
Studies on bimetallic Cu–Ag supported alumina catalysts for hydrodeoxygenation of 5-hydroxymethylfurfural to 2,5-dimethylfuran†‡
D. Dhana
Lakshmi
ab,
Yogita
 ab,
B.
Srinivasa Rao
a and
N.
Lingaiah
*ab
ab,
B.
Srinivasa Rao
a and
N.
Lingaiah
*ab
aDepartment of Catalysis and Fine Chemicals, CSIR-Indian Institute of Chemical Technology, Hyderabad 500007, India. E-mail: nakkalingaiah@iict.res.in; Tel: +91-40-27191722
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
First published on 7th November 2023
Abstract
Selective synthesis of 2,5-dimethylfuran (DMF) from 5-hydroxymethyl furfural (HMF) by hydrodeoxygenation (HDO) was studied over Ag-promoted Cu/γ-Al2O3 catalysts. A series of bimetallic Ag–Cu supported on γ-Al2O3 catalysts with varying Ag amounts were prepared and characterized using different techniques. The results indicated that the addition of Ag to Cu/γ-Al2O3 improved the dispersion of Cu in the catalyst, and the activity of Cu–Ag catalysts was dependent on the amount of Ag present. The catalyst with 2 wt% Ag-10% Cu exhibited a highest DMF yield of 93% with the complete conversion of HMF at 180 °C, and its activity was well correlated with its characteristics. The study also established the optimum reaction conditions for these catalysts and demonstrated their good reusability upon recycling. These findings provide valuable insights into the design and development of efficient and sustainable catalytic processes for the production of DMF from HMF.
Introduction
The overconsumption of fossil fuels creates several problems related to the environment and energy. Approximately, 80% of the world energy consumption is from fossil resources. This fact displays the world's heavy dependence on these limited fuel stocks. As a result, the reserved fossil raw materials are depleting very fast and will not be enough to compete with the increasing demands for energy of the upcoming world due to the growing population and usage. Heavy use of fossil fuels is alarming the world due to environmental ill-issues like greenhouse gas emissions and air pollution.1–4 Therefore, the use of fuels obtained from renewable resources instead of non-renewable resources has received remarkable attention globally. Biomass is receiving significant interest for the production of biofuels and value-added chemicals. Different chemicals, fuels, and fuel additives can be generated from biomass.5,6 Lignocellulose biomass is a rich source of carbohydrates, and different platform chemicals can be derived from them. Among the various valuable chemicals, 5-hydroxymethylfurfural (HMF), a good building block, can be synthesized from biomass-derived carbohydrates. Further, HMF can be converted to various beneficial fine chemicals by hydrogenation, esterification, etherification, and oxidation. Many pharmaceutical, fragrance, and polymer industries use the fine chemicals derived from HMF.7–102,5-Dimethylfuran, a product of HMF selective hydrogenation, has the potential to become an alternate biofuel. The first-generation biofuels, bioethanol, and biodiesel have been widely used in the transportation sector. Still, their production competes with the world's food security, and their drawbacks, like poor cold flow properties and destructive nature, limit their use in transportation fuels. On the other hand, biomass-derived DMF is uncompetitive with the food-supply chain and has beneficial physio-chemical properties, like high density and high research octane number, which make it far better than bio-ethanol and even gasoline. Also, its low oxygen-to-carbon ratio and low solubility in water enabled its use as a blended fuel, and its kinetic viscosity is almost similar to gasoline.11–14 These advantageous properties make DMF a promising alternative to fossil-based energy sources. Apart from this, DMF also finds its use as a precursor for the production of p-xylene, producing chemicals for pharmaceuticals, engineering polymers, and semiconductor manufacturing industries.15–17
Efficient metal catalysts, such as Ru, Pd, Ir, Pt, and Au, are commonly employed for the hydrogenation of HMF to DMF.18–23 However, these catalysts are expensive, making them economically impractical for industrial applications. To address this issue, researchers have explored alternative approaches. In a study conducted by Tzeng et al., Ru supported on carbon was utilized as a catalyst for converting HMF to DMF, achieving a DMF yield of 69% with complete conversion of HMF. This approach offers a more cost-effective solution.24 Deng's group also developed a promising strategy by synthesizing MOFs with Pd acid interfaces. These MOFs exhibited improved selectivity towards DMF yield compared to traditional Pd catalysts supported by MOFs.25 Researchers have also investigated the use of transition metals and noble metals to generate Lewis acidic sites that activate the hydroxyl and aldehyde groups of HMF, facilitating selective hydrogenation and dehydration of the –CH2OH group.26 For instance, a bimetallic catalyst composed of Pt and Fe supported on activated carbon demonstrated enhanced Pt dispersion and Lewis acidic density, resulting in a DMF yield of 91%.27 In a study led by Gan et al., Pt/Co single atom alloy catalysts were synthesized using a ball milling method. These catalysts were stable over five catalytic runs, offering potential for practical applications.28 Similarly, Pt supported on Ni3Fe single atom catalysts achieved an impressive 98% DMF yield within 1.5 h at 160 °C, with the Ni3Fe interface promoting C–OH bond cleavage and Pt contributing to hydrogenation activity.29 A dual metal acid catalyst, Pd–Br, obtained a high DMF yield of 96% at an ambient temperature of 30 °C.30 Although a sulphur-modified carbon nanotube catalyst with Pd–Co demonstrated successful DMF synthesis at 120 °C, the use of sulphur is undesirable due to its corrosive nature.31
Recently, researchers have focused on transition metals like Cu, Co, Ni, and Fe for the hydrogenation of HMF without using any precious metal to make it more economical. Zhao et al. developed a bimetallic Cox–Cu@C catalyst through the pyrolysis of MOFs. This catalyst demonstrated efficient activity for the hydrogenation of HMF to DMF. The reaction conditions were mild, with a temperature of 160 °C and a reaction time of 3 h, resulting in a high DMF yield of 85%. The catalyst activity tests revealed that the presence of CoOx in the catalyst effectively activated the C–O bond and enhanced the hydrogenation activity. The exceptional catalytic performance of the Co2O–Cu@C catalyst in HMF hydrogenation can be attributed to the synergistic effect between Cu and CoOx, which enhances its overall catalytic activity.32 Furthermore, bimetallic CuFe nanoparticles coated with thin carbon layers were developed as a potential alternative to noble metal catalysts. These nanoparticles have been successfully employed for the chemo-selective hydrogenation of HMF to 2,5-bis(hydroxymethyl)furan (BHMF). Compared to Cu catalysts supported on conventional solid carriers prepared through impregnation, the CuFe@C nanoparticles exhibited higher activity and improved stability, making them promising catalysts for the reaction.33 Z. An et al. reported Co catalyst derived from LDH for active hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds.34 Ni and Cu-based catalysts have emerged as an effective catalyst for synthesizing DMF effectively.35,36 The Cu metal is highly reactive to CO and possesses a poor affinity for CC bonds. It is also less expensive and more environmentally benign.37 L. M. Esteves et al. studied Cu-supported catalysts and observed that both metallic and Lewis acidic sites play a crucial role in activating hydrogenation sites and the oxygen atom of a hydroxyl group of HMF, respectively.38
O and C–O bonds.34 Ni and Cu-based catalysts have emerged as an effective catalyst for synthesizing DMF effectively.35,36 The Cu metal is highly reactive to CO and possesses a poor affinity for CC bonds. It is also less expensive and more environmentally benign.37 L. M. Esteves et al. studied Cu-supported catalysts and observed that both metallic and Lewis acidic sites play a crucial role in activating hydrogenation sites and the oxygen atom of a hydroxyl group of HMF, respectively.38
In this work, Ag-spromoted Cu supported on γ-Al2O3 catalysts were synthesized and used for selective hydrogenation of HMF to DMF. The synergy between Cu and Ag on the surface–structural properties and their influence on HMF hydrogenation was one of the aims of the study. The influence of reaction parameters is studied in detail to derive optimum reaction conditions.
Experimental
The chemicals DMF, DMTHF, 2-methyl, 5-hydroxymethylfuran (MFA), BHMF, MF, 2,5-hexane diol, 2,5-hexane dione, 2-hexanol, and THF were purchased from Sigma-Aldrich. HMF was acquired from Alfa-Aesar. All the chemicals were used as received without any further purification.Catalyst preparation
Silver containing Cu/γ-Al2O3 catalysts were prepared through the impregnation method. In brief, Cu(NO3)2·3H2O and AgNO3 were taken as metal precursors. The calculated amounts of aqueous solutions with the desired quantity of metal precursor were added drop by drop onto the support, γ-Al2O3. The resulting mixture was aged at 60 °C for 2 h, and a rotary evaporator was used to remove the excess water. Further, these dried catalyst masses were calcined at 450 °C for 4 h in atmospheric air. For all the catalysts, Cu loading was fixed to 10 wt%, and Ag loading was varied from 0.5 to 10 wt% The prepared catalysts were labelled as CAA-X, where X indicates the wt% of Ag loaded on γ-Al2O3. For relative analysis, 10%Cu/γ-Al2O3 (CA) and 2%Ag/γ-Al2O3 (AA) were also prepared by the same procedure as mentioned above. All the prepared catalysts were reduced at 350 °C for 2 h in the incidence of 35 mL min−1 hydrogen flow and the samples were cooled in the presence of N2 flow. These reduced catalysts are used for the evaluation of activity.Catalyst characterization
All the prepared catalysts were characterized with X-ray diffraction (XRD), Brunauer–Emmett–Teller surface area analysis (BET surface area), N2O chemisorption, temperature programmed desorption-NH3 (TPD-NH3), scanning electron microscopy-energy dispersive X-ray spectroscopy (SEM-EDX), transmission electron microscopy (TEM), H2-temperature programmed reduction (H2-TPR), thermo gravimetric analysis (TGA), inductively coupled plasma optical emission spectroscopy (ICP-OES), and X-ray photoelectron spectroscopy (XPS). The equipment used for these investigations and their detailed analytical conditions are provided in the ESI.†Catalytic activity measurement
All the reactions were carried out in batch mode using a Parr autoclave reactor. The reactor was fed with 2 mmol of HMF dissolved in 20 mL THF solvent and the required amount of freshly reduced catalyst (0.05–0.25 g). Before the reaction, the reactor was flushed with H2 gas two to three times to remove the air and pressurized to the required pressure from 0.5 to 2.5 MPa. The reactor was heated to mandatory reaction temperature (140–220 °C), and the reaction was performed for a particular time with a constant stirring rate of 600 rpm. After the reaction, the reactor temperature was brought to 30 °C and depressurized. The catalyst was filtered, washed with methanol, dried at 80 °C for at least 4 h, and then used for the next run. Gas chromatography equipped with an Inno-Wax column was used to analyze the reaction mixture. Three injections were conducted for each reaction, and the error was set to not more than 1%.HMF conversion, product yield/selectivity, and the carbon balance were calculated by using the equations below.
