
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning of TRAIL clustering on the surface of nanoscale liposomes by phase separation†
Zhenjiang
Zhang
and
Michael R.
King
 *
*
Department of Biomedical Engineering, Vanderbilt University, 2414 Highland Ave, Nashville, TN 37212, USA. E-mail: mike.king@vanderbilt.edu
First published on 29th December 2023
Abstract
Phase-separated liposomes were used to formulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), a protein that selectively kills cancer cells while sparing most healthy ones. By controlling the average number of TRAIL molecules per liposome, we demonstrate the ability to tune the formation of TRAIL clusters and their resulting apoptotic activity.
Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) was discovered as a type II transmembrane protein that specifically induces apoptosis in a broad spectrum of cancer cell lines while sparing most healthy ones.1 Extensive pre-clinical studies of TRAIL as a target of cancer yielded promising observations.2 However, the following >10 clinical trials to test TRAIL receptor agonistic antibodies, recombinant TRAIL, and other TRAIL based agents have showed little to no antitumor efficacy.3 It recently emerged that the resistance of human tumors to TRAIL-induced apoptosis is largely due to the poor agonistic activity of these agents.4 Efforts to increase the bioactivity of TRAIL-based agents have continued with the goal of developing new cancer-specific therapies.5
TRAIL activates the extrinsic apoptotic pathway upon binding to its cognate death receptors 4 and 5 (DR4 and DR5, also known as TRAIL-R1 and TRAIL-R2, respectively).6 Like other members of the TNF family, TRAIL must form a homotrimer to achieve its cancer cell-specific toxicity.7 DR4/5 receptors bind to the gaps between two neighboring monomers in the TRAIL trimer, one receptor to each of the three gaps.8 The binding results in receptor trimerization that leads to conformational changes in the TRAIL receptors.9 However, this conformation change alone is not always sufficient to transduce the intracellular signal. It seems that further multimerization of the receptors is required to complete the apoptotic signalling cascade.8 Previous studies have shown that some DRs need to be further cross-linked or oligomerized in high-order clusters on the cell surface to correctly induce the intracellular signal.10 It is possible that death ligands like TRAIL may achieve this by clustering into supra-molecular structures on the membranes of cells or cell-secreted vesicles. This idea is supported by the discovery that TRAIL is much more potent as a naturally expressed transmembrane protein than when in its free, soluble form.11
Different methods have been reported to increase the cancer-specific apoptotic activity of TRAIL. One of the earlier methods focused on increasing the stability of TRAIL homotrimer to optimize its pharmacokinetic profile.12 For this purpose, TRAIL has often been expressed in fusion with a peptide motif that is known to form trimers. Another way has been to increase TRAIL targeting by fusing TRAIL with domains or motifs that specifically target cancer cells or cells of the tumor stroma. Single-chain variable antibody fragments (scFv) have been the most popular domain explored for this purpose due to their equivalent targeting ability to antibody yet a much smaller size.13 Nanomaterials have also been extensively studied to increase TRAIL targeting due to the so called Enhanced Permeability and Retention (EPR) Effect in solid tumors.14 Nanomaterials also provide a surface on which TRAIL may be assembled into supra-molecular structures.15 Various types of nanomaterials including lipid nanoparticles, polymeric micelles, microspheres, and magnetic particles are available for TRAIL formulation and delivery.16 Among them, liposomes have been found to be the most versatile of these platforms due to well-established engineering methods and safety profile.17 Our lab previously developed leukocyte targeting liposomes by conjugating TRAIL, and another protein E-selectin (ES), to the surface of liposomes. The liposomes adhere to the surface of leukocytes following systemic administration since ES binds to sialyl LewisX, a characteristic surface glycan expressed by most leukocytes. We have demonstrated that such liposome formulations can block new formation of metastases by killing circulating tumor cells (CTCs) in mouse models of prostate cancer and breast cancer.18,19
In this study, we report a new method to increase the cancer-specific apoptotic activity of TRAIL by using phase separation of the surface of nanoscale liposomes. The liposomes were designed to have two distinct surface domains or phases and only the smaller phase allows conjugation of recombinant TRAIL. The density of the protein molecules can thus be controlled to form an optimal density of supramolecule clusters compared to even distribution on the liposome surface. By controlling the clustering of TRAIL on phase-separated liposomes, we can tune their apoptotic potential in a range higher than free TRAIL.
Preparation and characterization of phase-separated giant unilamellar vesicles (GUVs) and nanoscale liposomes
It is challenging to directly observe the phase separation on nanoscale liposomes using optical microscopy. We used giant unilamellar vesicles (GUVs) which are microsized vesicles as an alternative to liposomes to better visualize the phase separation.20 The three major lipids for GUVs and liposomes include DSPC, DOPC and cholesterol at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1.2. These lipids and their ratio were chosen because the phase separation behavior of GUVs and liposomes made of them has been well studied.21,22 The GUVs can be controlled to exhibit two distinct phases, liquid-ordered (Lα) phase formed mainly by DSPC and liquid-disordered (Lβ) phase mainly by DOPC. A small percentage of DGS-NTA-Ni was included for TRAIL conjugation and DSPE-PEG for stabilization of the vesicles.
:1:1.2. These lipids and their ratio were chosen because the phase separation behavior of GUVs and liposomes made of them has been well studied.21,22 The GUVs can be controlled to exhibit two distinct phases, liquid-ordered (Lα) phase formed mainly by DSPC and liquid-disordered (Lβ) phase mainly by DOPC. A small percentage of DGS-NTA-Ni was included for TRAIL conjugation and DSPE-PEG for stabilization of the vesicles.
The GUVs were made using a so-called gentle hydration method in which the lipid film was hydrated gently with PBS without vibration or stirring.23 The GUVs were directly visualized with confocal microscopy as the DSPC phase was labeled in green while DOPC was labeled in red. The diameter of large GUVs reached ∼15 μm with small Lβ phases ranging from 2 to 13 μm (Fig. 1A). The number of Lβ phases on an individual GUV varied. These phase separation results suggest the formation of smaller domains on the membrane of nanoscale liposomes that would be later prepared using the same lipid composition.
 | ||
| Fig. 1 Preparation and characterization of GUVs and liposomes. (A) Typical confocal microscopy images of GUVs showing separation of phases (green = DSPC, red = DOPC) to different extent. Scale bars = 10 μm. (B) Size distribution of naked liposomes and liposomes conjugated with varying numbers of TRAIL molecules. | ||
Nanoscale liposomes were prepared using an extrusion method following hydration of the lipid film with vortexing. The average size of the liposomes was measured to be ∼106 ± 3.7 nm. The number of liposomes was estimated using an equation on https://www.liposomes.org. Various amounts of TRAIL protein were added to a liposome suspension to conjugate different average numbers of TRAIL on an individual liposome, assuming the conjugation yield is 100%. The liposomes conjugated with 100, 200 and 400 TRAIL molecules on average on an individual liposome are designated as 100T-, 200T- and 400T-Lipo, respectively. The size of liposomes becomes larger with more TRAIL conjugated, suggesting success of TRAIL association with the liposome surface (Fig. 1B).
Cytotoxicity of phase-separated TRAIL liposomes
We used human prostate cancer PC3 and DU145 cells (both purchased from American Type Culture Collection) which exhibit high and medium sensitivity to TRAIL, respectively, to compare the apoptotic activity of the phase-separated liposomes conjugated with different numbers of TRAIL molecules.24 Our previous studies found that each of these two cell lines expresses both DR4 and DR5, with a higher expression of DR5 than DR4.25 In both of the cell lines, the TRAIL liposomes showed higher cytotoxic effects than free TRAIL after 24 h of treatment. More importantly, the liposomes conjugated with more TRAIL molecules are more potent that those conjugated with fewer TRAIL molecules at the same total concentration of TRAIL (Fig. 2A and B). In the treatment of DU145 cells, 400T-Lipo achieved more than twice the apoptotic activity of free TRAIL. It was reported that DR4 requires colocalization with lipid rafts on cell membranes to initiate the apoptotic signalling but doesn't need clustered TRAIL while DR5, on the contrary, strictly needs membrane bound/clustered TRAIL but are typically not localized in specific membrane domains.8 Therefore, these results are consistent with the idea that more TRAIL on the liposome surface, potentially forming larger TRAIL clusters, can more potently engage DR5 to initiate apoptotic signalling. It is also suggested that it is likely to control the clustering of TRAIL on the liposome surface with phase separation to achieve optimal apoptotic capability of TRAIL to specific cancer cell lines.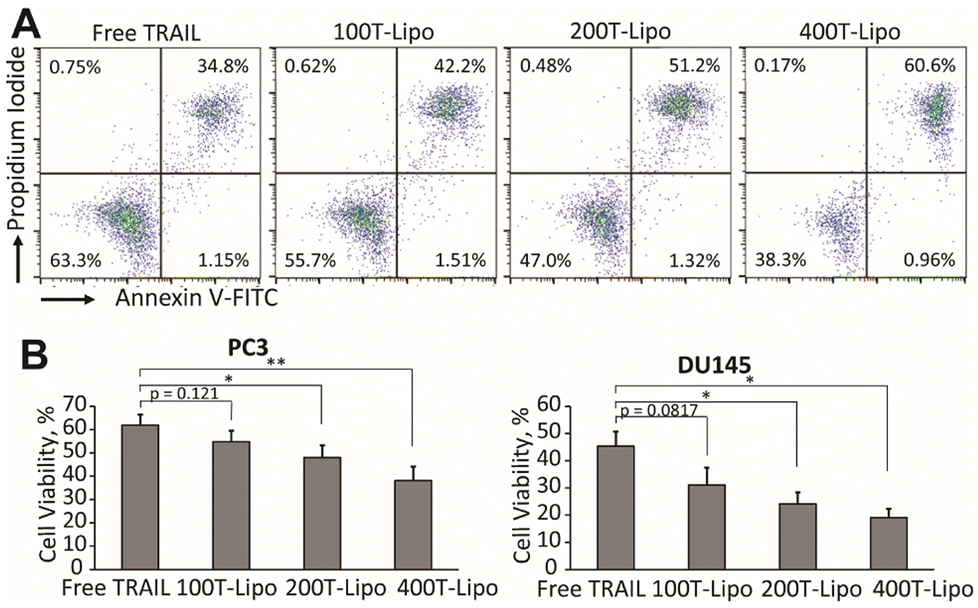 | ||
| Fig. 2 Apoptotic activity of TRAIL liposomes. (A) Representative flow cytometry plots of PC3 cells after treatment with TRAIL liposomes. Cell viability of PC3 cells and DU145 cells (B). Incubation concentrations of TRAIL liposomes are 100 ng mL−1 TRAIL for the treatment of PC3 and 250 ng mL−1 TRAIL for DU145 cells. *p < 0.05 and **p < 0.01. | ||
Cytotoxic effects of phase-separated TRAIL liposomes on Jurkat cells
We next compared the apoptotic activity of the TRAIL liposomes in human T lymphocyte immortalized Jurkat cells which are known to show a high expression of DR5 and are highly sensitive to TRAIL-induced apoptosis.26 Opposite to our expectation, the TRAIL liposomes with fewer TRAIL were more potent than those with more TRAIL, although all three formulations are more apoptotic than free TRAIL (Fig. 3A). One possible reason for this is that the density of TRAIL clusters of 100T-Lipo matched better with the distribution density of DR5 on the surface of Jurkat cells than the other two formulations. Therefore, TRAIL molecules of 200T-Lipo and 400T-Lipo were more than sufficient and partly wasted. We then used confocal microscopy to observe the binding of Jurkat cells with TRAIL liposomes, compared to naked liposomes as a control. The liposomes are labelled with two dyes in the DSPC and DOPC phases, respectively. The merging of the two colors indicates an intact structure of the liposomes. No binding of naked liposomes to the cell surface or internalization was observed within the first hour of incubation. The TRAIL liposomes were found to surround the membrane of Jurkat cells (Fig. 3B). Some of the TRAIL liposomes were internalized as supported by the two diffusive colors in the cytoplasm of the Jurkat cells (Fig. 3B). DR4/5 are known to translocate to the nucleus after activation by TRAIL. The rate of translocation of these death receptors induced by the TRAIL liposomes appears to be much faster than the endocytosis of naked liposomes. | ||
| Fig. 3 Apoptotic activity of the TRAIL liposomes to Jurkat cells. (A) Cell viability of Jurkat cells. TRAIL concentration was 50 ng mL−1 *p < 0.05 and ***p < 0.001. (B) Confocal microscopy images showing the binding of TRAIL liposomes to the membrane of Jurkat cells and their internalization at 1 h after incubation. TRAIL concentration was 10 μg mL−1. | ||
Conclusions
Phase separation naturally exists in biological membranes and is directly connected to various events in the life of a living cell.27 Many intriguing phase behaviors of biological membranes have been discovered and studied using GUVs as a model system.28 Although nanoscale phases cannot be visualized directly by optical microscopy, their existence and properties have been well documented with indirect methods such as neutron scattering.29It was previously reported that the cytotoxic potential of TRAIL can be enhanced by conjugating TRAIL to the surface of lipid-homogenous liposomes.30 It is hard to compare those formulations with those in this research since the average number of TRAIL molecules conjugated on the homogenous liposomes was not reported. In theory, it is possible to fully or nearly fully coat a homogenous liposome with TRAIL to promote the formation of TRAIL clusters. But this could compromise the stability of liposomes in vivo and the long-term stability in vitro. Our studies provide a facile method to prepare an optimal liposome formulation of TRAIL for a specific cell line by controlling the ratio of the two phases and average number of TRAIL per liposome.
In short, we prepared nanoscale liposomes that exhibit two distinct phases in their membrane. By conjugating TRAIL to only the smaller of the two phases, the density of TRAIL on the liposome surface was increased to promote cluster formation. Fine tuning of the TRAIL density on the smaller phase of an individual liposome yielded TRAIL liposomes with different levels of apoptotic activity. The relative apoptotic potential of the TRAIL liposomes with different numbers of TRAIL molecules was also found to be dependent on the types of tumor cells. These findings lay the foundation for the development of TRAIL liposomes with optimal activity for a specific type of tumor and thus may expedite their translation into the clinic.
Author contributions
Z. Z. and M. R. K. conceptualized the project. Z. Z. completed the experiments and wrote the original draft. M. R. K. supervised the work and edited the manuscript.Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This article was funded in part by National Institutes of Health, Grant No. CA256054 to M. R. K.References
- G. Pan, J. Ni, Y.-F. Wei, G.-l. Yu, R. Gentz and V. M. Dixit, Science, 1997, 277, 815–818 CrossRef CAS PubMed.
- D. W. Stuckey and K. Shah, Trends Mol. Med., 2013, 19, 685–694 CrossRef CAS.
- M. Snajdauf, K. Havlova, J. Vachtenheim Jr, A. Ozaniak, R. Lischke, J. Bartunkova, D. Smrz and Z. Strizova, Front. Mol. Biosci., 2021, 8, 628332 CrossRef CAS PubMed.
- R. Trivedi and D. P. Mishra, Front. Oncol., 2015, 5, 69 Search PubMed.
- D. de Miguel, J. Lemke, A. Anel, H. Walczak and L. Martinez-Lostao, Cell Death Differ., 2016, 23, 733–747 CrossRef CAS.
- X. Yuan, A. Gajan, Q. Chu, H. Xiong, K. Wu and G. S. Wu, Cancer Metastasis Rev., 2018, 37, 733–748 CrossRef CAS PubMed.
- A. Montinaro and H. Walczak, Cell Death Differ., 2023, 30, 237–249 CrossRef CAS.
- J. Naval, D. de Miguel, A. Gallego-Lleyda, A. Anel and L. Martinez-Lostao, Cancers, 2019, 11, 444 CrossRef CAS PubMed.
- C. C. Valley, A. K. Lewis, D. J. Mudaliar, J. D. Perlmutter, A. R. Braun, C. B. Karim, D. D. Thomas, J. R. Brody and J. N. Sachs, J. Biol. Chem., 2012, 287, 21265–21278 CrossRef CAS PubMed.
- H. Wajant, Cancers, 2019, 11, 954 CrossRef CAS.
- H. Wajant, D. Moosmayer, T. Wüest, T. Bartke, E. Gerlach, U. Schönherr, N. Peters, P. Scheurich and K. Pfizenmaier, Oncogene, 2001, 20, 4101–4106 CrossRef CAS PubMed.
- D. V. Rozanov, A. Y. Savinov, V. S. Golubkov, O. L. Rozanova, T. I. Postnova, E. A. Sergienko, S. Vasile, A. E. Aleshin, M. F. Rega and M. Pellecchia, Mol. Cancer Therap., 2009, 8, 1515–1525 CrossRef CAS PubMed.
- M. Siegemund, O. Seifert, M. Zarani, T. Džinić, V. De Leo, D. Göttsch, S. Münkel, M. Hutt, K. Pfizenmaier and R. E. Kontermann, mAbs, 2016, 8, 879–891 CrossRef CAS PubMed.
- Z. Zhang, S. B. Patel and M. R. King, Molecules, 2020, 26, 157 CrossRef PubMed.
- H. Li, J. Zhao, A. Wang, Q. Li and W. Cui, Colloids Surf., A, 2020, 590, 124486 CrossRef CAS.
- P. P. Guimarães, S. Gaglione, T. Sewastianik, R. D. Carrasco, R. Langer and M. J. Mitchell, ACS Nano, 2018, 12, 912–931 CrossRef PubMed.
- M. J. Mitchell, E. Wayne, K. Rana, C. B. Schaffer and M. R. King, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 930–935 CrossRef CAS PubMed.
- N. Jyotsana, Z. Zhang, L. E. Himmel, F. Yu and M. R. King, Sci. Adv., 2019, 5, eaaw4197 CrossRef CAS.
- E. C. Wayne, S. Chandrasekaran, M. J. Mitchell, M. F. Chan, R. E. Lee, C. B. Schaffer and M. R. King, J. Controlled Release, 2016, 223, 215–223 CrossRef CAS PubMed.
- O. Wesołowska, K. Michalak, J. Maniewska and A. B. Hendrich, Acta Biochim. Pol., 2009, 56, 33–39 CrossRef.
- J. Zhao, J. Wu, F. A. Heberle, T. T. Mills, P. Klawitter, G. Huang, G. Costanza and G. W. Feigenson, Biochim. Biophys. Acta, 2007, 1768, 2764–2776 CrossRef CAS PubMed.
- T. Q. Vu, J. A. Peruzzi, L. E. Sant'Anna, E. W. Roth and N. P. Kamat, Nano Lett., 2022, 22, 2627–2634 CrossRef CAS PubMed.
- K. Shohda, K. Takahashi and A. Suyama, Biochem. Biophys. Rep., 2015, 3, 76–82 CAS.
- K. A. Grayson, N. Jyotsana, N. Ortiz-Otero and M. R. King, PLoS ONE, 2021, 16, e0246733 CrossRef CAS PubMed.
- K. A. Grayson, J. M. Hope, W. Wang, C. A. Reinhart-King and M. R. King, Mol. Cancer Ther., 2021, 20, 833–845 CrossRef CAS PubMed.
- Y.-J. Jang, K. S. Park, H.-Y. Chung and H.-I. Kim, Cancer Lett., 2003, 194, 107–117 CrossRef CAS PubMed.
- F. A. Heberle and G. W. Feigenson, Cold Spring Harbor Perspect. Biol., 2011, 3, a004630 Search PubMed.
- S. F. Fenz and K. Sengupta, Integr. Biol. Macro, 2012, 4, 982–995 CrossRef CAS PubMed.
- J. D. Nickels, X. Cheng, B. Mostofian, C. Stanley, B. Lindner, F. A. Heberle, S. Perticaroli, M. Feygenson, T. Egami, R. F. Standaert, J. C. Smith, D. A. A. Myles, M. Ohl and J. Katsaras, J. Am. Chem. Soc., 2015, 137, 15772–15780 CrossRef CAS PubMed.
- D. De Miguel, A. Gallego-Lleyda, A. Anel and L. Martinez-Lostao, Leuk. Res., 2015, 39, 657–666 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00841j |
| This journal is © The Royal Society of Chemistry 2024 |