
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhen porphyrins meet 2D materials: spectroscopic and photocatalytic properties
Aleksandra
Lindner
 a,
Aleksandra
Lesniewicz
b,
Aleksander
Kolman
b,
Daria
Larowska-Zarych
bc,
Bronislaw
Marciniak
bd and
Anna
Lewandowska-Andralojc
*bd
a,
Aleksandra
Lesniewicz
b,
Aleksander
Kolman
b,
Daria
Larowska-Zarych
bc,
Bronislaw
Marciniak
bd and
Anna
Lewandowska-Andralojc
*bd
aHelmholtz-Zentrum Dresden-Rossendorf, Institute of Ion Beam Physics and Materials Research, Bautzner Landstraße 400, 01328 Dresden, Germany
bFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznanskiego 8, 61-614 Poznan, Poland
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland
dCenter for Advanced Technology, Adam Mickiewicz University, Uniwersytetu Poznanskiego 10, 61-614 Poznan, Poland. E-mail: alewand@amu.edu.pl
First published on 7th June 2024
Abstract
Since its discovery, graphene has gained considerable interest from scientists all over the world. For more than one decade, the scientific community has been spending notable amounts of intellectual and financial resources to study its properties, which paves a way towards the commercialization of graphene, other 2D materials and a manifold of their derivatives. In this review, the spectroscopic properties of porphyrin-functionalized 2D materials are comprehensively discussed, followed by an extensive presentation of state-of-the-art achievements in photocatalysis based on such composite/hybrid materials. The primary focus is on the fundamental understanding of the structure–property–performance relationship as well as its importance in the future target-oriented design and fabrication of photocatalysts with tailored properties. After a short introduction, different design strategies for the fabrication of porphyrin (Por)-functionalized graphene-based materials (GBMs) (covalent vs. non-covalent assemblies) are systematically summarized. Then, the photocatalysis-relevant properties of the composites are thoroughly discussed based on the experimental results provided by steady-state absorption spectroscopy (ground-state properties) and time-resolved absorption and emission spectroscopies (excited-state properties). The importance of appropriate data analysis, with particular respect to the photoemission processes, is brought to attention. Subsequently, the photocatalytic behavior towards hydrogen generation, CO2 reduction and pollutant degradation of Por and GBM hybrids are comprehensively reported and fundamental mechanisms of light-driven catalytic processes in such systems, together with efficiency-limiting steps, are highlighted. The role of spectroscopy as a very powerful tool that enables the determination of key photophysical properties essential for light-driven catalysis is emphasized. Finally, recent advances with respect to 2D materials beyond graphene and their assembly with Por as promising photocatalysts are presented. We believe that this review, which comprehensively presents the knowledge gained to date regarding composites based on 2D materials with porphyrins as promising photocatalysts, will stimulate further efforts of researchers to tackle the remaining challenges and contribute to taking a decisive step towards the commercialization of these photocatalysts in the future.
 Aleksandra Lindner | Aleksandra Lindner obtained her doctoral degree in chemistry in 2006, working within the Research and Training Network SULFRAD at the Leipzig University, Germany. Later, she moved to the Notre Dame Radiation Laboratory, IN, USA where her interests were focused on photochemistry of IR-absorbing dyes and dye-graphene nanohybrides. Since 2017 till now she is researcher at the Institute of Ion Beam Physics and Materials Research, Helmholtz-Zentrum Dresden-Rossendorf, Germany. Her current studies are concentrated on the modification of thin ferromagnetic films with metal nanoparticles and dyes and probing light-induced effects therein. |
 Aleksandra Lesniewicz | Aleksandra Lesniewicz received her BSc in Chemistry from Adam Mickiewicz University in Poznan, Poland, in 2019 and her MSc in Medicinal Chemistry from Jagiellonian University in Cracow, Poland, in 2021. She is a PhD student under the supervision of Prof. Anna Lewandowska-Andralojc at Adam Mickiewicz University in Poznan, Poland. Her primary research interests revolve around the applications of porphyrins and 2D materials in the photodegradation of dyes. |
 Aleksander Kolman | Aleksander Kolman earned his bachelor's degree in Chemistry (Materials Chemistry) from Adam Mickiewicz University in 2016. During the same year, he commenced his master's degree in Medicinal Chemistry at Jagiellonian University in Cracow. In 2021, he accomplished his master's degree. Starting from October 2021, he embarked on his PhD studies under the guidance of Prof. A. Lewandowska-Andralojc. His research is focused on the synthesis and characterization of nanohybrids involving photosensitizers and 2D materials, with potential applications in the field of phototherapy for cancer. |
 Daria Larowska-Zarych | Daria Larowska-Zarych obtained her PhD in Chemistry from Adam Mickiewicz University in 2023, under the guidance of esteemed scholar and supervisor, Prof. Bronislaw Marciniak. Her doctoral thesis was dedicated to the study of the photochemical and photophysical properties of nanostructures that were based on graphene oxide functionalized with porphyrin dyes. Following the completion of her PhD studies, she secured a postdoctoral position at the Institute of Physical Chemistry, Polish Academy of Sciences. Her current research focuses on investigating systems that exhibit photoinduced electron transfer and have potential applications in photocatalysis, particularly in the photodegradation of water pollutants. |
 Bronislaw Marciniak | Bronislaw Marciniak is a Full Professor at the Faculty of Chemistry of Adam Mickiewicz University (AMU), Poznan, Poland and the Director of the Center for Advanced Technology at AMU. He (2008–2016) held the position of Rector of Adam Mickiewicz University. He received Professor of Chemistry Title (1998) from the AMU and was a post-doctoral fellow at Simon Fraser University, Burnaby, Canada (1980–81) and a Fulbright Scholar in the Radiation Laboratory, University of Notre Dame, USA (1990–91). Professor Marciniak's research interests include photochemistry, photophysics and radiation chemistry of (1) organic compounds of biological interest, (2) silicon atom–containing polymers and their model compounds and (3) porphyrin-graphene oxide nanohybrids. |
 Anna Lewandowska-Andralojc | Anna Lewandowska-Andralojc is an Associate Professor at the Faculty of Chemistry of Adam Mickiewicz University in Poznan. She obtained her doctoral degree in 2011 in the field of chemistry. During her PhD studies, she participated in two research internships at Radiation Laboratory, Notre Dame University, USA. She completed a two-year postdoctoral fellowship at Brookhaven National Laboratory (USA) in the Artificial Photosynthesis group under the supervision of Dr Etsuko Fujita. Her principal research is focused on the photochemistry and photocatalysis of nanomaterials, synthesis and characterization of nanostructures based on 2D materials and mechanistic studies of protons and electrons for systems related to artificial photosynthesis. |
1. Introduction
There has been an exponentially growing demand for new functional materials that could contribute to: (i) solving the problem of energy generation and storage (e.g., solar cells, fuel cells, batteries, supercapacitors, and hydrogen production), (ii) resolving key environment-related issues (e.g., degradation of pollutants and desalination technologies), (iii) developing faster and low–energy consuming electronic devices and sensors and (iv) widening the scope of biomedical diagnostic and therapy technology. The investigations reached their turning point in 2004, when Andre Geim and Konstantin Novoselov from the University of Manchester successfully isolated a single layer of graphite – graphene – using a scotch tape.1 Even though the idea of a single layer of graphite has already been theoretically explored for at least 50 years, it was rather a general belief that the physical creation of such an atomically thin two-dimensional (2D) carbon material is not possible. Due to the extraordinary optical, thermal, mechanical and electrical properties of the isolated graphene monolayer as well as its versatile surface chemistry and high surface area, this topic has been attracting the interest of a broad scientific community already for more than a decade and simultaneously has triggered the steady progress in graphene commercialization.2 To date, it has been reported in a plethora of studies and reviews in the literature covering topics spanning from graphene synthesis, through graphene properties to its applications in fields such as energy harvesting, energy and smart material fabrication, photocatalysis, sensing and electronics.3–9The synthetic pathway used to fabricate a graphene monolayer as well as its related materials, which vary in terms of layer number, their lateral dimension and chemical modification, defines the structure and, in turn, the properties of the obtained materials. Various synthesis routes provide numerous graphene-based materials (GBMs) characterized by different properties and potential future applications, which can easily lead to a confusion when referring to the family of “graphene materials” or their individual members/counterparts. To avoid this, a clear and consistent system describing various derivatives of graphene is essential. A short introduction to the classification and nomenclature of GBMs can be found in various ref. 10 and 11. The rich variety of GBMs is visualized in Scheme 1. Note: graphene quantum dots (GQD) and graphene nanoribbons (GNR) are outside the scope of this review.
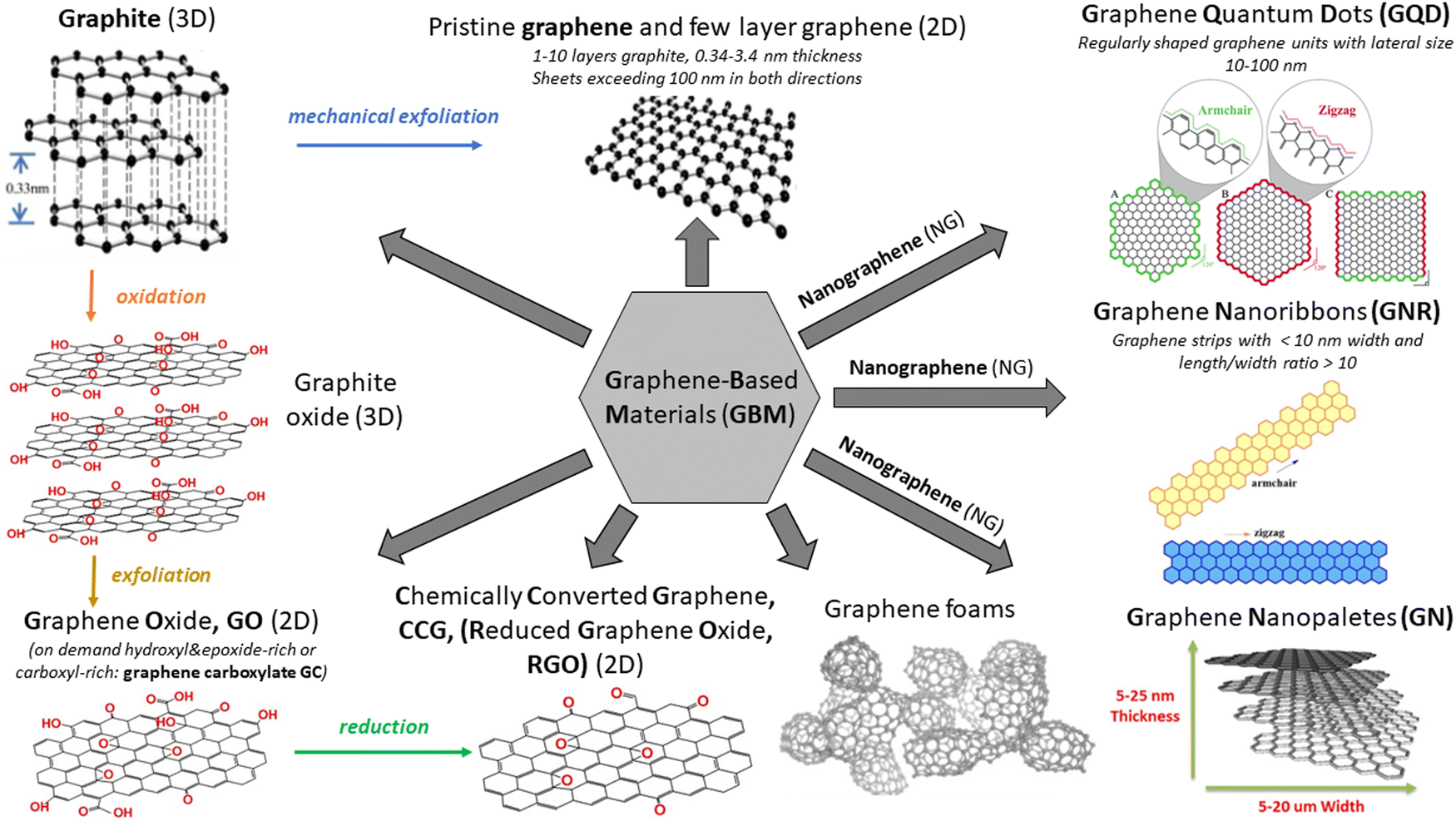 | ||
| Scheme 1 Various members of the GBM family. Adapted from ref. 12–16 with permission from the Royal Society of Chemistry (2020). | ||
The rise of graphene paved a way for 2D material research far beyond the carbon derivatives, which to this day evolved into a separate sub-discipline of materials sciences with a wide range of possible future applications.17,18 Since the physical properties of monolayers often dramatically change compared to those of their parent 3D materials, this leads to a new degree of freedom and allows for the exploration of novel physics. Noteworthy examples include a metal-free conjugated polymer graphitic carbon nitride (g-C3N4) broadly used in catalysis,19,20 2D transition metal dichalcogenides (TMDs, e.g., MoS2) with a direct bandgap in the visible range which renders them particularly interesting for optoelectronics,17,18 an analogue of graphene – hexagonal boron nitride (hBN),17 black phosphorus which exhibits great potential as a photocatalyst,21,22 a large family of 2D transition metal carbides and nitrides named MXenes which were initially investigated for energy storage applications23,24 and, last but not least, 2D perovskite materials with the crystal structure analogous to calcium titanate CaTiO3, which have meaningfully contributed to a rapid progress in solar cell efficiency within last years.25,26 Aside from the above-mentioned “conventional” 2D materials (2DMs), the so-called 2D framework materials such as 2D ordered polymers, covalent-organic frameworks and metal–organic frameworks synthetically constructed from molecular building blocks, which exhibit short- and long-range crystalline order and regular in-plane pores, have also gained much interest in the past decade.27 Various functional devices with superior performance and new features were developed based on 2D materials, and some have already been incorporated into the conventional integrated circuits.28 Note: the 2D framework materials are further excluded from this review.
The remarkable potential of the above-described 2DMs is closely related to their unique physiochemical properties resulting from the electronic structure and atomically thin dimensions, inseparably connected with the lateral size of the atomic layers, their number and stacking pattern, which allow for target-oriented fine-tuning of the materials’ desired properties. Furthermore, the chemical functionalization of 2DMs contributes to almost limitless possibilities for controlled tuning/boosting of the 2DMs’ electronic properties towards various applications of interest. Such molecular control over the electronic properties of 2DMs can be assured via two complementary approaches, i.e. covalent and non-covalent functionalization of the 2DM surface (see Section 2).29–31
Porphyrinoids are one of the most recognizable groups of organic molecules, with porphyrins (Por) constituting a very meaningful part of this family of organic compounds. Porphyrins (Scheme 2) attracted much attention from the scientific community due to their unique electronic, magnetic and optical properties.32,33
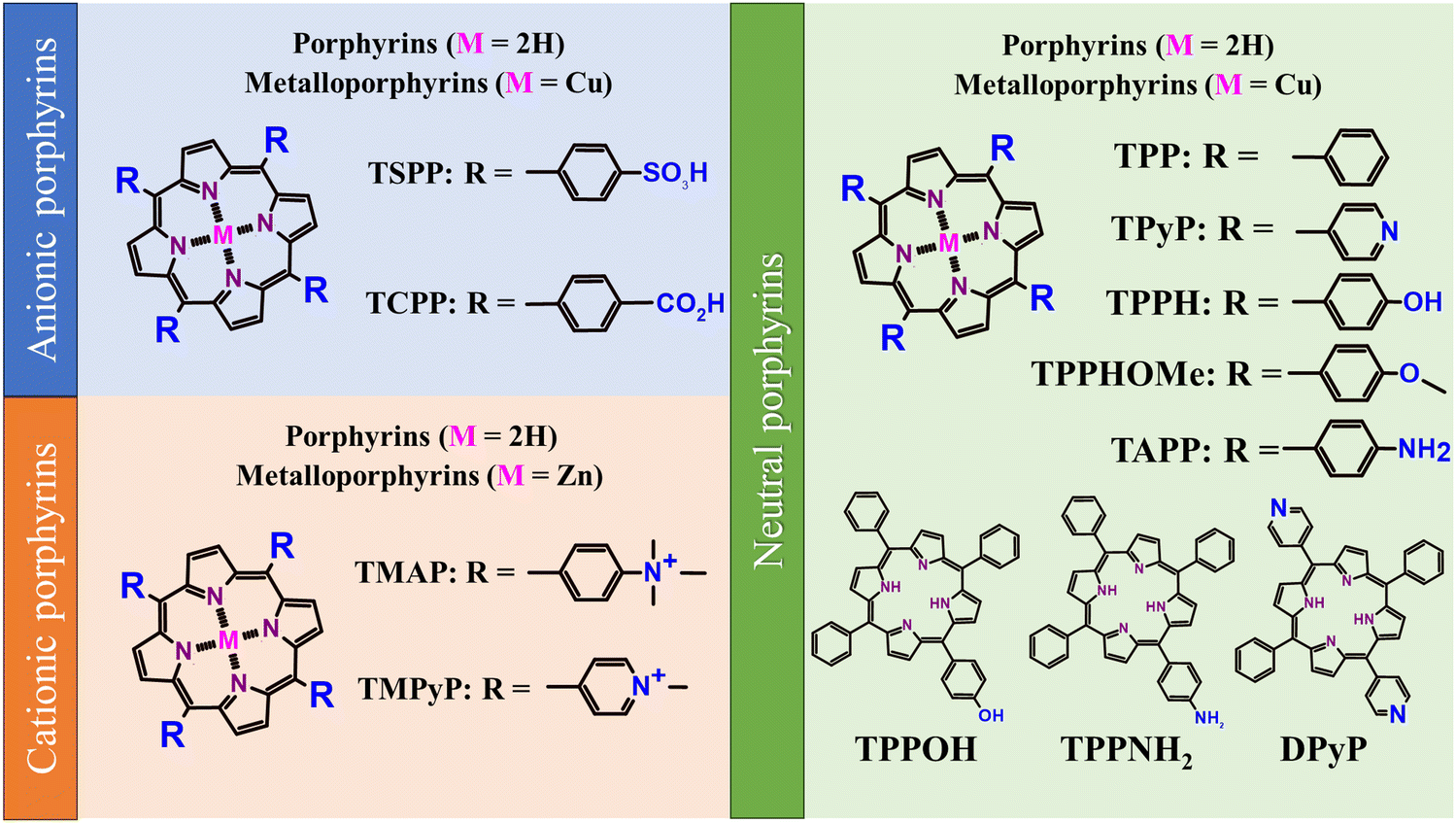 | ||
| Scheme 2 Structures of the most frequently used porphyrins together with the abbreviations of their names used within this study. | ||
They are characterized by an extended aromatic π–π structure, wherein various metal ions can be accommodated in the inner cavity of the macrocycle, which strongly affects their photophysical, photochemical and magnetic properties. Analogously, in a relatively uncomplicated manner, the properties of Por can be altered via a change in the substitution pattern of the pyrrole rings and methine bridges (meso carbons).34–37 This opens up almost limitless possibilities for tuning the properties of Por for specific applications, e.g. in photovoltaics, medicine, catalysis, sensing or information storage.32,38–40 For example, Por anchored to a wide-bandgap semiconductor are responsible for the absorption of solar radiation in dye-sensitized solar cells and their electrochemical, photophysical, ground- and excited-state properties essentially govern charge-transfer dynamics at the dye/semiconductor interface.41 Due to the efficient generation of long-lived triplet states, Por are broadly used as photosensitizers in photodynamic therapy (PDT) during cancer treatment.42,43 Homo- and heterogeneous tetrapyrrole-based catalysts mediate a plethora of chemical transformations such as oxidation of olefins, phenols, and thiols, rearrangement of epoxides to aldehydes or reduction of nitrite and nitrate, just to mention a few.44 Molecular electronics takes advantage of the tetrapyrroles’ unique properties as well. For instance, single-molecule magnets can be used to increase the density of the stored information45 and porphyrin-based devices can act as charge-storage memory elements.46 The broad spectrum of possible porphyrins’ applications ensures constant stimuli for the development of new synthetic pathways that enable the tailored fabrication of Por.47
Numerous technological and scientific applications of those dyes are inseparably connected to surface and interface phenomena.48,49 Therefore, the interaction of Por with diverse supports/substrates has been widely studied in various aspects, starting from the detailed characterization of the structure of molecular films on a given support to the technological applications of such hybrid systems.48,49 Por are known to adsorb onto metals as well as their oxides, either due to weak van der Waals forces (physisorption), or due to stronger interactions leading to the formation of chemical bonds between the adsorbate and the solid surface (chemisorption).50 The structure of such obtained molecular films (organic layers) depends not only on the type of interactions between the adsorbate and the substrate, but also on the interactions between the adsorbed molecules themselves (intermolecular interactions), e.g., hydrogen bonding, π–π stacking in the case of planar molecules and/or dipole–dipole interactions in the case of polar ones. Appropriate selection of the substrate and careful design/selection of the molecules allow us to tune the structure of the whole organic assembly at a given interface. The ultimate conformations of the adsorbed dye molecules, i.e. deformations of the macrocycles and/or reorientations of the substituents, are the result of all the above-mentioned interactions. These adsorption-induced deformations in turn affect the photophysical, photochemical and magnetic properties of Por immobilized on the surface of a solid sample.
Hybrid functional materials exhibit the properties of the individual counterparts to some extent but, at the same time, they are characterized by new, enhanced properties due to the synergy effect arising from the controlled attachment of carefully selected porphyrin macrocycles to the 2DM.30,31,51–56 The structures of the porphyrins most often employed as counterparts in hybrids with 2DM are depicted in Scheme 2, subdivided into three groups depending on their charge. The improved features of the 2DM hybrids as compared to the properties of their individual components (Por and 2DM) are visualized in Scheme 3, and will be referred to further on within the text.
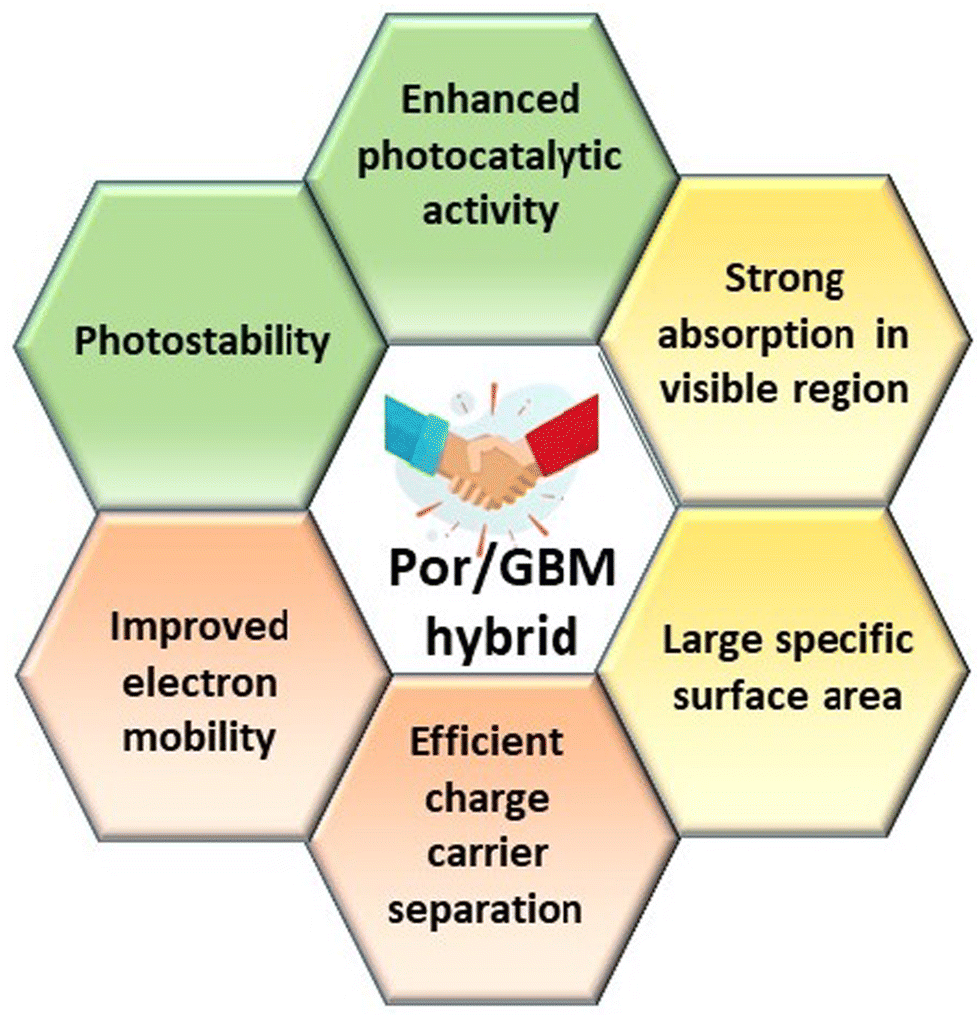 | ||
| Scheme 3 Synergistic effect resulting from the functionalization of 2DM with porphyrin derivatives. | ||
Numerous reports in the literature provide various information regarding the photochemical and photophysical characteristics of porphyrin-functionalized GBMs (note: significantly less is known about the assemblies of other 2DMs with porphyrins) as well as a wide range of their possible applications in the fields of photovoltaics, optoelectronics, photodynamic therapy (cancer treatment) and photocatalysis (H2 production, CO2 reduction, and degradation of pollutants). In recent years, substantial progress has been achieved, particularly in the lastly mentioned field. Therefore, the fact that only two review articles referring explicitly to GBM hybrids with porphyrins can be found in the literature is very surprising. The first one focuses on the synthetic routes used to fabricate the discussed hybrid materials, their structural characteristics and biomedical applications.30 The second concentrates on graphene hybrids with other low-dimensional nanomaterials, mentioning only three graphene hybrids with porphyrin molecules, and again putting emphasis on the synthetic methods.31 A. R. Monteiro et al. additionally devoted a few paragraphs in their review to 2DMs functionalized with porphyrins other than graphene. Taking all those into account, it becomes obvious that there is an urgent necessity for a comprehensive, detailed report focused on a short summary of the fabrication routes of 2DM and porphyrin hybrids and the discussion of their photochemical properties in reference to potential photocatalytic applications, with particular focus on the detailed analysis of their spectroscopic features enabling the target-oriented design of the hybrid structures. Our review aims to address this genuine gap. After a short section devoted to the fabrication of the discussed composites, the study concentrates on an extensive discussion of the spectroscopic data available for numerous hybrid materials and emphasizes the importance of their careful analysis, followed by a section that evaluates the photocatalytic performance of hybrids for hydrogen generation, CO2 reduction as well as pollutant degradation, and emphasizes the direct relation between the photocatalyst performance and its spectroscopic characteristics (Scheme 4).
 | ||
| Scheme 4 Schematic illustration of the framework and the sections of this review. | ||
Since most of the reports in the literature consider porphyrin-functionalized GBMs, whereas porphyrin functionalization of other 2D materials has received less attention to date, the above-mentioned sections of this review will be devoted to Por and GBM hybrids, followed by a separate section focused on 2DMs other than graphene, their functionalization with porphyrins and possible applications in photocatalysis. Ultimately, a short summary and further prospects regarding the use of porphyrin/2D composites will be presented.
The authors aim to provide the broad scientific community with comprehensive knowledge devoted to the spectroscopic properties of 2DM hybrids with Por and to highlight how these properties further determine the photocatalytic behavior of such composite materials. The timeline visualizing turning points within this field, discussed later on in detail in the text, is depicted in Scheme 5.
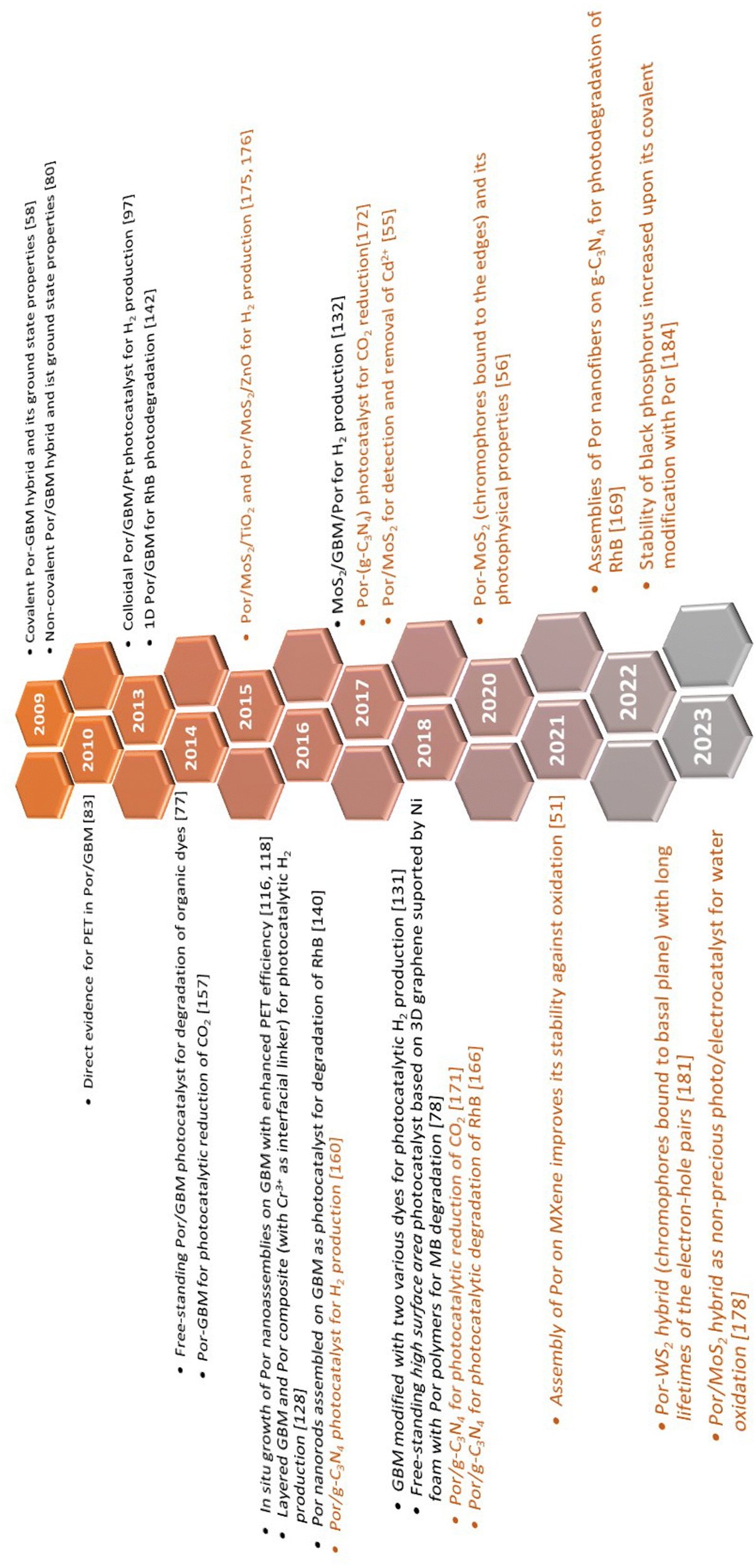 | ||
| Scheme 5 A timeline of selected events in the history of Por and 2DM hybrids focusing on their applicability in photocatalysis. Composites based on GBMs are highlighted in black font, whereas those with other 2DMs are presented in orange font. | ||
Note: the authors focus on quoting the most meaningful experimental results relevant to this review and presenting them as a starting point in the design of novel photocatalysts, rather than opening up a detailed discussion related to the interpretation of the results from each cited paper.
2. Synthesis routes for the functionalization of GBMs with porphyrins
The modification of GBMs with porphyrins in order to produce composite (hybrid) materials, in particular graphene integrated with porphyrin macrocycles, is achievable through both covalent and non-covalent functionalization methods, as illustrated in Fig. 1. The synthesis protocol of these composites, which may involve either covalent or non-covalent attachment of Por chromophores to graphene, significantly influences the characteristics of the resulting material. Consequently, both synthetic methodologies have been employed to fabricate these hybrids, with the choice depending on the desired properties of the final product. The non-covalent functionalization of GBMs relies on fundamental molecular interactions such as π–π-stacking, van der Waals forces, hydrogen bonding, and/or electrostatic forces between GBM and Por, and offers an easy synthesis route with high yields. Conversely, the covalent functionalization of GBMs with Por is a more demanding process that involves several synthesis steps. Nonetheless, the resulting composite materials are more stable and robust.57 The key features of both types of interactions are summarized in Table 1.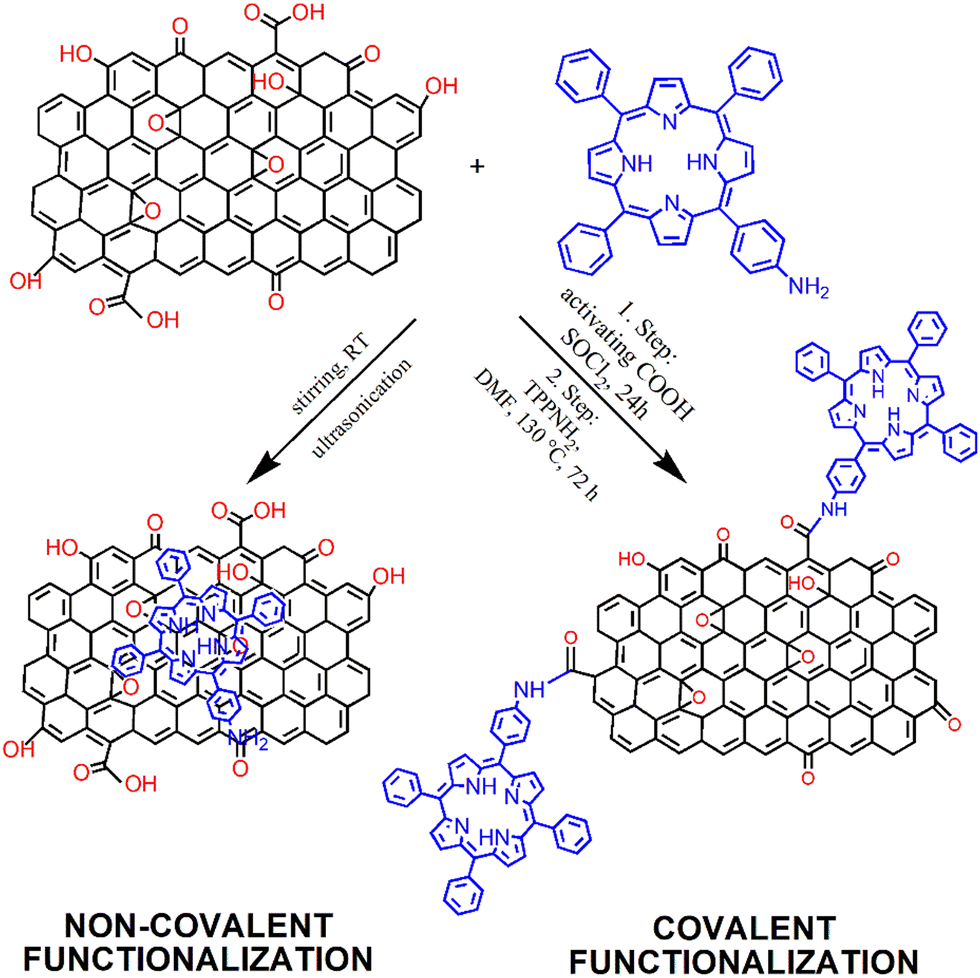 | ||
| Fig. 1 Schematic of the process of preparation of GO with covalently (Por–GO) and non-covalently (Por/GO) linked porphyrins. The conditions for covalent functionalization were taken from ref. 58. | ||
| Interactions | Mechanisms | Advantages | Drawbacks |
|---|---|---|---|
| Covalent | Covalent bonding between different groups of GBM and Por | (i) Strong interaction between hybrid components prevents desorption of Por from GBM sheets, | (i) Time-consuming reaction, |
| (ii) Low yield, | |||
| (ii) Well defined structure. | (iii) Limited to components that can form chemical bonds (e.g., amide, ester), | ||
| (iv) Low functionalization degree. | |||
| Non-covalent | π–π stacking, electrostatic interaction, van der Waals forces, hydrogen bonding | (i) Facile synthesis with high yields, | (i) Desorption and decomposition of Por from GBM sheets may occur during photocatalysis, |
| (ii) Self-assembly of Por on GBM, | |||
| (iii) Freedom in the selection of hybrid components, | (ii) Interaction between hybrid components is strongly solvent dependent, | ||
| (iv) Intrinsic properties of the material are retained, | (iii) Aggregation of Por on the GBM surface, | ||
| (v) Charge-transport characteristics depend only on the conjugating Por and GBM (no linker between Por and GBM). | (iv) Low weight content of Por in the hybrid. | ||
2.1 Covalent functionalization
The covalent methods of the GBM functionalization employ two primary approaches. The first concept involves alteration of the hydroxyl and carboxylic groups located at the periphery and defect sites of graphene sheets/nanostructures. The alternate strategy involves an addition reaction to the sp2-hybridized carbon network of the graphene surface. Taking advantage of the various functional groups present in GBMs (carboxyl, hydroxyl, and epoxy) and the straightforward modification of the meso-substituent in the porphyrin macrocycle, the most frequently used covalent functionalization technique involves the formation of a covalent bond between the oxygen-containing functional groups of GBMs and the meso-substituent in the porphyrin. A particularly popular synthetic approach involves the formation of an amide bond between the –COOH groups of GO and the –NH2 group(s) present in porphyrin molecules.57–60 In 2009, a seminal work reported the condensation reaction of TPPNH2 with thionyl chloride (SOCl2)-activated –COOH groups from GO for the first time (Fig. 1).58 The use of an activation agent like N,N′-dicyclohexylcarbodiimide (DCC) or converting the –COOH groups into more reactive entities (e.g., acyl chloride using SOCl2) is essential for the effective covalent bonding of the amine with GO.60,61 Using this method, Kumar et al. synthesized covalently functionalized GO with the metalated porphyrin Co-TAPP (Fig. 2A), which demonstrated the activity as a photocatalyst in the reduction of CO2 to formic acid.61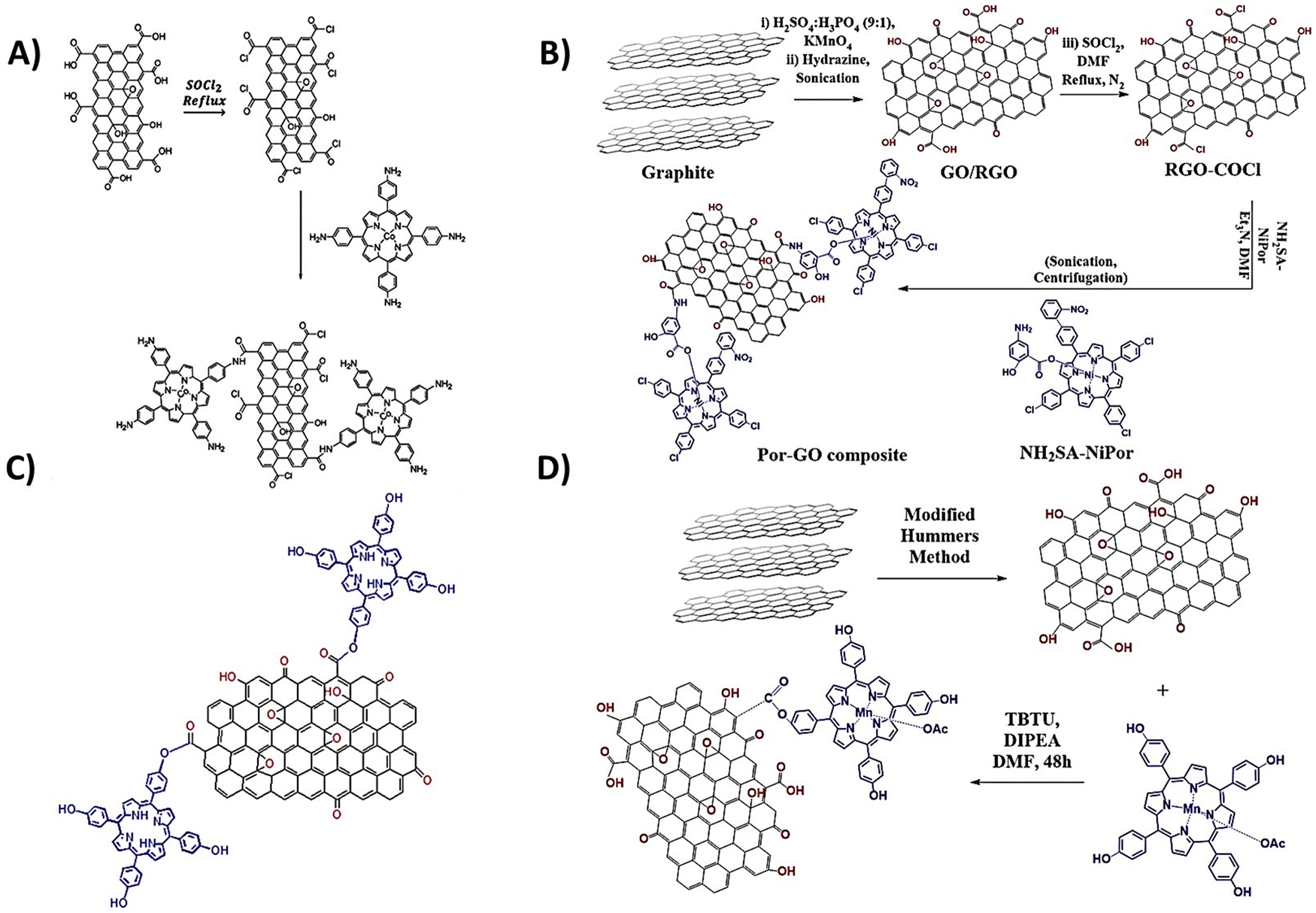 | ||
| Fig. 2 Schematic structure of covalent hybrids: (A) CoTAPP–GO, adapted from ref. 61 with permission from Elsevier (2018), (B) covalently linked axially ligated porphyrin–GO nanocomposite, based on ref. 62, and (C) TPPH–GO and (D) MnTPPH–GO, based on ref. 63. | ||
Ahmed et al. introduced a notable method for the covalent functionalization of GO with Por, as depicted in Fig. 2B.62 In their technique, the functionalization of GO was facilitated through an axial ligand on a Ni(II) metalloporphyrin, featuring an amide linker bridging the graphene oxide and the porphyrin unit. The amino group (–NH2) present on the axial salicylate ligand of the porphyrin macrocycle underwent reaction with the activated –COOH groups of GO, resulting in the establishment of an amide bond between Por and the GO surface. The synthesized hybrid was subsequently evaluated as a catalyst for the reduction of 4-aminophenol.