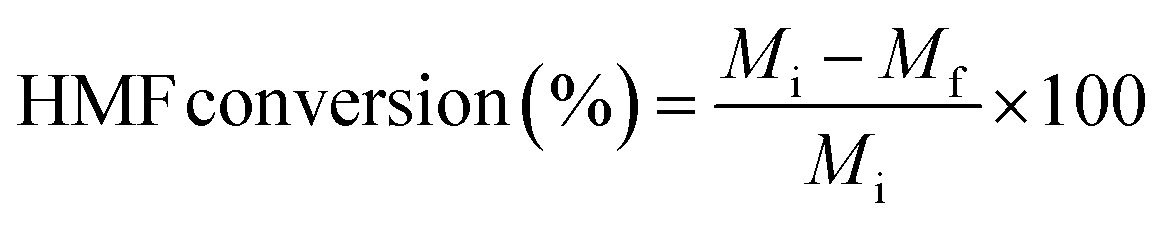
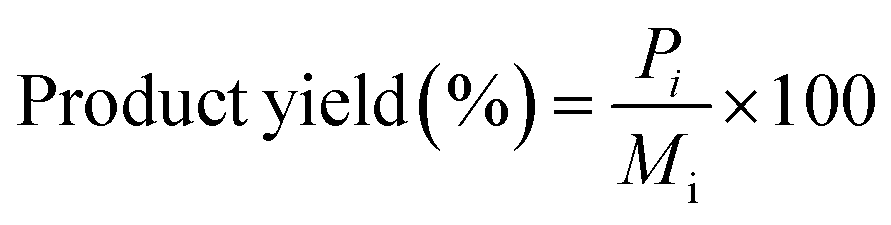

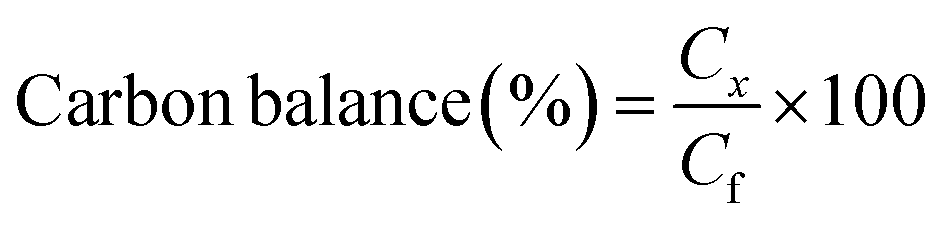
M
i is the initial moles of HMF, Mf is the final moles of HMF, Pi refers to moles of product i formed, Pt is the moles of total product formed, Cx is the number of mole carbons of product x, and Cf is the total number of carbon moles formed.
| ProductivityDMF = (mmol DMF formed/gram cat./time) |
Results and discussion
ICP-OES analysis
Table S1† presents the elemental compositions of the catalysts determined using inductively coupled plasma atomic emission spectroscopy (ICP-OES). It also includes a comparison between the theoretical and experimental Cu and Ag loadings. The results confirm that Cu and Ag were effectively impregnated onto γ-Al2O3, with the measured stoichiometries of Cu and Ag being slightly lower than their expected values, indicating a successful impregnation process.N2-physiosorption studies
The N2 adsorption–desorption isotherms of the prepared catalysts are exposed in Fig. 1, and the corresponding results of textural properties are revealed in Table 1. The BET method was utilized to calculate the total surface area of the catalysts based on the amount of N2 gas molecules adsorbed. The pore volume was also evaluated, which provided a better understanding of the metal occupation within the support. The isotherms of the γ-Al2O3 support and the bimetallic Cu–Ag supported on γ-Al2O3 catalysts exhibited a hysteresis loop of type IV owing to capillary condensation, suggesting a mesoporous structure. Specific surface area (SBET), pore volume (Vp), and average pore size diameter (Dp) of γ-Al2O3 were 213 m2 g−1, 0.57 cm3 g−1, and 16.06 nm, respectively. However, the values of these parameters contracted in the prepared catalysts due to the deposition of metal oxides on the surface of the support. The pores were found to be clogged with metal oxide species, thereby decreasing their surface area and pore volume. When 10% Cu was deposited on γ-Al2O3, the surface area decreased drastically to 162 m2 g−1 with only a marginal reduction in pore volume. On the other hand, the addition of 2% Ag to the support showed no significant effect on the surface area, but it decreased the pore volume to 0.28 cm3 g−1. This suggests that the Ag particles are more selectively deposited inside the pores, while most of the copper particles are coated on the surface. While adding Ag metal to the 10%Cu/γ-Al2O3 catalyst, the surface areas are increased up to 2 wt% of Ag. However, with an increase in Ag loading, the surface area and pore volume decreased. The pore diameters of 10%Cu/γ-Al2O3 and Ag-introduced catalysts were found to be up to 12.26 nm. The introduction of Ag into the pores of 10%Cu/γ-Al2O3 did not alter the pore diameter, indicating that the Ag species do not affect the number of pores in the catalyst but decrease the number of active sites in the pores. The pore size distribution of the prepared catalysts was calculated from the BJH curves. The most probable distribution of pore sizes across all catalysts was found to be less than 10 nm, suggesting that all the catalysts have uniform pore sizes. The increased particle size of the catalysts with above 2 wt% Ag could be due to the formation of agglomerated clusters.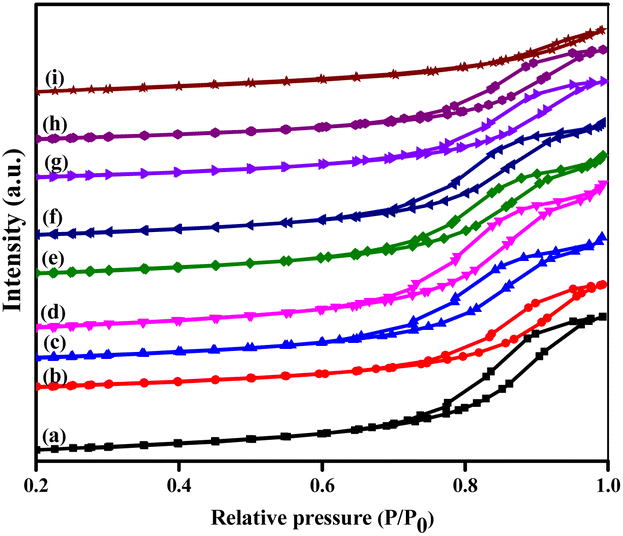 | ||
| Fig. 1 N2 adsorption–desorption isotherms of the prepared catalysts: (a) γ-Al2O3, (b) CA, (c) CAA-0.5, (d) CAA-1, (e) CAA-2, (f) CAA-5, (g) CAA-7, (h) CAA-10, and (i) AA. | ||
| Catalyst | S BET (m2 g−1) | V P (cm3 g−1) | D P (nm) | Cu particleb size (nm) | Ag particleb size (nm) |
|---|---|---|---|---|---|
| a BET surface area analysis. b XRD, nd-not done. | |||||
| γ-Al2O3 | 213 | 0.6 | 10.9 | nd | nd |
| AA | 201 | 0.3 | 6.2 | nd | 0.8 |
| CA | 162 | 0.4 | 11.0 | 5.6 | nd |
| CAA-0.5 | 168 | 0.5 | 12.2 | 6.8 | nd |
| CAA-1 | 197 | 0.6 | 12.2 | 6.9 | nd |
| CAA-2 | 167 | 0.5 | 11.9 | 7.2 | 0.9 |
| CAA-5 | 161 | 0.4 | 11.9 | 8.9 | 1.4 |
| CAA-7 | 153 | 0.40 | 11.0 | 9.2 | 1.5 |
| CAA-10 | 143 | 0.3 | 10.9 | 11.6 | 3.1 |
Powder XRD
Powder X-ray diffraction (XRD) was used to investigate the crystallinity of Cu–Ag/γ-Al2O3 catalysts. XRD diffractograms of the calcined and reduced samples are presented in Fig. 2a and b, respectively. The calcined catalysts (Fig. 2a) exhibited sharp diffraction peaks at 2θ = 35.6°, 38.7°, 48.6°, 61.5°, and 72.3°, corresponding to the crystalline copper oxide (JCPDS 80-1917, 45-0937).39 The broad peaks observed at 2θ = 37°, 46°, and 68° in both calcined and reduced samples represent the reflection of γ-Al2O3 (JCPDS: 86-1410). The XRD diffraction at 2θ = 33.76°, 38.12°, and 55° were assigned to the (111), (200), and (220) cubic planes of Ag2O, respectively. Furthermore, the XRD patterns of the reduced catalysts showed three peaks at 2θ = 44.3°, 50.4°, and 74.1°, corresponding to (111), (200), and (220) planes of metallic copper (JCPDS: 04-0836). The reflections at 2θ = 38.1°, 44.3°, 64.5°, and 78° indicated the existence of larger metallic Ag crystallites at Ag loading higher than 2 wt%. In addition, a shift in the lower angle of the diffraction peaks at 43.29° in reduced catalysts suggests the formation of Cu–Ag alloy.40 The formation of these phases could be influenced by the relative amounts of Cu and Ag in the catalyst. The XRD results suggest that the catalysts with Ag content up to 2 wt% only exhibited Cu metal, and with an increase in Ag content, the Cu–Ag alloy formed along with Ag metal. The absence of diffraction peaks attributed to Ag2O and metallic Ag at lower Ag loadings (0.5–2 wt%) suggests the presence of small crystal sizes beneath the XRD detection limit. The crystal size of the Cu and Ag metals was calculated using the Scherer equation and is presented in Table 1.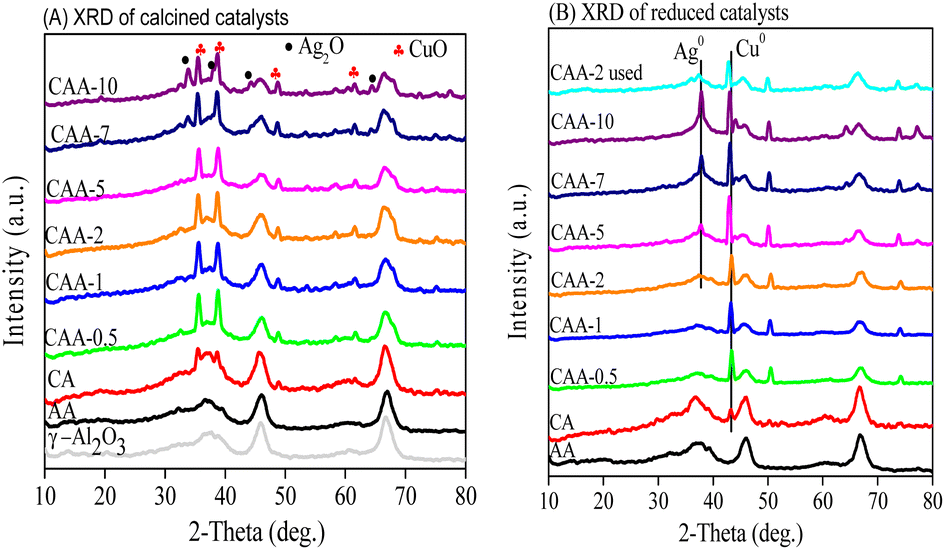 | ||
