
Sensitizing chemotherapy for glioma with fisetin mediated by a microenvironment-responsive nano-drug delivery system†
Wanyu
Wang‡
a,
Yuanyuan
Zhang‡
b,
Yue
Jian‡
a,
Shi
He
a,
Jiagang
Liu
a,
Yongzhong
Cheng
a,
Songping
Zheng
a,
Zhiyong
Qian
 a,
Xiang
Gao
a and
Xiang
Wang
*a
a,
Xiang
Gao
a and
Xiang
Wang
*a
aDepartment of Neurosurgery, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, PR China. E-mail: wangxiangtim@163.com
bKey Laboratory of Drug Targeting and Drug Delivery Systems, Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, China
First published on 22nd November 2023
Abstract
Drug resistance has become an obstacle to successful cancer chemotherapies, with therapeutic agents effectively traversing the blood–brain barrier (BBB) remaining a great challenge. A microenvironment responsive and active targeting nanoparticle was constructed to enhance the penetration of drugs, leading to improved therapeutic effects. Dynamic light scattering demonstrated that the prepared nanoparticle had a uniform size. The cRGD modification renders the nanoparticle with active targeting capabilities to traverse the BBB for chemotherapy. The disulfide-bond-containing nanoparticle can be disintegrated in response to a high concentration of endogenous glutathione (GSH) within the tumor microenvironment (TME) for tumor-specific drug release, resulting in more effective accumulation. Notably, the released fisetin further increased the uptake of doxorubicin by glioma cells and exerted synergistic effects to promote apoptosis, induce cellular G2/M cycle arrest, and inhibit cell proliferation and migration in vitro. Moreover, the nanoparticle showed favorable anti-glioma effects in vivo. Our study provides a new strategy to overcome drug resistance by utilizing a natural product to sensitize conventional chemotherapeutics with well-designed targeted nanodelivery systems for cancer treatment.
Introduction
In the global ranking of primary malignant brain tumors, glioblastoma (GBM) accounts for about 49%, with a high degree of aggression and heterogeneity. Moreover, GBM has a median survival of less than 2 years.1,2 The standardized therapeutics for GBM include surgical resection of tumors and simultaneous combination of radiation therapy and chemotherapy.3 However, the existence of the blood–brain barrier (BBB), poor solubility of the chemotherapeutic agents and non-specific drug diffusion and clearance in vivo seriously limit the treatment effect of the drugs.4,5 Even worse, residual glioblastoma is prone to recurrence, which becomes a greater difficulty in chemotherapy. Therefore, single treatment is often insufficient to achieve the desired treatment effect, and some researchers have reported drug combination treatment strategies. The basic principle and huge advantage of the drug combination strategy is to achieve better therapeutic effects upon co-administration of drugs with different mechanisms while reducing the occurrence of adverse drug reactions.6,7Doxorubicin (DOX) is a broad-spectrum antitumor antibiotic that is clinically used to treat breast,8 brain,9 lung,10 and other cancers by inducing cell cycle arrest and DNA damage. However, doxorubicin has some severe side effects, including cardiotoxicity and myelosuppression with considerable possibility of drug resistance, which greatly hinders its clinical application.11,12 In our previous studies, we designed a targeted nanoplatform for the codelivery of DOX and a BRD4-degrading proteolytic targeting chimera (PROTAC) ARV-825 to inhibit tumor growth, which reversed single-drug resistance and achieved chemosensitization.13 We have also proposed an efficient combination strategy of DOX and curcumin that could reduce the cardiotoxicity of DOX and drug resistance.14 Fisetin (3,7,3′,4′-tetrahydroxyflavone), a member of the polyphenols-flavonoid, can effectively inhibit tumor progression by enhancing intracellular anticancer drug accumulation, blocking the tumor cell cycle, inducing cell apoptosis and inhibiting proliferation and angiogenesis.15–17 Although fisetin has a variety of pharmacological activities, the bioavailability of fisetin is severely limited due to its hydrophobicity, and no clinical applications for fisetin combination therapy were reported. K. H. Lin et al. proved that flavonoids, including fisetin, could alleviate cardiotoxicity caused by doxorubicin.18 These findings indicate that fisetin has potential benefits for reducing DOX-induced cardiac toxicity, making this combinational strategy promising to increase therapeutic effects while decreasing adverse reactions. Therefore, the combination of chemotherapeutic drugs with fisetin could hopefully become a novel strategy in enhancing chemotherapy and solving drug resistance.
The nanomedicine-based tumor targeting strategy is currently applied in the study of a variety of cancer therapies including brain tumors to improve efficacy by decreasing toxicity and overcoming drug resistance. Research showed that GBM-cell membrane camouflaged and pH-sensitive biomimetic nanoparticles can efficiently co-load TMZ and CDDP and pass the BBB to target the GBM.19 Moreover, another research team designed a biocompatible nanoformulation to disrupt redox homeostasis in GBM cells and promote the simultaneous occurrence of efficient apoptosis and ferroptosis.20 Nanoscale drug carriers have the advantages of controlled drug release, increased drug solubility, preserved drug activity, and reasonable biosafety.21,22 To improve the permeability of the blood–brain barrier and achieve active targeting ability of drugs to brain tumors, nano-drug delivery systems are developed and functionally modified, such as connecting folic acid and polypeptides on the surface of the nanocarrier to facilitate targeted delivery and local accumulation.23 Integrin αvβ3, an important receptor, is overexpressed in cerebral microvascular endothelial cells and glioma cells.24,25 The tumor-targeting peptide cyclic arginine–glycine–aspartate acid (cRGD) specifically targets the cell attachment receptor integrin αvβ3.26 More impressively, cRGD decorated nanomaterials have demonstrated successfully delivery of therapeutics to the glioma.27 Thus, a promising targeted strategy modified by cRGD is expected to render the drug-loaded nanomaterials with a BBB penetration ability.28 After penetrating the blood–brain barrier, nanoparticles still need to face the complex intra-tumor environment. Glutathione (GSH) plays a significant role in tumor cell growth and function by modulating redox homeostasis, and it is reported that cancer cells have about fourfold higher GSH levels than normal cells.29 Based on this property, researchers have introduced reduction-responsive groups to nanocarriers so that high levels of GSH in tumor cells trigger disulfide bond breaking and nanocarrier disintegration, thereby achieving controlled release and targeted therapy.30,31
Herein, we designed a novel nano-delivery carrier with self-targeting capability and GSH-triggered drug release (Fis-DOX/cRGD-NPs). Firstly, fisetin and doxorubicin were loaded into the hydrophobic core through the thin film hydration method. Next, the surface of nanoparticles was functionalized with integrin αvβ3-targeting cRGD, and the disulfide bond-containing nanoparticles can respond to GSH within the tumor microenvironment. Finally, in vitro and in vivo experiments were performed to prove that the combination strategy enhanced the therapeutic effect of chemotherapy to eliminate tumors (Scheme 1).
 | ||
| Scheme 1 Schematic illustration of the system co-loading DOX and fisetin by cRGD-decorated nanoparticles, and the processes of drug delivery in an orthotopic glioma model. | ||
Materials and methods
Synthesis and characterization of the nanocomposite
To prepare PEG-SS-PLA with a disulfide bond structure, the terminal carboxyl group of mPEG-COOH was firstly modified with 2,2′-dithiodiethanol. mPEG-COOH, EDC, and NHS were added together into a dry round-bottomed flask. The reaction was stirred in an ice bath for 1 h, then the 2,2′-dithiodiethanol was added dropwise into the reaction system for 24 h. Next, the obtained products were dissolved in dichloromethane and transferred into a dialysis bag. They were subjected to lyophilization for 5 days. Next, the obtained PEG-SS-OH powder was dissolved in a round-bottomed flask with anhydrous toluene. Shortly, DL-lactide and the catalyzer Sn (Oct)2 were added when the temperature was increased up to 115 °C for 24 h under nitrogen gas protection. Finally, PEG-SS-PLA was obtained by dialysis and lyophilization (Fig. S1, ESI†).To obtain the block copolymer cRGD-PEG-PLA, MAL-PEG-OH was utilized to initiate the ring-opening polymerization of DL-lactide. The MAL-PEG-OH was dissolved in a round-bottomed flask with anhydrous toluene, then DL-lactide and catalyzer Sn (Oct)2 were added when the temperature was increased up to 115 °C for 24 h under nitrogen gas protection. The obtained reaction products were transferred to ice petroleum ether for precipitation. The resulting product was obtained through centrifugation, dialysis, and lyophilization. After that, MAL-PEG-PLA and cRGD were dissolved in DMSO, and the reaction was stirred overnight at room temperature. The obtained cRGD-PEG-PLA copolymer was dialyzed and subjected to lyophilization (Fig. S2, ESI†). The structures of the synthesized products were confirmed by 1H NMR (Fig. S3, ESI†).
