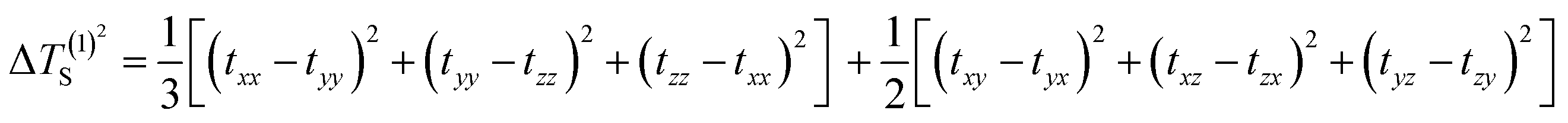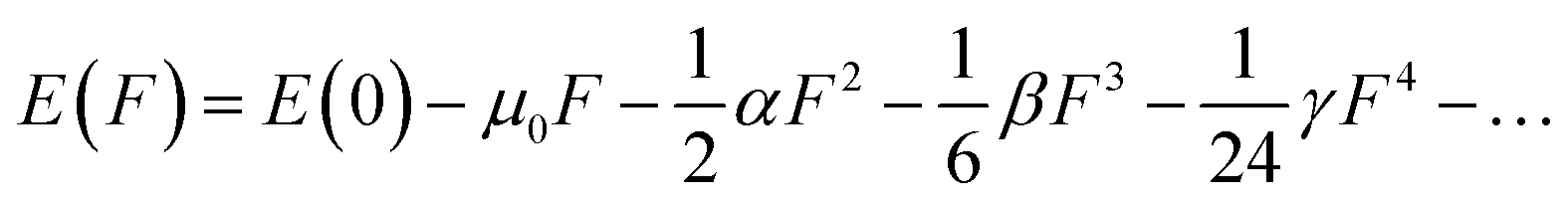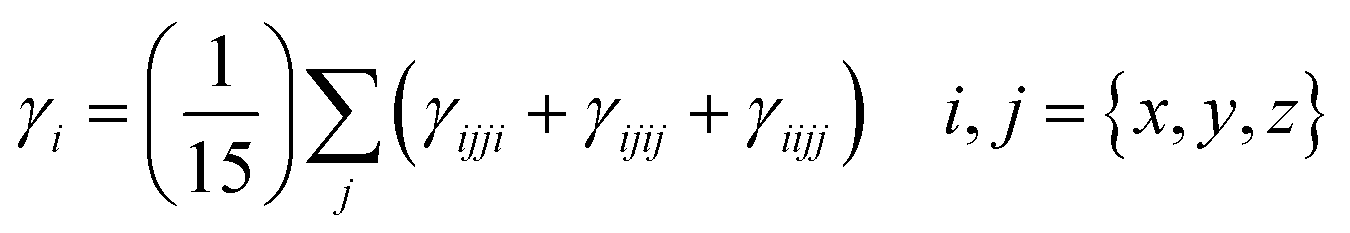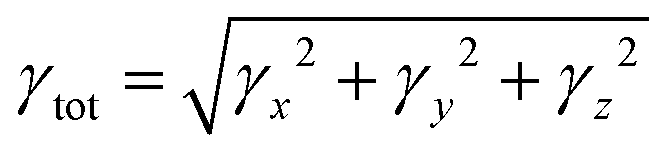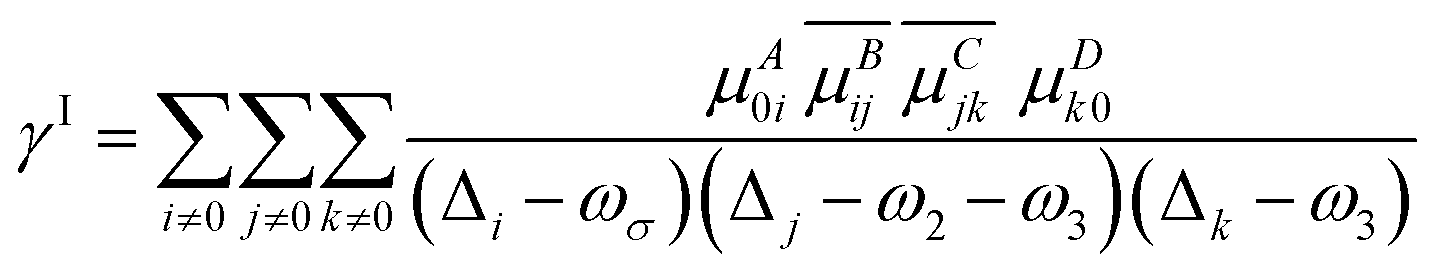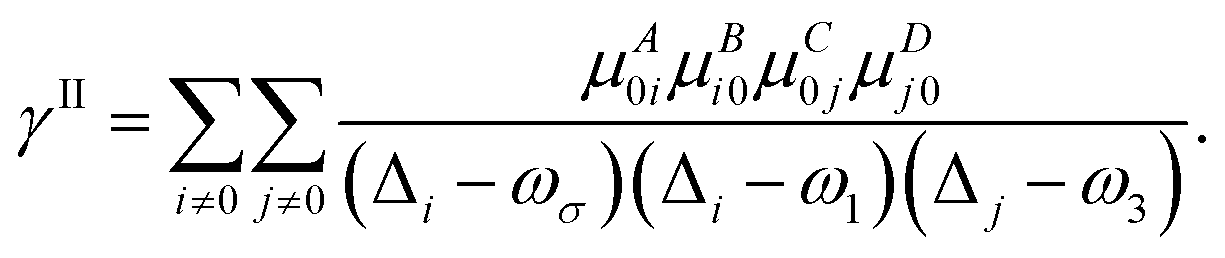DOI:
10.1039/D3MH01538F
(Communication)
Mater. Horiz., 2024,
11, 297-302
Designed metal–organic π-clusters combining the aromaticity of the metal cluster and ligands for a third-order nonlinear optical response†
Received
26th September 2023
, Accepted 1st November 2023
First published on 3rd November 2023
Abstract
The pivotal role of clusters and aromaticity in chemistry is undeniable, but there remains a gap in systematically understanding the aromaticity of metal–organic clusters. Herein, this article presents a novel metal–organic π-cluster, melding both metal–organic chemistry and aromaticity, to guide the construction of structurally stable Os-organic π-clusters. An in-depth analysis of these clusters reveals their bonding attributes, π-electronic composition, and origins of aromaticity, thereby confirming their unique metal–organic π-cluster properties. Furthermore, the Os5 cluster exhibits a promising third-order nonlinear optical (NLO) response, attributable to its narrow band gap and uniform electron/hole distribution, suggesting its potential as an optical switching material. This research introduces a fresh perspective on clusters, centered on delocalization, and broadens the domain of aromaticity studies. It also presents a novel method for designing efficient third-order NLO materials through consideration of the structure–activity relationship.
New concepts
Nonlinear optical (NLO) materials demonstrate broad application perspectives, as their applications in atomic cooling, quantum information storage, photonic flywheel, and optical switching. Combining the notable applications of π-electrons and cluster materials in third-order NLO materials, this article introduces a new concept called “metal–organic π-cluster”. Metal–organic chemistry and aromaticity are combined in this concept to guide the construction of structurally stable Os-organic π-clusters. Most existing theories mainly emphasis on geometric configurations, with relatively few descriptions of their performance characteristics. Therefore, it is important to explore the structure–activity relationship between structural characteristics and properties of clusters. The bonding attributes, π-electron composition, and origin of aromaticity of Os-organic π-clusters were revealed through this in-depth analysis, which confirmed their unique metal–organic π-cluster properties. The excellent NLO effect provides new insights into the feasibility of designing highly responsive optical materials at the electronic structure level, while demonstrating new directions for novel and practical NLO devices in materials science.
|
Introduction
Clusters are significant structural units in the field of chemistry that exist in sizes intermediate between molecules and bulk materials,1–5 consisting of core polyhedra and peripheral ligands. The origins of cluster structure theory can be traced back to Sidgwick's effective atomic number (EAN) rule of 1927.6 Subsequently, advancements like the Wade polyhedron skeleton bonding electron pair theory,7 Lu Jiaxi's “Unit construction” assumption in synthesizing transition metal cubane-like clusters,8,9 and Tang Aoqing's (9n − L) rule on bonding and non-bonding orbital numbers have enriched the theoretical framework of cluster structures.10 Nevertheless, these theories heavily emphasize the geometric configuration, neglecting performance attributes. Hence, it becomes imperative to investigate the structure–activity relationship to bridge this gap, emphasizing the link between the structural characteristics and performance of clusters.
Aromaticity, a vital concept in chemistry with about two centuries of extensive research, is associated with molecules possessing delocalized π-electrons.11–13 These structures exhibit a multitude of chemical and physical characteristics, excelling in applications like organic fuels, catalytic synthesis, and optical responses.14–16 Moreover, these π-electrons are crucial to the third-order nonlinear optical (NLO) properties of the clusters.17 NLO chromophores are divided into electron donors (D), electron acceptors (A), and π-electron bridges. Among them the π-electron bridges are important linkers connecting D and A, playing a role in electron transmission.18 However, reported π-electron bridges primarily consist of singular organic materials,19,20 with a few Zintl phase metal clusters also displaying aromaticity.21 Nevertheless, clusters that integrate metal d–d π-conjugation with organic ligand p–p π-conjugation, termed metal–organic π-clusters, have not been adequately defined or systematically studied. These metal–organic π-clusters comprise both cluster core and ligand π-electrons, forming a class of aromatic system. Their structural characteristics include metal involvement in the cluster core, a stable chemical bond linking the core and the ligand, and an aromatic ligand with strong delocalization effects. Such clusters should have both a clear cluster core and ligand π-molecular orbitals (MOs) and exhibit aromaticity. Instances of this include [Mo3S4]4+ clusters and [ZnI]8 with cubic aromaticity,9,22 though the research in this field remains limited.
Through the analysis of the electronic structure and aromaticity, a series of osmium (Os)-organic clusters, comprised of Os-organic units connected via Os–Os bonds, are identified as characteristic examples of metal–organic π-clusters. These clusters display exceptional third-order nonlinear optical responses. Notably, the Os5 cluster exhibits the most remarkable performance due to its narrow band gap and uniform electron–hole distribution. This study provides a thorough characterization of metal–organic π-clusters and delves into the intricacies of their third-order NLO properties and contributing factors.
Results and discussion
Aromaticity analysis of the Os-organic structural unit
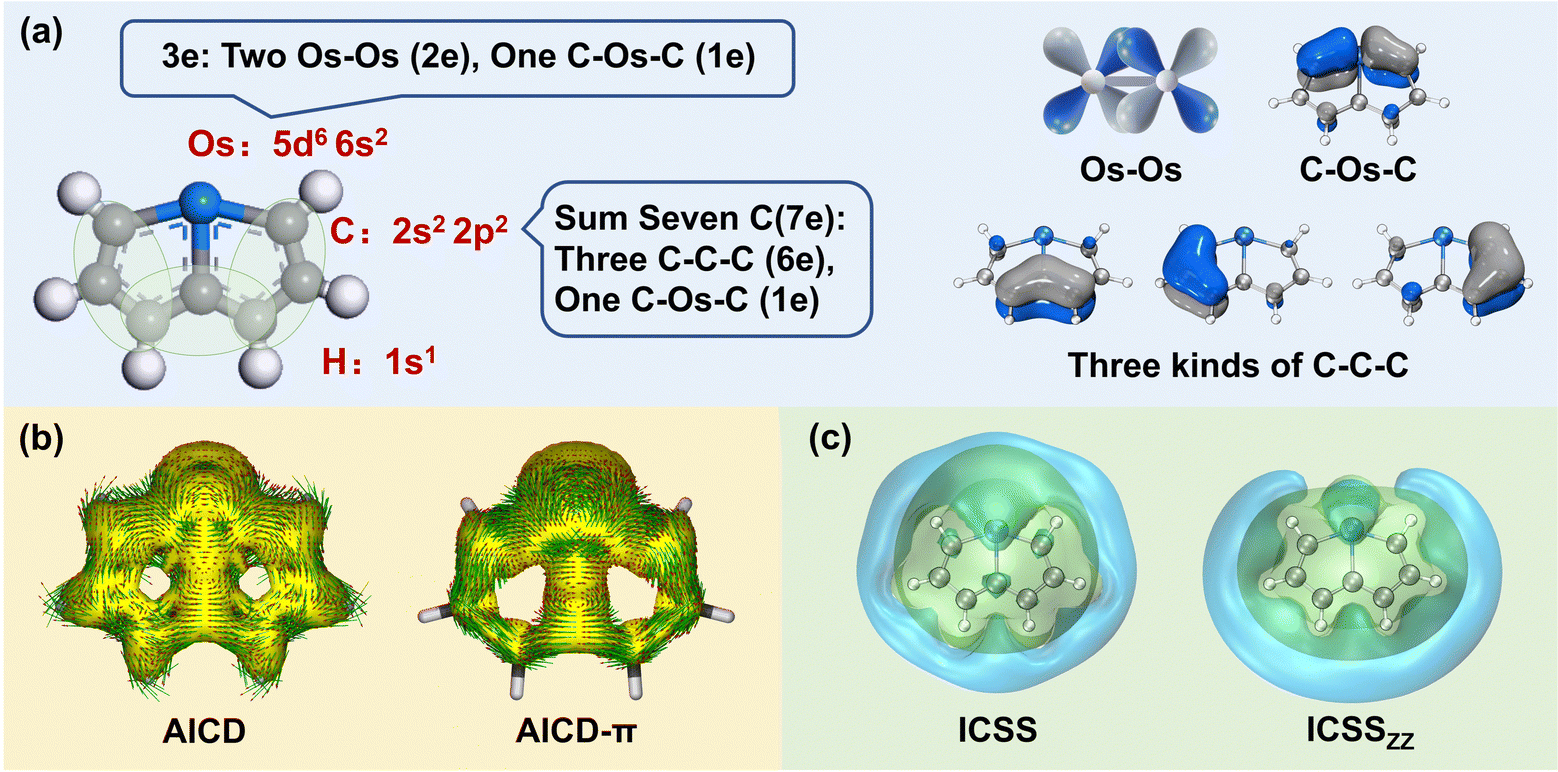
The selection of the Os-organic structural unit as the foundational element is based on experimental synthesis results.23 Each unit is composed of a single osmium atom, seven carbon atoms, and six hydrogen atoms. As depicted in Fig. 1a, the valence electron configuration of the involved atoms includes the 5d6 6s2 electron arrangement for osmium (Os), the 2s2 2p2 electron arrangement for carbon (C), and 1s1 electrons for hydrogenium (H). Together, the fragment carries 42 valence electrons. Of these, 12 are employed for C–H σ-bonding, 12 for C–C σ-bonding, 6 for Os–C σ-bonding, and 2 for Os–Os σ-bonding between units, with no lone pairs of electrons. Consequently, the residual 10 π-electrons contribute to three types of π-bonds: d–d orbital π-bonds between Os–Os, p–d–p orbital π-bonds between C–Os–C, and three p–p–p orbital π-bonds within the C–C–C chain (highlighted in green).
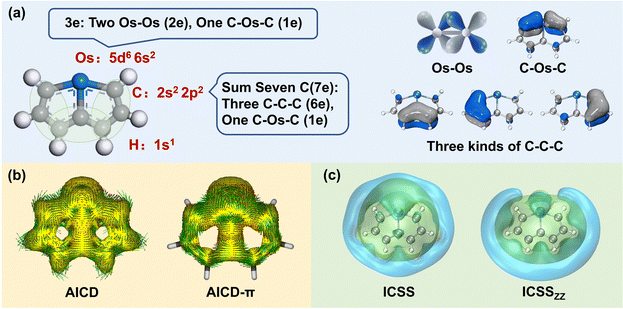 |
| | Fig. 1 Analysis of the distribution of the π-electrons and the aromaticity of the Os-organic structural unit: (a) distribution of π-electron composition. (b) Anisotropy of the induced current density (AICD, iso = 0.04 a.u.), and π contribution (AICD-π). (c) Iso-chemical shielding surfaces (ICSS, iso = 0.50 a.u.), and ICSS in the Z direction (ICSSZZ, iso = 1.50 a.u.). | |
In molecular systems exhibiting strong delocalization, electrons can move freely within a certain range, as observed with the π-electrons in benzene. When subjected to a magnetic field, a significant annular current is generated within the ring. The anisotropy of the induced current density (AICD) function is a useful tool to evaluate molecular aromaticity,24,25 indicative of the degree of system delocalization. AICD is defined as follows:
| | 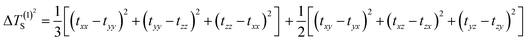 | (1) |
In particular, AICD-π focuses exclusively on all π-orbitals. As depicted in Fig. 1b, a notable ring current on the surface of the Os-organic structural unit mirrors the behavior seen in benzene (Fig. S1, ESI†). The direction of the induced current follows the left-hand rule, thus demonstrating the π-electron aromaticity of this structural component. The iso-chemical shielding surface (ICSS),26,27 a valuable analytical tool, enables a three-dimensional assessment of the shielding effect of structural units on the magnetic field, facilitating the determination of aromaticity. Fig. 1c presents a distinct isosurface perpendicular to the ring, which represents a shielding cone (green) surrounded by a deshielding loop (blue). These observations underscore strong magnetic shielding within the ring, attesting to the aromaticity of the Os-organic structural unit and aligning with the results from the AICD analysis.
Geometric structures and aromaticity analysis of Os-organic π-clusters
The Os-organic structural unit, linked through Os–Os bonds, exhibits the capability to form seven unique models with three to nine cores. After structural optimization, seven stable Os-organic clusters are obtained (Fig. 2a). To elucidate the structural characteristics of these clusters, bond length, and angle analysis are conducted. It is discernible from Fig. 2b that with an increasing number of core atoms, the average Os–Os bond length shows a dwindling trend (ranging from 2.527 Å to 2.415 Å) while the average Os–Os–Os bond angle increases (ranging from 60.0° to 120.8°). Moreover, even-kernel Os-organic clusters exhibit more uniform bond lengths and angles than their odd-kernel counterparts (Tables S1 and S2, ESI†). Natural population analysis (NPA) is utilized to examine the charge distribution of Os-organic clusters. The results revealed that the Os element carries a slight positive charge ranging from 0.10 to 0.40 (Fig. 2c and Table S3, ESI†), suggesting that the cluster core acts as a positively charged center of the structure. The negative formation energies further confirm the stability of the Os-organic clusters (Fig. 2d and Table S4, ESI†). Nucleus-independent chemical shift (NICS) is a prevalent measure of aromaticity,28,29 with negative values indicating aromaticity and positive values signifying antiaromaticity. To evaluate the aromaticity, we sifted through the occupied orbitals based on the total number of π-electrons and conducted NICS calculations on the π-MOs (Table S5, ESI†). NICS(0)πZZ and NICS(1)πZZ values, representing the Z component of NICSπ, are utilized to describe π-aromaticity. All seven Os-organic clusters exhibited negative values for both NICS(0)πZZ (from −59.40 ppm to −10.01 ppm) and NICS(1)πZZ (from −38.18 ppm to −6.15 ppm), corroborating their aromatic nature (Fig. 2e).
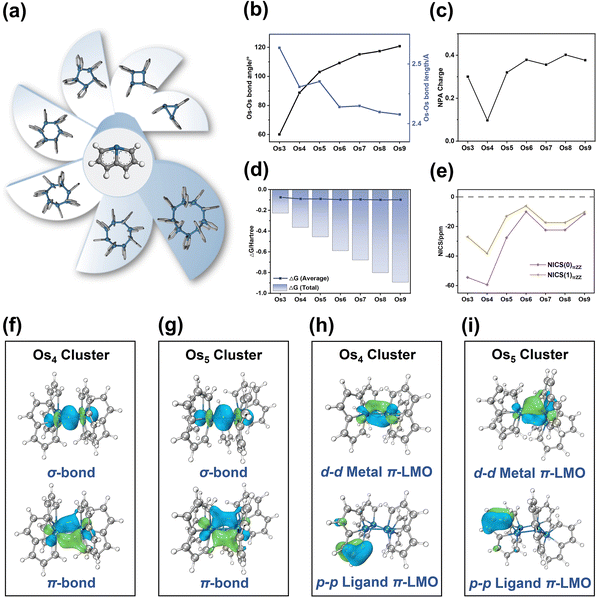 |
| | Fig. 2 Structural characteristics of Os-organic π-clusters. (a) Structures of seven clusters. (b) Average bond angle and average bond length of Os–Os. (c) The overall charge of the Os element. (d) The calculated formation free energy. (e) Nucleus independent chemical shift (NICS) values for key occupied π-MOs (NICSπ). (f) and (g) Natural adaptive orbital analysis (NAdO) of Os–Os bonds of Os4 and Os5 clusters, respectively. (h) and (i) Different π-LMOs of Os4 and Os5 clusters, respectively. | |
The Natural Adaptive Orbital (NAdO) method involves a linear transformation of the occupied MOs, paving the way for an intuitive exploration of orbital contributions to bonding. As shown in Fig. 2f and g, the Os–Os bonding within the Os4 and Os5 clusters encompasses both σ and π-bonds, demonstrating a stable covalent bond with pronounced delocalization. In non-planar systems, localized molecular orbitals (LMOs) provide valuable insights into both σ and π-electrons. These LMOs, like canonical molecular orbitals (CMO), are orthogonal sets with equal dimensions that can be interconverted via unitary transformation. The Os4 and Os5 clusters exhibit various types of π-orbitals, including d–d metal π-LMOs between Os–Os, d–p π-LMOs composed of Os and adjacent C, and p–p ligand π-LMOs composed of C–C. AICD and ICSS analyses are utilized to evaluate the aromaticity of these Os-organic clusters. As shown in Fig. S1 (ESI†), a clockwise ring current is observable on the Os ring, coupled with a pronounced shielding isosurface perpendicular to the ring, affirming its aromaticity. This type of Os-organic cluster comprises the metal that participates in the cluster core, and the aromaticity ligand via a stable covalent bond. Notably, it possesses distinct cluster core π-orbitals and ligand π-orbitals, both exhibiting aromaticity, therefore typifying a classic metal–organic π-cluster.
Third-order nonlinear optical properties and molecular orbital analysis
Considering the aforementioned findings, further investigations have proceeded to evaluate the third-order NLO performance of these Os-organic π-clusters. Under static conditions, both total and average third-order NLO coefficients are computed, as indicated in Table S6 (ESI†), and represented graphically in Fig. S2 (ESI†). The static third-order NLO response coefficient (γ) was calculated by the coupled perturbed Kohn–Sham method. The Taylor expansion of the energy (E) to the uniform external electric field (F) is as follows:| | 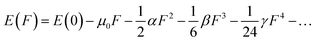 | (2) |
The i components of γ are defined as| | 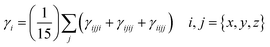 | (3) |
The total magnitude of γ is measured as| | 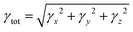 | (4) |
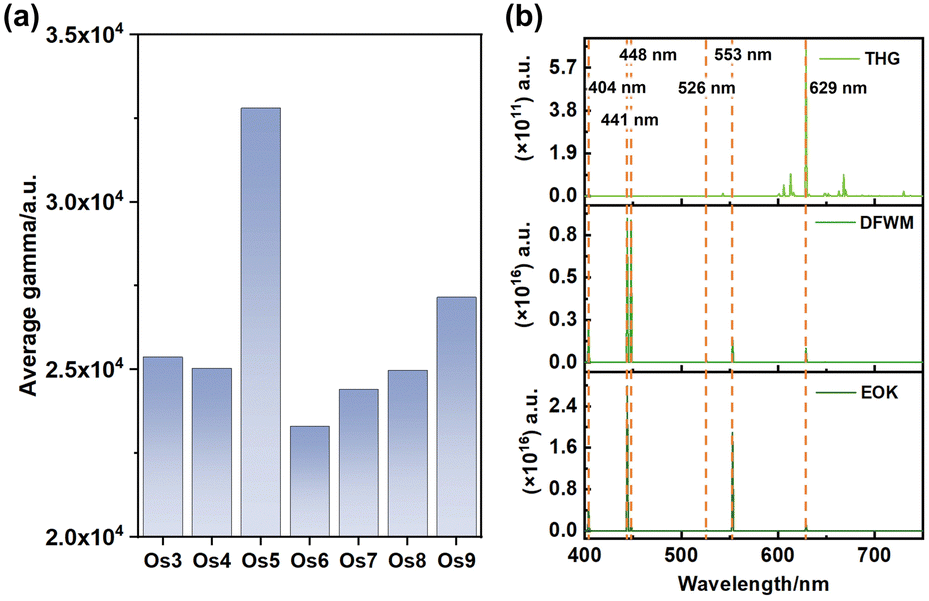
Particularly noteworthy is the exceptional third-order NLO response displayed by the Os5 cluster (Fig. 3a). Furthermore, the frequency-dependent second hyperpolarizability of the Os5 cluster in the visible light range is examined utilizing the sum-over-states (SOS) method as the following formula:30,31
| | γABCD(−ωσ; ω1, ω2, ω3) = ![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif) [A(−ωσ), B(ω1), C(ω2), D(ω3)](γI − γII) [A(−ωσ), B(ω1), C(ω2), D(ω3)](γI − γII) | (5) |
| | 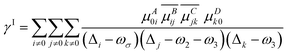 | (6) |
| |  | (7) |
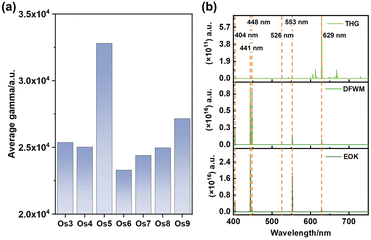 |
| | Fig. 3 Third-order nonlinear optical (NLO) properties of Os-organic π-clusters. (a) Average third-order NLO coefficients. (b) Frequency-dependent second hyperpolarizabilities in the visible-light range of the Os5 cluster. | |
A, B, C… denote one direction; ω is the energy of the external field; Δi stands for the excitation energy of state i concerning the ground state; μAij is a component of the transition dipole moment between state i and j.
Our analysis revealed prominent third harmonic generation (THG), degenerate four-wave mixing (DFWM), and electro-optical Kerr effect (EOK) responses at specific wavelengths, namely 404 nm, 441 nm, 448 nm, 526 nm, 553 nm, and 629 nm. These wavelengths correspond to the purple, green, and red light bands, hinting at the potent signal detection capabilities of these clusters.32
Understanding the exceptional third-order NLO response of the Os5 cluster demands a thorough analysis of the band gap and MO composition across the seven structures (Fig. 4). Compared to the original Os-organic structural unit, all seven Os-organic π-clusters show a decrease in Egap (Table S7, ESI†). There is a fluctuation in the Egap value with the number of nuclei, with the Os5 cluster presenting the narrowest band gap (1.47 eV), a result of an increase in the highest occupied molecular orbital (HOMO). The density of states (DOS) is utilized for analyzing the orbital contributions (Fig. S3, ESI†). The partial DOS (PDOS) is employed to discern the specific contribution of individual fragments to the total DOS (TDOS), revealing that the Os-organic π-clusters consist of two components: the Os cluster core and ligand, each contributing different proportions. Interestingly, the difference between the Os5 cluster from the other six Os-organic π-clusters is that it has a significant contribution (44.13%) from the 2p orbitals of carbon (C) in its HOMO. Moreover, the C 2p orbitals exhibit higher energy in occupied orbitals compared to the Os 5d and 6s orbitals (Fig. S4, ESI†). This greater presence of C 2p orbitals in the HOMO encourages its rise, subsequently leading to a narrower band gap. A smaller band gap promotes electronic transitions, thereby enhancing the third-order NLO response, a conclusion that aligns with reported experimental studies.17,33,34
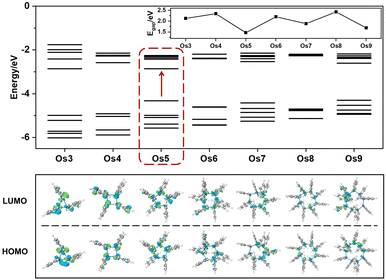 |
| | Fig. 4 Molecular orbitals and band gaps of Os-organic clusters. | |
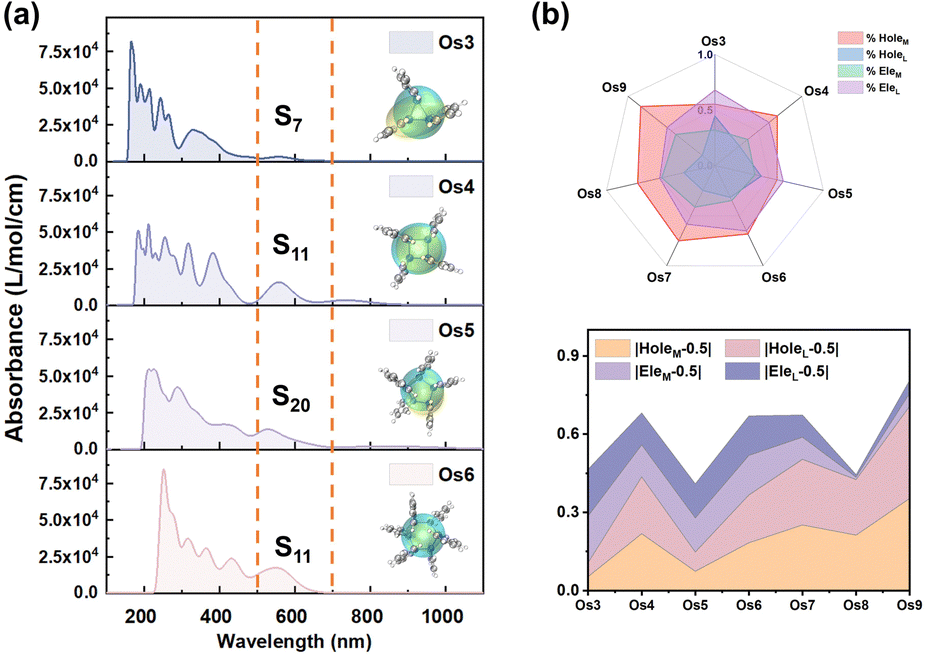
Excited state analysis
Employing UV-vis absorption spectra and electron–hole analysis, the superior third-order NLO response performance demonstrated by the seven Os-organic π-clusters under consideration is further elucidated (Table S8, ESI†). Fig. 5a highlights that the clusters' intense absorption in the low-energy section chiefly occurs within the 500–700 nm range. This corresponds to S0 → S7 (Os3, 557 nm), S11 (Os4, 564 nm), S20 (Os5, 526 nm), S11 (Os6, 567 nm), S6 (Os7, 654 nm), S15 (Os8, 546 nm), and S24 (Os9, 545 nm) excitations. The smoothed electron (Cele) and hole (Chole) contours profile the extent of electron–hole distribution in different directions.35–37 Notably, the Os5 cluster demonstrates an enhanced distance between the center of mass of holes and electrons (Table S9, D index = 0.868 Å, ESI†) when compared to the other Os-organic π-clusters. To further delineate the distribution, the holes and electrons of each Os-organic π-cluster are divided into cluster core and ligand components, as illustrated in Fig. 5b and Table S10 (ESI†). The line-fill radar map provides a comprehensive visualization of the proportion of different components. Among these, the Os5 cluster approximates more closely to the ideal average value of 50% regarding the proportion of cluster cores and ligands in electrons/holes. The distributional differences of metal and ligands (DDML) in holes and electrons are further defined as DDMLHole and DDMLEle, respectively, to refine our understanding of this process.| | | DDMLHole = |HoleM − 0.5| + |HoleL − 0.5| | (8) |
| | | DDMLEle = |EleM − 0.5| + |EleL − 0.5| | (9) |
| | | DDMLsum = DDMLHole + DDMLEle | (10) |
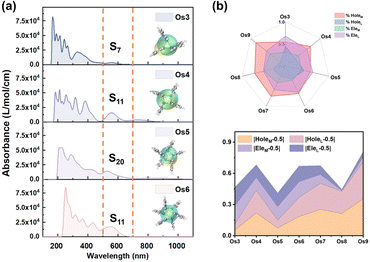 |
| | Fig. 5 (a) Normalized UV-vis absorption spectra and electron–hole distribution smoothing isosurface schematic (inset part) of Os-organic π-clusters. (b) Contribution ratio of cluster cores and ligands to electron–hole distribution. | |
The value of DDMLsum for the Os5 cluster, calculated at 0.41, highlights its relatively small size, implying a more balanced distribution of metal and ligand in the electron/hole components. This balance is critical in achieving optimal performance in third-order NLO responses. Thus, our findings suggest that enhancing the third-order NLO response of Os-organic π-clusters can be accomplished by extending the electron–hole distribution range and maintaining a consistent contribution ratio between the components. This nuanced understanding could prove pivotal in future developments and applications within the field of nonlinear optics.
Conclusions
In conclusion, this research introduces a concise and clear definition of metal–organic π-clusters that retain the aromaticity of both the cluster core and the ligand. Under this conceptual framework, a variety of stable Os-organic π-clusters, derived from Os-organic structural units through Os–Os bonds, have been identified and recognized as exemplary metal–organic π-clusters. Our analysis of electronic structure and aromaticity simultaneously verifies the origins of π-electrons in these clusters. Notably, the Os5 cluster manifests a robust third-order NLO response, with an average gamma magnitude of 3.28 × 104 a.u., attributable to its narrow band gap of 1.47 eV and relatively balanced electron/hole distribution, as indicated by a DDMLsum value of 0.41. This research thus expands the understanding of aromaticity and its connection to third-order NLO properties, offering a fresh perspective for the design and synthesis of high-response optical materials.
Author contributions
J. Z. and Q. H. L. conceived the project. Z. R. W. carried out the theoretical calculations. All the authors wrote and revised the manuscript. All the authors discussed and commented on the final manuscript preparation.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (92161105).
Notes and references
- W. Eberhardt, Surf. Sci., 2002, 500, 242–270 CrossRef CAS.
- J. Liu, J. Li, Z. Xu, X. Zhou, Q. Xue, T. Wu, M. Zhong, R. Li, R. Sun, Z. Shen, H. Tang, S. Gao, B. Wang, S. Hou and Y. Wang, Nat. Commun., 2021, 12, 1619 CrossRef CAS PubMed.
- M. Kepenekian, Y. Molard, K. Costuas, P. Lemoine, R. Gautier, S. Ababou Girard, B. Fabre, P. Turban and S. Cordier, Mater. Horiz., 2019, 6, 1828–1833 RSC.
- L. Liu, S. Liu, J. Liu, L. Lan, P. Cui, J. Yang, J.-F. Yin and P. Yin, Chin. J. Struct. Chem., 2023, 42, 100102 CrossRef.
- Y. Wu, Y. Yu, W. Shen, Y. Jiang, R. He and M. Li, Mater. Horiz., 2023 10.1039/D3MH01130E.
-
N. V. Sidgwick, The electronic theory of valency, Clarendon Press, 1927 Search PubMed.
- K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1–66 CrossRef CAS.
- J. Lu and B. Zhuang, Chin. J. Struct. Chem., 1989, 8, 233–248 CAS.
- J. Lu and Z. Chen, Acta Phys. -Chim. Sin., 1992, 8, 834–841 CAS.
- A. Tang and L. QianShu, Chin. Sci. Bull., 1983, 1, 25–27 CrossRef.
- B. Jin, J. Bian, X. Zhao, C. Yuan, J. Guo and Y. Wu, Chin. J. Struct. Chem., 2022, 41, 218–226 Search PubMed.
- A. M. Österholm, L. Nhon, D. E. Shen, A. M. Dejneka, A. L. Tomlinson and J. R. Reynolds, Mater. Horiz., 2022, 9, 252–260 RSC.
- R. Renner, M. Stolte, J. Heitmüller, T. Brixner, C. Lambert and F. Würthner, Mater. Horiz., 2022, 9, 350–359 RSC.
- S. H. Yoo, D. Jang, H.-I. Joh and S. Lee, J. Mater. Chem. A, 2017, 5, 748–755 RSC.
- X. Kong, P.-P. Zhou and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 9395–9400 CrossRef CAS PubMed.
- Z. Wang, W. Yan, G. Zhao, K. Wu, Z.-G. Gu, Q.-H. Li and J. Zhang, J. Phys. Chem. Lett., 2021, 12, 11784–11789 CrossRef CAS.
- M. Gao, Z. Wang, Q. Li, D. Li, Y. Sun, Y. H. Andaloussi, C. Ma, C. Deng, J. Zhang and L. Zhang, J. Am. Chem. Soc., 2022, 144, 8153–8161 CrossRef CAS.
- E. M. Breitung, C.-F. Shu and R. J. McMahon, J. Am. Chem. Soc., 2000, 122, 1154–1160 CrossRef CAS.
- T. Nishiuchi, K. Kisaka and T. Kubo, Angew. Chem., Int. Ed., 2021, 60, 5400–5406 CrossRef CAS PubMed.
- T. Nishiuchi, H. Sotome, K. Shimizu, H. Miyasaka and T. Kubo, Chem. – Eur. J., 2022, 28, e202104245 CrossRef CAS.
- C. Liu, I. A. Popov, Z. Chen, A. I. Boldyrev and Z.-M. Sun, Chem. Eur. J., 2018, 24, 14583–14597 CrossRef CAS.
- P. Cui, H. S. Hu, B. Zhao, J. T. Miller, P. Cheng and J. Li, Nat. Commun., 2015, 6, 6331 CrossRef CAS.
- C. Zhu, M. Luo, Q. Zhu, J. Zhu, P. V. R. Schleyer, J. I. C. Wu, X. Lu and H. Xia, Nat. Commun., 2014, 5, 3265 CrossRef.
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS.
- S. Klod and E. Kleinpeter, J. Chem. Soc., Perkin Trans. 2, 2001, 1893–1898 CAS.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 468–475 CrossRef CAS.
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. Van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- K. Sasagane, F. Aiga and R. Itoh, J. Chem. Phys., 1993, 99, 3738–3778 CrossRef CAS.
- F. Meyers, S. R. Marder, B. M. Pierce and J. L. Bredas, J. Am. Chem. Soc., 1994, 116, 10703–10714 CrossRef CAS.
- J. Kippenberg Tobias, L. Gaeta Alexander, M. Lipson and L. Gorodetsky Michael, Science, 2018, 361, eaan8083 CrossRef.
- C. C. Feng, Z. Zhang, Z. R. Wang, D. J. Li, Q. H. Li, L. Zhang and J. Zhang, Inorg. Chem., 2021, 60, 1885–1892 CrossRef CAS PubMed.
- Z. Wang, G. Zhao, W. Yan, K. Wu, F. Wang, Q. Li and J. Zhang, J. Phys. Chem. Lett., 2021, 12, 7537–7544 CrossRef CAS.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- X. Tang, L.-S. Cui, H.-C. Li, A. J. Gillett, F. Auras, Y.-K. Qu, C. Zhong, S. T. E. Jones, Z.-Q. Jiang, R. H. Friend and L.-S. Liao, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS PubMed.
- Z. Liu, T. Lu and Q. Chen, Carbon, 2020, 165, 461–467 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |

 ab,
Yayu
Yan
ac,
Jiali
Chen
ac,
Qiao-Hong
Li
ab,
Yayu
Yan
ac,
Jiali
Chen
ac,
Qiao-Hong
Li