
Synergistic design of g-C3N4-supported CNTs: experimental and DFT insights for enhanced electrochemical performance in flexible Li–S batteries†
Vijay K.
Tomer
 *a,
Rameshwar L.
Kumawat
b,
Otavio Augusto
Titton Dias
a,
Ritu
Malik
*a,
George C.
Schatz
b and
Mohini
Sain
a
*a,
Rameshwar L.
Kumawat
b,
Otavio Augusto
Titton Dias
a,
Ritu
Malik
*a,
George C.
Schatz
b and
Mohini
Sain
a
aDepartment of Mechanical & Industrial Engineering, University of Toronto, Toronto, Canada. E-mail: vj.kumar@utoronto.ca; ritu.kumar@utoronto.ca
bDepartment of Chemistry, Northwestern University, Illinois, USA
First published on 16th May 2024
Abstract
In addressing the shuttle effect and sluggish redox kinetics of lithium polysulfides (LiPSs) in lithium–sulfur batteries, we developed a novel heterostructure combining 1D carbon nanotubes (CNTs) grown on a 2D sheet of graphitic carbon nitride (g-C3N4) to improve conversion reaction kinetics and LiPS adsorption capacity. The high pyridine N content in g-C3N4 facilitates homogeneous Li ion deposition and enhances affinity between Li and N atoms. Extensive experimental characterization and density functional theory (DFT) calculations validated the interaction between g-C3N4-CNT/S and LiPSs. In pouch cell evaluation, the hybrid g-C3N4-CNT/S cathode, with ∼70% sulfur loading, demonstrated outstanding rate performance, delivering ∼895 mA h g−1 at 0.1C and retaining ∼500 mA h g−1 even at 1.5C under lean electrolyte conditions (E/S ∼5 μl mg−1). Long-term stability over 250 cycles, with a capacity retention of 86% and a coulombic efficiency (CE) of 90.4%, was achieved, even with an elevated sulfur loading of 6.2 mg cm−2. Post-mortem investigation using X-ray photoelectron spectroscopy (XPS) and electrochemical impedance spectroscopy (EIS) elucidated surface chemistry changes and elemental composition alterations, highlighting the formation of various reaction products during charge–discharge cycles. This study underscores the cost-effective heterostructure strategy's potential for advancing LiSBs in practical applications.
Introduction
Developing reliable energy storage and conversion technologies is essential in the face of the growing energy crisis and climate change.1,2 Meeting the increasing demand for portable electronic devices and electric vehicles necessitates advanced, affordable, lightweight, and durable rechargeable batteries.3 Lithium-ion batteries (LIBs) have long been favored because of their high power/energy density and extended service life, making them promising for various applications.4,5 However, conventional LIBs are nearing their theoretical energy density limits and cannot keep up with escalating energy demands.6 As a potential solution, lithium–sulfur batteries (LiSBs) offer significant cost reduction and a substantial boost in energy density, approximately 2600 W h kg−1, three to five times higher than that of commercial LIBs based on intercalation reactions.7,8 Sulfur, a low-cost and abundant cathode material, further enhances the appeal of LiSBs, showcasing advantages for large-scale applications compared to traditional LIB cathode materials such as LiFePO4, LiMn2O4, LiCoO2, etc.9 Despite these advantages, LiSBs face challenges such as rapid capacity loss and low coulombic efficiency (CE), hindering their commercialization.10 The poor conductivity of sulfur and its lithiation products (Li2S/Li2S2) results in sluggish reaction kinetics, leading to issues during charge–discharge cycles.11 Soluble lithium polysulfides (LiPSs) formed as reaction intermediates contribute to irreversible capacity loss, low CE, and Li anode corrosion.12 Additionally, the substantial volume expansion (∼80%) during cycling causes structural collapse and significantly reduces cathode activity.13 Addressing these challenges requires the design of suitable S-hosting materials to enhance cathode conductivity and limit polysulfide dissolution.Recently, cathode hosts with strong physical and chemical interactions toward LiPSs have improved capacity retention by suppressing the shuttle phenomenon and improving redox kinetics.13,14 A template-based method was employed to synthesize a composite host of macro/mesoporous carbon and defective TiO2 nanoparticles, showcasing a high capacity (1420 mA h g−1 at 0.2C) and cycling ability (65.6% retention at 0.2C over 60 cycles).15 Additionally, tin disulfide (SnS2) nanosheets were anchored onto nitrogen-doped hollow carbon with mesoporous shells, forming a bipolar dynamic host with a capacity of 947.4 mA h g−1 at 0.2C.16 Among the various host matrices explored in the past few years,17–19 graphitic carbon nitride (g-C3N4 or g-CN), a semiconductor material with a 2D structure akin to graphene, exhibits promising potential for improving Li–S battery performance.20 Recent studies highlight g-C3N4's catalytic activity, stability, cost-effectiveness, and environmental friendliness, making it an attractive additive for Li–S batteries. The heptazine units in g-C3N4, containing high levels of pyridinic N, enable homogenized Li ion deposition and a strong affinity between Li and N atoms.21,22 Benefiting from the inherent high charge polarity and abundant polysulfide binding site, g-C3N4 manifested remarkable electrocatalytic performance toward converting polysulfides during the charge/discharge process in LiSBs.23 Novel approaches, such as nitrogen-deficient g-C3N4 heterostructures developed through magnesiothermic denitriding technology,24 have demonstrated significant improvements. These structures achieved a discharge capacity of 650 mA h g−1 at 4C with low-capacity decay after 400 cycles. Similarly, g-C3N4/g-C3N4 heterojunctions25 adjusted the electron cloud structure, providing an initial discharge capacity of 1200 mA h g−1 and maintaining 464 mA h g−1 after 150 cycles. Other techniques, like the ethanol-assisted spray drying method, produced rGO/g-C3N4/CNT microspheres,26 offering enhanced electrode conductivity and stable cycling performance. 3D porous graphene@g-C3N4 hybrid sponges,27 as sulfur host materials, exhibited high specific capacity and excellent rate capability, thanks to their porous network structure. While these examples showcase g-C3N4's effectiveness as a sulfur host, many strategies have simply mixed carbon materials and g-C3N4 without considering their mutual impact. Consequently, there is a pressing need for a simple, efficient, and low-cost synthesis technique to produce a network structure hybrid with superior Li–S battery performance.
Leveraging a cost-effective melamine precursor and a straightforward pyrolysis process, we synthesized low-cost 2D sheet-like nitrogen-rich g-C3N4 and composited it with multi-walled 1D CNTs to form a heterostructure. This design not only accommodates sulfur volume expansion during lithiation but also acts as a physical barrier, preventing polysulfide diffusion from the cathode. The enriched N-sites in g-C3N4 serve as adhesion sites for polysulfides, establishing a ‘physical–chemical’ dual-confinement for enhanced confinement.20 The interlinked hierarchical network of 1D CNTs on 2D g-C3N4 promotes rapid electron/Li+ transport and structural integrity, ensuring swift reaction kinetics and long-term cycling stability. The g-C3N4-CNT/S composite exhibits outstanding performance, demonstrating a high specific capacity and exceptional cycling stability with a mere 0.053% capacity decay per cycle over 250 cycles and an impressive capacity retention of 90.4% at a 0.1C rate. Notably, the pouch cell has no sign of performance decay even at a folding angle of 180°, showcasing its potential for practical applications.
Experimental section
Materials
Melamine (99.99%), N-methyl-2-pyrrolidone (NMP), lithium bis(trifluoromethane) sulfonimide (LiTFSI), 1,3-dioxolane (DOL), 1,2-dimethoxyethane (DME) and lithium nitrate (LiNO3) were purchased from Sigma. Conducting carbon (CC, Super C45), polyvinylidene fluoride (PVDF) as the binder, Al foil (cathode current collector), Cu foil (anode current collector), a tri-layered (PP/PE/PP) 25 μm thick separator (Celgard 2325), and tabs (Al and Ni) were purchased from MTI (USA). Pure lithium foil (100 μm) as an anode, lithium sulfide (Li2S 99.99%), and CNTs (multi-walled) were purchased from Nanochemazone (Canada). All the materials were used as received without further treatment.Synthesis of cathode active materials
g-C3N4 powder was synthesized via thermal polymerization of melamine. In a typical process, the melamine (5 g) was heated to 600 °C at a ramp rate of 5 °C min−1 for 4 h in a covered ceramic crucible. After cooling to room temperature, the resulting light-yellow solid was ground to yield bulk g-C3N4 powder, which was further dispersed in deionized water (90 mL) and sonicated for 4 h to obtain delaminated g-C3N4 NSs. The g-C3N4 and CNTs (in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 weight ratio) were combined in a glass vial with 20 mL of deionized water and sonicated for 4 h. The resulting product was filtered and dried at 60 °C overnight, yielding g-C3N4-CNT. For sulfur-based composites, pure g-C3N4 and g-C3N4-CNT composites were individually ground with sulfur (3:7 weight ratio) for 1 h and heated at 160 °C for 15 h to produce g-C3N4/S and g-C3N4-CNT/S composites, respectively (Scheme 1).
:3 weight ratio) were combined in a glass vial with 20 mL of deionized water and sonicated for 4 h. The resulting product was filtered and dried at 60 °C overnight, yielding g-C3N4-CNT. For sulfur-based composites, pure g-C3N4 and g-C3N4-CNT composites were individually ground with sulfur (3:7 weight ratio) for 1 h and heated at 160 °C for 15 h to produce g-C3N4/S and g-C3N4-CNT/S composites, respectively (Scheme 1).
 | ||
| Scheme 1 Schematic illustration for the synthesis of the g-C3N4-CNT/S nanocomposite. | ||
Material characterization
Crystalline structures were analyzed using a Philips P.W. 1830 powder X-ray diffractometer with a CuKα-ray source (10–70° scan range). The microstructure and morphology were examined with a JEOL 2010 transmission electron microscope and QUANTA FEG 250 scanning electron microscope equipped with an energy-dispersive X-ray spectrometer (EDS). Fourier Transform Infrared (FTIR) spectroscopy was performed from 500 to 4000 cm−1 on a Tensor 27 instrument. Carbon defects were assessed using Raman spectroscopy (Bruker Senterra Infinity 1). The chemical and electronic states of the surface were investigated via X-ray photoelectron spectroscopy (XPS, PerkinElmer Phi 5500 ESCA spectrophotometer). The Brunauer–Emmett–Teller (BET) surface area and pore size were determined by N2 adsorption/desorption measurements (Quantachrome Nova 1200 Analyzer) after degassing the sample under vacuum at 150 °C for 2 h. The sulfur content in composite materials was measured by thermogravimetric differential scanning calorimetry (TGA, Q50) with a 5 °C min−1 heating rate from 25 to 600 °C. Conductivity measurements were performed on sheets (10 × 20 mm) using the Ossila four-point probe technique.Lithium–sulfur battery assembly
:1 vol. ratio of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME), had a volume maintained at 5 μL mg−1 sulfur. Sealed pouch cells were pressed for 2 h under an Ar atmosphere and then transferred to a vacuum oven for 12 h at 50 °C for aging.