Polymer micelles loaded with Fis and DOX were prepared by the thin film hydration method. Firstly, 60 mg of PEG-SS-PLA, 15 mg of cRGD-PEG-PLA, and 7.5 mg of fisetin were dissolved in 3 ml of acetone, then the solvent was rotary-evaporated under 55 °C at 100 rpm. Next, the dispersed film was dissolved in 7 ml of 5% glucose solution containing doxorubicin hydrochloride. The flask was shaken in a water bath at 55 °C to form Fis-DOX/cRGD-NPs. The blank NPs, fisetin/NPs, DOX/NPs, and Fis-DOX/NPs were prepared using the same method.
Dynamic light scattering (DLS) was used to measure the average size of the nanocomposites. The morphology of Fis-DOX/cRGD-NPs was observed by transmission electron microscopy. The polymer micelles were scattered in PBS buffer containing 0 mM, 0.01 mM, and 10 mM GSH, respectively. Next, they were placed in the incubator shaker at 37 °C and 100 rpm. The sizes of micelles were measured to evaluate the responsiveness of nano-micelles at 0 h, 1 h, 2 h, 4 h, 19 h, and 22 h.
Cell viability assay
GL261 or U87-MG cells were digested with trypsin and seeded in 96-well plates overnight (GL261 was seeded at a density of 2 × 103 and 3 × 103 cells per well, U87-MG was seeded at a density of 3 × 103 and 5 × 103 cells per well). GL261 or U87-MG cells were incubated with different concentrations of fisetin/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h and 48 h. Finally, CCK-8 was added and incubated for another 1–2 h. The absorbance value (OD: 450 nm) was determined by using a microplate reader.Cell uptake assay
GL261 or U87-MG cells were digested with trypsin and seeded at 2 × 105 cells per well of 6-well plates overnight. GL261 or U87-MG cells were incubated with different concentrations of fisetin/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs for 5 h. Then, the glioma cells were washed with PBS and collected for flow cytometry analysis.Cell apoptosis assay
GL261 or U87-MG cells were digested with trypsin and seeded at 2 × 105 cells per well of 6-well plates overnight. GL261 or U87-MG cells were incubated with different concentrations of fisetin/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h and 48 h. Then, each well was washed with pre-cooled PBS after incubation. After digestion with EDTA-free trypsin, the glioma cells were collected, resuspended and washed with pre-cooled PBS. Finally, the cells were stained with 100 μL binding buffer containing 5 μL annexin V-APC and 5 μL PI for 15 min, with an additional 400 μL of binding buffer to detect cell apoptosis by a flow cytometer.Cell cycle assay
GL261 or U87-MG cells were digested with trypsin and seeded at 2 × 105 cells per well of 6-well plates overnight. GL261 or U87-MG cells were incubated with different concentrations of fisetin/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h and 48 h. After incubation, the cells were washed with pre-cooled PBS and digested with EDTA-free trypsin. Then, the collected cell suspension was washed with pre-cooled PBS and fixed in 3 mL of 70% ethanol at 4 °C for 12–24 h. Next, the fixed cells were washed with pre-cooled PBS and stained with PI-containing RNase. Finally, the results were analyzed with a flow cytometer at a minimum speed.Colony formation assay
GL261 cells were digested with trypsin and seeded at 2 × 105 cells per well of 6-well plates overnight. GL261 cells were incubated with different concentrations of fisetin/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h. After incubation, the cells were washed with PBS and digested with trypsin. Then, the treated glioma cells were evenly seeded in six-well plates at 1.2 × 103 cells per well, with the complete medium replaced every 3 days. The culture medium was discarded, and the cells were washed with PBS when the cell colonies became visible. Then, 4% polymerized formaldehyde was added to fix cells at room temperature for 15 min, and the crystal violet staining solution was used to dye for 20 min. Next, each well was washed with water and left to dry. The level of proliferation was evaluated by calculating the cloning formation rate.Cell migration assay
GL261 were digested with trypsin, seeded at 2 × 105 cells per well of 6-well plates, and incubated to reach confluence. The monolayer was scratched using a sterile pipette tip and washed with serum-free medium to remove detached cells. Then the cells were cultured in DMEM with 1% FBS and different concentrations of micelles. Scratches were photographed at 0 h, 24 h, and 48 h. The closure area of the wound was calculated as follows: migration area (%) = (A0 − An)/A0 × 100, where A0 represents the initial wound area, and An represents the remaining area of the wound at the time point of measuring.Anti-tumor effects in subcutaneous glioma models
Female C57BL/6 mice (6–8 weeks) were purchased from Beijing Huafukang Biotechnology Co., Ltd. All animal experiments were carried out in accordance with the guidelines for Care and Use of Laboratory Animals of Sichuan University and approved by the Animal Experiment Ethics Committee of the State Key Laboratory of Biotherapy, Sichuan University (Chengdu, China). GL261 cells were subcutaneously inoculated into C57BL/6 female mice. When the tumor grew to 5 mm × 5 mm, these mice were randomly divided into six groups: (A) GS, (B) blank-NPs, (C) fisetin/NPs, (D) DOX/NPs, (E) Fis-DOX/NPs and (F) Fis-DOX/cRGD-NPs. The drug solution was injected through the tail vein at 200 μL per mouse. The dosages of fisetin and DOX were 2.5 mg per kg body weight. In addition, mice were injected three times every three days. The xenograft volume and body weight were recorded each time. After the treatment was completed, the blood sample was collected from the eyeballs for blood biochemical tests. The tumor tissues were harvested and photographed, and vital organs were taken for further study, including the heart, liver, spleen, lungs, and kidneys.Anti-tumor effects in an orthotopic implantation model
C57BL/6 mice anesthetized with Zoletil and xylazine were fixed with brain stereotactic instruments. After disinfection with povidone-iodine, the scalp was cut, and the anterior fontanelle was exposed. The injection point was localized 0.5 mm anterior and 2.0 mm right lateral to the bregma. Then, a hole was carefully made with a skull drill, and the microinjector was inserted vertically to 3.5 mm depth and retracted to 3 mm. Next, 3 μL of GL261-luc cell suspension (2 × 105 cells) was injected into the localized brain area. A microsyringe pump was used to control the flow rate and volume during the injection process. After 5 min of retention, the needle was slowly withdrawn for 1 min. Finally, bone wax was used to seal the hole, and the scalp was sutured with 3M vetbond and disinfected with iodophor. After the procedure, all mice were put on an electric blanket to preserve body temperature.These mice were randomly divided into six groups: (A) GS, (B) blank-NPs, (C) fisetin/NPs, (D) DOX/NPs, (E) Fis-DOX/NPs, and (F) Fis-DOX/cRGD-NPs. Each mouse was injected with 200 μL solution through the tail vein. The dosages of fisetin and DOX were 2.5 mg per kg body weight. In addition, mice were treated three times in total every three days. The body weight was recorded each time. After the treatment was completed, the mice were injected with 15 mg mL−1D-fluorescein potassium solution and anesthetized with isoflurane. After 10 min, the mice were placed in the small animal intravital imager to observe the luminescence. Finally, the mice were sacrificed, and the brain tissues were fixed in 4% paraformaldehyde, which was prepared for hematoxylin–eosin (HE) staining to assess the effect of drugs in vivo.
