvantageous compared to other synthesis methods. In summary, ultrasonic spray pyrolysis (USP) is a scalable method for the synthesis of yolk–shell nanostructures, involving precursor nebulization, decomposition in a furnace, and solvent evaporation. It offers versatility but has limitations including high equipment cost and complexity, with titanium-based materials showing promise for sodium-ion battery applications. Table 4 presents a summary of the electrochemical performances of yolk shell structures prepared via the ultrasonic spray pyrolysis process.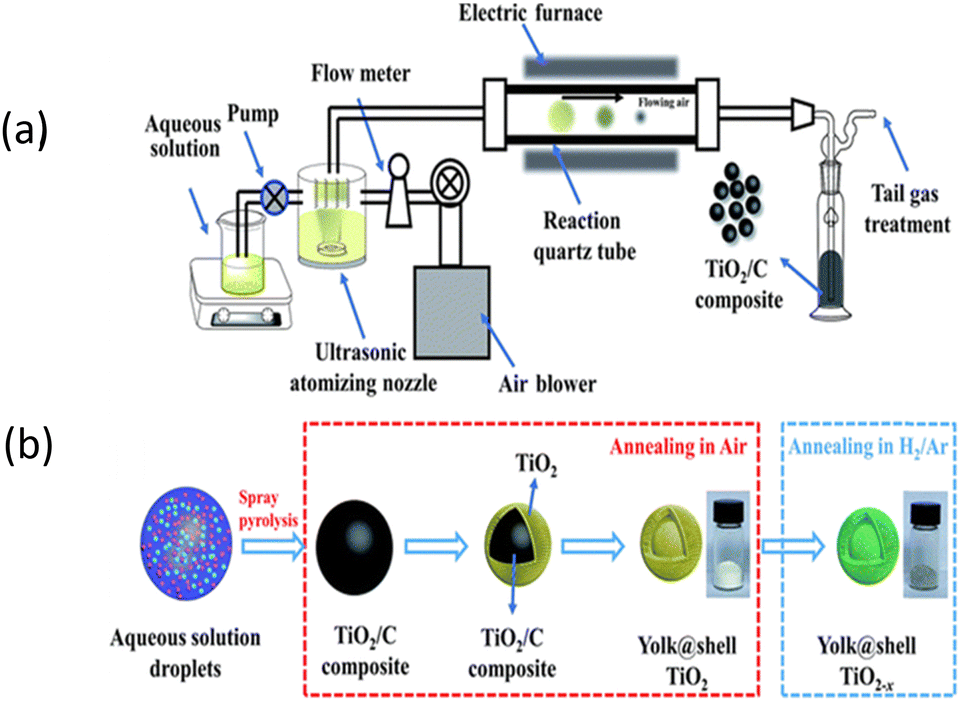 | ||
| Fig. 4 Schematic diagram of (a) ultrasonic spray pyrolysis process and (b) mechanism of yolk@shell-structured TiO2−x prepared using the ultrasonic pyrolysis method. Reprinted with permission from ref. 163. Copyright: 2019, the Royal Society of Chemistry. | ||
| Materials | Current density | Capacity (mA h g−1) | Cycle | Capacity retention | Method | Ref. |
|---|---|---|---|---|---|---|
| TiO2−x | 0.05 A g−1 | 230.7 | 200 | 91.7% after 1000 cycles | Spray pyrolysis | 163 |
| NiCoSe2/CNT | 0.2 A g−1 | 366 | 10000 |
85% after 2nd cycle | Spray pyrolysis | 165 |
| (Fe0.5Ni0.5)9S8 | 1 A g−1 | 527 | 100 | 99% after 2nd cycle | Spray pyrolysis | 166 |
| TiO2−x/C | 50 mA g−1 | 249 | 180 | 87.2% after 2nd cycle | Spray pyrolysis | 167 |
| FeSe2/graphitic carbon | 0.2 A g−1 | 510 | 200 | 88% after 2nd cycle | Spray pyrolysis | 168 |
3.5. The ship-in-bottle method
The Ostwald ripening or galvanic replacement process and Kirkendall effect-based methods have some disadvantages such as low efficiency given that the cores are captured inside shells.169 Many researchers have employed the “ship-in-bottle” approach for synthesizing yolk–shell nanostructures, wherein the core materials are protected by a mesoporous silica and/or zeolite-like framework. In material synthesis, particularly in nanotechnology, various methods are utilized to encapsulate or confine nanoparticles or nanostructures within a matrix or shell.170 Subsequently, these encapsulated entities are added in very small amounts into ship-like materials such as zeolites and metal–organic frameworks. The main driving force of this reaction is the electrostatic interaction between the porous template and the small nanoparticles, which are impregnated within the cavity and have dimensions smaller than the pore size of the cavity. In this method, porous templates such as zeolites, MCM-41, metal–organic frameworks, and (SAB)-16 are utilized as hosts to entrap the core particles.171 Numerous small nanoparticles are encaged via the self-assembly method through a chemical reaction in a porous cavity, forming a core-like structure. Various yolk–shell nanostructures such as Fe2O3/silica,172 Pt/silica,173 and SnO2/SiO2,174 have been prepared via the ship-in-bottle process. Likewise, Liu et al. prepared MOF crystals for creating porous channels to entrap various small nanoparticles.170 In the mesoporous UiO-66-NH2 MOF, nitric acid treatment created a hole in the centre-entrapped palladium salt (Na2PdCl4) in the cavity.170 The Qiu group also used zeolites, where the ions were assembled into the cavity by the ship-in-bottled method.170 Huang and co-workers175 synthesized new intermetallic NPs encapsulated with mesoporous silica via an easy facile method in the lab. However, intermetallic materials such as Pt–Sn and Pt–Cu could not be synthesized at higher temperature given that it affected their crystal structure. Hence, a silica template was chosen, where silica protected the intermetallic core during heat treatment. Firstly, they mixed 10 mM K2PtCl4 and tetradecyltrimethylammonium bromide in distilled water solution. Subsequently, 500 mM (3 mL) aqueous solution of sodium borohydride (NaBH4) was added to the clear solution of the resulting mixture and stirring continued for 15–20 h. The resultant dark brown colloidal solution of Pt nanoparticles was added to a sodium hydroxide solution, where the pH was kept between 11 and 12. While stirring, 500 μL of a 10% tetraethyl orthosilicate solution in methanol was added dropwise. After 24 h, the sample was centrifuged, and the coated particles (Pt@mSiO2) were redispersed in methanol. The surfactant was removed via an acidic methanol refluxing (6% hydrochloric acid solution) at 90 °C for 24 h.175 Sometimes, the cavity of porous carbon is also used for entrapping nanoparticles. Luo et al. reported the preparation of flower-like MoS2−x nanosheets (Fig. 5) confined with a carbon layer via the ship-in-bottle method, where the cavity of carbon was used as a closed space for reactants, which showed 85.2% capacity retention after 100 cycles.176Table 5 presents some of the performances obtained through the ship-in-bottle method. The “ship-in-bottle” method involves encapsulating nanoparticles within a porous template such as zeolites and MOFs. It enables the synthesis of yolk–shell nanostructures, offering protection of the core materials during reactions. Various nanoparticles have been entrapped using this approach, demonstrating promising performances in different applications.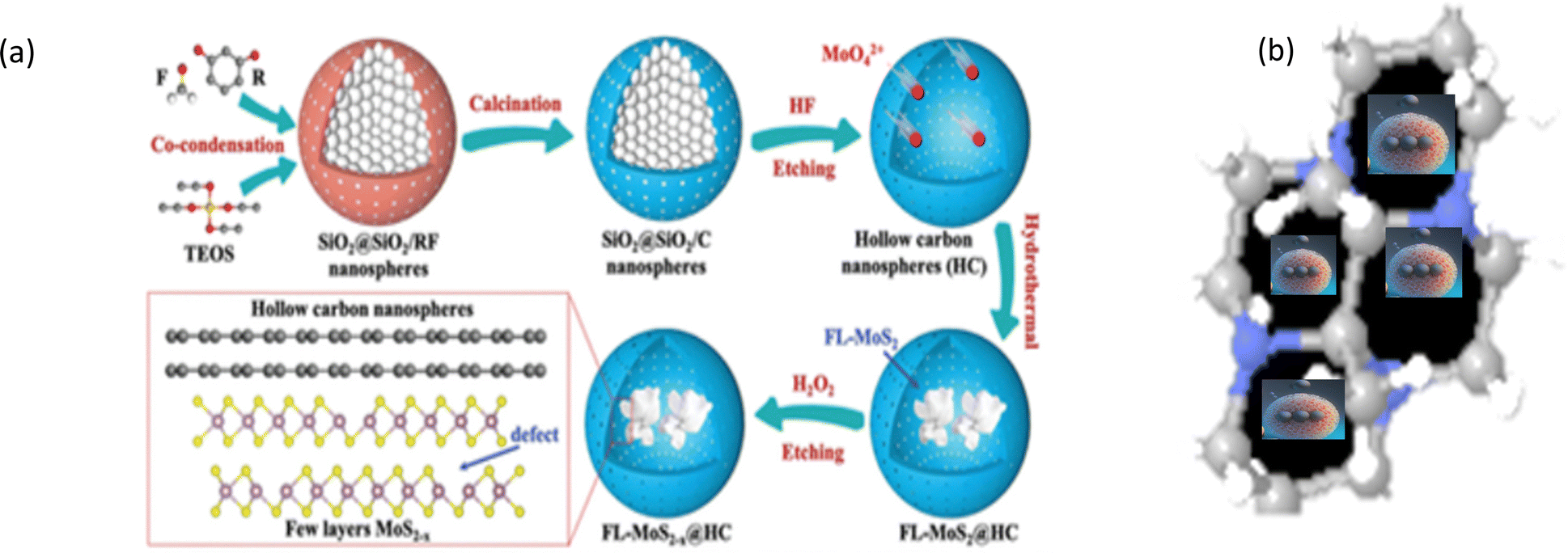 | ||
| Fig. 5 (a) Illustration of the preparation process of the FL-MoS2−x@HC matrix. (b) Molecular assembly into the molecular porous template. Reprinted with permission from ref. 176. ©2021, Wiley-VCH GmbH. | ||
3.6. Core shell synthesis
| Materials | Rate capability | Capacity | Method | Energy density (W h kg−1) | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| Na3V2(PO4)3 (NVP) | 94.9 mA h g−1 at 5C | — | Sol–gel method | 154.5@1.7 V | 96.1% after 700 cycles | 185 |
| Na3V2(PO4)3 | 70 mA h g−1 at 10C | — | Sol–gel method | — | 90% after 400 cycles | 186 |
| NaV3O8@10% PPy | 63 mA h g−1 at 3.68C | 99 mA h g−1 after 60 cycles at 80 mA g−1 | Sol–gel method | — | — | 187 |
| ZCS@S | 250 mA h g−1, 2000 cycles at 1.0 A g−1 | 570 mA h g−1, 1000 cycles at 0.2 A g−1 | Sol–gel method | 384 | 188 |
 000 cycles at 5 A g−1 and high specific capacity of 92.6 mA h g−1.198 Employing the Stöber method, products with versatile and scalable materials as well as monodisperse, uniform, and tunable products can be obtained. However, in this process, agglomeration and long duration cannot be neglected as disadvantages. In summary, the Stöber method is a sol–gel technique for producing monodisperse silica nanoparticles with a controllable morphology. It enables the synthesis of various core–shell structures, including different MOx@SiO2, with uniform shell thickness and ordered mesopore channels. However, although it is versatile and scalable, its challenges include agglomeration and the need for careful control of the reaction conditions and duration. Table 7 displays a summary of the electrochemical performances of the core shell structures prepared using the Stöber method.
000 cycles at 5 A g−1 and high specific capacity of 92.6 mA h g−1.198 Employing the Stöber method, products with versatile and scalable materials as well as monodisperse, uniform, and tunable products can be obtained. However, in this process, agglomeration and long duration cannot be neglected as disadvantages. In summary, the Stöber method is a sol–gel technique for producing monodisperse silica nanoparticles with a controllable morphology. It enables the synthesis of various core–shell structures, including different MOx@SiO2, with uniform shell thickness and ordered mesopore channels. However, although it is versatile and scalable, its challenges include agglomeration and the need for careful control of the reaction conditions and duration. Table 7 displays a summary of the electrochemical performances of the core shell structures prepared using the Stöber method.
 | ||
| Fig. 7 Schematic design of core–shell/yolk–shell formation via the Stöber method. | ||
| Materials | Rate capability | Cyclic capacity | Method | Energy density (Wh kg−1) | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| FeS@C | 452 mA hg−1 at 5C |
545 mA hg−1 over 100 cycles at 0.1 A g −1 |
Stöber method | 438 |
37.8% after 300 cycles | 30 |
| Sb@SiO2@C | 149 mA h g−1 at 8C | 271 mA h g−1 over 100 cycles at 0.1 A g−1 | Stöber method | 142 | 71.2% after 100 cycles | 75 |
| ZnS@S | 170 mA h g−1 at 5.0 A g−1 | 570 mA h g−1, 1000 cycles at 0.2 A g−1 | Stöber method | 384 | — | 188 |
| Na0.55Mn2O4·1.5H2O | 515.2 mA h g−1 at 4.0 A g−1 | 546.8 mA h g−1 at 2000 cycles at 4 A g−1 | Stöber method | — | — | 199 |
| HCM-Fe3O4@void@N-C | 196 mA h g−1 at a current density of 1200 mA g−1 | 522 mA h g−1 after 800 cycles at 160 mA g−1 | Stöber method | — | — | 200 |
| SnS2@C/CNF | 767.6 mA h g−1 at 0.2 A g−1 | 478.7 mA h g−1 at 2 A g−1 for 1000 cycles | Stöber method | 82.4% after 1000 cycle | 201 |
4. Performance based on sodium battery variants
4.1. Sodium-ion batteries (SIBs)
Yolk–shell and core–shell nanostructures offer enhanced performances in SIBs by providing a high surface area, shortened ion diffusion pathways, and improved structural stability during cycling. They can accommodate the volume changes associated with sodium intercalation and deintercalation, thus enhancing the cycling stability and rate capability. Generally, the use of a yolk structure as the anode improves the rate capacity and capacity retention of the material.202 Similarly, Fe3N@C115 and CoSe/C203 yolk–shell structures are employed as anodes, showing a high rate capability in SIB application. It has been shown that phosphorus, copper, and tin are promising materials for SIB applications. Sometimes metal sulphides also showed a higher contribution to the performance of SIBs such as nickel sulphide due to their ready dissociation and intercalation property.204 Various strategies and materials have been employed to enhance the performance of sodium-ion batteries (SIBs), aiming to overcome their challenges such as sluggish kinetics, substantial volume changes, and inadequate cycle life associated with specific electrode materials. Nickel sulfide-based materials (NiS, NiS2, Ni3S2, and Ni3S4) show promising potential as anode materials for SIBs due to their high theoretical capacity of 873 mA h g−1, as described by the four-electron conversion reaction NiS2 + 4Na+ + 4e− ↔ Ni + 2Na2S.205,206 However, their sluggish kinetics hamper conversion reactions and lead to significant volume changes during cycling, resulting in a limited capacity and poor cycle life.205,207 In this case, the addition of carbon has been proven to be effective in enhancing the kinetics and structural stability of the electrode. Yolk–shell NiS2 nanoparticles embedded in porous carbon fibers (denoted as NiS2⊂PCF) displayed an excellent electrochemical performance in flexible SIBs. Remarkably, compared to NiS2 nanoparticles coated on the surface of carbon fibers (NiS2@PCF), the NiS2⊂PCF electrode demonstrated a high reversible capacity (679 mA h g−1 at 0.1C), good rate performance (42% capacity retention from 0.1 to 10C), and excellent long-term cycling stability (76% retention over 5000 cycles at 5C).205 Further, the sodiation/desodiation process of the NiS2⊂PCF electrode was facilitated by the conductive carbon for rapid electron transfer, with its internal porous structure accelerating the penetration of the electrolyte in its channels. Further, the buffer layer expanded to accommodate the strain and volume changes during charge and discharge.205 Furthermore, the integration of Fe7Se8 with carbonaceous materials buffered the stress–strain and enhanced the conductivity. A newly developed Fe7Se8-based composite with a porous and intact conductive carbon coating demonstrated a reversible Na-storage capacity of 386 mA h g−1 at 100 mA g−1, remarkable rate capability (315 mA h g−1 at 5 A g−1), and ultra-long cycle life (no obvious capacity decay over 1000 cycles). Moreover, it exhibited an excellent full-cell performance when paired with a high-voltage Na3V2(PO4)2O2F (NVPOF) cathode, suggesting its potential applicability in the Fe7Se8@C/N NB//NVPOF full cell.208 Achieving dendrite-free plating and stripping of sodium, while maintaining high Coulombic efficiencies (CEs) at elevated current densities and capacities, remains a significant challenge. Yolk–shell structures consisting of MnSe/ZnSe heterojunctions embedded within one-dimensional carbon tubes (HCTs) functioned as anode materials for both SIBs and potassium ion batteries (KIBs), enabling efficient insertion and extraction of sodium ions, while establishing a chemical potential gradient. Furthermore, the ZnSe shell enhanced the stability and addressed concerns related to volume expansion.209 Efforts have been directed towards synergistically incorporating foreign atoms and carbon additives to enhance the performance of battery materials. An example of this is the utilization of nitrogen-doped/carbon-tuned TiO2 yolk-like materials (NC TiO2-Y), which demonstrated an exceptional initial coulombic efficiency, remarkable rate capability, and long-term cyclability in SIBs.210 However, antimony, tin and nickel cobalt integrate the overall performances required in SIB applications,52,211 and most of the SIB performances are discussed in this context. Hence, a summary of their electrochemical performances is presented in Table 10.4.2. Sodium metal batteries
In sodium metal batteries, yolk–shell and core–shell architectures are utilized to stabilize the sodium metal anode. These structures can mitigate issues related to dendrite formation and volume changes during cycling, leading to improved safety, cycle life, and energy density of the batteries.205,206 The formation of sodium dendrites is linked to the nucleation of sodium metal at the outset and its subsequent growth thereafter.212 Core–shell C@Sb nanoparticles (NPs) spatially control a physical barrier, which impedes dendrite growth, reduces the risk of short circuits and enhances the battery safety. Furthermore, porous Sb cores offer abundant nucleation sites due to their high surface area and low nucleation barrier, while outer carbon shells prevent agglomeration and dendritic formation.212 Similarly, C@Ag nanoparticles enable dendrite-free stripping/plating of Na metal together with sodiophilic Ag cores, providing abundant nucleation sites and outer carbon shells ensuring high efficiency during long-term cycling.2134.3. Sodium–sulfur batteries
Yolk–shell and core–shell configurations are also employed in sodium–sulfur batteries to enhance the performance of sulfur cathodes. These structures can confine the polysulfide species, prevent their dissolution, and improve the utilization of active sulfur, thereby increasing the energy density and cycling stability of sodium–sulfur batteries. However, the low conductivity of sulfur (5 × 10−30 S cm−1) and the solubility of lithium sulfide in organic electrolytes result in material loss and create a shuttle effect.214 However, materials such as MnSx exhibit a higher affinity to sulfur compared to oxides.214 Li et al. prepared MnS@N–C as the cathode of room-temperature sodium–sulfur (Na–S) batteries, achieving a high initial capacity of 893.9 mA h g−1 at 0.1C and demonstrating excellent cycling stability at 0.5C for 300 cycles with a capacity decay of only 0.16% per cycle.214 Moreover, the incorporation of conducting materials enhances the rate performance and cycle stability of the battery components. Cu nanoparticle-decorated mesoporous carbon spheres showed a promising rate performance and cycle stability.215 Similarly, the atomic dispersion of cobalt species on hollow carbon nanospheres encapsulated a high content of sulfur. The clustering of cobalt promoted the conductivity of sulfur, enabling a high storage capacity.215 Fe2N incorporated into NC yolk shell (YS-Fe2N@NC) also serves as a sulfur carrier.30 The hollow microporous structure with a void interior withstands volume strain and accommodated more sulfur particles and soluble polysulfides. Additionally, the nitrogen-doped carbon shell exhibited high conductivity, leading to an outstanding electrochemical performance in sodium–sulfur batteries.30 Furthermore, porous carbon shells enhanced the conductivity of the active materials, resulting in a high reversible capacity. The nanosized FeS cores provided large electrode/electrolyte contact areas and short diffusion paths for electrons and ions, which improved the sodium reaction rate, mitigated structural degradation, and shortened the Na diffusion paths. The suitable void space buffered the large volume variations of FeS during the sodiation/desodiation processes, maintaining the original nanoparticle morphology of FeS@C and ensuring prolonged cycling stability.30 Overall, yolk–shell and core–shell nanostructures play a vital role in advancing the performance of sodium-based battery technologies by addressing key challenges related to electrode stability, ion diffusion kinetics, and electrochemical reactions.5. Performances of core–shell structure in battery application
Core–shell structures have multiple functions such as enhancing the kinetics, effective protection from chemical attack, prevention of pulverisation, SEI layer growth and increasing the conductivity. Hence, core–shell structures can be used as active anodes in sodium ion batteries. It has been investigated that core–shell C@Sb nanoparticles showed extremely higher sodium plating/stripping cycles for 6000 h at a high areal capacity of 4 mA h cm−2 with an average Coulombic efficiency of 99.7%.216 This observation shows that antimony is the best-performing material for sodium metal batteries due to its long-term stable cyclic performance. TiO2 is another promising material for sodium storage performance. The 600–700 nm yolk-like TiO2 and 5–10 nm porous carbon structure showed a very high capacity retention of 95.5% after 3000 cycles at 25C.38The specific capacity of sodium ion batteries depends on the controlled thickness, nature, porosity and conductivity of the shell structure. The prevention of the growth of a thick SEI layer is another strategy for enhancing their overall performances. Polydopamine is the best example preventing the growth of a thick SEI layer. The Yu group prepared a phosphorus–CNT@polydopamine (P-CNT) composite, which exhibited excellent cycling stability (470 mA h g−1 after 5000 cycles at 5.2 A g−1). Moreover, the coating of polydopamine is the best example as a uniform ion transporter to the interface given that it inhibits the direct contact between the electrolyte and the active material. Furthermore, its large volumetric expansion accommodated a large amount of active materials.217 Among the transition elements and main group elements, antimony is the best-performing material towards SIB application.
Again, a carbon layer can be used as both a protecting agent and reducing agent of the core material. A resorcinol-formaldehyde layer produced a carbon coating towards different metal oxides and metal sulphides, which acted as a reducing agent and formed a mesoporous conducting carbon coating layer. Yin and co-workers prepared mesoporous hollow core–shell Sb/ZnS@C hybrid heterostructures, where resorcinol-formaldehyde reduced Sb2S3 to antimony. The hollow core–shell porous Sb/ZnS@C hybrid exhibited a reversible capacity of 602 mA h g−1 and maintained a capacity of about 200 mA h g−1 after 600 cycles. In addition, the conducting carbon layer and metallic Sb induced excellent performances.218
According to further investigations, it has been noted that iron and vanadium are promising materials for application in SIBs compared to nickel and cobalt. The simple strategy of Ostwald ripening produced FeS@C, which achieved an ultrahigh specific capacity of 632 mA h g−1 after 80 cycles at 100 mA g−1 and extraordinary cycling stability with a capacity retention of 97.9% after 300 cycles at 1 A g−1. Here, core–shell FeS and interconnected porous graphene greatly enhanced the performance of SIBs.219 The Chen group synthesized an NVP@C (Na3V2(PO4)3@C) core–shell structure, which exhibited a reversible capacity of 90.9 mA h g−1 at 2C and a specific energy density of about 154.5 W h kg−1 at the output voltage of 1.7 V. It also showed an initial capacity of 94.9 mA h g−1 at 5C and a noteworthy capacity retention of 96.1% after 700 cycles. The shell carbon originated from ascorbic acid and polyethylene glycol 400. However, this agglomerated core–shell structure contributed less specific capacitance than other materials.185 Wang et al. synthesized a hollow core–shell-type morphology in Zn/Ni–co-oxide synthesized from ZIF-67 (Co(mIm)2 (mIm = methylimidazole)). This type of MOF-based hollow shell exhibited a stable capacity of 300 mA h g−1 at 50 mA g−1 and it showed a stable capacity for 250 cycles.220
Morphology is also a vital factor influencing the effective Na-ion and Li ion storage capacity of core–shell structures. Su and co-workers prepared walnut-like porous core–shell TiO2, where oleylamine modulated the controlled porous morphology, which enabled higher cycle stability for lithium storage.221 Li et al. synthesized a core–shell lychee-like microsphere structure of FeS2@FeSe2 around a 2–3 μm core structure with a 4 nm pore size, which exhibited a reversible capacity of 350 mA h g−1 after 2700 cycles at 1 A g−1. It should be noted that the hydrazine hydrate reduced the precursors and controlled the lychee-like structure.222 Later, the Zhu group prepared core–shell-like Fe7S8@C nano-biscuits, delivering 530.8 mA h g−1 after 1000 cycles at a high current density of 5 A g−1. In addition, the thioacetamide solution successfully fabricated the desired morphology and provided the surface area of 39.22 m2 g−1 and micropore size of 0.56 nm, where the short and easy diffusion pathways accommodated the electrolyte ions.223 Between the nanorod and nanowire morphology, the nanorod morphology shows slightly greater advantage. The Kang group prepared a CoP@PPy NW/carbon paper 1D core–shell structure delivering a high areal capacity of 0.521 mA h cm−2 at 0.15 mA cm−2 after 100 cycles. The 1D core–shell-type nanowire on carbon fibre well-controlled the volume expansion during charge/discharge.224 Xu et al. prepared BiSbS3@N-doped carbon core–shell nano-rods, resulting in a high sodium storage capacity of 771.5 mA h g−1 in the 2nd cycle and excellent rate capacity at 1000 mA g−1.225
Later, we investigated the contribution of morphology such as nanofibers and nanoribbon structures. Lu and co-workers prepared Ge@graphene@TiO2 core–shell nanofibers via atomic layer deposition (ALD), which exhibited the reversible capacity of 182 mA h g −1 at 100 mA g−1 (250th cycle). The ultrathin thickness of the TiO2 coating protected the Ge@G (core–shell Ge@graphene@TiO2) from the electrolyte, where the SEI layer of Ge was less predominant than the new SEI layer by TiO2. The well-defined nanofiber architecture maintained its architecture after five cycles at the current density of 100 mA g−1.226 The Srinivasan group prepared a nano-ribbon-type core–shell structure (MWCNT@graphene oxide nanoribbon), which decreased the restacking ability of graphene and provided a capacity of 317 mA h g−1 at a current density of 50 mA g−1. The MWCNT shell material enhanced the rate capability owing to its high electronic conductivity and abundant functional groups on the graphene layer increasing the adsorption of Na+ on the surface.227
Besides, 2D- and 1D-like structures, polyhedron-like facet structures have also been investigated for their application in SIBs. Yin et al. prepared a ZnS–Sb2S3@C core-double-shell polyhedron, which exhibited a high reversible capacity of 630 mA h g−1 at 100 mA g−1 after 100 cycles.228 However, its double shell polyhedron-like structure helped to extend its reversible capacity compared to its cyclic stability, as discussed in the earlier reports. Further, rechargeable all-solid-state sodium batteries are one of the most promising high-safety energy storage devices compared to all sodium-ion batteries. Yao et al. prepared a rod-like core–shell structured Fe1–xS@Na2.9PS3.95Se0.05 electrode, which possessed an extremely high energy density of 910.6 W h kg−1.229 Moreover, acetonitrile controlled the higher conducting cubic morphology with a particle size of 500 nm. This a core–shell structure reduced the side reaction between Fe1–xS and Na2.9PS3.95Se0.05.