While the majority of studies focus on using edge groups in GO for amide bond formation, there are few reports that document the formation of the ester bond between the –COOH groups of GO and the –OH group of porphyrins (Fig. 2C and D).63–65 For instance, Zare-Dorabei et al. showcased the attachment of TPPH to GO via an esterification process. This TPPH–GO hybrid was then examined for its utility as an optical sensor in detecting mercury ions.64 Similarly, Rayati et al. documented the grafting of manganese porphyrin MnTPPH on graphene oxide also via ester bond formation.63 In a separate study, Mirzaeian et al. achieved the functionalization of GO using the Fe(III) complex TCPP via an ester linkage.65 The resulting material was investigated for its effectiveness as a catalyst in the removal of mercaptan from gas streams.
The covalent bonding of porphyrins to GBMs using functional groups at the edges of graphene sheets suffers from a significant limitation. The scarcity of edge carboxylic groups results in a relatively low functionalization rate, typically adding one functional porphyrin for every several hundreds of carbon atoms of the GO framework. Consequently, there is a pressing need for the development of new, simple, effective and robust functionalization approaches to create advanced graphene derivatives with a significantly enhanced degree of functionalization. In this context, the concept of attaching various functional groups directly onto graphene sheets is particularly promising, as it could foster more efficient interactions between the two components of the hybrid compared to merely attaching the organic chromophore at the graphene sheets' edges and/or defect sites.
This approach was effectively applied with two diazonium salt derivatives of TPP–NH2 (TPP1 and TPP2) (Fig. 3A) to form a covalent bond with the RGO basal plane.66 The XPS analyses revealed that the TPP1 and TPP2 contents in the hybrids reached as high as 26.28% and 35.24%, respectively. In a different strategy, Zhang et al. introduced an efficient two-step reaction for the covalent functionalization of GO.67 Initially, the –NH2 group of ethylenediamine reacted with GO, leading to the opening of epoxy rings on the GO surface and simultaneously engaging in Schiff base reactions with the ketone groups on GO, thus anchoring the ethylenediamine linker onto GO (Fig. 3B). Subsequently, the free –NH2 groups of ethylenediamine-modified GO further reacted with the porphyrin containing the –CHO group to produce the final product. This approach facilitated a much higher degree of functionalization than that of traditional techniques, such as covalent attachment of the porphyrin to GBM via functional groups at the edges of graphene sheets. XPS analysis confirmed that the resulting hybrid featured one porphyrin molecule for every 34 carbon atoms of the GO framework. This level of porphyrin incorporation significantly surpasses that achieved in the hybrids synthesized via the Suzuki coupling reaction (see next paragraph and Fig. 3C)68 or those obtained from porphyrin-modified liquid-phase exfoliated graphene via cycloaddition reactions, which amounted to one porphyrin molecule per 240 carbon atoms of the graphene sheet.69
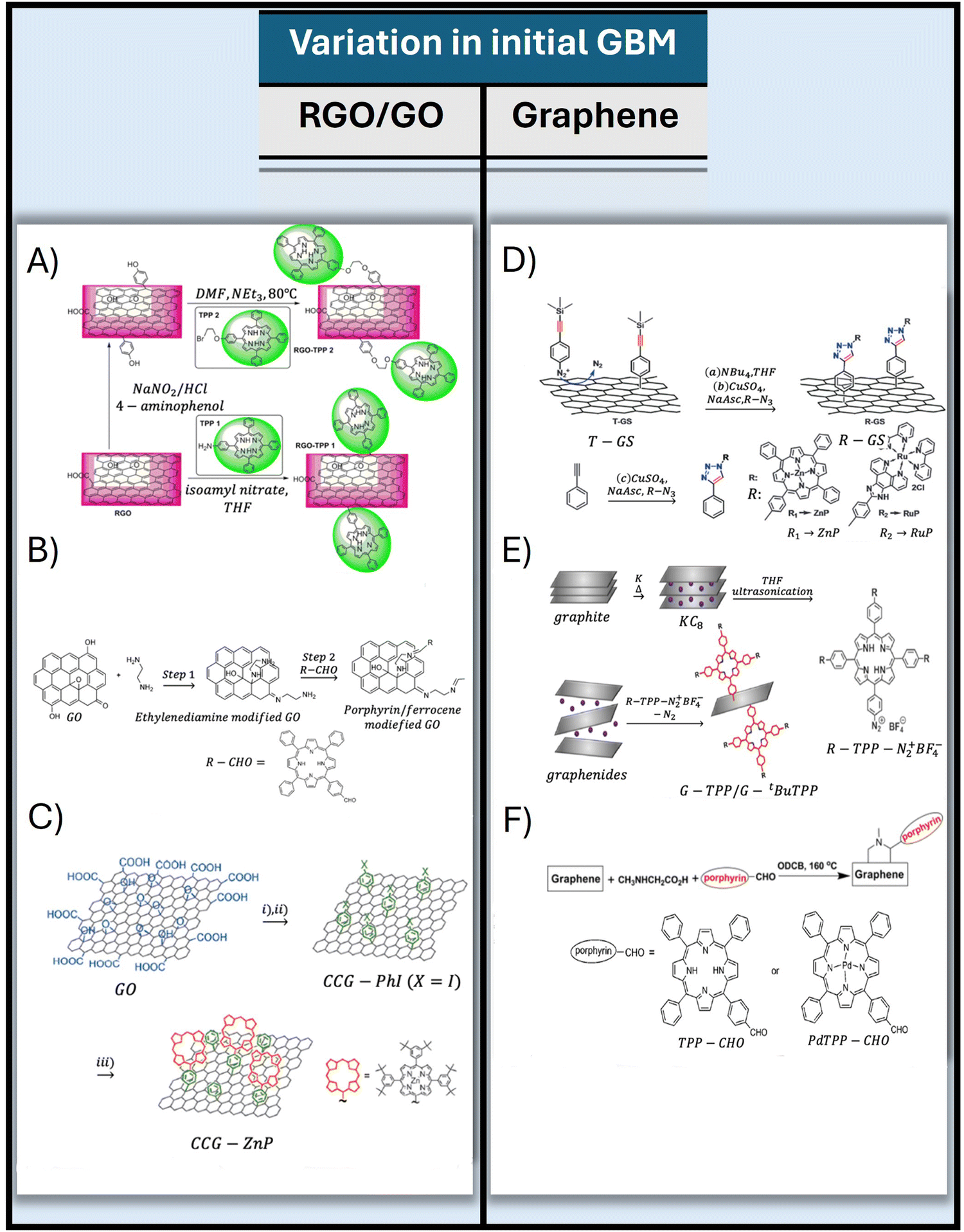 | ||
| Fig. 3 Illustration of various methods for covalently linking porphyrins to graphene and its derivatives through advanced chemical techniques: (A) scheme of the diazonium chemistry approach for attaching porphyrins to RGO, adapted from ref. 66, (B) scheme of the preparation of functionalized graphene derivatives with porphyrin-CHO via an epoxy ring-opening reaction, adapted from ref. 67 with permission from Wiley (2018), (C) description for the preparation of Por-CCG using a phenylene spacer achieved via the Suzuki coupling reaction, adapted from ref. 68 with permission from Wiley (2012), (D) presentation of the “click’’ chemistry method between the deprotected T-GS (graphene sheet modified with 4-(trimethylsilyl)ethynylaniline) and azide-terminated (zinc-porphyrin), adapted from ref. 70 with permission from the Royal Society of Chemistry (2012), (E) outline of the reductive synthesis approach for the Por-graphene hybrid via a direct σ-bond linkage of the functional components, adapted with permission from ref. 71. Copyright 2017, American Chemical Society, and (F) representation of the synthesis of graphene-TPP and graphene-PdTPP hybrid materials, adapted from ref. 69 with permission from Wiley (2011). | ||
Furthermore, Umeyama et al. established a protocol for the covalent attachment of porphyrin onto CCG using a short, rigid phenylene spacer.68 The synthesis involved a two-stage functionalization process for CCG, as illustrated in Fig. 3C. Initially, surfactant-encapsulated CCG was prepared to undergo a reaction with a p-iodobenzenediazonium salt (PhI). This step was followed by a Suzuki coupling reaction between CCG-PhI and a porphyrin boronic ester, resulting in the formation of a porphyrin-grafted CCG. The XPS analysis of the resulting Por-CCG hybrid indicated the presence of one porphyrin unit for every 400 carbon atoms of the CCG surface.
In most research focused on covalent Por-GBM composites, graphene oxide served as the foundational material, with its characteristics strongly pre-determined by the presence of oxygen-containing functional groups. These groups also affect the final properties of graphene-based hybrid nanomaterials. As a result, in recent years, considerable attention has been paid to the covalent functionalization of pristine graphene.69–71 Despite these efforts, the obtained level of functionalization is frequently modest, and the processes generally demand extended reaction periods.
Wang et al. demonstrated the application of “click” chemistry in the attachment of porphyrin molecules to the surface of graphene sheets (GS) (Fig. 3D).70 The GS were first altered by 4-(trimethylsilyl)ethynylaniline (T-GS) via an aryl diazonium salt reaction. This was followed by “click” chemistry involving the deprotected T-GS and azide-terminated zinc–porphyrin derivatives, resulting in a product with notable solubility in common organic solvents.
A one-pot covalent coupling method for attaching porphyrins to graphene via reductive diazotation (Fig. 3E) was explored by Dasler et al.71 The process began with a graphite potassium intercalation compound dispersed in tetrahydrofuran, which was then treated with porphyrin-diazonium salts, leading to the establishment of a single σ-bond between the porphyrin and graphene. This arrangement ensured perpendicular orientation of the porphyrin macrocycle relative to the graphene surface. Additionally, Zhang et al. reported a one-pot functionalization process of pristine graphene with TPP as well as PdTPP via 1,3-dipolar cycloaddition of azomethine ylides, as shown in Fig. 3F.69 In this case, graphite flakes underwent sonication in o-dichlorobenzene to promote exfoliation. In the following step, sarcosine and the respective aldehydes containing the TPP and PdTPP macrocycles were introduced. Stirring the mixture at 160 °C under N2 for one week resulted in the formation of structures shown in Fig. 3F.
2.2 Non-covalent functionalization
In contrast to the complex procedures involved in crafting covalent composites, the formulation of hybrid-based non-covalent interactions between GBMs and porphyrins presents several benefits, including enhanced yields, simpler synthetic paths, and the preservation of each component's primary characteristics within the hybrid.The non-covalent methodology relies on molecular interactions such as π–π stacking, hydrogen bonding, and electrostatic forces between the graphene and the porphyrin. Therefore, the structure of porphyrin (in particular the charge on the meso-substituent) along with the oxidation state of the GBM significantly affects the strength of the interaction between the two components of the hybrid. A major benefit of this approach is its straightforward synthesis, which in general simply involves mixing of a dye solution with a GBM dispersion and then centrifugation to separate the final product (Fig. 4A).
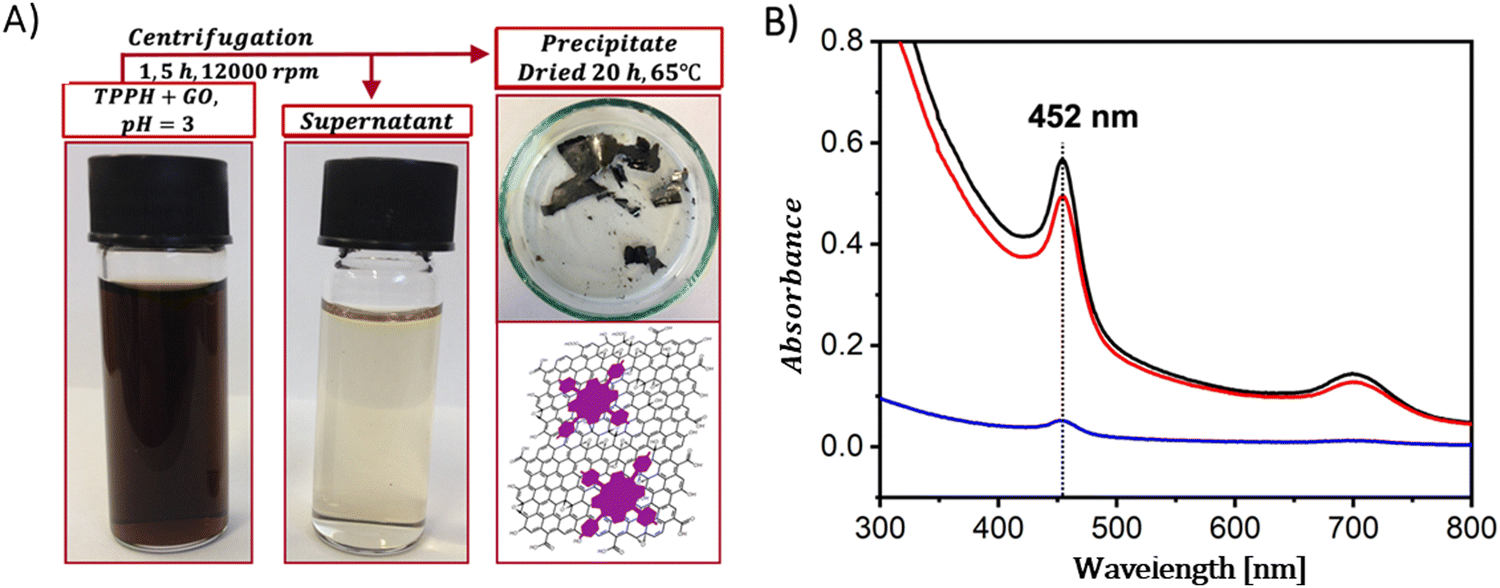 | ||
| Fig. 4 (A) Schematic summary of the formation process of the TPPH/GO nanohybrid. (B) Absorption spectra of the TPPH/GO hybrid suspension pre-centrifugation (black line), the spectrum of the supernatant post-centrifugation (blue line), and the re-suspended sediment (red line). Adapted with permission from ref. 72. Copyright 2019, American Chemical Society. | ||
A straightforward method to verify the effective adsorption of porphyrins on the GBM involves analyzing the UV-vis spectra of the supernatant following the centrifugation process (Fig. 4B). The detection of only a minor peak corresponding to the porphyrin in the supernatant indicates the successful isolation of the porphyrin/GMB as the sediment. However, the low concentration of the porphyrin within the hybrid material is a significant limitation of this technique, which is a particularly pronounced issue for neutral porphyrins that form assemblies primarily via π–π stacking.72–74 Recently, Fu and co-workers introduced an interesting strategy to enhance the π–π stacking interactions between porphyrin macrocycles and graphene.75 This method involves employing extensively π-conjugated chromophores such as porphyrin dimers75 for non-covalent attachment to the graphene surface (Fig. 5). The resulting composite demonstrated improved dispersibility in polar solvents owing to the strong van der Waals forces holding the porphyrin dimer on the graphene surface. Karousis et al. also reported a novel method for the preparation of Por/GBM composites.76 First, the authors synthesized positively charged imidazolium-modified graphene-oxide with Br− as the counter ion. Subsequently, these Br− ions were replaced with anionic protoporphyrin IX, illustrating another innovative approach to hybrid material preparation.76
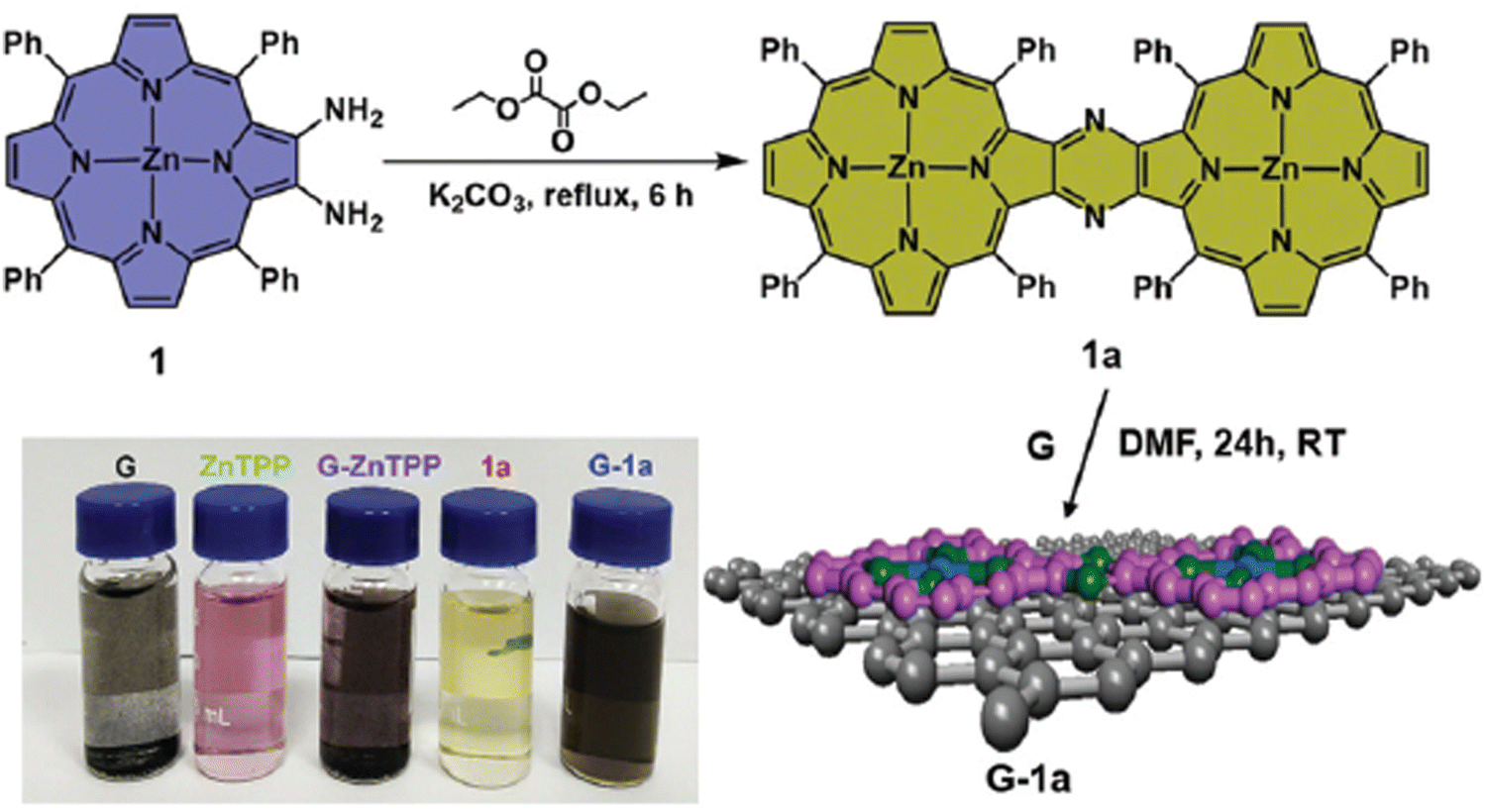 | ||
| Fig. 5 Schematic of the synthesis process of porphyrin dimer/graphene hybrids. Reproduced from ref. 75 with permission from the Centre National de la Recherche Scientifique (CNRS) and the Royal Society of Chemistry. | ||
In 2014, Chen et al. demonstrated the preparation of THPP/RGO hybrids incorporating porphyrin nanostructures and graphene sheets fashioned into free-standing films (Fig. 6A–D).77 This process entailed simple vacuum filtration of stable aqueous co-colloids comprising GO sheets and THPP nanoparticles, followed by reduction to produce the final hybrid material. Building on this innovative concept to Por/GBM material development, Ussia et al.78 delved further into the approach by fabricating free-standing hybrid nanostructured materials. These materials uniquely combined high-quality graphene 3D structures supported by a nickel foam and polyporphyrin (Fig. 6E).78 The methodology involved coating a nickel foam with graphene to provide high surface area for the subsequent attachment of the photosensitizer, with light sensitivity achieved through the use of polyporphyrins. The selection of cyclic polymeric porphyrins as the photoactive components was based on several considerations. First, the presence of cyclic porphyrin polymer structures on the graphene surface was anticipated to prevent the formation of non-photoactive dye aggregates. Second, polymers, as opposed to isolated molecules, are recognized for their ability to more effectively protect the co-catalyst surface.
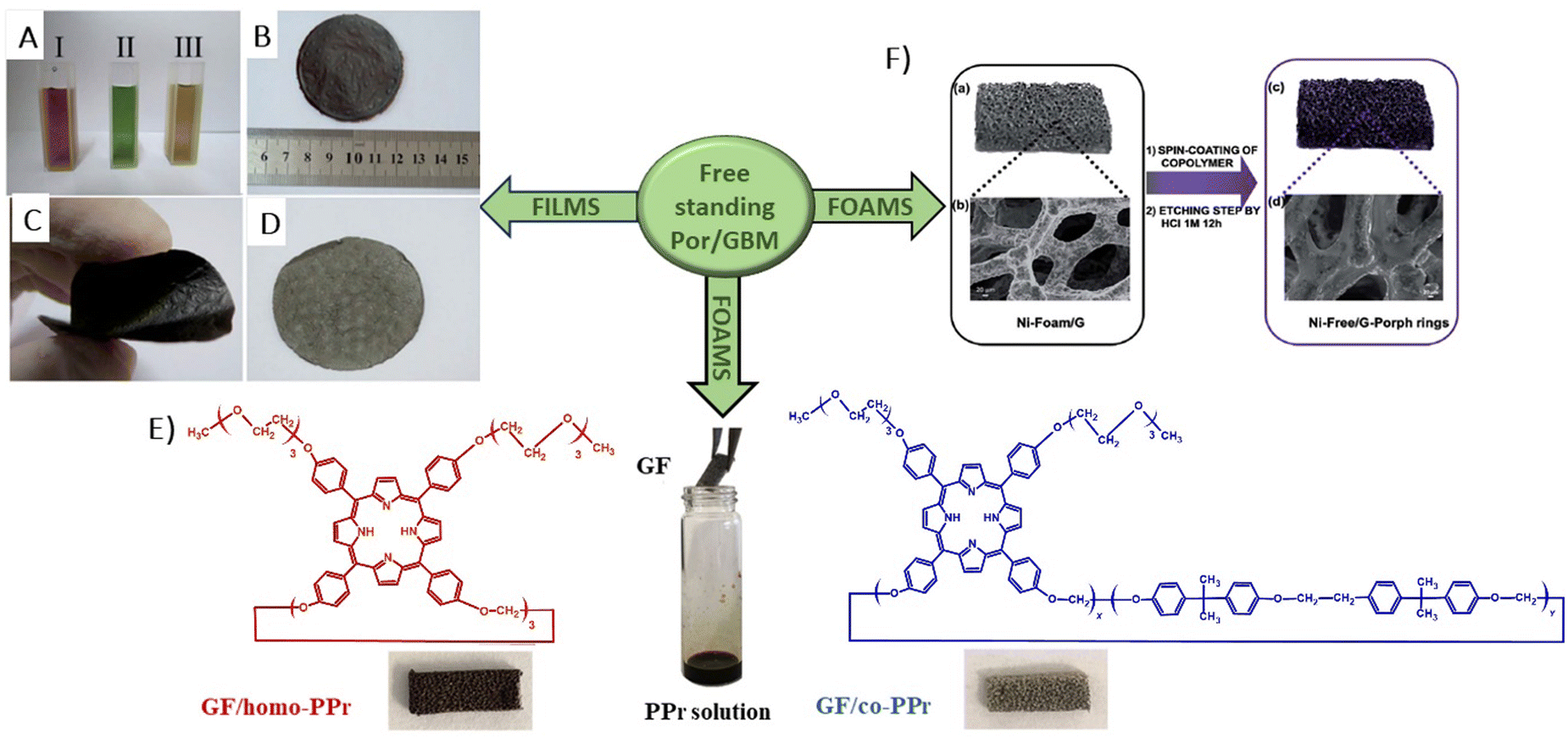 | ||
| Fig. 6 (A) Suspensions of GO (I), THPP (II) and THPP/GO (III) for vacuum filtration. The optical images of free-standing (B) GO, (C) RGO and (D) THPP/RGO films. Reproduced from ref. 77 with permission from the Royal Society of Chemistry, (E) structures of homo-PPr (red) and co-PPr (blue) used to coat a free-standing nickel foam covered with graphene and a formulation of graphene foam (GF)/polymer nanocomposites. Adapted from ref. 78, (F) Synthesis procedure of Ni free-G/Por rings. Adapted from ref. 79. | ||
Following their previous work, Ussia et al. demonstrated a novel technique for producing nickel-free hybrid materials, combining a graphene foam with porphyrin-based polymers.79 This efficient method effectively eliminates nickel, an element which is harmful for aquatic and human lives, from a graphene foam while preserving its 3D structure throughout the metal etching process. To accomplish this, water-insoluble polymeric materials featuring porphyrin rings were spin-coated onto the graphene foam. This coating successfully shielded the foam from defect formation and collapse during etching. The porphyrin rings functioned not only as a protective barrier against damage to the graphene foam, but also served as a photoactive component within the hybrid device.
Consequently, there was no need to remove them post-etching, which allowed to maintain the integrity of the 3D graphene architecture and to obtain the final product without any additional synthetic step required to solubilize the protective polymer layer. Moreover, this approach facilitated strong π–π interactions at the interface between the graphene foam and the porphyrin polymer, enhancing electron transfer processes vital for the efficient photocatalytic activity of the device. This enhancement was evidenced by a reduction in the emission spectrum intensity and through measurements of photocatalytic degradation.
3. Spectroscopic properties of Por and GBM hybrids
3.1 Ground-state properties
In 2009, the first report which presented data on the ground-state properties of CCG non-covalently functionalized with cationic porphyrin TMPyP was published.80 Xu et al. discovered that CCG, which is negatively charged in aqueous dispersions due to the presence of residual carboxyl groups, can be effectively combined with positively charged TMPyP via electrostatic and π–π stacking interactions.80 UV-vis absorption titration is a commonly applied technique to explore the ground-state interactions between the porphyrin and GBM, wherein the porphyrin concentration remains constant, while the GBM concentration varies (Fig. 7). Throughout such titrations, Xu et al. observed a progressive decrease in the intensity of the original Soret band (free porphyrin) at 421 nm, which decreased gradually with the increase in CCG concentration alongside the emergence of a new Soret band at 458 nm. Furthermore, the isosbestic point was identified at 429 nm, indicating a well-defined transition between free and adsorbed porphyrins (Fig. 7A). A spectrum that corresponds to TMPyP in the complex with CCG was identified by subtracting the contribution of the absorption from CCG dispersion from the UV-vis spectra of the porphyrin and CCG mixture. The UV-vis spectra of the TMPyP/CCG complex were characterized by a large bathochromic shift (37 nm), a broader half-bandwidth, and a lower molar extinction coefficient compared with that of free TMPyP. A bathochromic shift of the Soret band upon adsorption on GBM, the presence of the isosbestic point and the decreased molar extinction coefficient were also reported later for numerous other non-covalent as well as covalent assemblies of porphyrin with GBM (see Table 2). Xu et al. proposed three hypotheses for the observed large bathochromic shift of the Soret band: (1) protonation of the nitrogen atoms in the porphyrin macrocyclic ring, (2) J-aggregation of the porphyrin molecules and (3) flattening of the porphyrin molecule on the graphene sheet. They ruled out the first two possibilities based on the experimental conditions and observations. The protonation of the nitrogen atoms in the porphyrin ring can be achieved only in a highly acidic medium. Under the experimental conditions (pH 7–8), TMPyP existed as a free base bearing four positive charges at the meso substituents. Due to Coulombic repulsion among the porphyrin molecules themselves, their aggregation was not likely either. The clear isosbestic point observed during titration as well as the linear dependence of the absorbance at the maximum of the Soret band of nanohybrid on the CCG concentration suggested that the obtained material had a well-defined structure. The authors prepared a film of TMPyP with J-aggregated molecules for comparison, which exhibited only a 15 nm red-shift of the Soret band. This value was much lower than the one observed for the TMPyP/CCG complex (37 nm). Notably, AFM measurements corroborated their hypothesis, showing that TMPyP molecules were adsorbed onto CCG sheets as a monolayer. An increment of 0.6 nm was observed for the TMPyP/CCG hybrid in comparison to the thickness of CCG itself, which matched the thickness of the single TMPyP molecule.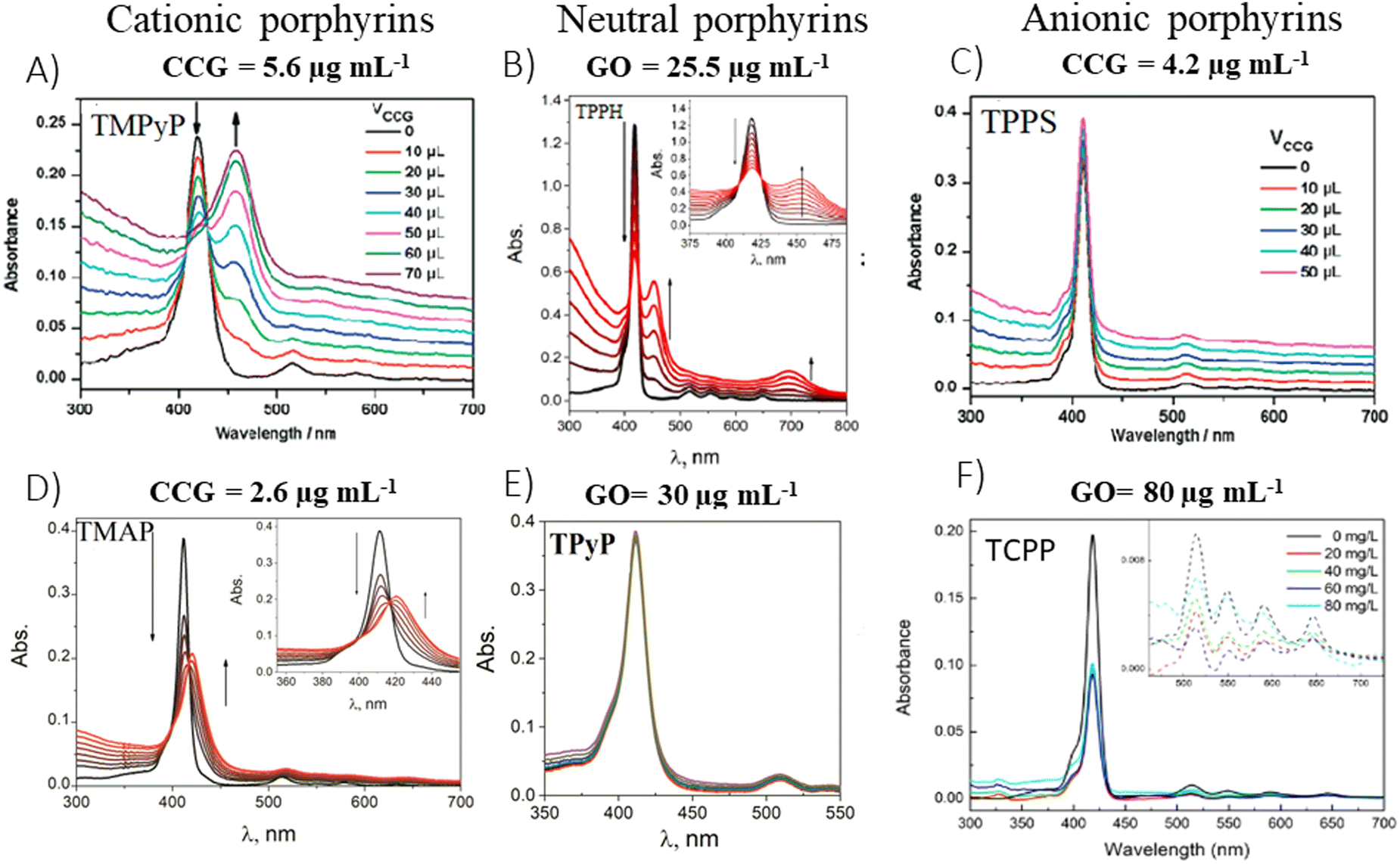 | ||
| Fig. 7 Titration-induced changes in the absorption spectra for various porphyrin/GBM: (A) TMPyP with CCG, (B) TPPH with GO, (C) TPPS with CCG, (D) TMAP with CCG, (E) TPyP with GO and (F) TCPP with GO. The concentrations listed above each panel indicate the final GBM concentration achieved upon completion of the porphyrin solution titration. Adapted from ref. 72, 80–82. Copyright 2019 American Chemical Society, Copyright 2009 American Chemical Society, Copyright 2019 John Wiley and Sons. | ||
| Porphyrin | GBM | Solvent | Soret band of free porphyrin (nm) | Soret band of porphyrin/GBM (nm) | Shift (nm) | Isosbestic point (nm) | Ref. |
|---|---|---|---|---|---|---|---|
| Non-covalent assemblies | |||||||
| TMPyP | CCG | H2O | 421 | 458 | 37 | 429 | 80 |
| TMAP | CCG | H2O | 411 | 436 | 35 | 416 | 80 |
| TMPyP | RGO | H2O | 422 | 452 | 30 | 442 | 83 |
| TMPyP | GO | H2O | 422 | 440 | 18 | 434 | 84 |
| ZnTMPyP | GO | H2O | 437 | 453 | 16 | 447 | 84 |
| TMAP | GO | H2O | 411 | 421 | 10 | 416 | 81 |
| TPPH | RGO | EtOH | 420 | 448 | 28 | — | 74 |
| TPPH | GO | EtOH-H2O | 418 | 452 | 34 | 428 | 85 |
| TPPH | RGO | EtOH-H2O | 418 | 442 | 24 | 429 | 85 |
| TAPP | GO | EtOH-H2O | 424 | 456 | 32 | 437 | 85 |
| TAPP | RGO | EtOH-H2O | 424 | 451 | 27 | 439 | 85 |
| CoTPP | RGO | DMF | 415 | 433 | 18 | 420 | 86 |
| Covalent assemblies | |||||||
| TPP-CHO-G | DMF | 419 | 421 | 2 | — | 69 | |
| PdTPP-CHO-G | DMF | 416 | 419 | 3 | — | 69 | |
| ZnP-CCG | DMF | 417 | 420 | 3 | — | 68 | |
| TPP-CHO-GO | EtOH | 415 | 425 | 10 | — | 67 | |
| TPPNH2-GO | DMF | 418 | 420 | 2 | — | 76 | |
| NH2 SA-NiPor-GO | DMF | 419 | 425 | 6 | — | 62 | |
| ZnP-GS | DMF | 426 | 430 | 4 | — | 70 | |
Therefore, the flattening phenomenon, driven by electrostatic and π–π stacking interactions, was responsible for the notable bathochromic shift of the Soret band in the TMPyP/CCG hybrid, highlighting the pivotal role of molecular orientation in the modulation of porphyrin's optical properties.87–89 The reorientation of the substituents into the same plane as the porphyrin ring, induced by combining TMPyP with CCG, extends the π conjugation leading to a notable bathochromic shift of the Soret band. Interestingly, the assembly of TMPyP on GO led to a less pronounced shift of only 15 nm.81 Furthermore, a smaller bathochromic shift (25 nm) was observed for another cationic porphyrin, TMAP, upon its interaction with CCG, compared to the shift observed with TMPyP/CCG (37 nm).84 The smaller shift for TMAP can be linked to its bulky cationic side groups, which interfere with the formation of a coplanar arrangement with the porphyrin core. Additionally, TMAP's interaction with GC (16 nm shift)90 and RGO (27 nm shift)91 lead to more significant shifts than this with TMAP/GO (10 nm shift), though all of them were lower than this with TMAP/CCG.81
To explore the impact of electrostatic attraction on hybrid formation, the cationic TMPyP was substituted with anionic TPPS. This change led to no noticeable alteration in the UV-vis spectra upon the addition of CCG (Fig. 7C). Bajjou et al. reported that the position of the Soret band of the TPPS was not affected by the presence of RGO either.92 Similar observations were made with neutral porphyrins like TPyP, as their absorption spectra remained largely unchanged in the presence of GBMs (Fig. 7E),81,93 underscoring the critical role of positive charge in facilitating effective Por interaction with GBM.
Siklitskaya et al. further validated these findings through theoretical studies regarding the interaction between neutral porphyrins, TPPH and TAPP, with GO and RGO (Fig. 8).85 Their calculations based on the Lerf–Klinowski model of GO and RGO suggested the flattening of these molecules upon adsorption, as evidenced by the reduced dihedral angle between the phenyl and porphyrin planes.85 The determined dihedral angle was equal to approx. 60° for the isolated porphyrins, while it ranged between 41° and 49° in the case of the porphyrin adsorbed on the GO surface.85 This study highlights the subtle yet significant molecular rearrangements that underpin these complex interactions.
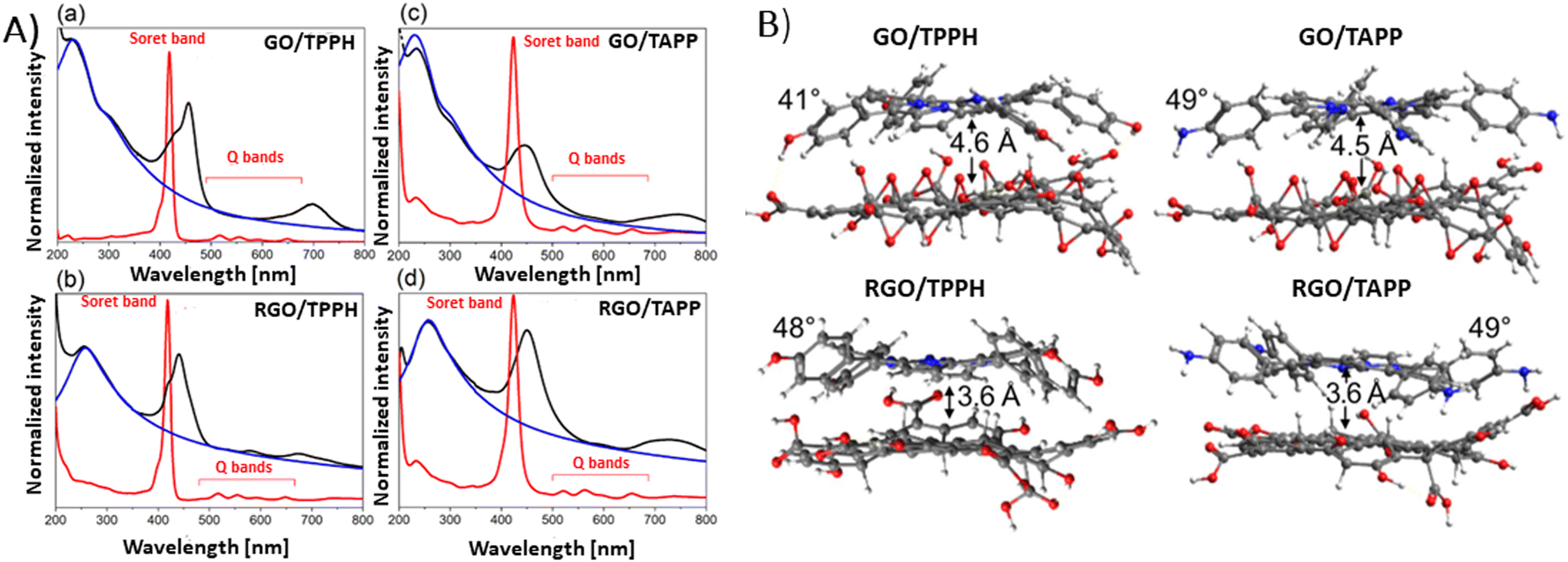 | ||
| Fig. 8 (A) Absorption spectra (black) obtained for (a) GO/TPPH, (b) RGO/TPPH, (c) GO/TAPP and (d) RGO/TAPP with the spectra of the unbound porphyrins TPPH and TAPP in red, and the spectra of isolated GO and RGO in blue. (B) Illustration of the optimized geometries for each porphyrin-graphene hybrid. Adapted from ref. 85. | ||
Theoretical analyses by Siklitskaya et al. also revealed that in complexes involving RGO, porphyrin rings were on average 1 Å nearer to the graphene plane than in those complexes with GO (Fig. 8B). Consequently, the interaction between RGO and porphyrin is anticipated to be more pronounced than in similar complexes with GO. This hypothesis was supported by higher interaction energies obtained for RGO than for GO, probably as a result of improved π–π stacking interactions.
Except for Xu et al.,80 numerous other authors81,83,84 have observed that the spectral alterations resulting from the incorporation of GBMs into aqueous solutions of cationic porphyrins were more distinct than those with TAPP and TPPH, since approximately tenfold less GBMs were required to induce noticeable spectral changes (Fig. 7). The authors concluded that the interaction of GBMs with neutral porphyrins is weaker than that with positively charged variants.
Gacka et al. explored how pH affects the interaction strength between TPPH and GO.72 The net charge of the TPPH molecule varied between negative (−4), neutral and positive (+2) depending on the solution's pH. These changes were related to the protonation of the imino nitrogen atoms and deprotonation of the –OH groups. This adjustment allowed for the on-demand tuning of the electrostatic interaction between TPPH and GO by selecting an appropriate pH level for the solution (Fig. 9). The findings revealed that both neutral TPPH and positively charged TPPH2+ molecules could attach to the surface of GO. However, the interaction was found to be stronger for TPPH2+, which corresponds well with the previous studies. In contrast, for the negatively charged TPPH4−, no significant changes in the UV-vis spectra of TPPH4− were registered in the presence of GO. Moreover, after the centrifugation of the suspension, almost all TPPH4− remained in the supernatant, indicating that the interaction between TPPH4− and GO was largely suppressed.
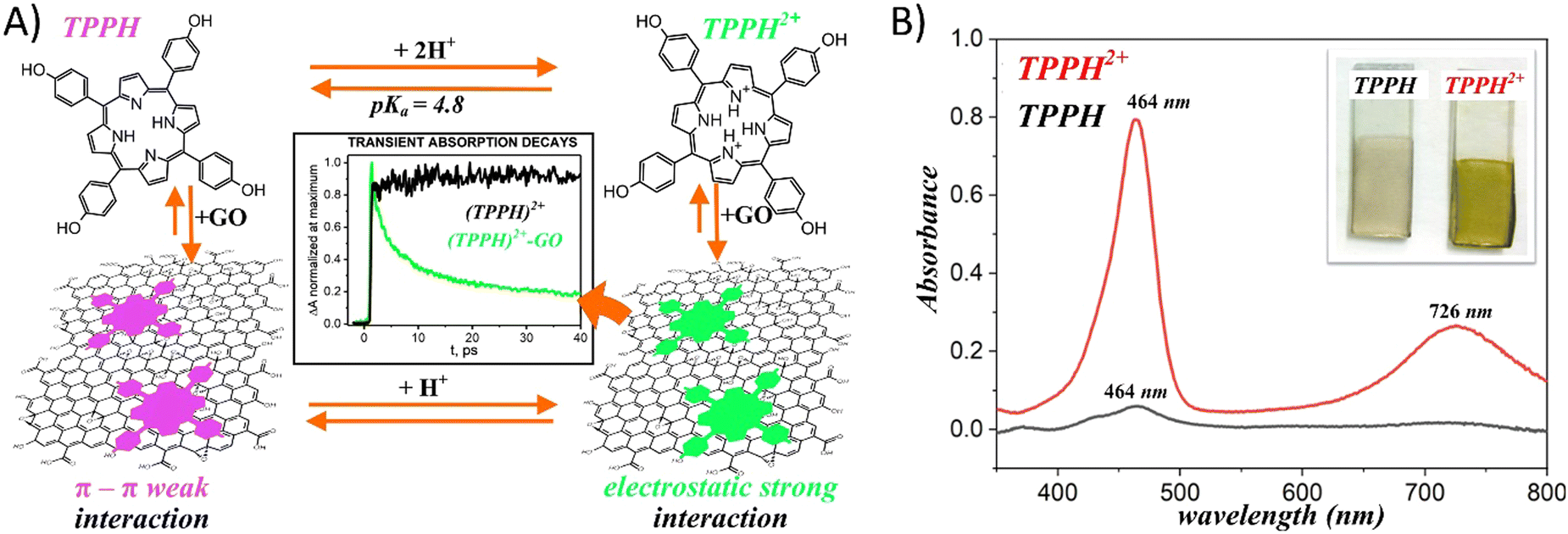 | ||
| Fig. 9 (A) Schematic of the pH influence on the interaction of TPPH with GO. (B) Comparison of the adsorption of porphyrins (TPPH (black) and cationic TPPH2+ (red)) from solutions on GO films monitored by UV-vis spectroscopy. Adapted with permission from ref. 72. Copyright 2019 American Chemical Society. | ||
The authors complemented their spectroscopic studies with theoretical calculations which provided further evidences for stronger interaction between cationic TPPH2+ and GO. Interaction energies were determined to be −22.4 kcal mol−1 and −58.0 kcal mol−1 for TPPH/GO and TPPH2+/GO, respectively. Furthermore, the calculated center-to-center distance between TPPH2+ and GO was equal to 4.4 Å, in contrast to 4.55 Å obtained for the TPPH/GO nanohybrid. Such a result indicated that the electronic coupling matrix element between graphene oxide and porphyrin was stronger in the former case.
As previously discussed, in the case of cationic porphyrins, the electrostatic interactions with GBMs have been identified as the primary mechanism, which facilitates the assembly of these porphyrins onto graphene sheets. In the case of neutral porphyrins, π–π stacking interactions and hydrogen bonding emerge as the leading forces driving the formation of composites with GBMs. Gacka et al. compared the interaction of the TPPH and meso-tetra(4-methoxyphenyl)porphyrin (TPPOMe) with GO. By blocking the OH groups with the methoxy group, the hydrogen bonding capability between TPPOMe and GO was effectively diminished.72
Through the use of absorption spectroscopy to observe the titration of porphyrin with GO, it was discovered that the interaction strength between TPPOMe and GO matched that between TPPH and GO. This finding underscored the necessity of π–π stacking interaction for the efficient formation of porphyrin nanoassemblies with GO, which is achievable simply by mixing the solutions of both components. Shu et al. demonstrated that the interaction between neutral porphyrin CoTPP and GO could be enhanced through ultrasonic treatment.94 Without ultrasonic assistance, no significant changes were noted in the absorption spectra of CoTPP following the addition of GO. However, interaction under ultrasonic oscillation led to the emergence of a new characteristic peak at 432 nm. Moreover, the intensity of this peak at 432 nm progressively increased with prolonged ultrasonic treatment, while the absorbance associated with unbound CoTPP at 416 nm gradually diminished. These results revealed that the ultrasonic oscillation accelerated the assembly of CoTPP on the GO sheets. The authors speculated that such ultrasonic oscillation aids the flattening of CoTPP molecules on the GO surface.
Ge et al. examined how functional groups attached to the edges of the porphyrin influence both the mode of binding to GO and the spectroscopic characteristics of the porphyrin/GO composites.95 Three porphyrins, each with a varying number of hydroxyl groups were selected for study, to understand the impact of peripheral substituents on the interaction of porphyrin with GO (Fig. 10).
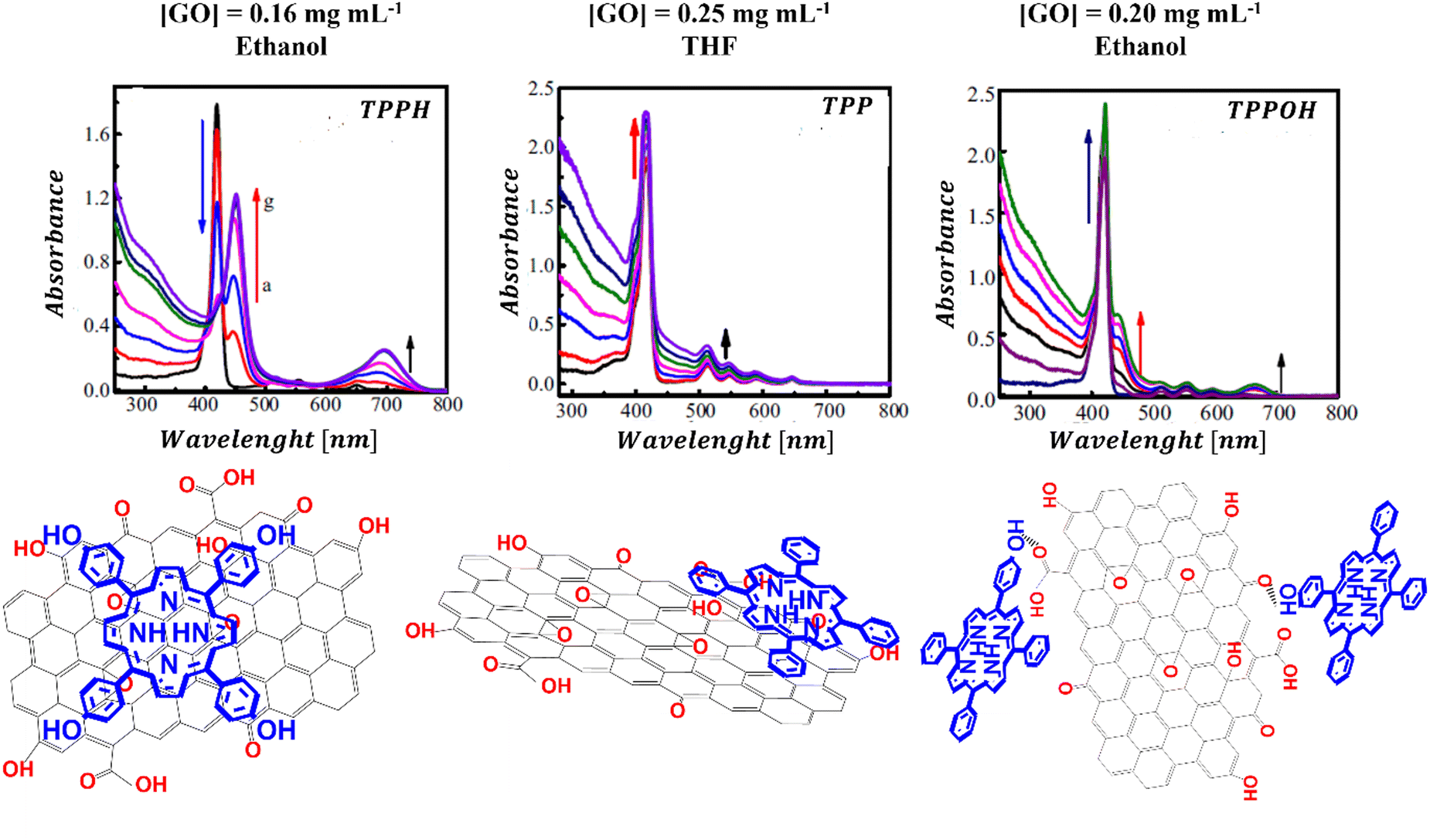 | ||
| Fig. 10 UV-vis absorption spectra and structural schemes of TPPH/GO, TPP/GO, and TPPOH/GO. Adapted with permission from ref. 95. Copyright 2015 Elsevier. | ||
The results revealed that the binding strength between GO and the porphyrins follows the order of TPPH/GO > TPPOH/GO > TPP/GO. It appears that the hydroxyl substituents alongside the TPPH core synergistically facilitate the assembly of the TPPH/GO hybrid allowing the TPPH molecules to flatten on the surface of GO. For the TPP porphyrin, π–π stacking interaction emerges as the sole driving force behind the hybrid formation. The observed weak interaction between TPP and GO was attributed to the presence of hydroxyl and epoxy functional groups on GO sheets, which hindered the efficient anchoring of TPP molecules on the surface of GO and prevented close contact between the graphene surface and the porphyrin macrocycle. The binding modes for the three porphyrins with GO that were deduced based on the cited study are depicted in Fig. 10.
In addition to the shifts in the Soret band of the porphyrin upon assembling with the GO, the location and number of Q-bands can be changed as well.72,83,84 For instance, in TMAP/GO composites, all four Q-band transitioned from 513, 550, 587, and 633 nm to 519, 554, 585 and 640 nm, respectively.81 For TPPH/GO hybrids, three out of the four Q-bands vanished following the nanohybrid formation, while a new broad band appeared at 699 nm.81 Similarly, the Q-bands’ disappearance and the emergence of a broad band at 750 nm were noted in TAPP hybrids with both GO and RGO, as well as in ZnTPPH with GO.85,96 This phenomenon can be attributed to a partial charge transfer from the TPPH to the GO sheet, leading to the formation of a positive charge on the porphyrin core, analogous to TPPH2+.
An interesting approach for assessing the maximal quantity of porphyrin, which can adhere to GBMs, involves reverse spectroscopic titration, in which the GBM concentration is held steady while porphyrin is successively introduced into the mixture. Initially, the UV-vis spectra predominantly display the Soret band corresponding to porphyrin attached to the GBM surface. As the concentration of porphyrin surpasses the highest chromophore amount that can be accommodated by the specified quantity of GBMs, the Soret band associated with the free porphyrin begins to emerge (Fig. 11). From such evaluations, it was deduced that the maximal quantities of TPPH, which could be adsorbed on RGO and GO, were equal to 0.11 and 0.036 mg per 1 mg of GBM, respectively (Fig. 11A and B).85,97 For cationic porphyrins such as ZnTMPyP, it was found that 0.15 mg of the chromophore can bind to 1 mg of GO.84
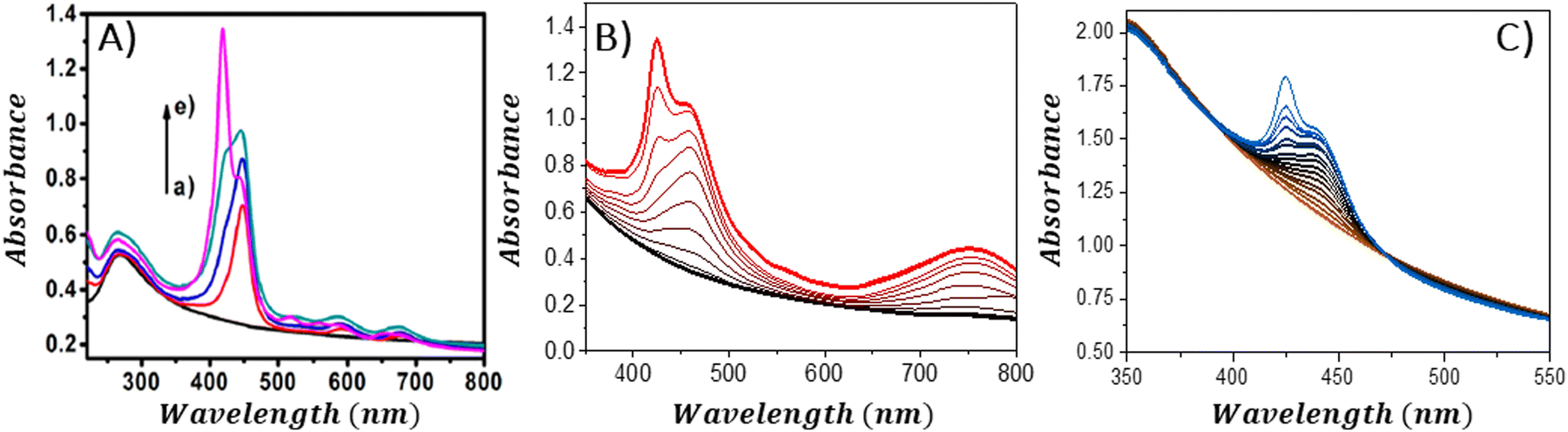 | ||
| Fig. 11 Absorption spectra obtained during the titration of the GBM solution (constant concentration) with the increase in the amount of porphyrin: (A) TPPH added to RGO; (B) TAPP to GO and (C) ZnTPPH to GO. Reproduced from ref. 85, 96 and 97. Copyright 2013, American Chemical Society. | ||
The UV-vis spectroscopy is also an easy method for the confirmation of the presence of porphyrin in the covalent Por–GBM hybrids. However, in contrast to non-covalent assemblies, the Soret band shift of the porphyrin covalently bound to GBMs is usually not observed (Table 2). For example, for GO covalently functionalized with TPPNH2, the porphyrin absorption band was broadened and bathochromically shifted by only 2 nm.59 In contrast, the shift observed for GO non-covalently functionalized with similar porphyrin (TAPP) was much more pronounced.85 Other covalent graphene-porphyrin composites which were formed via amide or ester linkage between –COOH at the edges of GO flakes and porphyrins possessing appropriate functional group(s) also exhibited a lack of significant change in the Soret band position as compared to the free chromophores.59,64 The absence of noticeable changes in porphyrin absorption upon covalent binding to GO might be explained by the rigid amide (or ester) bond preventing molecules from interacting via π–π stacking with the GBM surface they are linked to. Furthermore, it was reported that the Soret band position was red-shifted by 6 nm for covalent hybrids when porphyrin with modified longer amide axial ligands was used and by 11 nm when the porphyrin was attached directly to the GBM surface with the use of the epoxy groups present on the GO plane.62,67 These examples indicate that π–π stacking between hybrid components is essential to observe changes in the UV-vis spectra of porphyrin.
3.2 Excited-state properties
The porphyrin and graphene hybrids emerge as particularly attractive materials for light energy harvesting and offer almost limitless possibilities of on-demand tuning of their photophysical and photochemical properties essential for the efficient performance of future light-harvesting devices. Therefore, the ability of GBM to interact with the excited states of porphyrins has drawn tremendous attention of scientists in the last 10 years.98–103 Excited-state interactions between the components of the hybrids have important implications not only for the solar cells, but also for energy conversion applications such as photocatalytic water splitting, dye photodegradation and CO2 reduction.These interactions can conveniently be probed by monitoring the emission of radiation from the photoexcited porphyrin in the vicinity of graphene (covalently or non-covalently attached to graphene sheets). A common method employed for this purpose is steady-state emission spectroscopy, which is favored for its widespread availability of spectrofluorometers in many research facilities. The procedure typically involves a comparison of the fluorescence intensity of the free porphyrin against its intensity in the presence of GBM, which acts as the quencher. Even though in that case emission measurements allow relatively straightforward comparison of the porphyrin excited-state properties with and without graphene at first sight, one has to be aware that quantitative analysis of such fluorescence data is very demanding. When carried out inappropriately, it might result in wrong interpretation of the behavior of the studied hybrid in the excited state. For instance, fluorescence intensity can be reduced by the presence of any compound that is capable of absorbing a portion of the excitation energy.
Hence, correction for the absorption of part of the excitation light by the GBM (and for absorption of Por fluorescence by the GBM) – using appropriate equations for the inner filter effect – is crucial, particularly for hybrids which exhibit weak porphyrin–GBM interactions that necessitate higher concentrations of GBM for observable quenching of emission.104,105 Gacka and co-workers determined that the correction needed for the absorption of the excitation light by GO in their study on the non-covalent GO hybrid with neutral TPPH reached 24% at a GO concentration of 0.018 mg mL−1.72 In contrast, Larowska et al. found that during their investigation of emission quenching in a non-covalent GO hybrid with cationic TMAP, the correction necessary due to GO's absorption amounted to merely 0.5%, attributed to the notably low GO concentration (3.6 × 10−4 mg mL−1) required to effectively quench the emission.81 Beyond adjustment for the GBM's absorption of excitation light, it is essential to ensure the porphyrin's absorbance at the excitation wavelength matches between samples for an accurate quantitative analysis of emission data. If not properly accounted for, the measured reduction in the fluorescence intensity of porphyrin upon introducing GBMs could simply reflect changes in the absorbance of porphyrin at the excitation wavelength. As shown in Fig. 7, the absorbance at the Soret band of porphyrin can change dramatically upon addition of GBM. To maintain constant absorbance of porphyrin throughout the emission quenching process with the GBM, various researchers have suggested employing the isosbestic point for the excitation of both the isolated porphyrin and its GBM hybrid samples.72,73,81,83,84 A notable case involves the study of eosin Y (EY) emission quenching by GO, in which it was concluded that GO does not actually quench the EY emission in any significant way after adjusting for three key factors: (i) GO's absorption of the excitation light, (ii) GO's absorption of EY's emitted light, and (iii) small change of the EY absorbance at the excitation wavelength after binding to GO.104
A decrease in the porphyrins’ fluorescence intensity in the presence of GBM has been documented in several studies (Fig. 12). For instance, Masih et al.106 reported that the quenching efficiency for ZnTMPyP was equal to 50% at a graphene carboxylate concentration of 5.7 × 10−3 mg mL−1, and Wojcik et al.83 found a similar 50% emission quenching efficiency for TMPyP with RGO at a concentration of 8.0 × 10−4 mg mL−1. Additionally, it was determined that a GO concentration of 4.2 × 10−4 mg mL−1 was necessary to achieve 50% emission quenching of TMPyP.84 For neutral porphyrins, such as ZnTPPH, achieving a 39% reduction in emission required a GO concentration of 1.0 × 10−2 mg mL−1.73
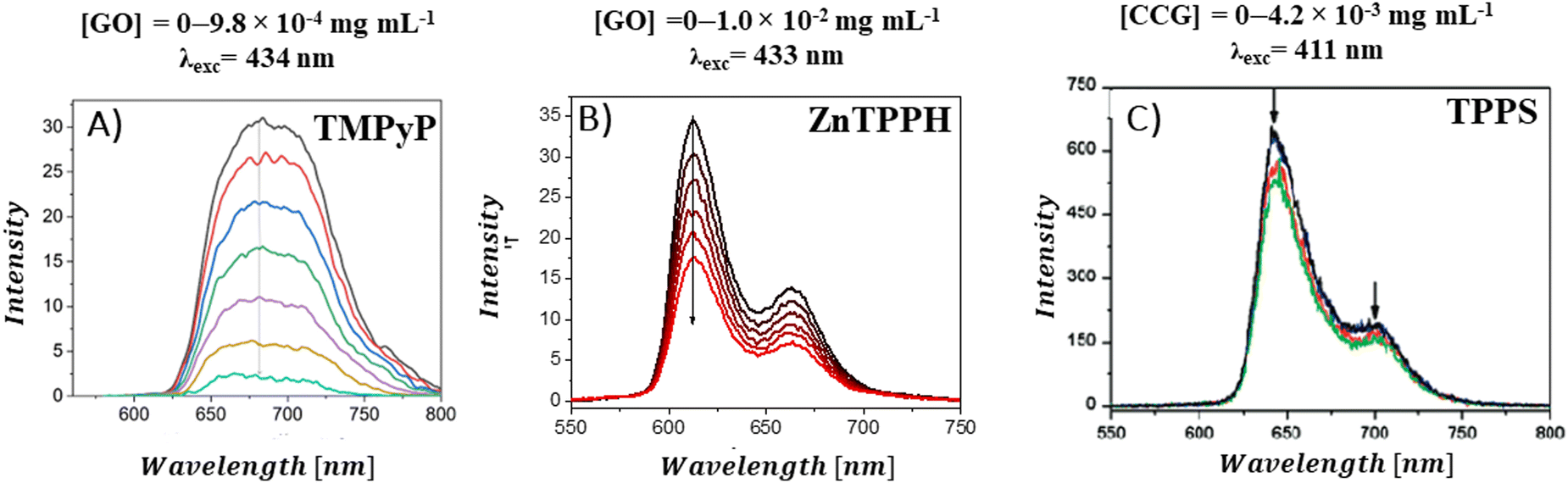 | ||
| Fig. 12 Quenching of the fluorescence of (A) TMPyP in the presence of GO, (B) ZnTPPH in the presence of GO and (C) TPPS in the presence of CCG. Adapted with permission from ref. 73, 80 and 84. Copyright 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim and Copyright 2009 American Chemical Society. | ||
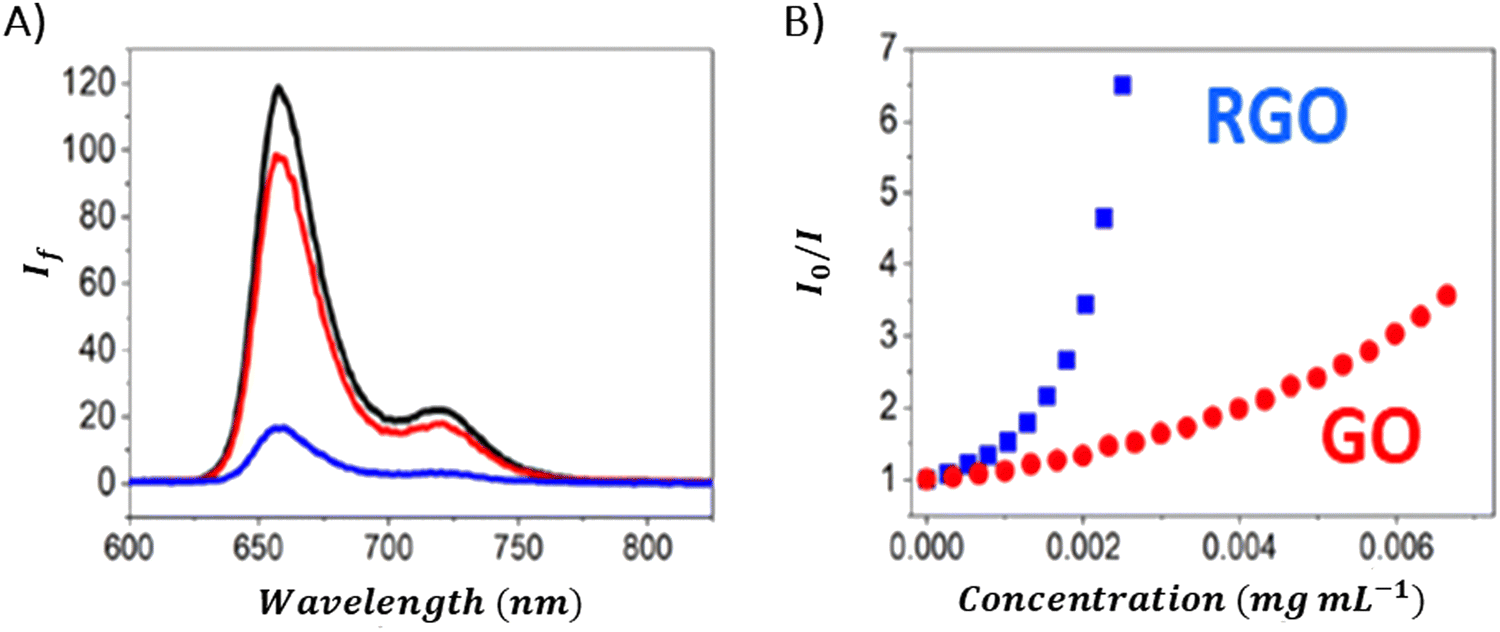
There are also reports in which no emission quenching for neutral porphyrins in the presence of GBM was observed: for example for TPyP composites with GC93 or TPyP composites with GO.95 For anionic TPPS, a slight decrease in emission intensity was observed in the presence of CCG (Fig. 12C).80 As evident from various reports published to date, significantly lower GBM concentrations are needed to efficiently quench the emission of cationic porphyrins compared to those required for neutral porphyrins. This difference is largely attributed to the electrostatic interaction between the cationic porphyrin and the negatively charged graphene material, which facilitates the quenching process,29,42,51 as opposed to neutral porphyrins in case of which the interaction with the graphene-based material is primarily through π–π stacking mechanisms.21,47,51,52 Careful comparison of the quenching efficiency under the same conditions allows for the assessment of the interaction strength between Por and GBM in various composites.
Ge and co-workers examined the interactions between certain neutral porphyrins (TPPH, TPPOH, and TPP) and GO, by determining the apparent binding constant using a Stern–Volmer-type equation.107 They found that the binding constant for TPPH reached 128.35 mL mg−1, surpassing those of TPPOH (KGO-TPPOH = 5.39 mL mg−1) and TPP (KGO-TPP = 0.39 mL mg−1), which indicated a stronger interaction with GO.107 Siklitskaya et al. conducted experiments on the steady-state emission to evaluate how GO and RGO affected the fluorescence of two neutral porphyrins: TPPH and TAPP.85 They found that RGO reduced the fluorescence of porphyrins (Fig. 13) more effectively, a phenomenon they linked to the improved π–π stacking interactions among the hybrid components. This suggests a more pronounced interaction mechanism facilitated by RGO's structural properties.
 | ||
| Fig. 13 (A) Quenching of the fluorescence of TPPH by GO (red) and RGO (blue) with the black curve corresponding to the emission of the porphyrin in the absence of graphene materials. Spectra were corrected for the inner filter effect. (B) Relationship between the fluorescence intensity and the GO (red) or RGO (blue) concentration. Adapted from ref. 85. | ||
The steady-state emission studies alone are insufficient since definite identification of the quenching mechanism which occurs in the presence of graphene material cannot be unambiguously determined. The observed reduction in emission intensity when graphene-based materials (GBM) are introduced is often mistakenly attributed to electron or energy transfer, neglecting the consideration of alternative quenching routes.108–111 As mentioned in several studies reporting emission quenching of various porphyrins (TMAP, TMPyP, ZnTMPyP, TPPH, TAPP and ZnTPPH) by graphene materials in solutions, two forms of porphyrin exist in equilibrium: free and adsorbed on the GBM. Both forms can contribute to the observed fluorescence. The alteration in the electronic structure of porphyrins upon adsorption onto GBM is evidenced by the changes in their UV-vis absorption spectra, distinct from those of free porphyrins.72,73,81,82,84,112 This suggest that the emission characteristics of porphyrin assembled with the GBM surface should also vary from those of unbound porphyrin. Despite this expectation, no measurable differences in both shape and band position of the peak in the emission spectra for corresponding porphyrins and their hybrids with graphene were detected.72,73,81,82,84,112 Furthermore, the fluorescence excitation spectra of these hybrids closely mirrored the UV-vis absorption spectra of the unbound porphyrins (Fig. 14). This led researchers to conclude that the porphyrins, namely, TMAP, TMPyP, ZnTMPyP, TPPH, TAPP, and ZnTPPH adsorbed on GO either do not emit light or their emission falls below detectable levels.72,73,81,82,84,112 This lack of measurable emission for porphyrin adsorbed on GO is thought to result from the rapid deactivation of the excited state of porphyrin molecules through various non-radiative processes, such as electron transfer, energy transfer or intersystem crossing, occurring once the porphyrin macrocycle is adsorbed onto a graphene sheet.
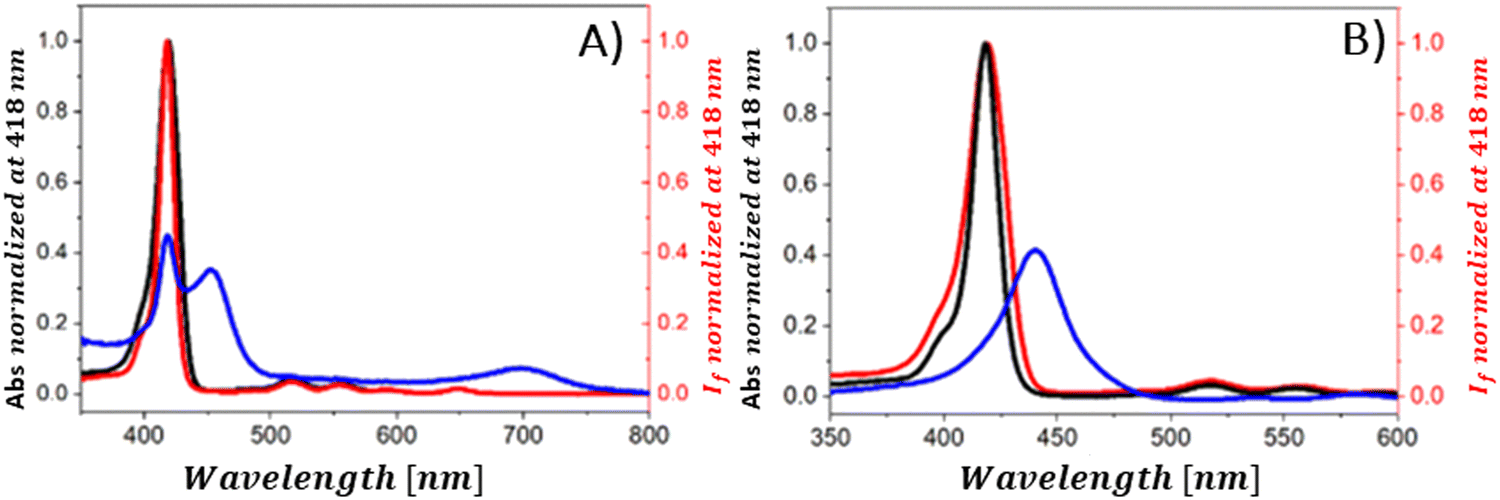 | ||
| Fig. 14 (A) Normalized fluorescence excitation spectrum of the mixture of TPPH and GO (red), absorption spectrum of this mixture (blue) and normalized absorption spectrum of TPPH in the absence of GO (black). (B) Normalized fluorescence excitation spectrum of the mixture of TPPH and RGO (red), absorption spectrum of this mixture (blue) and normalized absorption spectrum of TPPH in the absence of RGO (black). Reproduced from ref. 85. | ||
Shu et al. and Lewandowska et al., however, analyzed the steady-state emission quenching experiments using the Stern–Volmer equations, in order to elucidate whether static, dynamic or both quenching mechanisms contribute to the observed fluorescence quenching and to identify which mechanism was predominant.82,112 Static and dynamic quenching rate constants were determined based on eqn (1):
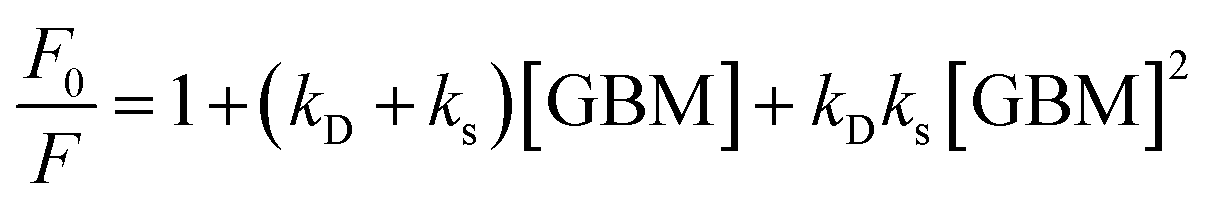 | (1) |
In the CoTPP/RGO,112 TPPH/GO82 and TCPP/GO82 systems, it was observed that kS significantly exceeded kD. As a result, the observed quenching of the emission was predominantly attributed to static quenching, i.e. formation of the non-fluorescent porphyrin/graphene complex. In other studies, the contribution of the static quenching to the decrease in the emission intensity was evaluated based on time-resolved emission experiments.72,73,81,84,85 In scenarios where dynamic quenching occurs, a reduction in the porphyrin fluorescence lifetime in the vicinity of graphene or, alternatively, the emergence of a second decay phase indicative of the nanohybrid's fluorescence would be anticipated. The reported fluorescence lifetimes of the non-covalent hybrids such as TPPH/GO,72 ZnTPPH/GO,73 TAPP/GO, TAPP/RGO,85 TMAP/GO,81 TMPyP/GO84 and ZnTMPyP/GO84 were, however, the same as for the free porphyrins. The time-correlated single-photon counting experiments carried out for the hybrids of the aforementioned porphyrins with GBMs did not reveal a second fluorescence decay either. This observation, coupled with the lack of significant alterations in the singlet excited-state lifetime of the porphyrins near graphene, rules out the likelihood of dynamic quenching by GO in these composites. Therefore, the observed decrease in the steady-state emission intensity can be solely attributed to the static quenching.
During the examination of covalent porphyrin-graphene hybrids, the emission spectra are commonly compared between the free porphyrin and the hybrid structure, adjusting their absorbances at the excitation wavelength to one and the same value. To achieve accurate emission measurements, it is crucial to adjust the absorbance of the porphyrin part specifically, instead of the whole hybrid material. This adjustment ensures that the absorbance matches that of the standalone reference porphyrin at the designated excitation wavelength. Most of the published research highlights efficient quenching of porphyrin emission following covalent linkage with GBMs.59,66–69 It was interpreted as an indication of the electronic interactions between the singlet excited state of the porphyrin and GBM. Zhang et al. showed that the fluorescence quantum yield of free TPP–CHO was equal to 4%, which dropped to 0.3% after covalently binding to graphene.69 Moreover, fluorescence decay of the graphene-TPP composite exhibited a biexponential pattern, with a rapid initial component of <500 ps and a subsequent longer component of 6.2 ns. According to the authors, the dominant short component was associated with the rapid quenching of the porphyrin singlet excited state by graphene, whereas the biexponential nature of the decay reflected the location of the porphyrin on graphene sheets (basal plane vs. edges). A similar electronic interaction was revealed for TPP–CHO when covalently bound to GO, showing a decrease in porphyrin fluorescence quantum yield from 5% to 0.2%.67 Karousis et al. also reported a decrease in the porphyrin fluorescence intensity upon formation of covalent GO–TPPNH2 hybrids (Fig. 15A).59 Analogously to the studies of Zhan et al.,69 in this case the biexponential fluorescence decays were also registered with lifetimes of 675 ps (50%) and 1600 ps (50%), both of which were significantly shorter than the monoexponential fluorescence lifetime of unbound TPPNH2 (2900 ps) (Fig. 15B). These outcomes prompted discussions on the potential roles of charge separation and/or energy transfer in the excited-state decay processes of the hybrid, specifically in how they contribute to the quenching of porphyrin emission when covalently bonded to graphene oxide (GO) sheets. Further analysis which relied on transient absorption spectroscopy was conducted to obtain direct evidence of charge separation (see further in text). Based on time-resolved emission measurements, the calculated quenching rate constant and quantum yield for charge separation for GO–TPPNH2 were equal to kCS = 1.14 × 109 s−1 and ϕCS = 0.77, respectively.
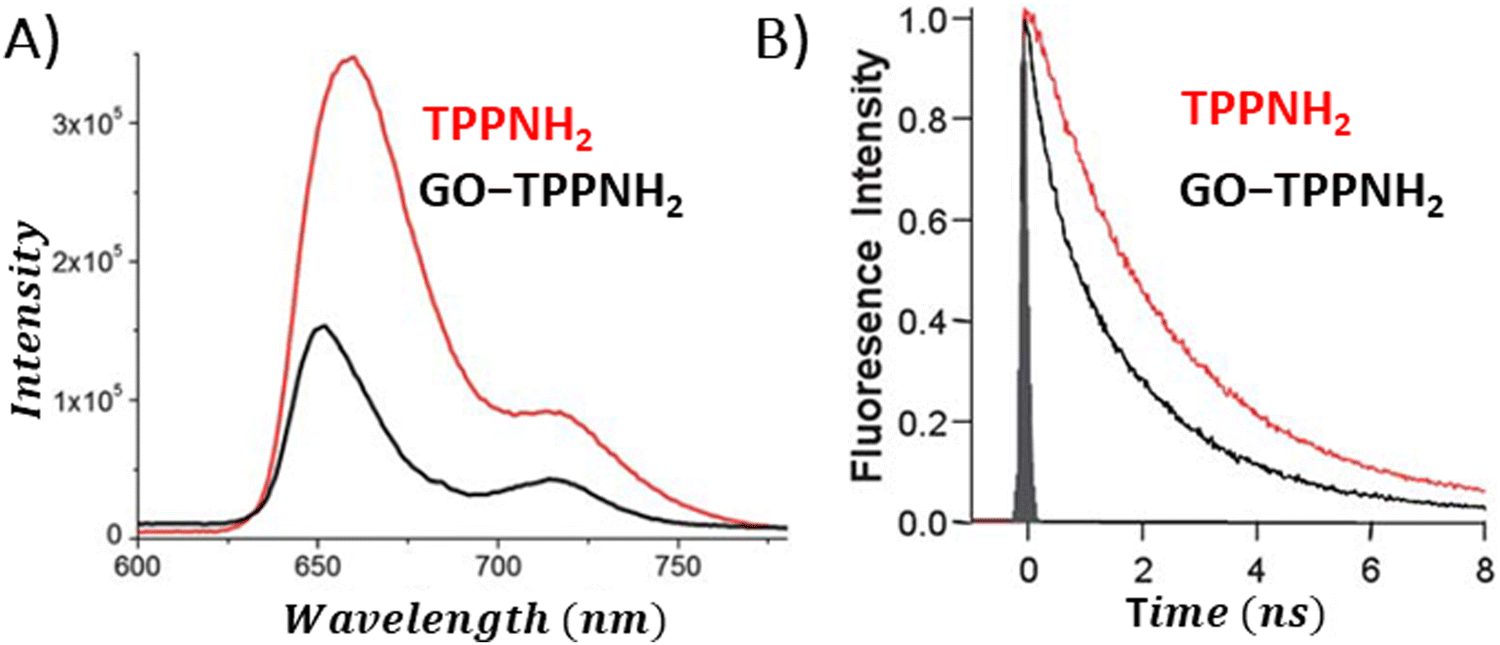 | ||
| Fig. 15 Comparison of the (A) fluorescence spectra and (B) fluorescence decay profiles of free porphyrin TPPNH2versus the porphyrin covalently bound to GO. Adapted with permission from ref. 59. Copyright 2011, Royal Society of Chemistry. | ||
The conversion of light energy into energy-rich molecules (solar fuel) or dye degradation via photocatalytic processes start with the light-triggered generation of energy-rich electrons, wherein photoinduced electron transfer (PET) plays a key role. Accordingly, an investigation of the generated transient species with suitable experimental methods, such as pump–probe absorption spectroscopy (transient absorption spectroscopy) (note: time-resolved emission typically gives a direct access only to the behavior of the singlet excited state of the chromophore and does not provide a direct evidence for electron transfer), is of utmost importance for unraveling the excited-state properties. Femtosecond transient absorption measurements, which are exceptionally well suited for examining light-induced phenomena in composites of porphyrins and graphene-based materials (GBM), have been effectively used in such research.72,73,81,83,84,93,106,113,114 In 2010, A. Wojcik and P. Kamat were the pioneers in demonstrating the actual evidence of the PET process in the porphyrin/GBM hybrid.83 The authors assembled non-covalently positively charged TMPyP with reduced graphene oxide via electrostatic and π–π stacking interactions. Femtosecond TA measurements revealed that the singlet excited state of TMPyP on a RGO film decays more rapidly than when TMPyP is adsorbed on SiO2 (Fig. 16A and B). Furthermore, the decay of the singlet excited state of TMPyP led to the emergence of a longer-living product, characterized by an absorption peak at approximately 515 nm, identified as the porphyrin radical cation. The oxidation potential of 1(TMPyP)* (−0.29 V vs. NHE) was found to be lower than the documented Fermi level of RGO (0 V vs. NHE), supporting the occurrence of PET. Photocurrent measurements offered further insights into the nature of the porphyrin-graphene interaction in the excited state. The photocurrent density observed for TMPyP/RGO, while modest, exceeded that of RGO alone. Low yield of photocurrent density was explained by low efficiency of the charge separation process within the TMPyP/RGO hybrid.