Biosafety evaluation
After the last treatment, blood samples from the subcutaneous GL261 tumor model mice were collected through the eyeball, placed in a refrigerator at 4 °C overnight, and centrifuged at 1500 rpm for 15 min at 4 °C to obtain serum. The serum was further used for serum biochemical analysis. In different treatment groups, HE staining was performed on sections of organs (heart, liver, spleen, lungs, kidneys) of mice.Statistical analysis
In this study, GraphPad Prism was used for statistical analysis. The T-test was used for comparison between two groups, and the one-way analysis of variance was used for multiple group comparison. The standard error of measurement was used in the statistical processing of the data. *p < 0.1, **p < 0.01, ***p < 0.001.Results and discussion
Preparation and characterization of Fis-DOX/cRGD-NPs
Glioblastoma is the most fatal, aggressive and malignant primary intracranial tumor.32,33 Although tremendous efforts have been made for glioma treatment in recent years, the therapeutic drugs accumulating within intracranial tumor sites remain limited due to the blood–brain barrier.34 The RGD-modified nanocarrier can actively target specific cells to achieve high-efficiency drug delivery.35 The active-targeting micelles were prepared by the thin film hydration method, which consisted of PEG-SS-PLA and cRGD-PEG-PLA. Fisetin and doxorubicin were loaded in the hydrophobic core (Fig. 1a). Fis-DOX/cRGD-NPs were spherical under transmission electron microscopy (Fig. 1b). As shown in Fig. 1c, the average diameter of Fis-DOX/cRGD-NPs was 126.02 ± 1.36 nm, and the PDI was 0.063 ± 0.027. Moreover, the zeta potential of Fis-DOX/cRGD-NPs was −12.48 ± 1.36 mV (Fig. S4, ESI†). Glutathione (GSH), as an antioxidant, maintains certain concentrations in normal cells. Studies have found that the content of GSH in tumor cells (about 2–10 mM) is much higher than in normal cells,36 utilizing GSH is a convincing strategy to enhance therapeutic efficacies. Different concentrations of GSH solutions were used to trigger the reduction reaction, and the particles disintegrated under 10 mM GSH (Fig. 1d). The S–S bond-containing Fis-DOX/cRGD-NPs not only consume GSH in the TME but also responsively release DOX and fisetin to exert chemotherapeutic effects. The results indicated that the Fis-DOX/cRGD-NPs featured a small diameter, exceptional stability, and effective responsiveness to GSH, making them eligible as brain-targeting drug delivery systems for subsequent research. | ||
| Fig. 1 Characteristics of Fis-DOX/cRGD-NPs micelles. (a) Structural diagram of Fis-DOX/cRGD-NPs. (b) TEM image of Fis-DOX/cRGD-NPs. (c) Particle size of Fis-DOX/cRGD-NPs. (d) Verification of redox responsive size distribution of micelles after different treatments. | ||
Fis-DOX/cRGD-NPs induced cytotoxicity and cell cycle arrest in vitro
Currently, clinical applications of DOX are limited due to chemoresistance acquisition that limits the antitumor efficacy of DOX and leads to chemotherapy failure. Therefore, different methods such as switching to another type of chemotherapeutic drug, combining with other treatment strategies like immunotherapy and using nanoplatforms to achieve targeted delivery and enhance therapeutic effects have been proposed to improve the efficacy of DOX.37,38 Fisetin is a bioactive flavonol that can alleviate cardiotoxicity caused by doxorubicin. Moreover, fisetin is a flavonoid compound that could inhibit P-glycoprotein (P-gp) to enhance drug concentration.39 Therefore, the drug combination of fisetin and DOX has certain advantages. We firstly detected the cytotoxicity of Fis-DOX/cRGD-NPs against glioma GL261 cells and U87 cells through CCK-8. When the DOX concentration was 0.0062 to 0.025 μg mL−1, the Fis-DOX/cRGD-NPs showed stronger cytotoxicity to GL261 cells (Fig. 2a and b). Meanwhile, the Fis-DOX/cRGD-NPs exerted stronger cytotoxicity when the DOX concentration ranged from 0.1 to 0.2 μg mL−1 in U87-MG cells at 48 h (Fig. S5, ESI†). In addition, the Fis/NPs showed cytotoxicity at high doses. The results demonstrated that the Fis-DOX/cRGD-NPs induced potent cytotoxicity even at low doses of DOX.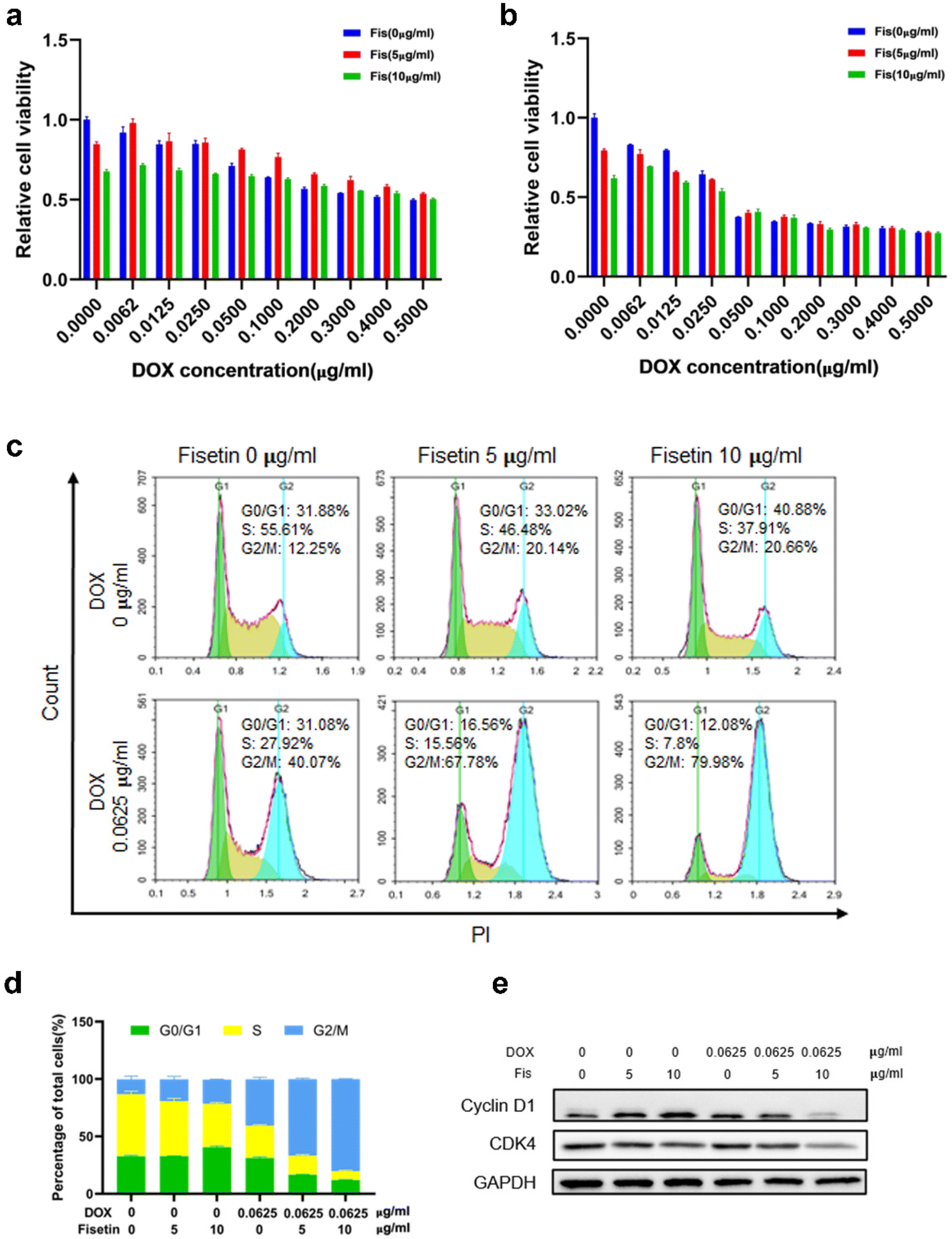 | ||