6. Performances of yolk shell structure in battery application
Yolk–shell structures have specially attracted interest for application in sodium batteries given that they prevent the pulverization process during charging and discharging.230 Similarly, in the case of core shell structures, it has been noted that tin, antimony and iron are promising materials for enhanced sodium storage capacity. Kim and co-workers prepared Fe3O4@N-doped carbon (NC) yolk–shell from Fe2O3 nanoparticles and dopamine hydrochloride followed by etching in an acidic medium and calcination in an argon atmosphere at 500 °C for 5 h. The NC provided numerous active sites and effective electronic conductivity. The distinctive voids in the Fe3O4@NC yolk- and yolk–shell structure exhibited a high reversible capacity of 594 mA h g−1 over 100 cycles at 100 mA g−1.231 The yolk–shell iron sulfide–carbon nanospheres exhibiting porous carbon shells (∼30 nm) and void space (∼20 nm) originating from the etching of the silica particles enabled an enhanced sodium storage capacity. In addition, this nanosized iron sulfide–carbon exhibited a capacity of 545 mA h g−1 over 100 cycles at 0.2C (100 mA g−1).96 However, decreasing the carbon shell thickness to 10 nm in Sn3P4@C yolk–shell materials resulted in a higher cycling performance (516 and 368 mA h g−1 after 500 cycles at 1.0 and 2.0 A g−1, respectively). Furthermore, the carbon nanocubes could buffer the volume expansion better than the fibre structure.128When the yolk shell is combined with carbon spheres, the cyclic stability increases. Kang et al. synthesized wrinkled nanosheets of yolk–shell-structured MoSe2 microspheres using hydrogen selenide vapour at 300 °C, exhibiting a sodium storage capacity of 433 mA h g−1 after 50 cycles at a current density of 0.2 A g−1. Here, the wrinkled nanosheets of yolk–shell-structured MoSe2 enabled the sodium storage capacity.232 Further, when it was modified with porous carbon, its cyclic stability increased. Chen and co-workers prepared yolk–shell NiS2 nanoparticles on porous carbon fibres from Ni(NO3)2via hydrophilic treatment on the channels of porous carbon fibres, exhibiting a high reversible capacity of 534 mA h g−1 after 300 cycles at 0.1C with 85% capacity retention. Additionally, the material exhibited a high-rate capacity of 245 mA h g−1 at a high rate of 10C. This excellent performance is attributed to its highly porous structure obtained from a longer duration of heating treatment, resulting in a surface area of 949 m2 g−1 and the presence of both micropores (<2 nm) and mesopores (5–15 nm).233
As another factor, the morphology of materials is another vital parameter enabling the sodium storage capacity. The modification of 1D-like structures with transition elements contributes an excellent performance towards SIB application. A necklace-like structure composed of Fe3N@C yolk–shell, obtained from polyacrylonitrile through the electrospinning method, exhibited good cycling stability and a capacity of 248 mA h g−1 at 2 A g−1.115 Its excellent performance emphasized that Fe3N or PAN fiber enlarged the performance than the previously discussed Fe3O4 or iron sulphide. Moreover, voids can be intentionally designed within the electrode structure to improve the ion diffusion pathways and enhance the accessibility of the active material to the electrolyte ions. Voids or empty spaces within a battery reduce the overall mass of the battery without sacrificing the active material. Given that the energy density is typically measured in terms of energy per unit mass, by reducing the mass, a high energy density is obtained.
Although it is difficult to compare the performances of different compounds with different morphologies, overall microspheres show a higher performance compared to wires. Likewise, the yolk–shell SnO2@void@C nanowires synthesised using polyethylene glycol-400, followed by etching of the SiO2 layer showed a capacity of 401 mA h g−1 at a current density of 50 mA g−1.126 Further, polyhedron compounds have attracted considerable interest in SIB application. The Zhang group prepared Sb@C yolk–shell microspheres, which revealed a reversible capacity of 400 mA h g−1 after 300 cycles with 1 A g−1.234
A yolk–shell mesoporous octahedron-like structure delivered a capacity of 597.2 and 361.9 mA h g−1 at 0.2 and 16 A g−1, respectively. Moreover, octahedron yolk–shell structures were formed by the Kirkendall effects between Se and Co on ZIF-67. Their pore size was found to be in the range of 2–10 nm, showing an excellent rate capability.203 Besides, yolk–shell nano-boxes have attracted considerable attention due to their sodium storage capacity.235 The Zhou group prepared yolk–shell Fe7S8@C nano-boxes and Fe3O4@C nano-cubes. The yolk–shell Fe7S8@C nano-boxes exhibited a higher cycle stability and higher coulombic efficiency than the cubic structure.236 Again, the double shell cube-like structure showed very high cycle stability. Recently, yolk–double-shell cube-like SnS@N–S co-doped carbon exhibited a capacity retention of 83.5% at 1 A g−1 after 1500 cycles.237 Mai and co-workers prepared a mesoporous CoS yolk with alveolus-like carbon shell, which exhibited the outstanding cyclic capacity of 532 mA h g−1 after 100 cycles at 0.2 A g−1.238 The alveolus-like carbon shell appeared from the resorcinol formaldehyde structure. However, the polydopamine coating box-like hollow yolk shell contributed a higher performance than the resorcinol formaldehyde coating. Among the transition-metal yolk–shell hollow spheres, NiCo2O4 yolk–shell,14 Ni2P@C yolk–shell nanocomposite,239 GF/FeS2@C,240 and yolk–shell CoMoO4 nanospheres241 have been found to be efficient sodium storage materials.
7. Comparative overview
Aerosol synthesis242 is a cost-effective, attractive, and scalable method for the preparation of materials. There are several aerosol spray methods such as spray-drying,243 aerosol-spray-freezing,244 spray pyrolysis245 and ultrasonic spray pyrolysis. However, all these techniques depend on the temperature, concentration and other parameters. Among these techniques, aerosol-spray pyrolysis has attracted considerable attention due to its industrial-scale production with a short time and high purity grade. Hence, this is a promising and economical technique for battery application.242 In the case of ultrasonic spray pyrolysis, an ultrasonic nebulizer coverts a homogeneous salt mixture into droplets at the hot zone of a tubular furnace, which upon cooling form solid particles. The obtained dried particles without calcination process form spherical porous particles.246 Zhang et al. described in detail the aerosol method for battery application.242,247 Hence, we focus on the materials generated through the aerosol pyrolysis method that contribute a higher performance. Among the elements, it has been shown that tin-based materials have comparatively higher performances and cycle stability compared to other transition-metal-based compounds due to the higher tendency of tin to form alloys with sodium. Recent research showed that an Sn@C nanocomposite with 46 wt% Sn delivered an initial reversible capacity of ≈493.6 mA h g−1 at the current density of 200 mA g−1 and a stable capacity of ≈ 415 mA h g−1 after 500 cycles at 1000 mA g−1,248 whereas other transition elements such as CoSex–rGO composite displayed a capacity of 420 mA h g−1 at a current density of 0.3 A g−1.249 Recently, Kang and co-workers described in detail the metal chalcogenides synthesized via the spray pyrolysis method employed as electrode materials. Among these materials, FeS prepared through the spray pyrolysis method showed the highest discharge capacity and cycle stability.250 The role of the diameter of spherical materials has also been considered for SIB applications. Wang and co-workers synthesized 200–300 nm NiS2 nanoparticles, which showed the capacity retention of 77% at 0.1C for 200 cycles.206 The Sb@S,N-doped-3D interconnected nanoporous carbon electrode with a cavity of 200 nm exhibited ∼93% of its initial discharge capacity after 500 cycles at 100 mA g−1.251 Further, a reduction in its diameter to less than 10 nm showed an enhanced capacity retention of 81% after 2000 cycles at 1 A g−1.252Several reports exhibited that the wrapping of materials with carbon increases their electrochemical performances. The thickness of the carbon coating depends on the reaction time, concentration of the reactants and temperature. A moderate thickness can lead to a significant enhancement in performances. In a few studies, it was observed that the outer conducting cell remarkably enhanced the performance. Feng et al. applied 200 mL pyrrole on 200 mg CuCo2S4 spheres, forming a 30 nm outer-layer coating, which exhibited remarkable cycle stability at 2 A g−1.253 It was observed that the addition of 5-times urea to ferric acetylacetonate formed a 10–15 nm outer carbon coating, delivering 500 cycles at 2 A g−1, which is comparatively less than that of the 30 nm polypyrrole coating.254 Further, keeping a 13–20 nm carbon layer on Sn2S3 spheres slightly reduced the cycle stability compared to the polypyrrole coating.255
The performances of yolk/core shell structures depend on the yolk and core as well as on the shell structure. Yolk–shell structures cannot only reduce the transmission resistance of lithium and sodium ions but also promote the diffusion dynamics of ions/electrons by shortening the transmission path. Importantly, the unique architecture provides a buffer space for volume changes caused by the insertion/extraction of Li+/Na+. Thus, the structural stability of the electrode material is guaranteed, confirming its good Li/Na-storage performance between the yolk–shell and core–shell structure.256 Also, lithiation depends on the void space in the yolk and shell structure. Given that the sodium ion is larger than the lithium ion, a higher void space is required in SIBs compared to the smaller void space for Li-systems.257 The commercialization of sodium batteries is mainly associated with the sodium ion battery. Hence, here, we discuss the stability and corresponding parameters in the sodium ion battery. It is assumed that the application of yolk–shell and core–shell structures in sodium ion batteries will be a major attraction in the near future.
Core–shell structures offer large electrode/electrolyte contact areas and increase the sodium diffusion rate by controlling the diffusion path.258 Further, the void space in yolk shell structures can suitably control the sodiation/desodiation processes by buffering the core in the large void space, which facilitate the cycling stability and enhance the energy density.258 A literature review also suggested that yolk shell structures have a higher surface area compared to core shell structures. Moreover, sodium sulphur batteries have recently become a popular area of research.
In sodium sulphur batteries, conversion occurs through the insertion of sodium into the core structure of Na2S or NaxS with multiple redox states. Thus, prolonged cycle stability can be achieved in core–shell or yolk–shell structures.259 The special contribution of yolk shell and core–shell structures is the electrolyte interface (SEI) on the electrode, which further increases the overall efficiency.260 An SEI layer is a crucial factor in the overall performances of batteries. A stable and well-formed SEI layer is essential for the battery performance, given that it improves the cycling stability, prevents capacity loss, and enhances safety by inhibiting further electrolyte decomposition and dendrite formation. The additional shell material for the yolk or core acts as a protective layer, but still there is a possibility of the formation of electrode and electrolyte interactions, forming an SEI layer. In this case, porous shell materials can form a uniform thin SEI layer, while a very thick SEI layer may be responsible for irreversible capacity loss.260 Further, the excessive growth of an SEI layer leads to increased internal resistance, capacity fading, and reduced battery performance over time.
It is difficult to draw a conclusion on efficacy of different yolk- and core–shell structures given that there are several factors to consider such as different metal centres, shell thickness, conductivity, and diameter of hollow structure. However, an overview of this topic can predict their net result and best performance for industrialization. It is well known that chalcogenide structures are interesting to achieve an enhanced sodium storage capacity due to their intercalation property.261 Among the heterostructures of SNS@MoS2, the yolk–shell structure showed a higher performance and comparatively low shifting of the SEI film compared to its dense structure.262,263 In addition, the dense structure showed lower charge transfer resistance compared to the yolk–shell structure, which also resulted in better performances.263
The EV market is dominated by Li-batteries. A new lithium titanium oxide anode showed direct contact with the electrolyte and a very thin SEI film of Li2O appeared, which would not impede the diffusion of lithium ion. Due to the absence of a very thick SEI film, the unchanged resistance as well as transport ability and stable structure over the electrolyte enhanced the prolonged cycle stabilty.264 Also, a few other factors such as safety, recycling ability, cost effectiveness, cobalt-free electrode, material supplier, and stoichiometry ratio of metal to Na-ions should be optimized. Henceforth, specifically designed yolk- and core–shell structures associated with these factors can substantially realize the future capacity goal.
8. Machine learning on sodium battery
Machine learning has a huge scope for the enhancement of battery technology and robust performance of sodium batteries. Machine learning algorithms are utilized for the improvement in overall battery health, i.e. cyclability, charging time and discharging time.265,266 It also improves the battery management system and the design of the electrolyte, cathode, and anode materials through the use of several existing experimental details. Hence, in a very rapid time, machine learning can predict the overall battery health. Furthermore, machine learning reduces the computational time and needs less data storage facilities given that the calculations are performed within a very short time. Again, it reduces the optimization steps of material preparation, and hence the material cost and overall experimental steps can be reduced. Specifically, it can be concluded that machine learning can predict materials design for fast charging capacity and highly durable battery technologies, which eventually benefit the overall production process. However, to achieve promising experimental conditions as well as the material details, a large dataset is needed, which is first pre-processed and cleaned through algorithms. This processed data is trained through a machine learning model and tested by different datasets.267 The model is chosen by matching the pattern of the dataset. Hence, after training of the datasets, the model predicts the accuracy score of the test data. The accuracy score could be evaluated through the root mean square value. The model is chosen by taking 90% accuracy of data prediction.268 The cycle stability of a battery continuously fed through time results in an increase in impedance and decrease in capacity. Hence, the machine learning model will develop after considering the cycle stability based on several commercial battery data. For dataset preparation, a large scale of commercial datasets is needed. However, sodium batteries are not as commercialized as lithium ion batteries. Hence, based on lithium battery datasets, the cycle stability and other parameters can be optimized. Zhang et al.269 prepared an advance dataset from the MIT database, where 124 commercial data were recorded within normal capacity of 1.1 A h. After collecting the data, namely feature extraction and feature selection were employed. A total of 42 features was extracted on battery degradation data from the first 100 cycles. Next, the feature dimensions were reduced by employing the filter, wrapper, embedded, and principal component analysis (PCA) methods. After, feature cleaning, different machine learning models such as elastic net, GPR, SVM, random forest (RF), GBRT, and NN were employed for the determination of the battery lifetime. They identified the SVM model as the best model for the accurate prediction of the battery lifetime.269 Recently, Yildirim et al. described the discharge capacity with a variation in the electrode preparation method, cathode material, and anode material. They arranged the data with the variation in different parameters from 355 experimental papers. Gradient boosting, support vector machines, random forest, (for regression), and decision tree (for classification) methods were used for the data analysis. They analyzed that the anode material and preparation methods are highly effective in determining the cycle stability.270 Chen et al.271 described over 160 NASICON materials through random forest (RF) and neural network (NN) models. They implied suitable NASICON electrolyte design for SIBs.272 Yildirim and co-workers also collected 1227 data points from 335 published papers and applied random forest (for discharge capacity prediction) and decision tree (classification).271 Chao et al. described different carbon-based materials as anodes for sodium batteries. They correlated the structure and synthesis of different carbon materials with their discharge capacity. The result shows that hard carbon with a high ID/IG value is the advanced adaptor for SIBs.273 However, manganese can be a promising material for improving the performance of SIBs, as it can achieve a high energy density of 133.1 W h kg−1, which can be further applicable for power grid applications.274 Zhang et al. prepared the manganese-based material P2-Na0.67Ni0.33Mn0.67O2, which showed record operating voltage windows beyond 3.5 V in a full cell and may be helpful for designing yolk/core shell-like structures with significant results.275 Recently, Dong et al. described a nickel–manganese material to enhance the discharge capacity of sodium batteries.276 They corelated different factors such as the amount of nickel and various dopants in the dataset. Their results showed better cycle stability depending on the nickel content. Hence, based on the overall literature analysis, it can be concluded that the dataset parameters are constructed using the FWHM value from XRD, Raman ID/IG value, and crystal system from XRD. The discharge capacity is summarised employing a particular cycle stability. Therefore, it can assumed that the discharge capacity with various parameters can be correlated by the correlation method and the prediction of discharge capacity can be done by regression analysis. Furthermore, the cycle stability could be determined by classification tests.9. Advantages of yolk–shell and core–shell structures in sodium batteries
Yolk–shell structure
Core–shell structure
10. Summary and conclusion
Researchers have devoted their efforts to developing sodium-ion batteries with the aim of achieving a higher capacity, while maintaining affordability compared to lithium batteries. They are anticipated to find widespread use, ranging from wearable electronics, laptops, and mobile phones to electric vehicles, due to the abundance of sodium resources compared to lithium. However, a recent article in Nature Communications suggests that alkaline medium is superior to aqueous or neutral medium for sodium batteries.277 Companies such as Tiamat, Faradion, and Novasis are further assembling Na ion batteries in 18650 form factor (18 mm diameter, 65 mm length and O indicates cylindrical cell) or as pouch cells.2 For comparison, 18650-size commercial Na-ion batteries with hard carbon as the anode showed a specific energy of 150 W h kg−1, which is close to that of LiFePO4 in 18650-size commercial Li-ion batteries. This energy density can be applied in small EV, solar cells, power grid, etc.278
In summary, novel single, multi-core–shell and yolk–shell structures have been designed with high facile surface area and conductive network towards sodium batteries. This perspective summarized the reports on core–shell and yolk–shell structures. The yolk shell and core shell structures showed energy storage and volumetric expansion during their charge discharge step. The capacity performance in sodium batteries also depends on the cell construction, type of electrolyte, cell binder, and temperature. Further investigation showed that the multi-shell cavity or porous channels of MOF-based structures increase the rate capability and capacity retention.279–281
However, the higher performances of yolk–shell structures are attributed to their adequate internal space, facilitating large volume expansion, shortened diffusion distance and more active sites of Na+.282 In this perspective, core–shell and yolk–shell structures both showed similar sodium storage capacity, but the cycle stability and rate capacity are slightly higher in the case of the yolk–shell structure. The sodium storage capacity further depends on the carbon architecture such as RF, polydopamine, and PEG. Alternatively, multichannel hollow porous core–shell structures may be beneficial for SIB applications. In hollow core–shell structures, sodiation and desodiation are much faster compared to their core–shell counterparts. The creation of voids between the core and outer shell prevents excessive expansion during the alloying or dealloying process. Further, the mesoporous size and void size also regulate the capacity performances. With a successive decrease in pore size, the reversible capacity increases. The best capacity is achieved with a 3–4 nm mesoporous channel. Further, with a decrease in the size of the core particle to ∼2.4 nm, the reversible capacity can be controlled was also noted that a conducting coating or metallic core and yolk-like structure show a lower resistance. Finally, regulating other factors such as low contact angle, maximum exposure of the electrolyte, and small size of the core particles will help attain a higher rate capability and stability. Tables 8–10 present a summary of all types performances in sodium batteries.
| Catalyst | Current density (mA g−1) | Capacity (mA h g−1) | Cycle stability | Energy density | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| P-doped NiS2/C | 500 | 766.8 | 400 | — | 283 | |
| FeS2@C | 100 | 616 | 100 | — | 284 | |
| Bi@Void@C | 20000 |
198 | 10000 |
— | 96% after 10 cycles | 50 |
| FeS@mesoporous carbon | 200 | 596 | 100 | — | 285 | |
| Bi2S3/C | 0.2C | 282.4 | 300 | — | 25 | |
| Sb@NS-3DPCMSs | 20000 |
331 | 10000 |
— | 286 | |
| NiCoSe2/CNT | 3000 | 366 | 10000 |
— | 26 | |
| NiCo2O4 | 100 | 314 | 100 | — | — | 14 |
| N-doped carbon coated FeS2 nanocages | 5000 | 375 | 1000 | 92% | 287 | |
| TiO2 microspheres | 50 | 230.7 | 200 | 91.7% after 1000 cycles at 1 A g−1 | 288 | |
| Sb@C yolk–shell microspheres | 5000 | 633 | 200 | 99% over 200 cycles | 289 | |
| Sb@C | 1000 | 400 | 300 | — | — | 234 |
| (Fe1−xS)@N-doped carbon (FS@NC) | 100 | 594 | 100 | — | — | 231 |
| Fe3N@C Yolk–Shell Particles | 2000 | 248 | 300 | 115 | ||
| SnO2 yolks and graphene shells | 1000 | 248.2 | 1000 | — | 86.9% | 202 |
| SnS2@C | 100 | 690 | 100 | — | 87% after 150 cycles at 1 A g−1 | 290 |
| Sb@C | 10C | 315 | — | 130 W h kg−1 | 92% over 200 cycles at 1C rate | 291 |
| Sn4P3@C | 1 A g−1 | 516 mA h g−1 | 500 | 55.3% | 128 | |
| Sb@C | 1000 | 280 | 200 | — | — | 114 |
| CoSe/C | 16000 |
361.9 | — | — | — | 203 |
| TiO2@C | 40C | 210 | 2000 | 85% | 292 |
| Catalyst | Capacity (mA h g−1) | Current density (A g−1) | Cycle stability | Energy density | Capacity retention | Ref. |
|---|---|---|---|---|---|---|
| NaTi2(PO4)3@polyaniline | 104.1 | 10C | 200 | — | 96.9% after 200 cycles | 293 |
| Sn@C | >500 mA h g−1 | 0.2C | 300 | — | 80.1% at 0.2C after 300 cycles | 294 |
| P–CNT@PD composite | 730 | 2.6 | 2000 | — | — | 217 |
| Sb2S3 | 604 | 2 A g−1 | — | — | — | 295 |
| Sb/ZnS@C | 613 | 0.1 | 100 | — | — | 218 |
| ZnS-Sb2S3@C | 630 | 0.1 | 120 | — | — | 228 |
| rGO/Sb2S3 | 306 | 0.1 | 60 | — | — | 296 |
| Na5V12O32@PPy | 202 | 0.1 | 100 | — | 97% after 100 cycles | 297 |
| CNF@NPC | 240 | 0.1 | 100 | — | — | 298 |
| SnO2@PANI | 213.5 | 0.3 | 400 | — | — | 299 |
| Ge@G@TiO2 | 182 | 0.1 | 250 | — | — | 226 |
| Ni2P@C/GA | 253.6 | 0.1 | 100 | — | — | 300 |
| Nb2O5@C NPs to rGO | 285 | 0.025 | — | 76 W h kg −1 | — | 301 |
| Fe1−xS@SC | 454.3 | 1.0 | 500 | — | — | 302 |
| Sb@Co(OH)2 | 555.9 | 0.5 | 180 | — | — | 303 |
| Co9S8@C/3DNCF | 400.4 | 1 | 1400 | — | — | 304 |
| Sample | Cyclic capacity | Coulombic efficiency | Reversible capacity | Rate capability | Capacity retention | Energy density | Full cell | Ref. |
|---|---|---|---|---|---|---|---|---|
| Sodium ion battery | ||||||||
| Yolk–shell P-doped NiS2/C | — | 72.9% | 1113.5 mA h g−1 at 0.1 A g−1 after 20 cycles | 766.8 mA h g− 1 at 0.5 A g−1 after 400 cycles | — | — | — | 283 |
| Nickel–cobalt selenide carbon yolk shell | 344 mA h g−1 over 200 cycles at 0.5 A g−1 | 237 mA h g−1 at 5 A g−1. | After 2nd cycle @98% | — | — | 305 | ||
| Yolk–Shell Sb@Ti–O–P | 760 mA h g−1 after 200 cycles at 500 mA g−1 | 94% | 360 mA h g−1 at 10 A g−1 | — | — | — | 392 mA h g−1 anode after 150 cycles at 1 A g−1 at high output voltage (∼2.7 V) | 306 |
| FeSe2@NDC-NBs | 403.3 mA h g−1 over 2000 loops at 5.0 A g−1 | 99% after 2nd cycle (full cell) | 106.4 mA h g−1 at 1C after 100 loops | 374.9 mA h g−1 at 10.0 A g−1 | — | — | Reversible discharge capacity of 312.5 mA h g−1 anode after 100 cycles at 2.1 V | 307 |
| ZnSe-NC@CoSe2-NC core shell | — | 56.9@150 cycles | 308.5 mA g−1 after 150 cycles at a current density of 0.1 A g−1 | — | — | — | — | 308 |
| CoSe2@NiCoSe4-NC yolk shell | Specific capacity of 341.3 mA h g−1 at a current density of 5 A g−1 | — | — | — | 94.8% after 400 cycles at 1 A g−1 | — | — | 309 |
| Sn/Cu6Sn5@N–C yolk shell | — | ICE = 84.9% | 440.1 mA h g−1 after 1000 cycles at 1 A g−1 | 486.6 mA h g−1@10C | 90.8% @10C | — | — | 52 |
| FeS/C yolk shell | 300.4 mA h g−1 after 10000 cycles at 10 A g−1 |
664.9 mA h g−1 at 0.1 A g−1 | 81.1% after 10000 cycles (half-cell)/71.9% after 1000 cycles at 2 A g−1 |
181.9 W h kg−1 at 0.05 A g−1 (full cell) | 27 | |||
| ZnCoSe@ NDC yolk shell | 344.5 mA h g−1 at 5.0 A g−1 over 2000 cycles | 99% after 200 cycles | 319.2 mA h g−1 at 10.0 A g−1 | 96.3% of its initial capacity at 0.5 A g−1 after 200 loops (full cell) | Reversible discharge/charge capacity of 340.6/335.9 mA h g−1 after 200 cycles @2.1 V | 310 | ||
| Fe1−x/SWNT@C yolk shell | 710 mA h g−1 after 100 cycles at 0.1 A g−1 | 99 – 100% | 317 mA h g−1 at 20 A g−1 | 311 | ||||
| YDSC-SnS@NSC yolk shell | — | — | — | 257 mA h g−1 at 8 A g−1 | 83.5%@1 A g−1 | — | — | 312 |
| NiS2/CuS yolk shell | 283.4 mA h g−1 even after 4200 cycles at 20.0 −1. | ICE of 94.0% | — | — | — | — | — | 313 |
| NiS2⊂PCF yolk shell | — | — | 679 mA h g−1 at 0.1C | 245 mA h g−1 at 10C | 76% @5000 cycles at 5C | — | — | 233 |
| Yolk–Shell MnSe/ZnSe | 408.5 mA h g−1 even after 1000 cycles at 0.1 A g−1 | — | — | 475 mA hg−1 at 0.1 A g−1 | 72% after 1000 cycles at 100 mA g−1 | 209 | ||
| Fe7Se8@C/N yolk shell | No decay in 1000 cycles | 316.0 mA h g−1 at 5 A g−1 | 98% after 100 cycles (full cell) | 216 W h kg−1 | 208 | |||
| Sodium metal battery | ||||||||
| Core–Shell_C@SbNP | 6000 h at a high areal capacity of 4 mA h cm−2 with an average CE 99.7% | 99.7% | — | — | — | — | 212 | |
| C@Ag yolk shell | — | 99.7% at 1 mA h cm−2 for more than 2000 cycles | 80.7@after 320 cycles (full cell) | 71 mA h g−1 after 320 cycles | 314 | |||
| Sodium sulphide battery | ||||||||
| S/YS–Fe2N@NC yolk shell | 467 mA h g−1 after 350 cycles | Specific capacity of 1123 mA h g−1 at the rate of 1C | 215 | |||||
| FeS@C yolk shell | 545 mA h g−1 over 100 cycles at 0.2C (100 mA g−1) | Capacity retention of 67.6% after 300 cycles | 438 W h kg−1 | 30 | ||||
Author contributions
Anurupa Maiti: writing – original draft, Visualization, review and editing. Rasmita Biswal: review. Soumalya Debnath: Drawing. Anup Bhunia: review and editing.Conflicts of interest
The authors declare no conflicts of interest.References
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- J.-M. Tarascon, Joule, 2020, 4, 1616–1620 CrossRef.
- J. Lihua Liu, R. Dong and M. Danilovic, Electrification of the transportation system in China: exploring battery technology for electrical vehicles in China 1.0, Sweden-China Bridge: Collaborative Academic Platform for the Electrification, 2021, Top 10 sodium ion battery manufacturers in China, L. Li, https://www.takomabattery.com/top-10-sodium-ion-battery-manufacturers-in-china/#CATL.
- H. Yadegari, Q. Sun and X. Sun, Adv. Mater., 2016, 28, 7065–7093 CrossRef CAS PubMed.
- C. Delmas, Adv. Energ. Mater., 2018, 8, 1703137 CrossRef.
- S. Chen, C. Wu, L. Shen, C. Zhu, Y. Huang, K. Xi, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1700431 CrossRef.
- C. B. Tabelin, J. Dallas, S. Casanova, T. Pelech, G. Bournival, S. Saydam and I. Canbulat, Miner. Eng., 2021, 163, 106743 CrossRef CAS.
- H. S. Hirsh, Y. Li, D. H. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- Y. Wang, D. Zhou, V. Palomares, D. Shanmukaraj, B. Sun, X. Tang, C. Wang, M. Armand, T. Rojo and G. Wang, Energy Environ. Sci., 2020, 13, 3848–3879 RSC.
- J. F. Peters, M. Baumann and M. Weil, In The importance of recyclability for the environmental performance of battery systems, Cascade Use in Technologies 2018: Internationale Konferenz zur Kaskadennutzung und Kreislaufwirtschaft–Oldenburg 2018, Springer, 2019, pp. 104–110 Search PubMed.
- Z.-X. Huang, X.-L. Zhang, X.-X. Zhao, Y.-Y. Zhao, V. Aravindan, Y.-H. Liu, H. Geng and X.-L. Wu, Inorg. Chem. Front., 2023, 10, 37–48 RSC.
- R. Chen, D. Bresser, M. Saraf, P. Gerlach, A. Balducci, S. Kunz, D. Schröder, S. Passerini and J. Chen, ChemSusChem, 2020, 13, 2205–2219 CrossRef CAS PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- Y. Zhang, X. Xiao, W. Zhang, Y. Liu, J. Zhong, M. Chen, X. Fan and L. Chen, J. Alloys Compd., 2019, 800, 125–133 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. Notten, Adv. Energy Mater., 2018, 8, 1800079 CrossRef.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13–24 CrossRef CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 RSC.
- Y. You and A. Manthiram, Adv. Energy Mater., 2018, 8, 1701785 CrossRef.
- Y. X. Wang, B. Zhang, W. Lai, Y. Xu, S. L. Chou, H. K. Liu and S. X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- L. Xiao, F. Ji, J. Zhang, X. Chen and Y. Fang, Small, 2023, 19, 2205732 CrossRef CAS.
- J. Tang, A. D. Dysart and V. G. Pol, Curr. Opin. Chem. Eng., 2015, 9, 34–41 CrossRef.
- S. Li, Z. Wang, J. Liu, L. Yang, Y. Guo, L. Cheng, M. Lei and W. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 19438–19445 CrossRef CAS.
- H. Xu, W. Wang, G. Yu, L. Qin, Y. Jiang, L. Ren and J. Chen, ACS Appl. Energy Mater., 2020, 3, 4738–4745 CrossRef CAS.
- Z. Zhang, Y. Huang, X. Liu, X. Wang and P. Liu, Electrochim. Acta, 2020, 342, 136104 CrossRef CAS.
- H. Kim, D. Kim, Y. Lee, D. Byun, H.-S. Kim and W. Choi, Chem. Eng. J., 2020, 383, 123094 CrossRef CAS.
- S. H. Oh and J. S. Cho, J. Alloys Compd., 2019, 806, 1029–1038 CrossRef CAS.
- M. Han, J. Liu, C. Deng, J. Guo, Y. Mu, Z. Zou, K. Zheng, F. Yu, Q. Li and L. Wei, Adv. Energy Mater., 2024, 2400246 CrossRef.
- Y. Liu, Y. Qing, B. Zhou, L. Wang, B. Pu, X. Zhou, Y. Wang, M. Zhang, J. Bai and Q. Tang, ACS Nano, 2023, 17, 2431–2439 CrossRef CAS.
- Z. Liu, T. Lu, T. Song, X.-Y. Yu, X. W. D. Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576–1580 RSC.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W.-X. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 8689 CrossRef CAS.
- Y. B. Kim, H. Y. Seo, S. H. Kim, T. H. Kim, J. H. Choi, J. S. Cho, Y. C. Kang and G. D. Park, Small Methods, 2023, 7, 2201370 CrossRef CAS.
- F. Xu, S. Li, S. Jing, X. Peng, L. Yuan, S. Lu, Y. Zhang and H. Fan, J. Colloid Interface Sci., 2024, 660, 907–915 CrossRef CAS PubMed.
- M. Song, D. Ye, W. Li, C. Lu, W. Wu and X. Wu, ACS Appl. Mater. Interfaces, 2024, 16, 16120–16131 CrossRef CAS PubMed.
- J. Fu, S. Wang, D. Wu, J. Luo, C. Wang, J. Liang, X. Lin, Y. Hu, S. Zhang and F. Zhao, Adv. Mater., 2024, 36, 2308012 CrossRef CAS PubMed.
- J. Wang, H. Jing, X. Wang, Y. Xue, Q. Liang, W. Qi, H. Yu and C. F. Du, Adv. Funct. Mater., 2024, 2315318 CrossRef.
- J. Li, W. Zhang and W. Zheng, Small, 2024, 20, 2305021 CrossRef CAS PubMed.
- X. Bai, T. Li, U. Gulzar, E. Venezia, L. Chen, S. Monaco, Z. Dang, M. Prato, S. Marras, P. Salimi, S. Fugattini, C. Capiglia and R. P. Zaccaria, Nanoscale, 2020, 12, 15896–15904 RSC.
- Y. Zhang, C. Wang, H. Hou, G. Zou and X. Ji, Adv. Energy Mater., 2017, 7, 1600173 CrossRef.
- X. Song, X. Li, Z. Chen and Z. Wang, Mater. Lett., 2020, 275, 128109 CrossRef CAS.
- H. Liu, B. Liu, H. Guo, M. Liang, Y. Zhang, T. Borjigin, X. Yang, L. Wang and X. Sun, Nano Energy, 2018, 51, 639–648 CrossRef CAS.
- L. Zhao, H.-H. Wu, C. Yang, Q. Zhang, G. Zhong, Z. Zheng, H. Chen, J. Wang, K. He and B. Wang, ACS Nano, 2018, 12, 12597–12611 CrossRef CAS PubMed.
- R. Li, J. Huang, J. Li, L. Cao, Y. Luo, Y. He, G. Lu, A. Yu and S. Chen, ChemElectroChem, 2020, 7, 604–613 CrossRef CAS.
- (a) H. Li, Y. He, Q. Wang, S. Gu, L. Wang, J. Yu, G. Zhou and L. Xu, Adv. Energy Mater., 2023, 13, 2302901 CrossRef CAS; (b) A. B. Peng, Y. Li, J. Gao, F. Zhang, J. Li and G. Zhang, J. Power Sources, 2019, 437, 226913 CrossRef.
- Y. Zhang, J. Wang, L. Wang, L. Duan, G. Zhang, F. Zhao, X. Zhang and W. Lü, J. Mater. Sci., 2020, 55, 13102–13113 CrossRef CAS.
- J. Ru, T. He, B. Chen, Y. Feng, L. Zu, Z. Wang, Q. Zhang, T. Hao, R. Meng and R. Che, Angew. Chem., Int. Ed., 2020, 59, 14621–14627 CrossRef CAS.
- X.-K. Wang, J. Shi, L.-W. Mi, Y.-P. Zhai, J.-Y. Zhang, X.-M. Feng, Z.-J. Wu and W.-H. Chen, Rare Met., 2020, 39, 1053–1062 CrossRef CAS.
- Z. Liu, C. Wang, W. Yuan and Y. Xu, Chin. J. Chem., 2024, 42, 301–316 CrossRef CAS.
- S. Dong, Q. Su, W. Jiao, S. Ding, M. Zhang, G. Du and B. Xu, J. Alloys Compd., 2020, 155888 CrossRef CAS.
- W. Mao, S. Zhang, F. Cao, J. Pan, Y. Ding, C. Ma, M. Li, Z. Hou, K. Bao and Y. Qian, J. Alloys Compd., 2020, 842, 155300 CrossRef CAS.
- H. Yang, L.-W. Chen, F. He, J. Zhang, Y. Feng, L. Zhao, B. Wang, L. He, Q. Zhang and Y. Yu, Nano Lett., 2019, 20, 758–767 CrossRef PubMed.
- W. Liu, L. Du, S. Ju, X. Cheng, Q. Wu, Z. Hu and X. Yu, ACS Nano, 2021, 15, 5679–5688 CrossRef CAS PubMed.
- K. Yang, H. Guo, M. Chen, B. Dong, C. Yan, J. Zai and X. Qian, ACS Mater. Lett., 2023, 5, 2683–2690 CrossRef CAS.
- J. Li, J. Cao, X. Li, H. M. K. Sari, L. Li, C. Lv, I. V. Zatovsky and W. Han, Electrochim. Acta, 2020, 332, 135446 CrossRef CAS.
- G. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609–4613 CrossRef CAS PubMed.
- S. Santangelo, Appl. Sci., 2019, 9, 1049 CrossRef CAS.
- X. Guo, Y. Xue, H. Zhou, Y. Weng and J. Zhou, ACS Appl. Mater. Interfaces, 2019, 12, 2407–2416 CrossRef PubMed.
- Q. Huang, P. He, L. Xiao, Y. Feng, J. Liu, Y. Yang, B. Huang, X. Cui, P. Wang and W. Wei, ACS Appl. Mater. Interfaces, 2019, 12, 2191–2198 CrossRef PubMed.
- T. Famprikis, O. U. Kudu, J. Dawson, P. Canepa, F. Fauth, E. Suard, M. Zbiri, D. Dambournet, O. Borkiewicz and H. Bouyanfif, J. Am. Chem. Soc., 2020, 142, 18422–18436 CrossRef CAS PubMed.
- Z.-Y. Gu, J.-M. Cao, J.-Z. Guo, X.-T. Wang, X.-X. Zhao, S.-H. Zheng, Z.-H. Sun, J.-L. Yang, K.-Y. Zhang, H.-J. Liang, K. Li and X.-L. Wu, J. Am. Chem. Soc., 2024, 146, 4652–4664 CrossRef CAS PubMed.
- G. Åvall and P. Johansson, J. Chem. Phys., 2020, 152, 234104 CrossRef.
- J. K. Kim and S.-K. Park, J. Alloys Compd., 2024, 174373 Search PubMed.
- X. Geng, X. Hou, X. He and H. J. Fan, Adv. Energy Mater., 2024, 14, 2304094 CrossRef CAS.
- J. Li, L. Shi, J. Gao and G. Zhang, Chem. – Eur. J., 2018, 24, 1253–1258 CrossRef CAS PubMed.
- P. Ge, S. Li, L. Xu, K. Zou, X. Gao, X. Cao, G. Zou, H. Hou and X. Ji, Adv. Energy Mater., 2019, 9, 1803035 CrossRef.
- Y. Zhong, X. Xia, F. Shi, J. Zhan, J. Tu and H. J. Fan, Adv. Sci., 2016, 3, 1500286 CrossRef PubMed.
- P. Santhoshkumar, N. Shaji, M. Nanthagopal, J. W. Park, C. Senthil and C. W. Lee, J. Power Sources, 2020, 470, 228459 CrossRef CAS.
- H. Jin, H. Wang, Z. Qi, D. S. Bin, T. Zhang, Y. Wan, J. Chen, C. Chuang, Y. R. Lu, T. S. Chan, H. Ju, A.-M. Cao, W. Yan, X. Wu, H. Ji and L.-J. Wan, Angew. Chem., Int. Ed., 2020, 59, 2318–2322 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, D. Li, Q. Sun, K. Lai, K. Li, Q. Yuan, X. Liu and L. Ci, J. Mater. Chem. A, 2020, 8, 22874–22885 RSC.
- J. Zhang, Y. Liu, H. Liu, Y. Song, S. Sun, Q. Li, X. Xing and J. Chen, Small, 2020, 16, 2000504 CrossRef CAS.
- J. Ye, H. Zhao, M. Kang, W. Song, Q. Kong, C. Chen, R. Wu, J. Mi and Z. Li, Chem. Commun., 2020, 56, 1089–1092 RSC.
- C. Wei, F. Luo, C. Zhang, H. Gao, J. Niu, W. Ma, Y. Bai and Z. Zhang, Ionics, 2019, 1–9 Search PubMed.
- W. Zhang, Z. Tu, J. Qian, S. Choudhury, L. A. Archer and Y. Lu, Small, 2018, 14, 1703001 CrossRef PubMed.
- G. Du, M. Tao, J. Li, T. Yang, W. Gao, J. Deng, Y. Qi, S. J. Bao and M. Xu, Adv. Energy Mater., 2020, 10, 1903351 CrossRef CAS.
- C. Luo, T. Shen, H. Ji, D. Huang, J. Liu, B. Ke, Y. Wu, Y. Chen and C. Yan, Small, 2020, 16, 1906208 CrossRef CAS PubMed.
- L. Dashairya, D. Das, S. Jena, A. Mitra and P. Saha, Nano Select, 2021, 2, 373–388 CrossRef CAS.
- A. Li, W. Zhu, C. Li, T. Wang and J. Gong, Chem. Soc. Rev., 2019, 48, 1874–1907 RSC.
- Y.-A. Chen, Y.-T. Wang, H. S. Moon, K. Yong and Y.-J. Hsu, RSC Adv., 2021, 11, 12288–12305 RSC.
- S. Liu, J. Feng, X. Bian, J. Liu, H. Xu and Y. An, Energy Environ. Sci., 2017, 10, 1222–1233 RSC.
- R. Güttel, M. Paul and F. Schüth, Catal. Sci. Technol., 2011, 1, 65–68 RSC.
- Y. Liu, J. Goebl and Y. Yin, Chem. Soc. Rev., 2013, 42, 2610–2653 RSC.
- L. Yi, H. Wu, Y. Xu, J. Yu, Y. Zhao, H. Yang and C. Huang, Nanoscale, 2023, 15, 127–143 RSC.
- T. Harada, S. Ikeda, F. Hashimoto, T. Sakata, K. Ikeue, T. Torimoto and M. Matsumura, Langmuir, 2010, 26, 17720–17725 CrossRef CAS.
- C. Chen, X. Fang, B. Wu, L. Huang and N. Zheng, ChemCatChem, 2012, 4, 1578–1586 CrossRef CAS.
- Y.-L. Min, Y. Wan and S.-H. Yu, Solid State Sci., 2009, 11, 96–101 CrossRef CAS.
- M. Kim, J. C. Park, A. Kim, K. H. Park and H. Song, Langmuir, 2012, 28, 6441–6447 CrossRef CAS.
- Y. Chen, H. Chen, D. Zeng, Y. Tian, F. Chen, J. Feng and J. Shi, ACS Nano, 2010, 4, 6001–6013 CrossRef CAS PubMed.
- Y. N. Ko, Y. C. Kang and S. B. Park, Chem. Commun., 2013, 49, 3884–3886 RSC.
- J. C. Park, J. Y. Kim, E. Heo, K. H. Park and H. Song, Langmuir, 2010, 26, 16469–16473 CrossRef CAS PubMed.
- C.-H. Kuo, Y. Tang, L.-Y. Chou, B. T. Sneed, C. N. Brodsky, Z. Zhao and C.-K. Tsung, J. Am. Chem. Soc., 2012, 134, 14345–14348 CrossRef CAS PubMed.
- D. Cheng, X. Zhou, H. Xia and H. S. O. Chan, Chem. Mater., 2005, 17, 3578–3581 CrossRef CAS.
- N. Zhang, X. Fu and Y.-J. Xu, J. Mater. Chem., 2011, 21, 8152–8158 RSC.
- Z. Teng, S. Wang, X. Su, G. Chen, Y. Liu, Z. Luo, W. Luo, Y. Tang, H. Ju and D. Zhao, Adv. Mater. Interfaces, 2014, 26, 3741–3747 CrossRef CAS.
- Z. Teng, X. Su, Y. Zheng, J. Zhang, Y. Liu, S. Wang, J. Wu, G. Chen, J. Wang and D. Zhao, J. Am. Chem. Soc., 2015, 137, 7935–7944 CrossRef CAS.
- W. Zhou, Y. Yu, H. Chen, F. J. DiSalvo and H. C. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- J. Lee, J. C. Park and H. Song, Adv. Mater. Interfaces, 2008, 20, 1523–1528 CrossRef CAS.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W.-X. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 1–9 CAS.
- H. Zhang, L. Zhou, O. Noonan, D. J. Martin, A. K. Whittaker and C. Yu, Adv. Funct. Mater., 2014, 24, 4337–4342 CrossRef CAS.
- S.-H. Wu, C.-T. Tseng, Y.-S. Lin, C.-H. Lin, Y. Hung and C.-Y. Mou, J. Mater. Chem. A, 2011, 21, 789–794 RSC.
- C.-H. Lin, X. Liu, S.-H. Wu, K.-H. Liu and C.-Y. Mou, J. Phys. Chem. Lett., 2011, 2, 2984–2988 CrossRef CAS.
- W. Dong, Y. Zhu, H. Huang, L. Jiang, H. Zhu, C. Li, B. Chen, Z. Shi and G. Wang, J. Mater. Chem. A, 2013, 1, 10030–10036 RSC.
- Z. Chen, J. Wang, G. Zhai, W. An and Y. Men, Appl. Catal., B, 2017, 218, 825–832 CrossRef CAS.
- J. Liu, S. Z. Qiao, S. Budi Hartono and G. Q. Lu, Angew. Chem., Int. Ed., 2010, 49, 4981–4985 CrossRef CAS PubMed.
- Y. Ren, H. Shui, C. Peng, H. Liu and Y. Hu, Fluid Phase Equilib., 2011, 312, 31–36 CrossRef CAS.
- G. A. Carlson, J. P. McReynolds and F. H. Verhoek, J. Am. Chem. Soc., 1945, 67, 1334–1339 CrossRef CAS.
- H. Wu, S. Zhang, J. Zhang, G. Liu, J. Shi, L. Zhang, X. Cui, M. Ruan, Q. He and W. Bu, Adv. Funct. Mater., 2011, 21, 1850–1862 CrossRef CAS.
- J. Li, X. Liu, Z. Sun and L. Pan, Ceram. Int., 2015, 41, 8592–8598 CrossRef CAS.
- Y. Li, J. Lu, X. Cheng, H. Shi and Y. Zhang, Nano Energy, 2018, 48, 441–447 CrossRef CAS.
- R. Purbia and S. Paria, Nanoscale, 2015, 7, 19789–19873 RSC.
- Y. Zhu, T. Ikoma, N. Hanagata and S. Kaskel, Small, 2010, 6, 471–478 CrossRef CAS PubMed.
- Q. Sun, C. Z. Guo, G. H. Wang, W. C. Li, H. J. Bongard and A. H. Lu, Chem. – Eur. J., 2013, 19, 6217–6220 CrossRef CAS PubMed.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Chem. Mater., 2008, 20, 6562–6566 CrossRef CAS.
- Y.-L. Min, Y. Wan, R. Liu and S.-H. Yu, Mater. Chem. Phys., 2008, 111, 364–367 CrossRef CAS.
- Z.-M. Cui, Z. Chen, C.-Y. Cao, L. Jiang and W.-G. Song, Chem. Commun., 2013, 49, 2332–2334 RSC.
- J. Liu, L. Yu, C. Wu, Y. Wen, K. Yin, F.-K. Chiang, R. Hu, J. Liu, L. Sun and L. Gu, Nano Lett., 2017, 17, 2034–2042 CrossRef CAS PubMed.
- Z. Li, Y. Fang, J. Zhang and X. W. Lou, Adv. Mater. Interfaces, 2018, 30, 1800525 CrossRef PubMed.
- Y. Zhu, Y. Fang and S. Kaskel, J. Phys. Chem. C, 2010, 114, 16382–16388 CrossRef CAS.
- W. Ni, Y. Wang and R. Xu, Part. Part. Syst. Charact., 2013, 30, 873–880 CrossRef CAS.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- Y. Zeng, X. Wang, H. Wang, Y. Dong, Y. Ma and J. Yao, Chem. Commun., 2010, 46, 4312–4314 RSC.
- W. Zhao and X. Ma, ACS Sustainable Chem. Eng., 2020, 8, 19040–19050 CrossRef CAS.
- A. Okada, D. Nagao, T. Ueno, H. Ishii and M. Konno, Langmuir, 2013, 29, 9004–9009 CrossRef CAS PubMed.
- K. Zhang, X. Zhang, H. Chen, X. Chen, L. Zheng, J. Zhang and B. Yang, Langmuir, 2004, 20, 11312–11314 CrossRef CAS PubMed.
- G. Li, E. Kang, K. Neoh and X. Yang, Langmuir, 2009, 25, 4361–4364 CrossRef CAS PubMed.
- G. Liu, H. Wang, X. Yang and L. Li, Eur. Polym. J., 2009, 45, 2023–2032 CrossRef CAS.
- H.-f Ji, X.-x Wang, X. Zhang and X.-l Yang, Chin. J. Polym. Sci., 2010, 28, 807–817 CrossRef CAS.
- H. Li, L. Yang, J. Liu, S. Li, L. Fang, Y. Lu, H. Yang, S. Liu and M. Lei, J. Power Sources, 2016, 324, 780–787 CrossRef CAS.
- Y. Zhao, J. Zhu, S. J. H. Ong, Q. Yao, X. Shi, K. Hou, Z. J. Xu and L. Guan, Adv. Energy Mater., 2018, 8, 1802565 CrossRef.
- L. Ma, P. Yan, S. Wu, G. Zhu and Y. Shen, J. Mater. Chem. A, 2017, 5, 16994–17000 RSC.
- Y. Zhang, G. Hu, Q. Yu, Z. Liu, C. Yu, L. Wu, L. Zhou and L. Mai, Mater. Chem. Front., 2020, 4, 1656–1663 RSC.
- Y. Zhang, C. Wang, H. Hou, G. Zou and X. Ji, Adv. Energy Mater., 2017, 7, 1600173 CrossRef.
- Y. Zhang, A. Pan, L. Ding, Z. Zhou, Y. Wang, S. Niu, S. Liang and G. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 3624–3633 CrossRef CAS PubMed.
- Z. Li, Y. Fang, J. Zhang and X. W. Lou, Adv. Mater., 2018, 30, 1800525 CrossRef PubMed.
- X. Xu, F. Li, D. Zhang, Z. Liu, S. Zuo, Z. Zeng and J. Liu, Adv. Sci., 2022, 9, 2200247 CrossRef CAS PubMed.
- J. Yang, T. Shao, C. Luo, J. Li, S. He, B. Meng, Q. Zhang, D. Zhang, Z. Xue and X. Zhou, J. Alloys Compd., 2020, 834, 155056 CrossRef CAS.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- J. Park, T. Kwon, J. Kim, H. Jin, H. Y. Kim, B. Kim, S. H. Joo and K. Lee, Chem. Soc. Rev., 2018, 47, 8173–8202 RSC.
- H. Cheng, C. Wang, D. Qin and Y. Xia, Acc. Chem. Res., 2023, 56, 900–909 CrossRef CAS PubMed.
- X. Kong, H. Wu, K. Lu, X. Zhang, Y. Zhu and H. Lei, ACS Appl. Mater. Interfaces, 2023, 15, 41205–41223 CrossRef CAS PubMed.
- S. Xie, M. Jin, J. Tao, Y. Wang, Z. Xie, Y. Zhu and Y. Xia, Chem. – Eur. J., 2012, 18, 14974–14980 CrossRef CAS PubMed.
- Q. Shi, P. Zhang, Y. Li, H. Xia, D. Wang and X. Tao, Chem. Sci., 2015, 6, 4350–4357 RSC.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- J. S. Cho and Y. C. Kang, Small, 2015, 11, 4673–4681 CrossRef CAS PubMed.
- J. Gao, G. Liang, J. S. Cheung, Y. Pan, Y. Kuang, F. Zhao, B. Zhang, X. Zhang, E. X. Wu and B. Xu, J. Am. Chem. Soc., 2008, 130, 11828–11833 CrossRef CAS PubMed.
- E. V. Shevchenko, M. I. Bodnarchuk, M. V. Kovalenko, D. V. Talapin, R. K. Smith, S. Aloni, W. Heiss and A. P. Alivisatos, Adv. Mater. Interfaces, 2008, 20, 4323–4329 CrossRef CAS.
- M. H. Oh, T. Yu, S.-H. Yu, B. Lim, K.-T. Ko, M.-G. Willinger, D.-H. Seo, B. H. Kim, M. G. Cho and J.-H. Park, Science, 2013, 340, 964–968 CrossRef CAS PubMed.
- Y. Liu, B. Zhou, S. Liu, Q. Ma and W.-H. Zhang, ACS Nano, 2019, 13, 5885–5892 CrossRef CAS PubMed.
- M. Kong, Y. Liu, B. Zhou, K. Yang, J. Tang, P. Zhang and W. H. Zhang, Small, 2020, 16, 2001976 CrossRef CAS PubMed.
- L. Zhou, Y. Cheng, Q. Sun, L. Sun, C. Wang, X. Wang, D. Yin, L. Wang and J. Ming, Chem. Commun., 2018, 54, 4049–4052 RSC.
- L. Yu, L. Zhang, J. Fu, J. Yun and K. H. Kim, Chem. Eng. J., 2021, 417, 129106 CrossRef CAS.
- G. Madras and B. J. McCoy, J. Chem. Phys., 2003, 119, 1683–1693 CrossRef CAS.
- A. Pan, H. B. Wu, L. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 2226–2230 CrossRef CAS PubMed.
- B. Liu and H. C. Zeng, Small, 2005, 1, 566–571 CrossRef CAS PubMed.
- W. Weng, J. Lin, Y. Du, X. Ge, X. Zhou and J. Bao, J. Mater. Chem. A, 2018, 6, 10168–10175 RSC.
- J. S. Chen, C. M. Li, W. W. Zhou, Q. Y. Yan, L. A. Archer and X. W. Lou, Nanoscale, 2009, 1, 280–285 RSC.
- Y. Ye, L. Kuai and B. Geng, J. Mater. Chem. A, 2012, 22, 19132–19138 RSC.
- L. Zhang, D. A. Blom and H. Wang, Chem. Mater., 2011, 23, 4587–4598 CrossRef CAS.
- W. Weng, J. Lin, Y. Du, X. Ge, X. Zhou and J. Bao, J. Mater. Chem. A, 2018, 6, 10168–10175 RSC.
- L. Zhao, X. Rong, Y. Niu, R. Xu, T. Zhang, T. Li, Y. Yu and Y. Hou, Small, 2020, 16, 2004925 CrossRef CAS PubMed.
- G. D. Park and Y. C. Kang, Small Methods, 2021, 5, 2100302 CrossRef CAS PubMed.
- S.-K. Park, J. K. Kim and Y. C. Kang, J. Mater. Chem. A, 2017, 5, 18823–18830 RSC.
- S. E. Skrabalak and K. S. Suslick, J. Am. Chem. Soc., 2005, 127, 9990–9991 CrossRef CAS PubMed.
- S. E. Skrabalak and K. S. Suslick, J. Am. Chem. Soc., 2006, 128, 12642–12643 CrossRef CAS PubMed.
- Z. Chen, L. Xu, Q. Chen, P. Hu, Z. Liu, Q. Yu, T. Zhu, H. Liu, G. Hu and Z. Zhu, J. Mater. Chem. A, 2019, 7, 6740–6746 RSC.
- J. Leng, Z. Wang, J. Wang, H.-H. Wu, G. Yan, X. Li, H. Guo, Y. Liu, Q. Zhang and Z. Guo, Chem. Soc. Rev., 2019, 48, 3015–3072 RSC.
- S. H. Oh and J. S. Cho, J. Alloys Compd., 2019, 806, 1029–1038 CrossRef CAS.
- J. H. Kim and Y. C. Kang, Nano Res., 2017, 10, 3178–3188 CrossRef CAS.
- D. Guan, Q. Yu, C. Xu, C. Tang, L. Zhou, D. Zhao and L. Mai, Nano Res., 2017, 10, 4351–4359 CrossRef CAS.
- J. H. Choi, S. K. Park and Y. C. Kang, Small, 2019, 15, 1803043 CrossRef PubMed.
- Z. A. Qiao, Q. Huo, M. Chi, G. M. Veith, A. J. Binder and S. Dai, Adv. Mater. Interfaces, 2012, 24, 6017–6021 CrossRef CAS PubMed.
- Y. Wang, L. Ling, W. Zhang, J. Guo, K. Ding, W. Duan and B. Liu, Chem. Mater., 2019, 31, 9546–9553 CrossRef CAS.
- A. Najafian, M. Rabbani, R. Rahimi, M. Deilamkamar and A. Maleki, Solid State Sci., 2015, 46, 7–13 CrossRef CAS.
- L. Guo, X. Cui, Y. Li, Q. He, L. Zhang, W. Bu and J. Shi, Chem. – Asian J., 2009, 4, 1480–1485 CrossRef CAS PubMed.
- X. W. Lou, C. Yuan, Q. Zhang and L. A. Archer, Angew. Chem., Int. Ed., 2006, 45, 3825–3829 CrossRef CAS PubMed.
- S. Ding, J. S. Chen, G. Qi, X. Duan, Z. Wang, E. P. Giannelis, L. A. Archer and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 21–23 CrossRef CAS PubMed.
- R. V. Maligal-Ganesh, C. Xiao, T. W. Goh, L.-L. Wang, J. Gustafson, Y. Pei, Z. Qi, D. D. Johnson, S. Zhang and F. Tao, ACS Catal., 2016, 6, 1754–1763 CrossRef CAS.
- S. Luo, J. Ruan, Y. Wang, J. Hu, Y. Song, M. Chen and L. Wu, Small, 2021, 17, 2101879 CrossRef CAS PubMed.
- J. Zhao, Y. Yang, C. Jiang, N. A. Khan, X. Jia, H. Zhao and S. Ding, Nanotechnology, 2021, 32, 505602 CrossRef CAS PubMed.
- K. Kaviyarasu, M. M. Maaza, E. Manikandan and J. Kennedy, Adv. Mater. Lett., 2016, 7, 684–696 CrossRef CAS.
- A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS PubMed.
- H. Chen, C. Deng and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 607–611 CrossRef CAS PubMed.
- M. H. R. Farimani, N. Shahtahmasebi, M. R. Roknabadi, N. Ghows and A. Kazemi, Phys. E, 2013, 53, 207–216 CrossRef CAS.
- G. S. Chaubey and J.-K. Kim, Bull. Korean Chem. Soc., 2007, 28, 2279–2282 CrossRef CAS.
- X. Ren, Y. Yao, P. Ren, Y. Wang and Y. Peng, Mater. Lett., 2019, 238, 286–289 CrossRef CAS.
- P.-W. Xiao, L. Zhao, Z.-Y. Sui and B.-H. Han, Langmuir, 2017, 33, 6038–6045 CrossRef CAS PubMed.
- W. Duan, Z. Zhu, H. Li, Z. Hu, K. Zhang, F. Cheng and J. Chen, J. Mater. Chem. A, 2014, 2, 8668–8675 RSC.
- S. Tao, P. Cui, W. Huang, Z. Yu, X. Wang, S. Wei, D. Liu, L. Song and W. Chu, Carbon, 2016, 96, 1028–1033 CrossRef CAS.
- H. Kang, Y. Liu, M. Shang, T. Lu, Y. Wang and L. Jiao, Nanoscale, 2015, 7, 9261–9267 RSC.
- H. Liu, W. Pei, W.-H. Lai, Z. Yan, H. Yang, Y. Lei, Y.-X. Wang, Q. Gu, S. Zhou and S. Chou, ACS Nano, 2020, 14, 7259–7268 CrossRef CAS PubMed.
- M. Mostafa, Z. Alrowaili, G. Rashwan and M. Gerges, Heliyon, 2020, 6, e03389 CrossRef CAS PubMed.
- J. Flores, V. Torres, M. Popa, D. Crespo and J. Calderón-Moreno, J. Non-Cryst. Solids, 2008, 354, 5435–5439 CrossRef CAS.
- A. Bai, H. Song, G. He, Q. Li, C. Yang, L. Tang and Y. Yu, Ceram. Int., 2016, 42, 7583–7592 CrossRef CAS.
- X. Guo, Y. Deng, D. Gu, R. Che and D. Zhao, J. Mater. Chem., 2009, 19, 6706–6712 RSC.
- J. Yang, Y. Deng, Q. Wu, J. Zhou, H. Bao, Q. Li, F. Zhang, F. Li, B. Tu and D. Zhao, Langmuir, 2010, 26, 8850–8856 CrossRef CAS PubMed.
- J. Yang, F. Zhang, Y. Chen, S. Qian, P. Hu, W. Li, Y. Deng, Y. Fang, L. Han and M. Luqman, Chem. Commun., 2011, 47, 11618–11620 RSC.
- W. Zhao, M. Lang, Y. Li, L. Li and J. Shi, J. Mater. Chem. A, 2009, 19, 2778–2783 RSC.
- F. Xie, L. Zhang, D. Su, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1700989 CrossRef PubMed.
- X. Chen, Y. Fang, H. Lu, H. Li, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Small, 2021, 17, 2102248 CrossRef CAS PubMed.
- W. Meng, Z. Dang, D. Li, L. Jiang and D. Fang, Adv. Energy Mater., 2022, 12, 2201531 CrossRef CAS.
- J. Zhang, T. He, W. Zhang, J. Sheng, I. S. Amiinu, Z. Kou, J. Yang, L. Mai and S. Mu, Adv. Energy Mater., 2017, 7, 1602092 CrossRef.
- Y. Zhao, F. Wang, C. Wang, S. Wang, C. Wang, Z. Zhao, L. Duan, Y. Liu, Y. Wu and W. Li, Nano Energy, 2019, 56, 426–433 CrossRef CAS.
- Z. Cui, S. A. He, J. Zhu, M. Gao, H. Wang, H. Zhang and R. Zou, Small Methods, 2022, 6, 2101484 CrossRef CAS PubMed.
- H. Xu, L. Qin, J. Chen, Z. Wang, W. Zhang, P. Zhang, W. Tian, Y. Zhang, X. Guo and Z. Sun, J. Mater. Chem. A, 2018, 6, 13153–13163 RSC.
- Y. Zhang, A. Pan, L. Ding, Z. Zhou, Y. Wang, S. Niu, S. Liang and G. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 3624–3633 CrossRef CAS PubMed.
- L. Wang, Z. Han, Q. Zhao, X. Yao, Y. Zhu, X. Ma, S. Wu and C. Cao, J. Mater. Chem. A, 2020, 8, 8612–8619 RSC.
- Q. Chen, S. Sun, T. Zhai, M. Yang, X. Zhao and H. Xia, Adv. Energy Mater., 2018, 8, 1800054 CrossRef.
- T. Wang, P. Hu, C. Zhang, H. Du, Z. Zhang, X. Wang, S. Chen, J. Xiong and G. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 7811–7817 CrossRef CAS PubMed.
- D. Zhang, W. Sun, Y. Zhang, Y. Dou, Y. Jiang and S. X. Dou, Adv. Funct. Mater., 2016, 26, 7479–7485 CrossRef CAS.
- Z. Sun, X. Wu, Z. Gu, P. Han, B. Zhao, D. Qu, L. Gao, Z. Liu, D. Han and L. Niu, Carbon, 2020, 166, 175–182 CrossRef CAS.
- X. Wang, J. Zhao, Y. Chen, X. Zhang, K. Zhu, Q. Wang, J. Yan, D. Cao and G. Wang, Small, 2024, 20, 2307747 CrossRef CAS PubMed.
- Y. Zhang, C. Wang, H. Hou, G. Zou and X. Ji, Adv. Energy Mater., 2017, 7, 1600173 CrossRef.
- X. Hu, X. Liu, K. Chen, G. Wang and H. Wang, J. Mater. Chem. A, 2019, 7, 11016–11037 RSC.
- G. Wang, Y. Zhang, B. Guo, L. Tang, G. Xu, Y. Zhang, M. Wu, H.-K. Liu, S.-X. Dou and C. Wu, Nano Lett., 2020, 20, 4464–4471 CrossRef CAS PubMed.
- N. Zhu, X. Mao, G. Wang, M. Zhu, H. Wang, G. Xu, M. Wu, H. K. Liu, S.-X. Dou and C. Wu, J. Mater. Chem. A, 2021, 9, 13200–13208 RSC.
- F. Ma, P. Hu, T. Wang, J. Liang, R. Han, J. Han and Q. Li, ACS Appl. Energy Mater., 2021, 4, 3487–3494 CrossRef CAS.
- M. K. Aslam, T. Hussain, H. Tabassum, Z. Wei, W. Tang, S. Li, S.-J. Bao, X. S. Zhao and M. Xu, Chem. Eng. J., 2022, 429, 132389 CrossRef CAS.
- G. Wang, Y. Zhang, B. Guo, L. Tang, G. Xu, Y. Zhang, M. Wu, H.-K. Liu, S.-X. Dou and C. Wu, Nano Lett., 2020, 20, 4464–4471 CrossRef CAS PubMed.
- W. Liu, X. Yuan and X. Yu, Nanoscale, 2018, 10, 16675–16682 RSC.
- S. Dong, C. Li, Z. Li, L. Zhang and L. Yin, Small, 2018, 14, 1704517 CrossRef PubMed.
- F. Bu, P. Xiao, J. Chen, M. F. A. Aboud, I. Shakir and Y. Xu, J. Mater. Chem. A, 2018, 6, 6414–6421 RSC.
- Y. Han, J. Li, T. Zhang, P. Qi, S. Li, X. Gao, J. Zhou, X. Feng and B. Wang, Chem. – Eur. J., 2018, 24, 1651–1656 CrossRef CAS PubMed.
- Y. Cai, H.-E. Wang, X. Zhao, F. Huang, C. Wang, Z. Deng, Y. Li, G. Cao and B.-L. Su, ACS Appl. Mater. Interfaces, 2017, 9, 10652–10663 CrossRef CAS PubMed.
- W. Zhao, C. Guo and C. M. Li, J. Mater. Chem. A, 2017, 5, 19195–19202 RSC.
- L. Shi, D. Li, J. Yu, H. Liu, Y. Zhao, H. Xin, Y. Lin, C. Lin, C. Li and C. Zhu, J. Mater. Chem. A, 2018, 6, 7967–7976 RSC.
- J. Zhang, K. Zhang, J. Yang, G. H. Lee, J. Shin, V. Wing-hei Lau and Y. M. Kang, Adv. Energy Mater., 2018, 8, 1800283 CrossRef.
- S. Wen, J. Zhao, J. Chen, J. Yang and J. Xu, Dalton Trans., 2019, 48, 10448–10454 RSC.
- X. Wang, L. Fan, D. Gong, J. Zhu, Q. Zhang and B. Lu, Adv. Funct. Mater., 2016, 26, 1104–1111 CrossRef CAS.
- H. Y. Chen, N. Bucher, S. Hartung, L. Li, J. Friedl, H. P. Liou, C. L. Sun, U. Stimming and M. Srinivasan, Adv. Mater. Interfaces, 2016, 3, 1600357 CrossRef.
- S. Dong, C. Li, X. Ge, Z. Li, X. Miao and L. Yin, ACS Nano, 2017, 11, 6474–6482 CrossRef CAS PubMed.
- H. Wan, J. P. Mwizerwa, X. Qi, X. Liu, X. Xu, H. Li, Y.-S. Hu and X. Yao, ACS Nano, 2018, 12, 2809–2817 CrossRef CAS PubMed.
- G. D. Moon, Nanomaterials, 2020, 10, 675 CrossRef CAS PubMed.
- G. K. Veerasubramani, Y. Subramanian, M.-S. Park, G. Nagaraju, B. Senthilkumar, Y.-S. Lee and D.-W. Kim, J. Mater. Chem. A, 2018, 6, 20056–20068 RSC.
- Y. N. Ko, S. Choi, S. Park and Y. C. Kang, Nanoscale, 2014, 6, 10511–10515 RSC.
- Q. Chen, S. Sun, T. Zhai, M. Yang, X. Zhao and H. Xia, Adv. Energy Mater., 2018, 8, 1800054 CrossRef.
- F. Sun, Q. Ma, M. Kong, X. Zhou, Y. Liu, B. Zhou, P. Zhang and W.-H. Zhang, RSC Adv., 2018, 8, 36826–36830 RSC.
- M. Wang, Y. Huang, Y. Zhu, N. Zhang, J. Zhang, X. Qin and H. Zhang, Electrochim. Acta, 2020, 335, 135694 CrossRef CAS.
- W. Weng, J. Xu, C. Lai, Z. Xu, Y. Du, J. Lin and X. Zhou, J. Alloys Compd., 2020, 817, 152732 CrossRef CAS.
- M. Chen, Z. Zhang, L. Si, R. Wang and J. Cai, ACS Appl. Mater. Interfaces, 2019, 11, 35050–35059 CrossRef CAS PubMed.
- Q. Li, L. Li, K. A. Owusu, W. Luo, Q. An, Q. Wei, Q. Zhang and L. Mai, Nano Energy, 2017, 41, 109–116 CrossRef CAS.
- J. Zheng, X. Huang, X. Pan, C. Teng and N. Wang, Appl. Surf. Sci., 2019, 473, 699–705 CrossRef CAS.