| Fig. 2 XRD patterns of the prepared catalysts (a) before and (b) after reduction. | ||
H2-TPR studies
The H2-TPR method was used to study the interaction between the metal support and the reduction temperature of catalysts. Fig. 3 shows the reduction profiles of different catalysts. The 10%Cu/γ-Al2O3 catalyst had two reduction peaks at 290 °C and 530 °C. The low-temperature peak is due to finely dispersed copper weakly interacting with the support, while the high-temperature peak is due to aggregated copper molecules strongly interacting with CuO on the support. The 2%Ag/γ-Al2O3 catalyst had almost no reduction peak due to the metallic state of Ag in the fresh catalyst, which was confirmed by XRD results. The TPR profiles of Ag–Cu/γ-Al2O3 catalysts also showed two reduction peaks, which were shifted to lower temperatures compared to the 10%Cu/γ-Al2O3 catalyst. With an increasing Ag loading, the lower reduction peak moved to even lower temperatures, and the higher reduction peak shifted to higher temperatures. The shift in the TPR peaks with an increasing Ag loading can be attributed to the interaction between Ag and Cu, as well as the interaction of both metals with the support. At lower Ag loadings, the Ag particles anchor Cu on the support surface, increasing the dispersion of Cu and its interaction with the support. As a result, the lower reduction peak shifts to lower temperatures due to the more finely dispersed Cu species. At the same time, the higher reduction peak shifts to higher temperatures because of the stronger interaction between Cu and the support, which requires more energy for reduction. As the Ag loading increases further, the Ag particles start to aggregate on the support surface, leading to a decrease in the dispersion of Cu species and their interaction with the support. At the highest Ag loading (10%), the Ag particles dominate the surface, leading to the formation of larger particles and a shift towards higher temperatures. Overall, the shift in the TPR peaks with increasing Ag loading can be attributed to changes in the dispersion of Cu species and their interaction with the support as a result of the presence of Ag particles.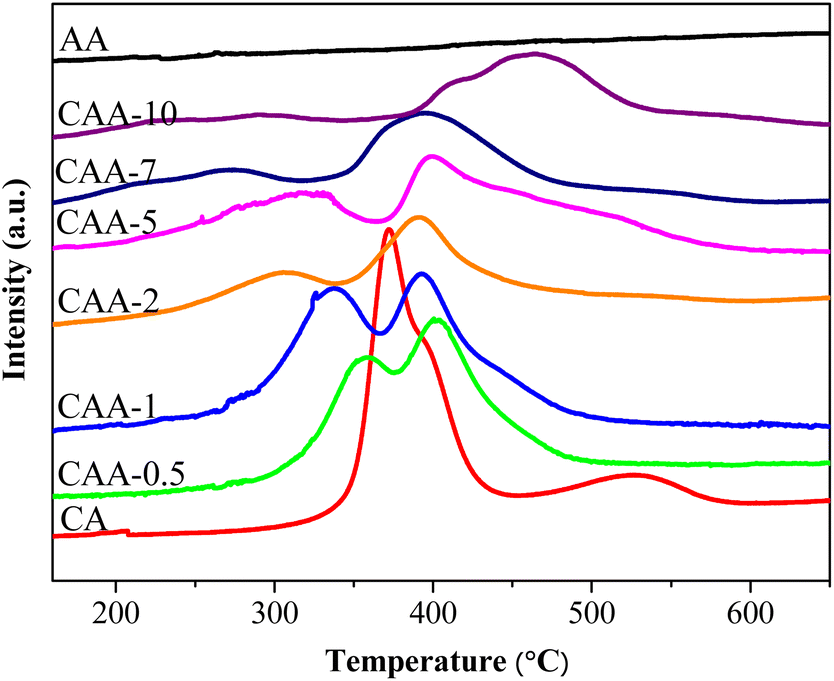 | ||
| Fig. 3 H2-TPR profiles of the prepared catalysts. | ||
NH3-temperature-programmed desorption
The surface acidity in Cu–Ag catalysts supported on γ-Al2O3 was measured using the NH3-TPD method, as shown in Fig. 4 and Table 2. The desorption temperature of NH3 was used to distinguish the strength of the acid site, with weaker acid sites resulting in desorption peaks at lower temperatures and stronger acid sites resulting in desorption peaks at higher temperatures.41 The NH3-TPD profiles of the catalysts exhibited two desorption peaks (Fig. 4), with one at approximately 200 °C indicating weak acidic sites, such as weak Lewis acid sites, and the other at 300–600 °C corresponding to moderate/strong acidic sites, such as strong Lewis acid/Brønsted acid sites. The observed peaks in the high-temperature range, spanning from the moderate acid region to the strong acid region, indicate the presence of strong acidic sites.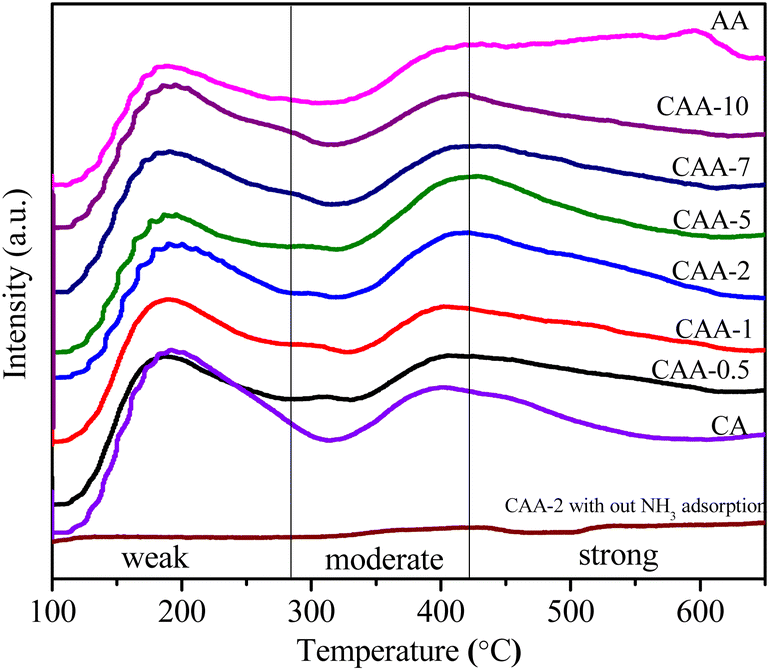 | ||
| Fig. 4 NH3-TPD profiles of the catalysts. | ||
| Catalyst | Weak acidity (mmol g−1) | Moderate/strong acidity (mmol g−1) | W/S | Total acidity (mmol g−1) |
|---|---|---|---|---|
| CA | 1.31 | 0.61 | 2.14 | 1.92 |
| AA | 0.58 | 0.45 | 1.28 | 1.03 |
| CAA-0.5 | 0.76 | 0.34 | 2.23 | 1.10 |
| CAA-1 | 0.71 | 0.42 | 1.69 | 1.13 |
| CAA-2 | 0.62 | 0.58 | 1.06 | 1.20 |
| CAA-5 | 0.61 | 0.73 | 0.83 | 1.34 |
| CAA-7 | 0.92 | 0.41 | 2.24 | 1.33 |
| CAA-10 | 1.12 | 0.38 | 2.94 | 1.50 |
| CAA-2 no NH3 | 0.008 | 0.012 | — | 0.02 |
The catalyst with 10%Cu/γ-Al2O3 displayed high acidity, but the addition of Ag resulted in decreased acidity in the Ag-containing catalysts. This decrease could be attributed to large Ag metal clusters covering the acid sites on the surface of the catalysts. Furthermore, the Ag content may influence the distribution and accessibility of active sites, potentially leading to changes in the distribution of weak/strong acid sites. The ratios of weak to strong acid site densities were also calculated and are presented in Table 2. These ratios showed that the density of weak to strong acid sites decreased with the increase in Ag content. Moreover, an equal portion of weak and strong acid sites were observed for the CAA-2 catalyst. Additionally, we performed a TPD analysis of CAA-2 catalyst in the absence of ammonia, where we observed almost a baseline profile with no desorption peaks. Overall, the NH3-TPD results provide insight into the surface acidity of the bimetallic Cu–Ag catalysts and suggest that the Ag content can significantly affect the acidic properties of the catalysts.
Morphology
The HR-TEM analysis of three different catalysts – CA, CAA-2, and CAA-10 revealed interesting observations regarding the morphology and Cu particle size (Fig. 5a–k). The HAADF image of the CAA-2 catalyst demonstrated a uniform distribution of Cu and Ag species over the γ-Al2O3 surface, where they overlapped with each other, while the parent CA catalyst had a rod-like morphology of the Cu–Al framework. The addition of 2 wt% Ag to the parent CA catalyst decreased the Cu particle size. This can be attributed to the formation of bimetallic nanoparticles, where Ag acted as a favourable nucleation site for Cu. This phenomenon occurred due to the smaller atomic radius of Ag, allowing for a tighter packing of atoms, leading to efficient nucleation of Cu particles on the Ag surface. The introduction of Ag can also enhance the mobility of Cu atoms, resulting in the formation of smaller particles. The XRD results showed that the catalysts with Ag content up to 2 wt% only exhibited Cu metal, indicating the formation of bimetallic nanoparticles. However, the Cu particle size increased at higher Ag loadings (10 wt%), possibly due to the agglomeration of Cu particles. As the Ag content increased, the Ag particles became larger, and the particle size distribution became broader. This could decrease the number of nucleation sites for Cu, leading to the agglomeration of Cu particles and an increase in their size. These observations were consistent with the XRD results, which showed that the catalysts with higher Ag content formed Cu–Ag alloys along with Ag metal.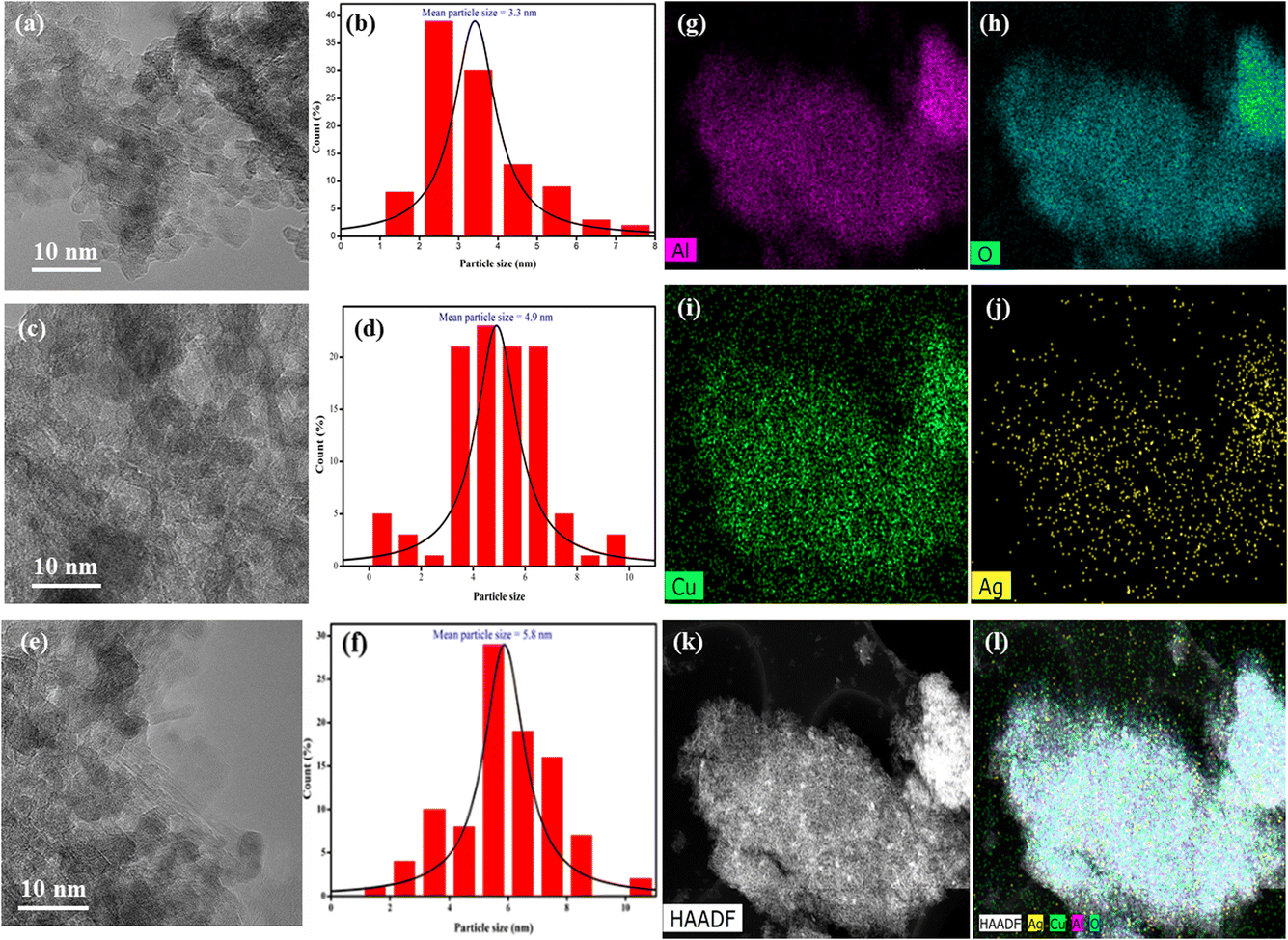 | ||
| Fig. 5 HR-TEM images and particle size of (a and b) CA, (c and d) CAA-2, (e and f) CAA-7, (g–l) elemental mapping of alumina (pink) oxygen (blue), copper (green), silver (yellow), and HADF image of CAA-2 catalyst. | ||
Hydrogenation of HMF to DMF
The hydrogenation of HMF to DMF is an essential step in the production of biofuels and fine chemicals from biomass. However, the selectivity of the reaction towards DMF is challenging to achieve, and the catalytic system's design is critical for this. In this study, we investigated the effect of adding Ag to a Cu/γ-Al2O3 catalyst on the selectivity and activity of the HMF hydrogenation reaction. The results in Table 3 showed that a Cu/γ-Al2O3 catalyst alone had poor activity and selectivity towards DMF, converting only 68% of HMF with a low DMF yield of 29%. However, adding Ag to the catalyst significantly improved its performance, with the 2%Ag–10%Cu/γ-Al2O3 catalyst showing the highest DMF yield of 93%. Further increase in Ag content led to a decrease in DMF yield. A catalyst containing Ag as a promoter showed much higher activity than a catalyst without Ag. The catalyst performance was evaluated by measuring the productivity of DMF, as shown in Table 3. Notably, only the Cu-containing Cu/γ-Al2O3 catalyst displayed a relatively low productivity of 0.9 mmol g−1 h−1. However, upon introducing Ag to the Cu/γ-Al2O3 catalyst, the productivity exhibited a noteworthy increase up to a 2% Ag addition. Subsequently, with further increases in Ag content, the productivity declined.| Catalyst | Conv. (%) | Yield (%) | Productivity (mmol g−1 h−1) | Carbon balance (%) | ||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|||
| a Reaction conditions: HMF: 2 mmol (0.252 g), catalyst (CAA-2): 0.15 g, temp.: 180 °C, time: 6 h, H2 pressure: 1.5 MPa, THF: 20 mL. | ||||||||
| CA | 68 | 30 | 3 | 6 | 29 | — | 0.94 | 100 |
| AA | 27 | 27 | — | — | — | — | — | 100 |
| CAA-0.5 | 100 | — | 29 | 7 | 64 | — | 1.42 | 100 |
| CAA-1 | 100 | — | 10 | 15 | 75 | — | 1.66 | 100 |
| CAA-2 | 100 | — | — | 7 | 93 | — | 2.06 | 100 |
| CAA-5 | 100 | — | — | 9 | 86 | 5 | 1.91 | 100 |
| CAA-7 | 100 | — | — | 16 | 81 | 3 | 1.80 | 100 |
| CAA-10 | 100 | — | — | 15 | 85 | — | 1.88 | 100 |
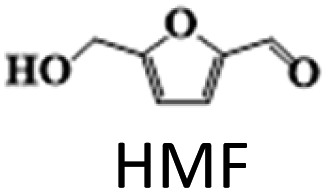
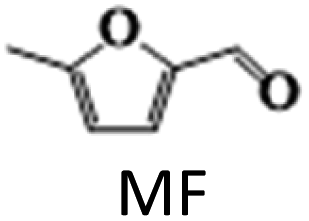
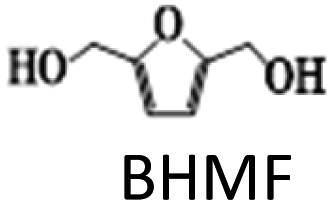

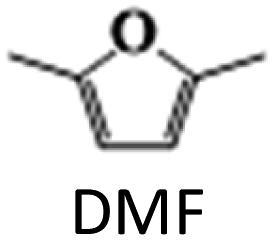
The above results highlight the importance of catalyst composition, particularly the addition of Ag to the Cu/γ-Al2O3 catalyst, in improving the activity and selectivity in HMF hydrogenation to DMF. The addition of small amounts of Ag (up to 2 wt%) was found to improve the dispersion of Cu nanoparticles on the support and increase the catalyst overall surface area. This is due to the higher affinity of Ag for the support than Cu, which helps to anchor Cu particles to the support, preventing their aggregation and increasing the available metal sites. However, increasing Ag content beyond 2 wt% led to the formation of larger particles on the support surface, which competed with Cu for surface sites, reducing the dispersion of Cu nanoparticles. This resulted in a decreased overall Cu metal availability and an increased Cu crystallinity size as Cu particles started to aggregate due to the reduced surface area available for dispersion. Catalysts with up to 2 wt% Ag exhibited higher activity due to the improved dispersion of Cu nanoparticles, while catalysts with more than 2 wt% Ag showed decreased activity towards DMF due to reduced Cu nanoparticle dispersion caused by the formation of larger Ag particles.