| Fig. 2 Synergistic effects on cytotoxicity and cell cycle arrest in GL261 cells. CCK-8 assay was used to assess the cytotoxicity of blank NPs, DOX/NPs, and Fis-DOX/NPs at 24 h (a) and 48 h (b). N = 3. (c) Flow cytometric analysis. GL261 cells were treated with blank NPs, DOX/NPs, and Fis-DOX/NPs for 24 h. (d) Populations of cells at G0/G1, S, and G2/M phases were displayed as percentages of the whole cell population. (e) Western blot of cell proliferation and cycle-related proteins. | ||
We further explored the effect of the combination therapy on cell cycle arrest, which could inhibit glioma cell proliferation. GL261 cells were treated with Fis/NPs for 24 h, and the cell cycle analysis results indicated an increase in the G2/M phase and a decrease in the S phase, with the result of the DOX/NP group showing the same trend. Compared with the DOX/NP group, more cells were arrested in the G2/M phase in the Fis-DOX/cRGD-NP group (Fig. 2c). When the fisetin concentration was 10 μg mL−1, the cycle arrest effect of Fis-DOX/cRGD-NPs was more significant; the percentage of cells in the S phase was as low as 7.8%, and the percentage of cells in the G2/M phase was as high as 79.98%, almost double that of the DOX/NP group (Fig. 2d). Similar results were also revealed in the U87 cells (Fig. S6, ESI†). Moreover, treatment with Fis-DOX/cRGD-NPs in GL261 and U87 cells led to the downregulation of cyclin D1 and CDK4 (Fig. 2e and Fig. S9, ESI†). It reported that fisetin is capable of inducing cell cycle arrest, including the G0/G1, S and G2/M phases.40 These results suggested that the G2/M phase was blocked by Fis-DOX/cRGD-NPs, thereby inhibiting glioma cell proliferation.
Fis-DOX/cRGD-NPs promoted cell apoptosis in vitro
GL261 cells were treated with Fis/NPs, DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h, and the effects of micelles on apoptosis were examined by Annexin V-APC/PI. The result showed that Fis/NPs could also induce pro-apoptotic effects at the concentration of 10 μg ml−1. What's more, the percentage of apoptotic cells in the Fis-DOX/cRGD-NP group was significantly higher than in the DOX/NP group when the fisetin concentration was 5 μg mL−1 and 10 μg mL−1, reaching 46.53% and 41.13% respectively (Fig. 3a and b). Analogously, the pro-apoptotic effect was observed in the U87 cells (Fig. S7, ESI†). Studies have proved that fisetin can promote apoptosis through both an exogenous pathway (dead receptor pathway) and an endogenous pathway (mitochondrial pathway), which is mainly achieved by promoting the activation of caspase-3, -8, -9 and adjusting the BAX/BCL-2 ratio.41 These results demonstrated that fisetin could promote apoptosis at high concentrations, while the combination with DOX further exerted robust synergistic effects to promote tumor cell apoptosis. | ||
| Fig. 3 Synergistic effects of DOX and fisetin in the NP carrier system on tumor cell apoptosis. GL261 cells were treated with blank NPs, DOX/NPs, and Fis-DOX/NPs at DOX concentrations of 0 μg mL−1 and 0.0625 μg mL−1 for 24 h. (a) and (b) Cells were collected and stained with annexin V and PI for flow cytometric analysis. (c) Western blot of apoptosis-related proteins. GL261 cells were exposed to blank NPs, DOX/NPs, and Fis-DOX/NPs at 0.0625 μg mL−1 for 24 h. | ||
The mechanism of cell apoptosis caused by the drug combination was further explored, and apoptosis-related proteins were detected. As shown in Fig. 3c, compared with the DOX/NP group, pro-caspase-3, which contributed to the pro-apoptotic effect in the caspase family, was activated by the Fis-DOX/cRGD-NPs. Fis-DOX/cRGD-NPs also induced the activation of pro-caspase-8 and pro-caspase-9 to initiate apoptosis. Moreover, the Fis-DOX/cRGD-NPs promoted cell apoptosis by upregulating BAX expression and downregulating BCL-2. The ratio of BAX and BCL-2 had a larger increase in the treatment group, indicating that the Fis-DOX/cRGD-NPs could accelerate cell apoptosis by promoting the release of cytochrome C. Similarly, this effect was observed in U87 cells (Fig. S8, ESI†). Therefore, the Fis-DOX/cRGD-NPs promoted cell apoptosis by activating the caspase cascade, downregulating the expression of BCL-2, and upregulating the expression of BAX.
Fis-DOX/cRGD-NPs inhibited cell proliferation and migration in vitro