- P. Jing, Q. Wang, B. Wang, X. Gao, Y. Zhang and H. Wu, Carbon, 2020, 159, 366–377 CrossRef CAS.
- J. Wang, L. Zhu, F. Li, T. Yao, T. Liu, Y. Cheng, Z. Yin and H. Wang, Small, 2020, 16, 2002487 CrossRef CAS PubMed.
- P. Nie, G. Xu, J. Jiang, H. Dou, Y. Wu, Y. Zhang, J. Wang, M. Shi, R. Fu and X. Zhang, Small Methods, 2018, 2, 1700272 CrossRef.
- A. B. D. Nandiyanto and K. Okuyama, Adv. Powder Technol., 2011, 22, 1–19 CrossRef CAS.
- T. Cheng, J. Xu, Z. Tan, J. Ye, Z. Tao, Z. Du, Y. Wu, S. Wu, H. Ji and Y. Yu, Energy Storage Mater., 2018, 10, 282–290 CrossRef.
- D. S. Jung, S. B. Park and Y. C. Kang, Korean J. Chem. Eng., 2010, 27, 1621–1645 CrossRef CAS.
- M. H. Aghaali and S. Firoozi, Int. J. Hydrogen Energy, 2021, 46, 357–373 CrossRef CAS.
- Y. Zhu, S. H. Choi, X. Fan, J. Shin, Z. Ma, M. R. Zachariah, J. W. Choi and C. Wang, Adv. Energy Mater., 2017, 7, 1601578 CrossRef.
- Y. Liu, N. Zhang, L. Jiao, Z. Tao and J. Chen, Adv. Funct. Mater., 2015, 25, 214–220 CrossRef CAS.
- G. D. Park and Y. C. Kang, Chem. – Eur. J., 2016, 22, 4140–4146 CrossRef CAS PubMed.
- J. K. Kim, S. Y. Jeong, S. H. Lim, J. H. Oh, S. K. Park, J. S. Cho and Y. C. Kang, Chem. – Asian J., 2019, 14, 3127–3140 CrossRef CAS PubMed.
- C. Yang, W. Li, Z. Yang, L. Gu and Y. Yu, Nano Energy, 2015, 18, 12–19 CrossRef CAS.
- J. Wang, J. Li, X. He, X. Zhang, B. Yan, X. Hou, L. Du, T. Placke, M. Winter and J. Li, J. Power Sources, 2020, 461, 228129 CrossRef CAS.
- Q. Li, Q. Jiao, W. Zhou, X. Feng, Q. Shi, Z. Dai, T. Gu, Y. Zhao, H. Li and C. Feng, Mater. Chem. Front., 2021, 5, 293–303 RSC.
- Z. Zhao, C. Li, Z. Liu and D. Li, Int. J. Hydrogen Energy, 2021, 46, 26457–26465 CrossRef CAS.
- G. Chen, X. Li, T. Zeng, R. Han and Q. Wang, Carbon, 2021, 171, 464–473 CrossRef CAS.
- B. Wang, Y. Cheng, H. Su, M. Cheng, Y. Li, H. Geng and Z. Dai, ChemSusChem, 2020, 13, 4078–4085 CrossRef CAS.
- J. Liu, L. Yu, C. Wu, Y. Wen, K. Yin, F.-K. Chiang, R. Hu, J. Liu, L. Sun and L. Gu, Nano Lett., 2017, 17, 2034–2042 CrossRef CAS PubMed.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W.-X. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6(8689), 1–9 CAS.
- W. Zhao, C. Guo and C. M. Li, J. Mater. Chem. A, 2017, 5, 19195–19202 RSC.
- H. Geng, J. Yang, Z. Dai, Y. Zhang, Y. Zheng, H. Yu, H. Wang, Z. Luo, Y. Guo and Y. Zhang, Small, 2017, 13, 1603490 CrossRef.
- J. Gao, Y. Li, Y. Liu, S. Jiao, J. Li, G. Wang, S. Zeng and G. Zhang, J. Mater. Chem. A, 2019, 7, 18828–18834 RSC.
- Y. Lin, X. Guo, M. Hu, B. Liu, Y. Dong, X. Wang, N. Li and H.-E. Wang, Nanoscale, 2020, 12, 14689–14698 RSC.
- S. H. Choi and Y. C. Kang, ACS Appl. Mater. Interfaces, 2015, 7, 24694–24702 CrossRef CAS PubMed.
- X. Han, M. Ouyang, L. Lu, J. Li, Y. Zheng and Z. Li, J. Power Sources, 2014, 251, 38–54 CrossRef CAS.
- Z. Wei, Q. He and Y. Zhao, J. Power Sources, 2022, 549, 232125 CrossRef CAS.
- Z. H. Shen, H. X. Liu, Y. Shen, J. M. Hu, L. Q. Chen and C. W. Nan, Interdiscip. Mater., 2022, 1, 175–195 CrossRef.
- D. Roman, S. Saxena, V. Robu, M. Pecht and D. Flynn, Nat. Mach. Intell., 2021, 3, 447–456 CrossRef.
- M. Berecibar, Nature, 2019, 568, 325–326 CrossRef CAS PubMed.
- Z. Fei, F. Yang, K.-L. Tsui, L. Li and Z. Zhang, Energy, 2021, 225, 120205 CrossRef.
- B. Oral, B. Tekin, D. Eroglu and R. Yildirim, Assessment of Na-Ion Battery Performance Using Machine Learning, Electrochemical Society Meeting Abstracts 243, The Electrochemical Society, Inc., 2023, pp. 882–882 Search PubMed.
- B. Oral, B. Tekin, D. Eroglu and R. Yildirim, J. Power Sources, 2022, 549, 232126 CrossRef CAS.
- Y. Zhang, T. Zhan, Y. Sun, L. Lu and B. Chen, ChemSusChem, 2023, e202301284 Search PubMed.
- X. Liu, T. Wang, T. Ji, H. Wang, H. Liu, J. Li and D. Chao, J. Mater. Chem. A, 2022, 10, 8031–8046 RSC.
- B. Peng, Z. Sun, S. Jiao, G. Wang and G. Zhang, Batteries Supercaps, 2020, 3, 147–154 CrossRef CAS.
- B. Peng, Z. Sun, L. Zhao, J. Li and G. Zhang, Energy Storage Mater., 2021, 35, 620–629 CrossRef.
- S. Yang, S. Hu, J. Zhao, H. Cui, Y. Wang, S. Zhao, C. Lan and Z. Dong, Energy Technol., 2022, 10, 2200733 CrossRef CAS.
- H. Wu, J. Hao, Y. Jiang, Y. Jiao, J. Liu, X. Xu, K. Davey, C. Wang and S.-Z. Qiao, Nat. Commun., 2024, 15, 575 CrossRef CAS PubMed.
- K. Abraham, ACS Energy Lett., 2020, 5, 3544–3547 CrossRef CAS.
- S. Fan, S. Huang, Y. Chen, Y. Shang, Y. Wang, D. Kong, M. E. Pam, L. Shi, Y. W. Lim and Y. Shi, Energy Storage Mater., 2019, 23, 17–24 CrossRef.
- Q. Chen, S. Sun, T. Zhai, M. Yang, X. Zhao and H. Xia, Adv. Energy Mater., 2018, 8, 1800054 CrossRef.
- G. Zhao, Y. Zhang, L. Yang, Y. Jiang, Y. Zhang, W. Hong, Y. Tian, H. Zhao, J. Hu and L. Zhou, Adv. Funct. Mater., 2018, 28, 1803690 CrossRef.
- Q. Zhou, Y. Wang, R. Ou, X. Ding, Y. Xin, F. Wu and H. Gao, Small, 2024, 2310699, DOI:10.1002/smll.202310699.
- L. Wang, Z. Han, Q. Zhao, X. Yao, Y. Zhu, X. Ma, S. Wu and C. Cao, J. Mater. Chem. A, 2020, 8, 8612–8619 RSC.
- Z. Man, P. Li, D. Zhou, Y. Wang, X. Liang, R. Zang, P. Li, Y. Zuo, Y. M. Lam and G. Wang, Nano Lett., 2020, 20, 3769–3777 CrossRef CAS PubMed.
- X. Chen, D. Wang and J. Chen, New J. Chem., 2019, 43, 10291–10296 RSC.
- B. Chen, H. Qin, K. Li, B. Zhang, E. Liu, N. Zhao, C. Shi and C. He, Nano Energy, 2019, 66, 104133 CrossRef CAS.
- R. Zang, P. Li, X. Guo, Z. Man, S. Zhang, C. Wang and G. Wang, J. Mater. Chem. A, 2019, 7, 14051–14059 RSC.
- Z. Chen, L. Xu, Q. Chen, P. Hu, Z. Liu, Q. Yu, T. Zhu, H. Liu, G. Hu and Z. Zhu, J. Mater. Chem. A, 2019, 7, 6740–6746 RSC.
- J. Song, D. Xiao, H. Jia, G. Zhu, M. Engelhard, B. Xiao, S. Feng, D. Li, D. Reed and V. L. Sprenkle, Nanoscale, 2019, 11, 348–355 RSC.
- X. Li, Y. Zhao, Q. Yao and L. Guan, Electrochim. Acta, 2018, 270, 1–8 CrossRef CAS.
- J. Song, P. Yan, L. Luo, X. Qi, X. Rong, J. Zheng, B. Xiao, S. Feng, C. Wang and Y.-S. Hu, Nano Energy, 2017, 40, 504–511 CrossRef CAS.
- S. Qiu, L. Xiao, X. Ai, H. Yang and Y. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 345–353 CrossRef CAS PubMed.
- H. Yan, Y. Fu, X. Wu, X. Xue, C. Li and L. Zhang, Solid State Ionics, 2019, 336, 95–101 CrossRef CAS.
- H. Tian, Y. Liang, J. Repac, S. Zhang, C. Luo, S.-C. Liou, G. Wang, S. H. Ehrman and W. Han, J. Phys. Chem. C, 2018, 122, 22232–22240 CrossRef CAS.
- F. Xie, L. Zhang, Q. Gu, D. Chao, M. Jaroniec and S.-Z. Qiao, Nano Energy, 2019, 60, 591–599 CrossRef CAS.
- S. Yao, J. Cui, Z. Lu, Z. L. Xu, L. Qin, J. Huang, Z. Sadighi, F. Ciucci and J. K. Kim, Adv. Energy Mater., 2017, 7, 1602149 CrossRef.
- Y. Cao, D. Fang, X. Liu, Z. Luo, G. Li, W. Xu, M. Jiang and C. Xiong, Compos. Sci. Technol., 2016, 137, 130–137 CrossRef CAS.
- Z. Zhang, J. Zhang, X. Zhao and F. Yang, Carbon, 2015, 95, 552–559 CrossRef CAS.
- X. Zhao, Z. Zhang, F. Yang, Y. Fu, Y. Lai and J. Li, RSC Adv., 2015, 5, 31465–31471 RSC.
- X. Miao, R. Yin, X. Ge, Z. Li and L. Yin, Small, 2017, 13, 1702138 CrossRef PubMed.
- E. Lim, C. Jo, M. S. Kim, M. H. Kim, J. Chun, H. Kim, J. Park, K. C. Roh, K. Kang and S. Yoon, Adv. Funct. Mater., 2016, 26, 3711–3719 CrossRef CAS.
- Q. Pan, F. Zheng, Y. Liu, Y. Li, W. Zhong, G. Chen, J. Hu, C. Yang and M. Liu, J. Mater. Chem. A, 2019, 7, 20229–20238 RSC.
- Y. Zhang, H. Gao, J. Niu, W. Ma, Y. Shi, M. Song, Z. Peng and Z. Zhang, ACS Nano, 2018, 12, 11678–11688 CrossRef CAS PubMed.
- X. Ma, X. Xiong, P. Zou, W. Liu, F. Wang, L. Liang, Y. Liu, C. Yuan and Z. Lin, Small, 2019, 15, 1903259 CrossRef CAS PubMed.
- H. Y. Seo, J. H. Choi, Y. B. Kim, J. S. Cho, Y. C. Kang and G. D. Park, J. Mater. Chem. A, 2023, 11, 24738–24753 RSC.
- N. Wang, Z. Bai, Y. Qian and J. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 447–454 CrossRef CAS PubMed.
- J. Feng, S.-H. Luo, Y. Zhan, S.-X. Yan, P.-W. Li, L. Zhang, Q. Wang, Y.-H. Zhang and X. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 51095–51106 CrossRef CAS PubMed.
- X. Hu, X. Liu, K. Chen, G. Wang and H. Wang, J. Mater. Chem. A, 2019, 7, 11016–11037 RSC.
- H. Liang, X. Li, Z. Liu, W. Yang, X. Liu, Y. Zhang and H. Fan, Mater. Chem. Front., 2022, 6, 194–202 RSC.
- J. Feng, S. h Luo, S. x Yan, Y. Zhan, Q. Wang, Y. h Zhang, X. Liu and L. j Chang, Small, 2021, 17, 2101887 CrossRef CAS PubMed.
- Y. Zhao, X. Shi, S. J. H. Ong, Q. Yao, B. Chen, K. Hou, C. Liu, Z. J. Xu and L. Guan, ACS Nano, 2020, 14, 4463–4474 CrossRef CAS PubMed.
- M. Chen, Z. Zhang, L. Si, R. Wang and J. Cai, ACS Appl. Mater. Interfaces, 2019, 11, 35050–35059 CrossRef CAS PubMed.
- W. Zhao, X. Wang, X. Ma, L. Yue, Q. Liu, Y. Luo, Y. Liu, A. M. Asiri and X. Sun, J. Mater. Chem. A, 2021, 9, 15807–15819 RSC.
- N. Zhu, X. Mao, G. Wang, M. Zhu, H. Wang, G. Xu, M. Wu, H. K. Liu, S.-X. Dou and C. Wu, J. Mater. Chem. A, 2021, 9, 13200–13208 RSC.
| This journal is © The Royal Society of Chemistry 2024 |