
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anodic dissolution of aluminum in non-aqueous electrolyte solutions for sodium-ion batteries†
Lars Olow Simon
Colbin
a,
Charles Aram
Hall
a,
Ahmed S.
Etman
 ab,
Alexander
Buckel
c,
Leif
Nyholm
a and
Reza
Younesi
*a
ab,
Alexander
Buckel
c,
Leif
Nyholm
a and
Reza
Younesi
*a
aDepartment of Chemistry-Ångström Laboratory, Uppsala University, Box 538, Uppsala SE-75121, Sweden. E-mail: reza.younesi@kemi.uu.se
bRISE Research Institutes of Sweden, Drottning Kristinas väg 61, Stockholm 114 28, Sweden
cAltris AB, Kungsgatan 70b, Uppsala 753 18, Sweden
First published on 23rd November 2023
Abstract
Anodic dissolution of aluminum (commonly called aluminum corrosion) is a potential issue in sodium-ion batteries. Herein, it is demonstrated how different sodium-ion battery electrolyte solutions affect this phenomenon. The type of electrolyte was critical for the presence of anodic dissolution, while the solvent appeared to alter the dissolution process.
Corrosion can be a critical issue in modern batteries. The aluminum current collector of the positive electrode is, for example, susceptible to anodic dissolution (sometimes referred to as aluminum corrosion) at sufficiently high potentials. This phenomenon has been extensively studied for lithium-ion batteries, where anodic dissolution is often seen when using lithium bis(fluorosulfonyl)imide (LiFSI)1,2 or lithium bis(trifluoromethanesulfonyl)imide (LiTFSI)3,4 electrolytes (at moderate concentrations, typically 1 M) in contact with aluminum at potentials beyond 3.5 V vs. Li+/Li.5 However, the dissolution may also depend on the solvent used.6 Moreover, the exact onset potential has also been proposed to depend on the general composition of the electrolyte solution,5 and morphology of the aluminum surface.7
Anodic dissolution of aluminum current collectors in sodium-ion batteries has received less attention; albeit, a few articles can be found in which the topic is addressed.8–10 It is, nevertheless, reasonable to assume that if aluminum dissolves anodically in a specific lithium-based electrolyte solution, then the phenomenon should persist for a corresponding sodium-based solution, at a comparable potential. For example, if aluminum starts to dissolve anodically at 3.5 V vs. Li+/Li when in contact with LiFSI in propylene carbonate (PC), one would expect a similar reaction to occur at 3.2 V vs. Na+/Na using sodium bis(fluorosulfonyl)imide (NaFSI) in PC. Yet, this will depend on how the electrochemical potential scales between the two corresponding electrolytes in a particular solvent system. The solvent is expected to largely influence the electrochemical potential of the electrolyte, which may complicate a direct comparison of anodic dissolution between different solvent systems.11,12 Essentially, this could theoretically cause the observed potential to differ by a few hundred millivolts. Moreover, a lack of data on activity coefficients in these solvent systems makes a comparison of electrochemical potentials between the systems even more difficult, due to an additional uncertainty in how a given concentration affects the electrochemical potential.
Studies on anodic dissolution of aluminum in lithium-ion batteries naturally tend to focus on solvent systems typical for such devices. It is, however, unclear whether these solvents are appropriate to use in electrolyte solutions for sodium-ion batteries.13 Here, glyme-based solvents are probably more interesting because of their apparent stability towards metallic sodium.14 Alternatively, alkyl organophosphate solvents could also be interesting because of their potential safety benefits.15
We have recently reported on the use of a new electrolyte solution, sodium bis(oxalato)borate (NaBOB) in triethyl phosphate (TEP), in sodium-ion batteries containing Prussian white and hard carbon electrodes.16 The results indicated that anodic aluminum dissolution could not be seen at potentials below 4.5 V vs. Na+/Na in ∼0.4 M NaBOB in TEP due to the formation of a passive layer. This effect was attributed to the use of NaBOB. Moreover, since it has been suggested in literature that lithium bis(oxalato)borate can passivate aluminum;17 it is reasonable to assume that NaBOB would have a similar effect in a sodium-ion battery. The present work investigates the possible anodic dissolution of aluminum in different sodium-based electrolyte solutions at concentrations well below the solubility limit, namely: (i) 0.284 m NaBOB in TEP, (ii) 1.00 m NaPF6 in TEP; (iii) 1.00 m NaFSI in TEP; (iv) 1.00 m NaFSI in PC; (v) 1.00 m NaPF6 in ethylene carbonate (EC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl carbonate (DEC) 1:1 vol; and (vi) 1.00 m NaPF6 in 1-methoxy-2-(2-methoxyethoxy)ethane (diglyme); note that the electrolyte concentrations are here presented in molal (m), i.e. mol kg−1. Here, NaPF6 in EC:DEC and NaPF6 in diglyme electrolyte solutions will serve as reference electrolyte solutions. According to literature describing the effect of using LiPF6,18–20 these solutions should passivate if the sodium-ions do not significantly alter the passivation process. Conversely, 1.00 m NaFSI in PC will serve as a single solvent electrolyte solution which is likely to facilitate anodic dissolution of aluminum.10 Together, these results will be compared with the responses for the TEP based electrolyte solutions to obtain a better understanding of how the anodic dissolution of aluminum is affected by the use of this solvent. The ultimate aim is to clarify whether; (i) the use of NaBOB in TEP prevents anodic dissolution of aluminum as previously suggested;16 and (ii) the passivation seen in this electrolyte solution should be attributed to NaBOB or TEP.
:diethyl carbonate (DEC) 1:1 vol; and (vi) 1.00 m NaPF6 in 1-methoxy-2-(2-methoxyethoxy)ethane (diglyme); note that the electrolyte concentrations are here presented in molal (m), i.e. mol kg−1. Here, NaPF6 in EC:DEC and NaPF6 in diglyme electrolyte solutions will serve as reference electrolyte solutions. According to literature describing the effect of using LiPF6,18–20 these solutions should passivate if the sodium-ions do not significantly alter the passivation process. Conversely, 1.00 m NaFSI in PC will serve as a single solvent electrolyte solution which is likely to facilitate anodic dissolution of aluminum.10 Together, these results will be compared with the responses for the TEP based electrolyte solutions to obtain a better understanding of how the anodic dissolution of aluminum is affected by the use of this solvent. The ultimate aim is to clarify whether; (i) the use of NaBOB in TEP prevents anodic dissolution of aluminum as previously suggested;16 and (ii) the passivation seen in this electrolyte solution should be attributed to NaBOB or TEP.
Anodic dissolution of aluminum was studied in a three-electrode configuration using polytetrafluoroethylene beaker cells containing approximately 2.5 ml electrolyte solution (see Fig. S1, ESI†). The use of a relatively large solution volume was intended to amplify the effects of any aluminum dissolution during the electrochemical experiments. Aluminum foil was used as the working electrode and two-separate Prussian blue electrodes were used as reference and counter electrodes. The cells were designed to give rise to a well-defined geometric surface area (i.e., 4.5 mm diameter) see Fig. S1 (ESI†).
Cyclic voltammetry (CV) experiments using the different electrolyte solutions were initially performed at a scan rate of 1 mV s−1. Each experiment started from the open circuit potential (OCP) (Table S1, ESI†), and the upper vertex potential was 5.3 V vs. Na+/Na (corresponding to 2.0 V vs. the potential of the Prussian blue reference), see Fig. 1. In addition, electrochemical impedance spectroscopy (EIS) experiments were performed at the OCP before each CV experiment (see Note S1, Fig. S2, and Table S2 in the ESI†). This was to make sure that each cell was representative. The CV results indicate that the aluminum electrode underwent passivation in electrolyte solutions containing NaBOB or NaPF6. For these solutions, the current gradually decreased as the scan direction was reversed, suggesting an increasing passivation. In essence, at highly oxidizing potentials the native passivating Al2O3 layer is not sufficient for preventing an electron transfer resulting in faradaic reactions. These reactions may enforce the passivation by either increasing the thickness or changing the composition of the native passivation layer, resulting in a decreasing current response at a given potential. This tendency for passivation was therefore further indicated by the significantly reduced currents during the consecutive cycles. Additionally, in the NaPF6 case, the choice of solvent did not seem to affect the outcome significantly (Fig. 1a, b, d, and e). In contrast, anodic dissolution of aluminum was found to take place in both of the solutions containing the NaFSI electrolyte (Fig. 1c and f). Here, a reversal of the scan-direction resulted in an increasing current until a distinct potential was reached where the oxidation ceased (see Fig. 1c and f). Similar to the foregoing scenario, at highly oxidizing potentials faradaic reactions will start to occur. However, rather than enforcing the passivation, these reactions may instead lead to a breakdown of the layer. This would then allow for an oxidation and dissolution of the previously protected aluminum. Here the current would gradually increase at a given potential as the surface becomes more activated. Yet, once the potential bias becomes less severe the current is expected to subside. The mechanism for this may vary and have implications for the continued stability, yet a detailed description of this is beyond the current scope. Still, in the NaFSI containing solutions, the choice of solvent also seemed to affect the current response. In the TEP solution, the current decreased from the first to the second cycle, whereas it increased in the PC-based electrolyte solution.
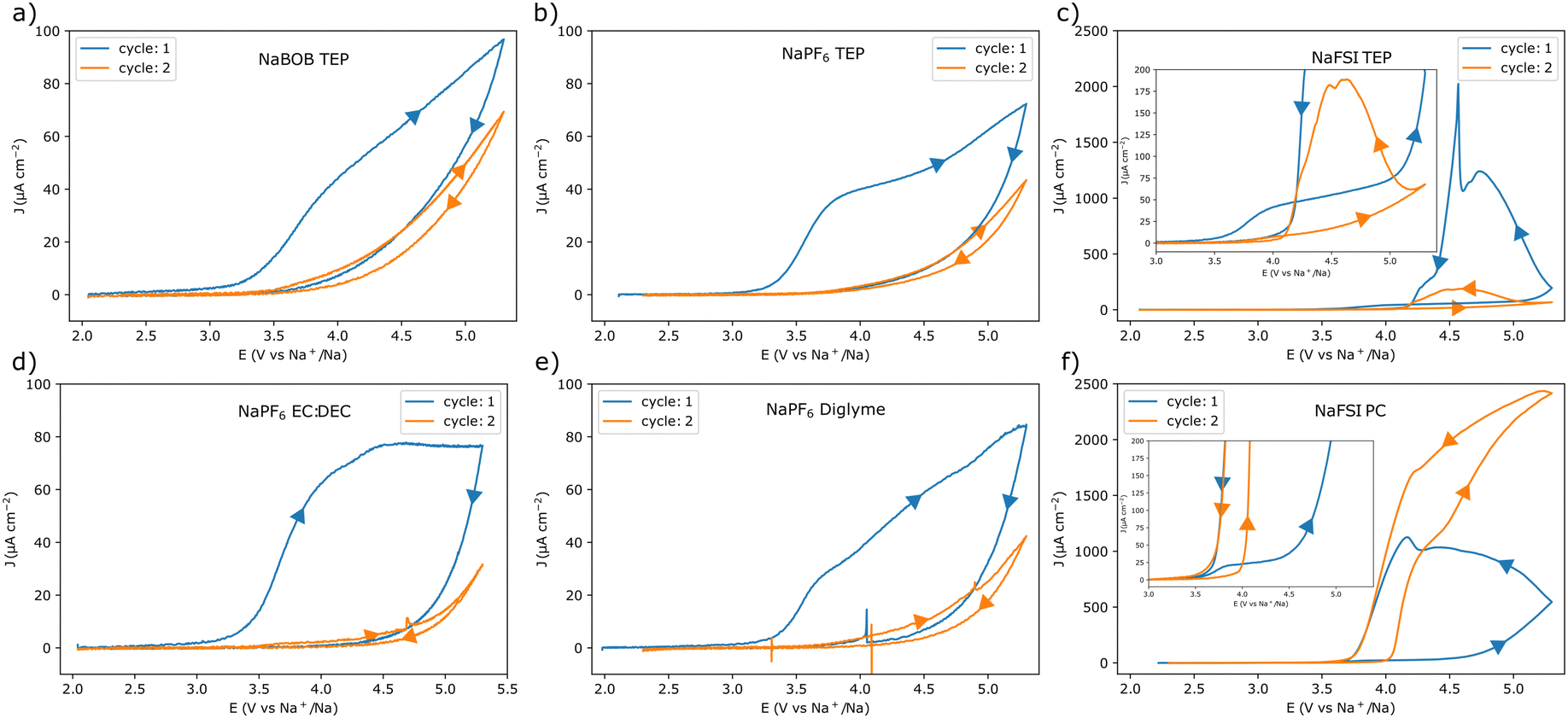 | ||
| Fig. 1 Cyclic voltammograms recorded with aluminum working electrodes at a scan rate of 1 mV s−1 in the following electrolyte solutions, (a) 0.284 m NaBOB in TEP, (b) 1.00 m NaPF6 in TEP, (c) 1.00 m NaFSI in TEP, (d) 1.00 m NaPF6 in EC:DEC 1:1 vol, (e) 1.00 m NaPF6 in diglyme, and (f) 1.00 m NaFSI in PC. The scan direction is indicated by the arrows. The insets in (c) and (f) show magnified parts of the parent graphs. Note also the different current density scales in (c) and (f). | ||
To obtain a better view of the current responses, the scan rate was varied from 10 mV s−1 to 1 V s−1 in an additional set of experiments (for each of the different scan rates, a fresh piece of aluminum foil was used). The first cycle voltammograms of these experiments are shown in Fig. 2. As expected, the electrolyte solutions that gave rise to passivation in the 1 mV s−1 experiment, also displayed a similar behavior at higher scan rates (Fig. 2a, b, d, and e). Likewise, the use of NaFSI in PC resulted in a clear anodic dissolution of aluminum at all tested scan rates (Fig. 2f). However, for NaFSI in TEP, the anodic dissolution was clearly dependent on the scan rate. In this case there was no indication of dissolution at a scan rate of 50 mV s−1 or higher (Fig. 2c). At a scan rate of 10 mV s−1, the current initially decreased, only to slightly increase at a lower potential, indicating that the anodic dissolution was taking place. The use of TEP thus seemed to obstruct the anodic dissolution process, as the current was gradually suppressed and delayed at scan rates ≥10 mV s−1.
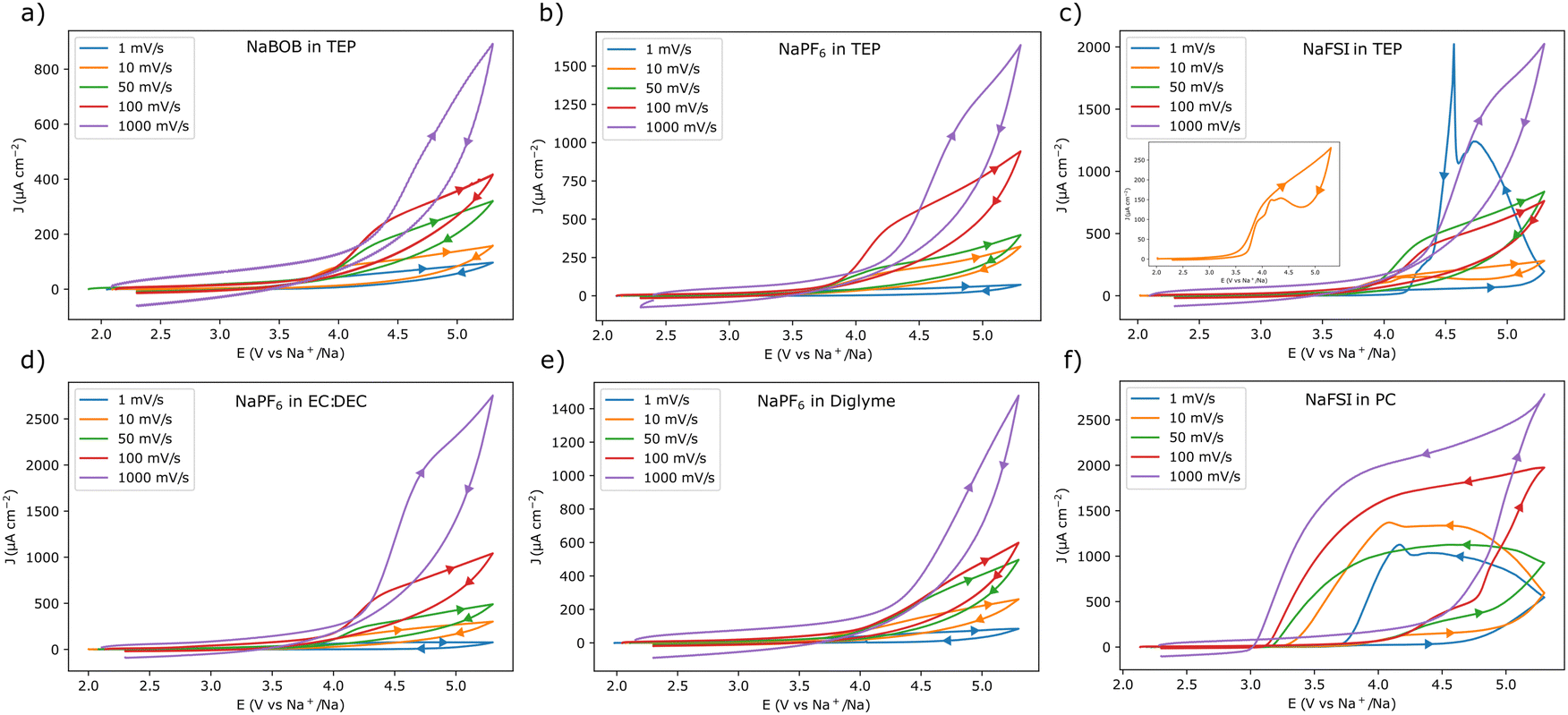 | ||
| Fig. 2 Cyclic voltammograms recorded with aluminum working electrodes at different scan rates in the following electrolyte solutions, (a) 0.284 m NaBOB in TEP, (b) 1.00 m NaPF6 in TEP, (c) 1.00 m NaFSI in TEP, (d) 1.00 m NaPF6 in EC:DEC 1:1 vol, (e) 1.00 m NaPF6 in diglyme, and (f) 1.00 m NaFSI in PC. Fresh aluminum electrodes were used for each of the different scan rates. Only the first cycle is shown for each scan rate. The scan direction is indicated by the arrows, note the change in directionality in (c) for the 1 mV s−1 scan rate. The inset in (c) shows a magnified view of the 10 mV s−1 voltammogram. The first three cycles for each experiment are shown as ESI† in Fig. S3. | ||
To facilitate the interpretation of the results, each system was also studied using chronoamperometry. The potential steps protocol used for each electrolyte solution was based on points of interest deduced from the cyclic voltammetry experiments. As a result, the exact step profile was intentionally made different for each electrolyte solution. After each measurement, the aluminum working electrode was extracted, washed in anhydrous methanol, and characterized using scanning electron microscopy (SEM). Both the chronoamperometry and SEM results suggested that the systems that employed NaBOB or NaPF6 as electrolyte did not undergo significant anodic dissolution below 5.3 V vs. Na+/Na (Fig. 3). For these systems, the current decayed exponentially after each potential step, and there were no clear indications of exfoliation or pitting of the surfaces. There remained, however, some organic residues on the electrode tested in the cell that contained NaPF6 dissolved in EC:DEC (Fig. 3d2). In contrast, the solutions containing NaFSI clearly resulted in anodic dissolution of the aluminum electrode at 5.3 V vs. Na+/Na (Fig. 3c2 and f2). The relatively large volume of electrolyte solution used in each experiment should have amplified the effects of this dissolution. In a cell that more closely resembles a battery, a corresponding dissolution process would likely be less pronounced. Nevertheless, long term tests are still required to fully assert the severity or absence of anodic dissolution in the investigated electrolyte solutions.
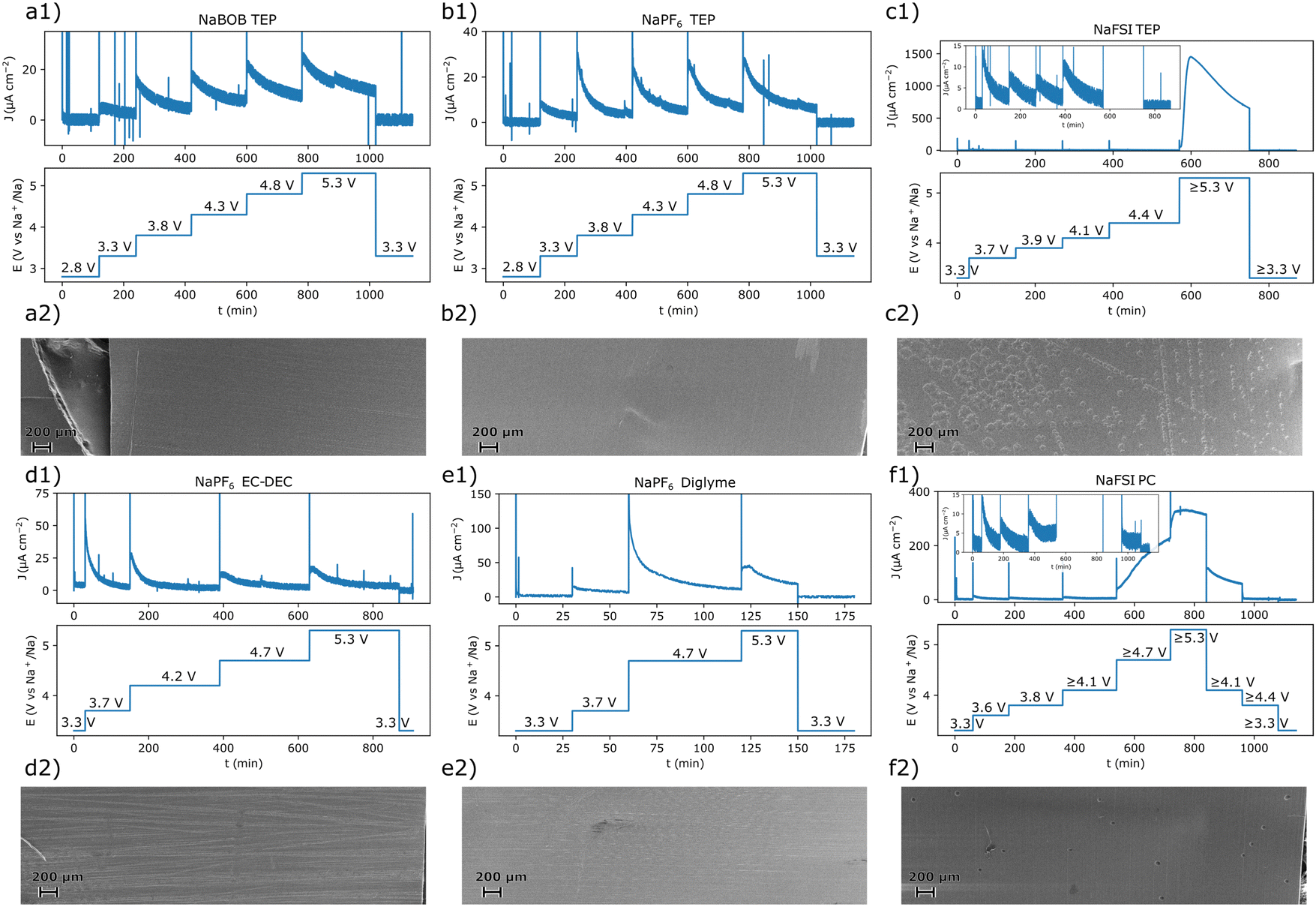 | ||
| Fig. 3 The chronoamperometric protocol and the resulting current–time curves for the experiment performed with an electrolyte solution composed of (a1) 0.284 m NaBOB in TEP, (b1) 1.00 m NaPF6 in TEP, (c1) 1.00 m NaFSI in TEP, (d1) 1.00 m NaPF6 in EC:DEC, (e1) 1.00 m NaPF6 in diglyme, and (f1) 1.00 m NaFSI in PC. The SEM images recorded for each corresponding reacted electrode after the chronoamperometric experiments are shown in (a2)–(f2). Magnified SEM images of the pristine aluminum foil and anodically dissolved samples are shown in Fig. S4 (ESI†) along with a short discussion in Note S3 in the ESI.† | ||
Comparing the chronoamperometry and CV results for the NaFSI electrolyte solutions, it appears that the current onset at 3.5 V vs. Na+/Na was not the starting point of the severe anodic aluminum dissolution seen at higher potentials (Fig. 1c and f). The chronoamperometry results (Fig. 3c1 and f1) suggest that the surface passivates at this potential in the NaFSI containing electrolyte solutions. Moreover, when using the NaFSI in TEP electrolyte solution, the current response in the chronoamperometry experiment approached a very small value even at 4.4 V vs. Na+/Na (Fig. 3c1, inset). This general behavior was also observed when NaFSI in PC was used. However, here a significant oxidation current seemed to persist at 4.1 V vs. Na+/Na. Still, for NaFSI in PC, the current response appeared to approach a very small value at 3.8 V vs. Na+/Na (Fig. 3fa, inset). This suggests that a 1.00 m NaFSI in PC electrolyte solution tends to anodically dissolve aluminum at a lower potential compared to a 1.00 M NaFSI in TEP solution. A higher tolerance to anodic dissolution of aluminum for NaFSI in TEP is also consistent with the cyclic voltammetry results (Fig. 1c and f). Here, the dissolution current diminished at a higher potential on the return scan for NaFSI in TEP compared to when PC was used. Nevertheless, long term experiments are also needed to pinpoint the onset potential for an intolerable anodic dissolution of aluminum. Even if the dissolution should be amplified in the employed experimental setup, there may still be a small amount of anodic dissolution that could grow significant with time. It should also be stressed that there are some uncertainties associated with the potential of the reference electrodes after the dissolution onset for the samples in Fig. 3c1 and f1, see Note S2 in the ESI.† This however, should not affect any conclusions regarding an absence of anodic dissolution, but care should be taken when interpreting the results obtained after the anodic dissolution had commenced.
The aluminum content in the electrolyte solutions was measured using inductively coupled plasma optical emission spectroscopy (ICP-OES). The solutions were taken from the cells held at 3.3 V vs. Na+/Na for 10 minutes, then kept at 5.3 V vs. Na+/Na for 4 h, thereafter allowed to relax at 3.3 V vs. Na+/Na for 1 h, these chronoamperometry results are shown in Fig. S5 (ESI†). Comparing the ICP-OES results (summarized in Table 1) with the charge passed during the chronoamperometry measurements suggest that a large portion of the current passed during the electrochemical experiment was caused by the aluminum dissolution, rather than a general solvent decomposition. For example, 1.83 C of charge passed during the 5.3 V vs. Na+/Na step using 1.00 mol kg−1 NaFSI in TEP. Assuming an unaltered density (1.16 g cm−3 at 27 °C for 1.00 mol kg−1 NaFSI in TEP), if the current was solely due to aluminum dissolution, then the charge passed would correspond to an aluminum concentration of 2.6 mol kg−1. Using the same density value, the ICP-OEC would translate to a concentration of 3.0 mol kg−1.
| Electrolyte solution | Aluminum content (ppm|mg kg−1) |
|---|---|
| 1.00 mol kg−1 NaPF6 in TEP | 3 |
| 1.00 mol kg−1 NaPF6 in diglyme | <1 |
| 1.00 mol kg−1 NaPF6 in EC:DEC | 3 |
| 0.284 mol kg−1 NaBOB in TEP | <1 |
| 1.00 mol kg−1 NaFSI in TEP | 67 |
| 1.00 mol kg−1 NaFSI in PC | 57 |
The aluminum electrodes used in the experiments shown in Fig. S5 (ESI†) were further analyzed using X-ray photoelectron spectroscopy (XPS). The working electrodes exposed to NaPF6 had the largest increase in fluoride content (Fig. 4b and d), whereas the downward shift of the high binding energy intensity suggest a formation of aluminum fluoride (Fig. 4a). A small amount of fluoride was detected on the pristine sample, and on the sample that was exposed to the NaBOB solution likely due to cross contamination during the XPS measurement. Interestingly, no significant amount of boron was detected on the aluminum exposed to the NaBOB in TEP solution (Fig. 4d and Fig. S6, ESI†). Instead, an increase in oxygen was observed for the NaBOB exposed sample, where the O1s spectra share similar shape, and position to the pristine sample. This could indicate a thickness increase of the aluminum oxide layer. Both aluminum samples exposed to NaFSI showed an increase in sulphur content. The S2p spectra (Fig. S6, ESI†) suggest the presence of sulphur compounds of low electron density (binding energies above 170 eV), which may be due to species resembling the FSI-anion. There were also signals coming from lower binding energies suggesting the prescience of variety of sulphur containing compounds.
 | ||
| Fig. 4 XPS deconvolution results of the spectra shown in Fig. S6–S8 (ESI†). The spectra shown here are energy calibrated to the “bulk”-signal of aluminum Al2p positioned to 72.7 eV, (a) Al2p, (b) F1s, and (c) O1s core levels. The relative atomic concentrations are shown in (d), this estimation is based on the models in Fig. S6–S8 (ESI†). The spectra in (a) have been area normalized to the intensity that was believed to be caused by the pure aluminium (“bulk” aluminium). | ||
In summary, anodic dissolution of aluminum was studied in different electrolyte solutions using cyclic voltammetry, chronoamperometry, and SEM imaging. Out of the tested solutions, severe anodic dissolution was not observed below 5.3 V vs. Na+/Na using: (i) 0.284 m NaBOB in TEP; (ii) 1.00 m NaPF6 in TEP; (iii) 1.00 m NaPF6 in EC:DEC; or (iv) 1.00 m NaPF6 in diglyme. Anodic dissolution was, however, detected with: (i) 1.00 m NaFSI in PC; and (ii) 1.00 m NaFSI in TEP. The nature of the dissolution appeared to be altered by the use of a different solvent; the use of TEP instead of PC, seemed to delay the initial anodic aluminum dissolution. Moreover, for the passivated electrodes the differences in the XPS results together with some minor differences in the cyclic, especially on the first forward scan (Fig. 1), indicate a difference in mechanism in how the passivation was maintained. The results emphasize the importance of further studies on the solvent's role during passivation or anodic dissolution of aluminum. To answer the stated questions: (i) the use of NaBOB in TEP electrolyte solution did not appear to result an anodic dissolution of aluminum below 5.3 V vs. Na+/Na. (ii) Here, the electrolyte was likely the essential component preventing anodic aluminum dissolution, since dissolution was seen in the TEP solvent when using a different electrolyte. However, considering that the use of TEP together with NaFSI resulted in a delayed dissolution, the passivation process by NaBOB in TEP could also have been significantly affected by the solvent. Future studies are required in two different areas: (i) focused studies describing the mechanisms behind the various electrochemical responses for the different electrolyte solutions, (ii) long term studies in more realistic battery systems. For real battery cells, both solution volume and the addition of a cathode material on the aluminum may affect the outcome significantly. Still, it is the authors' ambition that the results herein may stimulate such studies.
Conflicts of interest
The Prussian white electrodes used in this study are a commercial product provided by Altris A. B., a company co-founded by R. Y., and the current employer of A. B. Moreover, Altris A. B., holds a patent involving sodium bis(oxalato)borate in alkyl-organophosphates. The other authors in this paper declare no conflict of interest.Acknowledgements
The Swedish Energy Agency via project no. 50177-1, VINNOVA via projects no. 2022-01465 and 2019-00064 (Batteries Sweden), and H2020 research and innovation programme under Grant agreements no. 958174 and 963542 are acknowledged for financial support.Notes and references
- Y. Yamada, C. H. Chiang, K. Sodeyama, J. Wang, Y. Tateyama and A. Yamada, ChemElectroChem, 2015, 2, 1687–1694 CrossRef CAS.
- L. Nyholm, T. Ericson and A. S. Etman, Chem. Eng. Sci., 2023, 282, 119346 CrossRef CAS.
- K. Matsumoto, K. Inoue, K. Nakahara, R. Yuge, T. Noguchi and K. Utsugi, J. Power Sources, 2013, 231, 234–238 CrossRef CAS.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320–325 CrossRef CAS.
- A. Gabryelczyk, S. Ivanov, A. Bund and G. Lota, J. Energy Storage, 2021, 43, 103226 CrossRef.
- X. Wang, E. Yasukawa and S. Mori, Electrochim. Acta, 2000, 45, 2677–2684 CrossRef CAS.
- H. Yang, K. Kwon, T. M. Devine and J. W. Evans, J. Electrochem. Soc., 2000, 147, 4399–4407 CrossRef CAS.
- A. Bitner-Michalska, A. Krztoń-Maziopa, G. Żukowska, T. Trzeciak, W. Wieczorek and M. Marcinek, Electrochim. Acta, 2016, 222, 108–115 CrossRef CAS.
- J. Hwang, I. Aoyagi, M. Takiyama, K. Matsumoto and R. Hagiwara, J. Electrochem. Soc., 2022, 169, 080522 CrossRef CAS.
- L. Otaegui, E. Goikolea, F. Aguesse, M. Armand, T. Rojo and G. Singh, J. Power Sources, 2015, 297, 168–173 CrossRef CAS.
- R. A. Robinson and R. H. Stokes, Electrolyte Solutions, Dover Publications, Inc., Mineola, New York, 2nd rev edn, 2002 Search PubMed.
- J. S. Newman and K. E. Thomas-Alyea, Electrochemical systems, John Wiley & Sons, Inc., Hoboken, NJ, 2004, vol. 3 Search PubMed.
- R. Mogensen, S. Colbin and R. Younesi, Batteries Supercaps, 2021, 4, 791–814 CrossRef CAS.
- K. Westman, R. Dugas, P. Jankowski, W. Wieczorek, G. Gachot, M. Morcrette, E. Irisarri, A. Ponrouch, M. R. Palacín, J. M. Tarascon and P. Johansson, ACS Appl. Energy Mater., 2018, 1, 2671–2680 CrossRef CAS.
- X. Liu, X. Jiang, Z. Zeng, X. Ai, H. Yang, F. Zhong, Y. Xia and Y. Cao, ACS Appl. Mater. Interfaces, 2018, 10, 38141–38150 CrossRef CAS.
- L. O. S. Colbin, R. Mogensen, A. Buckel, Y. L. Wang, A. J. Naylor, J. Kullgren and R. Younesi, Adv. Mater. Interfaces, 2021, 8, 1–10 Search PubMed.
- X. Zhang and T. M. Devine, J. Electrochem. Soc., 2006, 153, B365 CrossRef CAS.
- D. Moosbauer, S. Zugmann, M. Amereller and H. J. Gores, J. Chem. Eng. Data, 2010, 55, 1794–1798 CrossRef CAS.
- S. S. Zhang and T. R. Jow, J. Power Sources, 2002, 109, 458–464 CrossRef CAS.
- M. Morita, T. Shibata, N. Yoshimoto and M. Ishikawa, Electrochim. Acta, 2002, 47, 2787–2793 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Investigating anodic dissolution of aluminum in non-aqueous electrolyte solutions relevant for sodium-ion batteries. XPS data can be obtained from figshare: see DOI: https://doi.org/10.6084/m9.figshare.2441229 electrochemical data can be obtained from figshare: See DOI: https://doi.org/10.6084/m9.figshare.24440998. see DOI: https://doi.org/10.1039/d3ya00233k |
| This journal is © The Royal Society of Chemistry 2024 |