GL261 cells were treated with Fis/NPs, DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h, and the effect of micelles on cell proliferation and migration was examined by the colony formation and scratch assay. As shown in Fig. 4a, compared with blank NPs and Fis/NPs, colony formation was inhibited by Fis-DOX/cRGD-NPs when the fisetin concentration increased to 5 μg mL−1, and the proliferation was almost blocked when the fisetin concentration increased to 10 μg mL−1. The inhibitory effects of DOX/NPs were much lower than that of Fis-DOX/cRGD-NPs. The results showed that the colony formation rate of the Fis-DOX/cRGD-NPs was reduced to about 16% when the fisetin concentration was 10 μg mL−1 (Fig. 4b).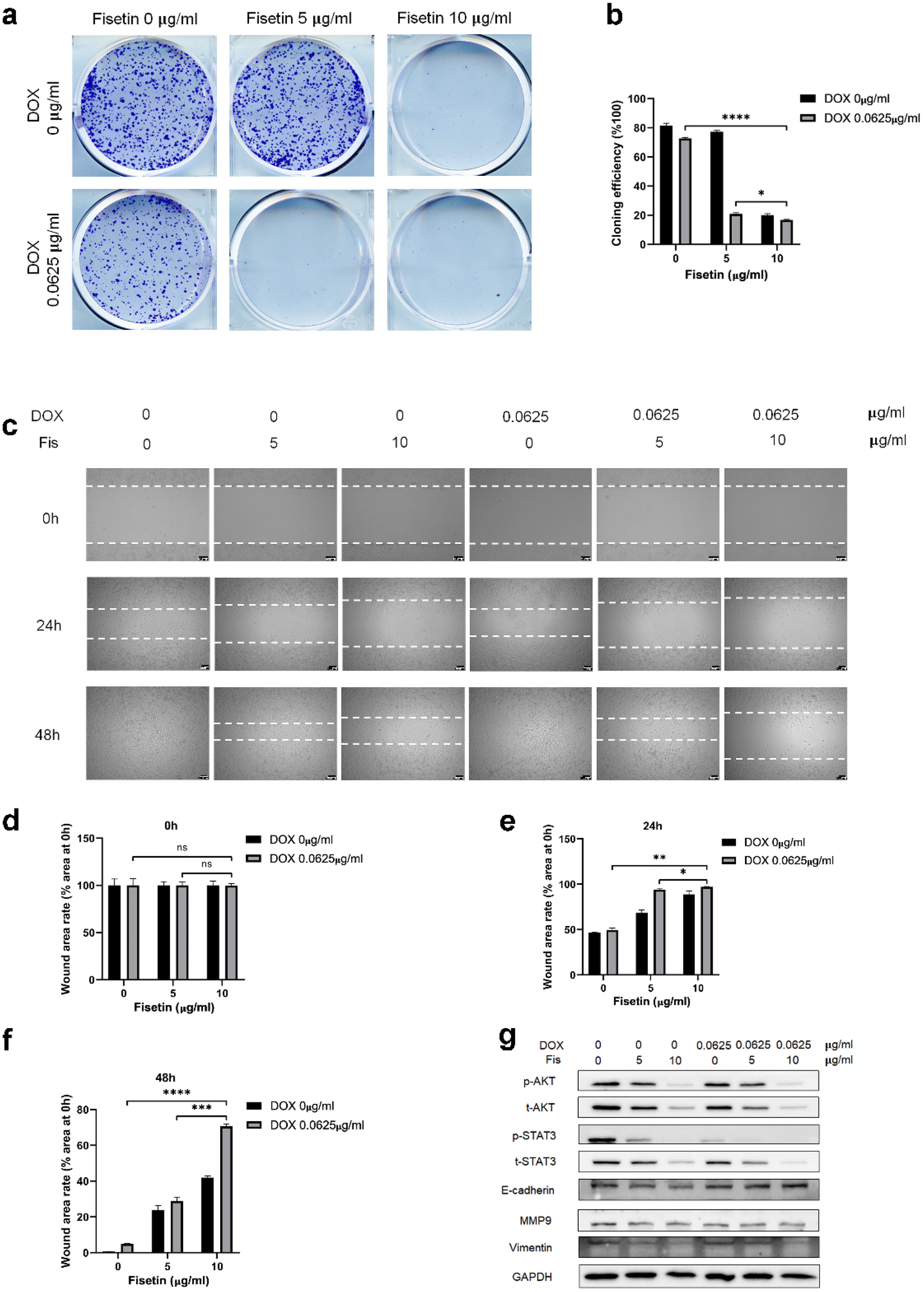 | ||
| Fig. 4 Synergistic effects of DOX and fisetin in the NP carrier system on tumor cell proliferation and migration. GL261 cells were treated with blank NPs, DOX/NPs and Fis-DOX/NPs at concentrations of 0 μg mL−1 and 0.0625 μg mL−1. (a) and (b) Cells were stained with crystal violet for analysis. (c)–(f) Cells were scratched on the dishes, then the wound healing was observed after 24 h and 48 h. (g) Western blot of proliferation-related and migration-related proteins. | ||
Through the cell scratch assay, we examined the effect of the combination therapy on the GL261 cell migration ability. There was no significant difference in the initial scratch area (Fig. 4d). As shown in Fig. 4c and e, compared with the blank NPs at 24 h, the inhibitory effect of Fis/NPs was slightly stronger than that of DOX/NPs. The cell migration area of Fis-DOX/cRGD-NPs was about 10% of the initial scratch area when the fisetin concentration was 10 μg mL−1. Next, the culture continued for 48 h, the scratches in blank NPs and DOX/NPs were almost completely healed, while the remaining scratch area of Fis-DOX/cRGD-NPs was about 70% of the initial scratch area with the fisetin concentration of 10 μg mL−1 (Fig. 4c and f). These results indicated that the Fis-DOX/cRGD-NPs could significantly reduce cell migration in vitro.
In addition, the expression levels of proliferation-related proteins were further detected. As shown in Fig. 4g and Fig. S9,† GL261 cells and U87-MG cells were treated with Fis/NPs, DOX/NPs, and Fis-DOX/cRGD-NPs for 24 h, and the levels of p-AKT/AKT and p-STAT3/STAT3 decreased obviously in the Fis-DOX/cRGD-NP group. Moreover, the Fis-DOX/cRGD-NPs inhibited cell metastasis by downregulating the expression of vimentin and MMP9 and upregulating the expression of E-cadherin. AKT and STAT3 play crucial roles in tumor cell proliferation, survival, angiogenesis, migration, differentiation, invasion and immunosuppression.42,43 AKT, a collective name of a set of serine/threonine kinases, is a small gene family composed of three highly homologous members: Akt1, Akt2 and Akt3.44 The Wnt/β-catenin and PI3K/Akt/mTOR (PAM) signaling pathways were key regulators of important biological functions, including cell proliferation, epithelial–mesenchymal transition (EMT), and cellular metabolism.45,46 STAT3 has two important biological functions, one is to transduce cytokine signals, and the other is to promote the transcription of specific genes, making it the most promising new target for cancer therapy.47 These results indicated that the drug combination could effectively inhibit proliferation by reducing the activation of AKT and STAT3. E-cadherin and vimentin are representative epithelial markers and mesenchymal markers in the process of epithelial–mesenchymal transition (EMT), which are associated with cell migration and invasion.48 It has been found that the 92 kDa gelatinase B/type IV collagenase (MMP-9) has high catalytic activity toward type IV collagen that promotes tumor growth, infiltration, and invasion.49 Therefore, the Fis-DOX/cRGD-NPs inhibited cell proliferation by reducing the activation of phospho protein kinase B (AKT) and signal transducer and activator of transcription-3 (STAT3), suppressed cell metastasis by downregulating the expression of vimentin and MMP9 and upregulating the expression of E-cadherin. These results showed that the drug combination of DOX and fisetin could inhibit cell proliferation and migration in vitro.