The experiments were conducted at lower conversion levels to determine the influence of Ag on the HMF hydrogenation to DMF. All the experiments were conducted at 180 °C, 1 MPa H2 pressure, and employing a catalyst amount of 0.075 g. We monitored the reactions over various time intervals. The outcomes of these experiments offer insightful observations (Fig. S4†). When utilizing the unmodified CA catalyst, we observed modest conversion levels and significantly extended reaction times, indicating limited effectiveness after 5 h.
However, upon introducing Ag to the catalyst system, we observed a notable improvement in both HMF conversion and DMF yield. This improvement was consistent as reaction times increased. These results strongly indicate that the presence of Ag enhances HMF conversion, leading to an overall increase in DMF yield.
These findings strongly suggest a synergistic effect, where the combined action of Ag and Cu exhibits enhanced catalytic activity, particularly in the production of DMF.
The results suggest that the hydrodeoxygenation of HMF can proceed through two distinct pathways, as shown in Scheme 1, influenced by the catalyst acidity and metal dispersion. Specifically, the formation of BHMF intermediate occurs when the CO group of HMF is adsorbed, whereas the formation of MF intermediate takes place when the alcohol group is adsorbed. Catalysts with weak acidity (Lewis acid sites) dominant have a higher propensity to generate BHMF intermediate. This is attributed to the ability of Lewis acid sites to readily accept electron pairs from the CO group, facilitating its adsorption and subsequent conversion to BHMF. Alternatively, when catalysts possess a more balanced distribution of weak and strong acid sites, the dehydration of HMF proceeds through the formation of MF intermediate. In this scenario, the catalyst's acidity, including both weak and strong acid sites, enables the adsorption of the alcohol group of HMF. This adsorption leads to the formation of the MF intermediate, which can further participate in subsequent hydrogenation and hydrodeoxygenation reactions.
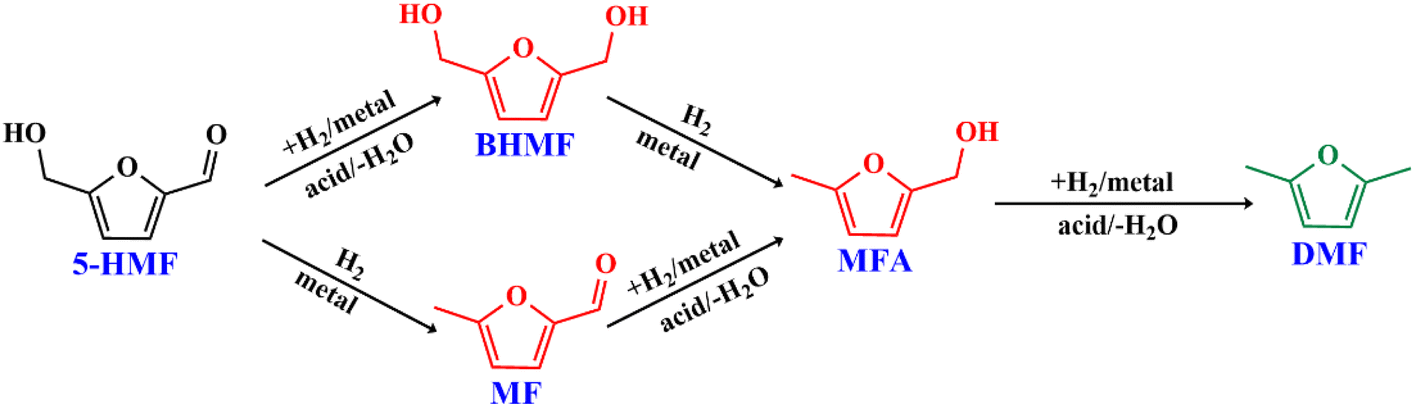 | ||
| Scheme 1 A schemaic representation of the reaction pathway of HMF hydrodeoxygenation to DMF. | ||
The size of the Cu particles and the catalyst acidity were found to play a critical role in the hydrogenation of HMF to DMF (Fig. 6). Optimal catalytic performance was observed when a balance was achieved between weak and strong acid sites. Notably, the addition of Ag to the Cu catalyst enhanced its performance in HMF hydrogenation to DMF by promoting Cu particle growth and improving the catalyst's acidity. The presence of Ag facilitated the growth of Cu particles, leading to larger particle sizes that positively impacted the catalytic activity. Additionally, the introduction of Ag contributed to the modification of the catalyst's acidity, creating an optimal balance between weak and strong acid sites, further enhancing the catalytic performance in the hydrogenation reaction.
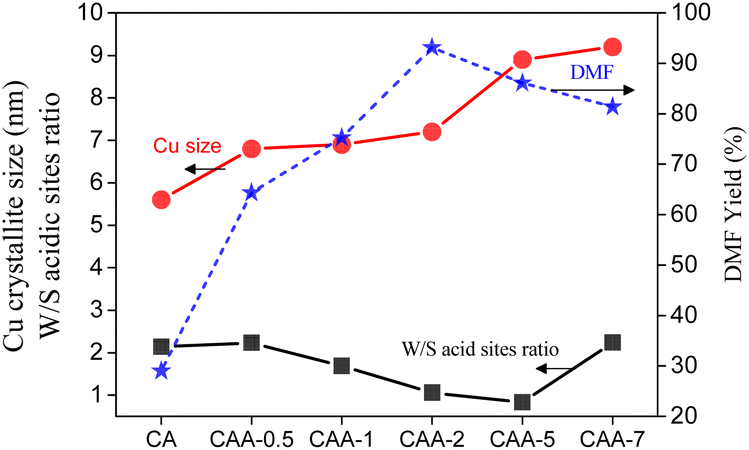 | ||
| Fig. 6 The catalyst activity correlation with Cu particle size and acidity. | ||
The CAA-2 catalyst, with fine dispersion of Cu metal and moderate particle size, as well as an equal portion of weak to moderate acidity, showed the best activity with 93% DMF yield. These findings demonstrate the importance of catalyst composition in controlling the Cu particle size and acidity for optimal performance in the hydrogenation of HMF to DMF, and provide insights into the role of Ag as a promoter in this reaction.
Previous research has demonstrated that Cu catalysts promoted with Ag exhibit high activity in hydrogenation reactions. In a recent study, bimetallic Cu–Ag catalysts supported on γ-Al2O3 were used for glycerol hydrogenolysis in a batch reactor. The addition of Ag was found to improve the dispersion of CuO on the support material, resulting in higher activity compared to a commercial copper chromite catalyst.42 However, another study reported a decrease in hydrogenation ability at high Ag loading in Cu–Ag/γ-Al2O3 catalysts for vapor-phase glycerol hydrogenolysis.43 Similarly, in the hydrogenation of dimethyl oxalate using silica-supported Cu–Ag catalysts, the addition of Ag facilitated the higher dispersion of Cu nanoparticles and promoted the formation of Cu–Ag alloys through clear interactions between the Cu and Ag species. The appropriate amount of Ag helped to maintain a balance of Cu+/Cu0 species, which were found to be highly responsible for the superior activity and stability of the Cu–Ag/SiO2 catalyst.44 Overall, these discussions strongly support the significant influence of Ag metal on the catalytic activity of Cu/γ-Al2O3 catalysts in the conversion of HMF to DMF.
Optimization of reaction conditions
As the CAA-2 catalyst showed better activity at the initial reaction conditions, all the reaction conditions were studied to optimize this catalyst. To corroborate the influence of catalyst loading on the hydrodeoxygenation reaction of HMF, experimental trials were led by varying catalyst loading between 0.05 and 0.25 g, and the outcomes are portrayed in Fig. 7a. The conversion of HMF was improved with the catalyst loading. The catalyst displayed less activity towards DMF (37% yield) with 0.05 g catalyst loading. With increasing catalyst loading, the yield of DMF also increased. It reached a maximum DMF yield of 93.1% at 0.15 g of catalyst. Further enhancing the catalyst loading beyond 0.15 g, DMF yield was decreased and observed an increase in DMTHF yield. The further hydrogenation of DMF forms this product due to the higher number of accessible Cu species.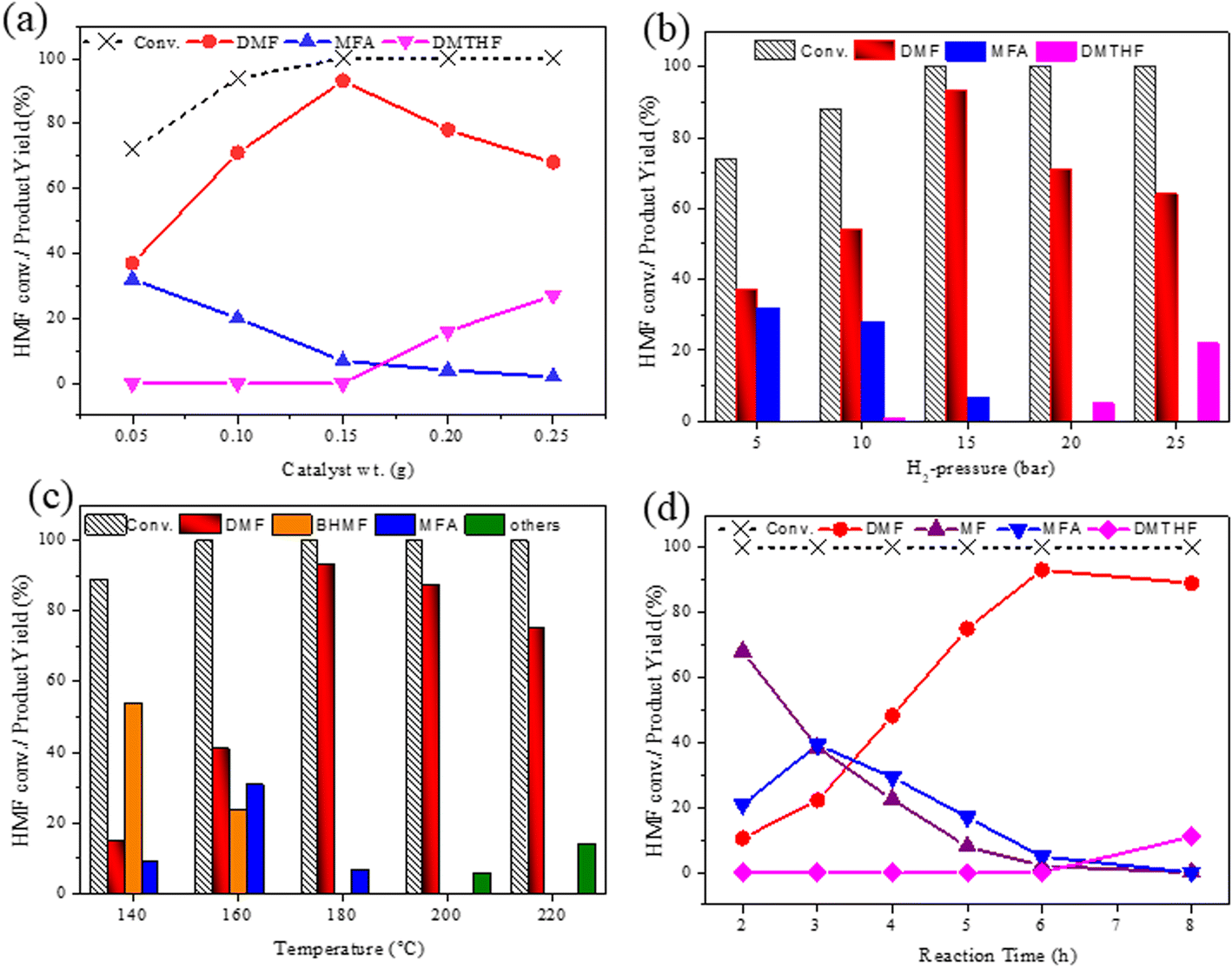 | ||
| Fig. 7 The effect of reaction parameters on HMF conversion over CAA-2 catalyst. (a) The effect of CAA-2 catalyst loading, HMF: 2 mmol, catalyst (CAA-2): 0.05–0.25 g, temperature: 180 °C, time: 6 h, H2 pressure: 1.5 MPa, and THF: 20 mL. (b) The influence of hydrogen pressure on product distribution over CAA-2 catalyst, HMF: 2 mmol, catalyst (CAA-2): 0.150 g, temp.: 180 °C, time: 6 h, H2 pressure: 0.5–2.5 MPa, and THF: 20 mL. (c) The effect of reaction temperature, HMF: 2 mmol, catalyst (CAA-2): 0.15 g, temp.: 140–220 °C, time: 6 h, H2 pressure: 1.5 MPa, THF: 20 mL. (d) The effect of reaction time, HMF: 2 mmol, catalyst (CAA-2): 0.15 g, temperature: 180 °C, time: 2–8 h, H2 pressure: 1.5 MPa, and THF: 20 mL. | ||
The effect of hydrogen pressure on the hydrogenation of HMF to DMF was investigated and the results are presented in Fig. 7b. The conversion of HMF was significantly increased from 74 to 100% with the raising of H2 pressure from 0.5 to 2.5 MPa. A complete transformation of HMF was observed at 1.5 MPa with the DMF and MFA yield of 93 and 17%, respectively. Further enhancing the H2 pressure of 2 MPa favoured ring hydrogenation producing DMTHF as a by-product at the cost of DMF yield. With the increase in hydrogen pressure, the solubility of H2 also increases in the reaction mixture, which directs more amount of hydrogen chemisorption on the catalyst surface. This could lead to over hydrogenation to yield DMTHF as a by-product. About 1.5 MPa was the optimum reaction pressure for HMF conversion to DMF.
The impact of reaction temperature on the conversion of HMF and DMF yield is shown in Fig. 7c. The experiments were conducted in the temperature range of 140 °C to 220 °C. The reaction temperature had a significant effect on the product distribution. HMF conversion was 100% at all temperatures with 6 h of reaction time except at 140 °C. At a lower temperature (140 °C), the primary product was BHMF, with a significant yield of 54%, along with a small amount of DMF (15%) and MFA (9%). Even at 160 °C, the yield of DMF was 41% only.
Further increasing the reaction temperature to 180 °C, the yield of DMF was drastically increased to 93.1%. Enhancing the temperature to 200 °C and 220 °C, DMF yields were declined to 87% and 75%, respectively. The reaction rate increased due to the cracking of the CO bond at higher reaction temperatures, which led to the formation of ring-opening products like hexane diols.45,46 Hence 180 °C was the optimum reaction temperature for this reaction.
In order to realize the reaction pathway and identify the possible reaction intermediates, the experiments were carried out at regular time intervals ranging from 2 to 8 h, and the respected activity results are presented in Fig. 7d. It was noticed that during the initial time (2 h) of reaction, the conversion of HMF reached 100%. The significant MF yield of 89.4% was observed as an intermediate with only 10.5% DMF yield. The formed MF was converted to FAL and DMF when prolonging the reaction time. The maximum DMF yield of 93.1% was observed at a reaction time of 6 h. However, when the reaction time was further increased to 8 h, the DMF yield dropped significantly to 89%. On further increasing the reaction time, the ring hydrogenated product DMTHF yield was increased. These results suggest that MF and MFA are observed as intermediate products and DMTHF as a by-product at these specified reaction conditions, which match the previous reports.31,47 The results indicate that 6 h was the optimum reaction time for the conversion of HMF to DMF over the present catalyst.
The stability and recyclability of a catalyst are crucial concerns in heterogeneous catalysis. In this study, we investigated the reusability of the catalyst for the conversion of HMF to DMF at low conversion level conditions (Fig. 8). The catalyst was recovered from the reaction mixture by filtration, washed with water and methanol, and then dried at 80 °C for about 12 h. The dried catalyst was then used for the next cycle, and the reusability tests were performed for five cycles. The fresh catalyst exhibited about 82% HMF conversion with over 52% yield towards DMF at 10 bar H2 pressure and 0.1 g catalyst in 5 h. The conversion and yield decreased by about 3% in the 5th-time reusability of the catalyst. This could be due to the strong interaction of the alcohol group in intermediate MFA with the catalyst, which could reduce the catalyst activity. To confirm the presence of organic moieties on the catalyst surface, a thermogravimetric analysis (TGA) was conducted (Fig. S2†). The TGA analysis of the used catalyst revealed a weight loss of approximately 5% at lower temperatures ranging from 260 to 340 °C, with an additional 4% loss observed in the temperature range of 340 to 430 °C. These weight losses indicate the presence of monomeric organic compounds, such as HMF and reaction products, adsorbed on the catalyst surface.
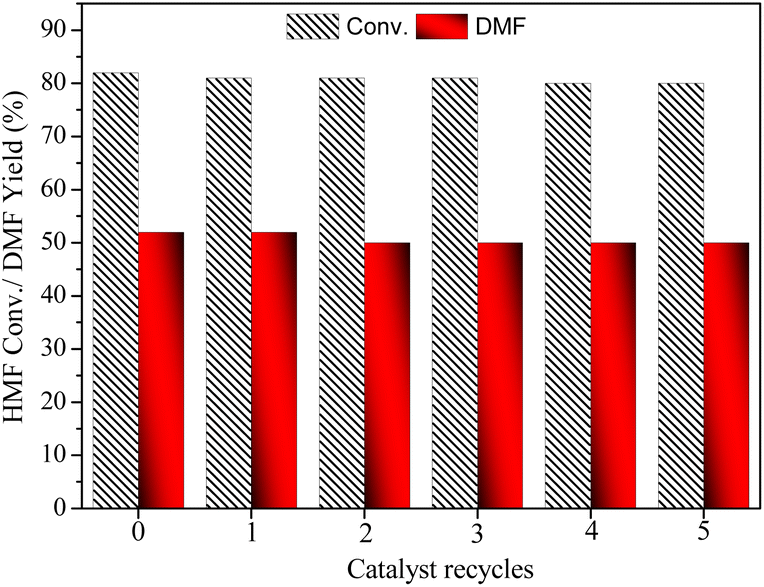 | ||
| Fig. 8 The reusability of CAA-2 catalyst. Reaction conditions: HMF: 2 mmol (0.252 g), catalyst (CAA-2): 0.1 g, temperature: 180 °C, time: 5 h, H2 pressure: 1 MPa, THF: 20 mL. | ||
The CAA-2 catalyst underwent various characterization techniques to evaluate its structural stability and potential for reuse. XRD analysis (Fig. 2b) showed that the structure of the used catalyst remained unchanged after its reuse, suggesting its stability. XPS analysis (Fig. S3†) was conducted to evaluate the changes on the surface of the catalyst. The analysis revealed that both Cu and Ag were present in metal and oxide forms, and the metal content of both elements increased after reuse, indicating that the catalyst remained active and suitable for reuse.
To assess the stability of the catalyst, a leaching test was conducted under the optimized reaction conditions (Fig. S5†). This involved removing the catalyst from the reaction mixture after 4 h while allowing the reaction to continue for an additional 4 h. The yield of DMF remained unchanged even after the catalyst was removed from the mixture. Therefore, it can be concluded that the active metal components of the catalyst remained stable throughout the reaction.
Overall, the results of the various characterization techniques confirmed the stability of the Cu–Ag/Al2O3 catalyst and its active metal components. The catalyst was found to be active and suitable for reuse even after multiple reaction cycles. These findings have important implications for the development of sustainable catalytic processes in the chemical industry.
The present catalyst activity was compared with the recently reported non-noble metal catalysts. For a better understanding along with the reaction parameters, productivity is also presented in Table 4. All the previously reported catalysts have shown almost the same conversions of HMF but the yields of DMF varied depending on their reaction parameters. Ni/ZrP reported moderate yield of DMF at high temperature 240 °C.48 D. Guo et al. studied Ni supported on ZSM and achieved 88% yield of DMF at high loading of Ni compared to CAA-2 catalyst.49 C. V. Rode reported bimetallic Cu–Fe (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) catalyst and achieved 93% yield of DMF with productivity of 3.69 mmol g−1. h−1.50 However, the reusability was poor and the conversion and DMF selectivities were continuously decreased and reached less than 80% after 4 cycles. Cu supported on Zn–Al mixed oxide showed 90% yield of DMF with improved productivity of 4.28 mmol g−1 h−1.51 However, the loading of Cu is over 20% in this case. Cobalt-supported catalysts were also studied for HMF HDO, such as Co/BN and 20Co/beta-DA-C723R that exhibited good yield of DMF at moderate reaction conditions but showed poor recyclability.52,53 N. Chen group developed FeCoNi-based catalyst and achieved 94% yield of DMF.54 In our previous study, we reported 15Cu/SBA-15, which exhibited 93% yield of DMF at 8 h of reaction time.55 In spite of good DMF yields, the above-mentioned catalysts had certain issues, including elevated metal loading, poor stability and recyclability, longer reaction times, and high hydrogen (H2) pressure requirements. Developing a heterogeneous catalyst that is both highly selective and durable while maintaining impressive productivity continues to be a challenge in the field of HMF hydrogenation to DMF. The present catalyst achieved an impressive 93% DMF yield at a reaction time of 6 h and a lower H2 pressure of 1.5 MPa. Notably, the catalyst exhibited a significantly improved productivity of 2.06 mmol g−1 h−1. As the addition of Ag improved the Cu metal dispersion, the present catalyst showed better stability and reusability. This underscores the exceptional performance of our present catalyst when compared to reported catalysts, effectively addressing concerns related to reaction conditions, productivity, stability, and reusability.
:2) catalyst and achieved 93% yield of DMF with productivity of 3.69 mmol g−1. h−1.50 However, the reusability was poor and the conversion and DMF selectivities were continuously decreased and reached less than 80% after 4 cycles. Cu supported on Zn–Al mixed oxide showed 90% yield of DMF with improved productivity of 4.28 mmol g−1 h−1.51 However, the loading of Cu is over 20% in this case. Cobalt-supported catalysts were also studied for HMF HDO, such as Co/BN and 20Co/beta-DA-C723R that exhibited good yield of DMF at moderate reaction conditions but showed poor recyclability.52,53 N. Chen group developed FeCoNi-based catalyst and achieved 94% yield of DMF.54 In our previous study, we reported 15Cu/SBA-15, which exhibited 93% yield of DMF at 8 h of reaction time.55 In spite of good DMF yields, the above-mentioned catalysts had certain issues, including elevated metal loading, poor stability and recyclability, longer reaction times, and high hydrogen (H2) pressure requirements. Developing a heterogeneous catalyst that is both highly selective and durable while maintaining impressive productivity continues to be a challenge in the field of HMF hydrogenation to DMF. The present catalyst achieved an impressive 93% DMF yield at a reaction time of 6 h and a lower H2 pressure of 1.5 MPa. Notably, the catalyst exhibited a significantly improved productivity of 2.06 mmol g−1 h−1. As the addition of Ag improved the Cu metal dispersion, the present catalyst showed better stability and reusability. This underscores the exceptional performance of our present catalyst when compared to reported catalysts, effectively addressing concerns related to reaction conditions, productivity, stability, and reusability.
| S. no | Catalyst | Reaction condition | HMF conv. (%) | DMF yield (%) | Productivity (mmol g−1 h−1) | Ref. |
|---|---|---|---|---|---|---|
| a P. W. – present work. | ||||||
| 1 | Ni/ZrP | 5 MPa H2, 240 °C, 5 h | 97 | 68 | 0.70 | 48 |
| 2 | 40%Ni/ZSM-5 | 0.25 MPa H2, 180, 7 h | 91.2 | 88 | 2.74 | 49 |
| 3 | Cu–Fe (1:2) |
2 MPa H2, 170, 4 h | 100 | 93 | 3.69 | 50 |
| 4 | Cu/ZnO–Al2O3 | 1.2 MPa H2, 180, 5 h | 100 | 90 | 4.28 | 51 |
| 5 | Co/BN | 2 MPa H2, 180, 4 h | 100 | 91 | 2.27 | 52 |
| 6 | 20Co/beta-DA-C723R | 1.5 MPa H2, 150, 3 h | 100 | 83 | 11.06 | 53 |
| 7 | Fe0.8-Co3.0-Ni1.9 | 2 MPa H2, 180, 4.5 h | 100 | 94 | 2.08 | 54 |
| 8 | 15Cu/SBA-15 | 2 MPa H2, 180, 8 h | 100 | 93 | 1.50 | 55 |
| 9 | 2Ag–10Cu/γ-Al2O3 | 1.5 MPa H2, 180, 6 h | 100 | 93 | 2.06 | P. W. |
Conclusions
In this study, silver-promoted copper supported on γ-Al2O3 catalysts were synthesized and used for the hydrodeoxygenation of HMF to DMF. The results indicated that the 2%Ag–10%Cu//γ-Al2O3 catalyst showed a high yield of DMF at 15 bar H2 pressure and 180 °C in 6 h. The presence of Ag played a crucial role in both HMF conversion and DMF yield. The Ag promoted catalysts with an equal portion of weak to strong acid sites were more effective for C–O bonds than CO and CC. The addition of Ag to the Cu/γ-Al2O3 catalyst was found to improve the Cu dispersion and overall surface area, which led to increased catalytic activity. However, the Ag content had to be balanced to avoid the formation of larger Ag particles on the support surface, which reduced the Cu dispersion and decreased overall activity. The optimal catalyst had a fine dispersion of Cu metal and moderate particle size, as well as an equal portion of weak to moderate acidity. The study also demonstrated the reusability of the catalyst for HMF to DMF conversion, with consistent activity upon reuse and stability confirmed by ICP, TGA, XRD, and leaching tests. The present study demonstrated that the addition of Ag to the Cu/γ-Al2O3 catalyst greatly improved its catalytic activity and selectivity. This finding could contribute to the development of an efficient and stable catalyst for the hydrodeoxygenation of biomass-derived compounds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University Grants Commission (UGC), New Delhi, India, for the financial support of a Senior Research Fellowship. We thank Director CSIR-IICT for permitting us to publish our results.References
- Z. Jiang, Y. Zeng, D. Hu, R. Guo, K. Yan and R. Luque, Green Chem., 2023, 25, 871–889 RSC.
- W. Zhang, H. Qian and Q. H. M. Ju, Green Chem., 2023, 25, 893–914 RSC.
- B. Zhang, T. Guo, Z. Li, F. E. Kühn, M. Lei, Z. K. Zhao, J. Xiao, J. Zhang, D. Xu, T. Zhang and C. Li, Nat. Commun., 2022, 13, 3365 CrossRef CAS PubMed.
- J. Gong and R. Luque, Chem. Soc. Rev., 2014, 43, 7466–7468 RSC.
- B. S. Rao, D. D. Lakshmi, P. K. Kumari, P. Rajitha and N. Lingaiah, Sustainable Energy Fuels, 2020, 4, 3428–3437 RSC.
- B. S. Rao, Y. Yogita, D. D. Lakshmi, P. K. Kumari and N. Lingaiah, Sustainable Energy Fuels, 2021, 5, 3719–3728 RSC.
- X. Zhang, D. Zhang, Z. Sun, L. Xue, X. Wang and Z. Jiang, Appl. Catal., B, 2016, 196, 50–56 CrossRef CAS.
- J. Slak, B. Pomeroy, A. Kostyniuk, M. Grilc and B. Likozar, Chem. Eng. J., 2022, 429, 132325 CrossRef CAS.
- M. Chatterjee, T. Ishizaka and H. Kawanami, Green Chem., 2014, 16, 4734–4739 RSC.
- D. K. Mishra, H. J. Lee, C. C. Truong, J. Kim, Y. W. Suh, J. Baek and Y. J. Kim, Mol. Catal., 2020, 484, 110722 CrossRef CAS.
- L. K. Zaitri, L. Negadi, I. Mokbel, N. Msakni and J. Jose, Fuel, 2012, 95, 438–445 CrossRef.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- H. Wang, C. Zhu, D. Li, Q. Liu, J. Tan, C. Wang, C. Cai and L. Ma, Renewable Sustainable Energy Rev., 2019, 103, 227–247 CrossRef CAS.
- Y. Qian, L. Zhu, Y. Wang and X. Lu, Renewable Sustainable Energy Rev., 2015, 41, 633–646 CrossRef CAS.
- G. P. Lu, B. Wang, Y. Li, Y. Lin, J. Hu, Z. Chen and F. Chen, Appl. Catal., A, 2023, 661, 119240 CrossRef CAS.
- Y. Shao, M. Guo, M. Fan, K. Sun, G. Gao, C. Li, F. Merime, B. Kontchouo, L. Zhang, S. Zhang and X. Hu, Renewable Energy, 2023, 208, 105–118 CrossRef CAS.
- Z. Zeng, L. Yang, X. Zhu, W. Zhao, X. Liu, Z. Huang, Q. Xu and W. Zhong, React. Chem. Eng., 2023, 8, 455–464 RSC.
- S. Li, M. Dong, M. Peng, Q. Mei, Y. Wang, J. Yang, Y. Yang, B. Chen, S. Liu, D. Xiao, H. Liu, D. Ma and B. Ha, Innovation, 2022, 3, 100189 CAS.
- B. Hu, L. Warczinski, X. Li, M. Lu, J. Bitzer, M. Heidelmann, T. Eckhard, Q. Fu, J. Schulwitz, M. Merko, M. Li, W. Kleist, C. H-ttig, M. Muhler and B. Peng, Angew. Chem., Int. Ed., 2021, 60, 6807–6815 CrossRef CAS PubMed.
- Z. Li, C. Zhu, H. Wang, Y. Liang, H. Xin, S. Li, X. Hu, C. Wang, Q. Zhang, Q. Liu and L. Ma, ACS Sustain. Chem. Eng., 2021, 9(18), 6355–6369 CrossRef CAS.
- B. Ledesmaa, J. Juáreza, J. Mazaríob, M. Domineb and A. Beltramone, Catal. Today, 2021, 360, 147–156 CrossRef.
- X. Wang, C. Zhang, B. Jin, X. Liang, Q. Wang, Z. Zhao and Q. Li, Catal. Sci. Technol., 2021, 11, 1298–1310 RSC.
- Q. Wang, X. Guan, L. Kang, B. Wang, L. Sheng and F. R. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 53712–53718 CrossRef CAS.
- T.-W. Tzeng, C.-Y. Lin, C.-W. Pao, J.-L. Chen, R. J. G. Nuguid and P.-W. Chung, Fuel Process. Technol., 2020, 199, 106225 CrossRef.
- Q. Deng, J. Zhu, Y. Zhong, X. Li, J. Wang, J. Cai, Z. Zeng, J.-J. Zou and S. Deng, ACS Sustainable Chem. Eng., 2021, 9, 11127–11136 CrossRef CAS.
- Y. Shang, C. Liu, Z. Zhang, S. Wang, C. Zhao, X. Yin, P. Zhang, D. Liu and J. Gui, Ind. Eng. Chem. Res., 2020, 59, 6532–6542 CrossRef CAS.
- Y. Xin, S. Li, H. Wang, L. Chen, S. Li and Q. Liu, Catalysts, 2021, 11, 915 CrossRef CAS.
- T. Gan, Y. Liu, Q. He, H. Zhang, X. He and H. Ji, ACS Sustain. Chem. Eng., 2020, 8, 8692–8699 CrossRef CAS.
- G. Meng, K. Ji, W. Zhang, Y. Kang, Y. Wang, P. Zhang, Y.-G. Wang, J. Li, T. Cui, X. Sun, T. Tan, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4139–4146 RSC.
- D. Wu, S. Zhang, W. Y. Hernandez, W. Baaziz, O. Ersen, M. Marinova, A. Y. Khodako and V. V. Ordomsky, ACS Catal., 2021, 11, 19–30 CrossRef CAS.
- W. Liao, Z. Zhua, N. Chen, T. Su, C. Deng, Y. Zhao, W. Ren and H. Lü, Mol. Catal., 2020, 482, 110756 CrossRef CAS.
- S. Srivastava, G. C. Jadeja and J. Parikh, Chin. J. Catal., 2017, 38699–38709 Search PubMed.
- G. H. Wang, J. Hilgert, F. H. Richter, F. Wang, H. J. Bongard, B. Splietho, C. Weidenthaler and F. Schuth, Nat. Mater., 2014, 13, 293–300 CrossRef CAS PubMed.
- Z. An, W. Wang, S. Dong and J. He, Catal. Today, 2019, 319, 128–138 CrossRef CAS.
- N. Viar, J. M. Requies, I. Agirre, A. Iriondo, C. G. Sancho and P. L. Arias, Energy, 2022, 255, 124437 CrossRef CAS.
- M. V. Morales, J. M. Conesa, A. J. Galvin, A. G. Ruiz and I. R. Ramos, Catal. Today, 2023, 423(114021) CAS.
- Y. Zhu, X. Kong, H. Zheng, G. Ding, Y. Zhu and Y. W. Li, Catal. Sci. Technol., 2015, 5, 4208–4217 RSC.
- L. M. Esteves, M. H. Brijaldo, E. G. Oliveira, J. J. Martinez, H. Rojas, A. Caytuero and F. B. Passos, Fuel, 2020, 270, 117524 CrossRef CAS.
- J. Zhou, J. Zhang, X. Guo, J. Mao and S. Zhang, Green Chem., 2012, 14, 156–163 RSC.
- H. He, C. Zhang and Y. Yu, Catal. Today, 2004, 90, 191–197 CrossRef CAS.
- T. Chetty, V. D. B. C. Dasireddy, L. H. Callanan and H. B. Friedrich, ACS Omega, 2018, 3, 7911–7924 CrossRef CAS PubMed.
- D. Sun, Y. Yamada and S. Sato, Appl. Catal., A, 2014, 475, 63–68 CrossRef CAS.
- J. Zhou, L. Guo, X. Guo, J. Mao and S. Zhang, Green Chem., 2010, 12, 1835–1843 RSC.
- Y. Huang, H. Ariga, X. Zheng, X. Duan, S. Takakusagi, K. Asakura and Y. Yuan, J. Catal., 2013, 307, 74–83 CrossRef CAS.
- A. J. Kumalaputri, G. Bottari, P. M. Erne, H. J. Heeres and K. Barta, ChemSusChem, 2014, 7, 2266–2275 CrossRef CAS PubMed.
- T. S. Hansen, K. Barta, P. T. Anastas, P. C. Ford and A. Riisager, Green Chem., 2012, 14, 2457–2461 RSC.
- S. Umasankar, P. Tamizhdurai, P. S. krishnan, S. Narayanan, V. L. Mangesh and K. Shanthi, Biomass Bioenergy, 2020, 143, 105868 CrossRef CAS.
- C. Zhu, Q. Liu, D. Li, H. Wang, C. Zhang, C. Cui, L. Chen, C. Cai and L. Ma, ACS Omega, 2018, 3, 7407–7417 CrossRef CAS.
- D. Guo, X. Liu, F. Cheng, W. Zhao, S. Wen, Y. Xiang, Q. Xu, N. Yu and D. Yin, Fuel, 2020, 274, 117853 CrossRef CAS.
- B. S. Solanki and C. V. Rode, Green Chem., 2019, 21, 6390 RSC.
- Q. Wang, Z. Yu, J. Feng, P. Fornasiero, Y. He and D. Li, ACS Sustainable Chem. Eng., 2020, 8, 15288–15298 CrossRef CAS.
- D. Bi, X. Chen, Z. Du, Z. Guo, Z. Liu, J. Lin, Y. Huang, C. Tang, G. Chen and Y. Fang, ChemistrySelect, 2022, 7, e202104043 CrossRef CAS.
- N. Chen, Z. Zhu, H. Ma, W. Liao and H. Lü, Mol. Catal., 2020, 486, 110882 CrossRef CAS.
- N. Chen, Z. Zhu, T. Su, W. Liao, C. Deng, W. Ren, Y. Zhao and H. Lü, Chem. Eng. J., 2020, 381, 122755 CrossRef CAS.
- D. D. Lakshmi, B. S. Rao, Y. Yogita and N. Lingaiah, Mater. Sci. Energy Technol., 2021, 4, 357–366 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3se01096a |
| ‡ IICT communication No: IICT/Pubs./2023/060. |
| This journal is © The Royal Society of Chemistry 2024 |