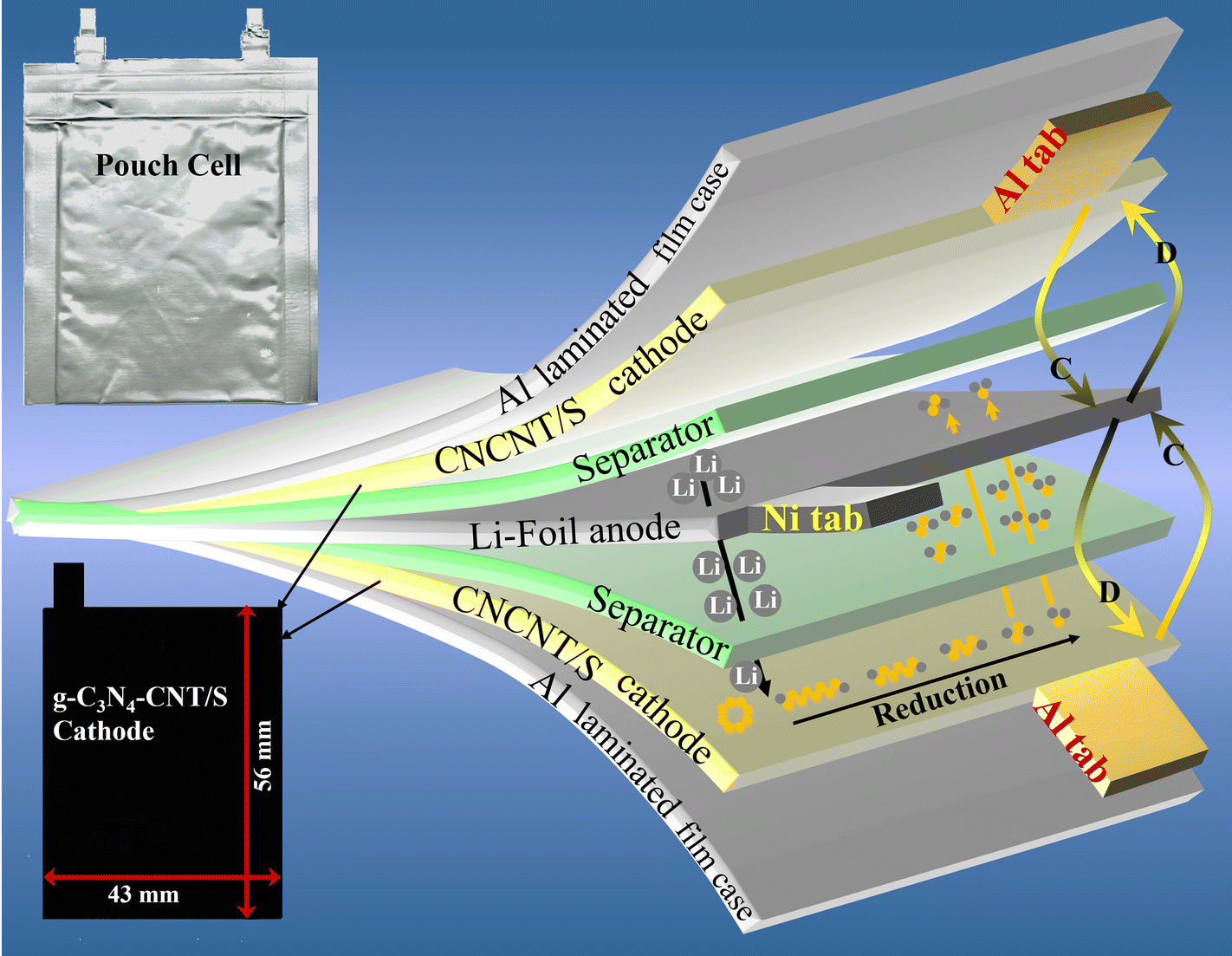 | ||
| Scheme 2 Schematic for the structure of the pouch cell demonstrating the arrangements of electrodes and separator. | ||
Polysulfide adsorption tests
The absorption behaviors of active materials towards polysulfides were assessed by immersing them in a Li2S6 solution, chosen as the representative of polysulfides. For Li2S6 solution preparation, Li2S and sulfur powders (1:5 molar ratio) were added to a DOL and DME solution (1:1, v/v) under vigorous stirring at 60 °C for 24 h in an Ar-filled glovebox. The concentration of the resulting Li2S6 solution was 5 mmol L−1. Before the polysulfide adsorption test, pure g-C3N4 and g-C3N4-CNT hybrids were dried at 60 °C under vacuum for 12 h. Subsequently, g-C3N4 and g-C3N4-CNT hybrids with an equal weight (20 mg) were immersed in a 5 mL Li2S6 solution for static adsorption over 12 h. The color variation of the supernatant over time was observed, and the adsorption capability of g-C3N4 and g-C3N4-CNT to Li2S6 was tested via UV-vis spectroscopy. A blank Li2S6 solution served as the reference.
Computational details
The electronic structure calculations used the Vienna ab initio Simulation Package (VASP) with projector-augmented wave (PAW) pseudopotentials.28–30 The Perdew–Burke–Ernzerhof generalized gradient approximation (GGA-PBE) is employed to capture the exchange–correlation potentials.31 A plane wave cutoff energy of 470 eV was set, enhancing the accuracy of our electronic structure calculations. To address van der Waals (vdW) interactions, the DFT-D3 method developed by Grimme was systematically incorporated into all calculations. The application of the DFT-D3 (ref. 32) method included the use of a zero-damping function, further contributing to the accuracy and reliability of our computational results. The geometry relaxation of all structures is meticulously conducted until the Hellmann–Feynman forces acting on each atom reach a value below 10−2 eV Å−1. Simultaneously, total energy convergence is rigorously pursued until it reaches 10−4 eV. To adequately sample the Brillouin zone, a regular Γ-centered k-point mesh is utilized with a grid that is 3 × 3 × 1.33 The initial optimization involved separate adjustments of the unit cells for the g-C3N4 and CNT surfaces. Subsequently, supercells of dimensions 2 × 1 × 1 are constructed for each system, accompanied by the introduction of a vacuum region of approximately 30 Å around each supercell in the z-direction. LiPSs and S8 molecules are also individually optimized before being placed on the surfaces. Further simulations involved the strategic placement of S8 and LiPS molecules on both g-C3N4 and the heterostructure formed by combining g-C3N4 and CNT (g-C3N4-CNT). A comprehensive relaxation of the S8/LiPSs + g-C3N4 and S8/LiPSs + g-C3N4-CNT configurations is then carried out using the above-mentioned computational parameters.In the pursuit of identifying the energetically most favorable configurations of S8/LiPSs, we theoretically investigated the adsorption of S8/LiPSs molecules on both g-C3N4 and g-C3N4-CNT surfaces. Our exploration encompassed a thorough examination of various possible orientations and configurations of the S8/LiPS molecules. The energetically most stable configurations for each LiPS and S8 molecule are considered for binding energy studies. The binding energy (Eb in eV) of each LiPS and S8 molecule on the g-C3N4 surface and g-C3N4-CNT composite surface is calculated using the following equation:
| Eb = ES + Eg-C3N4/g-C3N4-CNT − ES+g-C3N4/g-C3N4-CNT | (1) |
The Gibbs free energy (ΔG) of the sulfur reduction reaction: the reaction Gibbs free energy of S8 and Li2Sn on g-C3N4 and g-C3N4-CNT composite surfaces was calculated by using the following equation:
| ΔG = ΔEdft − (ΔZPE + TΔS) | (2) |
Results & discussion
Active material characterization
The XRD patterns in Fig. 1a depict the crystalline characteristics of as-prepared g-C3N4, g-C3N4/S, and g-C3N4-CNT/S. Pure g-C3N4 exhibits a low-angle reflection peak at ∼13°, indicative of lattice planes parallel to the c-axis.35,36 Additional peaks at 27.9° result from the periodic stacking of heptazine layers in the nanosheets.37,38 In the g-C3N4-CNT/S spectrum, peaks at 26.3° and 42.7° correspond to the hexagonal graphite structure in CNTs along the (002) and (100) directions.39 The well-aligned diffraction peaks of the orthorhombic sulfur phase (JCPDS card no. 08-0247) in both g-C3N4/S and g-C3N4-CNT/S confirm the crystalline nature of sulfur in the g-C3N4 or g-C3N4-CNT host. FTIR spectra in Fig. 1b reveal the characteristic features of g-C3N4, g-C3N4/S, and g-C3N4-CNT/S. The absorption band at 790 cm−1 signifies the out-of-plane skeletal bending modes of tri-s-triazine cycles, confirming the presence of g-C3N4.40,41 Bands at 1270 and 1540 cm−1 correspond to the C–N stretching and CN heterocycle stretching modes of g-C3N4, respectively.42 The broad band between 2950 and 3480 cm−1 indicates the stretching modes of terminal –NH groups at defect sites of the aromatic ring, and signals from S–H stretching modes (2347 cm−1) align with reported observations.43 The Raman spectra in Fig. 1c were recorded for g-C3N4, g-C3N4/S, and g-C3N4-CNT/S to assess the degrees of graphitization. Peaks around 706, 768, and 1230 cm−1 correspond to the breathing modes of the s-triazine ring in g-C3N4.44 Distinct Raman peaks at 1417 and 1577 cm−1 represent the D band and G band, indicative of defects in disordered carbon and the in-plane vibration of graphitic layers, respectively.45 The ID/IG intensity ratios decrease slightly with the addition of sulfur, which may be ascribed to the reduced disorder because of S particle encapsulation into void interlayers and defective sites of g-C3N4.46 The results further signify a higher degree of the crystalline graphitic structure with improved electronic conduction, aligning with the XRD pattern in Fig. 1a. The porosity of g-C3N4, g-C3N4/S, and g-C3N4-CNT/S was studied through N2 sorption curves (Fig. S1†), indicating a type-IV nitrogen adsorption–desorption process with an H3 type hysteresis loop, suggesting mesopores. The BET surface area decreases from 82 m2 g−1 for pure g-C3N4 to 64 and 49 m2 g−1 for g-C3N4/S and g-C3N4-CNT/S, respectively, indicating successful impregnation of CNT and sulfur in pure g-C3N4 pores. Given that sulfur and Li2S2/Li2S are insulators, good conductivity is crucial for the sulfur matrix. The conductivity of g-C3N4-CNT/S, probed by the four-point probe method, ranges between 10−2 and 10−3 S cm−1, significantly higher than that of g-C3N4 materials, which falls between 10−12 and 10−13 S cm−1. | ||
| Fig. 1 For g-C3N4, g-C3N4/S, and g-C3N4-CNT/S: (a) XRD patterns, (b) FTIR spectra and (c) Raman spectra; TEM images for (d) g-C3N4 and (e) g-C3N4-CNT/S materials; (f) SEM images for g-C3N4-CNT/S; (g) color mapping of g-C3N4-CNT/S demonstrating the uniform distribution of C, N, O, and S elements. | ||
The morphologies of the prepared active materials were investigated via TEM and SEM, complemented by elemental mapping. The TEM image in Fig. 1d displays g-C3N4 with loose morphology, featuring 2D nanosheets resulting from gas release during thermal polymerization. In contrast, the TEM image of g-C3N4-CNT/S (Fig. 1e) reveals 1D CNTs on 2D g-C3N4 sheets, with a clustered surface exhibiting wrinkles, curls, and sparse sulfur agglomerations. Abundant porosity benefits sulfur encapsulation, effectively accommodating volume variations during cycling. SEM images in Fig. 1f depict a well-maintained interlaced CNT network structure without noticeable sulfur agglomeration after sulfur diffusion into g-C3N4-CNT, indicating good sulfur dispersion in g-C3N4-CNT/S. Elemental mapping in Fig. 1g vividly illustrates uniform C, N, and S dispersion throughout g-C3N4-CNT/S, with mappings completely following the shapes of the samples confirming uniform sulfur deposition on g-C3N4-CNT surfaces. From these results, we conclude that in situ growth of 1D CNTs on 2D g-C3N4 sheets constructs a cross-linked 3D conductive network, chemically bonded in the g-C3N4-CNT composite. This hybrid system provides an efficient electron transport path via CNTs, acting as a loading platform with N-containing functional groups to trap sulfur particles. The highly integrated g-C3N4-CNT/S with N-containing functional groups exhibits strong LiPS confinement, ensuring excellent cycling performance. Sulfur loading, a crucial LiSB performance index,47 was calculated through the TGA of the g-C3N4-CNT/S composite. In Fig. S2,† the significant weight loss between 200 and 340 °C corresponds to sulfur evaporation, yielding a 68.6% sulfur content in g-C3N4-CNT/S, consistent with the experimentally added 70% sulfur.
XPS was utilized to delve deeper into the chemical bonding of the g-C3N4-CNT/S composite. In Fig. 2, XPS survey spectra exhibit peaks at approximately 161.9, 226.4, 283.6, 397.7 eV, and 532.1 eV, assigned to S 2p, S 2s, C 1s, N 1s, and O 1s binding energies, respectively. The fitted curve of C 1s spectra of the hybrid g-C3N4-CNT composite presents five peaks: –C–C– at 282.6 eV, conjugated –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N−/−CC– at 284.6 eV, S–C/C–N at 285.5 eV, and –S–CN at 288.4 eV.48 The red shifting of –C–C– from the usual 284 ± 0.5 eV could be due to the increase in electronic density around carbon, which causes the binding energy of –C–C– to shift to lower values. The computational simulations by Priyanga et al.49 suggest that the valence band (VB) originates from the Pz orbitals of nitrogen atoms, while the conduction band (CB) arises from the Pz orbitals of carbon atoms.49 Consequently, the process of reduction and oxidation occurs at carbon and nitrogen atoms, respectively. This phenomenon stems from nitrogen's higher electronegative potential compared to carbon.50 A 286.8 eV binding energy of g-C3N4-CNT is related to the CO groups and originates from the CNTs after thermal annealing.51 The N 1s spectra reveal four peaks at 397.5 eV, 398.5, 400.7, and 404.4 eV, attributed to pyridinic-like N in CN, –S–NC–, graphitic-like N (C3–N)/amino N (N–H), and oxidized N (N–O), respectively.52 Abundant pyridinic N content in g-C3N4-CNT/S enhances lithium polysulfide adsorption via Li–N interaction, thereby reducing the barrier height and improving conductivity and electron density.53,54 The S 2p spectrum of g-C3N4-CNT/S displays peaks at 162.5 (S 2p3/2) and 163.9 eV (S 2p1/2), assigned to –S–S– bonds (S8 molecules).55 Interestingly, the red shift in both the peaks (compared to XPS of pure S8: S 2p3/2 at 164 eV and S 2p1/2 at 165 eV)56 indicates a covalent link of sulfur chains to the host material.57 The S 2p peak at 167.02 eV is ascribed to sulfate species, formed through chemical interaction between surface oxygen-containing functional groups and sulfur atoms.58,59 Besides N, C, and S, oxygen (O) is detected, originating from oxygenated functional groups in g-C3N4 preparation at high temperatures. These oxygenated groups aid in chemically adsorbing sulfur and preventing polysulfides from dissolving into electrolytes effectively.60,61
N−/−CC– at 284.6 eV, S–C/C–N at 285.5 eV, and –S–CN at 288.4 eV.48 The red shifting of –C–C– from the usual 284 ± 0.5 eV could be due to the increase in electronic density around carbon, which causes the binding energy of –C–C– to shift to lower values. The computational simulations by Priyanga et al.49 suggest that the valence band (VB) originates from the Pz orbitals of nitrogen atoms, while the conduction band (CB) arises from the Pz orbitals of carbon atoms.49 Consequently, the process of reduction and oxidation occurs at carbon and nitrogen atoms, respectively. This phenomenon stems from nitrogen's higher electronegative potential compared to carbon.50 A 286.8 eV binding energy of g-C3N4-CNT is related to the CO groups and originates from the CNTs after thermal annealing.51 The N 1s spectra reveal four peaks at 397.5 eV, 398.5, 400.7, and 404.4 eV, attributed to pyridinic-like N in CN, –S–NC–, graphitic-like N (C3–N)/amino N (N–H), and oxidized N (N–O), respectively.52 Abundant pyridinic N content in g-C3N4-CNT/S enhances lithium polysulfide adsorption via Li–N interaction, thereby reducing the barrier height and improving conductivity and electron density.53,54 The S 2p spectrum of g-C3N4-CNT/S displays peaks at 162.5 (S 2p3/2) and 163.9 eV (S 2p1/2), assigned to –S–S– bonds (S8 molecules).55 Interestingly, the red shift in both the peaks (compared to XPS of pure S8: S 2p3/2 at 164 eV and S 2p1/2 at 165 eV)56 indicates a covalent link of sulfur chains to the host material.57 The S 2p peak at 167.02 eV is ascribed to sulfate species, formed through chemical interaction between surface oxygen-containing functional groups and sulfur atoms.58,59 Besides N, C, and S, oxygen (O) is detected, originating from oxygenated functional groups in g-C3N4 preparation at high temperatures. These oxygenated groups aid in chemically adsorbing sulfur and preventing polysulfides from dissolving into electrolytes effectively.60,61
 | ||
| Fig. 2 (a) XPS spectrum showing the complete scan and the corresponding spectra for the elements (b–e) present in the g-C3N4-CNT/S composite. | ||
The catalytic conversion process of LiPSs involves two steps: adsorption and catalytic reaction.58,62 To assess adsorption ability, g-C3N4, and g-C3N4-CNT materials were individually immersed in a Li2S6 solution with magnetic stirring for 30 minutes. After 24 hours, adsorbents were retrieved, and optical photos of solutions were captured (inset of Fig. 3). Notably, the original light-yellow color of the Li2S6 solution becomes colorless after immersing in g-C3N4-CNT, suggesting effective absorption of most polysulfides. In contrast, the Li2S6 solution color shows a slight change after immersing in g-C3N4. Moreover, UV-vis measurements for Li2S6 reveal a strong absorption band in the 240–300 nm region, assigned to S62− polysulfide species.63 g-C3N4 exhibits lower absorbance than the Li2S6 sample, while g-C3N4-CNT demonstrates the lowest absorbance, attributed to its high pyridinic/pyrrolic N content. This confirms the strong affinity between polysulfides and pyridinic/pyrrolic N, particularly effective in suppressing polysulfide dissolution in the electrolyte (Sx–Li⋯N interactions).64 This robust affinity contributes significantly to enhancing the cycling stability of the sulfur electrode.
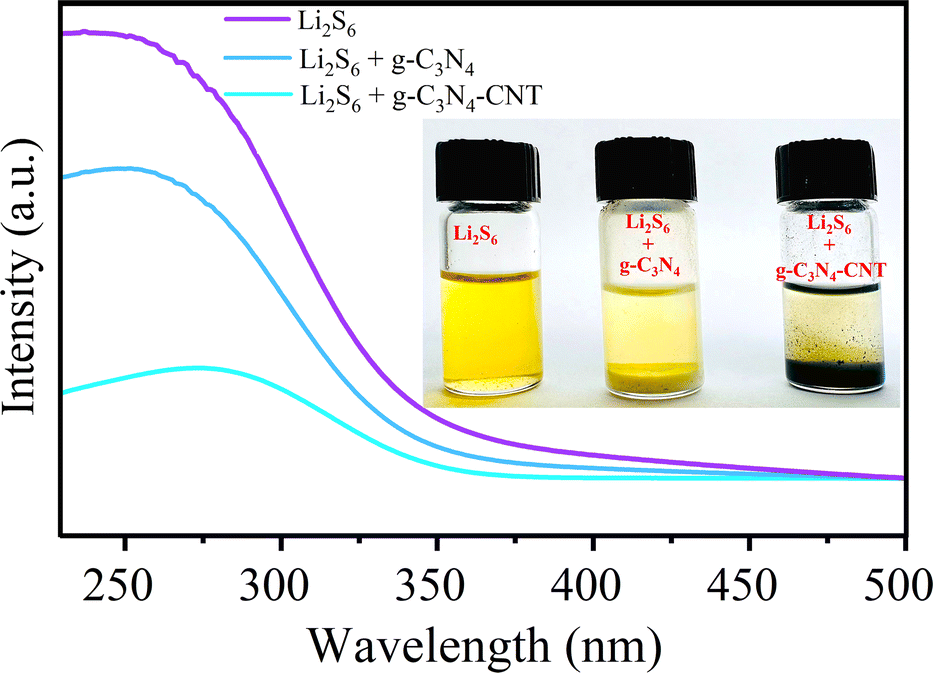 | ||
| Fig. 3 UV-vis spectra for blank Li2S6 and Li2S6 added with CN and g-C3N4-CNT. The inset shows the digital picture of the effect of the active material on the adsorption of LiPSs in Li2S6 solution. | ||
Theoretical calculations
We performed DFT calculations to assess and compare the binding strengths of S8/LiPSs on g-C3N4 and g-C3N4-CNT composite surfaces. The calculated binding energies (Eb in eV) of S8/LiPSs on the g-C3N4 surface align with prior study findings.65 Tables S1–S3† and Fig. 4 offer a comprehensive analysis of the electronic and binding properties of both g-C3N4 and g-C3N4-CNT composite surfaces when interacting with S8/LiPSs. The GGA-PBE bandgap for g-C3N4 and g-C3N4-CNT was determined to be approximately 1.195 and 0.049 eV, respectively (Table S1†). Regarding bandgap changes, notable decreases were observed, as seen in Li2S (1.7321 to 0.0152 eV) and Li2S2 (0.0383 to 0.0354 eV), indicating a potential enhancement in g-C3N4-CNT electronic conductivity. Conversely, an increase in the bandgap for Li2S and S8 when interacting with g-C3N4 implies a reduction in electronic conductivity, as a larger bandgap typically requires more energy for electron movement between the valence and conduction bands, hindering electronic transport. Binding energy changes, representing the strength of interactions, show varying values across the systems, with Li2S displaying a substantial binding energy decrease (8.309 to 1.618 eV). These findings suggest that the adsorption of S8/LiPSs onto g-C3N4 and g-C3N4-CNT composite surfaces leads to modifications in electronic and binding properties, with potential implications for the overall performance of the material. These outcomes suggest that the adsorption of S8/LiPSs onto g-C3N4-CNT results in favorable alterations in electronic and binding properties, potentially enhancing the material's overall performance. Notably, for all lithium contents (Li2Sn, n = 1, 2, 4, 6, 8), the LiPS molecules exhibit higher binding energies on the g-C3N4 surface compared to the g-C3N4-CNT surface (Fig. 4a–c). This observation suggests a more robust interaction between LiPSs and the g-C3N4 surface than with the g-C3N4-CNT surface. The structural impact becomes evident as the g-C3N4 layer undergoes distortion upon the adsorption of S8/LiPS molecules (Fig. 4a), while the geometry of the CNT (S8/LiPSs + g-C3N4-CNT) remains nearly unaltered (Fig. 4b). However, due to the weak van der Waals interaction between S8/LiPSs and g-C3N4-CNT, a small strain can be observed on the g-C3N4 surface as well. This discrepancy indicates that LiPSs are chemically and physically adsorbed on g-C3N4 and g-C3N4-CNT, respectively, providing a rationale for the experimental phenomena discussed in Fig. 3. A thorough discussion on binding energy is included in the ESI.†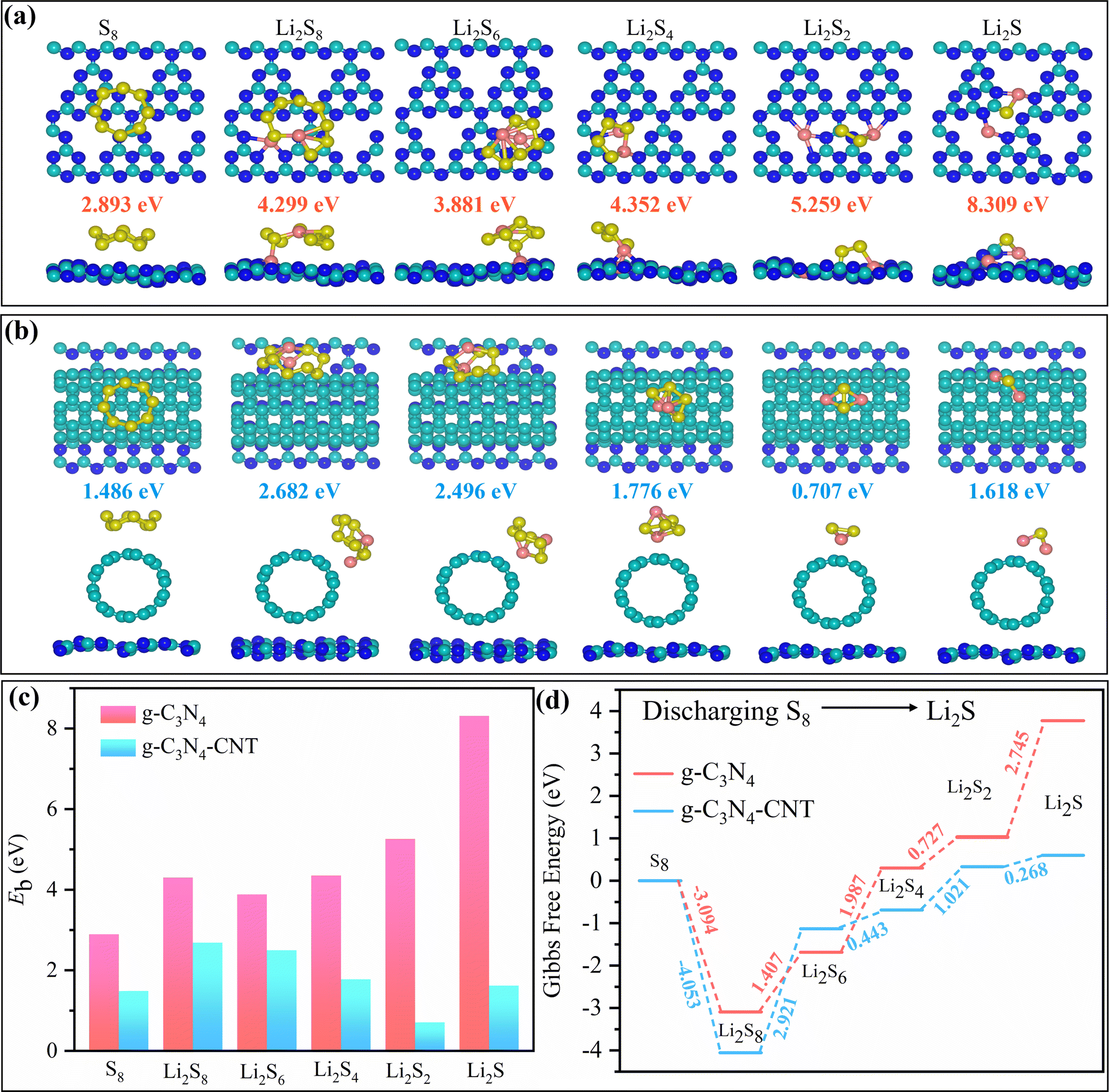 | ||
| Fig. 4 Optimized configurations of LiPSs on the surfaces of (a) g-C3N4 and (b) the heterostructure of g-C3N4-CNT. The yellow, pink, cyan, and blue spheres represent sulfur (S), lithium (Li), carbon (C), and nitrogen (N) atoms, respectively. Panel (c) displays the binding energies and panel (d) illustrates the relative free energy for the discharging process from S8 to Li2S on both g-C3N4 and g-C3N4-CNT surfaces. | ||
Upon closer examination of the structures, it is observed that all carbon (C) atoms and a quarter of the nitrogen (N) atoms in g-C3N4 are three-fold coordinated, with the remaining three-quarters of N atoms being two-fold coordinated. Analyzing the optimized structure of LiPS molecules on the g-C3N4 surface reveals that the interactions predominantly stem from chemical bonds between Li+ atoms and the two-fold coordinated N atoms (Fig. 4a). This underscores that the atomic under-coordination in g-C3N4 plays a pivotal role in stabilizing LiPSs. In the case of Li2S2 and Li2S, Li+ forms bonds with N atoms, and S2− binds with C on the polar g-C3N4 surface. Importantly, this explains the absence of chemical bonds between LiPSs and g-C3N4-CNT (Fig. 4b), where all C atoms are three-fold coordinated within the carbon nanotube. In-depth insights into the electrochemical reaction kinetics enhancement on both g-C3N4 and g-C3N4-CNT surfaces are sought by examining the complete reaction pathway involving the transformation of S8 to Li2S. In Fig. 4d, we show the landscape of relative Gibbs free energy (in eV) for the discharging process from S8 (solid) to Li2S (liquid) on both the g-C3N4 and the g-C3N4-CNT composite surfaces. It is noteworthy that the results of g-C3N4 differ significantly from those of g-C3N4-CNT. We hypothesize that in the case of g-C3N4, the transition from S8 → Li2S results in the generation of increasingly ionic structures where Li transforms into Li+ and the energy becomes highly negative, indicating the discharged structure. On the other hand, for the g-C3N4-CNT composite surface, the interaction between LiPSs and CNT is limited, and we do not observe the same extent of electron transfer. The composite structure of g-C3N4-CNT may be oversimplified, as some of the LiPSs interact with the CNT, while other sulfides interact with g-C3N4, resulting in a mixture of outcomes. A thorough discussion on the Gibbs free energy uphill process is included in the ESI.†
The initial phase of the discharge process involves the simultaneous double reduction of an S8 molecule and 2Li+ ions, resulting in the formation of a lengthy Li2S8 polysulfide chain. Subsequently, Li2S8 undergoes further reduction, sequentially forming three intermediate LiPSs (Li2Sn = 6, 4, 2), ultimately leading to the production of the end-product Li2S polysulfides. The obtained data unequivocally indicate a higher thermodynamic favorability of the discharging process on the g-C3N4-CNT composite surface compared to the g-C3N4 surface, providing a rationale for the experimental phenomena discussed in Fig. 5. Specifically, the reaction energy from solid S8 to liquid Li2S8 on the g-C3N4-CNT surface is more exothermic than that on the g-C3N4 surface (Fig. 4d). Notably, the subsequent four reduction stages from Li2S8 to Li2S exhibit endothermic behavior on the g-C3N4 as well as on the g-C3N4-CNT composite surface. The final two steps, from Li2S4 to Li2S2 and from Li2S2 to Li2S, exhibit substantial energy barriers (∼3.472 eV) compared to the other steps on both the g-C3N4 and g-C3N4-CNT composite substrates. This observation suggests that the precipitation process of Li2S2/Li2S is the rate-limiting step during discharging. In contrast, on the g-C3N4-CNT composite surface, the energy barrier for the endothermic precipitation process is effectively lowered (∼1.289 eV), signifying a more favorable discharge. Additionally, the mixed behavior in the subsequent reductions correlates with the bandgap values, suggesting that electronic properties play a crucial role in the energetics of the discharge process. The lower bandgap of g-C3N4-CNT may contribute to its overall more favorable Gibbs free energy values, suggesting better electrochemical performance compared to the g-C3N4 substrate. These findings contribute crucial insights into the kinetics of the discharge process on both g-C3N4 and g-C3N4-CNT substrates, highlighting key rate-determining steps and their corresponding energy landscapes. Furthermore, the electronically conductive nature of the g-C3N4-CNT surface facilitates rapid electron transfer between C atoms (from CNT) of g-C3N4-CNT and S atoms of LiPSs, thereby weakening the Li–S bonds of LiPSs. As a result, g-C3N4-CNT significantly reduces the reduction barrier of LiPSs, promotes Li2S precipitation, and enhances reaction reversibility, underscoring its potential superiority over g-C3N4 in terms of electrochemical performance.
 | ||
| Fig. 5 (a) CV profiles for g-C3N4/S and g-C3N4-CNT/S cathodes; CV curves at different scan rates for (b) g-C3N4-CNT/S and (c) g-C3N4/S; plots of the peak currents versus the square root of scan rates from the CV curves of g-C3N4-CNT/S and g-C3N4/S cathodes for (d) Peak A, (e) Peak C1 and (f) Peak C2. | ||
Electrochemical characterization
The electrochemical performance of g-C3N4/S and g-C3N4-CNT/S was assessed in a pouch cell configuration. In Fig. 5a, the CV curves of g-C3N4/S and g-C3N4-CNT/S composites are presented within a voltage range of 1.6 V to 2.8 V at a sweep rate of 0.05 mV s−1. During the cathodic scan, both g-C3N4/S and g-C3N4-CNT/S exhibit two main reduction peaks. The peak at approximately 2.27 V corresponds to the reduction of elemental sulfur to high-order polysulfides, Li2Sn (4 ≤ n ≤ 8), while the peak between 1.9 and 2.0 V signifies the formation of Li2S2/Li2S from low-order polysulfides.66 In the anodic scanning process, two oxidation peaks between 2.44 and 2.51 V are associated with the oxidation of Li2S2/Li2S to long-chain polysulfides (Li2Sn (n > 2)) and further to sulfur.67 Compared to g-C3N4/S, the main cathodic peaks of g-C3N4-CNT/S exhibit a slight shift to higher reduction potential, and the anodic peaks shift to lower oxidation potential, indicating reduced polarization in g-C3N4-CNT/S. This facilitates polysulfide conversion, indicating improved reversibility of the electrode during cycling.68 Furthermore, the smaller potential difference (ΔE) between cathodic and anodic peak current densities in g-C3N4-CNT/S reflects its remarkably stable tendency to effectively suppress the diffusion of liquid intermediate products during CV cycles.69 Additionally, during the scanning process for g-C3N4-CNT/S (Fig. S3†), the cathodic peak position, peak current, and peak area exhibit minimal change over the next 5 cycles, indicating superior capacity retention and good reversibility of the cell. CV curves at various current rates are depicted in Fig. 5b and c for g-C3N4-CNT/S and g-C3N4/S cathodes, respectively. The cathodic processes involve Peak C1 and Peak C2, attributed to S8 transformation to long-chain LiPSs (Li2Sn, 4 ≤ n ≤ 8) and the subsequent reduction of long-chain LiPSs to Li2S2/Li2S, respectively.66,70The anodic process is represented by Peak A, corresponding to the reverse transformation. The redox peaks at higher current rates appear broadened for both materials, indicating slow kinetics of LiPS conversion. Moreover, at increased scan rates, redundant peaks emerge after 2.6 V during the anodic scan for both materials, signifying sluggish cathode kinetics. Overall, g-C3N4-CNT/S exhibits higher peak current densities than g-C3N4/S, indicating enhanced Li+ transportability and superior rate capability. Additionally, both cathodic and anodic peak currents for both materials exhibit a linear relationship with the square root of the scan rate (ν0.5), suggesting diffusion-limited discharge/charge reactions (Fig. 5d–f).
Therefore, the diffusion coefficient of Li+ can be described by the Randles–Sevcik equation:71Ip = (2.69 × 105)n1.5AD0.5CLi+0.5v0.5, in which n is the number of charge transfers, A is the active electrode area, D is the Li+ diffusion coefficient, and CLi+ is the concentration of Li+ in bulk. Since n, A, and c0 are constants, 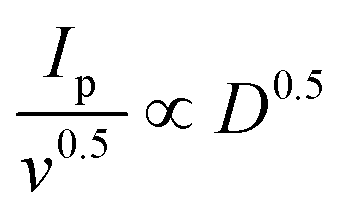 , a larger
, a larger  implies a higher D. The equation reveals a linear relationship between the peak current and the square root of the scan rate, with the slope indicating lithium-ion diffusion.72 Notably, in g-C3N4-CNT/S, the slopes for the two reduction peaks and one oxidation peak surpass those in g-C3N4/S at the same electrode area for each sulfur reduction and oxidation reaction. This suggests accelerated Li ion diffusivity and improved LiPS redox kinetics during discharge/charge processes attributed to CNT presence, promoting LiPS transformation and facilitating rapid Li+ migration.73 However, due to the formation of various polysulfides in the redox process, quantifying DLi+ is challenging due to an unknown number of electrons involved in polysulfide formation.
implies a higher D. The equation reveals a linear relationship between the peak current and the square root of the scan rate, with the slope indicating lithium-ion diffusion.72 Notably, in g-C3N4-CNT/S, the slopes for the two reduction peaks and one oxidation peak surpass those in g-C3N4/S at the same electrode area for each sulfur reduction and oxidation reaction. This suggests accelerated Li ion diffusivity and improved LiPS redox kinetics during discharge/charge processes attributed to CNT presence, promoting LiPS transformation and facilitating rapid Li+ migration.73 However, due to the formation of various polysulfides in the redox process, quantifying DLi+ is challenging due to an unknown number of electrons involved in polysulfide formation.
Galvanostatic charge–discharge tests were conducted to investigate the electrochemical performance of the pouch cells. In Fig. 6a, the 1st cycle charge–discharge voltage profiles of g-C3N4/S and g-C3N4-CNT/S cathodes at a 0.1C current density reveal two distinct discharging voltage plateaus and one major charging voltage plateau, consistent with the multistep reduction of sulfur indicated by CV curves. The polarization potential (ΔE), calculated as the gap between anode and cathode peaks, is lower for g-C3N4-CNT/S (190 mV) compared to g-C3N4/S (231 mV), reflecting reduced polarization in the g-C3N4-CNT/S electrode facilitated by CNT-N active site catalysis. Additionally, the capacity of the lower discharge plateau (QL) and higher discharge plateau (QH) is calculated from discharge curves. The higher QL/QH signifies superior electrocatalytic activity for polysulfides, where QH corresponds to sulfur conversion into soluble polysulfides, and QL represents efficient reduction of polysulfides to Li2S.74 In Fig. 6b, the capacity ratio for g-C3N4-CNT/S (1.95) surpasses that of CN/S (1.88), indicating enhanced sulfur utilization owing to the superior catalytic activity of Sx–Li⋯N in the g-C3N4-CNT/S electrode. The charge–discharge profiles of g-C3N4-CNT/S at 0.1C over 250 cycles (Fig. 6c) reveal sustained maintenance of the two discharge plateaus, demonstrating rapid mass transport and reaction kinetics. A comparison of long-term cycle performances between g-C3N4/S and g-C3N4-CNT/S (Fig. 6d) illustrates the initial capacity decay attributed to the activation process. Non-activated sulfur may aggregate on g-C3N4's surface initially leading to the formation of an unstable solid electrolyte interface (SEI) layer.75 The subsequent cycles witness enhanced sulfur utilization and uniform depreciation in capacity, with g-C3N4-CNT/S delivering an initial discharge capacity of 895.7 mA h g−1 and a capacity retention of 86% after 250 cycles, boasting a CE of 90.4%. In contrast, g-C3N4/S achieves a reversible initial discharge capacity of 679.6 mA h g−1. The improved cycle performance of g-C3N4-CNT/S is attributed to abundant anchor sites on nanosheets, influencing LiPS adsorption. Rate performance (Fig. 6e) indicates higher discharge capacity of g-C3N4-CNT/S than g-C3N4/S at different current rates, with g-C3N4-CNT/S recovering well at 0.1C, which is ascribed to the porous structures of g-C3N4 ensuring fast Li ion transportation and chemical confinement for lithium polysulfides.27,76 Charge–discharge curves at various rates (0.1C to 1.5C) for g-C3N4-CNT/S (Fig. 6f) demonstrate excellent maintenance of discharge plateaus at 1.5C, affirming swift mass transport and reaction kinetics. The cathode exhibits high reversibility, maintaining capacities of 847, 656, and 543 mA h g−1 at 0.2, 0.76, and 1.5C, respectively, corresponding to 95%, 73%, and 61% of the original capacity. High sulfur mass loading is crucial for achieving elevated energy density and facilitating lithium–sulfur battery commercialization. Therefore, pouch cells, featuring sulfur loadings of 4.3, 5.4, and 6.2 mg cm−2, underwent testing at 0.1C for the g-C3N4-CNT/S cathode. In Fig. 6g, the 6.2 mg cm−2 cell yields the lowest reversible discharge capacity of 566 mA h g−1 after 250 cycles, exhibiting a fading rate of 0.08% per cycle and a CE of 82%. This underscores g-C3N4-CNT/S's outstanding electrochemical performance under high-loading configurations, indicating robust LiPS inhibition and accelerated kinetic conversion for enhanced sulfur utilization.77 Table S4† provides a comparative overview of Li–S batteries, affirming the remarkable performance of the g-C3N4-CNT/S cathode in the pouch cell format (electrode size ∼22 cm2 and E/S ∼5 μl mg−1) as compared to the coin cells with a low electrode size (∼1.6 cm2) and flooded electrolyte (E/S > 15 μl mg−1). The large area pouch cells are more prone to uneven sulfur degradation,78 which would increase the electrode tortuosity and even block the electrolyte redistribution, thereby reducing the cycling performance and CE of the pouch cell.79 Therefore, the superior performance of our composite electrode further attests to its utilization for commercial applications.
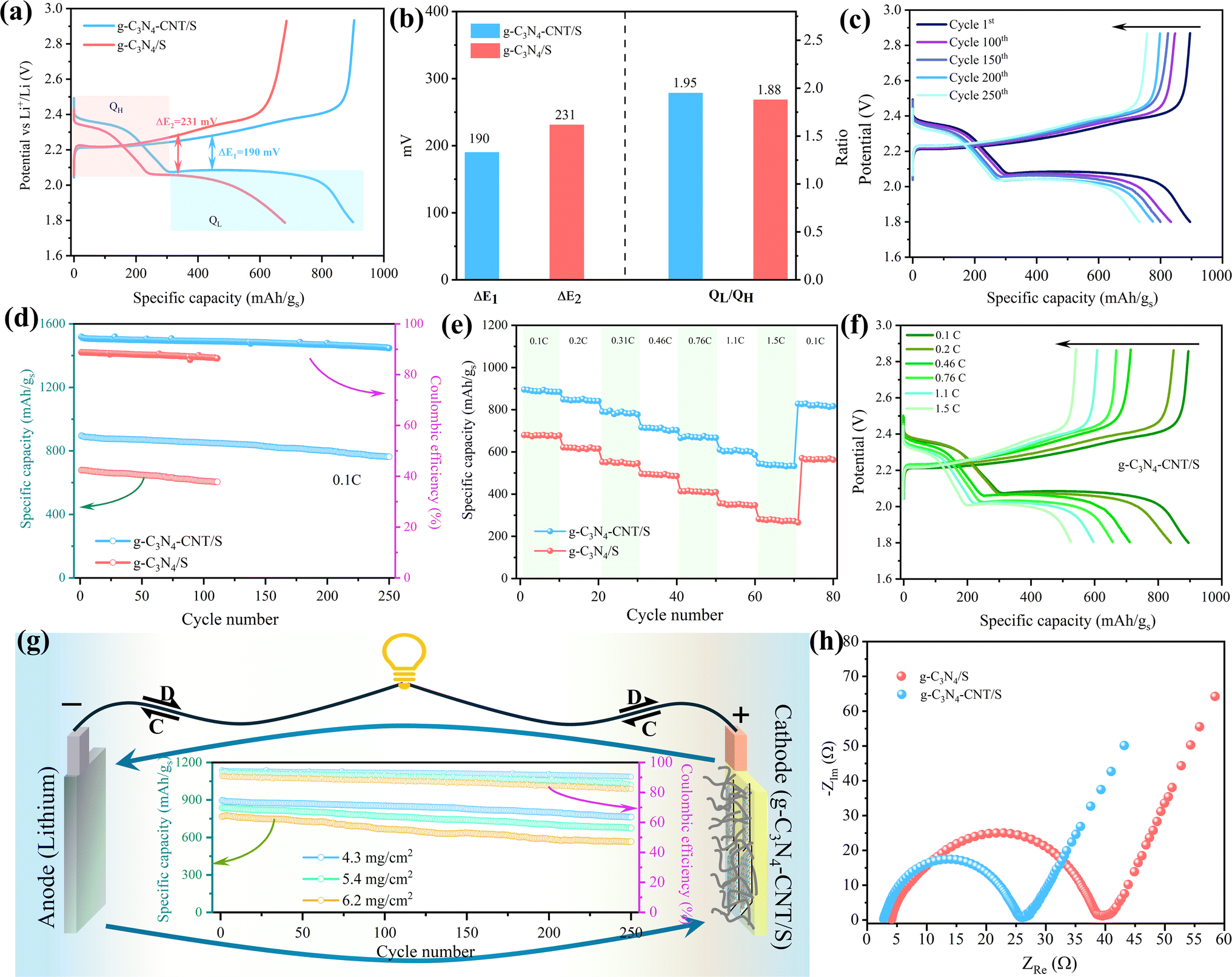 | ||
| Fig. 6 (a) Initial charge–discharge curves for the g-C3N4/S and g-C3N4-CNT/S cathodes at 0.1C, (b) the capacity contribution of QH, QL, and the QL/QH ratio of g-C3N4-CNT/S and g-C3N4/S at 0.1C, (c) galvanostatic charge–discharge profiles of the g-C3N4-CNT/S cathode for 250 cycles, (d) cycle performance and coulombic efficiency of g-C3N4/S and g-C3N4-CNT/S cathodes for 110 and 250 cycles, respectively, (e) rate capabilities of g-C3N4/S and g-C3N4-CNT/S cathodes under various current densities, (f) galvanostatic charge–discharge profiles of the g-C3N4-CNT/S cathode at different current densities, (g) cycle performance of the g-C3N4-CNT/S cathode with varied sulfur loading for 250 cycles at 0.1C and (h) EIS spectra of the freshly prepared uncycled cell. | ||
To further detail improved conductivity and enhanced polysulfide-trapping ability, EIS measurements for cells with g-C3N4/S and g-C3N4-CNT/S cathodes were conducted (Fig. 6h) in the frequency range of 0.5 Hz–2 mHz, with an excitation potential of 10 mV and 20 points per decade. Nyquist plots reveal three frequency regions: a high-frequency segment indicating electrolyte resistance (Re), a middle-frequency semicircle corresponding to charge transfer impedance (Rct), and a low-frequency spike representing Warburg impedance (W) associated with lithium-ion diffusion in the cathode.80,81 As can be seen, the semicircle diameter and Rct value for the g-C3N4-CNT/S cathode cell (22.6 Ω) are notably lower than those of the g-C3N4/S cathode cell (35.6 Ω), indicating reduced charge transfer resistance due to enhanced electrical conductivity and improved electrical contact upon CNT incorporation into g-C3N4. Additionally, the Re for g-C3N4-CNT/S is merely 2.7 Ω, affirming outstanding cell conductivity. This outcome underscores lower electrochemical impedance in the g-C3N4-CNT/S host, attributed to the presence of CNTs and abundant pyridine N acting as chemically active sites that robustly adsorb polysulfides, facilitate charge and ion transfer, and diminish electrode–electrolyte interface resistance and charge transfer resistance. Parameters from the Nyquist plot are detailed in Table S5.† To showcase the superior mechanical flexibility of the designed g-C3N4-CNT/S electrodes, various folding angles for pouch cells were tested. Fig. 7 reveals impressive open circuit voltage (OCV) stability at 2.36 V, successfully powering an LED bulb at varying bending angles. Remarkably, no discernible change in LED brightness or OCV readings occurred even at nearly 180° bending angles. The g-C3N4-CNT/S-based Li–S pouch cells demonstrate favorable electrical performance, emphasizing their practical utility in flexible energy systems, owing to excellent cycling stability achieved through effective polysulfide shuttling suppression.
 | ||
| Fig. 7 Upper row: digital pictures of the OCV for the pouch cell consisting of the g-C3N4-CNT/S cathode at different folding angles and bottom row: pictures of LEDs lit up by the pouch cell at different folding angles demonstrating the excellent flexibility of the developed pouch cell. | ||
Postmortem analysis
The lithiation mechanism of the g-C3N4-CNT/S cathode was explored through XPS analysis after the 250th discharge cycle. The pouch cell was dismantled in an argon-filled glovebox, and a 10 mm × 10 mm segment of the g-C3N4-CNT/S cathode was cut and dried before XPS measurements. Fig. 8 exhibits well-defined peaks for C, N, S, and O, consistent with the pure active material, revealing some new bond formations. Notably, additional elements (Li and F) were observed. In the C 1s spectra, peaks at 282.6 eV, 284.4, 286.6, and 288.3 eV corresponded to –C–C–, –CN–/–CC–, CO, and –S–CN, mirroring the active material's C 1s spectra in Fig. 2. However, a new peak at 292.5 eV, representing C–F due to electrolyte passivation, emerged. Deconvoluting the N 1s spectra revealed peaks at 397.5 eV for pyridinic-like N (–CN–) in the CNT structure, 399.1 eV for –S–NC–, and 403.6 for oxidized N (N–O).82 The 396.7 eV peak confirmed Li–N bond formation during cathode lithiation, contributing to lithium storage.83 Additionally, the 407.2 eV peak indicated nitrate (NO3−) presence on the cathode, formed by LiNO3 reduction during cycling and hindering active material reversibility.84 For S 2p, various reduction products and intermediates were identified after cycling including Li2S2–S at 164.5 eV, thiosulfates at 167.2 eV, sulfites at 168.0 eV, and S–F bonds at 169.1 eV.85 Compared to the S-spectra in Fig. 2, more sulfates were observed in the cycled cathode, which could result from oxidized S-species from the reaction between LiPSs and oxygen species. The O–S bond emerged due to sulfate species interacting with residual oxygen-containing species, and the S–F bond indicated strong covalent interaction between F-containing electrolytes and polysulfides.86 Interestingly, the doublet peaks at around 163 eV representing S 2p3/2 and S 2p1/2 for elemental sulfur were absent in the cycled cell. This agrees with the fact that a fully discharged cathode after 250 cycles should have all sulfur in the reduced state (Li2S/Li2S2) and the absence of a doublet peak confirms that no ‘dead’ sulfur was present on the cathode even after 250 cycles.87 F 1s peaks at 684.3 and 688.1 eV reflected electrolyte and PVDF passivation, suggesting g-C3N4-CNT/electrolyte interaction during cycling. In Li 1s spectra, peaks at 54.0, 55.0, 55.6, and 56.3 denoted Li–C, Li–S, Li–N, and Li–O bond formations, signifying CN bond involvement in lithium storage in addition to Li–S bonds during discharge.12,64,88 Overall, the XPS analysis of the cycled cell affirmed polysulfide adsorption on the g-C3N4-CNT surface.
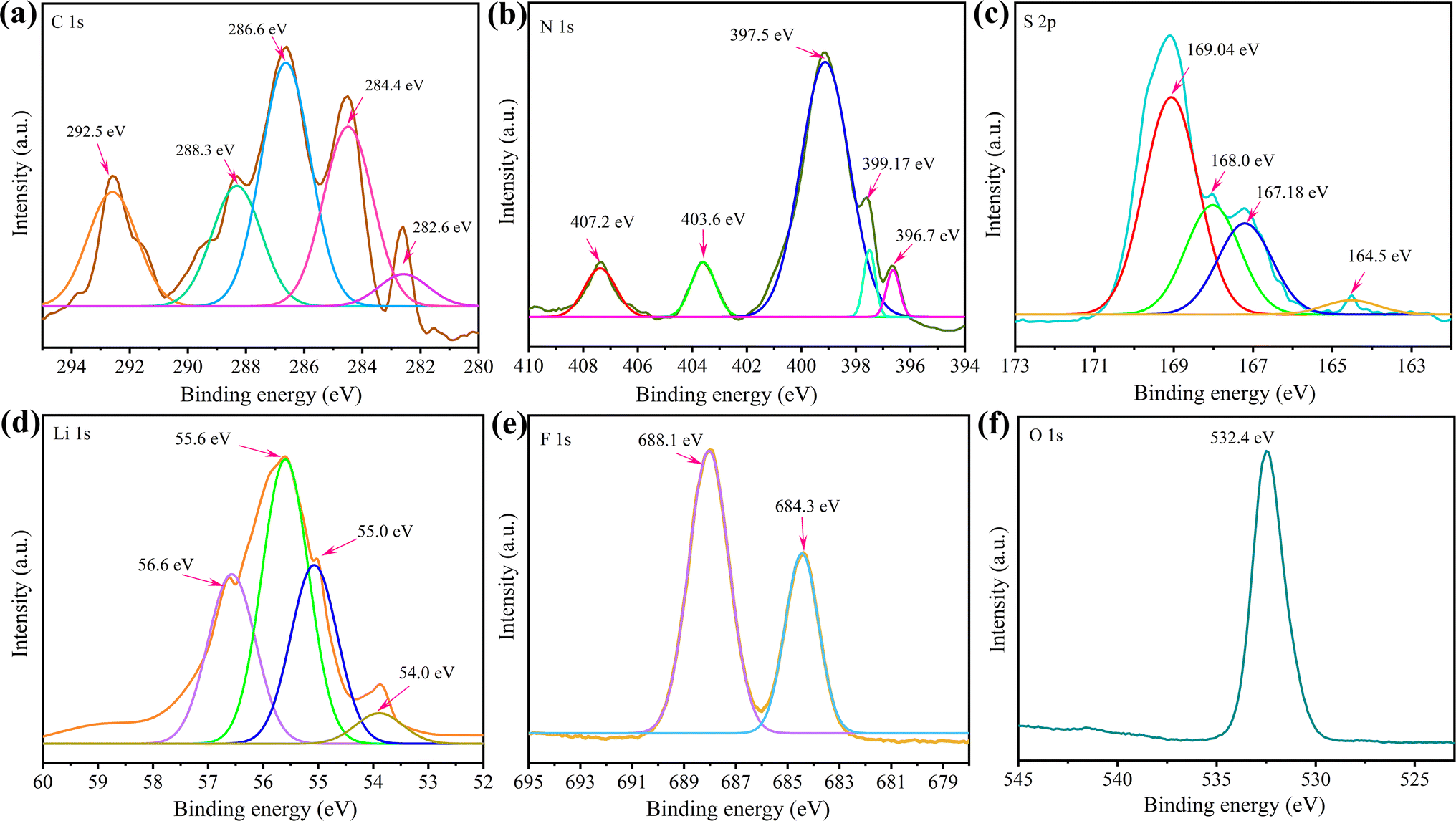 | ||
| Fig. 8 (a–f) XPS spectra of the g-C3N4-CNT/S cathode obtained from the pouch cell cycled for 250 charge–discharge cycles. | ||
To delve into the electrochemical kinetics of the cathodes, Fig. S5† displays the EIS results from the pouch cell cycled for 250 cycles (g-C3N4-CNT/S) and 110 cycles (g-C3N4/S) at 0.1C. As can be seen, after cycling, the Re of the cell with g-C3N4-CNT/S as the cathode increases from 2.7 to 5.2 Ω, notably lower than the cell with g-C3N4/S as the cathode (rising from 4.2 to 11.1 Ω), signifying restricted polysulfide diffusion from g-C3N4-CNT/S. Moreover, the reduced Rct for both cells (Table S5†) post-cycling suggests enhanced electrolyte penetration into the composite, shortening charge and ion transport paths.84,89 Another observed semicircle at low frequency for the g-C3N4/S cathode (the second semicircle) may indicate resistance from an insulating layer like Li2S (denoted as Rg), which incompletely converts to sulfur during charging.90 In contrast, the cell with g-C3N4-CNT/S as the cathode exhibits a single semicircle, signifying an efficient redox reaction. The impedance spectra affirm that the well-designed g-C3N4-CNT/S not only enhances sulfur cathode conductivity but also mitigates the shuttle effect by retaining more polysulfides within the cathode.
Conclusion
In summary, a hierarchical architecture, featuring 1D CNTs grown on 2D g-C3N4 sheets, is synthesized as a host matrix for a sulfur cathode in Li–S pouch cells. Experimental analyses reveal the g-C3N4-CNT/S composite's high pyridine N content, promoting robust interactions with LiPSs and inhibiting polysulfide shuttling, thereby enhancing electrochemical performance in Li–S batteries. DFT studies provide valuable insights through a comprehensive examination of the electronic bandgap, binding energy, and relative Gibbs free energy alterations upon the interaction of S8/LiPSs with both g-C3N4 and g-C3N4-CNT composite surfaces. While g-C3N4 exhibits increased bandgap values for Li2S and S8, indicative of reduced electronic conductivity, g-C3N4-CNT demonstrates a decrease in the bandgap, suggesting improved electronic properties. Additionally, changes in binding energy highlight distinct interactions between g-C3N4 and g-C3N4-CNT with S8/LiPSs. The relative Gibbs free energy landscape further highlights differences, with g-C3N4-CNT showcasing higher thermodynamic favorability in the discharging process compared to g-C3N4. These findings collectively contribute to a nuanced understanding of the material dynamics, emphasizing the potential benefits of g-C3N4-CNT in enhancing the electrochemical performance in S8/LiPS systems. As a result, the g-C3N4-CNT/S electrode achieves a capacity of 895 mA h g−1 at 0.1C with excellent high-rate, long-term cycling stability (0.053% capacity decay per cycle over 250 cycles). The electrode also attains a high discharge capacity of 566 mA h g−1 under a high sulfur loading of 6.2 mg cm−2 after 250 cycles, exhibiting a fading rate of 0.08% per cycle and flexibility during repeated bending. Postmortem XPS and EIS analyses further substantiate the excellent performance of the Li–S pouch cell by elucidating new bond formation and changes in cell resistance, respectively. The postmortem investigations from XPS show that the binding energy peaks of elemental S8 are missing from the cycled cell, which means that no dead sulfur on the cathode side was present even after the 250th cycle, thus revealing the excellent potential of the g-C3N4-CNT host matrix for the sulfur cathode. This work lays the foundation for the rational design of an ideal S host, enabling cathodes with multifunctional adsorption-catalytic sites for Li–S batteries with prolonged cycling life and high-rate capability.Author contributions
VKT: methodology, investigation, data curation, validation, and writing – original draft. RLK: DFT calculation and analysis. OTD: formal analysis and conceptualization. RM: methodology, data curation, and visualization. GCS: project administration and supervision. MS: conceptualization, review & editing, supervision, and project administration.Conflicts of interest
All the authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank MITACS (Canada), Ms Denise Byrne at the Powertrain Engineering Research & Development Centre (PERDC), and Ford Motor Company of Canada for providing financial and in-kind support for this project. RLK and GCS were supported by the Center for Molecular Quantum Transduction, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under award no. DE-SC0021314.References
- A. G. Olabi and M. A. Abdelkareem, Renewable Sustainable Energy Rev., 2022, 158, 112111 CrossRef CAS.
- R. Xu, L. Du, D. Adekoya, G. Zhang, S. Zhang, S. Sun, Y. Lei, R. Xu, Y. Lei, L. Du, G. Zhang, S. Sun, D. Adekoya and S. Zhang, Adv. Energy Mater., 2021, 11, 2001537 CrossRef CAS.
- M. Fichtner, K. Edström, E. Ayerbe, M. Berecibar, A. Bhowmik, I. E. Castelli, S. Clark, R. Dominko, M. Erakca, A. A. Franco, A. Grimaud, B. Horstmann, A. Latz, H. Lorrmann, M. Meeus, R. Narayan, F. Pammer, J. Ruhland, H. Stein, T. Vegge and M. Weil, Adv. Energy Mater., 2022, 12, 2102904 CrossRef CAS.
- A. G. Olabi, Q. Abbas, P. A. Shinde and M. A. Abdelkareem, Energy, 2023, 266, 126408 CrossRef CAS.
- C. P. Grey and D. S. Hall, Nat. Commun., 2020, 11, 1–4 CrossRef PubMed.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- G. Zhou, H. Chen and Y. Cui, Nat. Energy, 2022, 7, 312–319 CrossRef CAS.
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- Z. Han, S. Li, Y. Wu, C. Yu, S. Cheng and J. Xie, J. Mater. Chem. A, 2021, 9, 24215–24240 RSC.
- L. Huang, J. Li, B. Liu, Y. Li, S. Shen, S. Deng, C. Lu, W. Zhang, Y. Xia, G. Pan, X. Wang, Q. Xiong, X. Xia and J. Tu, Adv. Funct. Mater., 2020, 30, 1910375 CrossRef CAS.
- F. Zhao, J. Xue, W. Shao, H. Yu, W. Huang and J. Xiao, J. Energy Chem., 2023, 80, 625–657 CrossRef CAS.
- G. Gao, X. Yang, J. Bi, W. Guan, Z. Du and W. Ai, J. Mater. Chem. A, 2023, 11, 26318–26339 RSC.
- J. Wang and W. Q. Han, Adv. Funct. Mater., 2022, 32, 2107166 CrossRef CAS.
- D. Wang, J. Liu, X. Bao, C. Qing, T. Zhu and H. E. Wang, ACS Appl. Energy Mater., 2022, 5, 2573–2579 CrossRef CAS.
- Q. Zhao, X. Bao, L. Meng, S. Dong, Y. Zhang, C. Qing, T. Zhu and H. E. Wang, J. Colloid Interface Sci., 2023, 644, 546–555 CrossRef CAS PubMed.
- V. K. Tomer, O. A. T. Dias, A. Gouda, R. Malik and M. M. Sain, Mater. Horiz., 2024 10.1039/D4MH00200H.
- H. E. Wang, X. Li, N. Qin, X. Zhao, H. Cheng, G. Cao and W. Zhang, J. Mater. Chem. A, 2019, 7, 12068–12074 RSC.
- R. Zhe, T. Zhu, X. Wei, Y. Ren, C. Qing, N. Li and H. E. Wang, J. Mater. Chem. A, 2022, 10, 24422–24433 RSC.
- W. Sun, Z. Song, Z. Feng, Y. Huang, Z. J. Xu, Y. C. Lu and Q. Zou, Nano-Micro Lett., 2022, 14, 1–23 CrossRef PubMed.
- S. A. Thomas, M. R. Pallavolu, M. E. Khan and J. Cherusseri, J. Energy Storage, 2023, 68, 107673 CrossRef.
- Y. Wang, L. Liu, T. Ma, Y. Zhang and H. Huang, Adv. Funct. Mater., 2021, 31, 2102540 CrossRef CAS.
- W. Sun, L. Xu, Z. Song, H. Lin, Z. Jin, W. Wang, A. Wang and Y. Huang, Adv. Funct. Mater., 2023, 2313112 Search PubMed.
- Y. Li, Y. Deng, J. L. Yang, W. Tang, B. Ge and R. Liu, Adv. Funct. Mater., 2023, 33, 2302267 CrossRef CAS.
- H. Zou, Y. Zou, Y. Lv, Z. Ao, N. Chen and Y. Huang, ACS Appl. Energy Mater., 2022, 5, 10067–10075 CrossRef CAS.
- J. Wang, Z. Meng, W. Yang, X. Yan, R. Guo and W. Q. Han, ACS Appl. Mater. Interfaces, 2019, 11, 819–827 CrossRef CAS PubMed.
- J. Zhang, J. Y. Li, W. P. Wang, X. H. Zhang, X. H. Tan, W. G. Chu and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1702839 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- V. Wang, N. Xu, J. C. Liu, G. Tang and W. T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- D. Nepak, V. K. Tomer and K. Kailasam, in Metal-free Functionalized Carbons in Catalysis, ed. A. Villa and N. Dimitratos, Royal Society of Chemistry, UK, 2018, pp. 67–102 Search PubMed.
- R. Malik, N. Joshi and V. K. Tomer, Coord. Chem. Rev., 2022, 466, 214611 CrossRef CAS.
- R. Malik, V. K. Tomer, T. Dankwort, Y. K. Mishra and L. Kienle, J. Mater. Chem. A, 2018, 6, 10718–10730 RSC.
- V. K. Tomer, R. Malik and V. Chaudhary, Appl. Mater. Today, 2019, 16, 193–203 CrossRef.
- L. P. Yu, L. Xu, C. Shearer, X. H. Zhou and L. Lu, Adv. Sustainable Syst., 2023, 7, 2300061 CrossRef CAS.
- R. Malik, V. K. Tomer, N. Joshi, T. Dankwort, L. Lin and L. Kienle, ACS Appl. Mater. Interfaces, 2018, 10, 34087–34097 CrossRef CAS PubMed.
- R. Malik, V. K. Tomer, V. Chaudhary, M. S. Dahiya, A. Sharma, S. P. Nehra, S. Duhan and K. Kailasam, J. Mater. Chem. A, 2017, 5, 14134–14143 RSC.
- R. Malik and V. K. Tomer, Renewable Sustainable Energy Rev., 2021, 135, 110235 CrossRef CAS.
- Y. Dong, H. Pang, H. Bin Yang, C. Guo, J. Shao, Y. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- V. K. Tomer, N. Thangaraj, S. Gahlot and K. Kailasam, Nanoscale, 2016, 8, 19794–19803 RSC.
- Z. Li, L. Deng, I. A. Kinloch and R. J. Young, Prog. Mater. Sci., 2023, 135, 101089 CrossRef CAS.
- X. Wu, S. Li, B. Wang, J. Liu and M. Yu, Renewable Energy, 2020, 158, 509–519 CrossRef CAS.
- M. Wang, Z. Bai, T. Yang, C. Nie, X. Xu, Y. Wang, J. Yang, S. Dou and N. Wang, Adv. Energy Mater., 2022, 12, 2201585 CrossRef CAS.
- P. Chen, Z. Wu, T. Guo, Y. Zhou, M. Liu, X. Xia, J. Sun, L. Lu, X. Ouyang, X. Wang, Y. Fu and J. Zhu, Adv. Mater., 2021, 33, 2007549 CrossRef CAS PubMed.
- G. S. Priyanga, G. Pransu and S. Sampath, Chem. Phys. Impact, 2024, 8, 100408 CrossRef.
- X. Ma, Y. Lv, J. Xu, Y. Liu, R. Zhang and Y. Zhu, J. Phys. Chem. C, 2012, 116, 23485–23493 CrossRef CAS.
- R. Li, H. Peng, Q. Wu, X. Zhou, J. He, H. Shen, M. Yang and C. Li, Angew. Chem., Int. Ed., 2020, 59, 12129–12138 CrossRef CAS PubMed.
- Y. Yan, P. Zhang, Z. Qu, M. Tong, S. Zhao, Z. Li, M. Liu and Z. Lin, Nano Lett., 2020, 20, 7662–7669 CrossRef CAS PubMed.
- G. Wang, F. Li, D. Liu, D. Zheng, C. J. Abeggien, Y. Luo, X. Q. Yang, T. Ding and D. Qu, Energy Storage Mater., 2020, 24, 147–152 CrossRef.
- H. Wu, H. Jiang, Y. Yang, C. Hou, H. Zhao, R. Xiao and H. Wang, J. Mater. Chem. A, 2020, 8, 14498–14505 RSC.
- M. A. Weret, C. F. J. Kuo, W. N. Su, T. S. Zeleke, C. J. Huang, N. A. Sahalie, T. A. Zegeye, Z. T. Wondimkun, F. W. Fenta, B. A. Jote, M. C. Tsai and B. J. Hwang, J. Power Sources, 2022, 541, 231693 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, 1992 Search PubMed.
- Y. Xu, Y. Wen, Y. Zhu, K. Gaskell, K. A. Cychosz, B. Eichhorn, K. Xu and C. Wang, Adv. Funct. Mater., 2015, 25, 4312–4320 CrossRef CAS.
- Y. Hu, A. Hu, J. Wang, X. Niu, M. Zhou, W. Chen, T. Lei, J. Huang, Y. Li, L. Xue, Y. Fan, X. Wang and J. Xiong, J. Mater. Chem. A, 2021, 9, 9771–9779 RSC.
- C. Zhao, B. Jiang, Y. Huang, X. Sun, M. Wang, Y. Zhang and N. Zhang, Energy Environ. Sci., 2023, 16, 5490–5499 RSC.
- S. D. S. Fitch, G. E. Moehl, N. Meddings, S. Fop, S. Soulé, T. L. Lee, M. Kazemian, N. Garcia-Araez and A. L. Hector, ACS Appl. Mater. Interfaces, 2023, 15, 39198–39210 CrossRef CAS PubMed.
- Z. Wang, J. Shen, J. Liu, X. Xu, Z. Liu, R. Hu, L. Yang, Y. Feng, J. Liu, Z. Shi, L. Ouyang, Y. Yu and M. Zhu, Adv. Mater., 2019, 31, 1902228 CrossRef PubMed.
- C. Tian, P. Li, X. Hu, W. Yan, X. Xiang and L. Lu, Small, 2023, 2306928 Search PubMed.
- S. F. Ng, M. Y. L. Lau and W. J. Ong, Adv. Mater., 2021, 33, 2008654 CrossRef CAS PubMed.
- M. Zhang, W. Chen, L. Xue, Y. Jiao, T. Lei, J. Chu, J. Huang, C. Gong, C. Yan, Y. Yan, Y. Hu, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
- K. Liao, P. Mao, N. Li, M. Han, J. Yi, P. He, Y. Sun and H. Zhou, J. Mater. Chem. A, 2016, 4, 5406–5409 RSC.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- A. Hu, M. Zhou, T. Lei, Y. Hu, X. Du, C. Gong, C. Shu, J. Long, J. Zhu, W. Chen, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2002180 CrossRef CAS.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 RSC.
- M. Liu, Q. Li, X. Qin, G. Liang, W. Han, D. Zhou, Y. B. He, B. Li and F. Kang, Small, 2017, 13, 1602539 CrossRef PubMed.
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- S. J. Kim, Y. Jeoun, J. Park, S. H. Yu and Y. E. Sung, Nanoscale, 2020, 12, 15466–15472 RSC.
- J. Shen, X. Xu, J. Liu, Z. Liu, F. Li, R. Hu, J. Liu, X. Hou, Y. Feng, Y. Yu and M. Zhu, ACS Nano, 2019, 13, 8986–8996 CrossRef CAS PubMed.
- L. Ding, Q. Lu, A. D. C. Permana, S. Oswald, M. Hantusch, K. Nielsch and D. Mikhailova, Energy Technol., 2021, 9, 2001057 CrossRef CAS.
- Y. Xie, J. Ao, L. Zhang, Y. Shao, H. Zhang, S. Cheng and X. Wang, Chem. Eng. J., 2023, 451, 139017 CrossRef CAS.
- D. Wang, K. Ma, J. Hao, W. Zhang, C. Wang, C. Xu, H. Shi, Z. Ji, X. Yan and Y. Gu, Nano Energy, 2021, 89, 106426 CrossRef CAS.
- H. Zhang, Q. Liu, S. Ruan, C. Ma, X. Jia, W. Qiao, L. Ling and J. Wang, Appl. Surf. Sci., 2022, 578, 152022 CrossRef CAS.
- Y. Hu, W. Chen, T. Lei, Y. Jiao, J. Huang, A. Hu, C. Gong, C. Yan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- L. Shi, S. M. Bak, Z. Shadike, C. Wang, C. Niu, P. Northrup, H. Lee, A. Y. Baranovskiy, C. S. Anderson, J. Qin, S. Feng, X. Ren, D. Liu, X. Q. Yang, F. Gao, D. Lu, J. Xiao and J. Liu, Energy Environ. Sci., 2020, 13, 3620–3632 RSC.
- S. Dörfler, H. Althues, P. Härtel, T. Abendroth, B. Schumm and S. Kaskel, Joule, 2020, 4, 539–554 CrossRef.
- D. Qu, G. Wang, J. Kafle, J. Harris, L. Crain, Z. Jin, D. Zheng, D. Qu, G. Wang, J. Kafle, J. Harris, L. Crain, D. Zheng and Z. Jin, Small Methods, 2018, 2, 1700342 CrossRef.
- J. T. Kim, A. Rao, H. Y. Nie, Y. Hu, W. Li, F. Zhao, S. Deng, X. Hao, J. Fu, J. Luo, H. Duan, C. Wang, C. V. Singh and X. Sun, Nat. Commun., 2023, 14, 1–10 Search PubMed.
- T. Pakki, E. H. Mohan, N. Y. Hebalkar, J. Adduru, S. V. Bulusu, A. Srinivasan, K. M. Mantravadi and N. R. Tata, J. Mater. Sci., 2019, 54, 9075–9087 CrossRef CAS.
- R. Li, Y. Zeng, L. Song, J. Lv, C. Wang, C. Zhou, S. Cai, T. Chen, S. Yue, K. Ma and H. Yue, Small, 2023, 2305283 Search PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A920–A923 CrossRef CAS.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Electrochem. Soc., 2012, 159, A1816–A1821 CrossRef CAS.
- S. Ponnada, M. S. Kiai, D. B. Gorle and A. Nowduri, Nanoscale Adv., 2021, 3, 4492–4501 RSC.
- X. Gao, C. Zheng, Y. Shao, V. R. Shah, S. Jin, J. Suntivich and Y. L. Joo, ACS Appl. Mater. Interfaces, 2023, 15, 19011–19020 CrossRef CAS PubMed.
- Y. Hu, W. Chen, T. Lei, Y. Jiao, J. Huang, A. Hu, C. Gong, C. Yan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- H. Pan, K. S. Han, M. H. Engelhard, R. Cao, J. Chen, J.-G. Zhang, K. T. Mueller, Y. Shao, J. Liu, H. Pan, K. S. Han, R. Cao, J. Chen, J.-G. Zhang, K. T. Mueller, Y. Shao, J. Liu and M. H. Engelhard, Adv. Funct. Mater., 2018, 28, 1707234 CrossRef.
- Z. Meng, S. Li, H. Ying, X. Xu, X. Zhu, W.-Q. Han, Z. Meng, H. Ying, X. Xu, X. Zhu, S. Li and Q. W. Han, Adv. Mater. Interfaces, 2017, 4, 1601195 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Gibbs free energy of the sulfur reduction reaction (SRR); tables illustrating bandgap and binding energy values for S8/LiPSs calculated from DFT analysis, electrochemical performance comparison with previously published studies; BET sorption isotherms; TGA; CV cycling performance; complete XPS scan and EIS spectra for the cycled cell. See DOI: https://doi.org/10.1039/d4ta00918e |
| This journal is © The Royal Society of Chemistry 2024 |