Cellular uptake assay
We further investigated the cellular uptake of Fis/NPs, DOX/NPs and Fis-DOX/cRGD-NPs with different concentrations. Fis/NPs had almost no fluorescence signal at 2.5, 5, and 10 μg mL−1, which indicated that fisetin did not affect the fluorescence signal of DOX. Some research suggests that reduced intracellular accumulation of DOX in cancer cells is responsible for developing DOX resistance.50 Therefore, it is a critical point to enhance the tumor cell uptake of DOX in solving chemotherapy resistance. Fisetin is a flavonoid compound that acts as a P-gp inhibitor to prevent drug efflux.39 It is worth noting that the cellular uptake of DOX was enhanced with fisetin. The results showed that the cellular uptake of DOX was enhanced with the increase of fisetin concentration. Similar results were also obtained in the U87-MG cells (Fig. S9, ESI†).Fis-DOX/cRGD-NPs exerted anti-tumor effects in a subcutaneous glioma model
The anti-cancer activity of the smart-responsive nanoparticle was evaluated in a subcutaneous GL261 model. Mice were treated with normal saline (NS), blank NPs, Fis/NPs, DOX/NPs, Fis-DOX/NPs, and Fis-DOX/cRGD-NPs with tumor volume and body weight recorded. According to the tumor size, tumor growth curve and tumor weight, the tumor growth in the NS group and the blank NP group was not inhibited, and the inhibitory effect of DOX/NPs was slightly better than that of Fis/NPs. The tumor growth curves of Fis-DOX/NPs and Fis-DOX/cRGD-NPs tended to be gentle, and the most critical thing is that the anti-tumor effect in the Fis-DOX/cRGD-NP group was superior to that in other groups (Fig. 5a–c). Moreover, the Fis-DOX/cRGD-NP group mice had no significant weight change during the treatment (Fig. 5d). The results suggested that Fis-DOX/cRGD-NPs possessed better anti-tumor efficacy than other treatments, likely attributed to active targeting ability and GSH responsiveness. | ||
| Fig. 5 Combination therapy improved the anti-tumor activity in the subcutaneous GL261 model. C57BL/6 mice were subcutaneously inoculated with GL261 cells and received the following treatments by intravenous injection: NS; blank NPs; Fis/NPs; DOX/NPs; Fis-DOX/NPs or Fis-DOX/cRGD-NPs. (a) Photos of the tumors from different groups. (b) Tumor growth curves. (c) Tumor weights. (d) Body weights (n = 5; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001). | ||
Fis-DOX/cRGD-NPs exerted anti-tumor effects in an orthotopic glioma model
An orthotopic GL261-luc glioma mouse model was established to further verify the anti-glioma effect of Fis-DOX/cRGD-NPs in vivo. Similarly, mice were treated with normal saline (NS), blank NPs, Fis/NPs, DOX/NPs, Fis-DOX/NPs and Fis-DOX/cRGD-NPs. After the treatment, in vivo imaging showed that the fluorescence signal in the Fis-DOX/cRGD-NP group was weaker than in other groups, indicating that the Fis-DOX/cRGD-NPs could inhibit tumor growth significantly (Fig. 6a). Moreover, the brain tissue slices were stained with hematoxylin–eosin (HE) dye, and the results showed that the tumor volume in Fis-DOX/cRGD-NPs was the smallest (Fig. 6b and c). These results showed that the nanoparticles disintegrated in the presence of GSH, and the nanoparticles relied on the surface-loaded cRGD to penetrate the BBB and target the GBM, exhibiting outstanding targeted delivery ability and robust antitumor effects in vivo. | ||
| Fig. 6 Combination therapy improved anti-tumor activity in an orthotopic implantation glioma model. C57BL/6 mice were intracranially inoculated with GL261-Luc cells and received the following treatments by vein injection: NS; blank NPs; Fis/NPs; DOX/NPs; Fis-DOX/NPs or Fis-DOX/cRGD-NPs. (a) Optical in vivo imaging. (b) H&E staining of the brain. (c) Enlargement of the corresponding position of the tumor. | ||
Fis-DOX/cRGD-NPs promoted tumor cell apoptosis in vivo
Next, we evaluated the pro-apoptotic effect of the Fis-DOX/cRGD-NPs in vivo. Tumor tissues from the subcutaneous glioma mouse model were stained with TUNEL immunofluorescence. As shown in Fig. 7, few TUNEL-positive cells were observed in the NS, blank NP and Fis/NP groups, and the apoptosis rate of GL261 cells in the DOX/NP group was about 20%. In contrast, the Fis-DOX/cRGD-NPs significantly increased TUNEL-positive cells. The results showed that Fis-DOX/cRGD-NPs could effectively induce tumor cell apoptosis in vivo.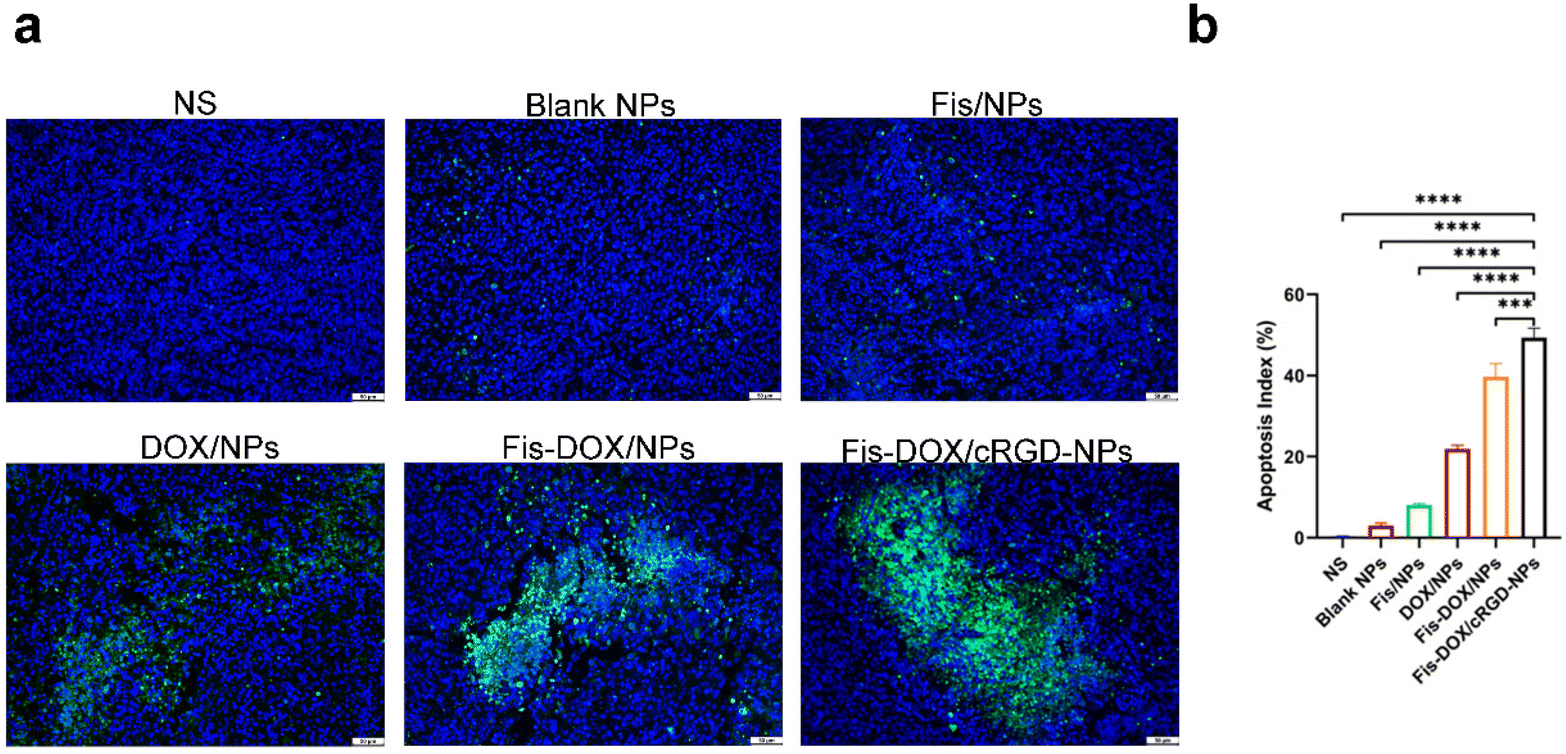 | ||
| Fig. 7 Combination therapy promoted tumor cell apoptosis in the subcutaneous GL261 glioma model. (a) TUNEL assay was applied to detect cell apoptosis in tumor sections. The photos present randomly selected field in tumor sections from different groups. (b) The average percentage of TUNEL-positive cells in each group was counted and analyzed (mean ± SEM, n = 5, ****p < 0.001). | ||
Fis-DOX/cRGD-NPs inhibited cell proliferation and angiogenesis in vivo
To further explore the anti-tumor mechanisms of Fis-DOX/cRGD-NPs in vivo, tumor tissues from the subcutaneous glioma mouse model were stained with Ki67 and CD31. As shown in Fig. 8, the Ki67-positive rate of cells was as high as 70% in NS and blank NPs, whereas the Fis-DOX/cRGD-NP treatment decreased Ki67 expression. The CD31-positive rate of cells was also reduced in the Fis-DOX/cRGD-NP group. The results indicated that Fis-DOX/cRGD-NPs could inhibit tumor cell proliferation and angiogenesis in vivo. | ||
| Fig. 8 Combination therapy inhibited tumor cell proliferation and vascular generation in a subcutaneous GL261 glioma model. (a) Ki67 staining was applied to detect proliferation in tumor tissues. (b) The average percentage of Ki67-positive cells in each group. (c) CD31 staining was performed to visualize the tumor vascular generation. (d) The numbers of CD31-positive vessels in each group. Scale bars is 50 μm (n = 5, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001). | ||
Biosafety evaluation
The body weight changes of mice were continuously monitored during the treatment. As shown in Fig. 5d, Fis-DOX/cRGD-NPs administration had no significant impact on body weight. H&E staining of the vital organs was performed and showed no abnormality in the Fis-DOX/cRGD-NP group (Fig. S10, ESI†). The serum samples from different treatment groups were obtained for biochemical testing, and the values were within normal limits (Fig. S11, ESI†). Therefore, the targeted nanodelivery systems proved to possess reasonable biosafety in the animal experiment.Conclusion
In this study, we developed an active-targeting and stimulus-responsive nanoplatform coloaded with fisetin and DOX (Fis-DOX/cRGD-NPs) to cross the BBB for glioma targeting and combinational chemotherapy. The results demonstrate that Fis-DOX/cRGD-NPs can be efficiently loaded with fisetin and DOX through the thin film rehydration method and acquire desired stability, BBB crossing ability and glioma cancer cell targeting specificity upon polypeptide modification. The Fis-DOX/cRGD-NPs could result in obvious tumor suppression by inducing tumor cell cycle arrest, promoting tumor cell apoptosis, inhibiting tumor cell proliferation and migration, and enhancing the uptake of DOX in vitro. Furthermore, the micelles exerted excellent chemotherapeutic effects on the subcutaneous GL261 model and the orthotopic glioma model in vivo. Collectively, the created Fis-DOX/cRGD-NPs with an active-targeting ability and GSH-responsiveness may be developed as a promising theranostic nanomedicine formulation to cross the BBB and sensitize chemotherapies.Author contributions
Wanyu Wang and Xiang Wang conceived and supervised the work. Wanyu Wang, Yuanyuan Zhang and Yue Jian participated in the design and implementation of the experiment. Shi He, Jiagang Liu, Yongzhong Cheng, Songping Zheng carried out data processing and analysis. Xiang Wang, Zhiyong Qian and Xiang Gao revised the manuscript. All authors discussed and approved the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no. 32222046, 82172630, 82170844 and 82270613, China) and the Sichuan Science and Technology Program (no. 2023YFS0271, China).References
- L. R. Schaff and I. K. Mellinghoff, J. Am. Med. Assoc., 2023, 329, 574–587 CrossRef PubMed
.
- S. Lapointe, A. Perry and N. A. Butowski, Lancet, 2018, 392, 432–446 CrossRef PubMed
- R. Stupp, W. P. Mason, M. J. van den Bent, M. Weller, B. Fisher, M. J. Taphoorn, K. Belanger, A. A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R. C. Janzer, S. K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J. G. Cairncross, E. Eisenhauer, R. O. Mirimanoff, European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups and National Cancer Institute of Canada Clinical Trials Group, N. Engl. J. Med., 2005, 352, 987–996 CrossRef CAS PubMed
- O. van Tellingen, B. Yetkin-Arik, M. C. de Gooijer, P. Wesseling, T. Wurdinger and H. E. de Vries, Drug Resistance Updates, 2015, 19, 1–12 CrossRef CAS PubMed
- Z. Zhao, A. R. Nelson, C. Betsholtz and B. V. Zlokovic, Cell, 2015, 163, 1064–1078 CrossRef CAS PubMed
- J. Ding, G. Lu, W. Nie, L. L. Huang, Y. Zhang, W. Fan, G. Wu, H. Liu and H. Y. Xie, Adv. Mater., 2021, 33, e2005562 CrossRef PubMed
- G. Chen, R. Jaskula-Sztul, C. R. Esquibel, I. Lou, Q. Zheng, A. Dammalapati, A. Harrison, K. W. Eliceiri, W. Tang, H. Chen and S. Gong, Adv. Funct. Mater., 2017, 27, 1604671 CrossRef PubMed
- T. Cui, S. Zhang and H. Sun, Oncol. Rep., 2017, 37, 1253–1260 CrossRef CAS PubMed
- H. Wu, X. Gao, Y. Luo, J. Yu, G. Long, Z. Jiang and J. Zhou, Adv. Sci., 2022, 9, e2203894 CrossRef PubMed
- M. Hu, J. Zhang, L. Kong, Y. Yu, Q. Hu, T. Yang, Y. Wang, K. Tu, Q. Qiao, X. Qin and Z. Zhang, ACS Nano, 2021, 15, 3123–3138 CrossRef CAS PubMed
- L. Gao, Z.-X. Wu, Y. G. Assaraf, Z.-S. Chen and L. Wang, Drug Resistance Updates, 2021, 57, 100770 CrossRef CAS PubMed
- M. Ozcan, Z. Guo, C. Valenzuela Ripoll, A. Diab, A. Picataggi, D. Rawnsley, A. Lotfinaghsh, C. Bergom, J. Szymanski, D. Hwang, A. Asnani, M. Kosiborod, J. Zheng, R. J. Hayashi, P. K. Woodard, A. Kovacs, K. B. Margulies, J. Schilling, B. Razani, A. Diwan and A. Javaheri, Cell Metab., 2023, 35, 928–942 CrossRef CAS PubMed
- Y. He, X. Zan, J. Miao, B. Wang, Y. Wu, Y. Shen, X. Chen, H. Gou, S. Zheng, N. Huang, Y. Cheng, Y. Ju, X. Fu, Z. Qian, P. Zhou, J. Liu and X. Gao, Mater. Today Bio, 2022, 16, 100423 CrossRef CAS PubMed
- Y. Zhang, T. Li, Y. Hu, J. Chen, Y. He, X. Gao and Y. Zhang, Chin. Chem. Lett., 2022, 33, 2507–2511 CrossRef CAS
- K. Sundarraj, A. Raghunath and E. Perumal, Biomed. Pharmacother., 2018, 97, 928–940 CrossRef CAS PubMed
- M. J. Yousefzadeh, Y. Zhu, S. J. McGowan, L. Angelini, H. Fuhrmann-Stroissnigg, M. Xu, Y. Y. Ling, K. I. Melos, T. Pirtskhalava, C. L. Inman, C. McGuckian, E. A. Wade, J. I. Kato, D. Grassi, M. Wentworth, C. E. Burd, E. A. Arriaga, W. L. Ladiges, T. Tchkonia, J. L. Kirkland, P. D. Robbins and L. J. Niedernhofer, EBioMedicine, 2018, 36, 18–28 CrossRef PubMed
- D. Kashyap, V. K. Garg, H. S. Tuli, M. B. Yerer, K. Sak, A. K. Sharma, M. Kumar, V. Aggarwal and S. S. Sandhu, Biomolecules, 2019, 9, 174 CrossRef CAS PubMed
- K. H. Lin, S. Ramesh, S. Agarwal, W. W. Kuo, C. H. Kuo, M. Y. Chen, Y. M. Lin, T. J. Ho, P. C. Huang and C. Y. Huang, Phytother. Res., 2023, 37, 3964–3981 CrossRef CAS PubMed
- Y. Zou, Y. Wang, S. Xu, Y. Liu, J. Yin, D. B. Lovejoy, M. Zheng, X. J. Liang, J. B. Park, Y. M. Efremov, I. Ulasov and B. Shi, Adv. Mater., 2022, 34, e2203958 CrossRef
- Y. Zhang, K. Xi, X. Fu, H. Sun, H. Wang, D. Yu, Z. Li, Y. Ma, X. Liu, B. Huang, J. Wang, G. Li, J. Cui, X. Li and S. Ni, Biomaterials, 2021, 278, 121163 CrossRef CAS PubMed
- M. Zhao, D. van Straten, M. L. D. Broekman, V. Préat and R. M. Schiffelers, Theranostics, 2020, 10, 1355–1372 CrossRef CAS PubMed
- J. Cui, Y. Xu, H. Tu, H. Zhao, H. Wang, L. Di and R. Wang, Acta Pharm. Sin. B, 2022, 12, 1100–1125 CrossRef CAS PubMed
- T. Tian, R. Liang, G. Erel-Akbaba, L. Saad, P. J. Obeid, J. Gao, E. A. Chiocca, R. Weissleder and B. A. Tannous, ACS Nano, 2022, 16, 1940–1953 CrossRef CAS PubMed
- N. Qi, S. Zhang, X. Zhou, W. Duan, D. Gao, J. Feng and A. Li, J. Nanobiotechnol., 2021, 19, 446 CrossRef CAS PubMed
- D. Arosio and C. Casagrande, Adv. Drug Delivery Rev., 2016, 97, 111–143 CrossRef CAS PubMed
- E. Ruoslahti and M. D. Pierschbacher, Science, 1987, 238, 491–497 CrossRef CAS PubMed
- W. Niu, Q. Xiao, X. Wang, J. Zhu, J. Li, X. Liang, Y. Peng, C. Wu, R. Lu, Y. Pan, J. Luo, X. Zhong, H. He, Z. Rong, J.-B. Fan and Y. Wang, Nano Lett., 2021, 21, 1484–1492 CrossRef CAS PubMed
- P. Zhao, Y. Tian, Y. Lu, J. Zhang, A. Tao, G. Xiang and Y. Liu, J. Nanobiotechnol., 2022, 20, 525 CrossRef CAS PubMed
- M. H. Lee, Z. Yang, C. W. Lim, Y. H. Lee, S. Dongbang, C. Kang and J. S. Kim, Chem. Rev., 2013, 113, 5071–5109 CrossRef CAS PubMed
- F. Shen, D. Tao, R. Peng, Y. He, Z. Liu, J. Ji and L. Feng, Biomaterials, 2022, 283, 121428 CrossRef CAS PubMed
- X. Ma, S. Yang, T. Zhang, S. Wang, Q. Yang, Y. Xiao, X. Shi, P. Xue, Y. Kang, G. Liu, Z. J. Sun and Z. Xu, Acta Pharm. Sin. B, 2022, 12, 451–466 CrossRef CAS PubMed
- D. N. Louis, A. Perry, G. Reifenberger, A. von Deimling, D. Figarella-Branger, W. K. Cavenee, H. Ohgaki, O. D. Wiestler, P. Kleihues and D. W. Ellison, Acta Neuropathol., 2016, 131, 803–820 CrossRef PubMed
- D. N. Louis, A. Perry, P. Wesseling, D. J. Brat, I. A. Cree, D. Figarella-Branger, C. Hawkins, H. K. Ng, S. M. Pfister, G. Reifenberger, R. Soffietti, A. von Deimling and D. W. Ellison, Neuro-Oncology, 2021, 23, 1231–1251 CrossRef CAS PubMed
- E. P. W. Jenkins, A. Finch, M. Gerigk, I. F. Triantis, C. Watts and G. G. Malliaras, Adv. Sci., 2021, 8, e2100978 CrossRef PubMed
- M. Li, Y. Wang, M. Li, X. Wu, S. Setrerrahmane and H. Xu, Acta Pharm. Sin. B, 2021, 11, 2726–2737 CrossRef CAS PubMed
- Z. Zhou, H. Liang, R. Yang, Y. Yang, J. Dong, Y. Di and M. Sun, Angew. Chem., Int. Ed., 2022, 61, e202202843 CrossRef CAS PubMed
- Z. Li, Y. Zhu, H. Zeng, C. Wang, C. Xu, Q. Wang, H. Wang, S. Li, J. Chen, C. Xiao, X. Yang and Z. Li, Nat. Commun., 2023, 14, 1437 CrossRef CAS PubMed
- S. Shen, T. Li, J. Fan, Q. Shao, H. Dong, X. Xu and R. Mo, Acta Pharm. Sin. B, 2023, 13, 1262–1273 CrossRef CAS PubMed
- H. M. Abdallah, A. M. Al-Abd, R. S. El-Dine and A. M. El-Halawany, J. Adv. Res., 2015, 6, 45–62 CrossRef CAS PubMed
- G. Ding, X. Xu, D. Li, Y. Chen, W. Wang, D. Ping, S. Jia and L. Cao, Cell Death Dis., 2020, 11, 893 CrossRef CAS PubMed
- Q. Ren, F. Guo, S. Tao, R. Huang, L. Ma and P. Fu, Biomed. Pharmacother., 2020, 122, 109772 CrossRef CAS PubMed
- D. E. Johnson, R. A. O'Keefe and J. R. Grandis, Nat. Rev. Clin. Oncol., 2018, 15, 234–248 CrossRef CAS PubMed
- H. Sun, C. Wang, B. Hu, X. Gao, T. Zou, Q. Luo, M. Chen, Y. Fu, Y. Sheng, K. Zhang, Y. Zheng, X. Ren, S. Yan, Y. Geng, L. Yang, Q. Dong and L. Qin, Signal Transduction Targeted Ther., 2021, 6, 187 CrossRef CAS PubMed
- D. M. Hyman, L. M. Smyth, M. T. A. Donoghue, S. N. Westin, P. L. Bedard, E. J. Dean, H. Bando, A. B. El-Khoueiry, J. A. Pérez-Fidalgo, A. Mita, J. H. M. Schellens, M. T. Chang, J. B. Reichel, N. Bouvier, S. D. Selcuklu, T. E. Soumerai, J. Torrisi, J. P. Erinjeri, H. Ambrose, J. C. Barrett, B. Dougherty, A. Foxley, J. P. O. Lindemann, R. McEwen, M. Pass, G. Schiavon, M. F. Berger, S. Chandarlapaty, D. B. Solit, U. Banerji, J. Baselga and B. S. Taylor, J. Clin. Oncol., 2017, 35, 2251–2259 CrossRef CAS PubMed
- J. Liu, Q. Xiao, J. Xiao, C. Niu, Y. Li, X. Zhang, Z. Zhou, G. Shu and G. Yin, Signal Transduction Targeted Ther., 2022, 7, 3 CrossRef CAS PubMed
- R. J. Shaw and L. C. Cantley, Nature, 2006, 441, 424–430 CrossRef CAS PubMed
- H. Yu, H. Lee, A. Herrmann, R. Buettner and R. Jove, Nat. Rev. Cancer, 2014, 14, 736–746 CrossRef CAS PubMed
- J. Yang, P. Antin, G. Berx, C. Blanpain, T. Brabletz, M. Bronner, K. Campbell, A. Cano, J. Casanova, G. Christofori, S. Dedhar, R. Derynck, H. L. Ford, J. Fuxe, A. Garcia de Herreros, G. J. Goodall, A. K. Hadjantonakis, R. Y. J. Huang, C. Kalcheim, R. Kalluri, Y. Kang, Y. Khew-Goodall, H. Levine, J. Liu, G. D. Longmore, S. A. Mani, J. Massague, R. Mayor, D. McClay, K. E. Mostov, D. F. Newgreen, M. A. Nieto, A. Puisieux, R. Runyan, P. Savagner, B. Stanger, M. P. Stemmler, Y. Takahashi, M. Takeichi, E. Theveneau, J. P. Thiery, E. W. Thompson, R. A. Weinberg, E. D. Williams, J. Xing, B. P. Zhou, G. Sheng and E. M. T. I. Association, Nat. Rev. Mol. Cell Biol., 2020, 21, 341–352 CrossRef CAS PubMed
- Q. X. Sang, Cell Res., 1998, 8, 171–177 CrossRef CAS PubMed
- X. Wang, Y. Zhao, Y. Hu, Y. Fei, Y. Zhao, C. Xue, K. Cai, M. Li and Z. Luo, Small, 2021, 17, e2102269 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr05195a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |