
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorine-free “solvent-in-salt” sodium battery electrolytes: solvation structure and dynamics†
Yanqi
Xu
 a,
Andrei
Filippov
a,
Sourav
Bhowmick
a,
Patrik
Johansson
*bc and
Faiz Ullah
Shah
*a
a,
Andrei
Filippov
a,
Sourav
Bhowmick
a,
Patrik
Johansson
*bc and
Faiz Ullah
Shah
*a
aChemistry of Interfaces, Luleå University of Technology, SE-971 87 Luleå, Sweden. E-mail: faiz.ullah@ltu.se
bDepartment of Physics, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden. E-mail: patrik.johansson@chalmers.se
cALISTORE-European Research Institute, FR CNRS 3104, Hub de l’Energie 15, Rue Baudelocque, 80039 Amiens, France
First published on 9th February 2024
Abstract
The solvation structure, dynamics, and transport properties, as well as thermal and electrochemical stabilities of “solvent-in-salt” (SIS) electrolytes, also known as highly concentrated electrolytes, are far from fully understood. Furthermore, these special types of electrolytes are almost without exception based on fluorinated salts. In contrast, here we report on fluorine-free SIS electrolytes comprising ambient temperature liquid sodium bis(2-(2-ethoxyethoxy)ethyl)phosphate (NaDEEP) salt and tris(2-(2-ethoxyethoxy)ethyl)phosphate (TEOP) solvent, for which the ionic conductivities and ion diffusivities are altered profoundly as the salt concentration is increased. A careful molecular level analysis reveals a microstructure with a “solvent-rich” phase with almost an order of magnitude faster ion diffusion than in a “salt-rich” phase. Aggregated ionic structures in these SIS electrolytes lead to higher ionic conductivities alongside lower glass transition temperatures, <−80 °C, but also agreeable thermal stabilities, up to 270 °C, and improved anodic stabilities, possibly up to 7.8 V vs. Na/Na+ and at least >5 V vs. Na/Na+. Altogether, this provides a foundation for both better understanding and further development of fluorine-free SIS electrolytes for sodium batteries.
Introduction
Driven by the growing market of electric vehicles, the interest in R&D on technologies beyond lithium-ion batteries (LIBs) has increased drastically in recent years. This is not the least true for sodium-ion batteries (SIBs) with large research efforts traceable back to the 1980's and earlier, but with renewed interest due to the abundant resources,1,2 relatively low cost of sodium,2,3 and also improved power performance as compared to LIBs.4 While most SIB research still focuses on electrode materials,5–8 work on understanding the properties of SIB electrolytes and electrode–electrolyte interfaces, including the solid electrolyte interphase (SEI), is rapidly picking up.9,10Conventional liquid electrolytes for SIBs are often based on flammable organic solvents and fluorinated salts,11,12 adversely affecting the overall battery performance in some cases and causing safety issues.13–16 In addition, simple adoption of the most studied and rather thoroughly understood LIB electrolyte designs might anyhow not provide the necessary performance for SIBs. As an example, electrolytes based on NaPF6 dissolved in carbonates were found to be more reactive towards sodium inserted hard carbon (Na/HC) than the corresponding LiPF6-based electrolyte vs. Li/HC.17 As for interfaces/interphases, Iermakova et al.18 examined the SEIs formed on Li and Na metal anodes and demonstrated the former to be more stable. Therefore, the development of more performant SIB electrolytes is an open avenue of battery research.9,19–22
For both aqueous and non-aqueous electrolytes, the concept of “super-concentration” has attracted attention and been proposed to reduce electrolyte volatility23 and improve the cell cycling stability.24,25 Already in 1985 McKinnon and Dahn26 reported that a saturated solution of LiAsF6 in propylene carbonate (PC) could eradicate the co-intercalation of PC for LIB anodes, which later has been followed by numerous studies.27 Somewhat similarly, Angell et al.28 strived to decouple the mechanical properties from the ionic conductivity of polymer electrolytes, and developed and coined “polymer-in-salt” electrolytes, which in terms of ionic conductivity outperformed conventional “salt-in-polymer” concepts.28 Much later, the concept of “solvent-in-salt” (SIS) electrolytes, today also known as highly concentrated electrolytes (HCEs) was coined and popularized, and is most commonly defined as electrolytes in which either the mass or volume ratio of salt to solvent exceeds unity.29
The most commonly studied SIS electrolytes are based on heavily fluorinated salts, such as lithium bis(trifluoromethanesulfonyl)imide (LiTFSI)29 and lithium bis(fluorosulfonyl)imide (LiFSI),30 and the corresponding sodium salts including NaFSI,31,32 NaTFSI33 as well as sodium hexafluorophosphate (NaPF6).34 The high salt concentration renders them enhanced thermal and redox stabilities,29,35–37 but lower ionic conductivities and higher viscosities than their corresponding traditional ca. 1 M electrolytes.24 Despite their promising properties, these heavily fluorinated electrolytes can, however, also induce safety problems at all levels from the production to the usage phase and the recycling.38,39
To counter this, Scheers et al.40 suggested totally fluorine-free LIB electrolytes, Jonsson et al.41 proposed a number of novel fluorine-free Li based anions/salts, and recently Colbin et al.42 investigated sodium bis(oxalato)borate (NaBOB) dissolved in triethyl phosphate (TEP), resulting in promising long-term cycling and rate performance. Similarly, Mogensen et al.43 found that NaBOB dissolved in a mixture of N-methyl-2-pyrrolidone (NMP) and trimethyl phosphate (TMP) provides high ionic conductivity over a wide temperature range and enhanced SIB performance.
Here we combine the two strategies above and report on the very creation, the solvation structure, the dynamics, and the transport properties of a new class of fluorine-free sodium battery SIS electrolytes composed of our newly designed ambient temperature liquid salt: sodium bis(2-(2-ethoxyethoxy)ethyl)phosphate (NaDEEP) and its structurally analogous solvent: tris(2-(2-ethoxyethoxy)ethyl)phosphate (TEOP)44 (Fig. 1). Apart from being fluorine-free, both the salt and the solvent contain oligoether chains that are expected to bring the beneficial properties of glymes45–48 to the resulting SIS electrolytes/HCEs.
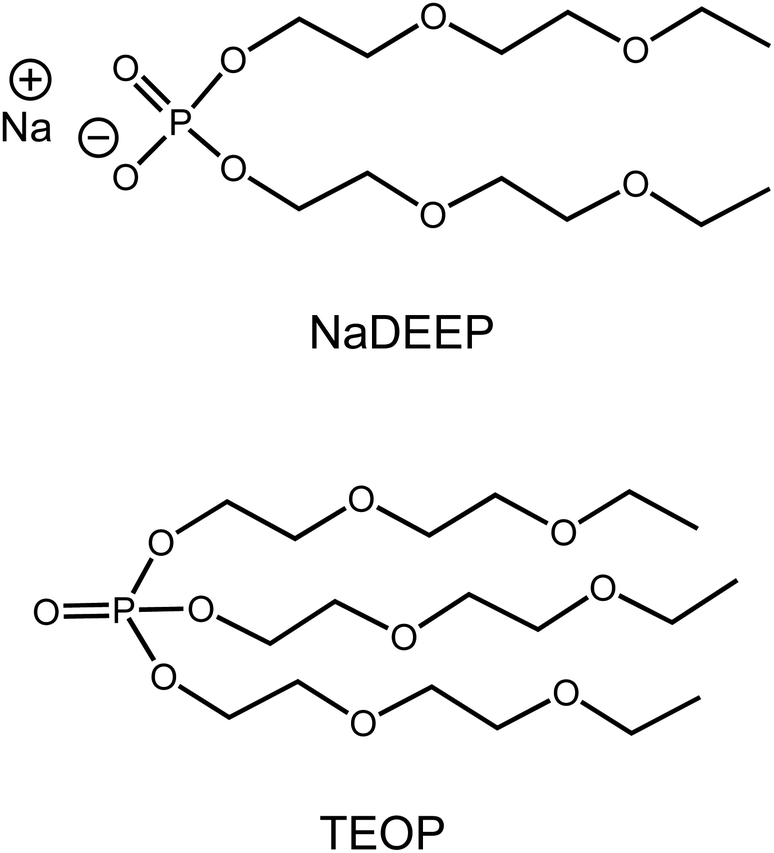 | ||
| Fig. 1 Chemical structures of NaDEEP and TEOP. | ||
Experimental
Preparation of electrolytes
Both the synthesis and structural characterization of the NaDEEP salt and the TEOP solvent were recently reported.44 All the SIS electrolytes were prepared by mixing stoichiometric amounts of NaDEEP in TEOP, rendering 50 ≤ XX ≤ 90 mol% (xNaDEEP) electrolytes being denoted “SISXX” (Table 1). Briefly, both salt and solvent were added to a 25 mL vial and ultrasonicated for 2 hours to make homogenous solutions, followed by drying in a vacuum oven at 75 °C for 3–5 days and then transferred to a nitrogen-filled glovebox with water and oxygen contents <1 ppm. The water contents of the electrolytes were <100 ppm, as determined by Karl–Fischer titration using a 917 coulometer (Metrohm), placed inside the glovebox.| Acronym | x NaDEEP (%) | NaDEEP concentration (mol kg−1) | Salt-to-solvent molar ratio |
|---|---|---|---|
| SIS50 | 50 | 2.2 | 1.0 |
| SIS60 | 60 | 3.3 | 1.5 |
| SIS70 | 70 | 5.2 | 2.3 |
| SIS80 | 80 | 8.9 | 4.0 |
| SIS90 | 90 | 20.2 | 9.0 |
NMR spectroscopy
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker Ascend Aeon WB 400 (Bruker BioSpin AG) spectrometer. The working frequencies were 400.21 MHz for 1H, 162.011 MHz for 31P and 105.86 MHz for 23Na. The samples were placed in standard 5 mm glass tubes that were sealed with a plastic stopper to avoid direct contact with air. Prior to each test, the samples were equilibrated at the specific temperature for 30 min. NMR spectra were obtained by Fourier transformation of free induction decay followed by a 90° pulse.NMR diffusometry
Pulsed gradient spin echo-nuclear magnetic resonance (PGSE-NMR) experiments were done on 1H and on 31P with a PGSE-NMR probe Diff50 (Bruker). The diffusional decays (DDs) were recorded using the stimulated echo (StE) and the spin-echo pulse trains. For single-component diffusion, the form of the DD can be described by eqn (1):49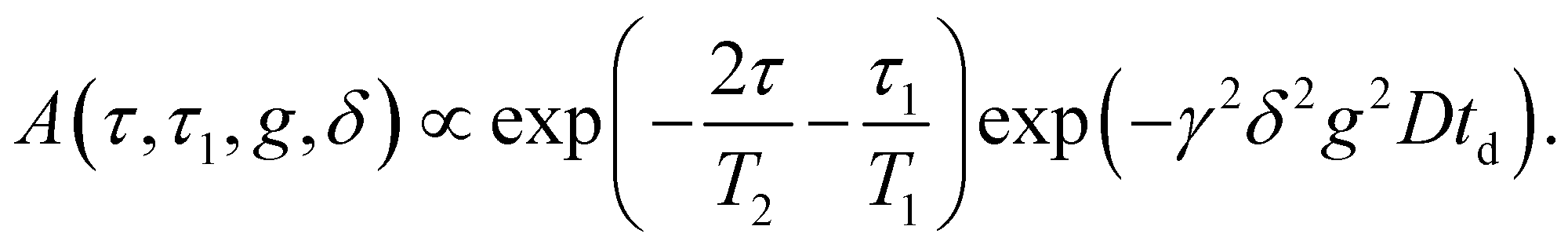 | (1) |
The 1H diffusivity data is analyzed by a Vogel–Fulcher–Tammann (VFT) equation (eqn (2)), with the fitting parameters tabulated in Table S5 (ESI†):
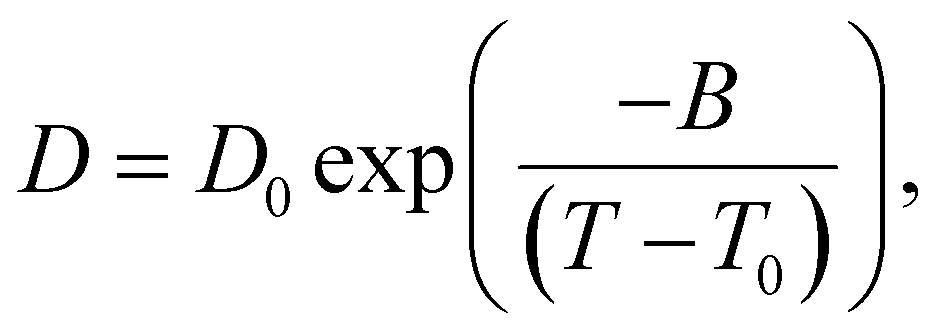 | (2) |
FT-IR spectroscopy
Fourier transform infrared (FT-IR) spectroscopy was carried out on a Bruker Tensor 27 FT-IR spectrometer using KBr pellets created using a manual hydraulic press (Specac, UK). The FT-IR spectra were recorded in the range from 400 to 4000 cm−1 for 64 scans and with a resolution of 4 cm−1. The spectra were deconvoluted using the Origin software.Thermal characterization
Thermogravimetric analysis (TGA) was performed using a PerkinElmer 8000 TGA apparatus under nitrogen gas atmosphere, with a heating rate of 10 °C min−1, in the temperature range from 30 to 600 °C, using ca. 2–4 mg of sample. Differential scanning calorimetry (DSC) was carried out using a PerkinElmer DSC 6000 apparatus. For each experiment, ca. 2–5 mg of sample was sealed in an aluminum pan and data were recorded from −140 °C to 0 °C at a scan rate of 10 °C min−1 or −80 °C to 100 °C at a scan rate of 5 °C min−1. Nitrogen gas was applied at a constant flow rate of 20 mL min−1 to preserve a dry environment and the lower temperatures were achieved by using liquid nitrogen. By using the Pyris software, the intersection of the baseline and the tangent was determined to obtain the onset of the decomposition temperature (Tdecomp) and the glass transition temperature (Tg), respectively.Electrochemical assessment
A Metrohm Autolab PGSTAT302N electrochemical workstation with an FRA32M module and the Nova 2.02 software was employed to assess the electrochemical stability windows (ESWs) and the ionic conductivities. 70 μL of sample was packed in a closed TSC 70 cell coupled to a temperature-controller Microcell HC (RHD Instruments, Germany). For the linear sweep voltammetry (LSV), performed at 20 °C and at a scanning rate of 1 mV s−1, a three-electrode configuration was used; a platinum (Pt) wire (diameter = 0.25 mm) or a glassy carbon (GC, diameter = 2 mm) as the working electrode (WE), a 70 μL Pt crucible as sample container as well as counter electrode (CE), and an Ag wire coated with AgCl (Ag/AgCl) as pseudo reference electrode (RE). Ferrocene (Fc/Fc+) was used as an internal reference and the EFc/Fc+ potential was converted to ENa/Na+ by +3.07 V.52 A cut-off current density of ±0.1 mA cm−2 was used to define the anodic and cathodic stabilities.Ionic conductivities were determined using electrochemical impedance spectroscopy (EIS) from 1 Hz to 1 MHz frequency with an AC voltage amplitude of 10 mVrms in the temperature range of −20 to 100 (±0.1) °C. A two-electrode configuration was used: Pt wire as WE and Pt crucible as RE (see above). For all of the above electrochemical characterization, both the Pt based electrodes were polished by using a Kemet diamond paste (average diameter = 0.25 μm) and a 100 μS cm−1 KCl standard solution from Metrohm was applied to determine the cell constant (Kcell = 18.5396 cm−1). The cell was thermally equilibrated for 10 min prior to each data recording. The ionic conductivity data were further analyzed using a VFT equation (eqn (3)):
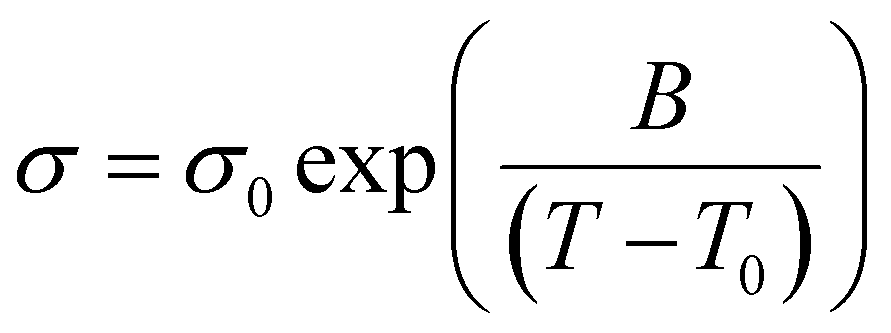 | (3) |
Results and discussion
We start by first evaluating the thermal and phase behavior, and thereafter we assess the electrochemical properties. Subsequently, FT-IR and NMR spectroscopies are employed to explain some of the macroscopic observations by a thorough study of the ion–ion and ion–solvent interactions and hence local structure. Finally, NMR diffusometry is used to gain insight into the dynamic properties of the electrolytes including the ion transport.Thermal properties
The NaDEEP salt, the TEOP solvent, and the SISXX electrolytes all exhibit decomposition temperatures >200 °C (Fig. 2a and Table S1, ESI†), which for the electrolytes increase as a function of the salt content. The NaDEEP salt and the SIS90 electrolyte both reveal two-step decomposition paths: the first step at 225 °C and 200 °C, and the second step at 283 °C and 289 °C, respectively.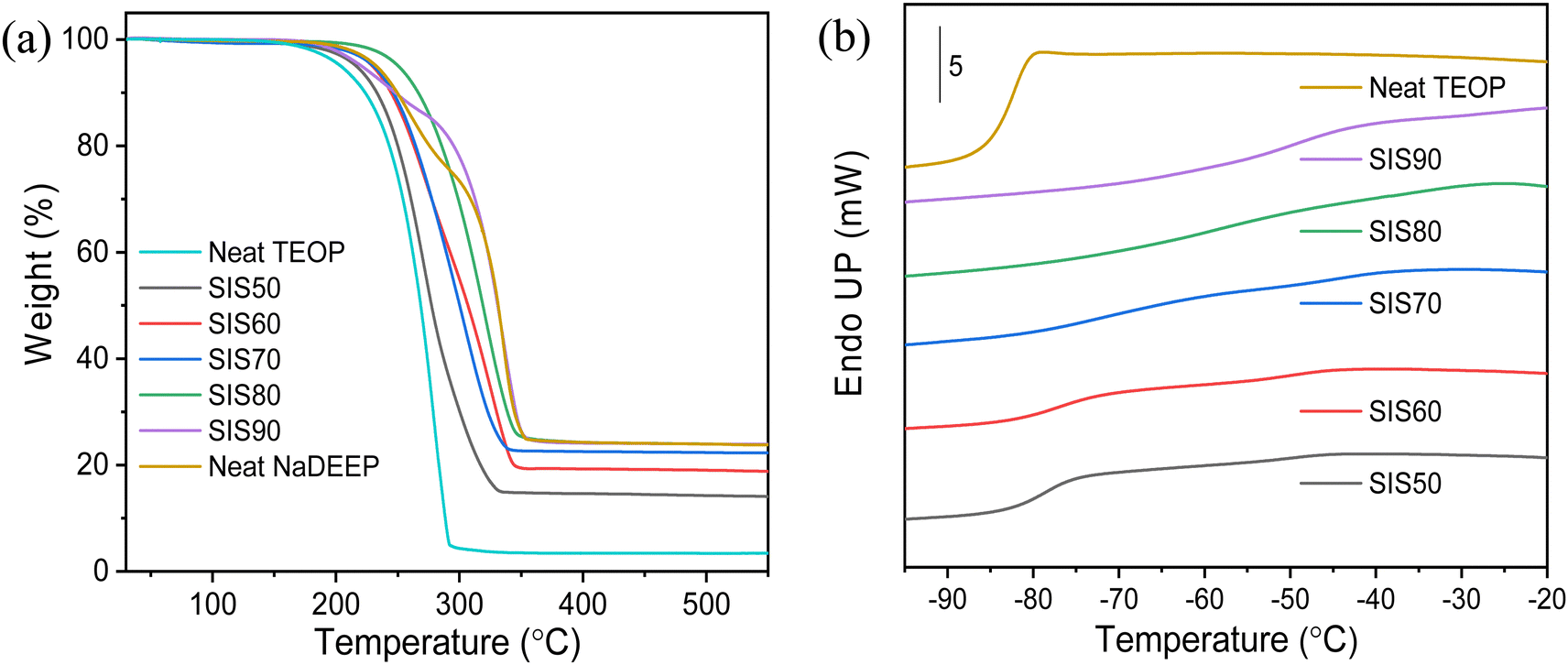 | ||
| Fig. 2 (a) TGA thermograms of the NaDEEP salt, the TEOP solvent and the SISXX electrolytes, and (b) DSC traces of the SISXX electrolytes and the TEOP solvent. | ||
The DSC traces of the TEOP solvent and the SIS50 electrolyte reveal glass transition temperatures of −85 °C and −83 °C, respectively. The difference between the rubbery state and the glassy state becomes less obvious with increased salt concentration (Fig. 2b), why it is difficult to determine the exact Tg for both SIS80 and SIS90. To exclude the effect of scan rate and determine more precisely Tg, different heating rates were applied to SIS80, but no clear glass transition was anyhow observed (Fig. S1a, ESI†). These electrolytes display a glass transition behavior over a broad temperature range (Fig. 2b and Fig. S1b, ESI†), which might be due to a cooperative segmental mobility of the structurally flexible oligoether chains of the DEEP anion, something that is well known for polymeric materials.53 While the ethylene oxide units enable low energy rotations, addition of NaDEEP to create SISXX electrolytes could be expected to result in dynamic cross-linking, but only negligible changes are observed in the Tg (Table S1, ESI†). Moreover, no other distinct phase transitions were detected <100 °C (Fig. S1b, ESI†).
Electrochemical properties
The ionic conductivities of the electrolytes display some unusual behaviour; the first heating and subsequent cooling cycle data do not match well (Fig. S2, ESI†) and there is no linear increase during the first heating cycle, which taken together suggest some structural changes, despite that no transitions were detected in the DSC data. Applying a heating–cooling-heating cycle to the SIS50 electrolyte render the second heating cycle data to match very well with the cooling cycle data (Fig. S3, ESI†) and we therefore report ionic conductivity data only from the latter.In contrast to much of the Na-conducting SIS electrolyte/HCE literature,31,33,54 the ionic conductivities here increase as a function of salt concentration, emphasizing the special solvation structure of oligoether based electrolytes.48 The most concentrated electrolyte, SIS90, displays the highest ionic conductivity over the whole temperature range, but while all the SISXX electrolytes show somewhat similar temperature dependencies, the increase as a function of temperature is much larger for the neat NaDEEP salt, to finally, at 100 °C, be on par with SIS90 (Fig. 3b).
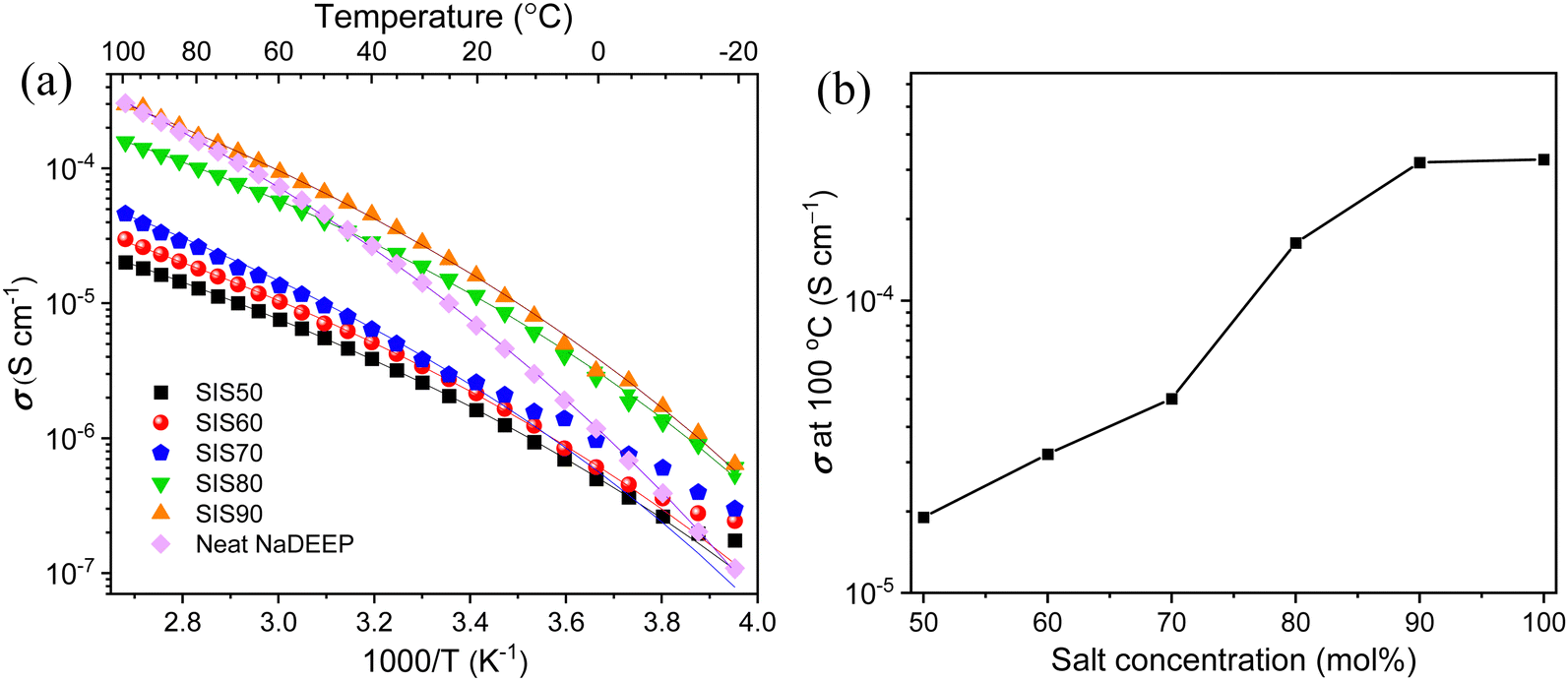 | ||
| Fig. 3 Ionic conductivity as a function of (a) temperature for the neat NaDEEP salt and the SISXX electrolytes, and (b) salt concentration at 100 °C. | ||
The VFT analysis shows that while there is no linear correlation between Eσ and the salt concentration, Eσ is higher for the SIS90 electrolyte and the neat NaDEEP salt, indicating that more energy is required, likely due to the stronger ion–ion interactions (Table S2, ESI†). Reassuringly, the T0 values are ca. 33 to 40 °C lower than the Tg values obtained from the DSC data and furthermore the T0/Tg ratios are ∼0.75 for the electrolytes, as is common for ionic liquid based electrolytes.55
Turning to the electrochemical stabilities the oxidation potentials are quantitatively, using the current density limit we have set, >5 V vs. Na/Na+ for all the electrolytes (Fig. 4a and Table S3, ESI†), and the highest oxidation potential is a remarkable 7.82 V vs. Na/Na+ achieved for SIS50, and thus, rather unexpectedly, the addition of TEOP solvent lead to an increase in the oxidation stability of the SISXX electrolytes. To further validate this, LSV of the TEOP solvent itself was performed and revealed an excellent oxidation potential of 7.51 V vs. Na/Na+, much higher than for the liquid NaDEEP salt. By the eye, however, we do note that all NaDEEP containing systems have a feature starting at ca. 5.1 V vs. Na/Na+, while it starts first >6 V vs. Na/Na+ for the TEOP solvent (Fig. 4a). While no apparent reduction peaks are observed for SIS80, SIS90 and neat NaDEEP salt, the “less concentrated” SISXX electrolytes exhibit reduction stabilities of <2 V vs. Na/Na+ (Fig. 4b and Table S3, ESI†) – and again, by the eye there are several small features present, making any conclusive statement on the reductive stability very problematic (Fig. 4). The oxidative stability is anyhow significantly higher than that reported for 5 M NaFSI in DME on a Pt electrode: 4.7 V vs. Na/Na+.31 To further validate the electrochemical stabilities of these systems, LSV using GC as WE was performed – resulting in a similar trend; TEOP displays a much higher oxidative stability than NaDEEP, while the reduction current density did not reach the set 0.1 mA cm−2 limit even by scanning down to −6 V vs. Na/Na+ (Fig. S4, ESI†). For comparison, the corresponding anodic and cathodic stabilities using GC and Pt electrodes at a current density limit of ±0.05 mA cm−2 are presented in Table S4 (ESI†). Overall, the “less concentrated” electrolytes exhibit higher electrochemical stabilities than the HCEs.
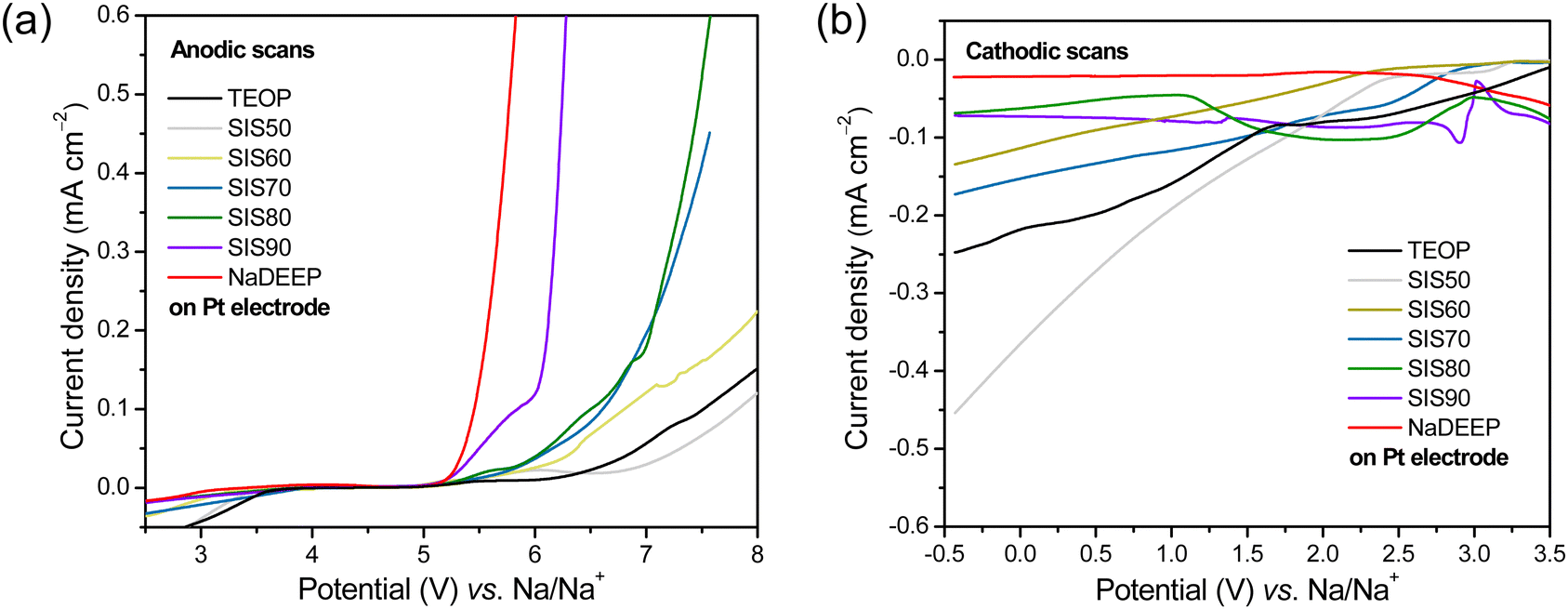 | ||
| Fig. 4 (a) Anodic and (b) cathodic LSV scans of the TEOP solvent, the SISXX electrolytes, and the NaDEEP salt using Pt as the WE. | ||
Local structure
In the FT-IR spectra the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration mode changes upon addition of salt; from ∼1280 cm−1 for the neat TEOP solvent to ∼1247 cm−1 confirming an increasing content of NaDEEP salt as well as Na+−PO interactions36 (Fig. 5a), semi-quantified by deconvolution (Fig. 5b and Fig. S5, ESI†) to increase almost linearly as a function of the salt concentration (Fig. 5c). This suggests an increase in aggregate formation with increasing salt concentration, as previously indicated by spectroscopy and molecular dynamics simulations for HCEs of sodium triflate dissolved in monoglyme and diglyme.48 Similarly, NaFSI in dimethoxyethane electrolytes show the ratio of free solvent molecules to anions to significantly decrease with increased salt concentration, and only 10.5% and 0.5% of free DME and free FSI, respectively, remained in the HCE.56
O stretching vibration mode changes upon addition of salt; from ∼1280 cm−1 for the neat TEOP solvent to ∼1247 cm−1 confirming an increasing content of NaDEEP salt as well as Na+−PO interactions36 (Fig. 5a), semi-quantified by deconvolution (Fig. 5b and Fig. S5, ESI†) to increase almost linearly as a function of the salt concentration (Fig. 5c). This suggests an increase in aggregate formation with increasing salt concentration, as previously indicated by spectroscopy and molecular dynamics simulations for HCEs of sodium triflate dissolved in monoglyme and diglyme.48 Similarly, NaFSI in dimethoxyethane electrolytes show the ratio of free solvent molecules to anions to significantly decrease with increased salt concentration, and only 10.5% and 0.5% of free DME and free FSI, respectively, remained in the HCE.56
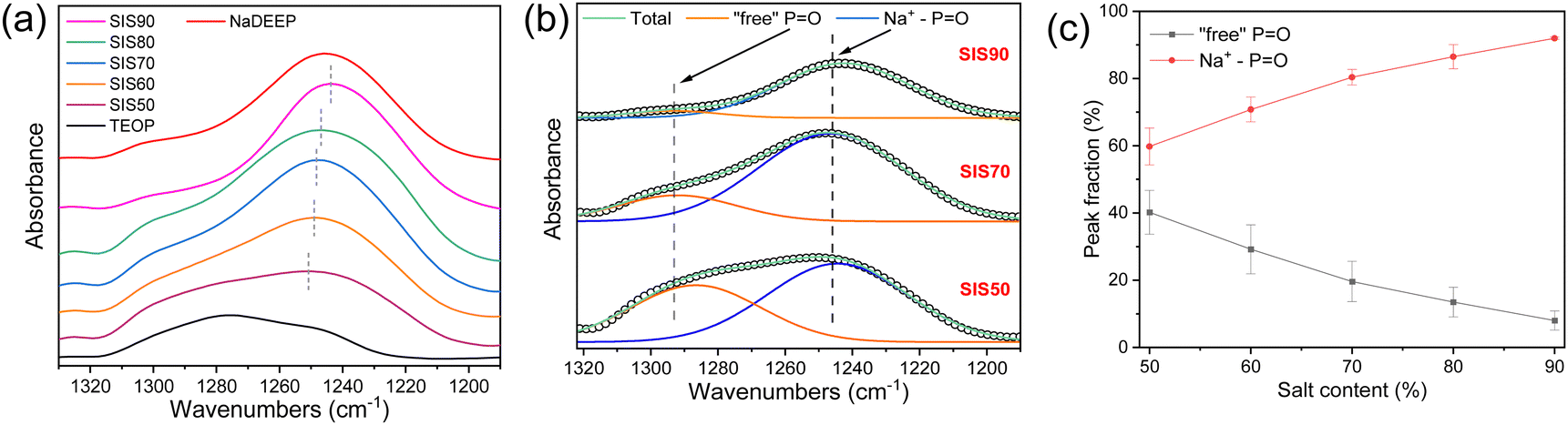 | ||
| Fig. 5 (a) FT-IR spectra of the NaDEEP salt, the TEOP solvent, and the SISXX electrolytes in the PO stretching vibrational mode region, (b) deconvolution of this region for SIS50, SIS70 and SIS90, and (c) the resulting peak fractions of “free” PO and PO interacting with Na+ (Na+−PO) as a function of salt content. | ||
Moving to the NMR spectroscopy, the 31P NMR spectra of the electrolytes show two resonance lines with different line broadening centered at ∼2 and ∼4 ppm, while single resonance lines are found at 2.4 ppm and 3.9 ppm, for the TEOP solvent and NaDEEP salt, respectively (Fig. 6a). Indeed, the electrolytes contain two phases: a “solvent-rich” phase and a “salt-rich” phase, as discussed in more detail in the next section. For the SIS90 electrolyte, the two resonance lines merge into a single broad line due to the fast and/or intermediate regimes of exchange between the 31P nuclei of the salt and the solvent. The 31P resonance line corresponds to the NaDEEP in the “less concentrated” electrolytes gets narrower with increasing temperature and shifts slightly towards higher ppms, while the line width and chemical shift of the TEOP solvent both remain unchanged (Fig. 6b and Fig. S6, ESI†).
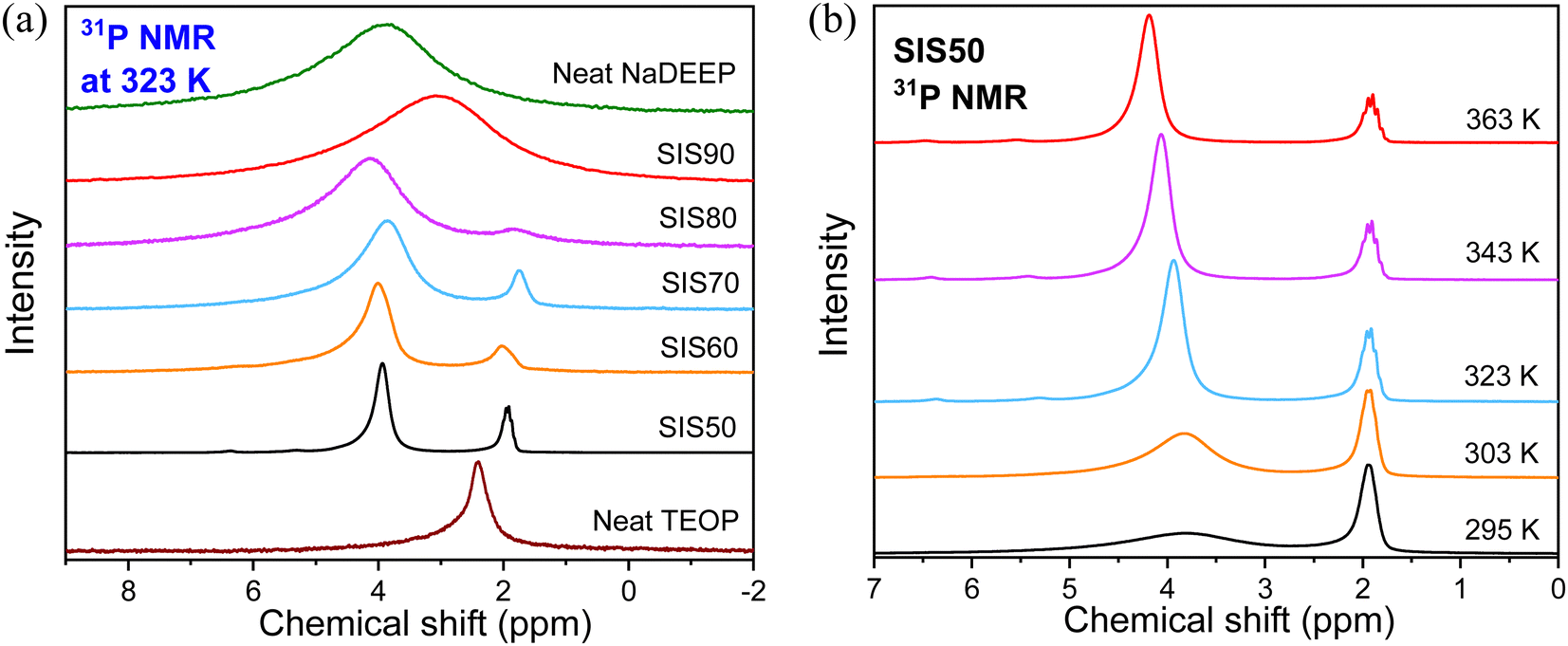 | ||
| Fig. 6 (a) 31P NMR spectra of the NaDEEP salt, the TEOP solvent, and the SISXX electrolytes at 323 K, and (b) 31P NMR spectra of SIS50 at various temperatures. | ||
The significant changes in line broadening and chemical shift of the 31P resonance line of the NaDEEP salt, and in particular for the HCEs, reveal a higher anion mobility at higher temperatures and agrees well with the ionic conductivity data (Fig. 7a and b). The shift in 31P resonance lines towards lower magnetic field with increasing salt content suggests the existence of stronger electron-withdrawing effect induced by the Na+–PO interactions, similar to the CO–Li+ interactions57 – and is in agreement with the FTIR spectroscopic analysis as discussed above. At higher temperatures (>313 K), the chemical shifts remain unchanged revealing a faster anion exchange around Na+ and weaker ionic interactions between the phosphate anion and Na+. Unlike the 31P spectral lines, the line broadening of the 23Na NMR resonance lines increases together with a change in the chemical shift as a function of temperature (Fig. 8a, b and Fig. S7, ESI†). Altogether, these changes imply variations in the microstructures and dynamics of the electrolytes with increasing salt concentration and temperature.
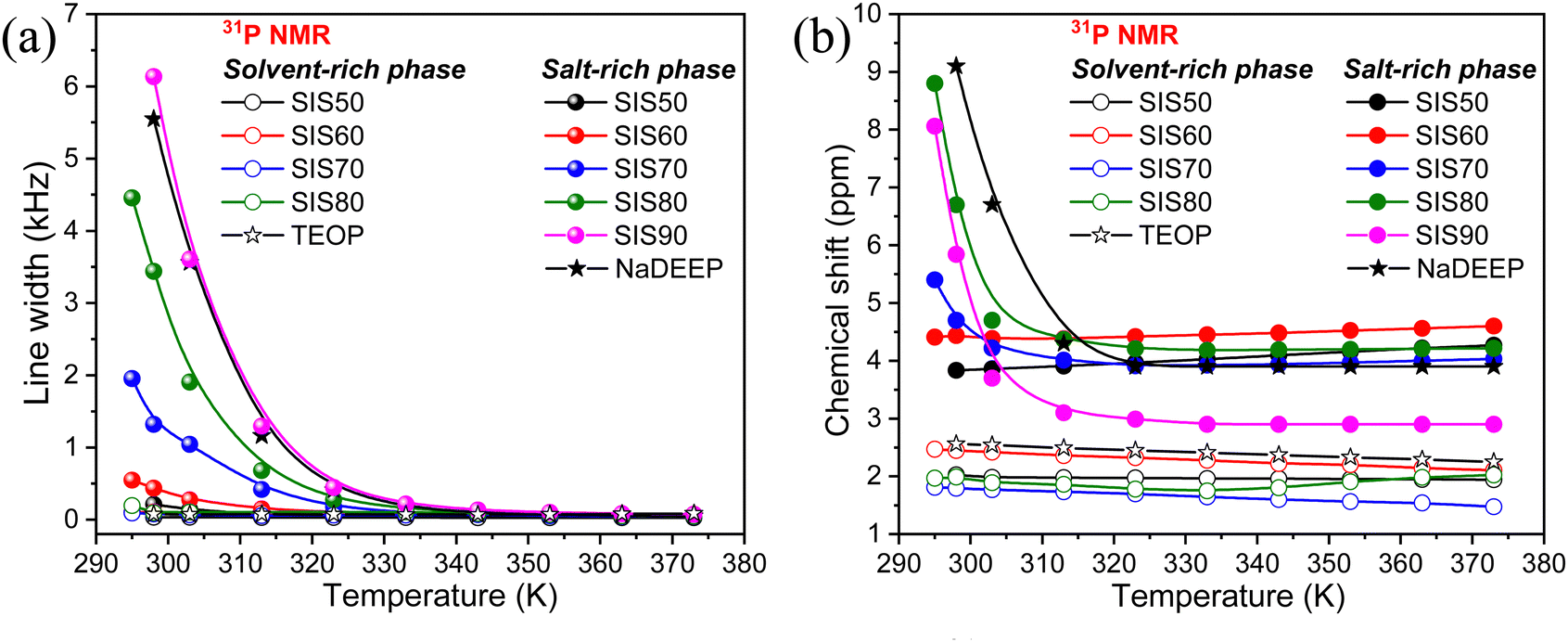 | ||
| Fig. 7 (a) Line width and (b) chemical shift of 31P NMR spectra of the NaDEEP salt, the TEOP solvent, and the SISXX electrolytes as a function of temperature. | ||
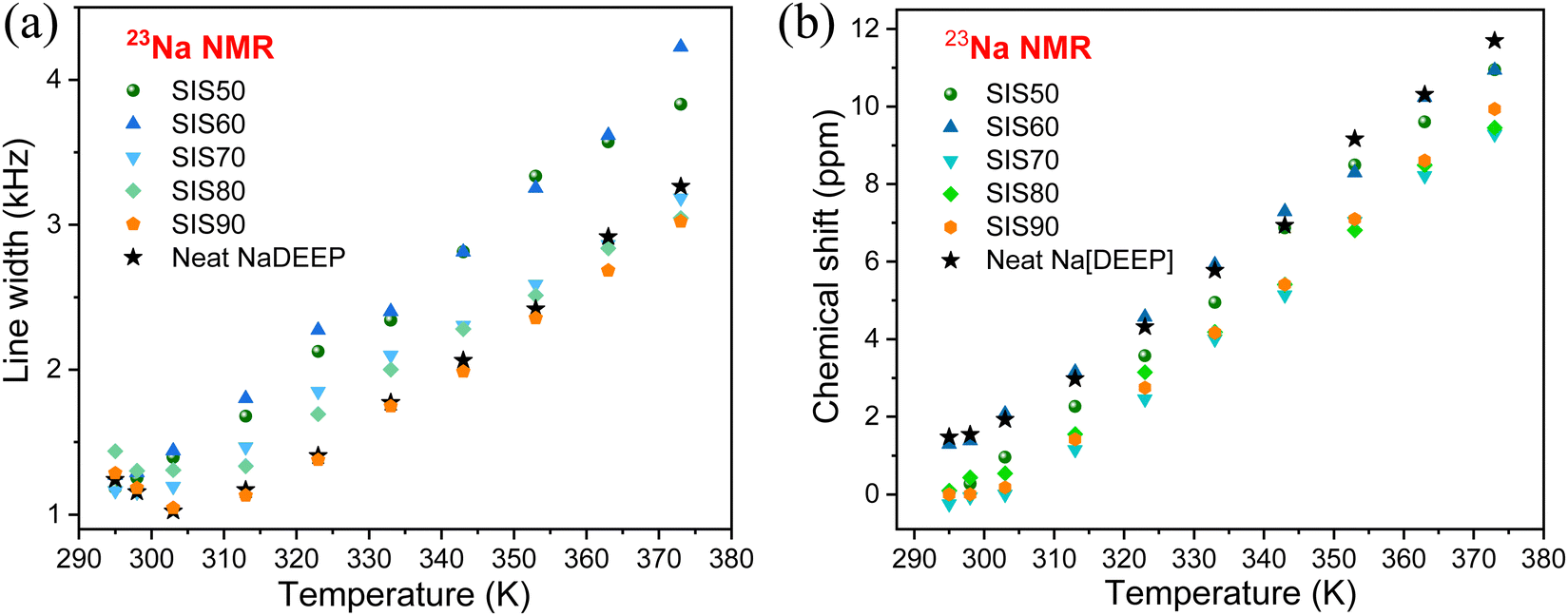 | ||
| Fig. 8 (a) Line width and (b) chemical shift of 23Na NMR spectra as a function of temperature. | ||
In contrast to Li-based electrolytes, the 23Na resonance line shape is strongly influenced by quadrupolar interaction because 23Na has a larger quadrupole moment than 7Li58 and this leads to a more efficient relaxation contribution as the dynamic time scale decreases with increasing temperature. The changes in both 31P and 23Na resonance lines further confirm that the Na+−PO interactions are influenced by the Na-salt content, agreeing well with the FT-IR spectroscopic data. This also agrees well with the previous findings for NaPF6/G2 electrolytes,20 where a significant reduction of “free” solvent species with increased salt concentration from 0.05 M to 3 M is observed by Raman and FT-IR spectroscopy and also the 23Na NMR resonance lines shift to a lower magnetic field, which is explained by increased Na+–PF6− contact, resulting in a lower electron density around Na+. The shift of the resonance lines to a lower magnetic field for Na+ and/or Li+ in solution is expected as the solvation shell has strong electron donating ability.59,60 In our case, the changes in chemical shift of the 23Na NMR resonance lines are also concentration-dependent and the phosphate anion is able to alter the solvation shell structure to promote the ionic interactions with the Na+ cation.61,62
Dynamics and ion transport
In 1H and 31P PGSE-NMR diffusometry two diffusion components are observed; the fast-diffusion and slow-diffusion components do not exist independently, why both the salt-rich and the solvent-rich phases contribute to the 1H NMR signals of all resonance lines. Furthermore, these have no single-component decay but a decomposition using the typical DD for SIS50 showed that the total DD is the sum of two components (Fig. S8, ESI†). The fast-decaying DD component is observed only for the less concentrated electrolytes: SIS50, SIS60 and SIS70. The 31P NMR DD analysis reveal that the fast-decaying component belongs to the solvent-rich while the slow-decaying component corresponds to the salt-rich phase. The manifestation of a single diffusion coefficient for a thermodynamic phase demonstrates that the system forms a homogeneous phase with strong interactions between the diffusing components and all the components are diffusing in a concerted manner.For the SISXX electrolytes, this is true for the single-phase in the electrolytes with higher salt concentrations, >SIS70, and for both the phases of the less concentrated electrolytes <SIS70. Therefore, there is no noticeable difference in the diffusivities of the salt and the solvent inside the salt-rich and the solvent-rich phases at concentrations >SIS70, while at lower salt concentrations, <SIS70, there is a prominent difference, suggesting that there is no distinct ion and/or solvent exchange between the two phases (Fig. 9). Since the diffusion-time of the 1H NMR diffusometry is in the range from 4 to 100 ms, the phases might be stable for at least hundreds of milliseconds. The SIS70 is an exception: above 353 K two diffusion coefficients are merged, which might be due to the intensification of exchange between the phases as a result of thermal motion at elevated temperatures (Fig. 9a). The 1H NMR diffusometry data is further complemented by 31P NMR diffusometry and the data match very well (Fig. 9 and Fig. S9, ESI†). The 31P NMR diffusometry data are presented for TEOP and SIS50 over the whole temperature range, while only at elevated temperatures for the concentrated systems (Fig. 9b). It was not possible to measure diffusivity due to the fast-decaying diffusion component caused by the lower sensitivity of 31P diffusometry − a 2.5 times lower gyromagnetic ratio of 31P than 1H. It was not possible to measure 23Na diffusion coefficients because of its too short T2 relaxation.
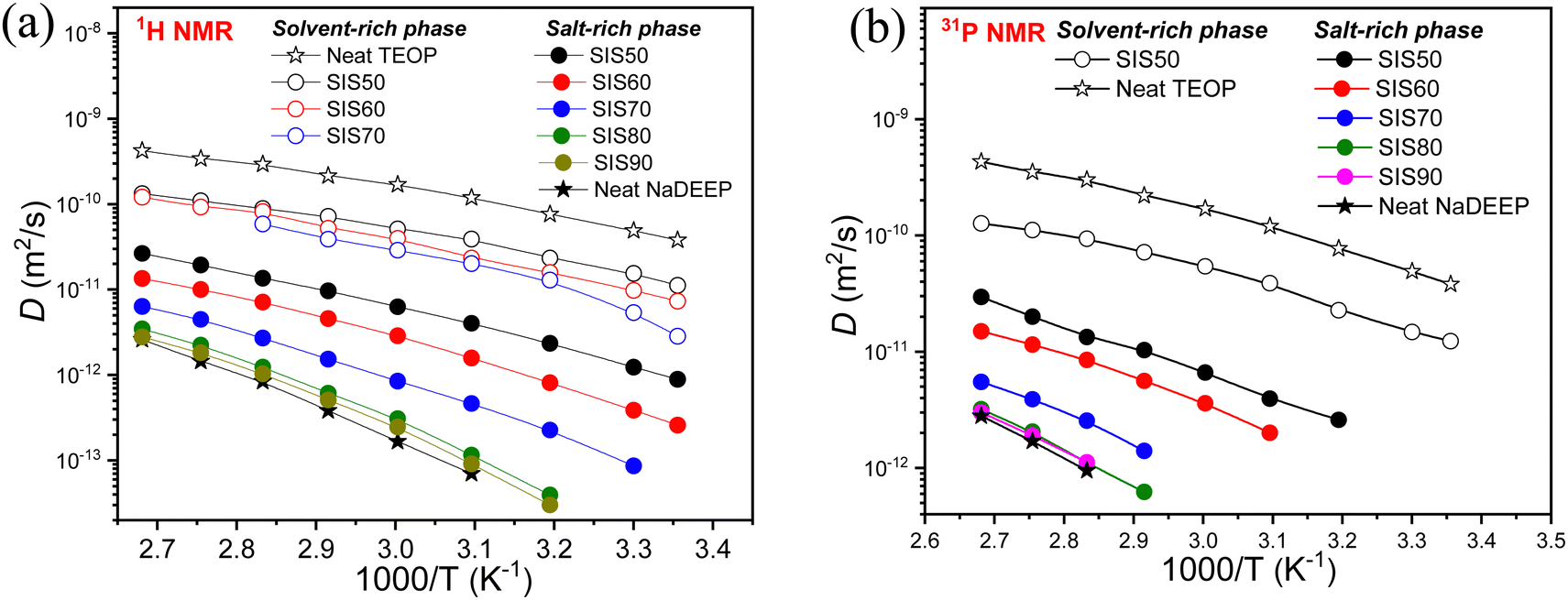 | ||
| Fig. 9 Diffusion coefficients as a function of temperature for the NaDEEP salt, the TEOP solvent, and the SISXX electrolytes obtained by (a) 1H NMR and (b) 31P NMR spectroscopy. | ||
In comparison, the diffusivity of the TEOP solvent is significantly faster than of the phosphate anion in the NaDEEP salt, implying that the diffusivity is controlled by electrostatic interactions rather than molecular size (as the solvent has one more oligoether chain than the salt). As expected, the diffusivity of all components increases with increasing temperature and decrease with salt concentration. Among the electrolytes, SIS50 displays the highest diffusion coefficients followed by SIS60 and SIS70, while SIS80 and SIS90 are more or less comparable with the NaDEEP salt. For the “solvent-rich” phase increased salt content led to an increase in T0, while the apparent activation energy remained unchanged up to SIS70 followed by an abrupt increase. For the “salt-rich” phase, T0 remained in the range from 175 to 229 K and the activation energy in the range 8–10.6 kJ mol−1 (Table S5, ESI†). Again, these data suggest a high degree of aggregation induced by the Na+ cation, as also confirmed by the NMR and FT-IR spectroscopies.
Altogether, the opposite trend in ionic conductivity vs. ion diffusivity indicates extensive ionic interactions in the HCEs fostering structures similar to polymeric networks with shorter residence times of the Na+ cations at their coordination sites – as recently outlined by different simulation approaches.63,64 Indeed, the diffusometry data average over less mobile large neutral aggregates and fast diffusing small charged species, and therefore the higher ionic conductivities of the HCEs are not captured, as the Na+ mobility is (likely) facilitated through a sequential series of movements between nearby sites. All of this has large similarities to inorganic solid-state ion conductors65 and polymer-in-salt electrolytes.66
Concluding remarks
New structurally flexible “solvent-in-salt” sodium battery electrolytes comprising the ambient temperature liquid NaDEEP salt and the structural analogous TEOP solvent have been created. The solvation structure and ion transport are systematically investigated and an unusual relationship between ionic conductivities and ion diffusivities has been established. That both the ionic conductivities and the Na+–PO interactions increase with increasing salt concentration suggest to us that some kind of polymeric highly viscous networks may be formed within the electrolytes. In addition, although the diffusion in HCEs are underestimated by NMR diffusometry, due to electrostatic interactions, the rapid exchange between the TEOP solvent and the DEEP anion in the aggregates, is likely what result in improved ionic conductivities. Overall, this work provides fundamental understanding of new fluorine-free sodium-ion conducting electrolytes in general, and HCEs in particular, and this for systems with high oxidation stabilities.
Author contributions
Y. X. performed all the experiments except organic synthesis and NMR diffusivity, employed data visualization and analysis, wrote the manuscript, and revised the manuscript under supervisions. S. B. synthesized the salt and the solvent. A. F. performed PGSE-NMR experiments and analyzed data. P. J. and F. U. S. conceived the idea and supervised the work. All authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Swedish Research Council for Sustainable Development (Grant number: 2020-00969) is gratefully acknowledged.References
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Research Development on Sodium-ion Batteries, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Sodium-ion Batteries: Present and Future, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Sodium-Ion Batteries, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, From Li-Ion Batteries toward Na-Ion Chemistries: Challenges and Opportunities, Adv. Energy Mater., 2020, 10, 2001310 CrossRef CAS.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Recent Progress in Electrode Materials for Sodium-Ion Batteries, Adv. Energy Mater., 2016, 6, 1600943 CrossRef.
- Y. Fang, X.-Y. Yu and X. W. Lou, Nanostructured Electrode Materials for Advanced Sodium-Ion Batteries, Matter, 2019, 1, 90–114 CrossRef.
- H. Wang, F. Liu, R. Yu and J. Wu, Unraveling the Reaction Mechanisms of Electrode Materials for Sodium-Ion and Potassium-Ion Batteries by In Situ Transmission Electron Microscopy, Interdiscip. Mater., 2022, 1, 196–212 CrossRef.
- Z.-X. Huang, X.-L. Zhang, X.-X. Zhao, Y.-L. Heng, T. Wang, H. Geng and X.-L. Wu, Hollow Na0.62K0.05Mn0.7Ni0.2Co0.1O2 polyhedra with exposed stable {001} facets and K riveting for sodium-ion batteries, Sci. China: Mater., 2023, 66, 79–87 CrossRef CAS.
- G. G. Eshetu, G. A. Elia, M. Armand, M. Forsyth, S. Komaba, T. Rojo and S. Passerini, Electrolytes and Interphases in Sodium-based Rechargeable Batteries: Recent Advances and Perspectives, Adv. Energy Mater., 2020, 10, 2000093 CrossRef CAS.
- Y.-T. Liu, H.-J. Liang, M. Du, J.-L. Yang, Z.-Y. Gu, X.-T. Wang, Y.-Z. Tang, J.-Z. Guo and X.-L. Wu, Ester-Based Anti-Freezing Electrolyte Achieving Ultra-Low Temperature Cycling for Sodium-Ion Batteries, J. Mater. Sci. Technol., 2024, 182, 111–118 CrossRef.
- E. de la Llave, V. Borgel, K.-J. Park, J.-Y. Hwang, Y.-K. Sun, P. Hartmann, F.-F. Chesneau and D. Aurbach, Comparison between Na-Ion and Li-Ion Cells: Understanding the Critical Role of the Cathodes Stability and the Anodes Pretreatment on the Cells Behavior, ACS Appl. Mater. Interfaces, 2016, 8, 1867–1875 CrossRef CAS PubMed.
- A. Bhide, J. Hofmann, A. Katharina Dürr, J. Janek and P. Adelhelm, Electrochemical Stability of Non-aqueous Electrolytes for Sodium-Ion Batteries and Their Compatibility with Na0.7CoO2, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- L. Xia, S. Lee, Y. Jiang, Y. Xia, G. Z. Chen and Z. Liu, Fluorinated Electrolytes for Li-Ion Batteries: The Lithium Difluoro(oxalato)borate Additive for Stabilizing the Solid Electrolyte Interphase, ACS Omega, 2017, 2, 8741–8750 CrossRef CAS PubMed.
- R. Jung, M. Metzger, D. Haering, S. Solchenbach, C. Marino, N. Tsiouvaras, C. Stinner and H. A. Gasteiger, Consumption of Fluoroethylene Carbonate (FEC) on Si-C Composite Electrodes for Li-Ion Batteries, J. Electrochem. Soc., 2016, 163, A1705 CrossRef CAS.
- K. Kim, I. Park, S.-Y. Ha, Y. Kim, M.-H. Woo, M.-H. Jeong, W. C. Shin, M. Ue, S. Y. Hong and N.-S. Choi, Understanding the Thermal Instability of Fluoroethylene Carbonate in LiPF6-Based Electrolytes for Lithium Ion Batteries, Electrochim. Acta, 2017, 225, 358–368 CrossRef CAS.
- N. P. Lebedeva and L. Boon-Brett, Considerations on the Chemical Toxicity of Contemporary Li-Ion Battery Electrolytes and Their Components, J. Electrochem. Soc., 2016, 163, A821 CrossRef CAS.
- X. Xia and J. R. Dahn, Study of the Reactivity of Na/Hard Carbon with Different Solvents and Electrolytes, J. Electrochem. Soc., 2012, 159, A515 CrossRef CAS.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, On the Comparative Stability of Li and Na Metal Anode Interfaces in Conventional Alkyl Carbonate Electrolytes, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- J. He, A. Bhargav, W. Shin and A. Manthiram, Stable Dendrite-free Sodium–sulfur Batteries Enabled by a Localized High-concentration Electrolyte, J. Am. Chem. Soc., 2021, 143, 20241–20248 CrossRef CAS PubMed.
- Z.-L. Xu, G. Yoon, K.-Y. Park, H. Park, O. Tamwattana, S. Joo Kim, W. M. Seong and K. Kang, Tailoring Sodium Intercalation in Graphite for High Energy and Power Sodium Ion Batteries, Nat. Commun., 2019, 10, 2598 CrossRef PubMed.
- H.-J. Liang, Z.-Y. Gu, X.-X. Zhao, J.-Z. Guo, J.-L. Yang, W.-H. Li, B. Li, Z.-M. Liu, W.-L. Li and X.-L. Wu, Ether-Based Electrolyte Chemistry Towards High-Voltage and Long-Life Na-Ion Full Batteries, Angew. Chem., Int. Ed., 2021, 60, 26837–26846 CrossRef CAS PubMed.
- Z. Zhang, P.-N. Roy, H. Li, M. Avdeev and L. F. Nazar, Coupled Cation–Anion Dynamics Enhances Cation Mobility in Room-Temperature Superionic Solid-State Electrolytes, J. Am. Chem. Soc., 2019, 141, 19360–19372 CrossRef CAS PubMed.
- Y. Yamada and A. Yamada, Review—Superconcentrated Electrolytes for Lithium Batteries, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, Unusual Stability of Acetonitrile-Based Superconcentrated Electrolytes for Fast-Charging Lithium-Ion Batteries, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS PubMed.
- S.-K. Jeong, M. Inaba, Y. Iriyama, T. Abe and Z. Ogumi, Interfacial Reactions between Graphite Electrodes and Propylene Carbonate-Based Solutions: Electrolyte-Concentration Dependence of Electrochemical Lithium Intercalation Reaction, J. Power Sources, 2008, 175, 540–546 CrossRef CAS.
- W. R. McKinnon and J. R. Dahn, How to Reduce the Cointercalation of Propylene Carbonate in LixZrS2 and Other Layered Compounds, J. Electrochem. Soc., 1985, 132, 364–366 CrossRef CAS.
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, New Concepts in Electrolytes, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS PubMed.
- C. A. Angell, C. Liu and E. Sanchez, Rubbery Solid Electrolytes with Dominant Cationic Transport and High Ambient Conductivity, Nature, 1993, 362, 137–139 CrossRef CAS.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, A New Class of Solvent-in-Salt Electrolyte for High-Energy Rechargeable Metallic Lithium Batteries, Nat. Commun., 2013, 4, 1481 CrossRef PubMed.
- M. Martinez-Ibañez, N. Boaretto, A. Santiago, L. Meabe, X. Wang, O. Zugazua, I. Raposo, M. Forsyth, M. Armand and H. Zhang, Highly-Concentrated Bis(fluorosulfonyl)imide-Based Ternary Gel Polymer Electrolytes for High-Voltage Lithium Metal Batteries, J. Power Sources, 2023, 557, 232554 CrossRef.
- J. Lee, Y. Lee, J. Lee, S.-M. Lee, J.-H. Choi, H. Kim, M.-S. Kwon, K. Kang, K. T. Lee and N.-S. Choi, Ultraconcentrated Sodium Bis(fluorosulfonyl)imide-Based Electrolytes for High-Performance Sodium Metal Batteries, ACS Appl. Mater. Interfaces, 2017, 9, 3723–3732 CrossRef CAS PubMed.
- R. Cao, K. Mishra, X. Li, J. Qian, M. H. Engelhard, M. E. Bowden, K. S. Han, K. T. Mueller, W. A. Henderson and J.-G. Zhang, Enabling room temperature sodium metal batteries, Nano Energy, 2016, 30, 825–830 CrossRef CAS.
- S. Terada, H. Susa, S. Tsuzuki, T. Mandai, K. Ueno, Y. Umebayashi, K. Dokko and M. Watanabe, Dissociation and Diffusion of Glyme-Sodium Bis(trifluoromethanesulfonyl)amide Complexes in Hydrofluoroether-Based Electrolytes for Sodium Batteries, J. Phys. Chem. C, 2016, 120, 23339–23350 CrossRef CAS.
- C. Geng, D. Buchholz, G.-T. Kim, D. V. Carvalho, H. Zhang, L. G. Chagas and S. Passerini, Influence of Salt Concentration on the Properties of Sodium-Based Electrolytes, Small Methods, 2019, 3, 1800208 CrossRef CAS.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, High Rate and Stable Cycling of Lithium Metal Anode, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, Oxidative-Stability Enhancement and Charge Transport Mechanism in Glyme–Lithium Salt Equimolar Complexes, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Concentrated Electrolytes: Decrypting Electrolyte Properties and Reassessing Al Corrosion Mechanisms, Energy Environ. Sci., 2014, 7, 416–426 RSC.
- Y. Li, W. Lv, H. Huang, W. Yan, X. Li, P. Ning, H. Cao and Z. Sun, Recycling of Spent Lithium-ion Batteries in View of Green Chemistry, Green Chem., 2021, 23, 6139–6171 RSC.
- S. Dühnen, J. Betz, M. Kolek, R. Schmuch, M. Winter and T. Placke, Toward Green Battery Cells: Perspective on Materials and Technologies, Small Methods, 2020, 4, 2000039 CrossRef.
- J. Scheers, D.-H. Lim, J.-K. Kim, E. Paillard, W. A. Henderson, P. Johansson, J.-H. Ahn and P. Jacobsson, All Fluorine-free Lithium battery Electrolytes, J. Power Sources, 2014, 251, 451–458 CrossRef CAS.
- E. Jónsson, M. Armand and P. Johansson, Novel Pseudo-delocalized Anions for Lithium Battery Electrolytes, Phys. Chem. Chem. Phys., 2012, 14, 6021–6025 RSC.
- L. O. S. Colbin, R. Mogensen, A. Buckel, Y.-L. Wang, A. J. Naylor, J. Kullgren and R. Younesi, A Halogen-Free and Flame-Retardant Sodium Electrolyte Compatible with Hard Carbon Anodes, Adv. Mater. Interfaces, 2021, 8, 2101135 CrossRef CAS.
- R. Mogensen, A. Buckel, S. Colbin and R. Younesi, A Wide-Temperature-range, Low-cost, Fluorine-free Battery Electrolyte Based On Sodium Bis(Oxalate)Borate, Chem. Mater., 2021, 33, 1130–1139 CrossRef CAS.
- S. Bhowmick, M. Ahmed, A. Filippov, L. C. Loaiza, F. U. Shah and P. Johansson, Ambient temperature liquid salt electrolytes, Chem. Commun., 2023, 59, 2620–2623 RSC.
- C. Wang, L. Wang, F. Li, F. Cheng and J. Chen, Bulk Bismuth as a High-Capacity and Ultralong Cycle-Life Anode for Sodium-Ion Batteries by Coupling with Glyme-Based Electrolytes, Adv. Mater., 2017, 29, 1702212 CrossRef PubMed.
- K. Westman, R. Dugas, P. Jankowski, W. Wieczorek, G. Gachot, M. Morcrette, E. Irisarri, A. Ponrouch, M. R. Palacín, J. M. Tarascon and P. Johansson, Diglyme Based Electrolytes for Sodium-Ion Batteries, ACS Appl. Energy Mater., 2018, 1, 2671–2680 CrossRef CAS.
- B. Jache, J. O. Binder, T. Abe and P. Adelhelm, A Comparative Study on the Impact of Different Glymes and Their Derivatives as Electrolyte Solvents for Graphite Co-intercalation Electrodes in Lithium-ion and Sodium-ion Batteries, Phys. Chem. Chem. Phys., 2016, 18, 14299–14316 RSC.
- S. R. Galle Kankanamge, K. Li, K. D. Fulfer, P. Du, R. Jorn, R. Kumar and D. G. Kuroda, Mechanism behind the Unusually High Conductivities of High Concentrated Sodium Ion Glyme-Based Electrolytes, J. Phys. Chem. C, 2018, 122, 25237–25246 CrossRef CAS.
- E. O. Stejskal and J. E. Tanner, Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- J. Hallett, The Temperature Dependence of the Viscosity of Supercooled Water, Proc. Phys. Soc., 1963, 82, 1046 CrossRef CAS.
- G. S. Fulcher, Analysis of Recent Measurements of the Viscosity of Glasses, J. Am. Ceram. Soc., 1992, 75, 1043–1055 CrossRef.
- R. Wibowo, L. Aldous, S. E. Ward Jones and R. G. Compton, The Group I Alkali Metals in Ionic Liquids: Electrodeposition and Determination of Their Kinetic and Thermodynamic Properties, ECS Trans., 2019, 33, 523–535 CrossRef.
- E. Vidal Russell and N. E. Israeloff, Direct Observation of Molecular Cooperativity near the Glass Transition, Nature, 2000, 408, 695–698 CrossRef PubMed.
- S. Terada, T. Mandai, R. Nozawa, K. Yoshida, K. Ueno, S. Tsuzuki, K. Dokko and M. Watanabe, Physicochemical Properties of Pentaglyme–Sodium Bis(trifluoromethanesulfonyl)amide Solvate Ionic Liquid, Phys. Chem. Chem. Phys., 2014, 16, 11737–11746 RSC.
- M. Galiński, A. Lewandowski and I. Stępniak, Ionic Liquids as Electrolytes, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef.
- M. Okoshi, C.-P. Chou and H. Nakai, Theoretical Analysis of Carrier Ion Diffusion in Superconcentrated Electrolyte Solutions for Sodium-Ion Batteries, J. Phys. Chem. B, 2018, 122, 2600–2609 CrossRef CAS PubMed.
- K. Kimura, J. Motomatsu and Y. Tominaga, Correlation between Solvation Structure and Ion-Conductive Behavior of Concentrated Poly(ethylene carbonate)-Based Electrolytes, J. Phys. Chem. C, 2016, 120, 12385–12391 CrossRef CAS.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, John Wiley & Sons, 2013 Search PubMed.
- E. G. Bloor and R. G. Kidd, Solvation of Sodium Ions Studied by 23Na Nuclear Magnetic Resonance, Can. J. Chem., 1968, 46, 3425–3430 CrossRef CAS.
- T. Umecky, K. Suga, E. Masaki, T. Takamuku, T. Makino and M. Kanakubo, Solvation Structure and Dynamics of Li+ in Lewis-Basic Ionic Liquid of 1-octyl-4-aza-1-azoniabicyclo[2.2.2]octane Bis(trifluoromethanesulfonyl)amide, J. Mol. Liq., 2015, 209, 557–562 CrossRef CAS.
- Y. M. Cahen, P. R. Handy, E. T. Roach and A. I. Popov, Spectroscopic Studies of Ionic Solvation. XVI. Lithium-7 and Chlorine-35 Nuclear Magnetic Resonance Studies in Various Solvents, J. Phys. Chem., 1975, 79, 80–85 CrossRef CAS.
- H. A. Berman and T. R. Stengle, Contact Ion Pairing of the Perchlorate Ion. Chlorine-35 Nuclear Magnetic Resonance Study. I. Solutions in Pure Solvents, J. Phys. Chem., 1975, 79, 1001–1005 CrossRef CAS.
- G. Åvall and P. Johansson, A Novel Approach to Ligand-Exchange Rates Applied to Lithium-Ion Battery and Sodium-Ion Battery Electrolytes, J. Chem. Phys., 2020, 152, 234104 CrossRef PubMed.
- F. Årén, R. Andersson, A. A. Franco and P. Johansson, Global and Local Structure of Lithium Battery Electrolytes: Origin and Onset of Highly Concentrated Electrolyte Behavior, J. Electrochem. Soc., 2023, 170, 060506 CrossRef.
- Z. Zhang, Y. Shao, B. Lotsch, Y.-S. Hu, H. Li, J. Janek, L. F. Nazar, C.-W. Nan, J. Maier, M. Armand and L. Chen, New Horizons for Inorganic Solid State Ion Conductors, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- M. A. Ratner, P. Johansson and D. F. Shriver, Polymer Electrolytes: Ionic Transport Mechanisms and Relaxation Coupling, MRS Bull., 2000, 25, 31–37 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00002a |
| This journal is © The Royal Society of Chemistry 2024 |