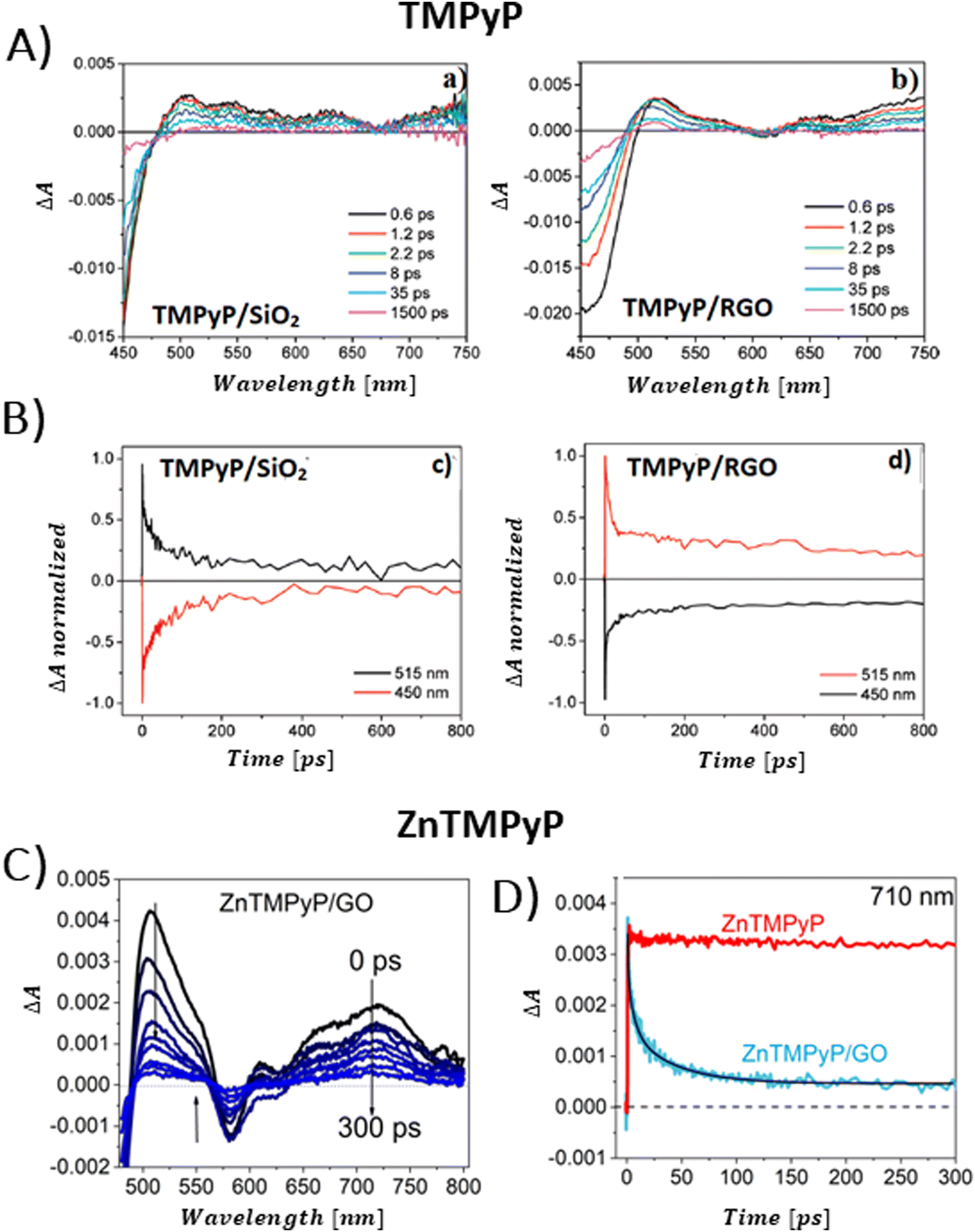 | ||
| Fig. 16 (A) TAS recorded for TMPyP adsorbed on (a) SiO2 and (b) RGO films following 387 nm laser excitation. (B) Normalized transient absorption time profiles taken at 450 and 515 nm for TMPyP adsorbed on (c) SiO2 and (d) RGO films. Adapted from ref. 83. Copyright 2010 American Chemical Society. (C) TAS recorded for ZnTMPyP in the presence of GO following 453 nm laser excitation. (D) Transient absorption time profiles at 710 nm measured for ZnTMPyP (red) and ZnTMPyP/GO (blue). Adapted with permission from ref. 84. | ||
Larowska et al. also delved into the interactions between two cationic porphyrins (ZnTMPyP and TMPyP), however with GO instead of RGO.84 The TAS recorded immediately after exciting ZnTMPyP/GO differed from those obtained for unbound ZnTMPyP. While only the excited-state absorption was evident for free porphyrin, the hybrid showcased the formation of an additional band within the 650–800 nm range, which was identified as the porphyrin radical cation ZnTMPyP˙+ (Fig. 16C). The authors adjusted the TAS of the hybrids by subtracting TA of the GO itself. Fast disappearance of the signal from ZnTMPyP˙+, within tens of picoseconds, was explained by fast back electron transfer (charge recombination). Notably, all decay profiles exhibited a persistent residual signal, at approx. 14%, which did not participate in back electron transfer within the 3 ns observation period (Fig. 16D). The authors rationalized this observation with varying geometries of the ZnTMPyP/GO nanoassemblies having different back electron transfer rate constants. When GO was added to TMPyP, the corrected TAS measurements showed negligible differences from the spectra of free TMPyP, yet the excited state of TMPyP/GO decayed much quicker than in the case of TMPyP alone in the absence of GO. The absence of clear spectral indications of the porphyrin radical cation, which would have served as definitive evidence of photoinduced electron transfer (PET), was reasoned to be due to comparable rates of electron transfer and back electron transfer. The inclusion of Zn in the porphyrin core was suggested to facilitate charge separation in ZnTMPyP/GO composites, a conclusion supported by their photocatalytic activity: ZnTMPyP/GO demonstrated a superior Rhodamine B (RhB) photodegradation efficiency to TMPyP/GO.
Aly et al. conducted a study on three porphyrins, each with different meso substituents and redox properties, to explore how these variations influence the efficiency of light-driven electron injection into carboxylate graphene (GC) within porphyrin/graphene hybrids.93 In case of the neutral porphyrin, TPyP, its excited state remained unaffected by the presence of GC (Fig. 17A). In contrast, the TAS for the two cationic porphyrins, TMPyP and TMAP, exhibited significantly faster dynamics and alterations in the spectral characteristics in the presence of GC. Interestingly, a new band within 500–800 nm range, assigned to the porphyrin radical cation, emerged immediately following the femtosecond laser pulse (Fig. 17A). This indicated that a very fast PET process occurred from the excited state of TMPyP and TMAP to GC. The efficiency of this electron transfer process is probably due to immediate proximity between GC and porphyrins, established in their ground state via strong electrostatic interaction between cationic porphyrin and GC. Through global fitting analysis, two time constants were identified for the charge recombination, spanning a few to tens of picoseconds, which is highly undesirable for photocatalytic applications. In the follow-up study, the same group expanded their investigation to include the non-covalent functionalization of GC with Zn-metallated cationic porphyrin (ZnTMPyP), aiming to create a hybrid material capable of sustaining a long-lived charge separation state.106 Ultrafast TA measurements performed with constant ZnTMPyP and the varying GC concentrations showed that higher GC concentrations altered the dynamics of the TA signal, particularly in the range of 650–800 nm, where spectral features indicative of the ZnTMPyP˙+ radical cation were observed (Fig. 17B). The detection of the ZnTMPyP radical cation, within temporal limits of the equipment, pointed to an ultrafast electron transfer from photoexcited Zn–porphyrin to GC. However, similar to the observations conducted with TMPyP and TMAP, the radical ion pair formed between ZnTMPyP˙+ and GC reverted to the ground state, characterized by a recombination time constant of 20.18 ps (Fig. 17C) (Scheme 6).
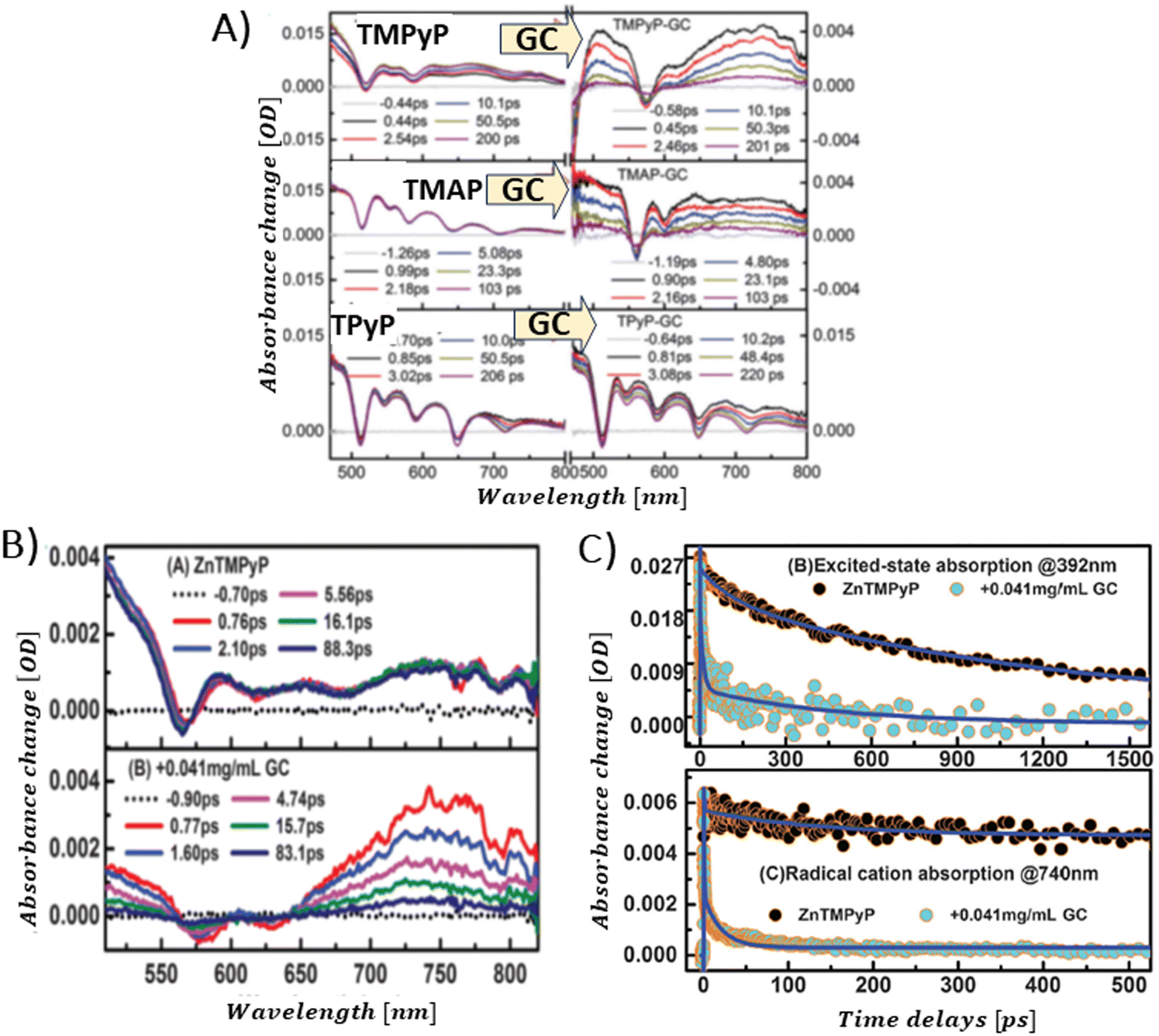 | ||
| Fig. 17 (A) TAS recorded after 425 nm pulsed laser excitation (120 fs) for TPyP, TMAP and TMPyP prior to (left) and following (right) the introduction of GC. Reprinted with permission from ref. 93. Copyright 2014, Royal Society of Chemistry. (B) TAS observed at different time delays for free ZnTMPyP and in the presence of GC (0.04 mg mL−1). (C) Normalized transient absorption time profiles taken at 392 nm and 740 nm for ZnTMPyP and ZnTMPyP/GC. Adapted from ref. 106 with permission from the PCCP Owner Societies. | ||
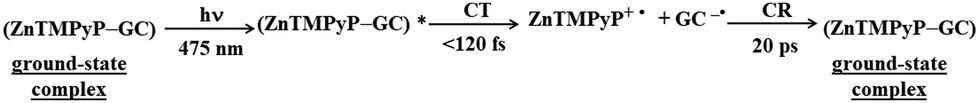 | ||
| Scheme 6 Excited-state dynamics of the ZnTMPyP–GC hybrid. CT denotes charge transfer (here analogous with PET and ET from the photoexcited state), CR charge recombination (here analogous with radical ion pair recombination and back electron transfer). Based on ref. 106. | ||
Garrido et al. synthesized free base Zn-incorporated porphyrins that were covalently bound to substituents, featuring a varying number of pyrene units (either one or three), which acted as anchor points.115 These porphyrins were then used to facilitate the exfoliation of nanographene through liquid phase sonication.115 Excited-state dynamics of the thus-prepared composites was probed by time-resolved absorption spectroscopy. TA measurements revealed that efficient PET occurred predominantly in NG composites that contained Zn–porphyrin linked to a single pyrene unit (Fig. 18). The deconvolution of the TAS using global analysis was carried out using a four-component model, enabling the identification of spectra for individual transient species and their concentration dynamics. The shortest-lived transient (component 1) was recognized as the one-electron-oxidized version of the porphyrin-graphene composite, a determination supported by the spectroelectrochemical studies. Charge recombination was observed to take place within 1000 ps. A component exhibiting a 2 ns lifetime was linked to the ISC transition. Meanwhile, the longest-lived species (>2 μs) was identified as the porphyrin triplet state once it had attached to graphene. Molecular dynamics simulations demonstrated that the monopodal porphyrin forms interact with NG via both their pyrene and porphyrin components, facilitated by π–π stacking interactions. In contrast, simulations for tripodal porphyrin/NG showed that the porphyrin macrocycle remained distant from the NG surface, hindering any effective interaction between the porphyrin and NG. This spatial separation effectively prevents photoinduced PET, a phenomenon corroborated by transient absorption spectroscopy measurements.
 | ||
| Fig. 18 Porphyrin derivative featuring (A) mono- and (B) tripodal pyrene units alongside the molecular dynamics simulation snapshots for porphyrin systems assembled with NG in a THF solvent, (C) temporal evolution of the transient spectra from femtosecond pump–probe experiments (λex = 387 nm) for NG/monopodal porphyrin in THF, including respective concentration profiles. Adapted with permission from ref. 115. Copyright 2020 American Chemical Society. | ||
Umeyama et al. discovered that the fluorescence decay of 10,15-tris(3,5-di-tert-butylphenyl)-20-(4,4,5,5-tetramethyl[1,3,2]dioxaborolan-2-yl)porphyrinatozinc(II) (ZnP) covalently bonded to CCG via a stiff phenylene spacer was too fast to be captured by their time-correlated single-photon counting setup, with a resolution of 60 ps. This rapid decay contrasted with the 2.0 ns fluorescence decay of free ZnP.68 This phenomenon was rationalized by the ultrafast quenching of the singlet excited state of the porphyrin 1ZnP* by the CCG.68 To delve deeper into the quenching mechanism, the authors conducted femtosecond TA experiments, revealing that the TA signal from the ZnP–CCG hybrid, after pulsed excitation, showed decay with two time constants of 0.3 ps and 38 ps. These were notably shorter than the 2 ns monoexponential decay rate of the free 1ZnP*. The shorter decay constant of 0.3 ps was attributed to the decay of the excited state of CCG. Considering the absence of a porphyrin radical cation, the study inferred that the 38 ps quenching of 1ZnP* by CCG occurred through energy transfer. The TA measurements of CCG alone were not presented in the study. The authors did not discuss the possibility of fast back electron transfer either.
In contrast, the earlier mentioned work of Karousis et al. concerning GO-TPPNH2 also provides solid evidence for the photoinduced charge transfer in the studied hybrid obtained using TA spectroscopy in the nanosecond domain, except for the observed fluorescence quenching.59 Photoexcitation at the porphyrin's Q-band resulted in the TA signal in the visible range (610 nm), which was identified as the porphyrin radical cation. Concurrently, bands in the near-infrared (NIR) spectrum indicative of electrons in the reduced graphene sheets were observed (Fig. 19A and B). All TA signals exhibited similar decay kinetics, which suggested a charge recombination process (CR). The CR rate constant derived from the monoexponential fit of the TA decay at 610 nm was equal to kCR = 1.8 × 107 s−1, corresponding to a radical ion pair lifetime of 56 ns. Clearly, a higher charge separation rate constant (as determined from the time-resolved emission measurements, see earlier in text) than the recombination rate constant enables efficient separation of charges under light illumination of the hybrid. This feature is a prerequisite for any photovoltaic applications. The authors further investigated the charge separation capability of the composite by constructing a photoelectrochemical cell using GO-TPPNH2 as the photoanode. Incident photon-to-photocurrent-efficiency (IPCE) for the designed cell was equal to 1.3%. The thermodynamically favorable formation of (GO)˙−−(TPPNH2)˙+ radical ion pairs was verified by a negative free energy change during photoinduced electron transfer in the hybrid, ascertained through cyclic voltammetry.
 | ||
| Fig. 19 Nanosecond TAS for: (A) free TPPNH2 (inset: TA decay at 450 nm) and (B) covalent hybrid GO-TPPNH2 (inset: TA decay at 610 nm). Adapted from ref. 116. Copyright 2011, Royal Society of Chemistry. | ||
In 2022, Arellano et al. reported studies of N-doped graphene (N-G) that was covalently bonded with Zn–porphyrin (N-G-ZnP(TPA)3) (TPA = triphenylamine) using a “click” chemistry approach.113 Interestingly, for N-G-ZnP(TPA)3, the singlet excited state of porphyrin decayed rapidly without any evidence of the triplet state formation (Fig. 20A and B). Instead, a new band appeared in the range of 625–775 nm, identified as the ZnP(TPA)3˙+ radical cation (Fig. 20B). The rate constants for charge separation and charge recombination were derived from the fitting of growth (kCS) and decay (kCR) of the TA signal acquired at 650 nm, and were found to be approximately ∼1010 and 4.56 × 109 s−1, respectively (Fig. 20D). The similar values of kCS and kCR indicate a challenge in achieving efficient light-driven charge separation, suggesting that additional structural modifications of the hybrid are necessary to stabilize the charge-separated states.
 | ||
| Fig. 20 Femtosecond TAS at indicated delay times for (A) ZnP(TPA)3 control and (B) its hybrid with N-G, (C) TA decays for 1ZnP(TPA)3*: control at 495 nm (free porphyrin) and hybrid at 498 nm (porphyrin bound to N-G), (D) time profile for the 650 nm peak corresponding to ZnP(TPA)3˙+. Adapted from ref. 113. | ||
Moreover, in 2022, Fu et al. described the covalent bonding of GO with meso-substituted formylporphyrins via imidazole condensation, resulting in a novel nanohybrid with enhanced optoelectronic properties.117 The nanohybrid displayed absorption with a pronounced 7 nm red shift, a significant 95% reduction in fluorescence, and substantially enhanced nonlinear optical (NLO) properties, as observed in the Z-scan experiments at reduced input energies, suggesting a more effective photoinduced energy/electron transfer within the hybrid components. The authors of the research concluded that the developed method of covalently functionalizing the edges of GO through imidazole linkages paves the way for the development of various GO nanohybrids that could exhibit efficient energy/electron transfer and stronger nonlinear optical responses, offering new insights into the creation of versatile optoelectronic devices.
Further support for the photoinduced charge transfer in the porphyrin and GBM is also provided by photoelectrochemical measurements.116,118,119 In 2016, Bera et al. demonstrated effective photoinduced charge separation in a composite combining a one-dimensional (1D) nanostructure of TCPP nanorods (TCPP-NR) with RGO (Fig. 21A).118 Femtosecond fluorescence upconversion spectroscopic measurements revealed a rapid decay of TCPP-NR fluorescence in the TCPP-NR/RGO hybrid (Fig. 21B). The fluorescence decay observed in the TCPP-NR/RGO composite was fitted by triexponential function with the fastest component linked to the PET process. The PET rate, deduced from the variance in the emission decay kinetics between TCPP-NR and the TCPP-NR/RGO composite, was determined to be 1.0 × 109 s−1, given an RGO content of 240 μg.118 Furthermore, a notable increase (1.9 time) in the photocurrent was observed for the TCPP-NR/RGO composite under visible-light illumination compared to the TCPP-NR alone, providing additional evidence of PET within the composite.
 | ||
| Fig. 21 (A) Schematic of the TCPP-NR/RGO system exposed to light irradiation. (B) Comparative fluorescence decays of free TCPP-NR and TCPP-NR/RGO composites. Adapted from ref. 118. Copyright 2016, American Chemical Society. (C) Photocurrent generation with pure porphyrin and TPPOH-NR/RGO hybrid as active materials on the photoanode. (D) Schematic energy-level diagram and charge separation path in the tested device. Adapted from ref. 116 with permission from the Royal Society of Chemistry. | ||
On the other hand, Mondal et al. reported that the non-radiative decay rate constant increased with both the aging time of the hybrids and the concentration of RGO due to electron transfer from the photoexcited TPPOH nano-aggregates to RGO within non-covalent hybrids.116 In particular, for TPPOH J-aggregates that had aged for 48 h on the RGO surface, with an RGO concentration of 0.02 mg mL−1 the electron transfer process decay rate was measured at 6.43 × 109 s−1. The enhancement of the PET mediated by the porphyrin aggregates nanoassemblies with RGO was also evident due to the 4-fold amplification of the photocurrent generated under visible-light irradiation, as compared to the free porphyrin itself (Fig. 21C). Energy level diagram and charge separation pathway in the tested device are presented in Fig. 21D.
The authors concluded that the in situ formation of porphyrin nanoassemblies on the surface of the RGO surface leads to enhanced electronic interaction between the hybrid components. This enhancement results from the reduction in the interfacial distance between graphene and porphyrin, which in turn boosts the efficiency of PET.
In 2018, Yao et al. published a report which focused on studying the influence of the morphology of porphyrin aggregates assembled with RGO on the efficiency of PET.119 First, they employed a surfactant-assisted self-assembly approach to generate porphyrin aggregates with distinct structures (TCPP-AGG) in a selected solvent. RGO was then electrochemically deposited onto glass substrates coated with indium-doped tin oxide (ITO). Subsequently, porphyrin aggregates were spin-coated onto the graphene support. The final step involved the photodeposition of Pt nanoparticles (Pt NPs) (Fig. 22A). The composites consisting of rod-shaped TCPP aggregates turned out to yield the highest photocurrent values (Fig. 21B). The authors used a combinatorial technique of UV-vis spectroscopy and scanning electrochemical microscopy (UV-vis/SECM) to investigate local PET kinetics at the heterogeneous interface for RGO composites with various porphyrin aggregates (Fig. 22B and C). They simulated the SECM profiles using a model formulated by the Bard group.120 This approach enabled the calculation of PET rate constants for TCPP composites with RGO, differentiated by their morphologies. Notably, the composites featuring rod-like porphyrin aggregates with RGO showed a superior PET rate constant with a value of 8.547 × 102 cm s−1.
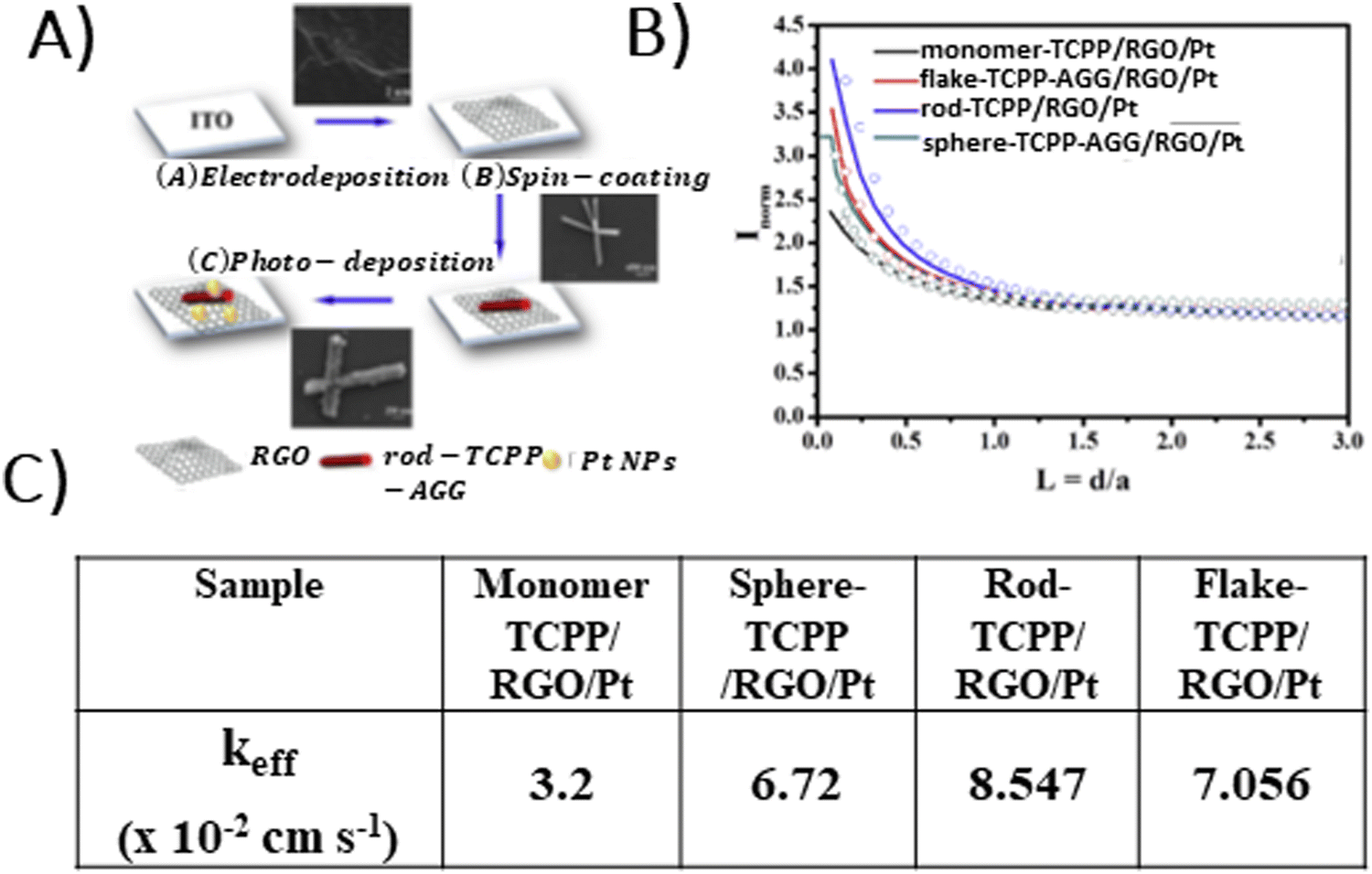 | ||
| Fig. 22 (A) Scheme of the TCPP-AGG/RGO/Pt fabrication procedure on the ITO substrate, (B) normalized SECM feedback curves recorded for different TCPP-AGG/RGO/Pt films on ITO, and (C) morphology-dependent apparent/effective PET rate constants (keff) for samples with various porphyrin aggregates, obtained from simulating the experimental SECM curves presented in B. Adapted with permission from ref. 119. Copyright 2018 Elsevier Ltd. | ||
In the studies described up to this point, the focus was placed on the interaction of the porphyrin singlet excited state with GBMs. Nonetheless, Masih and co-workers expanded the scope to include the potential interaction of the triplet excited state of ZnTMPyP with GC.106 Using nanosecond flash photolysis, the authors determined that the successive addition of GC to the ZnTMPyP solution under irradiation led only to a reduced amplitude of the TA signal, without altering the lifetime of the porphyrin triplet state.106 This proved that electron transfer observed in the femtosecond TA measurements indeed occurred only from the porphyrin singlet excited state. The photoinduced electron transfer process is so efficient that it competes with intersystem crossing, effectively diminishing the formation of the triplet state. These findings align with those obtained by Larowska et al.81 for the TMAP/GO system, in case of which the authors found that the development of TMAP's triplet state was completely inhibited at higher concentrations of GO. Such an observation can be rationalized by fast electron transfer process from the singlet excited state that precludes intersystem crossing, which consequently reduces the triplet-state population. Similarly to ZnTMPyP/GC, no change in the lifetime of the TMAP triplet state was observed upon addition of GO. In contrast, for the ZnTPPH/GO and TPPS/GO hybrids, it was reported that the lifetime of the triplet excited state of the porphyrins increases in the presence of GO.73,121 Gacka et al.73 elucidated that the increase in the triplet state's lifetime can be attributed to the reduced rate of nonradiative decay. This reduction is due to the restricted rotation of the hydroxyphenyl rings that occurs when the porphyrin is anchored to the graphene plane, hindering their free movement. Such an explanation is well justified in light of other reports which relate radiationless decay of the triplet excited state to the ground state with the ease of flipping and twisting of conformationally free meso-phenyl rings around the core of porphyrin.122,123 As is evident from the research results presented in Section 3 of this review, absorption and emission spectroscopy methods are an extremely powerful tool that enables detailed characterization of the photophysical and photochemical properties of Por and GBM hybrids, and in turn allows for the comprehensive understanding of interactions between hybrid components in both ground and excited states. These interactions determine the potential applicability of selected hybrids in photocatalysis (see Sections 4 and 5) and are the key to target-oriented design of future devices based on Por and GBM composites. To facilitate the understanding of the role of spectroscopic methods in the determination of the essential properties of Por and GBM hybrid materials, the most important information, accessible based on the appropriate analysis of spectroscopic data, is summarized in Table 3.
| Technique | Observation upon assembly of Por with GBM | Information obtained |
|---|---|---|
| UV-vis spectroscopy | (i) For non-covalent hybrids: red-shifted Soret and Q-bands with decreased intensity as compared to the free porphyrin, | (i) Probes the interaction between components in the ground state, |
| (ii) For covalent hybrids: presence of the Soret and Q-bands being fingerprint of the porphyrin chromophore. | (ii) Senses changes of the electronic structure of porphyrin occurring upon adsorption on the GBM, e.g. due to flattening of the Por chromophore, | |
| (iii) Provides information about the strength of the interaction between hybrid components in the ground state, | ||
| (iv) Presence of the Soret band for covalent hybrids confirms successful functionalization of graphene sheets with Por. | ||
| Fluorescence spectroscopy (steady-state) | Decrease of the fluorescence intensity (quenching). | (i) May be an indication of the electronic interaction between singlet excited state of Por and GBM (dynamic quenching) and/or |
| (ii) Formation of a non-emissive complex between Por and GBM (static quenching) which suggests the occurrence of a fast deactivation process of Por chromophore once bound to GBM other than fluorescence, | ||
| (iii) Differentiation between the static and dynamic quenching is required by complementary time-resolved methods. | ||
| Note: in some reports the observed fluorescence quenching was attributed solely to static quenching! | ||
| (iv) Quenching mechanism (energy vs. electron transfer) cannot be clearly defined without using complementary time-resolved methods. | ||
| Fluorescence spectroscopy (time-resolved) | (i) No changes in the fluorescence lifetime, | (i) Static quenching if a decrease of the fluorescence intensity is observed in parallel, |
| (ii) Shortening of the fluorescence lifetime in comparison to free porphyrin. | (ii) An electron transfer and/or energy transfer between components of the hybrid initiated by the absorption of light. | |
| Femtosecond transient absorption spectroscopy | (i) Shortening of the Por singlet excited state lifetime, as compared to the free Por, | (i) Direct evidence for the interaction between the singlet excited state of Por and GBM, |
| (ii) Detection of the porphyrin radical cation and/or bands in NIR associated with electrons trapped between graphene sheets. | (ii) Direct proof for the electron transfer mechanism between hybrid components, | |
| (iii) PET rate constant can be determined. | ||
| Nanosecond transient absorption spectroscopy | (i) No change in the triplet excited state lifetime of the porphyrin, | (i) Lack of interaction between triplet excited state of Por and GBM, |
| (ii) Detection of the PET products and their decay kinetics. | (ii) Direct evidence for PET, | |
| (iii) Recombination rate constants of the PET products can be determined. | ||
It should be emphasized that the differences in the distance and orientation between the π-conjugated graphene sheets and the porphyrin macrocycles as well as the orientation of porphyrin substituents with respect to the macrocycle plane and plane of the graphene sheets together with porphyrin aggregation state/morphology significantly affect the excited-state interaction between the GBM and the organic chromophores. Moreover, the properties of the GBM itself (e.g., GO, RGO, CCG, and GC) also determine the behavior of the hybrid under light illumination to some extent.
The molecular structures/morphologies of the GBM and Por hybrids discussed up to this point, as well as those addressed further in Paragraph 4 from the perspective of the photoactive component (Por), are summarized in Scheme 7.
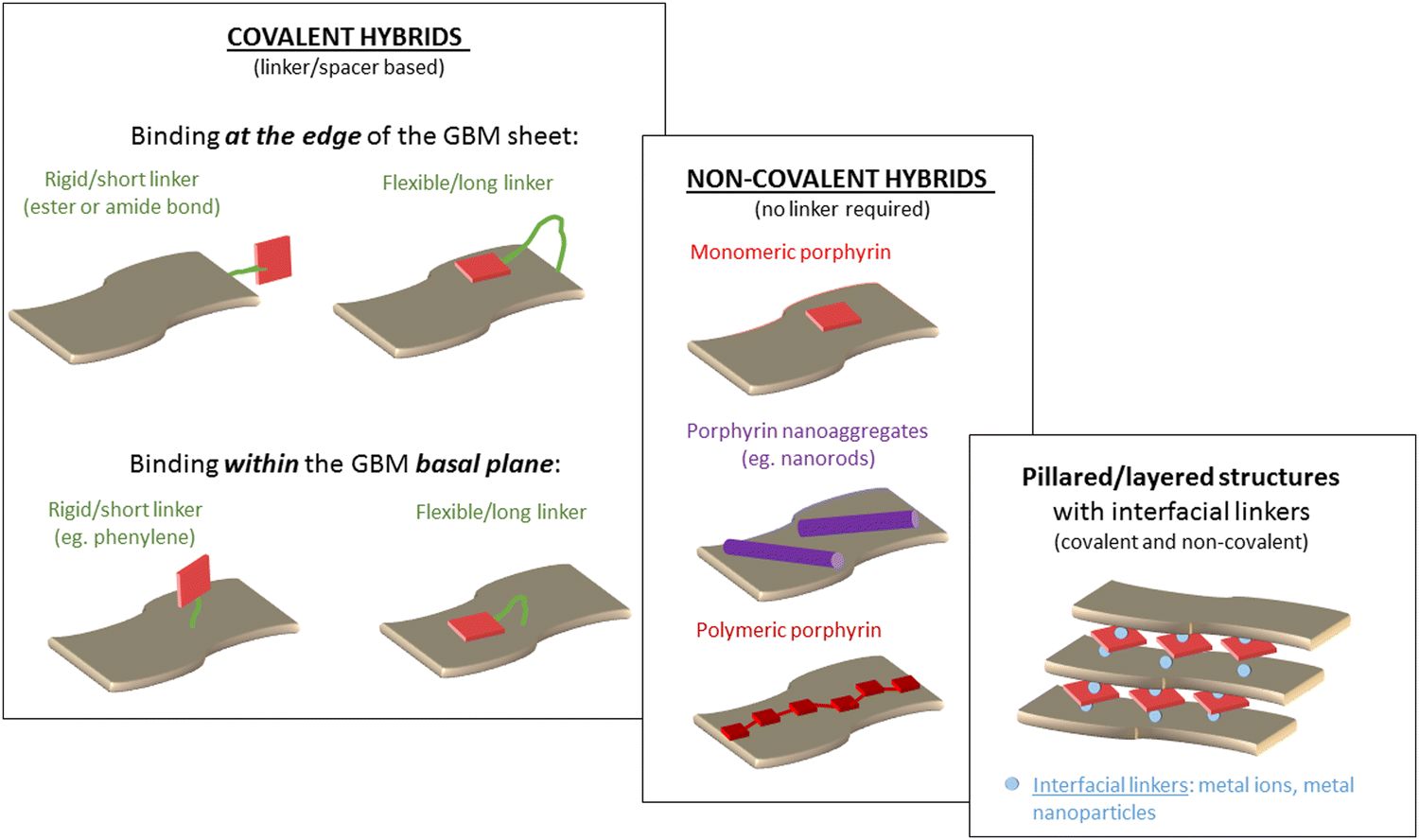 | ||
| Scheme 7 Schematic illustration of the possible binding modes in GBMs and Por hybrids. Note: various binding modes are presented from the perspective of the photoactive component of the hybrid. The possible variation of GBMs is intentionally not taken into account. | ||
In case of covalent hybrids, the selected linker mostly determines whether the porphyrin macrocycle will closely interact with the graphene surface: short and rigid linkers, such as amide bonds, will promote structures with the Por plane pointing away from the graphene surface, whereas longer and more flexible linkers will allow more intimate interaction between Por and graphene. Stronger coupling between Por and GBMs will promote more efficient PET, which is one of the prerequisites for a good photocatalyst. Therefore, even though the covalent linkage of Por to GBMs offers numerous advantages (see Table 1), the vast majority of the published reports dealing with the photocatalytic properties of Por and GBM hybrids concern non-covalent composites, in case of which the nature of the GBM and Por bonding assures very close contact between the photoactive component and the graphene surface. This trend is also clearly visible in Tables 4–6. It is evident that among these non-covalent composites, the assemblies of GBMs with monomeric porphyrins prevail. Such an observation is surprising, taking into account the fact that well-organized porphyrin assemblies (nanoaggregates) are known to exhibit superior photocatalytic properties to their monomeric counterparts, due to their better charge separation properties.124–126 Similar observations for Por hybrids with graphene were reported only three times in the literature to date,116,118,119 with clear indication that the morphology of the Por aggregates strongly affects the PET process as well as the general photocatalytic performance of such materials. Unfortunately, a comprehensive study focusing on detailed understanding of this direct linkage between the Por nanoaggregate morphology and PET process/photocatalytic activity in the discussed systems is still missing. Similarly to Por nanoassemblies, Por polymers used instead of monomeric Por macrocycles as photoactive components of hybrids with GBM exhibited better photocatalytic efficiency than Por monomers, which also depended on the morphology of the tested system.78,79,127 The formation of layered/pillared hybrid structures using interfacial linkers such as metal cations and metal nanoparticles also enhanced the photocatalytic performance of the hybrids due to the strengthened interaction between Por and GBMs.128–130 Nonetheless, also in the latter two cases, no further detailed studies were conducted to date.
| Photocatalyst | Synthetic procedure | Experimental conditions | H2 (μmol g−1 h−1) | Ref. |
|---|---|---|---|---|
| TPPH/RGO/Pt | Mixing of suspensions and stirring for 12 h | Xe lamp 150 W, TEOA | 1060 AQE = 1.7% | 74 |
| TPPH/RGO/Pt | Mixing of suspensions and stirring for 12 h | Xe lamp 150 W, CTAB, TEOA | 2240 AQE = 3.6% | 74 |
| TPPH/RGO/Pt | Mixing of suspensions and stirring for 12 h | Xe lamp 150 W (λ > 420 nm) TEOA | 138 | 74 |
| TPPH/PSA/GO | Mixing of suspensions | 300 W Xe lamp, TEOA | 44.3 AQE = 0.74% at 450 nm | 131 |
| GO–Cr3+–DPyP | Mixing and stirring of GO and Cr3+, removing the excess of Cr3+ followed by mixing with DPyP | 300 W Xe lamp, TEOA | 116 | 128 |
| GO–Co–DPyP | Preparation of GO–Co2+–DPyP via mixing and subsequent stirring of the components followed by photoreduction of Co2+ | 300 W Xe lamp, TEOA | 546 | 129 |
| GO–Sm3+–DPyP | Mixing and stirring of GO and Sm3+, removing the excess of Sm3+ followed by mixing with DPyP | 300 W Xe lamp TEOA | 550 | 130 |
| ZnTMPyP/MoS2/RGO | Preparation of MoS2/RGO by hydrothermal reaction of graphene oxide nanosheets dispersed in an aqueous solution with sodium molybdate dehydrate and thioacetamide. Subsequent mixing of MoS2/RGO with ZnTMPyP solution | 300 W Xe lamp (λ > 420 nm) TEOA | 2560 AQE = 15.2% at 420 nm | 132 |
| TPP–GO–Ni molecular complex | Covalent GO functionalization via amide bond formation with TPP and then mixing with Ni catalyst solution | 450 W Hg lamp (λ > 380 nm) ascorbic acid | 1.2 μL | 133 |
Therefore, further work is required to understand the mechanisms governing photoinduced charge separation processes in composites of graphene and porphyrin chromophores in detail and to gain comprehensive knowledge which allows us to directly relate the structural features of such composites with their ability to promote light-driven charge separation. Only then will the targeted design and synthesis of hybrid systems based on graphene and porphyrin macrocycles exhibiting light sensitivity in pre-defined spectral regions and capable of generating longer-lived charge separation states upon light illumination be possible. This would be a milestone in the on-demand fabrication of such composites and their effective usage as light-harvesting systems and photocatalysts.
4. Photocatalytic activity of Por and GBM hybrids
4.1 Photocatalytic hydrogen production
The first report on the application of porphyrin and graphene-based materials for photocatalytic hydrogen production was published in 2013.74 Zhu et al. observed light-driven photocatalytic hydrogen evolution from the TPPH/RGO composite–integrated Pt nanoparticle as a co-catalyst (Fig. 23A).74 The efficiency of hydrogen production by the TPPH/RGO/Pt nanostructure was notably superior to systems in which TPPH was linked with Pt colloids or when RGO was modified with Pt. The TPPH/RGO/Pt nanocomposite produced H2 achieving AQE = 1.7% under UV-vis irradiation (Fig. 23B). The addition of cetyltrimethylammonium bromide (CTAB) surfactants, which prevented the aggregation of the TPPH/RGO/Pt nanocomposite, further improved the catalytic performance (AQE = 3.6%) and stability of the tested photocatalyst. Under visible-light irradiation (λ > 400 nm) for the TPPH/RGO/Pt nanocomposite in the presence of surfactants, the measured hydrogen evolution rate was equal to 1060 μmol g−1 h−1. It was proposed that the light-driven proton reduction by the TPPH/RGO/Pt nanocomposite occurs in the following steps: (1) excitation of the TPPH molecules adsorbed on the RGO surface upon UV-vis light irradiation, (2) PET from the excited state of TPPH to RGO, (3) electron transfer from the RGO to the Pt NPs adsorbed on the RGO surface where the water molecules accept the electrons to form hydrogen and (4) restoration of the TPPH ground state by accepting electrons from the triethanolamine (TEOA).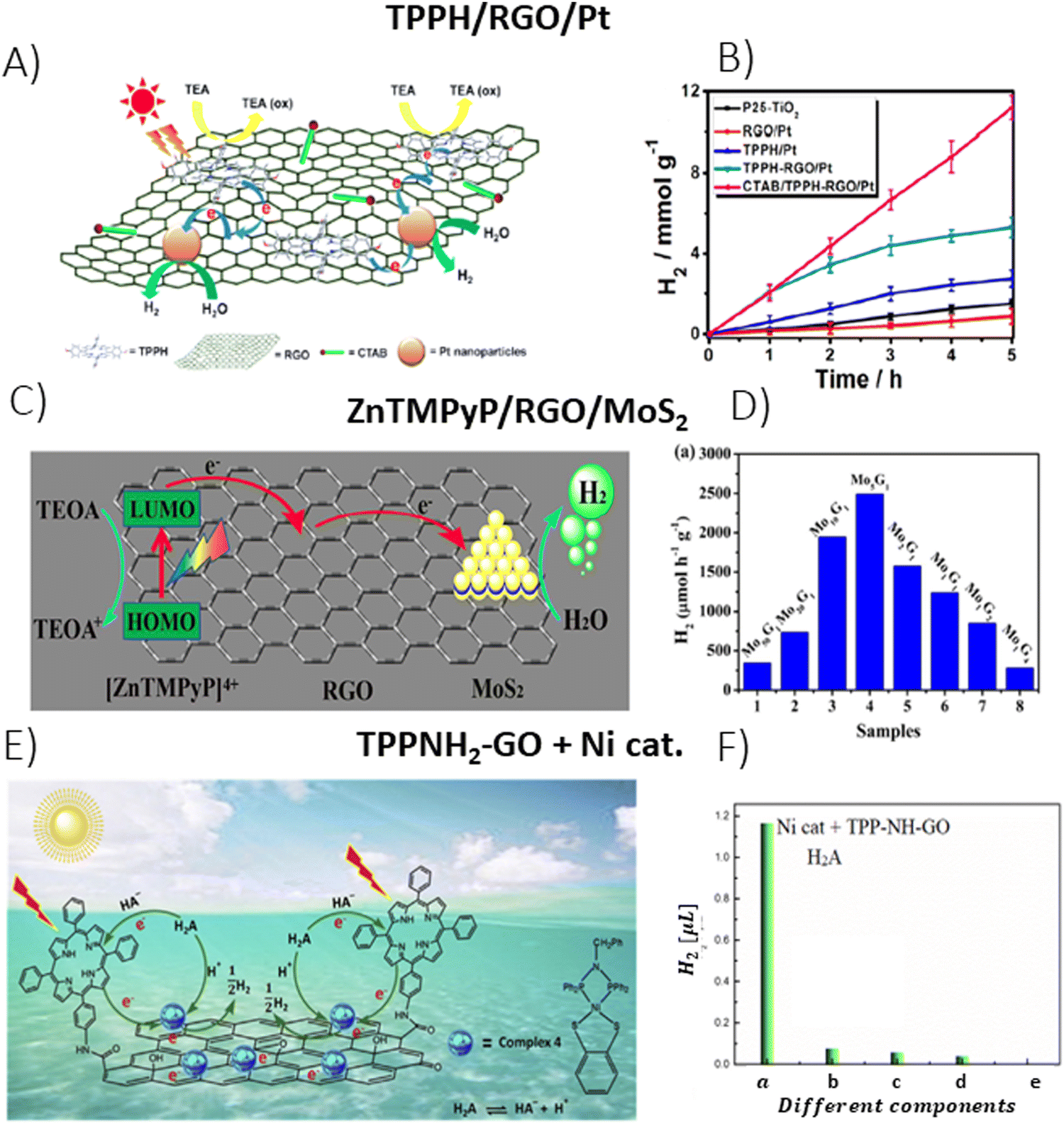 | ||
Fig. 23 (A) Depiction of the photocatalytic H2 generation using RGO/TPPH/Pt NPs in the presence of CTAB under light irradiation, (B) comparative data on H2 evolution from different photocatalysts (P25-TiO2, RGO/Pt, TPPH/Pt, TPPH/RGO/Pt, and CTAB/TPPH/RGO/Pt) under UV-vis light irradiation, reproduced with permission from ref. 74. Copyright 2013 American Chemical Society, (C) schematic of the process for photocatalytic H2 generation in ZnTMPyP/MoS2/RGO under UV-vis irradiation, (D) rates of photocatalytic H2 production upon visible-light irradiation of aqueous solutions containing ZnTMPyP, TEOA and MoS2/RGO at different mass ratios. Reproduced with permission from ref. 132. Copyright 2017, American Chemical Society, (E) mechanism of H2 production in a bio-inspired photocatalytic system composed of a mononuclear nickel complex and a graphene nanohybrid covalently bonded with porphyrin macrocycles and (F) the amount of photocatalytic H2 evolution using various combinations: (a) Ni complex, TPPNH2–GO, and ascorbic acid; (b) TPPNH2–GO and ascorbic acid; (c) Ni complex and TPPNH2–GO; (d) Ni complex and ascorbic acid, in an aqueous ethanol solution (VEtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VH2O = 1:24) with 5 h illumination period; and (e) Ni complex, TPPNH2–GO, and ascorbic acid without light exposure. Reproduced with permission from ref. 133. Copyright 2016 Elsevier Inc. :VH2O = 1:24) with 5 h illumination period; and (e) Ni complex, TPPNH2–GO, and ascorbic acid without light exposure. Reproduced with permission from ref. 133. Copyright 2016 Elsevier Inc. | ||
Luo et al. sought to enhance the light absorption capabilities of GO and facilitate the separation of electron/hole pairs by attaching two distinct dyes, TPPH and 1-pyrenesulfonic acid (PSA), to GO via non-covalent modifications.131 For such hybrids, the measured average rate of hydrogen production was equal to 44.3 μmol h−1 g−1 and the quantum yield for hydrogen evolution was equal to 0.74% at a wavelength of 450 nm. Notably, this material was utilized without the addition of a co-catalyst, which is commonly included in hydrogen production systems. The photocurrent generation by GO itself was improved dramatically after functionalization with TPPH/PSA, which was attributed to high utilization of light by the two chosen dyes, which were both characterized by complementary absorption spectra and very strong interaction with GO.
Yuan et al. developed a hybrid material consisting of a MoS2/RGO catalyst and cationic porphyrin ZnTMPyP as a photosensitizer, which was tested toward photocatalytic H2 production under visible-light irradiation (Fig. 23C).132 The ZnTMPyP/MoS2/RGO achieved a photocatalytic H2 evolution rate of 2.56 mmol h−1 g−1 at pH 7 when the mass ratio of MoS2 to graphene was 5/1 (Fig. 23D). An apparent quantum yield of 15.2% at 420 nm was achieved under optimized conditions. Investigations by means of photoluminescence decay indicated a reduction in the excited state lifetime of ZnTMPyP with the increased presence of RGO within the MoS2/RGO composites. A comprehensive analysis, which incorporated both steady-state and time-resolved luminescence tests, led the researchers to deduce that electron transfer to the RGO surface was initiated upon the photoexcitation of ZnTMPyP. Given that the oxidation potential of the excited state of ZnMPyP* (−1.09 V vs. NHE) is more negative than the RGO/RGO˙− redox potential (−0.16 V vs. NHE), such electron transfer is thermodynamically feasible. In addition, the redox potential of the RGO/RGO˙− couple is more negative than the conduction band of MoS2 nanosheets (−0.13 V vs. NHE), which indicates a sufficient driving force for further electron transfer from RGO to MoS2. The edges of MoS2 crystallites can act as the active sites for the H2 evolution reaction once they are reduced with the electrons from RGO. The oxidized ZnTMPyP can be regenerated by reduction with TEOA (Fig. 23C). In summary, RGO in the ZnTMPyP/MoS2/RGO composite acted as a conductive electron transport bridge, which enhanced the efficiency of the electron transfer from the photoexcited photosensitizer (porphyrin) to MoS2.
Liu et al. conducted research on a biomimetic system in which a mononuclear nickel complex, serving as a hydrogen evolution catalyst, was adsorbed onto the surface of GO nanohybrids covalently functionalized with TPPNH2 (TPPNH2–GO) (Fig. 23E).133 The photocatalytic experiments for evaluating H2 production using a TPPNH2–GO photosensitizer and a Ni catalyst in an aqueous ethanol solution were performed in the presence of ascorbic acid as a sacrificial electron donor under irradiation with λ > 380 nm (Fig. 23F).
Wang et al. developed a donor–acceptor-type ternary nanohybrid material, which was synthesized by covalently binding TPyP and tetraaminophthalocyanine (ZnPc) with GO. The hybrid demonstrated a superior optical limiting and electrocatalytic activity toward hydrogen evolution reactions (HERs) due to effective charge transfer and strong electronic interactions, and thus, outperformed its binary counterparts TPyP–GO and ZnPc–GO. Characterization through various spectroscopic techniques confirmed the enhanced redox and optoelectronic properties of TPyP–GO–ZnPc, and indicated that the concurrent functionalization of GO with porphyrins and phthalocyanines could lead to advanced electrocatalytic systems and optical limiting devices. Therefore, such ternary systems could also find potential applications in photocatalysis.134
In 2016, a novel methodology for synthesizing porphyrin and GBM hybrids was introduced. This technique relied on the incorporation of metal ions as interfacial linkers between GO and DPyP (5,15-diphenyl-10,20-di(4-pyridyl)porphyrin) (Fig. 24A and B).128 The authors elucidated how Cr3+ ions affect the morphology and structure of these nanocomposites, as well as their photocatalytic activity in generating hydrogen production and the dynamics of electron transfer involved. It was found that without Cr3+, the DPyP molecules would spread out and form J-aggregates on the GO surface, spanning 100–150 nm in length. Differently, a layered (sandwich-like) nanostructure was formed after introducing Cr3+ ions between the GO and the DPyP. Metal ions anchored on the surface of GO coordinated with two pyridyl groups within the DPyP molecule served as the interfacial linker, which enabled the formation of a pillared GO–Cr3+–DPyP composite. The fabricated material exhibited enhanced photocatalytic activity toward H2 production. Without the use of any co-catalysts and upon exposure to light for 8 h, the total H2 yield achieved by GO–DPyP and GO–Cr3+–DPyP reached 686 and 928 μmol g−1, respectively. The study also evaluated the performance of other metal ions such as K+, Ca2+, Zn2+ and Cu2+, among which Cr3+ showed the highest boost in H2 production and the photocurrent response. This enhanced performance was credited to the stronger coordination between the Cr3+ metal ions and DPyP than between other tested metal ions and the porphyrin of interest.
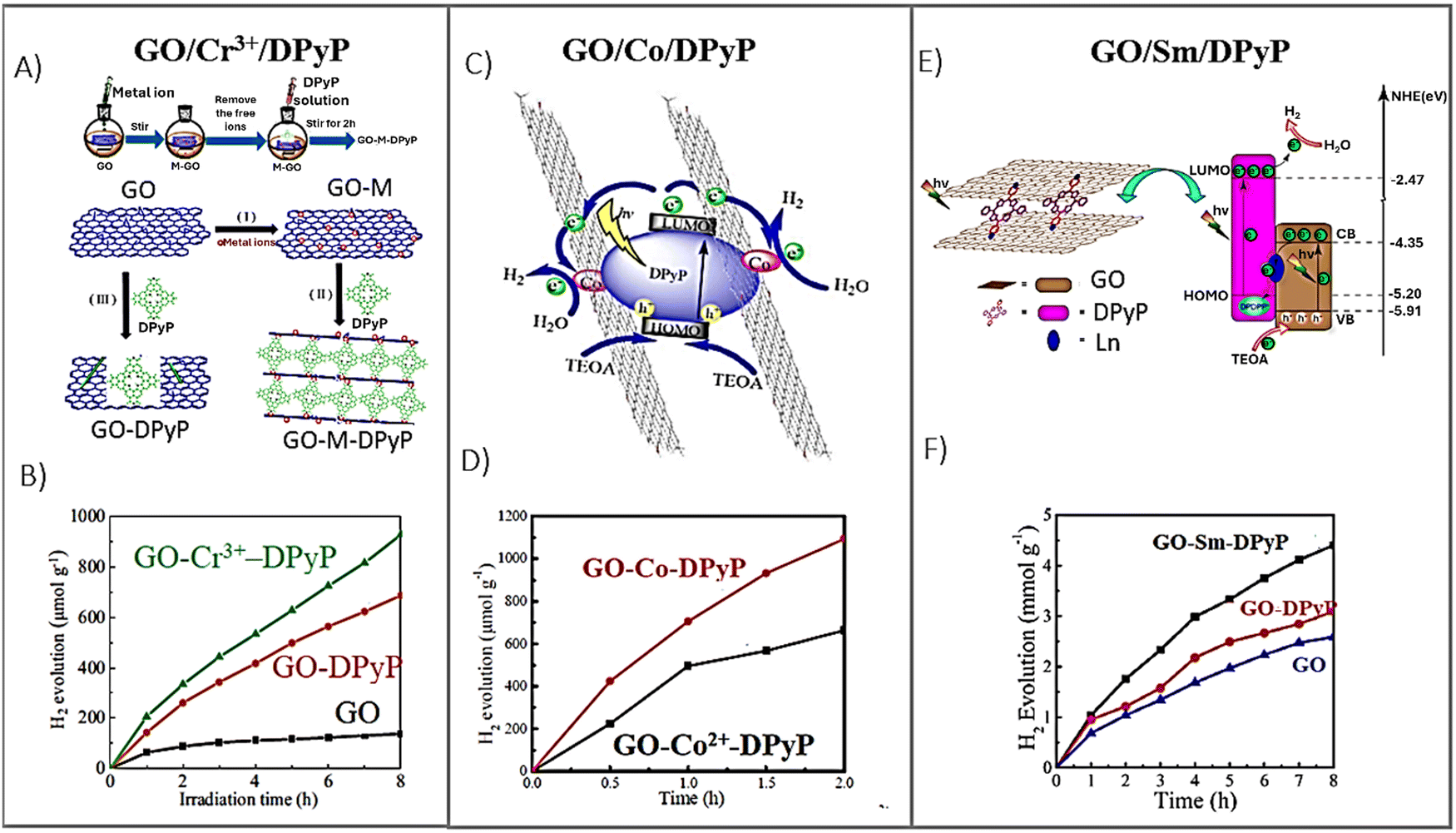 | ||
| Fig. 24 (A) Synthesis of the GO–Cr3+–DPyP composite, (B) time evolution of photocatalytic hydrogen generation using various photocatalysts, adapted with permission form ref. 128. Copyright 2016 Elsevier B.V., (C) diagram showing the electron transfer and hydrogen evolution over the GO–Co–DPyP nanohybrid under light irradiation, (D) comparison of the H2 yield from the GO–Co2+–DPyP and GO–Co–DPyP photocatalysts, adapted from ref. 129 with permission from the Royal Society of Chemistry, (E) outline of the photocatalytic mechanism of GO–Sm3+–DPyP and (F) H2 production measurements from GO–Sm2\3+–DPyP (black), GO–DPyP (red) and GO (blue), Adapted from ref. 130. Copyright 2019, American Chemical Society. | ||
In the follow-up paper, the authors shifted their focus towards replacing metal ions with metal nanoparticles, which could serve as a more active catalyst.129 Novel GO–Co–DPyP composites were synthesized by introducing metallic cobalt nanoparticles (Co NPs) as the interfacial linker (Fig. 24C).129 A similar material employing Co2+ ions instead of the Co nanoparticles was utilized for comparison as a benchmark. The hybrid featuring cobalt nanoparticles demonstrated enhanced interactions between GO and DPyP compared to its counterpart, leading to better light absorption and photoinduced electron transfer characteristics. Furthermore, its photocatalytic activity toward hydrogen evolution was superior to that of GO–Co2+–DPyP (Fig. 24D). In particular, the quantity of hydrogen produced by the GO–Co–DPyP composite within 2 h of irradiation (1093 μmol g−1) was approximately twice greater than that generated by the analogous GO–Co2+–DPyP composite.
In 2019, Zhang et al. employed a similar approach to fabricate graphene/pyridylporphyrin hybrids, which were interconnected via rare earth ions.130 Unlike the assembly of DPyP with GO, which led to the formation of porphyrin nanorods (J-aggregates) because of dominant π–π interactions among the porphyrin macrocycles, the combination of DPyP with GO pre-treated with Sm3+ ions resulted in a layered structure, depicted in Fig. 24E. The Sm3+ ions are bound to the GO surface in solutions due to the electrostatic attraction. Modified GO flakes are capable of coordinating with DPyP, which significantly reduces the aggregation of DPyP and facilitates the assembly between GO and DPyP, with Sm3+ ions acting as interfacial linkers. The measured H2 production over photocatalysts with and without Sm3+ ions (4.39 mmol g−1 for GO–Sm3+–DPyP and 3.09 mmol g−1 for GO–DPyP) illustrates that the inclusion of the Sm3+ ions is beneficial for the overall photocatalytic performance of the porphyrin and GBM composites (Fig. 24F). Since PET and subsequent efficient separation of the generated electrons and holes are the key processes for the photocatalytic activity of any catalyst, the photoelectric response and electrochemical resistance of GO–Sm3+–DPyP were evaluated to provide a direct experimental rationale for the enhanced activity of this hybrid. The photoresponse of GO–Sm3+–DPyP exceeded that of GO–DPyP, suggesting a higher availability of photogenerated electrons for the photocatalytic reaction in the Sm3+ ion-enhanced composite. Furthermore, the electrochemical measurements for the GO−Sm3+–DPyP composite indicated that the interfacial charge transfer resistance turned out to be lower than that for the reference GO–DPyP sample, further boosting the photocatalytic performance of the hybrid. Detailed information about the porphyrin- and graphene-based photocatalysts used for hydrogen production to date is compiled in Table 4.
4.2 Photodegradation of pollutants
Well-organized porphyrin nanostructures show enhanced photocatalytic performance compared to amorphous (monomeric) porphyrins.40,124–126 This improvement is due to the fact that porphyrin aggregates with a distinct molecular arrangement offer better charge separation capabilities than the monomeric forms. The self-assembly of porphyrins was proven to be an effective method for the fabrication of nanostructured porphyrin-based materials with adjustable morphologies and functionalities.135 Various self-assembly protocols were reported for the synthesis of porphyrin nanostructures including ionic self-assembly,136 surfactant-assisted self-assembly (SAS)137 and reprecipitation.138 Notably, porphyrin nanostructures can be precisely engineered to achieve specific sizes, shapes, and functions via meticulous molecular and synthetic design. These tailored porphyrin assemblies have been thoroughly investigated in terms of their catalytic properties under visible light.126,135,139,140 For example, Mandal et al. reported the formation of TCPP nanostructures via a surfactant-assisted self-assembly, which resulted in a variety of morphologies such as spheres, rods, flakes, and flowers, all of which were active in the photodegradation of the pollutant Rhodamine B (RhB) under visible-light irradiation.141 Notably, the rod-shaped TCPP nanostructures showed the greatest efficacy in degrading RhB. In a separate study, La et al. detailed the production of TCPP nanostructures prompted by arginine, which displayed improved photocatalytic activity for degrading RhB under visible-light conditions.140Although the photocatalytic capabilities of porphyrin aggregates with various morphologies have been effectively demonstrated, their efficiency is often curtailed by the rapid recombination of the photo-induced electron–hole pairs. To address this issue, the concept of enhancing the photocatalytic performance of porphyrin nanostructures—improving light-harvesting efficiency, surface area, recyclability, and charge separation—was conceived by integrating them with graphene-based materials (GBM). The use of graphenes as a support material, which is known for its large surface area and high electron mobility, helps increase the number of catalytically active sites. Moreover, it suppresses the recombination of electrons and holes generated during the irradiation of porphyrin aggregates, which, in turn, increases the charge carrier lifetime and results in enhanced photocatalytic performance. Additionally, graphene effectively transfers electrons and holes to the adsorbed agents for photocatalytic reactions.135 Consequently, binding porphyrin nanostructures with GBM has gained considerable interest over the past few years, offering new routes to design and fabricate hybrids with enhanced light-harvesting and charge transfer capabilities. The activities of the reported Por- and GBM-based materials in degrading pollutants through photocatalysis are compiled in Table 5. However, one has to be aware that any comparison between the reported systems needs to be done with caution since the photocatalytic activity of these systems depends not only on the intrinsic catalytic activity of the photocatalyst, but also on the experimental variables such as the concentrations of RhB and the photocatalyst, or the intensity of the light source, all of which show considerable variation across the referenced studies (Table 5).
| Photocatalysts | Preparation | Pollutant | Experimental conditions | Activity | Ref. |
|---|---|---|---|---|---|
| ZnTPyP nanorods/GO | SAS in which GO plays the role of surfactant | RhB | Xe lamp 500 W (λ > 400 nm) | k = 2.6 × 10−2 min−1 | 142 |
| [RhB] = 10 mg L−1 | |||||
| 1D ZnTPyP = 10 mg L−1 | |||||
| TCPP nanorods/GNPs | CTAB-mediated self-assembly | RhB | 1500 W Xe lamp (λ > 400 nm) | k = 7.3 × 10−3 min−1 | 143 |
| [RhB] = 5 mg L−1 | |||||
| Photocatalyst: 5 mg L−1 | |||||
| TCPP nanofibers/GNPs | Arginine-mediated self-assembly | RhB | 1500 W Xe lamp (λ > 400 nm) | k = 15.8 × 10−3 min−1 for MB | 144 |
| MO | [RhB] = 5 mg L−1 | k = 6.5 × 10−3 min−1 for MO | |||
| Photocatalyst: 5 mg L−1 | |||||
| 5,15-Bisdodecyl porphyrin NPs/RGO | Sonication followed by sedimentation and filtration | Congo red | UV and UV-vis irradiation | 78% removal of Congo red after 90 min | 145 |
| [Congo red] = 10 mg L−1 | |||||
| Photocatalyst = 200 mg L−1 | |||||
| TMPyP/GO | Re-precipitation self-assembly | Mixed MB and MO | 150 W Xe lamp (λ > 420 nm) | k = 3.9 × 10−2 min−1 for MO | 146 |
| [Mixed dyes] = 9.0 × 10−4 M | k = 1.1 × 10−2 min−1 for MB | ||||
| Photocatalyst = 500 mg L−1 | |||||
| THPP NPs/RGO (free-standing) | THPP NPs/GO colloid was vacuum filtrated followed by gaseous reduction | MB, RhB | 500 W xenon lamp (λ > 400 nm) | 90% MB removal after 60 min, | 77 |
| [MB] = [RhB] = 0.01 mM | 80% of RhB removal after 100 minutes | ||||
| Photocatalyst: 100 mg L−1 | |||||
| TCPP/graphene/Pt | Layer-by-layer self-assembly | MO | Xe lamp 300 W (λ > 400 nm) | k = 4.8 × 10−3 min−1 | 119 |
| TMPyP/GO | Mixing of solutions | RhB | 150 W halogen (λ > 400 nm) | k = 1.0 × 10−3 min−1 | 84 |
| [RhB] = 1.66 mg L−1 | |||||
| Photocatalyst: 16.8 mg L−1 | |||||
| ZnTMPyP/GO | Mixing of solutions | RhB | 150 W halogen (λ > 400 nm) | k = 1.9 × 10−3 min−1 | 84 |
| [RhB] = 1.66 mg L−1 | |||||
| Photocatalyst: 16.4 mg L−1 | |||||
| BiTPPH–RGO | Covalent functionalization via solvothermal reaction | RhB | Visible light irradiation | 94% removal of RhB after 90 min | 147 |
| [RhB] = 4.8 mg L−1 | |||||
| Photocatalyst: 1 g L−1 | |||||
| BiTCPP–RGO | Covalent functionalization via solvothermal | RhB | Visible light irradiation | 94% removal of RhB after 90 min | 147 |
| [RhB] = 4.8 mg L−1 | |||||
| Photocatalyst: 1 g L−1 | |||||
| FeTPPH–GO | Covalent functionalization via solvothermal and reflux heating | Crystal violet | LED bulbs, 18 W | k = 3.2 × 10−2 min−1 | 148 |
| [CV] = 4 mg L−1 | |||||
| Photocatalyst: 0.4 mg L−1 | |||||
| TPPNO2/RGO/(TiO2 nanowire) | Electrochemical deposition of RGO on TiO2 nanowire followed by reflux with porphyrin | MB | 200 W Xenon lamp (λ > 400 nm) | 85% removal of MB after 7 h | 149 |
| [MB] = 9.2 mg L−1 | |||||
| Photocatalyst: 180 mg L−1 | |||||
| TCPP/RGO/(TiO2 nanotubes) | Hydrothermal and reflux heating | MB | Halogen lamp (λ > 390 nm) | 90% removal of MB after 2 h | 150 |
| [MB] = 10 mg L−1 | |||||
| Photocatalyst: 100 mg L−1 | |||||
| CuTCPP/RGO/(TiO2 nanotubes) | Two-step hydrothermal method and heating reflux | MB | Halogen lamp (λ > 390 nm) | 90% removal of MB after 2 h | 151 |
| [MB] = 10 mg L−1 | |||||
| Photocatalyst: 100 mg L−1 | |||||
| SnTCPP/RGO/TiO2 | Hydrothermal method plus heating reflux | MO | 400 W Xe lamp (λ > 420 nm) | 100% removal of MO after 3 h | 152 |
| [MO] = 10 mg L−1 | |||||
| Photocatalyst: 600 mg L−1 | |||||
| TiO2/G/TCPP aggregates | One-pot surfactant-assisted acid–base neutralization | RhB | Xe lamp 1500 W | k = 9.4 × 10−3 min−1 | 153 |
| [RhB] = 5 mg L−1 | |||||
| Photocatalyst: 5 mg L−1 | |||||
| BiVO4/G/TCPP | Two-step hydrothermal method and heating reflux | MO | 40 W white LED lamp | 100% removal of MO after 3 h | 154 |
| [MO] = 10 mg L−1 | |||||
| Photocatalyst: 1 g L−1 | |||||
| Fe2O3–TiO2/G/TCPP | Precipitation self-assembly | RhB | 350 W xenon lamp | k = 1.12 × 10−2 min−1 | 155 |
| [RhB] = 5 ppm | |||||
| Photocatalyst: 40 mg L−1 | |||||
| Fe3O4/S-RGO/TCPP | Coprecipitation Fe3O4/S-RGO and mixing with TCPP | MB | LED | 98% removal of MB after 3 h | 156 |
| MB = 10 mg L−1 | |||||
| Photocatalyst: 1 g L−1 | |||||
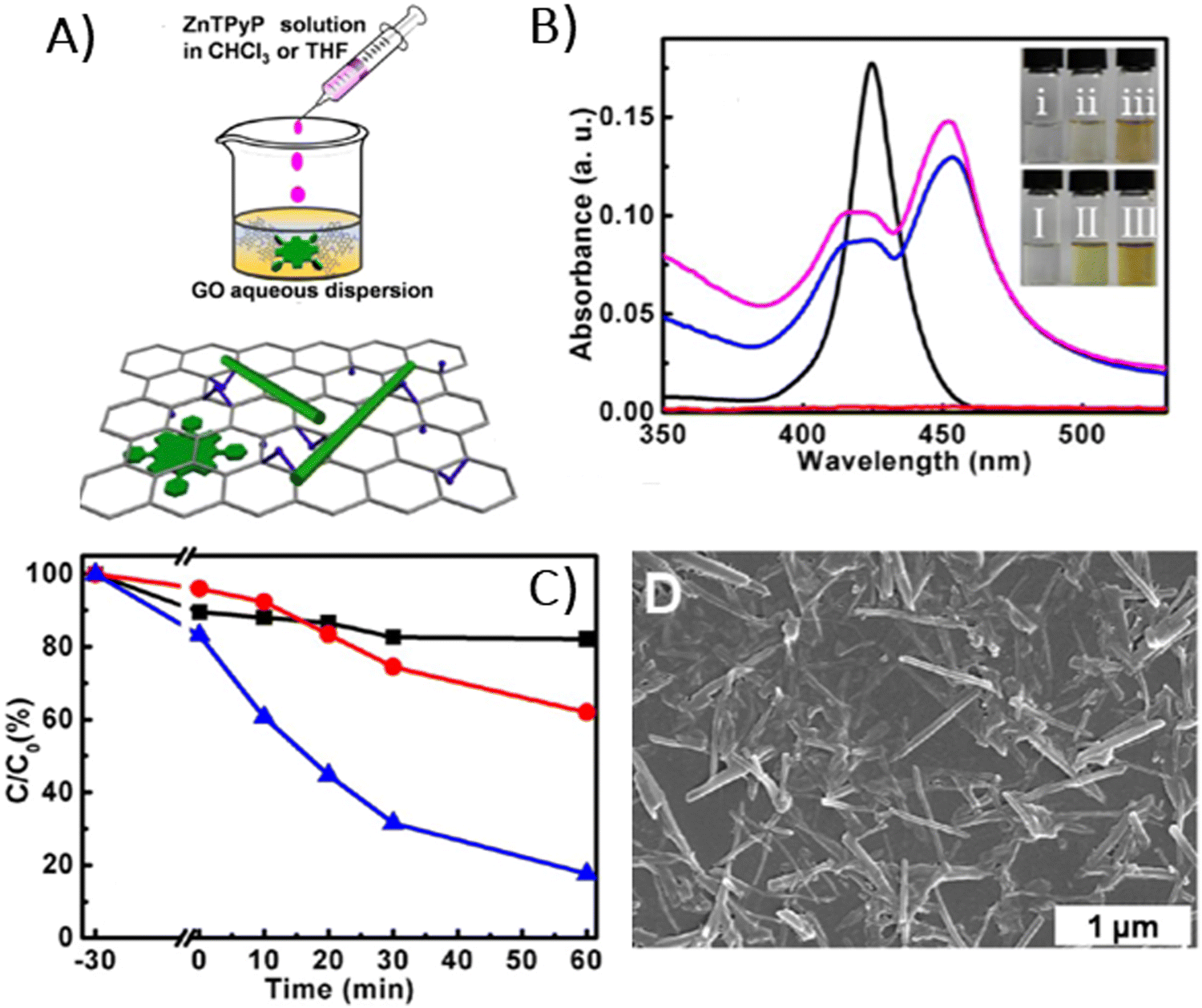 | ||
| Fig. 25 (A) Formation of ZnTPyP 1D nanostructures via a GO-assisted self-assembly, (B) UV-vis spectra of ZnTPyP in THF (black curve), ZnTPyP nanostructures self-assembled in the chloroform/water system with different GO concentrations: 0 mg mL−1 (red curve), 0.01 mg mL−1 (blue curve) and 0.02 mg mL−1 (magenta curve), (C) photocatalytic performance of the GO sample alone (black curve), 1D ZnTPyP nanoassemblies produced via the CTAB-assisted (red curve) and GO-assisted (blue curve) self-assembly towards the photodegradation of RhB under visible-light irradiation and (D) representative TEM image of 1D ZnTPyP/GO complexes. Reprinted with permission from ref. 142. Copyright 2013, American Chemical Society. | ||
In 2016 La et al. reported a straightforward fabrication of well-dispersed porphyrin nanorods on the surface of graphene nanoplates (GNPs) via direct self-assembly of monomeric TCPP molecules on graphene, aided by the CTAB surfactant (Fig. 26A).143 The resulting TCPP nanorods, measuring approximately 50 nm in diameter and ∼250 nm in length, were uniformly distributed over the GNPs, as verified by SEM (Fig. 26B) and TEM. Following their self-assembly, the hybrid materials displayed noticeable changes in the UV-vis spectrum. Compared to the Soret band of monomeric TCPP molecules (416 nm), the TCPP/GNP hybrids exhibited a distinct (8 nm) bathochromic shift of the Soret band to 424 nm. This was accompanied by a slight red-shift in the Q-bands. These spectral alterations led to the suggestion that most of the TCPP molecules in the supramolecular assemblies created with the aid of CTAB (surfactant) and GNPs tend to adopt a J-type aggregation pattern. Consequently, emission spectra also revealed changes, including variations in the number of peaks and their positions, along with a significant reduction in emission intensity following the self-assembly of TCPP on GNPs. The hybrid material showed improved visible-light photocatalytic activity for degrading RhB compared to the standalone TCPP nanorods. In the presence of GNPs-supported TCPP aggregates, the RhB degradation reached 80%, with a photodegradation rate constant of ca. 7.3 × 10−3 min−1 (Fig. 26C).
 | ||
| Fig. 26 (A) Diagram depicting the self-assembly of porphyrin aggregates on GNPs mediated by arginine and CTAB, (B) SEM image showing porphyrin nanorods on GNPs, (C) photocatalytic efficacy of GNP/TCPP assemblies obtained with the assistance of CTAB in degrading RhB: (a) control without catalyst, (b) GNPs, (c) free standing TCPP nanorods, and (d) GNP-supported TCPP nanorods, (D) SEM image of porphyrin nanofibers on GNPs, and (E) photocatalytic performance of GNP/TCPP assemblies obtained with the assistance of D-arginine during RhB degradation: (a) control without catalyst, (b) GNPs, (c) free-standing TCPP nanofibers, and (d) GNP/TCPP nanofibers. Adapted from ref. 143 and 144. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In 2017, La et al. reported that the fabrication of graphene/TCPP nanofiber hybrids can also be achieved without the use of CTAB, instead using arginine (Fig. 26A).144 SEM and TEM analyses revealed that the porphyrin nanofibers were densely and uniformly distributed across the surface of the GNPs, with diameters ranging from 50 to 200 nm and length extending to several micrometers (Fig. 26D). Unlike the CTAB-assisted self-assembly of TCPP with GNPs, the presence of arginine facilitated the formation of elongated aggregates of TCPP on graphene, producing nanofibers on the micron scale. The UV-vis spectra of the GNPs/TCPP nanofibers prepared in this manner displayed a Soret peak at 420 nm, which was 4 nm red-shifted in comparison to the Soret absorption band of monomeric TCPP. Moreover, a prominent peak at 666 nm along with three relatively weaker peaks in the Q-band region was registered. The emission spectra of GNP/TCPP nanofibers displayed weak and broad emission peaks at 686 nm and 730 nm, in contrast to the TCPP monomer, which emitted peaks at 655 and 714 nm. The authors explained the emergence of new emission peaks by the coupling effects from the spatial arrangement of the TCPP porphyrins, and they explained the reduction in emission intensity as a result of the quenching effect of graphene, indicating an efficient photoinduced electron transfer process. The GNP/TCPP nanofibers exhibited a remarkable visible-light photocatalytic performance towards degradation of RhB, which was completely removed after 150 minutes of light exposure (Fig. 26E). The hybrid material also exhibited substantial photocatalytic effectiveness towards MO degradation, achieving a degradation rate of 80% after 180 minutes. The authors proposed a potential mechanism for the enhanced photocatalytic activity of GNP-supported TCPP aggregates. In particular, when irradiated with visible-light, electrons in the valence band of TCPP crystals are excited across the bandgap to the conduction band (CB), leading to creation of e−/h+ pairs. These photoexcited electrons in the CB are then able to migrate freely to the graphene sheets. As a consequence, the generated electron–hole pairs are efficiently separated. This transfer ensures efficient separation of the electron–hole pairs, enhancing charge separation due to the presence of graphene. The reaction of holes with H2O or OH− produces hydroxyl radicals OH˙, which are capable of oxidizing the dye molecule. Simultaneously, oxygen can be reduced by the electrons accumulated on the graphene sheets to form superoxide radicals (O2˙−), which further react with the Rhodamine B dye.
In a different study, Bera et al. prepared TCPP aggregates (TCPP-AGGs) with various morphologies by a surfactant-assisted self-assembly method, which were then used to develop composites with RGO and Pt NPs.118 This research revealed that the morphology of the TCPP aggregates not only influences the photocurrent generation by the TCPP-AGGs/RGO/Pt, but also affects its photocatalytic activity toward MO degradation. The composite featuring rod-shaped TCPP aggregates exhibited the highest degradation rate constant k = 4.8 × 10−3 min−1 for MO. El-Shafai et al. fabricated another hybrid material composed of cationic TMPyP and GO by the self-assembly method.146 The fabricated GO/TMPyP displayed substantial photocatalytic activity for the concurrent degradation of MB and MO in water under visible light irradiation. The degradation rate constants for MO and MB with the GO/TMPyP were equal to 0.039 and 0.011 min−1, respectively.
Raveena et al. developed a nanocomposite crafted by covalently binding GO with FeTPPH through an ester linkage.148 The resulting nanocomposite demonstrated a strong affinity for crystal violet (CV) dye, with a significant detection limit and binding constant, enhancing the concentration of the dye on the photocatalyst surface. Exhibiting a high photodegradation efficiency of 93.45% for CV dye under visible light within 80 minutes at a low catalyst dosage, the nanocomposite proved to be stable and reusable, showing potential for wastewater treatment applications. In 2024, Yasmeen et al. similarly synthesized nanocomposites by covalently attaching bismuth porphyrins (Bi-TCPP, Bi-TPPH) to thermally reduced graphene oxide via ester bond formation. The nanocomposites showed promising photocatalytic efficiency, degrading RhB under visible light, with BiTCPP-RGO and BiTPPH-RGO achieving degradation rates of 94.58% and 93.26%, respectively.147
 | ||
| Fig. 27 (A) Picture of the TPPH NP/RGO film. (B) Top view of the photocatalysis setup for degrading the RhB aqueous solution. (C) UV-vis absorption spectra and (D) fluorescence spectra of the fabricated films. Photocatalytic degradation of (E) RhB and (F) MB in the presence of various catalysts under visible light irradiation. (G) Photocatalytic mechanism of the free-standing TPPH NP/RGO nanohybrid film. (H) Fluorescence decay profiles of TPPH and TPPH NPs/RGO hybrids in H2O. Adapted from ref. 77 with permission from the Royal Society of Chemistry. | ||
The emission intensity of the TPPH NPs adsorbed on the GO surface was significantly diminished relative to that of free TPPH NPs (Fig. 27D). This reduction in emission intensity was even more pronounced in the case of TPPH NPs/RGO films (Fig. 27D and H). The observed luminescence quenching was attributed to a robust interaction between the singlet excited state of TPPH and GO (RGO) in the hybrid. To substantiate the charge transfer mechanism responsible for the observed emission quenching, the authors conducted electrochemical impedance spectra. The charge transfer resistance during photoirradiation of the TPPH NP/RGO film (RCT ≈ 46.7 Ω) was significantly lower than that of the RGO film alone (RCT ≈ 176.2 Ω). This photoresponsivity was related to the presence of extensive donors/acceptors, facilitating effective charge separation. Most importantly, the free-standing TPPH NPs/RGO films exhibited enhanced photocatalytic activity during RhB and MB degradation compared to RGO standalone films and the TPPH NPs (Fig. 27E and F), which was explained by the mechanism depicted in Fig. 27G.
A few years later, Ussia et al. introduced a new concept of a free-standing graphene-based device characterized by a high surface area capable of binding photosensitizers via π–π interactions, which displayed improved stability and photocatalytic efficiency compared to traditional colloidal photocatalysts.78 The used photosensitizers included two types of cyclic porphyrin polymers: homo-PPrs and co-PPrs (Fig. 6E). The free standing nanocomposites were prepared using high-quality 3D graphene supported by a nickel foam, which was then immersed in the porphyrin solutions. The fabricated materials exhibited a decreased shielding effect (presence of high content of GBMs limits light absorption by the photosensitizer) as well as radical scavenger activity (graphene-based materials are effective scavengers of OH˙ radicals). SEM analysis of the GF/homo-PPr and GF/co-PPr revealed that both polymers thoroughly coated the surface of the foams (Fig. 28A and B).
 | ||
| Fig. 28 (A) Dried GF/co-PPr sample along with its SEM image, (B) dried GF/homo-PPr sample along with its SEM image, (C) photocatalytic performance of GF/co-PPr and GF/homo-PPr in degrading MB, (D) recyclability after re-deposition of co-PPr on degraded GF/co-PPr and (E) mechanism of the dye photodegradation in water facilitated by GF/polyporphyrin hybrids. Adapted from ref. 78. | ||
Yet, their morphologies varied: the graphene-supported co-PPr displayed a relatively smooth surface, whereas the graphene-supported homo-PPr exhibited greater surface roughness. The photocatalytic efficiencies of both nanocomposites for the photodegradation of the MB dye in water under visible light irradiation were evaluated and compared to those of graphene hybrids with monomeric porphyrin. The latter, when applied onto a Ni foam, did not show significant photocatalytic activity, probably due to the rapid recombination of the photogenerated electron–hole pairs. However, when the polymeric porphyrins were integrated with a graphene/Ni foam, the interface between the hybrid components effectively prevented charge recombination, resulting in enhanced photocatalytic activity of these composites (Fig. 28C). The authors also conducted the recyclability tests, which are essential for any practical applications. It was discovered that the percentage of MB degradation for the GF/co-PPr device decreased from 71% in the first cycle to 22% by the third cycle (Fig. 28D). SEM analysis carried out after the third run indicated that the reduction in photocatalytic activity was primarily due to the degradation of porphyrin macromolecules on the graphene surface. The device's performance was restored when the graphene foam was re-embedded again with the co-PPrs polymers prior to undergoing another MB photodegradation process. Based on these experimental findings, a possible pathway for the MB photodegradation using the GF/polyporphyrin device was suggested (Fig. 28E). Upon exposure to visible light, the polyporphyrin is excited from the ground state to the singlet excited state. From this state, several scenarios could unfold: (1) the photogenerated electrons from the singlet excited state of polyporphyrin are transferred to the graphene foam, (2) these electrons are utilized to reduce molecular oxygen, forming superoxide radical anions, (3) the singlet excited state of polyporphyrin undergoes intersystem crossing to triplet state, which then transfers energy to molecular oxygen to produce singlet oxygen, and (4) the surplus holes in the porphyrin move to the surface and react with OH− to generate reactive oxygen species.
In a follow-up study, Ussia et al. prepared a free-standing material composed of graphene foam and porphyrin-based polymers (Porph rings), this time excluding nickel.79 The photocatalytic efficacy of the resulting Ni-free material was assessed against specific pollutants: 2,4-dichlorophenoxyacetic acid (2,4-D) herbicide, MB dye and water-soluble polyethylene–glycol (PEG). The Ni-free device managed to degrade the 2,4-D herbicide, although it did not achieve complete mineralization. Conversely, the same photocatalyst was able to degrade and mineralize PEG, as confirmed by total organic carbon measurements. Observations after 120 minutes of light exposure revealed that the (Ni-free foam)/G/(Porph rings) composite degraded 93.4% of MB (Fig. 29A and B), while the degradation efficiency reached only 60% with the Ni-foam/G/(Porph rings). As anticipated, the Ni-foam/G used as a control did not show any degradation of MB. These results underscored the significance of removing Ni to enhance the photodegradation efficiency. To identify which reactive oxygen species were pivotal in the MB photodegradation process, the authors of the referenced study conducted further tests using radical-trapping agents. It is well documented that upon photocatalyst irradiation with light, various reactive oxygen species including hydroxyl radicals (OH˙), superoxide radicals (O2˙−) and the singlet oxygen (1O2) can be generated. These reactive species are capable of degrading pollutants through multiple redox reactions.
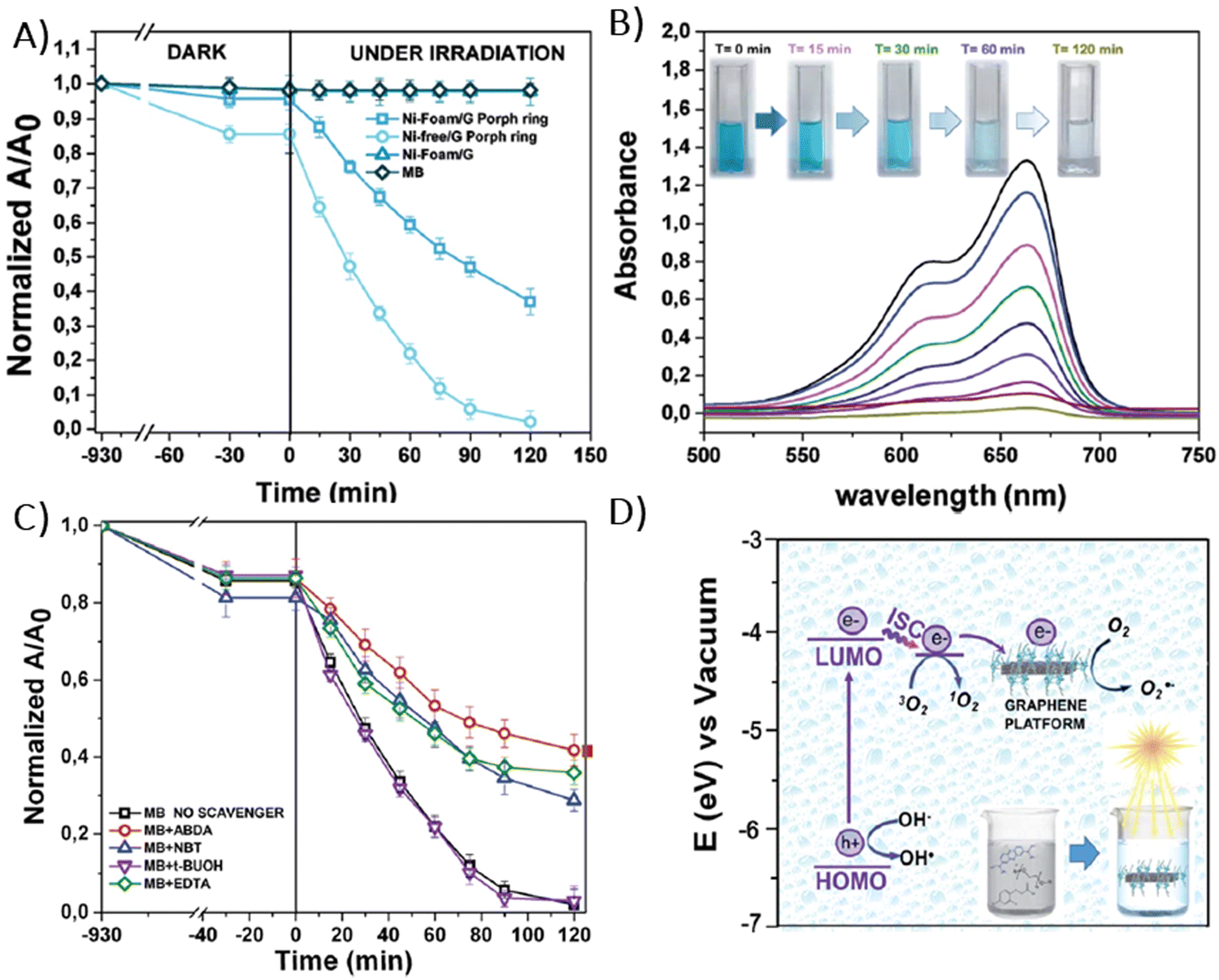 | ||
| Fig. 29 (A) Photocatalytic activity of Ni-foam/G, Ni-foam/G/(Porph rings), (Ni-free foam)/G/(Porph rings) in the degradation of MB, (B) variations in the MB absorption spectra following visible light irradiation in the presence of (Ni-free foam)/G/(Porph ring), (C) influence of ABDA, NBT, EDTA and t-BuOH radical scavengers on the MB degradation rate of MB with (Ni-free foam)/G/(Porph rings) and (D) energy diagram illustrating the proposed mechanism for the photocatalytic degradation of MB by using (Ni-free foam)/G/(Porph rings). Adapted from ref. 79. | ||
Differentiation among the various produced reactive oxygen species can be achieved by assessing the photocatalytic activity in the presence of specific radical-trapping agents. For example, ethylene diamine tetraacetic acid (EDTA), tetra-butanol (t-BuOH), nitro-blue tetrazolium (NBT) and 9,10-anthracene-diyl-bis(methylene)dimalonic acid (ABDA) are utilized to effectively scavenge photogenerated holes, hydroxyl radicals, superoxide radicals and singlet oxygen, respectively. Radical trapping experiments conducted with an MB solution confirmed that singlet oxygen plays a primary role in the photocatalytic activity of the studied hybrid (Fig. 29C), though other oxygen species also contribute to some extent in the degradation of the pollutant. Additionally, the HOMO and LUMO energy levels of the photosensitizer, determined through electrochemical methods, allowed the authors to construct an energy diagram and propose a mechanism of the photodegradation process, which is detailed in Fig. 29D.
Ussia et al. also explored the impact of the solvent used in the preparation of copolyporphyrin and graphene hybrids.127 The porphyrin-based polymer was dissolved in different solvents including chloroform, tetrahydrofuran and dimethylformamide (DMF), and then applied to the graphene surface via bath deposition. The nature of the solvent influenced how the polymer coated the graphene surface, as evidenced by SEM and AFM imaging. Crucially, upon exposure to visible light irradiation, the graphene coated with copolyporphyrin dissolved in DMF demonstrated the most effective photocatalytic activity for degrading MB.
 | ||
| Fig. 30 Ternary nanostructures of various porphyrin/RGO/TiO2 composites and their photocatalytic performance. Adapted with permission from ref. 149, 151 and 153. Copyright 2014 Elsevier B.V, 2016 Hydrogen Energy Publications LLC, 2015 Elsevier Inc. | ||
 | ||
| Fig. 31 Diagram depicting the photocatalytic activity of the TCPP/RGO/(TiO2 nanotubes = TNT) during the degradation of MB pollutants, including the light-initiated reactions within this photocatalytic system. Adapted with permission from ref. 150. Copyright 2016 Hydrogen Energy Publications LLC. | ||
Wei et al.151 prepared analogous ternary photocatalysts using TiO2 nanotubes and RGO, but opted for copper(II) porphyrin, CuTCPP, as the photosensitizer instead of the non-metallated form used by Wan et al.150,151 These composites showed improved photocatalytic activity toward MB degradation, achieving more than 95% decomposition of the pollutant over 2 hours of irradiation (compared to 92% for the photocatalyst with TCPP as in ref. 151). For context, the amount of MB decomposed by TiO2 nanotubes alone under identical experimental conditions was 5 times lower.
In contrast to the earlier-discussed photocatalysts using TiO2 nanorods/nanotubes, the photocatalytic system for RhB degradation examined by La et al. utilized TiO2 nanoparticles (Fig. 30C).153 Graphene nanoplates were decorated with TiO2 nanoparticles ranging from 15 to 30 nm in diameter and porphyrin nanorods, 50–60 nm wide and several hundreds of nanometers in length, synthesized via a CTAB-assisted self-assembly process. The hybrid material, when exposed to light irradiation, facilitated RhB degradation with a rate constant of ca. 9.4 × 10−3 min−1. For comparison, when free-standing GNPs, TiO2, and TCPP aggregates were employed in photocatalytic tests, RhB decomposed with rate constants of ca. 1.7, 2.2 and 3.9 × 10−3 min−1, respectively.
Zargari et al. also fabricated graphene sheets decorated with TiO2 nanoparticles, but uniquely pillared with SnTCPP. This chemical functionalization helped to maintain appropriate separation of the graphene nanosheets, resulting in a material with innovative electronic and optical properties (Fig. 32).152
 | ||
| Fig. 32 Diagram depicting the charge transfer process in the prepared SnTCPP/graphene/TiO2 hybrid exposed to visible light irradiation. Adapted with permission from ref. 152. Copyright © 2015 Elsevier Inc. | ||
The intercalation of SnTCPP within the structure of the TiO2-decorated graphene sheets was confirmed through a series of analyses including FT-IR spectroscopy, Raman spectroscopy, BET, and EDX. Upon exposure to visible light irradiation, the degradation of MO over SnTCPP/graphene/TiO2 was completed within a span of 180 min. The resulting photocatalytic performance of this hybrid material exhibited a remarkable enhancement, showing a fivefold increase compared to that of pristine TiO2. In addition, a substantial 23-fold boost in the photocurrent response was noted for the hybrid in comparison to pure TiO2. The significant improvement in photocatalytic activity was attributed to the incorporation of SnTCPP within the graphene sheets. This intercalation not only augmented the available adsorption sites for the porphyrin photosensitizer within the hybrid structure, but also facilitated the charge transfer channel. In particular, the SnTCPP molecules acted as effective channels for charge transfer, aiding in the injection of electrons through the axial ligand of the excited SnTCPP into the conduction band of TiO2.
In addition to TiO2, various other inorganic semiconductors were successfully integrated into hybrids with porphyrin and graphene, aiming to fabricate materials with broad spectral responsiveness and superior charge separation properties, thereby improving their photocatalytic activity. For instance, Aghakhaninejad et al. described the synthesis of ternary composites, where BiVO4 replaced TiO2.154 This resulted in the formation of a BiVO4/G/TCPP hybrid material, which exhibited a distinct pineapple slab-like morphology. Following 30 minutes of visible light exposure, the composite displayed 78% efficiency in the photodegradation of MO, marking a 3.5-fold improvement compared to its binary counterpart, BiVO4/G. Among the various nanocomposites prepared with varying graphene contents, the BiVO4/G nanocomposite featuring 0.5% graphene exhibited the highest observed photocatalytic activity. Furthermore, the optimum photocatalytic activity was achieved for BiVO4/G/TCPP with a 1/20 weight ratio of porphyrin to the nanocomposite. Subsequent examinations utilizing radical scavengers revealed that the photocatalytic reaction involved not only O2−˙ and OH˙ but also holes.
Moshari et al. developed a composite material by the initial preparation of sulfur-doped RGO (S-RGO), which was then functionalized with TCPP and Fe3O4 nanoparticles to form a TCPP/S-RGO/Fe3O4 composite (Fig. 33A).156 This material was further tested for its efficacy in MB photodegradation. The results showed that after 3 h of visible light irradiation, the removal efficiencies of MB in the presence of RGO, S-RGO, Fe3O4/S-RGO and TCPP/S-RGO/Fe3O4 composites were found to be 60%, 86%, 75%, and 98%, respectively. Notably, the TCPP/S-RGO/Fe3O4 composite exhibited superior photocatalytic activity when compared to the other tested materials. In contrast, La et al. reported the fabrication of graphene sheets decorated with Fe2O3–TiO2 particles and TCPP aggregates, achieved through the self-assembly of porphyrin monomers in the presence of Fe2O3–TiO2 nanoparticles and graphene (Fig. 33B).155 The aim of this magnetic photocatalyst construction was to develop a material that could be easily separated from the solution. The Fe2O3–TiO2 binary oxides and porphyrin aggregates were evenly dispersed on the graphene surface. The TCPP formed fiber-like nanostructures with a diameter of 100 nm and lengths in the micrometer range, while the diameters of Fe2O3–TiO2 nanoparticles ranged from 100 to 150 nm. The resulting composite displayed remarkable photocatalytic performance during the degradation of RhB, with a rate constant for RhB decomposition of 1.12 × 10−2 min−1. This performance was significantly improved compared to the TCPP aggregates alone and graphene/Fe2O3–TiO2 composite. In the reusability tests, only a negligible loss of photocatalytic activity (less than 8%) was registered after 4 runs, indicating the material's stability and its potential for multiple catalytic uses.
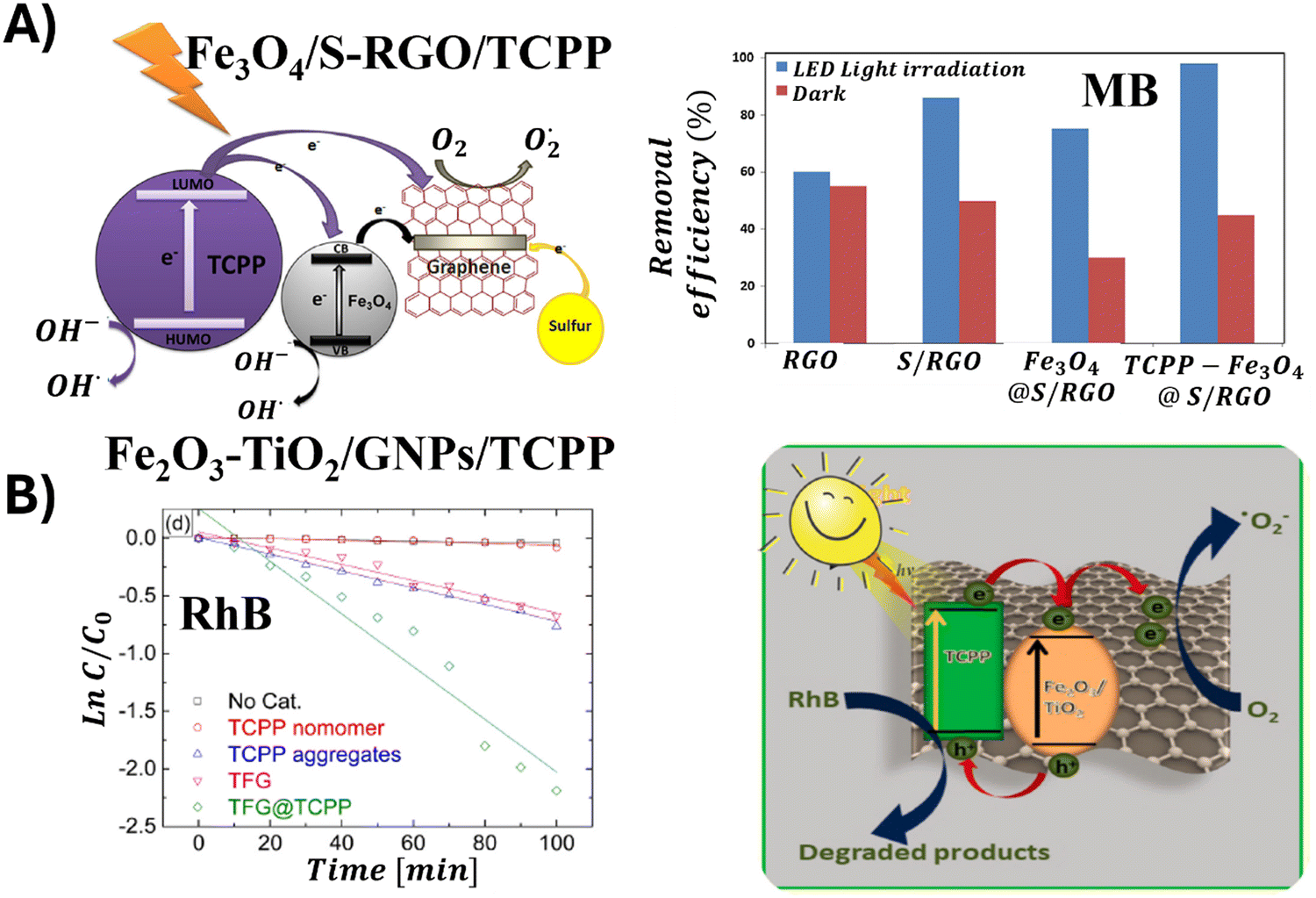 | ||
| Fig. 33 (A) Mechanism of MB photodegradation catalyzed by Fe3O4/S-RGO/TCPP. Adapted with permission from ref. 156. Copyright 2015, Springer Science Business Media Dordrecht. (B) Mechanism of RhB photodegradation catalyzed by Fe2O3–TiO2/GNPs/TCPP. Adapted with permission from ref. 155. Copyright 2021 Elsevier Ltd. | ||
For a comprehensive overview of the performance of porphyrin and graphene hybrid photocatalysts in pollutant decomposition discussed in Section 4.2, please refer to Table 5.
4.3 Photocatalytic CO2 reduction
To date, only a few research groups have demonstrated the activity of porphyrin and graphene-based materials (GBM) during the photocatalytic reduction of CO2. In 2014, Yadav et al. synthesized a photocatalyst by integrating two units of isatin and porphyrin (IP), which were subsequently covalently bonded to CCG.157 The resulting CCG-IP hybrid acted as a photocatalyst, which efficiently utilized visible light to catalyze multi-electron reduction of CO2 to methanol under ambient conditions, particularly when integrated with a series of sequentially coupled enzymes (Fig. 34A). The performance of this system was assessed under visible light irradiation of the CCG-IP photocatalyst combined with a rhodium complex, β-NAD+, three enzymes and TEOA. After 60 minutes of irradiation of this integrated photocatalyst/biocatalyst system in the presence of CO2, a methanol concentration of 11.21 μM was detected. The notably enhanced performance of the graphene-based photocatalyst CCG-IP during CO2 reduction to methanol was ascribed to the efficient charge separation, evident from the significantly higher photocurrent response of the hybrid compared to IP alone, as well as from time-resolved fluorescence measurements. These measurements provided insights into the charge separation and charge recombination rate constants for the photoinduced electron transfer between IP and CCG: 1.9 × 108 s−1 (charge separation) and 0.7 × 108 s−1 (charge recombination). Furthermore, Kumar et al. non-covalently modified graphene oxide with cobalt-metallated TAPP porphyrin (GO/CoTAPP) and investigated the potential of this hybrid during the conversion of CO2 into formic acid under visible light irradiation.61 The photocatalytic efficiency in generating formic acid from CO2 was evaluated to be 96.49 μmol after 2 h of irradiation.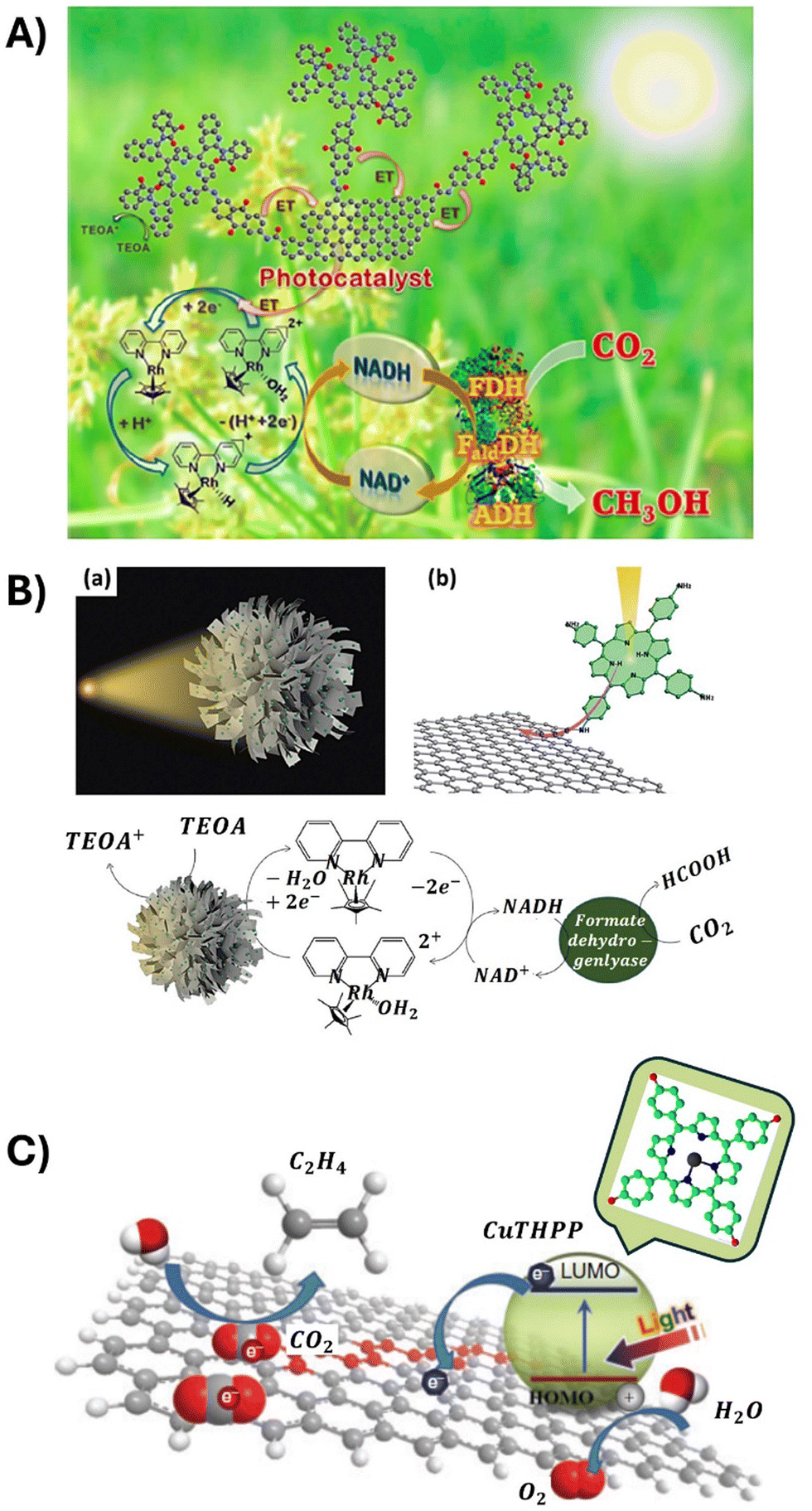 | ||
| Fig. 34 (A) Schematic illustration of the CCG-IP photocatalyst/biocatalyst integrated system for the photocatalytic reduction of CO2 to methanol. Reproduced from ref. 157. Copyright 2014 American Chemical Society. (B) Schematic illustration of the urchin-like structure of porphyrin/graphene microspheres and the working mechanism of the photocatalytic and biocatalytic cycles. Reproduced from ref. 158 with permission from the Royal Society of Chemistry (2017). (C) Illustration of the generation and transfer of photogenerated electrons in the illuminated CuTHPP/graphene composite. Reproduced from ref. 159 with permission from CSIRO. | ||
A new approach towards the fabrication of a photocatalyst active in the visible spectrum for CO2 reduction was developed by Li et al., who designed an urchin-like morphology by covalently linking TAPP with graphene (Fig. 34B).158 In brief, the authors outlined a method for the in situ assembly of TAPP/G into a micron-sized urchin-like spherical structure. This unique morphology was achieved via an evaporation-assisted self-assembly process using organic solvents at elevated temperatures. Using a concentration of 0.32 mg mL−1 of the urchin-like TAPP/G microspheres, the production of formic acid during a 2.5 h irradiation with visible light reached 215.28 mmol. In comparison, for the same concentration of TAPP/G and TAPP used as photocatalysts, the production of formic acid was equal to only 42.80 and 14.25 mmol, respectively. These findings clearly revealed the superior performance of the urchin-like TAPP/G microspheres compared to other photocatalytic materials. This study also emphasized that the photocatalytic performance is not solely an intrinsic property of the materials, but also arises from the carefully designed structure of the whole catalytic system.
Synthesis of the free-standing films composed of graphene sheets modified with CuTPPH photoactive toward the reduction of CO2 to C2H4 was reported by Piao et al.159 (Fig. 34C). The SEM analysis of the as-prepared thin films revealed the presence of nano-sized CuTHPP crystals growing on the surface of graphene sheets. To assess the photocatalytic properties of this material, the authors utilized a double-sided photocatalytic thin film placed within a chamber filled with CO2 and subsequently irradiated with a solar light simulator. The resulting production rates of ethylene and methane were calculated to be 23.17 mmol g−1 h−1 and 14.94 mmol g−1 h−1, respectively. The authors suggested that the photocatalytic activity and selectivity of this material can be optimized by altering the central metallic ion and grafting branch.
5. Por hybrids with other 2D materials
As already mentioned in the introduction, in recent years, there has been a rapidly increasing number of studies focused on non-graphene layered materials, including graphitic nitride (g-C3N4), transition metal dichalcogenides (TMDs) or transition metal carbides and nitrides (MXenes), just to mention a few. Nonetheless, there are only a limited number of reports devoted to composites of these materials with porphyrins, their photophysical and/or photoelectronic properties as well as their performance as photocatalysts. Recent advances within that field will be presented in the following paragraphs.To date, there have been successful reports of synthesizing various porphyrin/g-C3N4 composite materials, which consistently demonstrate superior photocatalytic performance to pure g-C3N4. Analogously to the composites of GBMs with porphyrins, the interaction between the components of the hybrids, i.e. porphyrin (Por) and other two-dimensional materials (2DM), significantly affects both the properties and photocatalytic capabilities of the composites.
Both porphyrins and g-C3N4 feature conjugated structures with numerous surface functional groups, which make their binding via non-covalent interactions straightforward. Using this method, Wang et al. successfully fabricated an organic heterostructure consisting of g-C3N4 and μ-oxo dimeric iron(III) porphyrin [(FeTPP)2O].160 Their synthesis relied on π–π stacking and Fe–amine interactions, which were enabled by the flexible 2D structure of g-C3N4 and its abundant amine groups. The experiments demonstrated that (FeTPP)2O porphyrin not only served as a photosensitizer but also promoted charge separation, effectively reducing the recombination of photoinduced electrons and holes within g-C3N4. Additionally, Wang et al. employed two asymmetric zinc porphyrins (ZnMT3PyP and ZnMTPP) as photosensitizers, attaching them non-covalently to the g-C3N4 surface (Fig. 35).161 When attached to Pt/g-C3N4, porphyrin molecules featuring three 3-pyridine substituents (ZnMT3PyP) displayed notably improved visible light–driven photocatalytic H2 production and stability compared to their counterparts with three phenyl groups (ZnMTPP). The photoactivity of the ZnMT3PyP/Pt/g-C3N4 catalyst was equal to 437 μmol g−1, while that of ZnMTPP/Pt/g-C3N4 was only equal to 293 μmol g−1, under light irradiation with λ > 420 nm (Table 6).
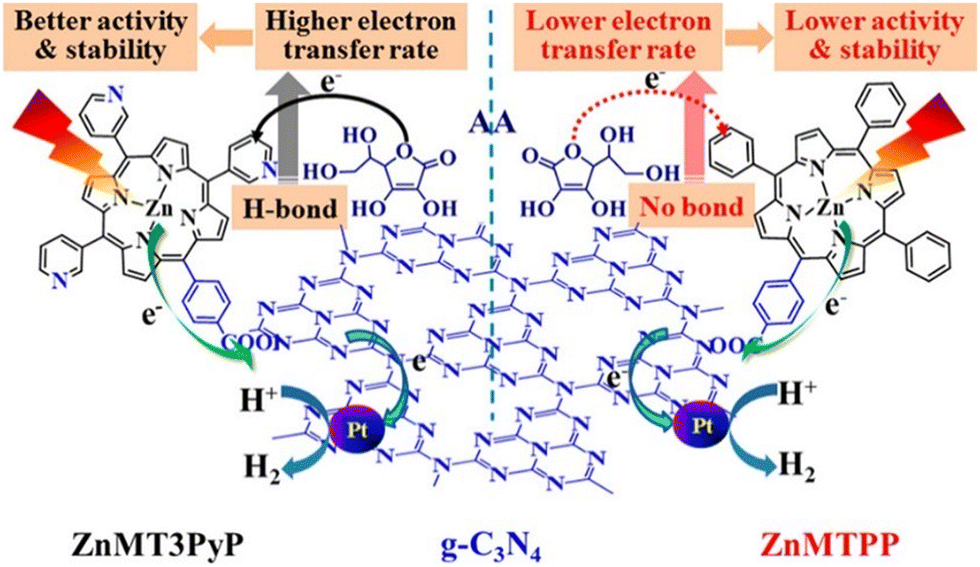 | ||
| Fig. 35 ZnMT3PyP and ZnMTPP sensitized graphitic carbon nitride for efficient visible light-driven H2 production. Reproduced form ref. 161. Copyright 2017, American Chemical Society. | ||
| Photocatalyst | Experimental conditions | Activity | Ref. |
|---|---|---|---|
| H 2 production | |||
| g-C3N4/[(FeTPP)2O] | Xe lamp 300 W, 10 vol% TEOA | 187.5 μmol g−1 h−1 | 160 |
| AQE = 0.0415% (420 nm) | |||
| ZnMT3PyP–Pt/g-C3N4 | Xe lamp 300 W, ascorbic acid (50 mM) | 2.28 mmol g−1 h−1 | 161 |
| AQE = 25.1% (420 nm) | |||
| TCPP/Pt/g-C3N4 | Hg lamp 450 W (λ > 380 nm) 10 vol% TEOA | 1.21 mmol g−1 h−1 | 162 |
| mTCPP–Pt/g-C3N4 | Mercury vapour lamp 20 mM EDTA | 2.28 mmol g−1 h−1 | 163 |
| g-C3N4–Cu–TCPP | Xe lamp 300 W, 10 vol% TEOA | 1.67 mmol g−1 h−1 | 164 |
| g-C3N4–Cu–THPP | Xe lamp 300 W, 10 vol% TEOA | 7.5 μmol g−1 h−1 | 165 |
| Pollutant degradation | |||
| CoTPP/g-C3N4 | Xe lamp 300 W, [RhB] = 25 μmol L−1 | Decomposition rate: 99.79% in 90 min | 166 |
| ZnTCPP/g-C3N4 | Xe lamp 300 W, [tetracycline] = 30 mg L−1 | Decomposition rate: 80.3% in 120 min | 167 |
| SA-TCPP/g-C3N4-O | Xe lamp 500 W, [bisphenol] = 10 ppm | Decomposition rate: 0.15 h−1 | 168 |
| Nanofibers-TCPP/g-C3N4 | Xe lamp 350 W, [RhB] = 10 ppm | Decomposition rate: 0.033 min−1 | 169 |
| Nanofibers-TCPP/g-C3N4/Ag NPs | Xe lamp 350 W, [RhB] = 10 ppm | Decomposition rate: 97% in 120 min | 170 |
| CO 2 reduction | |||
| 0.75% FeTCPP/g-C3N4 | CH3CN:H2O = 3:1, Xe lamp 300 W, 20 vol% TEOA |
CO: 1.09 mmol g−1 h−1 | 171 |
| CoTCPP/g-C3N4 | CH3CN Xe lamp 300 W, 20 vol% TEOA | CO: 17 μmol g−1 h−1 | 172 |
| CH4: 0.7 μmol g−1 h−1 | |||
| Selectivity: 80% | |||
| 3% TCPP NS/g-C3N4 | Xe lamp 300 W, H2O | CO: 16.8 μmol g−1 h−1 | 173 |
| O2: 10 μmol g−1 h−1 | |||
| ZnTPCN/g-C3N4 | Xe lamp 300 W, λ ≥ 400 nm | CO: 19.4 μmol g−1 h−1 | 174 |
| H2O | Selectivity: 95.8% | ||
The differences noted between the two investigated photocatalysts were mainly attributed to the rapid electron transfer from porphyrin molecules containing pyridine groups to g-C3N4, a process that did not occur with porphyrin molecules that only contained phenyl groups and g-C3N4.
In the research conducted by Mei et al., TCPP was deposited onto the Pt/g-C3N4 surface using a simple adsorption technique, resulting in the non-covalent composite TCPP/Pt/g-C3N4.162 The inclusion of TCPP into Pt/g-C3N4 markedly enhanced its capability for visible light–driven photocatalytic production of H2 from water. With a 1 wt% loading of TCPP, the TCPP/Pt/g-C3N4 composite displayed superior photoactivity, outperforming Pt/g-C3N4 by a factor of 2.1. The improvement in the photocatalytic activity of this organic heterostructure was not only due to better utilization of visible light by the porphyrin chromophore but also due to enhanced electron transfer, which helped to minimize the recombination of photoinduced charge carriers (Fig. 36A).
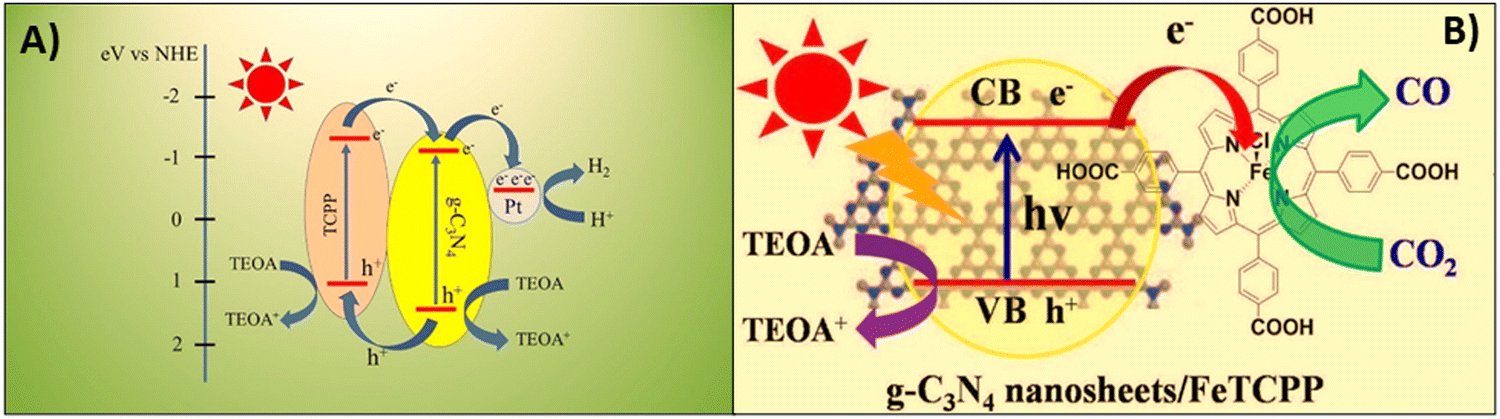 | ||
| Fig. 36 (A) Suggested mechanism for photocatalytic H2 evolution over TCPP/g-C3N4/Pt under visible light irradiation (λ > 380 nm). Reproduced from ref. 162. Copyright 2017 Elsevier. (B) Structure of the g-C3N4/FeTCPP heterogeneous catalyst. Adapted from ref. 171, Copyright 2018 Elsevier. | ||
The FeTCPP/g-C3N4 heterogeneous catalyst was synthesized by the He group via a facile self-assembly approach. This involved not only π–π stacking interactions but also hydrogen bonding between the carboxyl group of FeTCPP and the abundant amino groups on g-C3N4, as depicted in Fig. 36B.171 The resulting photocatalyst displayed excellent performance in reducing CO2, with a CO2 evolution rate of 6.52 mmol g−1 h−1 and a selectivity of 98%. Additionally, Da Silva et al. conducted a study which examined how the carboxyl groups and their positions on the porphyrin macrocycle substituents influence H2 generation.163 Three free-base porphyrins (TPP, m-TCPP, and p-TCPP) were used to sensitize g-C3N4via non-covalent interactions, creating three hybrid photocatalysts (Fig. 37). Each exhibited improved photocatalytic performance for hydrogen evolution over pure g-C3N4, with the m-TCPP yielding the highest amount of H2.
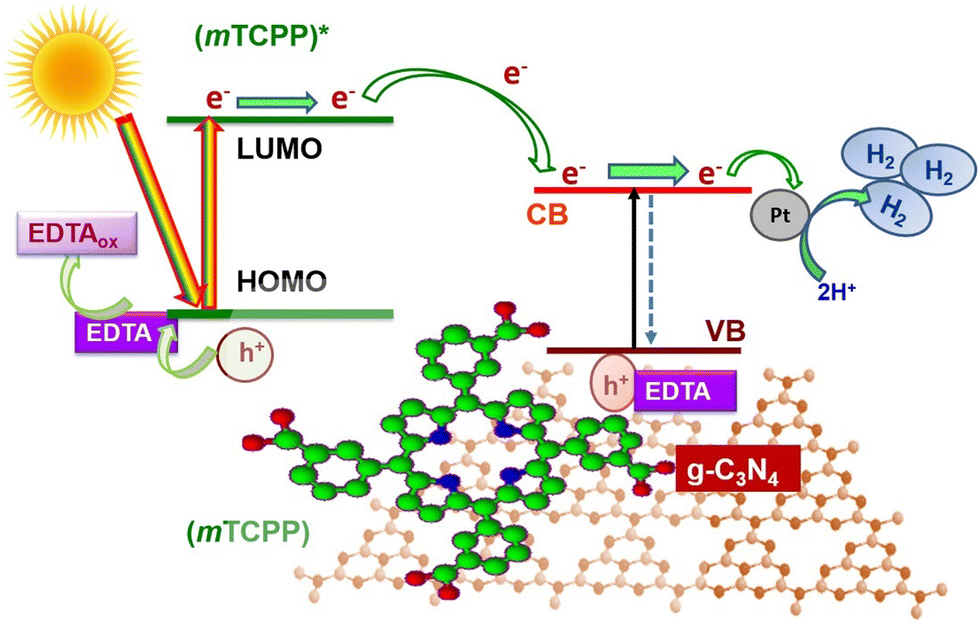 | ||
| Fig. 37 Mechanism of photocatalytic hydrogen production from water over g-C3N4 sensitized with m-TCPP porphyrin. Reproduced from ref. 163. Copyright 2018 Elsevier. | ||
Furthermore, Zhang et al. explored the effect of specific metal–porphyrins (MTPPs, M(II) = Co, Ni, and Cu) on the photocatalytic performance of g-C3N4.166 The composites with porphyrins were synthesized via a straightforward solvothermal approach. Notably, the CoTPP/g-C3N4 composite demonstrated exceptional efficiency during the photodegradation of RhB compared to pristine g-C3N4 and its hybrids with NiTPP and CuTPP. This improved photocatalytic activity was ascribed to more effective interfacial charge transfer occurring between CoTPP and g-C3N4.
In addition to the two-component hybrids and those requiring a Pt co-catalyst already mentioned, successful syntheses of other tricomponent composites involving porphyrin and g-C3N4 have already been achieved. For example, Zhu et al. and Zhang et al. developed copper-bridged porphyrin/g-C3N4 nanocomposites (Fig. 38) aimed at boosting photocatalytic hydrogen generation.164,165 Within these composites, porphyrin functioned as a visible light absorption antenna, with g-C3N4 functioning as the substrate. The addition of copper at the interface of g-C3N4 and porphyrin not only affects the morphology and structure of the nanocomposite but also strengthens the interaction between the two components.
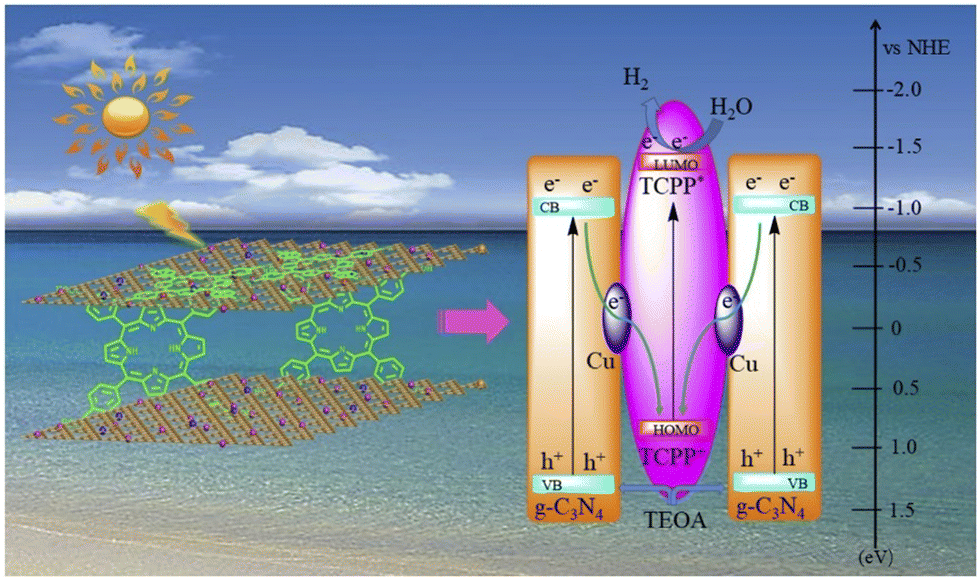 | ||
| Fig. 38 Scheme of the TCPP–Cu–g-C3N4 composite and illustration of the separation and transfer of photo-produced charges promoted by this photocatalyst. Reproduced from ref. 164. Copyright 2019 Elsevier. | ||
The hydrogen production rate over TCPP–Cu–g-C3N4 was determined to be 10.3 times greater than that of pure g-C3N4, and 9.6 times higher than that of TCPP/g-C3N4. The authors attributed this significant increase in photocatalytic hydrogen production activity to the enhanced electron transfer between g-C3N4 and TCPP, achieved without the need for additional co-catalysts.
As repeatedly noted throughout this review, the efficiency of electron separation in Por and 2DM hybrids can be remarkably improved by enhancing the interaction between the macrocyclic Por chromophores and the surface of the 2DM sheets, the same being true for g-C3N4. One way to achieve this is by forming a covalent bond between g-C3N4 and porphyrin. A few years ago, in the pioneering work of Zhu et al.,164 the fabrication of such a hybrid, linking Co–porphyrin and low-molecular weight g-C3N4 through a covalent bond, was reported. Therein, porphyrin acted as the reaction center, while g-C3N4 functioned as a visible-light-harvesting antenna. The effective transmission and accumulation of electrons by the active Co centers, combined with Co-porphyrin's affinity for CO2, led to an exceptional photocatalytic performance of the hybrid. The Ma group conducted successful covalent functionalization of g-C3N4 with Por (ZnTCPP) using a straightforward thermal polycondensation method to prepare a composite material.167 The effective attachment of ZnTCPP molecules onto the g-C3N4 surface was enabled by the amide groups acting as bridging units. The optimized 10% ZnTCPP–g-C3N4 composite showed remarkable photocatalytic activity, decomposing both methylene blue (MB) and tetracycline (TC) under visible light irradiation, with long-term reusability and elimination rates of 96% and 80.3%, respectively. This superior photocatalytic performance primarily stemmed from the highly efficient separation of electron–hole pairs and the enhanced harnessing of the solar light, as evidenced by photoluminescence measurements, electrochemical impedance spectra and photocurrent responses.
As depicted in Fig. 39, Xu et al. developed a heterojunction photocatalyst made up of self-assembled TCPP (SA-TCPP) and oxidized g-C3N4 (g-C3N4-O) by an in situ electrostatic method, resulting in a 0D/2D heterostructure (Fig. 39).168 Compared to g-C3N4-O and SA-TCPP alone, the SA-TCPP/g-C3N4-O hybrid exhibited greatly improved visible photocatalytic oxidation performance for the degradation of pollutants, oxygen evolution and disinfection. This improvement was primarily due to well-matched band structures and the π–π stacking interaction between SA-TCPP and g-C3N4-O, which effectively facilitated interfacial charge transfer.
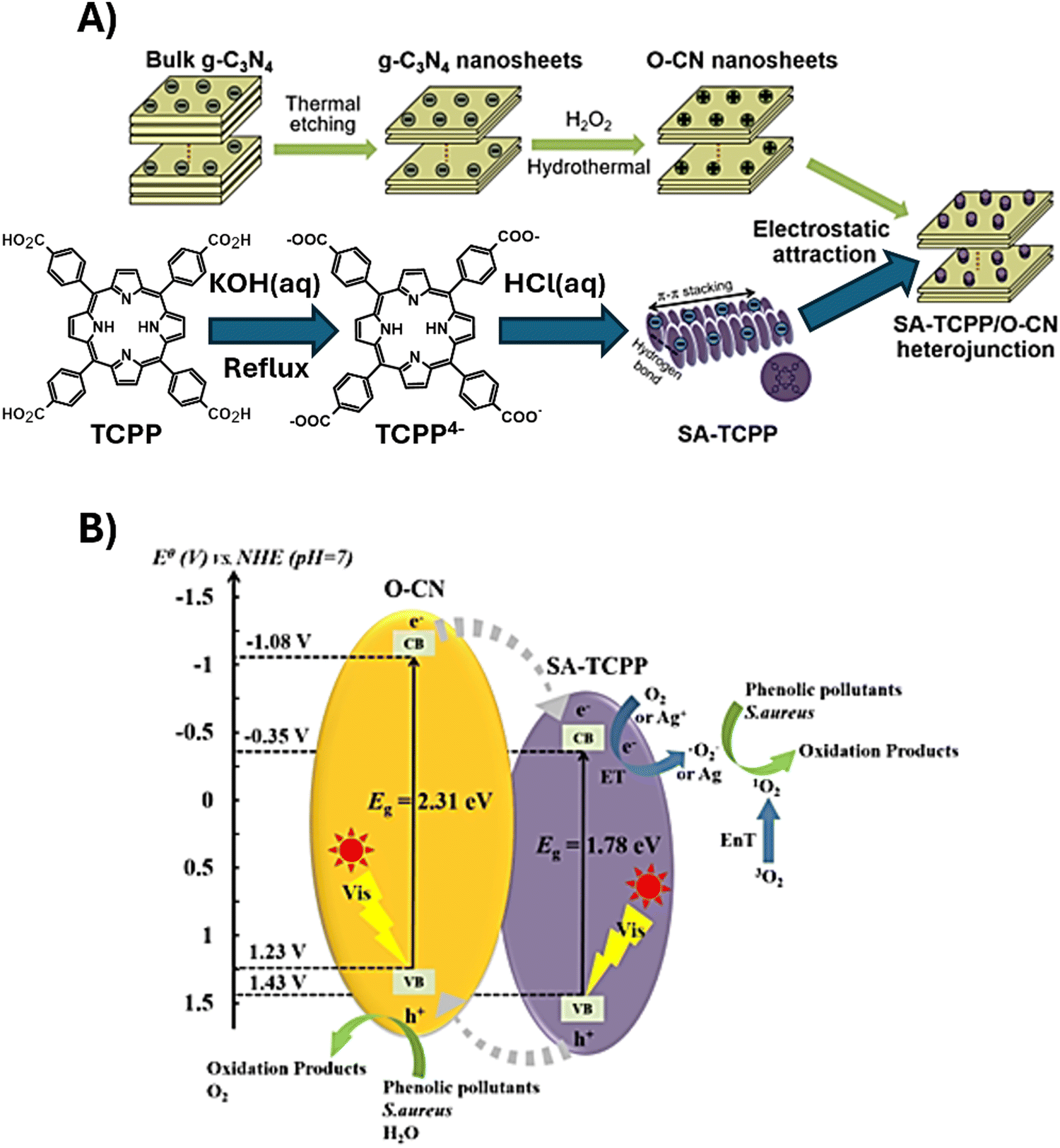 | ||
| Fig. 39 (A) Synthesis of g-C3N4-O, SA-TCPP and SA-TCPP/g-C3N4-O heterojunctions and (B) schematic representation of the proposed carrier separation and photocatalytic processes under visible light irradiation in the SA-TCPP/g-C3N4-O heterojunction catalyst. Reproduced from ref. 168 Copyright 2021 Elsevier. | ||
In another study, TCPP nanofibers with a diameter lower than 100 nm and lengths of several micrometers were successfully integrated onto the g-C3N4 nanoflakes.169 The obtained non-covalent hybrids possessed two bandgap energies of 2.38 and 2.7 eV, facilitating efficient photon capture from the visible light range. The photodegradation efficiency of these hybrids in decomposing Rhodamine B dyes was notably high, achieving a RhB removal rate of 3.3 × 10−2 min−1.
In 2023, Jia et al. introduced a novel heterojunction photocatalyst consisting of porous g-C3N4 nanosheets combined with zinc(II) tetra(4-cyanophenyl)porphyrin (ZnTPCN). Nanoassemblies were designed to act as a photocatalyst for the reduction of CO2 with H2O into valuable chemicals. Synthesized via a simple self-assembly process, this catalyst exhibited enhanced CO2 adsorption capacity and efficient charge carrier separation. Significantly outperforming pristine g-C3N4 nanosheets, the ZnTPCN/g-C3N4 composites achieved a CO generation rate of 19.4 μmol g−1 h−1 with a selectivity of 95.8%. By applying theoretical calculations and in situ Fourier transform infrared spectra, the authors suggested that the zinc sites in the porphyrin facilitate CO2 activation and CO production.174
As is apparent based on the above-discussed literature data, hybrids of porphyrin and g-C3N4 turn out suitable for solar energy utilization and conversion, including photocatalytic hydrogen production, CO2 reduction, and pollutant degradation. The advancements in photocatalysis using porphyrin-functionalized g-C3N4 are summarized in Table 6.
Compared to the graphene family or g-C3N4, TMDs are still in their infancy in the realm of visible light–driven photocatalysis and target-oriented assemblies with porphyrin chromophores. Among 2D TMDs, Mo- and W-based chalcogenides (MX2, M = Mo or W and X = S, Se or Te) are most intensively investigated materials with respect to their envisaged photocatalytic applications. As presented in the following paragraphs, thorough exploration of their spectroscopic characteristics as well as photophysical properties of their porphyrin-functionalized analogues may shed light on the utility of such hybrids in various energy conversion applications.
Two recent studies detail the integration of zinc porphyrin onto the MoS2 nanosheets, which are heterostructured with TiO2 and ZnO.175,176 The resulting composites, ZnTCPP/MoS2/TiO2, demonstrated activity in visible light–induced hydrogen evolution. Remarkably, even in the absence of noble metals, the ZnTCPP/MoS2/TiO2 photocatalyst with a MoS2 loading of 1.00 wt% showed the highest H2 production rate of 10.2 μmol h−1, and the turnover number (TON) relative to ZnTCPP dye reached 261 after 12 h of visible light irradiation.175 In a similar manner, for the ZnTCPP/MoS2/ZnO heterostructure with 0.5 wt% of MoS2, a H2 production rate of 75 μmol h−1 g−1 was reported (Fig. 40A). The authors suggested that the ZnTCPP complex, when anchored on the TiO2 or ZnO surface, allows the electrons from the excited ZnTCPP molecules to be injected into the conduction band of TiO2 or ZnO, and then transferred to MoS2 for the reduction of protons. Thereby, TiO2 or ZnO act as intermediaries in efficiently transferring electrons from photoexcited dye molecules to MoS2 (Fig. 40B). The quantity of MoS2 loaded on the TiO2 or ZnO surface is critical for trapping the conduction band electrons injected by the photoexcited zinc porphyrin, which helps suppress the recombination of photogenerated charge carriers.
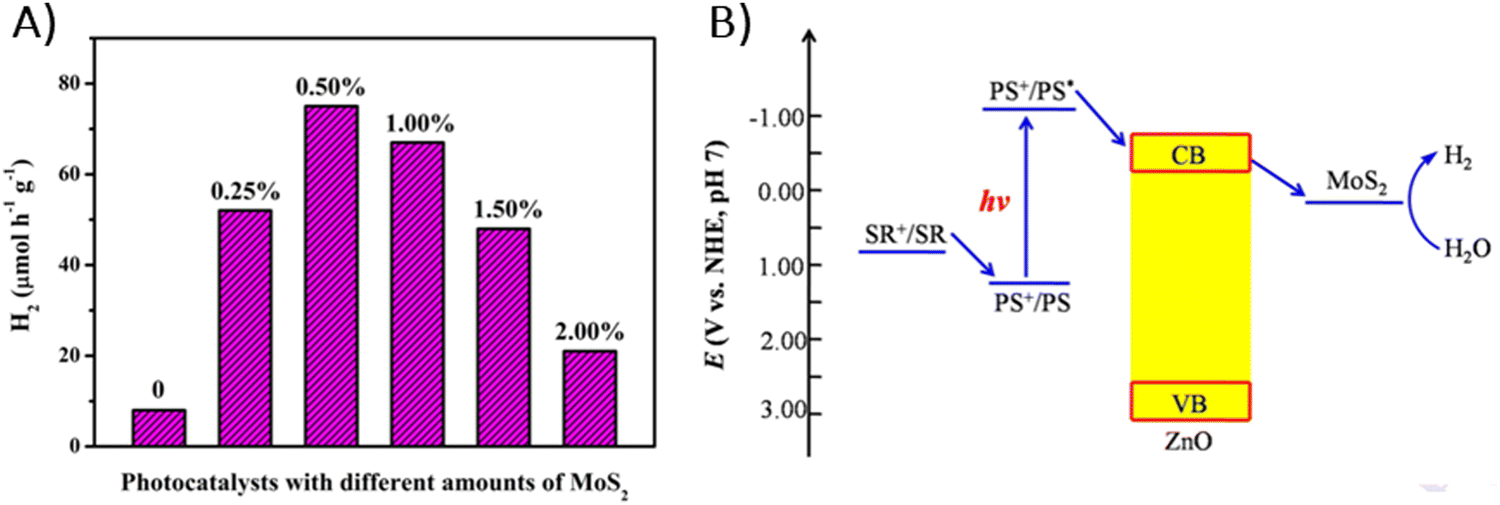 | ||
| Fig. 40 (A) Photocatalytic H2 evolution over ZnTCPP/MoS2/ZnO photocatalysts with different co-catalyst loadings in a 0.2 M TEOA aqueous solution under visible light irradiation (λ > 420 nm). (B) Suggested mechanism for visible-light-induced hydrogen production using ZnTCPP/MoS2/ZnO, photosensitizer (PS): ZnTCPP, sacrificial reagent (SR): TEOA. Reproduced with permission from ref. 176. Copyright 2015 Elsevier. | ||
Zhang et al. experimented with eight different porphyrins and found that the modification of the exfoliated large area 2D MoS2 sheets with these chromophores via physisorption generally led to the enhancement of photocurrent, which was highly dependent on the structure of the porphyrin molecule.54 After immobilizing porphyrins on MoS2, only minor changes were noted in the chromophore's absorption spectra. The faintness of the corresponding Por absorption bands suggests a minimal amount of the adsorbed Por. Nevertheless, emission measurements for some of porphyrins displayed a significant increase in photoemission, indicating the successful functionalization of MoS2 with Por. Through extensive photochemical experiments, the authors deduced that while adsorbed porphyrins act as photosensitizers contributing to the photocurrent via charge transfer from the photoexcited Por to the conduction band of MoS2, the bulk of the photocurrent is derived from the MoS2 itself. The greatest photocurrent enhancement was registered for porphyrins with a higher HOMO level, and the proportion of MoS2 flakes to Por was crucial for optimal photocurrent output. Karantanais et al. developed a nanoassembly composed of exfoliated MoS2 nanosheets interfaced with a quaternized diblock copolymer, which electrostatically captures anionic TPPS porphyrin (Fig. 41). The authors evaluated the emergence of photoinduced charge-transfer within this hybrid material by performing absorption and fluorescence (steady-state and time-resolved). The obtained data confirmed that the porphyrin units attached via the ammonium side groups in the copolymer block participate in the charge-transfer processes with the MoS2 lattice.177
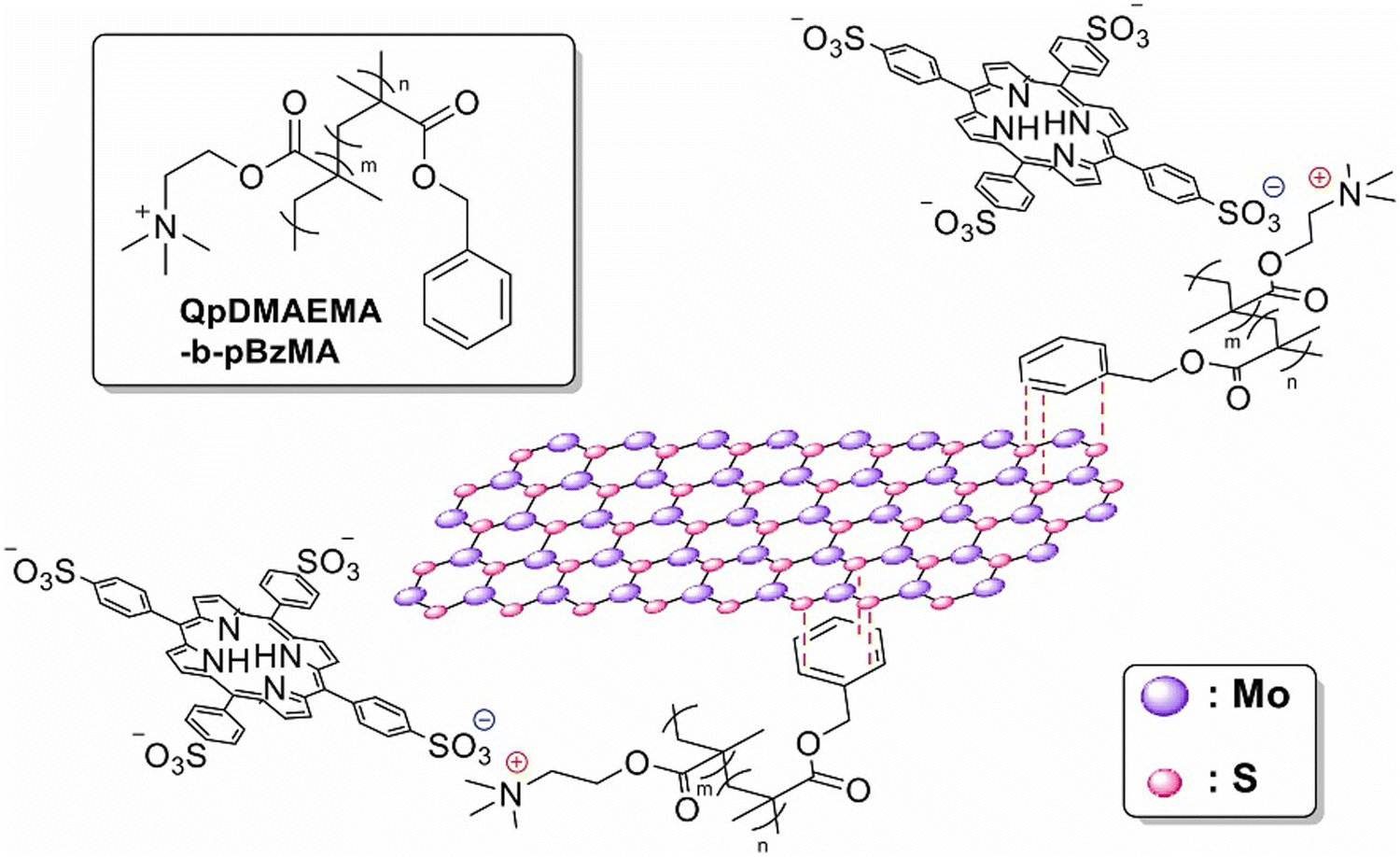 | ||
| Fig. 41 Illustrative structure of the MoS2/QpDMAEMA-b-pBzMA/TPPS nanohybrid. Reproduced with permission from ref. 177. Copyright 2024 Elsevier Inc. | ||
In 2023, Perivoliotis et al. assembled a composite by combining anionic cobalt porphyrin (CoP) with a positively charged modified molybdenum disulfide (MoS2), creating the CoP/MoS2 complex, which was then used as a non-precious photo/electrocatalyst for water oxidation reaction and selective H2O2 production.178 Their methodology relied on robust ion–pair interactions between the imidazolium-based MoS2 and the carboxylated CoP, which helped prevent the aggregation of chromophores, thereby enhancing the accessibility of the reactants to the cobalt active sites. A similar level of photocatalytic activity for water oxidation was observed in another study, in which WS2 was covalently linked to nickel porphyrin using a flexible long alkyl chain as a linker.179 These specially designed composites exhibited significant activity in electrochemically driven water oxidation, attributed to the well-dispersed Ni centers and improved electronic processes. The propensity of Ni to form high-valent complexes with oxygen moieties also aided in facilitating O–O bond formation. Moreover, the photoenhanced electrocatalytic properties of Por-WS2 were credited to the porphyrins, which functioned as photoreceptors. While fewer photogenerated holes are produced in the hybrid when illuminated, these holes are more readily transferred to the electrolyte solution, reducing charge recombination phenomena. The research conducted by Canton-Vitoria et al. shed more light on the spectroscopic properties of the 1,2-dithiolane-based porphyrin covalently linked to the edges of MoS2 sheets via addition reaction.56 Detailed photophysical studies using both steady-state and time-resolved measurements disclosed a complex decay dynamic of the photoexcited porphyrin when attached to MoS2, featuring a ping-pong energy transfer between porphyrin and MoS2. The research presented by Wei et al. focused on covalently functionalized MoS2 with Por obtained by a thiol–ene “click” reaction.180 Optical experiments revealed that the MoS2–Por nanohybrid shows reverse saturable absorption in response to femtosecond pulsed laser light at 800 nm, a distinct behavior compared to the saturable absorption observed with standard MoS2 nanosheets. This notable change in the optical response was attributed to an increase in two-photon absorption in the MoS2 nanosheets, which can be the result of effective charge transfer processes in the excited state of the MoS2–Por hybrid. This research demonstrated that the MoS2–Por nanohybrid could be a candidate for optical limiting in the infrared region towards femtosecond pulses. Similarly, WS2 covalently functionalized with 1,2-dithiolane-based zinc porphyrin ZnP (with chromophores primarily attached to the basal plane of TMD sheets through axial organometallic bonds between zinc and sulfur, where the linker only restricts surface mobility) exhibited a new emission band around 885 nm with a 3.5 ns lifetime when excited at 77.5 K. This emission band was attributed to charge-separated states (an interlayer exciton) between the porphyrin and WS2. In addition to the improved photoresponse of the hybrid due to the generation of long-lifetime electron–hole separation, the stability of ZnP–WS2 was also remarkably improved compared to WS2 alone.181
In 2022, few-layer TiS2 was covalently functionalized with tetraphenylporphyrin TPP diazonium salts (Fig. 42A) and underwent extensive morphological and spectroscopic analysis.182 Photoluminescence measurements were conducted to gain a better insight into the dynamics of photoexcited TPP-TiS2 (Fig. 42B and C). It was observed that the intensity of TPP emission bands significantly decreased upon its assembly with TiS2, suggesting that TiS2 acts as a quencher of the porphyrin's emission. Time-resolved fluorescence measurements revealed a monoexponential fluorescence decay for free TPP, characterized by a lifetime of 10.21 ns. Conversely, the decay for the TPP–TiS2 nanohybrid displayed a biexponential pattern, with a slower component at 9.18 ns and a faster one at 558 ps. The slow component was ascribed to the decay of TPP's singlet excited state not interacting with TiS2 sheets, while the faster component was linked to the quenching of the singlet excited-state of TPP by TiS2. Given the substantial energy difference between the Fermi level of TiS2 and LUMO of TPP, direct photoexcited electron transfer was deemed unlikely. Considering the strong overlap between TPP's emission spectra and TiS2's absorbance spectra, a non-radiative energy transfer process was suggested as the likely mechanism for the fluorescence quenching within the nanohybrid.
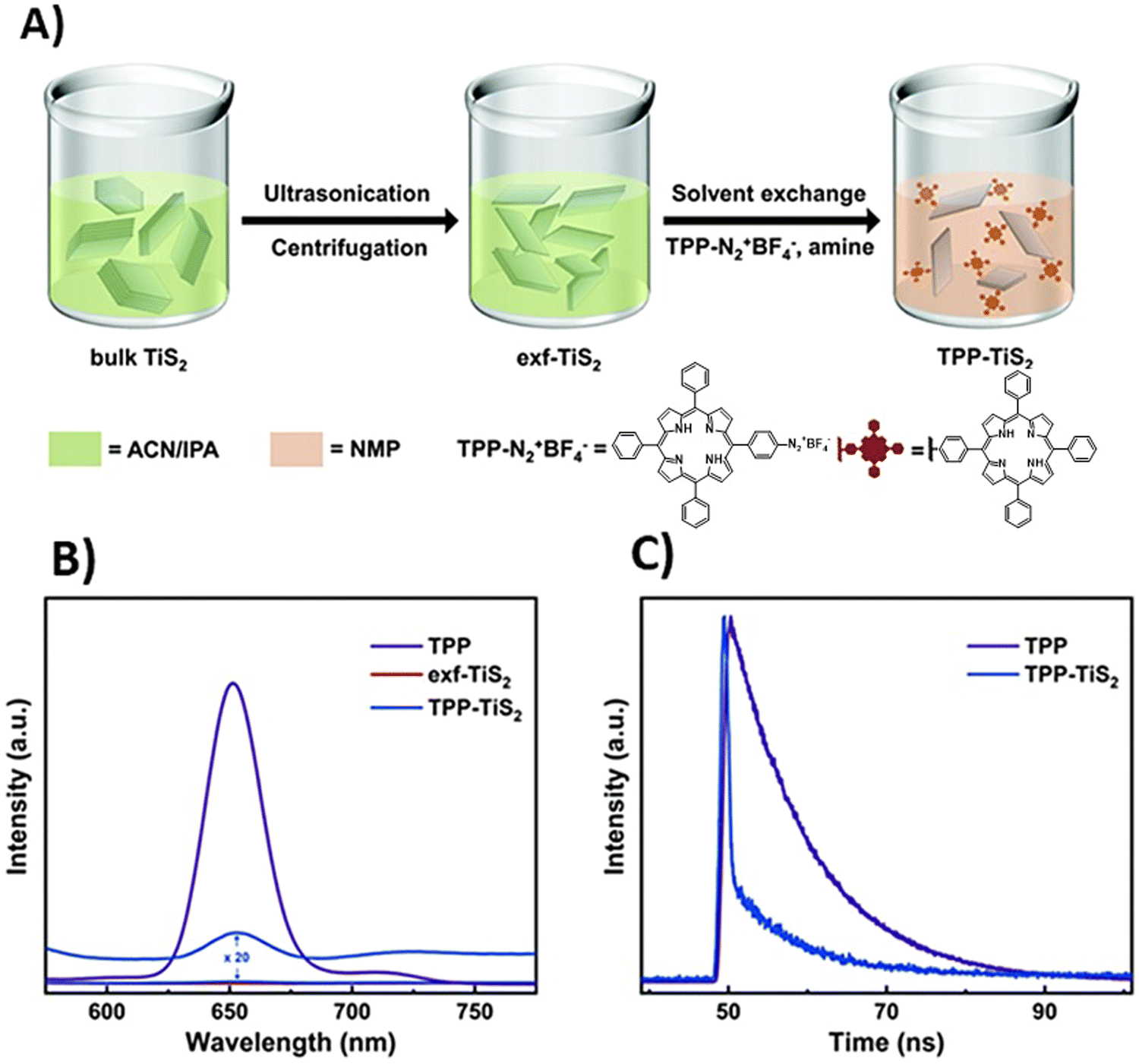 | ||
| Fig. 42 (A) Diagram illustrating the preparation process of TPP-modified TiS2 sheets from bulk TiS2, (B) photoluminescence spectra of free TPP, exfoliated (exf)-TiS2, and TPP-TiS2 hybrid, and (C) fluorescence decay profiles (excitation at 405 nm) of free TPP and TPP-TiS2. Reproduced from ref. 182. | ||
Yin et al. reported the self-assembly of cationic TMPyP with negatively charged MoS2 nanosheets in an aqueous solution by a straightforward mixing technique (Fig. 43A).55 This MoS2/TMPyP nanocomposite was evaluated for its ability to detect and remove Cd2+ ions. The introduction of MoS2 accelerated the coordination reaction between TMPyP and Cd2+ 200 times, as evidenced by absorption and emission spectroscopy (Fig. 43B–E).55
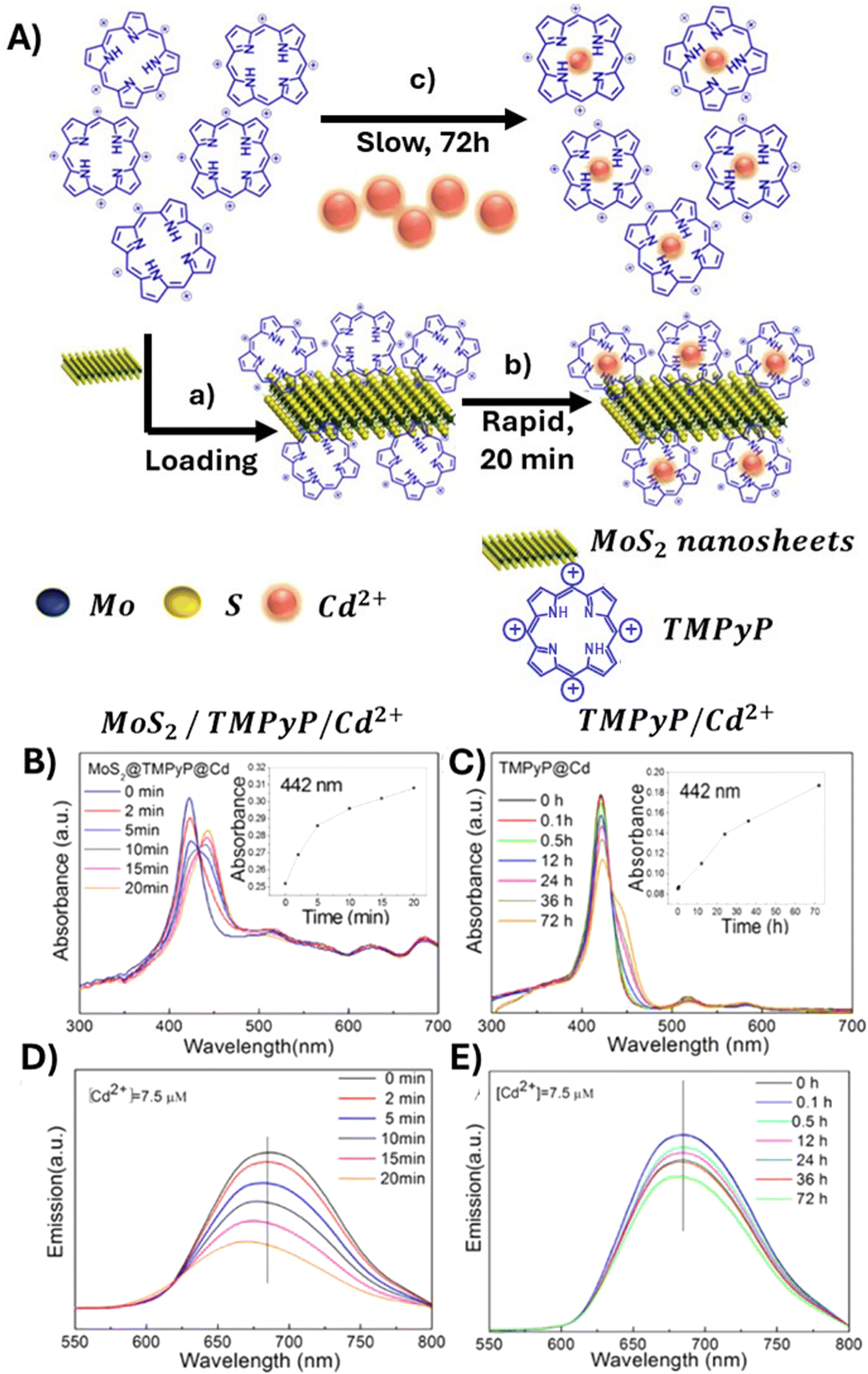 | ||
| Fig. 43 (A) Schematic showing the strategy for removing Cd2+ using TMPyP self-assembled with MoS2 nanosheets. (B) Time-dependent UV-vis absorption spectra of MoS2/TMPyP/Cd2+ and (C) TMPyP with Cd2+; insets: plots of the absorbance at 442 nm versus reaction time. (D) Time-dependent emission spectra of MoS2/TMPyP/Cd2+ and (E) TMPyP/Cd2+. Reproduced from ref. 55. Copyright 2017 American Chemical Society. | ||
Tagmatarchis group constructed novel donor–acceptor assemblies by electrostatically bonding modified MoS2 and WS2, both equipped with ammonium units to an anionic porphyrin with a carboxylate group.183 To delve into the properties of these hybrids, they utilized electronic absorption spectroscopy along with steady-state and time-resolved photoluminescence titration measurements. The UV-vis absorption spectra revealed only minor electronic interactions between the porphyrin and MoS2 or WS2 in the ground state. However, the interactions in the photoexcited state appeared much stronger, evidenced by the efficient quenching of the porphyrin's emission by MoS2 and WS2 in the nanoassemblies, as well as the reduction in porphyrin luminescence lifetimes. The authors deduced that there was an energy transduction from the photoexcited porphyrin to MoS2 or WS2.
The only known study which documents the functionalization of MXene Ti3C2Tx with porphyrins was conducted by S. Thurakkal and X. Zhang.184 This research detailed the non-covalent interaction of two different cationic porphyrins (TMPyP and TMAP) with Ti3C2Tx, focusing extensively on their structural and photophysical properties. The absorption spectra of these hybrids exhibited a pronounced red-shift in the Soret band, which was attributed to the porphyrin molecules flattening against the MXene sheets. A completely quenched fluorescence emission was an indication of efficient energy and/or electron transfer within these composites. Since any practical application of MXenes remains a challenge due to their degradation into TiO2 nanoparticles, a notable aspect of the Por/Ti3C2Tx hybrids was their improved stability against oxidation. To complete their studies, the authors conducted spectroscopic evaluation of the hybrid's response to pH changes, which is crucial for potential biological uses. Considerable release of porphyrins was noted under acidic conditions.
Similarly, there is only one known study on the covalent functionalization of two-dimensional black phosphorus nanosheets (BPNSs) with porphyrins.51 In this study, TPP was covalently bonded to BPNSs via direct P–C bonds using diazonium chemistry.51 Steady-state emission measurements indicated a decrease in the fluorescence quantum yield from 5.9% for free TPP to 3.5% for the BPNS-TPP composite. Fluorescence lifetimes analysis supported the occurrence of an electron and/or energy transfer between the photoexcited porphyrin molecules and the BPNSs. Additionally, the functionalization with TPP enhanced the ambient stability of BPNSs, suggesting that these composites might be useful for solar energy harvesting and optoelectronic devices.
Likewise, only one report deals with the modification of the hBN sheets with porphyrin molecules.52 In this study, chromophores were non-covalently deposited from a solution onto an hBN surface, resulting in two distinct phases stabilized by hydrogen bonding, one with square and the other with hexagonal symmetry, the latter being less stable. TCPP molecules adopted a stable planar configuration, evident from their fluorescence spectrum, which was strongly altered by the hBN substrate as well as the red-shift of their absorption bands when assembled with the 2D material, in analogy to the interaction of these chromophores with GBMs and TMDs. These findings imply that such structures have potential in photonic applications.
6. Summary and outlook
Herein, we provide a comprehensive overview of the literature data available to date on the variety of 2D materials functionalized with porphyrin chromophores (hybrids of porphyrins and 2DMs). Particular emphasis is put on their spectroscopic characteristics determined by the selected synthetic pathway (covalent vs. non-covalent hybrids) and the direct relation between their photophysical properties and photocatalytic performance towards hydrogen generation, CO2 reduction and pollutant degradation. The assemblies of Por and 2DMs offer not only unique features observed for their individual components, but also exhibit new interesting properties due to the synergy effect. Thorough understanding of the synthesis–structure relationships and the behavior of the discussed composites under light irradiation (photophysical characteristics) are essential for their target-specific design and fabrication. We hope that this review will inspire and evoke even more interest in the goal-oriented fabrication of Por and 2DM composites for photocatalysis, by showing their great potential within that field, referring to the significant advancements witnessed during recent years as well as still unexplored themes related to them.At the beginning, we discuss various strategies for the fabrication of those materials with the focus on the comparison of the non-covalent and covalent approaches to preparation of Por and GBM composites. Subsequently, we move on to present the spectroscopic properties of these composites. Initial focus is given to the monitoring of ground-state interactions between the hybrid components by means of steady-state absorption spectroscopy. Further, the excited-state properties of the composites are presented, as explored by time-resolved absorption and emission spectroscopies. Both types of measurements allow us to gain insights into the structure–property relationships. Since the conversion of light energy into energy-rich molecules (solar fuel) via photocatalytic processes starts with the photoinduced generation of energy-rich electrons, the PET process in the hybrids plays a key role in converting solar light into chemical energy or electricity. Therefore, the occurrence of PET in composite materials consisting of porphyrins and graphene is discussed in detail for several systems. Subsequently, an overview of the photocatalytic activity of the hybrids toward H2 generation, CO2 reduction and pollutant degradation is provided. Finally, recent advances and important new developments concerning porphyrin composites with 2D materials beyond graphene are outlined, all of which are discussed in the light of their future applications in photocatalysis.
Despite the mentioned tremendous progress made in the development and use of Por and 2DM hybrids as photocatalysts, much work remains to be done to attain a comprehensive understanding and to realize the full potential of such composites for light-driven catalytic applications. A fundamental understanding of the structure–property–performance relationship is still lacking in most of the cases, which makes it challenging to control material fabrication and its photocatalytic behavior. Main obstacles faced by the design and preparation of photocatalytically active 2DM assemblies with porphyrins are summarized in Scheme 8.
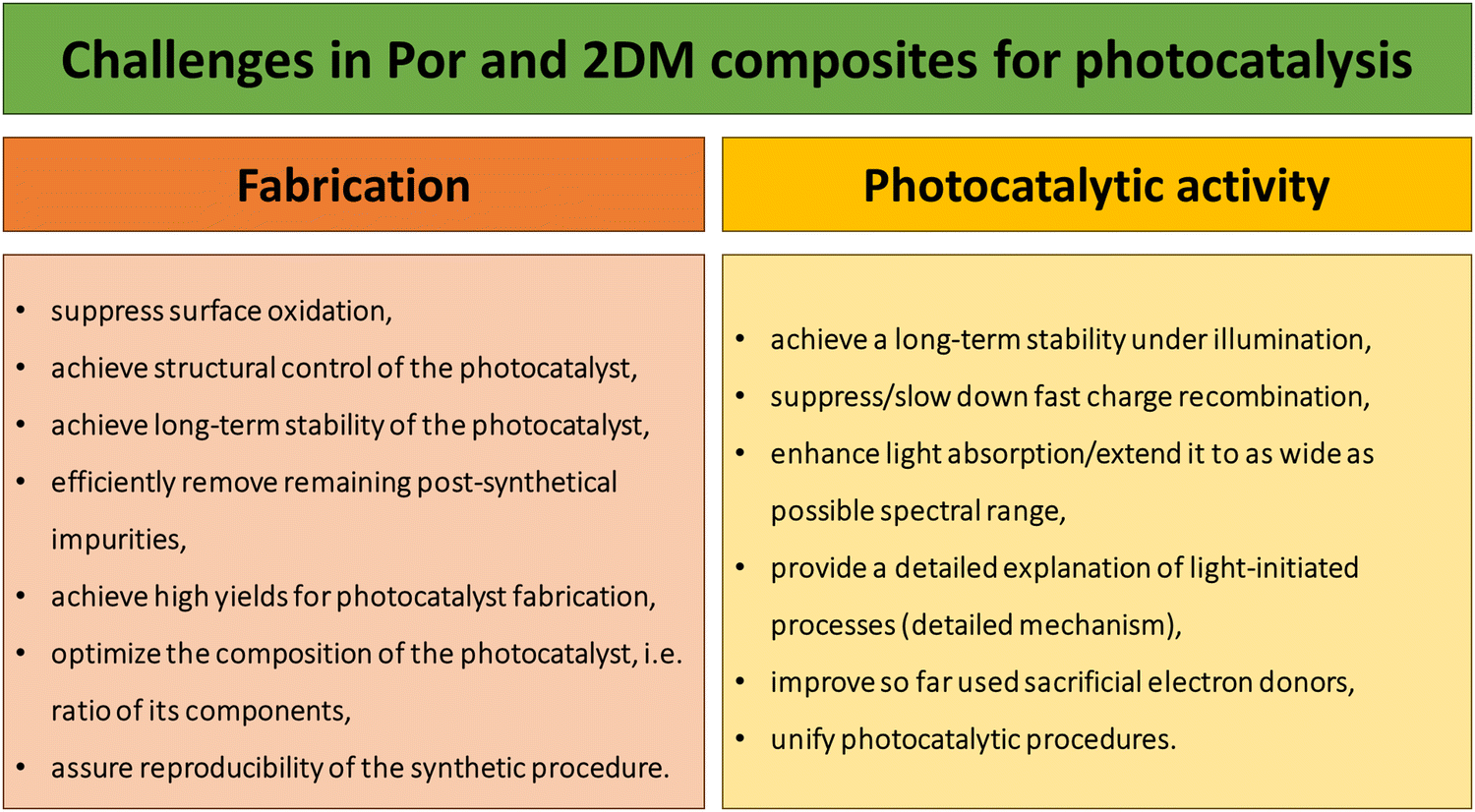 | ||
| Scheme 8 Challenges faced during the fabrication of Por and 2DM composites for photocatalysis. | ||
With respect to the fabrication process, several critical aspects still require additional attention. These include, but are not limited to, optimizing synthesis protocols to achieve high synthesis yields, controlling the structure of the materials, ensuring their long-term stability, minimizing the presence of impurities and mitigating surface oxidation. For the non-covalent functionalization, achieving an exquisite control over the process of molecular self-assembly on 2D surfaces is of paramount importance. When working with 2D materials, ensuring the reproducibility of synthesis is also a vital and non-trivial concern, since even a small modification of the reaction conditions can very strongly affect the properties of the final composite product. The structure of the 2DM (one of the reactants) is essential, since not only number and location of the functional groups are important, but also the lateral size of the 2D sheets and their orientation with respect to each other.
When considering photocatalytic performance, there are also numerous challenges that still need to be tackled. Most importantly, the efficiency of photoinduced charges’ separation should be substantially enhanced. Moreover, although the presence of porphyrin chromophores in the vicinity of 2DM sheets assures efficient light absorption over a broad (including visible) spectral range, there is a need to further increase the utilization of higher-wavelength solar energy (>500 nm). The detailed mechanism of the photocatalytic activity of Por and 2DM composites remains unclear and requires additional exploration and research. The stability and recyclability of Por- and 2DM-based photocatalysts are essential in order to meet the industrial requirements and should be optimized for practical applications in the future. Furthermore, any comparison of various photoactive materials should take into account that the parameters directly related to the photocatalytic performance, such as quantum yields, fuel generation or pollutant degradation efficiency and reaction rates, are significantly influenced by factors such as photon flux of the incoming radiation, its wavelength, and degree of scattering (as compared to its absorption by the photocatalyst). Surprisingly, this critical light-related information as well as concentration of the photocatalyst itself are often omitted when reporting the photocatalytic behavior of Por and 2DM assemblies. This omission undoubtedly hinders the possibility to reproduce the reported experiments by other researchers and, in turn, slows down the progress in the area of Por- and 2DM-based photocatalysts.
As already shortly mentioned in the Introduction, the importance of Por and 2DM composites is certainly not limited to the field of photocatalysis. They are also very attractive for a wide range of biomedical applications, such as cancer therapies (photodynamic and photothermal therapies (PDT and PTT), respectively) and early cancer diagnostics30,31 as well as energy harvesting concepts (e.g. dye-sensitized solar cells),185 photodetectors31 or sensors.186–188 More detailed information regarding the use of porphyrin-functionalized graphene for these purposes is outside the scope of our review and can be found in the above-cited literature. Nonetheless, we would like to point out that the photophysical properties of the hybrids and their direct relation to the hybrids’ structure and/or morphology remain a central point of interest, which enables further progress also for these application fields.
To date, the potential of Por and 2DM composites for optospintronics is completely unexplored. This class of hybrid materials might turn out to be a very attractive alternative to the molecular systems studied so far. Spintronics deals with generation, manipulation and detection of spin-polarized electrons and pure spin-currents, i.e. it uses the fundamental property of electrons, their spins, for information processing.189 Furthermore, chiral organic molecules are capable of spin filtering, i.e. favoring either spin up or spin down electrons during the electron transfer/transport across the molecular frame, depending among others on the handedness of the chiral molecule – the effect known as chiral-induced spin selectivity (CISS).190,191 The spin filtering efficiency of chiral molecular layers, i.e. generation of spin current due to the CISS effect, reaches values as high as 80% and more.192 This feature renders chiral molecules/molecular layers an attractive alternative to the so far fully inorganic components of spintronic devices. Nonetheless, surprisingly little has been done so far to combine this potential with the advantages offered by light-induced processes – photoinduced CISS effect193 – in case of which light could be used as an external stimulus affecting/controlling the magnetic properties of matter (optospintronics). In this respect, Por and 2DM hybrids seem to be perfect candidates since: (i) the presence of porphyrin macrocycle assures tunable and efficient light absorption, enabling PET; (ii) the vicinity of graphene sheets facilitates charge separation/promotes electron transfer; and (iii) chirality can be introduced in numerous ways, e.g. via chiral substituents in Por, formation of chiral porphyrin assemblies,194 and use of chiral graphene.195,196 Recent studies of S. Pramanik et al. carried out for RGO flakes functionalized with chiral dipeptide molecules confirm that the CISS effect is active also in such hybrid systems.197 Due to the plethora of structural variations as well as the tunability of spectral and electronic properties, these composites are extremely promising elements of future CISS-based optospintronic devices.
As already emphasized on numerous occasions, in order to further expand the spectral response of Por and 2DM hybrids and simultaneously improve the separation efficiency of photogenerated charge carriers as well as the recovery and stability of the hybrids themselves, it is imperative to develop a comprehensive understanding of their structure–property relationship. This review aims to facilitate such an approach and promote further studies of Por and 2DM composites for harnessing solar energy into fuels, with 2D materials being undoubtedly an ideal platform to construct high-performance photocatalytic systems and porphyrins perfectly suited to additionally boost the performance of 2DM by efficient absorption of solar radiation in a wide spectral range, promotion of PET and charge separation. A variety of different combinations of 2D materials and porphyrins, both tunable with respect to their individual properties and unique features arising from the synergy effect accompanying the assembly of 2DM and Por, still await exploration and may pave the way toward novel composites with superior photophysical and photochemical properties in long term.
List of abbreviations
| 2DMs | 2D materials |
| AGGS | Aggregates |
| AQE | Apparent quantum yield |
| BET | Brunauer–Emmett–Teller isotherm |
| CCG | Chemically converted graphene |
| CR | Charge recombination |
| CT | Charge transfer |
| CTAB | Cetyltrimethylammonium bromide |
| CV | Crystal violet |
| ET | Electron transfer |
| EDX | Energy-dispersive X-ray |
| GC | Graphene carboxylate |
| GF | Graphene foam |
| GBM | Graphene-based materials |
| GNPs | Graphene nanoplates |
| GO | Graphene oxide |
| G | Graphene |
| GSs | Graphene sheets |
| hBN | Hexagonal boron nitride |
| MB | Methylene blue |
| MO | Methyl orange |
| MXene | Transition metal carbides or nitrides |
| N-G | Nitrogen-doped graphene |
| NG | Nanographene |
| NP | Nanoparticle |
| NRs | Nanorods |
| NS | Nanosheet |
| PDT | Photodynamic therapy |
| PET | Photoinduced electron transfer |
| Por | Porphyrin |
| Por-GBM | Covalent assemblies |
| Por/GBM | Non-covalent assemblies |
| RGO | Reduced graphene oxide |
| RhB | Rhodamine B |
| SAS | Surfactant-assisted self-assembly |
| SEM | Scanning electron microscopy |
| TA | Transient absorption |
| TAS | Ttransient absorption spectra |
| TEM | Transmission electron microscopy |
| TMDs | Transition metal dichalcogenides |
| XPS | X-ray photoelectron spectroscopy |
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This research was financially supported by the National Science Centre (project no. 2020/39/I/ST5/00597) and the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft, project no. 464974971).References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- W. Kong, H. Kum, S.-H. Bae, J. Shim, H. Kim, L. Kong, Y. Meng, K. Wang, C. Kim and J. Kim, Nat. Nanotechnol., 2019, 14, 927 CrossRef CAS PubMed.
- K. A. Madurani, S. Suprapto, N. I. Machrita, S. L. Bahar, W. Illiya and F. Kurniawan, ECS J. Solid State Sci. Technol., 2020, 9, 093013 CrossRef CAS.
- Q. Abbas, P. A. Shinde, M. A. Abdelkareem, A. H. Alami, M. Mirzaeian, A. Yadav and A. G. Olabi, Materials, 2022, 15, 7804 CrossRef CAS PubMed.
- S. Das, P. Sudhagar, Y. S. Kang and W. Choi, J. Mater. Res., 2014, 29, 299 CrossRef CAS.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 RSC.
- X. Li, J. Yu, S. Wageh, A. A. Al-Ghamdi and J. Xie, Small, 2016, 12, 6640 CrossRef CAS PubMed.
- R. Wang, X.-G. Ren, Z. Yan, L.-J. Jiang, W. E. I. Sha and G.-C. Shan, Front. Phys., 2018, 14, 13603 CrossRef.
- X. Yu, H. Cheng, M. Zhang, Y. Zhao, L. Qu and G. Shi, Nat. Rev. Mater., 2017, 2, 17046 CrossRef CAS.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1 CrossRef CAS.
- P. Wick, A. E. Louw-Gaume, M. Kucki, H. F. Krug, K. Kostarelos, B. Fadeel, K. A. Dawson, A. Salvati, E. Vázquez, L. Ballerini, M. Tretiach, F. Benfenati, E. Flahaut, L. Gauthier, M. Prato and A. Bianco, Angew. Chem., Int. Ed., 2014, 53, 7714 CrossRef CAS PubMed.
- Y.-C. Chiou, H.-Y. Chou and M.-Y. Shen, Mater. Des., 2019, 178, 107869 CrossRef.
- S. N. Tripathi, G. S. S. Rao, A. B. Mathur and R. Jasra, RSC Adv., 2017, 7, 23615 RSC.
- M. H. M. Facure, R. Schneider, L. A. Mercante and D. S. Correa, Environ. Sci.: Nano, 2020, 7, 3710 RSC.
- A. Adetayo and D. Runsewe, Open J. Compos. Mater., 2019, 9, 207 CAS.
- T. M. Paronyan, A. K. Thapa, A. Sherehiy, J. B. Jasinski and J. S. D. Jangam, Sci. Rep., 2017, 7, 39944 CrossRef CAS PubMed.
- V. Shanmugam, R. A. Mensah, K. Babu, S. Gawusu, A. Chanda, Y. Tu, R. E. Neisiany, M. Försth, G. Sas and O. Das, Part. Part. Syst. Charact., 2022, 39, 2200031 CrossRef.
- S. Das, D. Pandey, J. Thomas and T. Roy, Adv. Mater., 2019, 31, 1802722 CrossRef PubMed.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449 CrossRef CAS PubMed.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406 RSC.
- Z.-K. Shen, Y.-J. Yuan, L. Pei, Z.-T. Yu and Z. Zou, Chem. Eng. J., 2020, 386, 123997 CrossRef CAS.
- G.-Q. Zhao, J. Hu, X. Long, J. Zou, J.-G. Yu and F.-P. Jiao, Small, 2021, 17, 2102155 CrossRef CAS PubMed.
- Y. Gogotsi and Q. Huang, ACS Nano, 2021, 15, 5775 CrossRef CAS PubMed.
- U. U. Rahman, M. Humayun, U. Ghani, M. Usman, H. Ullah, A. Khan, N. M. El-Metwaly and A. Khan, Molecules, 2022, 27, 4909 CrossRef CAS PubMed.
- J. Y. Kim, J.-W. Lee, H. S. Jung, H. Shin and N.-G. Park, Chem. Rev., 2020, 120, 7867 CrossRef CAS PubMed.
- T. L. Leung, I. Ahmad, A. A. Syed, A. M. C. Ng, J. Popović and A. B. Djurišić, Commun. Mater., 2022, 3, 63 CrossRef.
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600 RSC.
- L. Jia, J. Wu, Y. Zhang, Y. Qu, B. Jia, Z. Chen and D. J. Moss, Small Methods, 2022, 6, 2101435 CrossRef PubMed.
- A. R. Brill, E. Koren and G. de Ruiter, J. Mater. Chem. C, 2021, 9, 11569 RSC.
- A. R. Monteiro, M. G. P. M. S. Neves and T. Trindade, ChemPlusChem, 2020, 85, 1857 CrossRef CAS PubMed.
- C. Anichini and P. Samorì, Small, 2021, 17, 2100514 CrossRef CAS PubMed.
- M. V. Martínez-Díaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090 RSC.
- J. Jiang and D. K. P. Ng, Acc. Chem. Res., 2009, 42, 79 CrossRef CAS PubMed.
- A. Cidlina, V. Novakova, M. Miletin and P. Zimcik, Dalton Trans., 2015, 44, 6961 RSC.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163 CrossRef CAS.
- F. Odobel, E. Blart, M. Lagrée, M. Villieras, H. Boujtita, N. El Murr, S. Caramori and C. Alberto Bignozzi, J. Mater. Chem., 2003, 13, 502 RSC.
- Y. Rio, M. Salomé Rodríguez-Morgade and T. Torres, Org. Biomol. Chem., 2008, 6, 1877 RSC.
- J. M. Park, K.-I. Hong, H. Lee and W.-D. Jang, Acc. Chem. Res., 2021, 54, 2249 CrossRef CAS PubMed.
- R. Paolesse, S. Nardis, D. Monti, M. Stefanelli and C. Di Natale, Chem. Rev., 2017, 117, 2517 CrossRef CAS PubMed.
- D. A. Gkika, K. Ladomenou, M. Bououdina, A. C. Mitropoulos and G. Z. Kyzas, Sci. Total Environ., 2024, 908, 168293 CrossRef CAS PubMed.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330 CrossRef CAS PubMed.
- L. B. Josefsen and R. W. Boyle, Theranostics, 2012, 2, 916 CrossRef CAS PubMed.
- L. M. Moreira, F. Vieira dos Santos, J. P. Lyon, M. Maftoum-Costa, C. Pacheco-Soares and N. Soares da Silva, Aust. J. Chem., 2008, 61, 741 CrossRef CAS.
- J. C. Barona-Castaño, C. C. Carmona-Vargas, T. J. Brocksom and K. T. de Oliveira, Molecules, 2016, 21, 310 CrossRef PubMed.
- F. Habib and M. Murugesu, Chem. Soc. Rev., 2013, 42, 3278 RSC.
- J. S. Lindsey and D. F. Bocian, Acc. Chem. Res., 2011, 44, 638 CrossRef CAS PubMed.
- M. O. Senge, N. N. Sergeeva and K. J. Hale, Chem. Soc. Rev., 2021, 50, 4730 RSC.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105 CrossRef PubMed.
- E. Goiri, P. Borghetti, A. El-Sayed, J. E. Ortega and D. G. de Oteyza, Adv. Mater., 2016, 28, 1340 CrossRef CAS PubMed.
- S. Thurakkal and X. Zhang, Mater. Chem. Front., 2021, 5, 2824 RSC.
- V. V. Korolkov, S. A. Svatek, A. Summerfield, J. Kerfoot, L. Yang, T. Taniguchi, K. Watanabe, N. R. Champness, N. A. Besley and P. H. Beton, ACS Nano, 2015, 9, 10347 CrossRef CAS PubMed.
- D. Molina, J. Follana-Berná and Á. Sastre-Santos, J. Mater. Chem. C, 2023, 11, 7885 RSC.
- H. Zhang, J. Choi, A. Ramani, D. Voiry, S. N. Natoli, M. Chhowalla, D. R. McMillin and J. H. Choi, ChemPhysChem, 2016, 17, 2854 CrossRef CAS PubMed.
- W. Yin, X. Dong, J. Yu, J. Pan, Z. Yao, Z. Gu and Y. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 21362 CrossRef CAS PubMed.
- R. Canton-Vitoria, T. Scharl, A. Stergiou, A. Cadranel, R. Arenal, D. M. Guldi and N. Tagmatarchis, Angew. Chem., Int. Ed., 2020, 59, 3976 CrossRef CAS PubMed.
- A. Stergiou, G. Pagona and N. Tagmatarchis, Beilstein J. Nanotechnol., 2014, 5, 1580 CrossRef PubMed.
- Y. Xu, Z. Liu, X. Zhang, Y. Wang, J. Tian, Y. Huang, Y. Ma, X. Zhang and Y. Chen, Adv. Mater., 2009, 21, 1275 CrossRef CAS.
- N. Karousis, A. S. D. Sandanayaka, T. Hasobe, S. P. Economopoulos, E. Sarantopoulou and N. Tagmatarchis, J. Mater. Chem., 2011, 21, 109 RSC.
- Z.-B. Liu, Y.-F. Xu, X.-Y. Zhang, X.-L. Zhang, Y.-S. Chen and J.-G. Tian, J. Phys. Chem. B, 2009, 113, 9681 CrossRef CAS PubMed.
- S. Kumar, R. K. Yadav, K. Ram, A. Aguiar, J. Koh and A. J. F. N. Sobral, J. CO2 Util., 2018, 27, 107 CrossRef CAS.
- A. Ahmed, G. Devi, A. Kapahi, S. Kundan, S. Katoch and G. D. Bajju, J. Mater. Sci.: Mater. Electron., 2019, 30, 19738 CrossRef CAS.
- S. Rayati, S. Rezaie and F. Nejabat, C. R. Chim, 2018, 21, 696 CrossRef CAS.
- R. Zare-Dorabei, R. Rahimi, A. Koohi and S. Zargari, RSC Adv., 2015, 5, 93310 RSC.
- M. M. Mirzaeian, L. Eskandari and A. M. Rashidi, J. Nanostruct. Chem., 2019, 9, 19 CrossRef CAS.
- A. Wang, W. Yu, Z. Huang, F. Zhou, J. Song, Y. Song, L. Long, M. P. Cifuentes, M. G. Humphrey, L. Zhang, J. Shao and C. Zhang, Sci. Rep., 2016, 6, 23325 CrossRef CAS PubMed.
- X. Zhang, L. Hou, F. Richard and P. Samorì, Chem. – Eur. J., 2018, 24, 18518 CrossRef CAS PubMed.
- T. Umeyama, J. Mihara, N. Tezuka, Y. Matano, K. Stranius, V. Chukharev, N. V. Tkachenko, H. Lemmetyinen, K. Noda, K. Matsushige, T. Shishido, Z. Liu, K. Hirose-Takai, K. Suenaga and H. Imahori, Chem. – Eur. J., 2012, 18, 4250 CrossRef CAS PubMed.
- X. Zhang, L. Hou, A. Cnossen, A. C. Coleman, O. Ivashenko, P. Rudolf, B. J. van Wees, W. R. Browne and B. L. Feringa, Chem. – Eur. J., 2011, 17, 8957 CrossRef CAS PubMed.
- H.-X. Wang, K.-G. Zhou, Y.-L. Xie, J. Zeng, N.-N. Chai, J. Li and H.-L. Zhang, Chem. Commun., 2011, 47, 5747 RSC.
- D. Dasler, R. A. Schäfer, M. B. Minameyer, J. F. Hitzenberger, F. Hauke, T. Drewello and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 11760 CrossRef CAS PubMed.
- E. Gacka, A. Wojcik, M. Mazurkiewicz-Pawlicka, A. Malolepszy, L. Stobiński, A. Kubas, G. L. Hug, B. Marciniak and A. Lewandowska-Andralojc, J. Phys. Chem. C, 2019, 123, 3368 CrossRef CAS.
- E. Gacka, G. Burdzinski, B. Marciniak, A. Kubas and A. Lewandowska-Andralojc, Phys. Chem. Chem. Phys., 2020, 22, 13456 RSC.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732 CrossRef CAS PubMed.
- L. Fu, Y. Fang, R. Yang, Z. Guan, Z. Wei, N. Shan, F. Liu, Y. Zhao, M. G. Humphrey and C. Zhang, New J. Chem., 2022, 46, 10433 RSC.
- N. Karousis, S. P. Economopoulos, E. Sarantopoulou and N. Tagmatarchis, Carbon, 2010, 48, 854 CrossRef CAS.
- Y. Chen, Z.-H. Huang, M. Yue and F. Kang, Nanoscale, 2014, 6, 978 RSC.
- M. Ussia, E. Bruno, E. Spina, D. Vitalini, G. Pellegrino, F. Ruffino, V. Privitera and S. C. Carroccio, Sci. Rep., 2018, 8, 5001 CrossRef PubMed.
- M. Ussia, M. Urso, M. Miritello, E. Bruno, G. Curcuruto, D. Vitalini, G. G. Condorelli, M. Cantarella, V. Privitera and S. C. Carroccio, RSC Adv., 2019, 9, 30182 RSC.
- Y. Xu, L. Zhao, H. Bai, W. Hong, C. Li and G. Shi, J. Am. Chem. Soc., 2009, 131, 13490 CrossRef CAS PubMed.
- D. Larowska, A. Wojcik, M. Mazurkiewicz-Pawlicka, A. Malolepszy, L. Stobiński, B. Marciniak and A. Lewandowska-Andralojc, ChemPhysChem, 2019, 20, 1054 CrossRef CAS PubMed.
- K. Lewandowska, N. Rosiak, A. Bogucki, J. Cielecka-Piontek, M. Mizera, W. Bednarski, M. Suchecki and K. Szaciłowski, Molecules, 2019, 24, 688 CrossRef PubMed.
- A. Wojcik and P. V. Kamat, ACS Nano, 2010, 4, 6697 CrossRef CAS PubMed.
- D. Larowska, J. M. O’Brien, M. O. Senge, G. Burdzinski, B. Marciniak and A. Lewandowska-Andralojc, J. Phys. Chem. C, 2020, 124, 15769 CrossRef CAS PubMed.
- A. Siklitskaya, E. Gacka, D. Larowska, M. Mazurkiewicz-Pawlicka, A. Malolepszy, L. Stobiński, B. Marciniak, A. Lewandowska-Andrałojć and A. Kubas, Sci. Rep., 2021, 11, 7977 CrossRef CAS PubMed.
- L. Zheng, D. Ye, L. Xiong, J. Xu, K. Tao, Z. Zou, D. Huang, X. Kang, S. Yang and J. Xia, Anal. Chim. Acta, 2013, 768, 69 CrossRef CAS PubMed.
- F. J. Vergeldt, R. B. M. Koehorst, A. van Hoek and T. J. Schaafsma, J. Phys. Chem., 1995, 99, 4397 CrossRef CAS.
- M. Aydin and D. L. Akins, in Applications of Molecular Spectroscopy to Current Research in the Chemical and Biological Sciences, ed. M. T. Stauffer, InTech, Rijeka, 2016, p. 10 DOI:10.5772/64583.
- M. Aydin, Molecules, 2014, 19, 20988 CrossRef PubMed.
- S. M. Aly, M. R. Parida, E. Alarousu and O. F. Mohammed, Chem. Commun., 2014, 50, 10452 RSC.
- Z. D. Liu, H. X. Zhao and C. Z. Huang, PLoS ONE 7, 2012, 7, 50367 CrossRef PubMed.
- O. Bajjou, A. Bakour, M. Khenfouch, M. Baitoul, B. M. Mothudi, M. Maaza and E. Faulques, J. Mater. Sci.: Mater. Electron., 2018, 29, 8594 CrossRef CAS.
- S. M. Aly, M. R. Parida, E. Alarousu and O. F. Mohammed, Chem. Commun., 2014, 50, 10452 RSC.
- J. Shu, Z. Qiu, Q. Wei, J. Zhuang and D. Tang, Sci. Rep., 2015, 5, 15113 CrossRef CAS PubMed.
- R. Ge, X. Wang, C. Zhang, S.-Z. Kang, L. Qin, G. Li and X. Li, Colloids Surf., A, 2015, 483, 45 CrossRef CAS.
- E. Gacka, G. Burdzinski, B. Marciniak, A. Kubas and A. Lewandowska-Andralojc, Phys. Chem. Chem. Phys., 2020, 22, 13456 RSC.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732 CrossRef CAS PubMed.
- S. Cao and J. Yu, J. Photochem. Photobiol., C, 2016, 27, 72 CrossRef CAS.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242 CrossRef CAS.
- I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2013, 46, 2235 CrossRef CAS PubMed.
- Y. Sun, Y. Sun, X. Meng, Y. Gao, Y. Dall'Agnese, G. Chen, C. Dall'Agnese and X.-F. Wang, Catal. Sci. Technol., 2019, 9, 310 RSC.
- X. Zhang, T. Peng and S. Song, J. Mater. Chem. A, 2016, 4, 2365 RSC.
- Y. Zhang, F. Mao, H. Yan, K. Liu, H. Cao, J. Wu and D. Xiao, J. Mater. Chem. A, 2015, 3, 109 RSC.
- A. Lewandowska-Andralojc and B. Marciniak, ACS Energy Lett., 2019, 4, 1898 CrossRef CAS.
- B. Marciniak, J. Chem. Educ., 1986, 63, 998 CrossRef CAS.
- D. Masih, S. M. Aly, A. Usman, E. Alarousu and O. F. Mohammed, Phys. Chem. Chem. Phys., 2015, 17, 9015 RSC.
- R. Ge, X. Wang, C. Zhang, S.-Z. Kang, L. Qin, G. Li and X. Li, Colloids Surf., 2015, 483, 45 CrossRef CAS.
- M. Zhu, Z. Li, B. Xiao, Y. Lu, Y. Du, P. Yang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 1732 CrossRef CAS PubMed.
- Z. Mou, Y. Dong, S. Li, Y. Du, X. Wang, P. Yang and S. Wang, Int. J. Hydrogen Energy, 2011, 36, 8885 CrossRef CAS.
- W. Zhang, Y. Li and S. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 15187 CrossRef CAS PubMed.
- S. Min and G. Lu, Int. J. Hydrogen Energy, 2012, 37, 10564 CrossRef CAS.
- J. Shu, Z. Qiu, Q. Wei, J. Zhuang and D. Tang, Sci. Rep., 2015, 5, 15113 CrossRef CAS PubMed.
- L. M. Arellano, H. B. Gobeze, Y. Jang, M. Barrejón, C. Parejo, J. C. Álvarez, M. J. Gómez-Escalonilla, Á. Sastre-Santos, F. D'Souza and F. Langa, Chem. – Eur. J., 2022, 28, e202200254 CrossRef CAS PubMed.
- D. Li, X. He, X. Zhang, J. Chen, Q. Jin and J. Xu, ChemPhotoChem, 2024, e202300271 CrossRef.
- M. Garrido, M. K. Volland, P. W. Münich, L. Rodríguez-Pérez, J. Calbo, E. Ortí, M. Á. Herranz, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2020, 142, 1895 CrossRef CAS PubMed.
- B. Mondal, R. Bera, S. K. Nayak and A. Patra, J. Mater. Chem. C, 2016, 4, 6027 RSC.
- L. Fu, M. G. Humphrey and C. Zhang, Nano Res., 2023, 16, 25 CrossRef CAS.
- R. Bera, S. Mandal, B. Mondal, B. Jana, S. K. Nayak and A. Patra, ACS Sustainable Chem. Eng., 2016, 4, 1562 CrossRef CAS.
- M. Yao, Y. Meng, X. Mao, X. Ning, Z. Zhang, D. Shan, J. Chen and X. Lu, Electrochim. Acta, 2018, 282, 575 CrossRef CAS.
- H. S. Park, K. C. Leonard and A. J. Bard, J. Phys. Chem. C, 2013, 117, 12093 CrossRef CAS.
- M. de Miguel, M. Álvaro and H. García, Langmuir, 2012, 28, 2849 CrossRef CAS PubMed.
- A. Lukaszewicz, J. Karolczak, D. Kowalska, A. Maciejewski, M. Ziolek and R. P. Steer, Chem. Phys., 2007, 331, 359 CrossRef CAS.
- U. Tripathy, D. Kowalska, X. Liu, S. Velate and R. P. Steer, J. Phys. Chem. A, 2008, 112, 5824 CrossRef CAS PubMed.
- L. Wang, H. Fan and F. Bai, MRS Bull., 2020, 45, 49 CrossRef.
- N. Zhang, L. Wang, H. Wang, R. Cao, J. Wang, F. Bai and H. Fan, Nano Lett., 2018, 18, 560 CrossRef CAS PubMed.
- Y. Chen, A. Li, Z.-H. Huang, L.-N. Wang and F. Kang, Nanomaterials, 2016, 6, 51 CrossRef PubMed.
- M. Ussia, F. Ruffino, E. Bruno, E. Spina, I. Conticello, V. Privitera and S. C. Carroccio, Polym. Bull., 2020, 77, 2073 CrossRef CAS.
- R. Ge, X. Li, S.-Z. Kang, L. Qin and G. Li, Appl. Catal., B, 2016, 187, 67 CrossRef CAS.
- K. Zhu, Q. Luo, S.-Z. Kang, L. Qin, G. Li and X. Li, Nanoscale, 2018, 10, 18635 RSC.
- L. Zhang, L. Qin, S.-Z. Kang, G.-D. Li and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 8358 CrossRef CAS.
- Q. Luo, R. Ge, S.-Z. Kang, L. Qin, G. Li and X. Li, Appl. Surf. Sci., 2018, 427, 15 CrossRef CAS.
- Y.-J. Yuan, D. Chen, J. Zhong, L.-X. Yang, J.-J. Wang, Z.-T. Yu and Z.-G. Zou, J. Phys. Chem. C, 2017, 121, 24452 CrossRef CAS.
- X.-F. Liu, R.-X. Li, X.-T. Ren, Y.-B. Yin, S.-K. Mei, T. Liu and J. Yan, J. Catal., 2017, 348, 314 CrossRef CAS.
- A. Wang, X. Shen, Q. Wang, L. Cheng, W. Zhu, D. Shang and Y. Song, Dyes Pigm., 2021, 187, 109142 CrossRef CAS.
- D. D. La, H. H. Ngo, D. D. Nguyen, N. T. Tran, H. T. Vo, X. H. Nguyen, S. W. Chang, W. J. Chung and M. D.-B. Nguyen, Coord. Chem. Rev., 2022, 463, 214543 CrossRef CAS.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954 CrossRef CAS PubMed.
- P. Guo, P. Chen, W. Ma and M. Liu, J. Mater. Chem., 2012, 22, 20243 RSC.
- S. J. Lee, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 9632 CrossRef CAS PubMed.
- Y. Zhong, Z. Wang, R. Zhang, F. Bai, H. Wu, R. Haddad and H. Fan, ACS Nano, 2014, 8, 827 CrossRef CAS PubMed.
- D. D. La, S. V. Bhosale, L. A. Jones and S. V. Bhosale, Photochem. Photobiol. Sci., 2017, 16, 151 CrossRef CAS PubMed.
- S. Mandal, S. K. Nayak, S. Mallampalli and A. Patra, ACS Appl. Mater. Interfaces, 2014, 6, 130 CrossRef CAS PubMed.
- P. Guo, P. Chen and M. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 5336 CrossRef CAS PubMed.
- D. D. La, A. Rananaware, M. Salimimarand and S. V. Bhosale, ChemistrySelect, 2016, 1, 4430 CrossRef CAS.
- D. D. La, R. V. Hangarge, S. V. Bhosale, H. D. Ninh, L. A. Jones and S. V. Bhosale, Appl. Sci., 2017, 7, 643 CrossRef.
- A. M. El-Khawaga, H. Tantawy, M. A. Elsayed and A. I. A. Abd El-Mageed, Sci. Rep., 2022, 12, 17075 CrossRef CAS PubMed.
- N. El-Shafai, M. E. El-Khouly, M. El-Kemary, M. S. Ramadan and M. S. Masoud, Photochem. Photobiol. Sci., 2019, 18, 2071 CrossRef CAS PubMed.
- R. Yasmeen, G. D. Bajju and H. N. Sheikh, J. Porphyrins Phthalocyanines, 2024, 28, 121 CrossRef CAS.
- Raveena, M. P. Singh, M. Sengar and P. Kumari, ChemistrySelect, 2023, 8, e202203272 CrossRef CAS.
- C. Ruan, L. Zhang, Y. Qin, C. Xu, X. Zhang, J. Wan, Z. Peng, J. Shi, X. Li and L. Wang, Mater. Lett., 2015, 141, 362 CrossRef CAS.
- J. Wan, M. Wei, Z. Hu, Z. Peng, B. Wang, D. Feng and Y. Shen, Int. J. Hydrogen Energy, 2016, 41, 14692 CrossRef CAS.
- M. Wei, J. Wan, Z. Hu, Z. Peng and B. Wang, Appl. Surf. Sci., 2016, 377, 149 CrossRef CAS.
- S. Zargari, R. Rahimi, A. Ghaffarinejad and A. Morsali, J. Colloid Interface Sci., 2016, 466, 310 CrossRef CAS PubMed.
- D. D. La, S. V. Bhosale, L. A. Jones, N. Revaprasadu and S. V. Bhosale, ChemistrySelect, 2017, 2, 3329 CrossRef CAS.
- S. Aghakhaninejad, S. Zargari and R. Rahimi, Appl. Sci., 2020, 2, 625 CAS.
- D. D. La, T. A. Nguyen, X. S. Nguyen, T. N. Truong, H. P. Nguyen T, H. D. Ninh, H. T. Vo, S. V. Bhosale, S. W. Chang, E. R. Rene, T. H. Nguyen, S. M. Lee, L. D. Tran and D. D. Nguyen, J. Environ. Chem. Eng., 2021, 9, 106034 CrossRef CAS.
- M. Moshari, M. Rabbani and R. Rahimi, Res. Chem. Intermed., 2016, 42, 5441 CrossRef CAS.
- R. K. Yadav, G. H. Oh, N.-J. Park, A. Kumar, K.-J. Kong and J.-O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728 CrossRef CAS PubMed.
- Q. Li, Q. Zeng, L. Gao, Z. Ullah, H. Li, Y. Guo, W. Li, Y. Shi, G. Tao and L. Liu, J. Mater. Chem. A, 2017, 5, 155 RSC.
- M. Piao, N. Liu, Y. Wang and C. Feng, Aust. J. Chem., 2016, 69, 27 CrossRef CAS.
- D. H. Wang, J. N. Pan, H. H. Li, J. J. Liu, Y. B. Wang, L. T. Kang and J. N. Yao, J. Mater. Chem. A, 2016, 4, 290 RSC.
- J. Wang, Y. Zheng, T. Peng, J. Zhang and R. Li, ACS Sustainable Chem. Eng., 2017, 5, 7549 CrossRef CAS.
- S. Mei, J. Gao, Y. Zhang, J. Yang, Y. Wu, X. Wang, R. Zhao, X. Zhai, C. Hao, R. Li and J. Yan, J. Colloid Interface Sci., 2017, 506, 58 CrossRef CAS PubMed.
- E. S. Da Silva, N. M. M. Moura, M. G. P. M. S. Neves, A. Coutinho, M. Prieto, C. G. Silva and J. L. Faria, Appl. Catal., B, 2018, 221, 56 CrossRef CAS.
- K. Zhu, M. Zhang, X. Feng, L. Qin, S.-Z. Kang and X. Li, Appl. Catal., B, 2020, 268, 118434 CrossRef CAS.
- M. Zhang, K. Zhu, L. Qin, S.-Z. Kang and X. Li, Catal. Sci. Technol., 2020, 10, 1640 RSC.
- J. Zhang, A. Wang, W. Zhao, C. Li, X. Chen, Y. Wang, W. Zhu and Q. Zhong, Dyes Pigm., 2018, 153, 241 CrossRef CAS.
- Z. Ma, C. Zeng, L. Hu, Q. Zhao, Q. Yang, J. Niu, B. Yao and Y. He, Appl. Surf. Sci., 2019, 484, 489 CrossRef CAS.
- J. Xu, Q. Gao, Z. Wang and Y. Zhu, Appl. Catal., B, 2021, 291, 120059 CrossRef CAS.
- H. T. Lai, G. T. Nguyen, N. T. Tran, T. T. Nguyen, C. Van Tran, D. K. Nguyen, S. W. Chang, W. J. Chung, D. D. Nguyen, H. P. N. Thi and D. D. La, Catalysts, 2022, 12, 1630 CrossRef CAS.
- T. T. Nguyen, H. T. Bui, G. T. Nguyen, T. N. Hoang, C. Van Tran, P. H. Ho, P. T. Hoai Nguyen, J. Y. Kim, S. W. Chang, W. J. Chung, D. D. Nguyen and D. D. La, Environ. Res., 2023, 231, 115984 CrossRef CAS PubMed.
- L. Lin, C. Hou, X. Zhang, Y. Wang, Y. Chen and T. He, Appl. Catal., B, 2018, 221, 312 CrossRef CAS.
- G. Zhao, H. Pang, G. Liu, P. Li, H. Liu, H. Zhang, L. Shi and J. Ye, Appl. Catal., B, 2017, 200, 141 CrossRef CAS.
- Q. Chen, Y. Zhang, E. You, Q. Jiang, X. Chen, Y. Wang, Z. Song, K. Chang, Z. Xie and Q. Kuang, Small, 2022, 18, 2204924 CrossRef CAS PubMed.
- Z. Jia, Y. Xiao, S. Guo, L. Xiong, P. Yu, T. Lu and R. Song, ACS Appl. Mater. Interfaces, 2023, 15, 47070 CrossRef CAS PubMed.
- Y. Yuan, H. Lu, Z. Ji, J. Zhong, M. Ding, D. Chen, Y. Li, W. Tu, D. Cao, Z. Yu and Z. Zou, Chem. Eng. J., 2015, 275, 8 CrossRef CAS.
- Y.-J. Yuan, J.-R. Tu, Z.-J. Ye, H.-W. Lu, Z.-G. Ji, B. Hu, Y.-H. Li, D.-P. Cao, Z.-T. Yu and Z.-G. Zou, Dyes Pigm., 2015, 123, 285 CrossRef CAS.
- G. Karantanais, M. P. Minadakis, V. Chrysostomou, H. J. Ojeda-Galvan, M. Quintana, S. Pispas and N. Tagmatarchis, Colloids Surf., A, 2024, 682, 132755 CrossRef CAS.
- D. K. Perivoliotis, C. Stangel, Y. Sato, K. Suenaga and N. Tagmatarchis, 2D Mater., 2023, 10, 014007 CrossRef CAS.
- M. P. Minadakis, R. Canton-Vitoria, C. Stangel, E. Klontzas, R. Arenal, J. Hernández-Ferrer, A. M. Benito, W. K. Maser and N. Tagmatarchis, ChemSusChem, 2023, 16, e202202322 CrossRef CAS PubMed.
- Z. Wei, Z. Guan, N. Shan, H. Li, Y. Fang, Y. Zhao, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Alloys Compd., 2023, 934, 167902 CrossRef CAS.
- R. Canton-Vitoria, T. Hotta, Y. Tanuma, I. K. Sideri, N. Tagmatarchis, C. Ewels and R. Kitaura, J. Phys. Chem. C, 2023, 127, 10699 CrossRef CAS.
- Y. Fang, H. Li, Z. Wei, Z. Guan, N. Shan, L. Fu, Z. Huang, M. G. Humphrey and C. Zhang, J. Mater. Chem. C, 2022, 10, 10876 RSC.
- R. Canton-Vitoria, C. Stangel and N. Tagmatarchis, ACS Appl. Mater. Interfaces, 2018, 10, 23476 CrossRef CAS PubMed.
- S. Thurakkal and X. Zhang, Mater. Chem. Front., 2022, 6, 561 RSC.
- B. Mao, B. Hodges, C. Franklin, D. G. Calatayud and S. I. Pascu, Front. Chem., 2021, 9, 727574 CrossRef CAS PubMed.
- G. Magna, F. Mandoj, M. Stefanelli, G. Pomarico, D. Monti, C. Di Natale, R. Paolesse and S. Nardis, Nanomaterials, 2021, 11, 997 CrossRef CAS PubMed.
- A. S. Khune, V. Padghan, R. Bongane, V. N. Narwade, B. N. Dole, N. N. Ingle, M.-L. Tsai, T. Hianik and M. D. Shirsat, J. Electron. Mater., 2023, 52, 8108 CrossRef CAS.
- A. S. Khune, V. N. Narwade, B. N. Dole, N. N. Ingle, M.-L. Tsai, T. Hianik and M. D. Shirsat, Appl. Phys. A: Mater. Sci. Process., 2024, 130, 60 CrossRef CAS.
- A. Hirohata, K. Yamada, Y. Nakatani, I.-L. Prejbeanu, B. Diény, P. Pirro and B. Hillebrands, J. Magnet. Magn. Mater., 2020, 509, 166711 CrossRef CAS.
- R. Naaman and D. H. Waldeck, J. Phys. Chem. Lett., 2012, 3, 2178 CrossRef CAS PubMed.
- B. P. Bloom, Y. Paltiel, R. Naaman and D. H. Waldeck, Chem. Rev., 2024, 124, 1950 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250 CrossRef CAS.
- J. M. Abendroth, D. M. Stemer, B. P. Bloom, P. Roy, R. Naaman, D. H. Waldeck, P. S. Weiss and P. C. Mondal, ACS Nano, 2019, 13, 4928 CrossRef CAS PubMed.
- L. Zhang, T. Wang, J. Jiang and M. Liu, Aggregate, 2023, 4, e198 CrossRef CAS.
- B. Zhao, S. Yang, J. Deng and K. Pan, Adv. Sci., 2021, 8, 2003681 CrossRef CAS PubMed.
- Q. Cheng, H. Pei, Q. Ma, R. Guo, N. Liu and Z. Mo, Chem. Eng. J., 2023, 452, 139499 CrossRef CAS.
- S. Firouzeh, S. Illescas-Lopez, M. A. Hossain, J. M. Cuerva, L. Álvarez de Cienfuegos and S. Pramanik, ACS Nano, 2023, 17, 20424 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |