
A skeletal randomization strategy for high-performance quinoidal-aromatic polymers†
Quanfeng
Zhou‡
a,
Cheng
Liu‡
a,
Jinlun
Li‡
a,
Runze
Xie
a,
Guoxiang
Zhang
a,
Xiang
Ge
 a,
Zesheng
Zhang
f,
Lianjie
Zhang
f,
Junwu
Chen
f,
Xiu
Gong
b,
Chen
Yang
c,
Yuanyu
Wang
*a,
Yi
Liu
*de and
Xuncheng
Liu
*a
a,
Zesheng
Zhang
f,
Lianjie
Zhang
f,
Junwu
Chen
f,
Xiu
Gong
b,
Chen
Yang
c,
Yuanyu
Wang
*a,
Yi
Liu
*de and
Xuncheng
Liu
*a
aCollege of Materials and Metallurgy, Guizhou University, Guiyang 550025, China. E-mail: xcliu3@gzu.edu.cn; yuanyuwang0216@163.com
bCollege of Physics, Guizhou University, Guiyang 550025, China
cCollege of Big Data and Information Engineering, Guizhou University, Guiyang 550025, China
dThe Molecular Foundry and Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, California 94720, USA. E-mail: yliu@lbl.gov
eMaterials Sciences Division, Lawrence Berkeley National Laboratory, One Cyclotron Road, Berkeley, California 94720, USA
fInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou 510640, China
First published on 2nd November 2023
Abstract
Enhancing the solution-processability of conjugated polymers (CPs) without diminishing their thin-film crystallinity is crucial for optimizing charge transport in organic field-effect transistors (OFETs). However, this presents a classic “Goldilocks zone” dilemma, as conventional solubility-tuning methods for CPs typically yield an inverse correlation between solubility and crystallinity. To address this fundamental issue, a straightforward skeletal randomization strategy is implemented to construct a quinoid-donor conjugated polymer, PA4T-Ra, that contains para-azaquinodimethane (p-AQM) and oligothiophenes as repeat units. A systematic study is conducted to contrast its properties against polymer homologues constructed following conventional solubility-tuning strategies. An unusually concurrent improvement of solubility and crystallinity is realized in the random polymer PA4T-Ra, which shows moderate polymer chain aggregation, the highest crystallinity and the least lattice disorder. Consequently, PA4T-Ra-based OFETs, fabricated under ambient air conditions, deliver an excellent hole mobility of 3.11 cm2 V−1 s−1, which is about 30 times higher than that of the other homologues and ranks among the highest for quinoidal CPs. These findings debunk the prevalent assumption that a random polymer backbone sequence results in decreased crystallinity. The considerable advantages of the skeletal randomization strategy illuminate new possibilities for the control of polymer aggregation and future design of high-performance CPs, potentially accelerating the development and commercialization of organic electronics.
New conceptsThis paper introduces an innovative concept in the design of high mobility conjugated polymer research, challenging the conventionally perceived trade-off between solubility and crystallinity that has long constrained the practicality of conjugated polymers. Our study disrupts this dichotomy with the successful application of a skeletal randomization approach. By introducing randomized sequences in a quinoid-aromatic polymer system, a fine balance between solubility and interchain aggregation strength was achieved, leading to enhanced solubility, increased crystallinity, and a much higher carrier transport mobility compared to its regioregular counterparts. Our method offers a remarkable divergence from traditional solubility-tuning methods by simultaneously fostering solubility and thin-film crystallinity. In a counterintuitive fashion, the introduced “disorder” along the backbone has resulted in notably improved film crystallinity and a hole mobility of 3.11 cm2 V−1 s−1, 50 times higher than that of the regioregular homologues. This also places the resulting polymer among the highest performing quinoidal CPs, which additionally demonstrates superior solution-processibility and device stability. The insights provided by our work present a ground-breaking departure from established norms, illuminating a novel path for designing solution processed high performing conjugated polymers that may find wider applications in organic electronics. |
1. Introduction
The pursuit of high-performing conjugated polymers (CPs) has been central to the advancement of organic solar cells (OSCs),1–3 organic field-effect transistors (OFETs)4–6 and other electronic devices.7–9 Recently, OFETs have shown promising potential for their use as driving circuits for large-area flexible displays and other types of flexible electronics, the performance of which is largely determined by the CPs utilized in the active layer.10–12 Understanding the correlation between the molecular structure and material properties in high mobility CPs is critical to empower the design of CPs with enhanced performance.13The structural design of CPs involves careful consideration of the conjugated structure of the repeat unit, the number of the repeat unit (i.e., molecular weight), and the structure and density of sidechains. Typically, the conjugation of repeat units is deterministic to intrinsic optoelectronic properties of CPs, while the molecular weight14,15 and alkyl sidechains16–18 can be tailored to achieve sufficient solubility in organic solvents. This solubility facilitates necessary solution-processibility for the fabrication of high performance thin-film OFET devices.17,19 However, sidechain engineering can substantially affect interchain interactions and self-assembly of CPs, compromising film crystallinity and charge carrier transport in devices,18,20–22 not to mention the increased synthetic complexity towards controlling the structure and density of side chains and the polymerization conditions.23 This balance between the solubility, film morphology and carrier mobility presents one of the fundamental challenges for developing high mobility CPs. Therefore, novel solubility-tuning strategies that simultaneously enhance solubility and ordered molecular packing, thereby boosting carrier mobility, are in high demand for achieving high-performance OFETs.24,25
Mixed quinoidal-aromatic CPs containing quinoidal units in the conjugated polymer backbone have exhibited distinctive optoelectronic properties and impressive semiconducting properties, inspiring complementary design principles to the classic donor–acceptor (D–A) type CPs.26–28 The introduction of a quinoidal unit with oligothiophene in the repeat unit in the polymer backbone often leads to high mobility due to the reinforced backbone, strong interchain interaction and aggregation, high crystallinity, and dense molecular packing.29–33 As previously demonstrated by us, a planar quinoidal building block, para-azaquinodimethane (p-AQM), is easily accessible and has been successfully employed in organic semiconductors for OFETs and various applications.34–40 The PA4T conjugated polymer, containing one p-AQM and one quaterthiophene in the repeat unit, exhibited superb crystallinity in both as-cast and annealed films.40 However, only a mediocre hole mobility of 0.084 cm2 V−1 s−1 was obtained from PA4T-based OFETs, due to poor solubility and processibility arising from excessive interchain interaction and high aggregation strength. Further efforts of introducing longer side chains onto the backbone of PA4T showed limited improvement in solubility and hole mobility.41 Given that PA4T represents a typical kind of quinoidal-aromatic CP featuring a simple structure, strong aggregation and high film crystallinity, it serves as a great system to exercise novel strategies to address the solubility-performance dilemma.
Regioregularity control has recently emerged as a novel approach to construct high-performance D–A CPs, which has a substantial impact on solubilities, backbone conformations, interchain interactions, solid state microstructures, and the corresponding device performance.42–48 Traditionally, regioregular D–A polymers tend to show higher mobilities and more ordered microstructures than their regiorandom counterparts.49–51 Recent advances, however, have demonstrated that regiorandom D–A polymers could show impressive charge transport.52,53 Despite the performance improvement, there lacks a detailed study to properly address the structure–property–performance relationship. For instance, there are several cases where regiorandom polymers exhibit both high mobilities and decreased thin film crystallinity,54–56 which contradicts the general relationship that high crystallinity typically translates to high mobility. More often, previous studies mainly focused on electronic properties as a result of regioregularity control, while little effort has been devoted to understanding its influence on the solubility and crystallization behavior of CPs.57,58 Clearly, a systematic study of the randomization effect in a well-defined conjugated polymer system would help achieve more rational molecular design strategies.
Herein, regioregularity control is pioneered in the p-AQM-based quinoidal-aromatic CP setting by converting the regular backbone of PA4T into the polymer PA4T-Ra with random p-AQM-oligothiophene sequences. This skeletal randomization strategy, which features remarkable synthetic and structural simplicity, is compared against two other commonly adapted solubilization methods, i.e., molecular weight control and sidechain engineering, in its capacity to tune solubility, aggregation strength, and thin film properties (Scheme 1a and b). The systematic studies of the five quinoidal PA4T homologues revealed unconventional insights into the structure–property–performance relationships of conjugated polymers. As a result, the skeletal randomization strategy has been validated as the most effective method in balancing solubility, fine-tuning the interchain aggregation strength, and achieving the highest crystallinity in thin films, which have a substantial influence on charge carrier transport properties. The corresponding air-processed OFET devices based on PA4T-Ra showed remarkable operational stability and a reliable hole mobility of up to 3.11 cm2 V−1 s−1, reflecting a more than 30-fold enhancement over the parent PA4T.
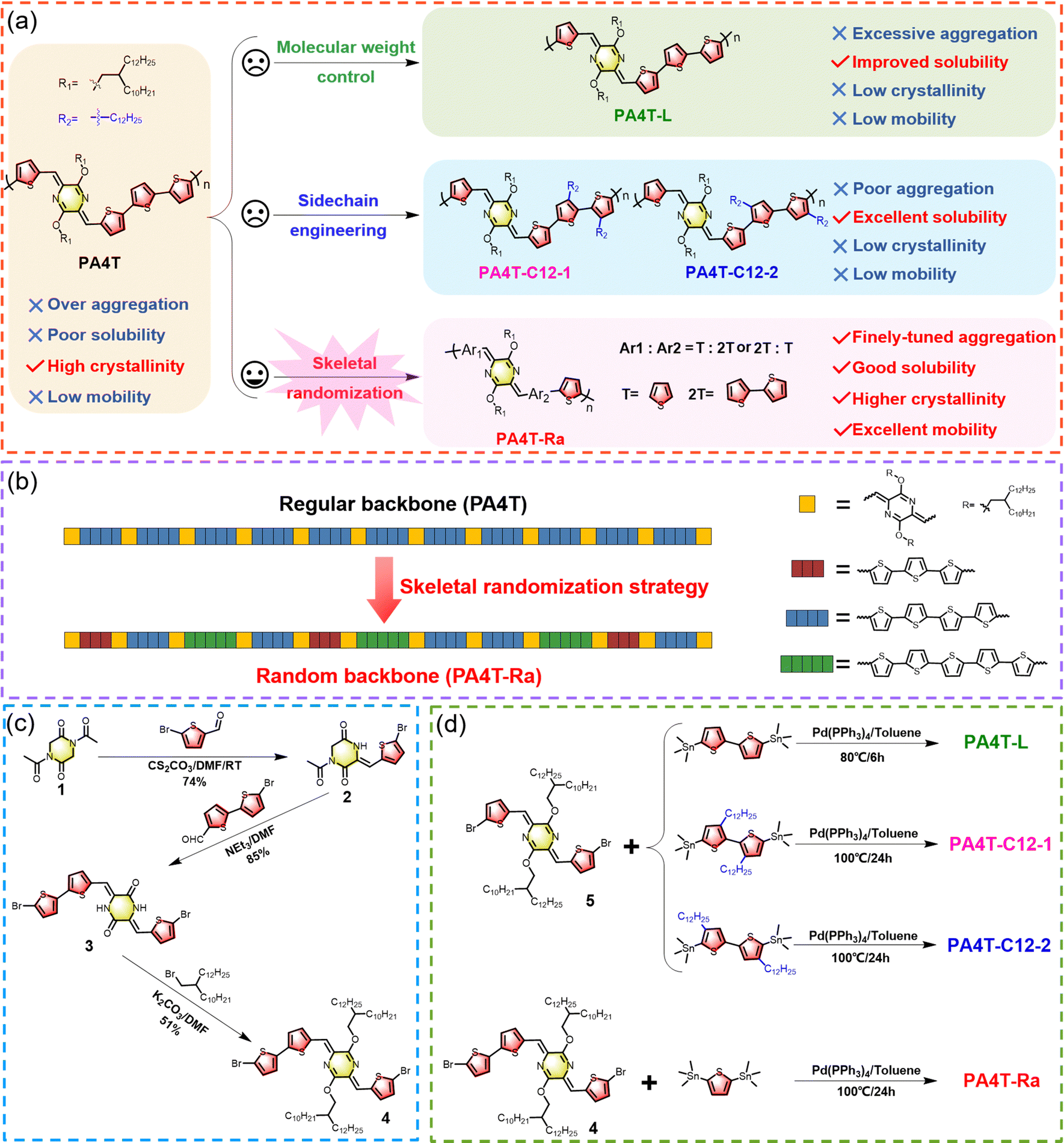 | ||
| Scheme 1 (a) Structures of the conjugated polymer PA4T and four PA4T homologues designed for comparative validation of the solubilization strategies. (b) Schematic illustration of the skeletal randomization strategy and backbone sequence of PA4T and PA4T-Ra. Synthetic routes to (c) the dissymmetric dibromo-p-AQM-based monomer and (d) the designed polymers. | ||
2. Results and discussion
2.1. Polymer synthesis
The synthetic routes to dissymmetric dibromo-p-AQM-based monomer 4 and its derived regiorandom polymer PA4T-Ra are depicted in Scheme 1c and d, respectively. The synthesis of three regioregular PA4T polymer homologues is also illustrated in Scheme 1d. Detailed procedures and characterization are provided in the ESI.† The preparation of monomer 4 was accomplished in three straightforward steps from readily available starting materials in a high overall yield. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Knoevenagel condensation between 1,4-diacetyl-2,5-diketopiperazine 1 and 5-bromothiophene-2-carbaldehyde gave rise to monobromide 2 in 74% yield. Another 1:1 Knoevenagel condensation of compound 2 with 5′-bromo-[2,2′-bithiophene]-5-carbaldehyde afforded dibromide 3 in excellent yield. Subsequent alkylation afforded the dibromo AQM monomer 4. The other dibromo monomer 5 was synthesized in a similar fashion following previously reported procedures.40 All polymers were prepared via Stille-coupling polymerization between the p-AQM-based dibromides and distannylated-thiophene monomers, while the molecular weight variations were controlled by altering the reaction temperature and duration.59,60 For the PA4T polymer with a lower molecular weight, PA4T-L, the polymerization was carried out at 80 °C for 6 h, while for keeping the molecular weights of PA4T-Ra, PA4T-C12-1 and PA4T-C12-2 to be comparable with that of PA4T, all of the reactions were carried out at 100 °C for 24 h. PA4T-C12-1 and PA4T-C12-2 were fractionated using chloroform (CF) while PA4T-L and PA4T-Ra were extracted with chlorobenzene using a Soxhlet extractor, and the respective molecular weights were determined using high temperature size exclusion chromatography (SEC) at 140 °C with 1,2,4-trichlorobenzene as the eluent. As shown in Table 1, the number-average molecular weight (Mn) of PA4T-L was 13.9 kDa, lower than that of the previously reported PA4T (26.4 kDa), which was in a similar range to that of the other three polymers (23.9–27.0 kDa). Additionally, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out to probe the thermal properties of all five polymers. As shown in Fig. S1 (ESI†), all polymers presented decent thermal stability with a decomposition temperature (Td) at 5% weight loss in the range of 361–382 °C, and exhibited no distinct phase transition in the temperature range from 25 to 250 °C. The random backbone sequence of PA4T-Ra was confirmed by FTIR spectroscopic analysis.55 As shown in Fig. S2 (ESI†), PA4T-Ra shared many similar characteristic peaks for most of the spectra with the regular PA4T, because they have the same chemical composition and proportion of primary units in the backbone. However, the fingerprint region around 1051 cm−1, which corresponds to C–C skeletal vibrations between two adjacent aromatic units, was distinctly different. PA4T-Ra showed multiple and featureless peaks, indicative of various sequences of aromatic units existing in the resulting random backbone, which correlates well with the expected chemical structure of PA4T-Ra in Scheme 1.
:1 Knoevenagel condensation between 1,4-diacetyl-2,5-diketopiperazine 1 and 5-bromothiophene-2-carbaldehyde gave rise to monobromide 2 in 74% yield. Another 1:1 Knoevenagel condensation of compound 2 with 5′-bromo-[2,2′-bithiophene]-5-carbaldehyde afforded dibromide 3 in excellent yield. Subsequent alkylation afforded the dibromo AQM monomer 4. The other dibromo monomer 5 was synthesized in a similar fashion following previously reported procedures.40 All polymers were prepared via Stille-coupling polymerization between the p-AQM-based dibromides and distannylated-thiophene monomers, while the molecular weight variations were controlled by altering the reaction temperature and duration.59,60 For the PA4T polymer with a lower molecular weight, PA4T-L, the polymerization was carried out at 80 °C for 6 h, while for keeping the molecular weights of PA4T-Ra, PA4T-C12-1 and PA4T-C12-2 to be comparable with that of PA4T, all of the reactions were carried out at 100 °C for 24 h. PA4T-C12-1 and PA4T-C12-2 were fractionated using chloroform (CF) while PA4T-L and PA4T-Ra were extracted with chlorobenzene using a Soxhlet extractor, and the respective molecular weights were determined using high temperature size exclusion chromatography (SEC) at 140 °C with 1,2,4-trichlorobenzene as the eluent. As shown in Table 1, the number-average molecular weight (Mn) of PA4T-L was 13.9 kDa, lower than that of the previously reported PA4T (26.4 kDa), which was in a similar range to that of the other three polymers (23.9–27.0 kDa). Additionally, thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out to probe the thermal properties of all five polymers. As shown in Fig. S1 (ESI†), all polymers presented decent thermal stability with a decomposition temperature (Td) at 5% weight loss in the range of 361–382 °C, and exhibited no distinct phase transition in the temperature range from 25 to 250 °C. The random backbone sequence of PA4T-Ra was confirmed by FTIR spectroscopic analysis.55 As shown in Fig. S2 (ESI†), PA4T-Ra shared many similar characteristic peaks for most of the spectra with the regular PA4T, because they have the same chemical composition and proportion of primary units in the backbone. However, the fingerprint region around 1051 cm−1, which corresponds to C–C skeletal vibrations between two adjacent aromatic units, was distinctly different. PA4T-Ra showed multiple and featureless peaks, indicative of various sequences of aromatic units existing in the resulting random backbone, which correlates well with the expected chemical structure of PA4T-Ra in Scheme 1.
| Polymer | M n [kDa] | PDI | Solubilityb [mg mL−1] | Solution | Film | HOMOd [eV] | LUMOe [eV] | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ max1 [nm] | λ max2 [nm] | E g [eV] | λ max1 [nm] | λ max2 [nm] | E g [eV] | ||||||
| a Molecular weight by high temperature SEC. b In chlorobenzene at 80 °C. c Optical bandgaps estimated on the basis of the absorption onset. d Measured by cyclic voltammetry. e Calculated by subtraction of the film's optical bandgap from the HOMO level. f Reported previously. | |||||||||||
| PA4T | 26.4 | 2.7 | 5.2 | 660 | 731 | 1.54 | 660 | 731 | 1.53 | −5.07 | −3.54 |
| PA4T-L | 13.9 | 1.5 | 8.4 | 651 | 720 | 1.57 | 655 | 724 | 1.51 | −5.10 | −3.59 |
| PA4T-C12-1 | 27.0 | 2.3 | 53.1 | 590 | 672 | 1.72 | 563 | 603 | 1.67 | −5.23 | −3.56 |
| PA4T-C12-2 | 23.9 | 2.9 | 50.5 | 601 | — | 1.69 | 664 | 739 | 1.53 | −5.09 | −3.56 |
| PA4T-Ra | 25.1 | 2.4 | 17.8 | 667 | 731 | 1.56 | 667 | 734 | 1.50 | −5.03 | −3.53 |
2.2. Solubility test
The solubility of polymers is critical for achieving excellent charge carrier mobility, which has a dramatic influence on the film formation, morphology and microstructure. Solubility tests were carried out to study the effectiveness of each solubility-tuning approach outlined in Scheme 1a. The previously reported polymer PA4T was insoluble in CB at room temperature (RT) and partially soluble in hot (80 °C) CB, while PA4T-L was solubilized more easily in CB. In addition, PA4T-C12-1 and PA4T-C12-2 were readily soluble in both CB and chloroform (CF) at RT; however, PA4T-Ra was only partially soluble in RT CB (Fig. S3, ESI†). Such observations indicated that the solubilities of all four polymers were improved over PA4T. For quantitative comparison, the respective solubility limits for each polymer in 80 °C CB were determined using UV-vis absorption (Fig. S4, ESI†) and are summarized in Fig. 1a and Table 1.61,62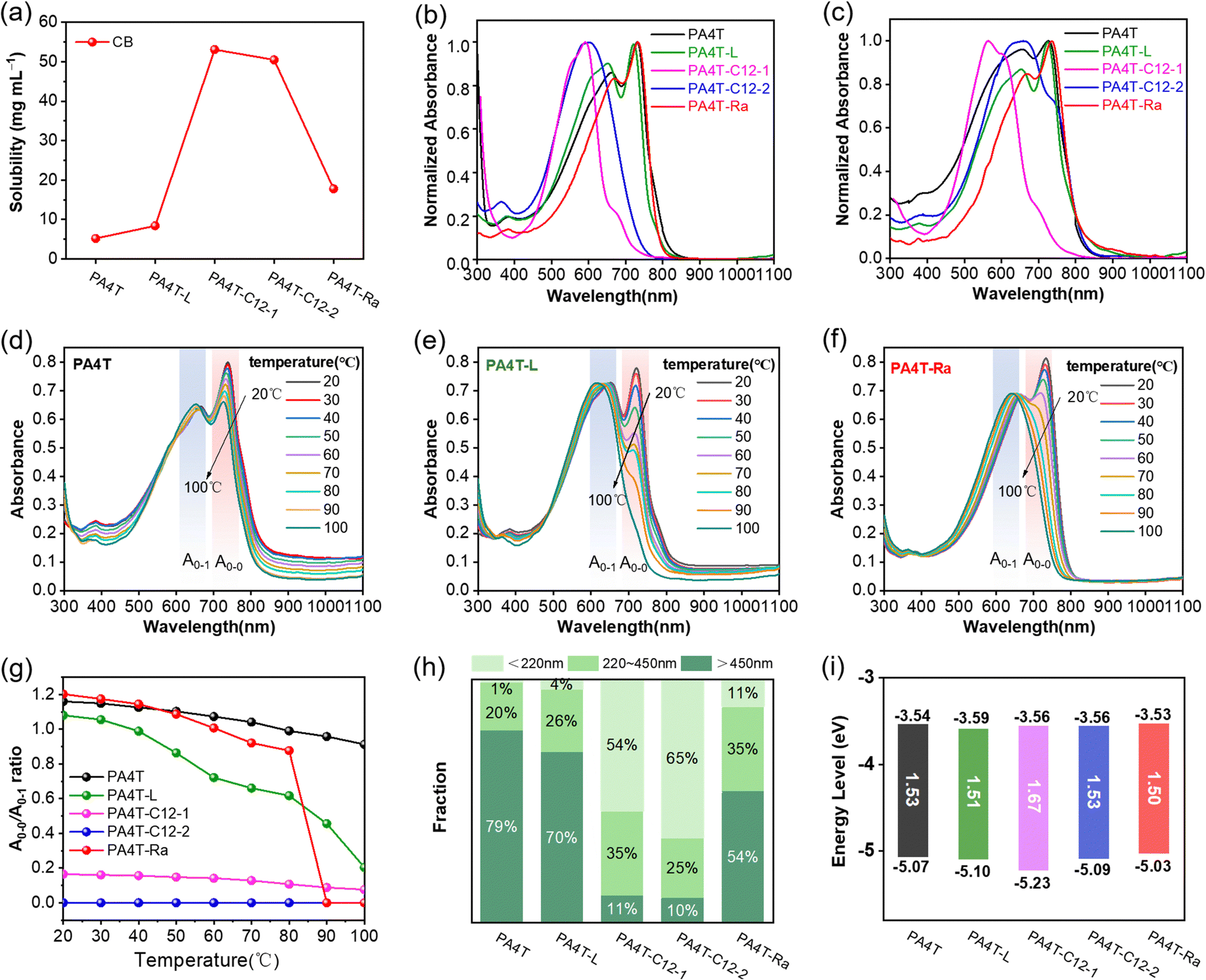 | ||
| Fig. 1 (a) The solubilities of five polymers in 80 °C chlorobenzene. Normalized UV-vis absorption spectra of five polymers (b) in chlorobenzene and (c) thin film at room temperature. (d)–(f) The evolution of UV-vis absorption spectra of PA4T, PA4T-L and PA4T-Ra in chlorobenzene during the heating process. (g) A0–0/A0–1 ratio of five polymers extracted from varying temperature UV-vis absorption spectra. (h) Size distributions of solution aggregates at room temperature and (i) diagram of energy levels of five polymers. | ||
The solubilities of those polymers follow the trend of PA4T-C12-1 ≈ PA4T-C12-2 > PA4T-Ra > PA4T-L > PA4T. The trend indicates that attaching more sidechains to the polymer backbone results in excellent solubilities of up to above 50.5 mg mL−1, while decreasing the molecular weight of the polymer works less effectively, resulting in a limited improvement of the solubility (8.4 mg mL−1). In addition, skeletal randomization transpires to a better solubility of 17.8 mg mL−1 in the resulting polymer PA4T-Ra, which is twice that of PA4T and sufficient for facile solution-based film formation. Among the three solubilization strategies, skeletal randomization is more effective than controlling the molecular weight and less than introducing more solubilizing side chains. As shown in the following sections, this medium solubility endowed the resulting polymer with sufficient processability and desirable aggregation in CB, allowing the fabrication of high mobility OFET devices.
2.3. Optoelectronic properties and aggregation behavior
The UV-vis absorption spectra of all the PA4T polymers in dilute CB at RT and thin films are depicted in Fig. 1b and c, respectively, and summarized in Table 1. In CB, PA4T-L with a lower molecular weight displayed a similar but slightly blue-shifted absorption to that of PA4T, a result of decreased effective conjugation length as previously reported.63 In cases of PA4T-C12-1 and PA4T-C12-2, a substantial blue shift of the absorption maxima of around 100 nm was observed when compared to that of PA4T, implying that the polymer backbones became more twisted after attaching two additional octyl sidechains to the repeat unit. Such conformational differences consequently led to weaker interchain interactions in PA4T-C12-1 and PA4T-C12-2 and corroborated their improved solubilities. In contrast, PA4T-Ra shared almost identical absorption behavior to PA4T, suggesting that randomization of the backbone imposed little influence on the chain coplanarity or interchain packing. Moreover, distinct 0–0 and 0–1 peaks were observed in the solution spectra of PA4T-L, PA4T-Ra and PA4T, indicating strong interchain aggregation in polymer solutions.64,65 The ratios of the peak intensities, A0–0/A0–1, were in the order of PA4T-Ra > PA4T > PA4T-L. As reported previously,66,67 a high A0–0/A0–1 ratio of above 1 corresponds to more ordered interchain packing. The highest A0–0/A0–1 ratio revealed a somewhat counterintuitive result that instead of disrupting the interchain packing, skeletal randomization reinforced such interactions and promoted the formation of more ordered aggregates. In contrast, the absence of the obvious double-peak feature in the spectra of PA4T-C12-1 and PA4T-C12-2 suggested that the two solubility-tuning motifs severely disrupted the degree of interchain ordering in solution. When moving from the solution to a thin film, PA4T-C12-2 showed a pronounced red shift in its absorption while only slight spectrum broadening was observed for PA4T-C12-1, indicating a significant enhancement of chain stacking for PA4T-C12-2 in the solid state. In clear contrast, the absorption features of PA4T-L, PA4T-Ra and PA4T were almost identical to their respective solution spectra, implying that strong pre-aggregated states have been established in solutions and remained after drying. Moreover, a similar trend in A0–0/A0–1 ratios was observed in thin films, where PA4T-Ra retained the highest A0–0/A0–1 ratio among all the polymers, correlating with its highest degree of order in interchain packing in the solid state. To gain further insight into the relative strength of interchain aggregation in solution, which is decisive to the quality and morphology of corresponding thin films, temperature-dependent absorption spectra of those polymers in CB were acquired. As shown in Fig. 1d–f and Fig. S5 (ESI†), upon heating the solutions from 20 to 100 °C, 0–1 peaks of all polymers were progressively blue-shifted and the intensity of 0–0 peaks gradually decreased because of the enhanced backbone torsion and reduced effective conjugation length at a higher temperature. For PA4T, a distinct 0–0 peak at ∼726 nm was still retained with a high A0–0/A0–1 ratio even at 100 °C, which implied the persistence of ultra-strong interchain aggregates that were in accordance with its high film roughness (see later sections). PA4T-L with a lower molecular weight exhibited an obviously diminished 0–0 peak, though the shoulder peak at ∼720 nm remained distinguishable at even 100 °C with a greatly reduced A0–0/A0–1 ratio. In contrast, PA4T-Ra displayed the largest variation of the A0–0/A0–1 ratio across the temperature window, with the 0–0 peak disappearing at ∼90 °C (Fig. 1g). The highest temperature responsiveness for PA4T-Ra indicated that PA4T-Ra is more adaptive than PA4T and PA4T-L for assembly and reorganization towards ordered microstructures.65 To gain quantitative insight into interchain aggregates in solution, filtration experiments were conducted to quantify the aggregate size of different polymers.68–70 As shown in Fig. 1h and Fig. S6 (ESI†), these PA4T polymers possessed distinctly varied size distributions of solution aggregates. The amounts of large aggregates (diameter >450 nm) follow the trend of PA4T > PA4T-L > PA4T-Ra > PA4T-C12-1 > PA4T-C12-2, while small aggregates (diameter < 220 nm) are in the order of PA4T < PA4T-L < PA4T-Ra < PA4T-C12-1 < PA4T-C12-2. The above results evidence that the order of intermolecular interactions and the “relative aggregation strength” of those polymers is PA4T > PA4T-L > PA4T-Ra > PA4T-C12-1 > PA4T-C12-2, illustrating the impressive effectiveness of skeletal randomization in fine-tuning interchain aggregation with modest strength and a high degree of structural order.With the exception of PA4T-C12-1 of which the absorption edge was located at ∼742 nm, all other polymers showed similar absorption edges of ∼810 nm. The optical band gap (Eg) estimated from the film absorption edge is ∼1.67 eV for PA4T-C12-1 and ∼1.52 eV for the rest of other polymers. Cyclic voltammetry (CV) was carried out to estimate the energy levels of the PA4T-based polymers. The highest occupied molecular orbital (HOMO) energy levels were determined by the oxidation onsets while the lowest unoccupied molecular orbital (LUMO) levels were calculated from the HOMO level and the corresponding optical band gap. As shown in Fig. 1i and Fig. S7 (ESI†), and Table 1, all polymers showed comparable LUMO levels of around −3.55 eV and HOMO levels of around −5.05 eV, with the exception of PA4T-C12-1 which exhibited a lower HOMO level of −5.23 eV as a result of decreased coplanarity (see discussions below).
2.4. Theoretical calculations
In general, high backbone coplanarity can enhance intermolecular interactions, induce dense interchain packing and benefit charge carrier transport.71 To gain a deeper understanding of the difference in interchain aggregation strength and illustrate the backbone conformation of those polymers, density functional theory (DFT) calculations employing the B3LYP/6-311G(d,p) basis set were conducted based on a tetramer segment of each of the polymers.36 For simplicity, sidechains were substituted with methyl groups in all segments. It is to be noticed that except for PA4T-Ra, the donor moiety between two adjacent p-AQM units in the polymer backbone is quaterthiophene (4T), while PA4T-Ra comprises terthiophene (3T), 4T and quinquethiophene (5T) as its donor moiety owing to the dissymmetric design. Here, one of the possible bonding modes involving all three oligothiophenes was modelled to represent the tetramer segment of PA4T-Ra. For comparison, the coplanarity of each segment of the polymers was quantified by the calculated standard deviation of atoms in Y coordinates (denoted as Y_StDev).50 As depicted in Fig. 2a, the Y_StDev values of PA4T/PA4T-L, PA4T-C12-1, PA4T-C12-2 and PA4T-Ra were 0.589, 1.103, 0.800 and 0.994 Å, respectively. It can be seen that, except for PA4T-C12-2, the coplanarity of the rest of the polymers followed the order of PA4T/PA4T-LPA4T-L > PA4T-Ra > PA4T-C12-1, in good agreement with the above-mentioned trend of intermolecular interaction and “relative aggregation strength” in those polymers. However, in the case of PA4T-C12-2, despite its moderate coplanarity, the large steric hindrance and relatively large lamellar d-spacing (vide infra) caused by high density of sidechains in the backbone may give rise to weak interchain aggregation strength. Moreover, intrachain charge transport of those polymers was evaluated by the theoretical calculations of effective hole masses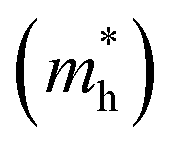 utilizing the Vienna ab initio simulation package (VASP) with the Perdew–Burke–Ernzerhof (PBE) functional.72,73 Band structures, partial densities of states (DOS) and calculated
utilizing the Vienna ab initio simulation package (VASP) with the Perdew–Burke–Ernzerhof (PBE) functional.72,73 Band structures, partial densities of states (DOS) and calculated 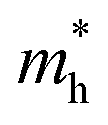 values of polymers are illustrated in Fig. 2b and Fig. S8 (ESI†). The theoretically predicted band gaps of those polymers were in the approximate order of PA4T-C12-1 > PA4T/PA4T-L ≈ PA4T-C12-2 ≈ PA4T-Ra, consistent with the experimental results. Moreover, PA4T/PA4T-L, PA4T-C12-1 and PA4T-C12-2 had comparable
values of polymers are illustrated in Fig. 2b and Fig. S8 (ESI†). The theoretically predicted band gaps of those polymers were in the approximate order of PA4T-C12-1 > PA4T/PA4T-L ≈ PA4T-C12-2 ≈ PA4T-Ra, consistent with the experimental results. Moreover, PA4T/PA4T-L, PA4T-C12-1 and PA4T-C12-2 had comparable 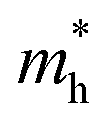 values in the range of 0.143 to 0.169 me, while a considerably smaller
values in the range of 0.143 to 0.169 me, while a considerably smaller 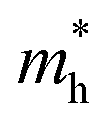 value of 0.115 me was obtained for PA4T-Ra. The smallest
value of 0.115 me was obtained for PA4T-Ra. The smallest 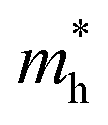 value for PA4T-Ra presumably originated from its smallest degree of bond length alternation (BLA) among all the polymers (Fig. S9, ESI†), which is consistent with a previous report.74 The small
value for PA4T-Ra presumably originated from its smallest degree of bond length alternation (BLA) among all the polymers (Fig. S9, ESI†), which is consistent with a previous report.74 The small 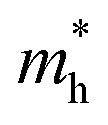 value implies efficient intrachain hole transport along the backbone of PA4T-Ra,74 which correlates well with its remarkably high hole mobility achieved in the PA4T-Ra based OFET device (vide infra).
value implies efficient intrachain hole transport along the backbone of PA4T-Ra,74 which correlates well with its remarkably high hole mobility achieved in the PA4T-Ra based OFET device (vide infra).
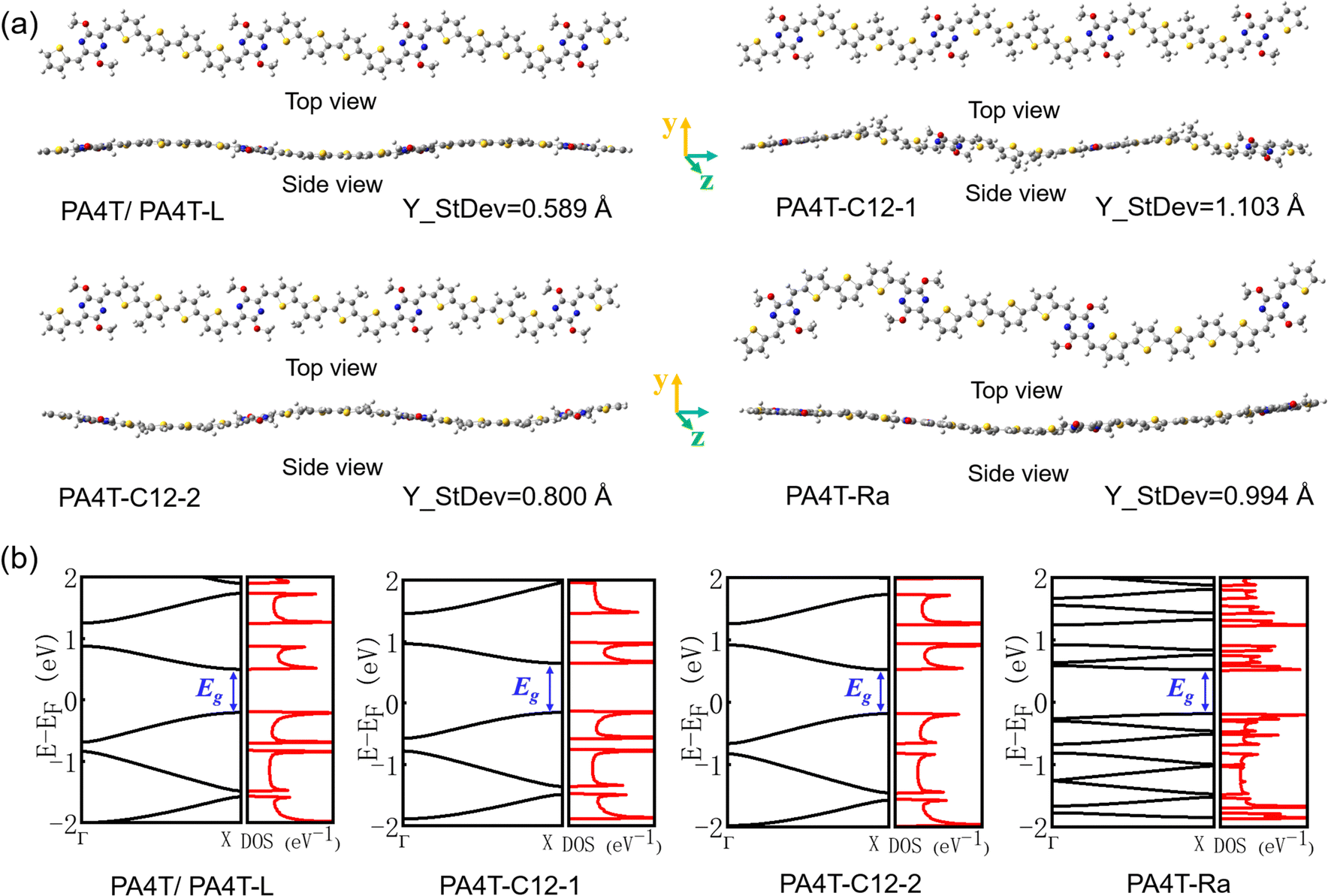 | ||
| Fig. 2 (a) Optimized geometries and standard deviations of each atom in Y coordinates for the tetramer segments of PA4T homologues, where sidechains are replaced by methyl groups for simplicity. (b) Band structures and partial densities of states (DOS) of PA4T homologues. | ||
2.5. Thin film microstructures and morphologies
Solution-state aggregation behavior has a critical influence on the solid-state microstructures and morphologies of CPs.75 To gain insight into the morphological properties of these polymer films, grazing-incidence wide-angle X-ray scattering (GIWAXS) measurement and tapping-mode atomic force microscopy (AFM) characterization were performed (Fig. 3 and Fig. S10, ESI†). As previously reported, PA4T exhibited obvious (h00) diffraction peaks (h up to 4) and a feeble (010) diffraction in the out-of-plane (OOP) direction with a lamellar d-spacing of 22.0 Å, indicating a predominant edge-on orientation. The spectrum of PA4T-L showed similar OOP diffraction peaks albeit with a weaker intensity, suggesting its slighter lower crystallinity. In cases of PA4T-C12-1 and PA4T-C12-2, not only the OOP diffraction peak intensity was greatly diminished, commensurate with the disappearance of higher order (300) and (400) peaks, but also the (100) diffraction in the in-plane (IP) direction showed up, which indicated a bimodal texture due to the emergence of face-on oriented crystallites. The lamellar d-spacings of PA4T-C12-1 and PA4T-C12-2 increased to 24.2 and 26.0 Å, respectively, in accordance with the less dense packing due to the additional octyl sidechains attached to the polymer backbone. As for PA4T-Ra, the diffraction pattern was similar to that of PA4T while the peak intensity was higher and a smaller lamellar distance of 20.8 Å was observed, indicative of better crystallinity, tighter chain packing and the preservation of the edge-on orientation preference in the random polymer. Crystalline coherence lengths (CCLs) and paracrystalline disorder (g factor), which represent the crystalline domain size and packing dislocation, respectively, were calculated to quantitatively compare the crystallinity and degree of crystalline perfectness in those polymers.76,77 As shown in Fig. 3e and summarized in Table 2, all polymers except PA4T-Ra exhibited a small CCL in the range of 190–223 Å and large paracrystalline disorder in the range of 12.1–13.7%, corresponding to relatively lower film crystallinity and higher structural disorder in the solid state. In sharp contrast, fine tuning the aggregation to medium strength via skeletal randomization resulted in a significantly larger CCL of 400 Å and a lower paracrystalline disorder of 8.1% in the thin film of PA4T-Ra, corresponding to a highly ordered packing structure desirable for better charge carrier transport in OFET devices. It should be noted that there is no (010) diffraction in the IP direction for all the polymer films, a peak that is typically expected from π–π stacking in lamellar conjugated polymers. Instead, the scattering signal in the shape of a concentrated arc at q ∼1.6 Å−1 at an azimuth angle of ∼30°, as observed in the GIWAXS spectra of PA4T-L and PA4T-Ra (Fig. 3a), presumably originated from π–π stacking. The atypical location of the π–π stacking peak, which is unique to AQM and oligothiophene-based conjugated polymers, is ascribed to the formation of a structure that is distinct from the commonly observed orthorhombic structure in crystalline conjugated polymers.21 The exact structure however remains unassigned and requires a future in-depth study.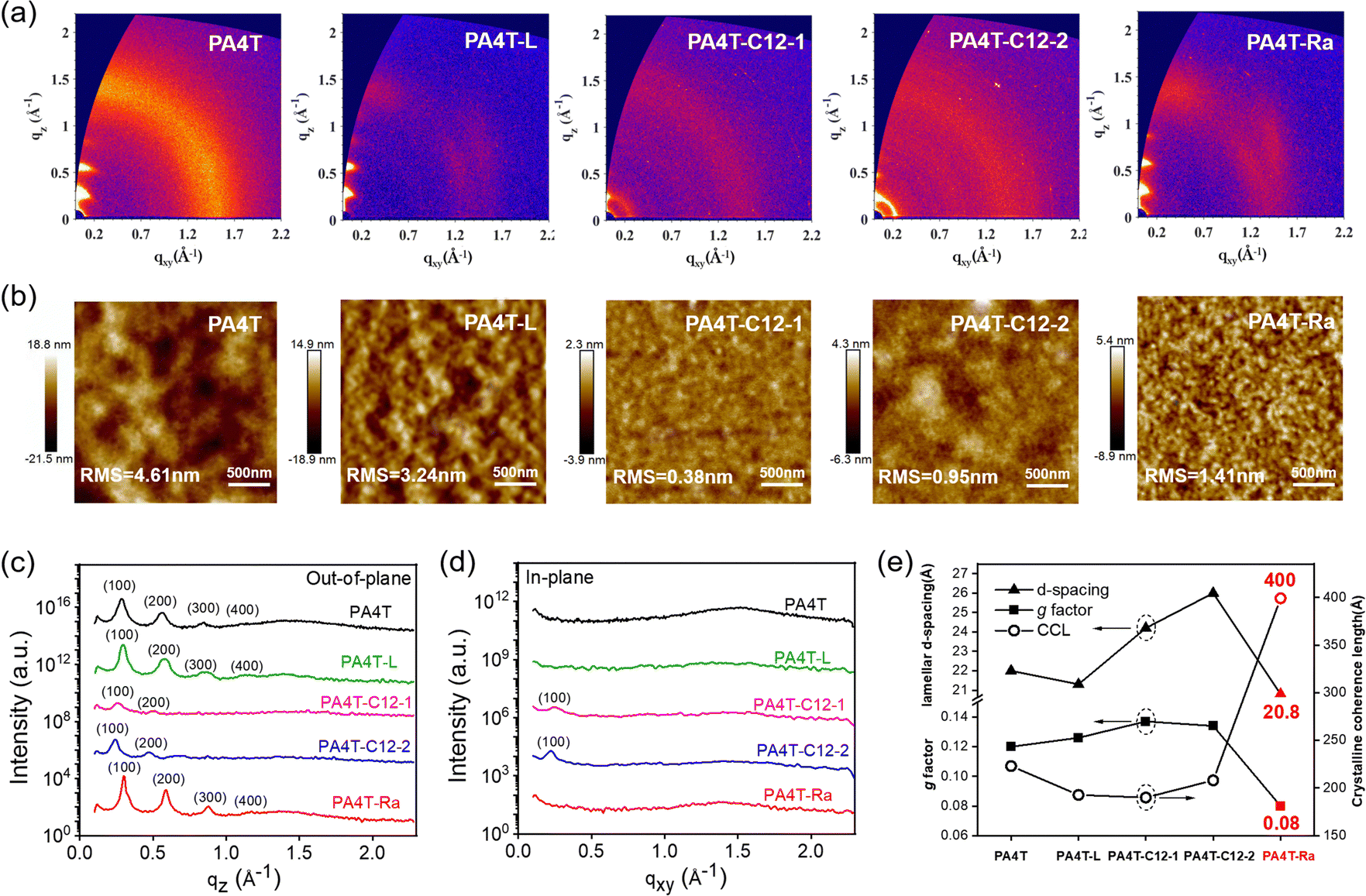 | ||
| Fig. 3 (a) GIWAXS patterns and (b) AFM topography images of five annealed polymer films. Line-cut profiles along (c) out-of-plane and (d) in-plane directions for five annealed polymer films. (d) Comparison of lamellar d-spacing, crystalline coherence length, and g factor of five annealed polymer films. | ||
| Polymer | Lamellar d-spacinga (Å) | CCLb (Å) | g factorb (%) |
|---|---|---|---|
| a Lamellar d-spacing was calculated according to OOP (100) diffraction peaks. b Crystalline coherence length and g factor were calculated according to OOP (200) diffraction peaks. c Reported previously. | |||
| PA4T | 20.5c | 223 | 12.1 |
| PA4T-L | 21.3 | 193 | 12.6 |
| PA4T-C12-1 | 24.2 | 190 | 13.7 |
| PA4T-C12-2 | 26.0 | 208 | 13.4 |
| PA4T-Ra | 20.8 | 400 | 8.1 |
In addition, the as-cast films of those polymers showed decreased crystallinity compared to their annealed films, concomitant with a smaller RMS surface roughness of as-cast films as seen in AFM images (Fig. S11, ESI†). In comparison to PA4T, the four new polymers exhibited surface roughness in the range of 0.38–3.24 nm, lower than that of PA4T (4.61 nm). The surface roughness of those five polymers followed the order of PA4T > PA4T-L > PA4T-Ra > PA4T-C12-2 > PA4T-C12-1, in close agreement with the trends in aggregation strengths and solubilities. In particular, discontinuous and large domains of aggregates were observed in both PA4T and PA4T-L films. On the other hand, PA4T-C12-1 and PA4T-C12-2 films displayed featureless and smooth surface morphologies with an RMS roughness of below 1 nm. In contrast, the annealed film of PA4T-Ra showed an appropriate RMS value of 1.4 nm and interconnected grains. The uniform surface and formation of an interconnected network are expected to favor charge carrier transport across crystalline grain boundaries.
It is generally believed that strong interchain aggregation would be advantageous to realize high crystallinity and ordered packing in CPs.78 Nevertheless, the relative crystallinity observed in those five polymers, in the order of PA4T-Ra > PA4T > PA4T-L > PA4T-C12-1 ≈ PA4T-C12-2, is inconsistent with their “relative aggregation strength” which follows the order of PA4T > PA4T-L > PA4T-Ra > PA4T-C12-1 > PA4T-C12-2. A plausible transition from solution aggregates to thin-film microstructures is outlined in Fig. 4 to enlighten the seemingly contradictory results whereas PA4T-Ra with modest aggregation strength could deliver unusually high crystallinity and ordered structure.
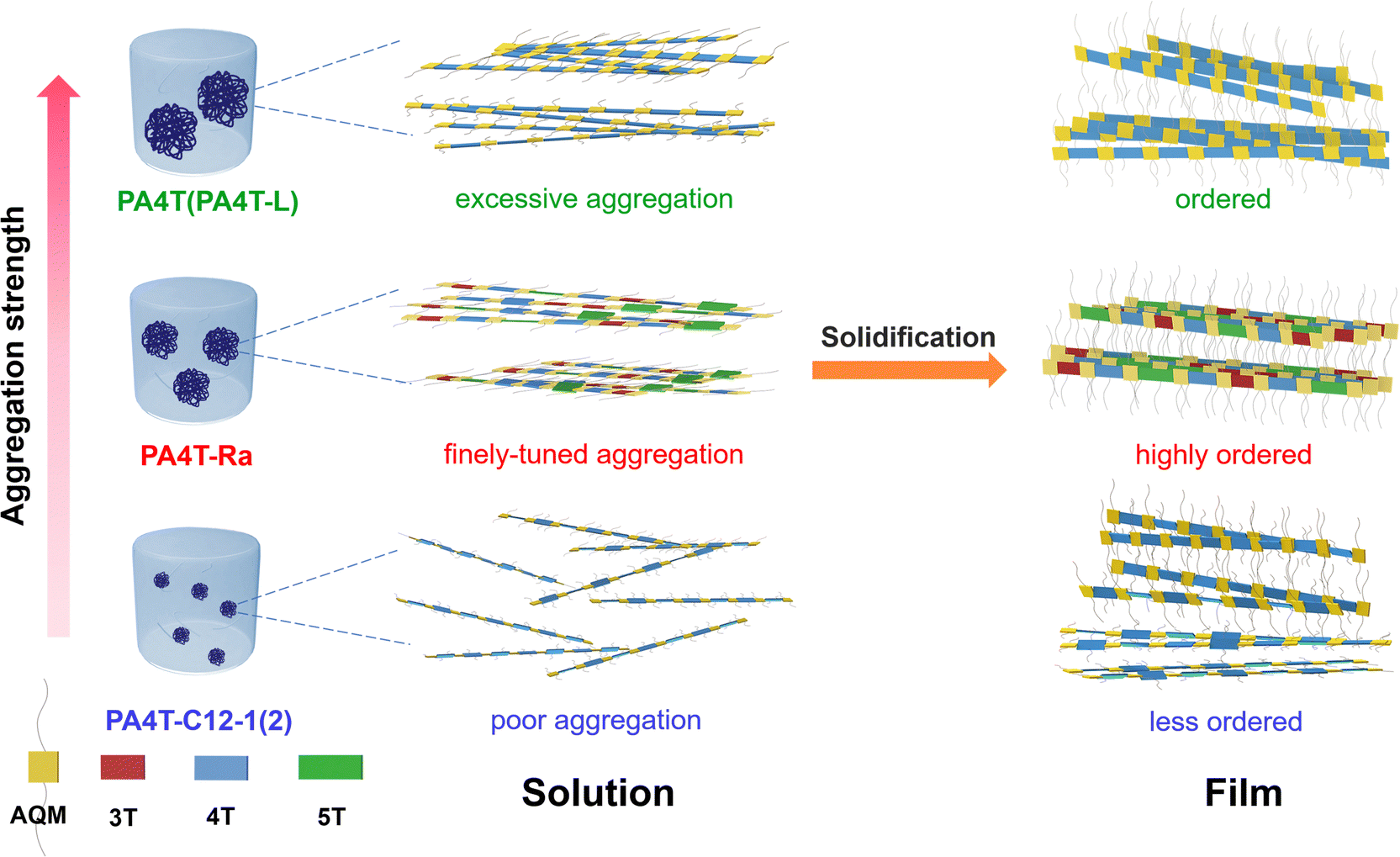 | ||
| Fig. 4 Schematic illustration of the proposed transitions from solution aggregates to thin-film microstructures for polymers with different aggregation strengths. | ||
While lowering the molecular weight of PA4T improves the solubility and slightly modulates its aggregation strength, both PA4T and PA4T-L displayed excessive aggregation in solution. The overly strong interchain interactions and intensively tight chain entanglements may limit the polymers’ ability to reorganize and reassemble into more ordered packing during the film-forming process. On the other hand, the conventional solubilization strategy of introducing alkyl chains was too disruptive to interchain interactions, resulting in much reduced aggregation strength and poorly ordered structures in the thin film. Notably, the skeletal randomization strategy stands out as a counterintuitive method that endows a Goldilocks zone for the aggregation strength in solution, which facilitates the self-assembly of polymer chains into orderly packed structures in highly crystalline films. Such correlations are consistent with previous findings by Pei and coworkers who have demonstrated that solution-state aggregation critically determines solid-state microstructures and corresponding charge transport.69
In order to obtain an in-depth understanding of the counterintuitive result, we further included a regioregular polymer PA3T, containing one p-AQM and one terthiophene (3T) in the repeat unit, in the comparisons against PA4T and PA4T-Ra. Polymer PA3T comprising the smaller-sized terthiophene moiety was reported by us previously as an analogue to PA4T.38,40 It exhibits reduced solution-state aggregation strength in comparison with PA4T, which is comparable to that of PA4T-Ra (Fig. S12, ESI†). However, A0–0/A0–1 ratios in RT solution and thin film are both below 1, implying less ordered interchain packing in PA3T. This can be attributed to the increased side chain density and the upraised steric hindrance effect in PA3T when compared to PA4T and PA4T-Ra, endowing an improved solubility of 23.1 mg mL−1 for PA3T (Table S1, ESI†).79 Compared to PA4T, PA3T film shows a larger lamellar d-spacing of 22.3 Å, a smaller CCL of 205 Å and a higher g factor of 12.8%, while PA4T-Ra delivers a larger CCL of 400 and a smaller g factor of 8.1% (Table S1, ESI†). Consequently, PA3T and PA4T-Ra analogues share comparable solution-state aggregation strength but possess distinctly different film crystallinity and lattice disorder. We accordingly propose that the difference in film microstructures may be related to the density and distribution of side chains in the polymer backbone. Despite that moderate aggregation strength is favorable for the reorganization and reassembly of polymer chains into more ordered packing during solidification, the increased side chain density in PA3T would impede interchain interaction and packing, resulting in decreased crystallinity and increased lattice disorder. Randomizing the backbone sequence can not only give rise to moderate aggregation strength, but also maintain the average side chain density along the PA4T-Ra backbone because of the distributed arrangement of side chains. Such an arrangement would not impede dense interchain packing in film, but can instead facilitate more ordered packing owing to the favorable interchain interactions. On the other hand, the presence of a longer quinquethiophene (5T) segment in PA4T-Ra compared to 4T and 3T may facilitate adjacent interchain contact, which also possibly contributes to the dense interchain packing during solidification and/or interchain charge transport.80 Overall, the two factors, namely moderate aggregation strength and the distributed arrangement of side chains, together account for the optimal solid-state microstructure and efficient charge transport observed in PA4T-Ra (vide infra).
2.6. OFET fabrication and characterization
OFET devices with a bottom-gate top-contact (BGTC) configuration were fabricated using chlorinated solvents (a mixture of CB and CF) to investigate the charge transport properties of those CPs. Both the deposition of active layers and the measurement of OFET characteristics were conducted in an ambient environment, a condition that is desirable for low-cost processing and scale-up. Representative transfer and output characteristics are shown in Fig. 5a, b and Fig. S13 (ESI†), and device performance was summarized in Fig. S14 (ESI†) and Table 3 and Table S2 (ESI†). Despite the improved solubility for PA4T-L, PA4T-C12-1 and PA4T-C12-2, they showed only modest hole mobilities in the range of 0.041 to 0.062 cm2 V−1 s−1. The mobilities were slightly lower than that of PA4T and correlated well with the polymers’ relatively small CCL and large paracrystalline disorder. It is worth noting that alkylated bithiophene with head-to-head (HH) linkage tends to have a large dihedral angle, endowing the resulting polymer with a twisted backbone, amorphous film morphology and poor charge carrier mobility according to previous literature.25 Lower carrier mobility is expected for the HH linkage-based PA4T-C12-1 than the non-alkylated bithiophene-based PA4T and tail-to-tail (TT) linkage-based PA4T-C12-2. While this is the case, the observed mobilities are in the same order and essentially comparable, suggesting that there are other contributing factors to the hole transporting properties, for instance, surface roughness and lamellar d-spacings. Impressively, PA4T-Ra delivered an excellent hole mobility of up to 3.11 cm2 V−1 s−1, which is more than 30 times higher than that of PA4T and 50 times higher than that of the other three polymers (Fig. 5c). Moreover, the device behavior was characterized by a high reliability coefficient of 88%.81,82 This mobility, to the best of our knowledge, is among the highest for quinoidal-donor polymer-based, spuncast OFETs with a satisfying reliability factor (Fig. 5d, Fig. S15 and Table S3, ESI†). Additionally, the dependence of mobilities on gate voltages was studied for all four polymers to further evaluate the validity of the extracted mobility. As shown in Fig. S16 (ESI†), PA4T-L and PA4T-Ra-based OFET devices showed a low gate voltage dependence (i.e., small mobility variations) in the range of −30 to −80 V, while in the case of PA4T-C12-1 and PA4T-C12-2, low gate voltage dependence was only observed in a narrower range between −60 and −80 V. Such contrast in gate voltage dependence is in accordance with the high reliability factors observed in PA4T-L and PA4T-Ra based devices. To evaluate the batch-to-batch reproducibility of the resulting polymer based on the skeletal randomization strategy, a second batch of the polymer was prepared under the same polymerization condition, which also exhibited similar charge transport properties with a hole mobility of 2.98 cm2 V−1 s−1 (Fig. S17 and Table S4, ESI†). The results confirmed that skeletal randomization could be able to impart the resulting random polymer with highly reproducible properties, which is essential for large-scale synthesis.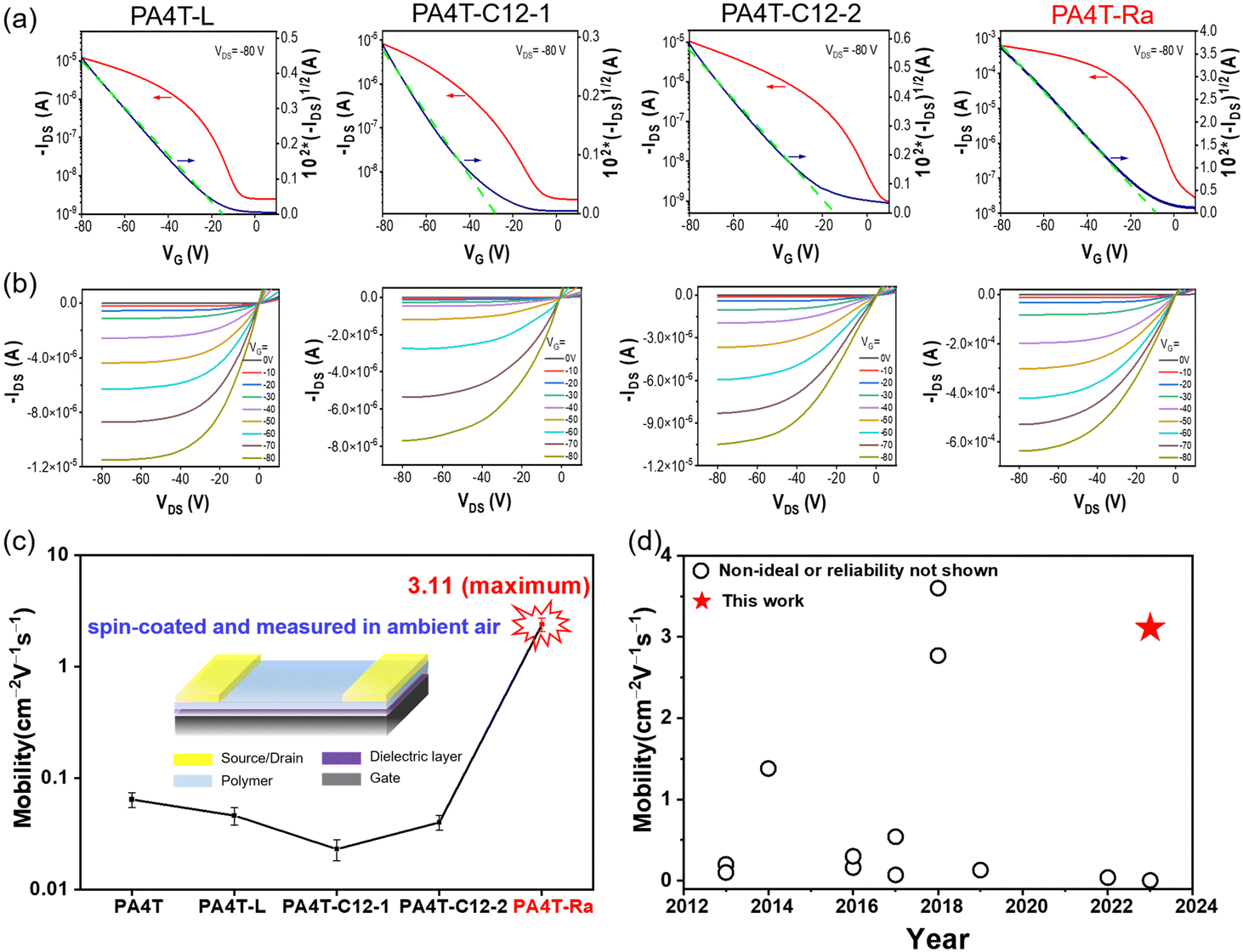 | ||
| Fig. 5 Typical (a) transfer and (b) output characteristics of annealed OFETs based on four new polymers using chlorinated solvents. (c) Comparison of maximum hole mobilities of the PA4T homologues. (d) Hole mobilities of OFETs based on quinoid-donor polymers characterized by conventional spin-coating deposition method in recent 10 years. | ||
| Polymer | μ h,max [μh,avg]a [cm2 V−1 s−1] | V th [V] | I on/off | γ [%] |
|
|---|---|---|---|---|---|
a Maximum mobility under optimized annealing conditions, average mobilities were calculated based on 10 independent devices and are listed in parentheses.
b Reliability factor γ calculated according to ref. 77 and 78.
c Effective hole mass 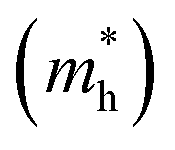 extracted from the theoretical calculations. me represents the mass of an electron.
d Reported previously.
e Not available. extracted from the theoretical calculations. me represents the mass of an electron.
d Reported previously.
e Not available.
|
|||||
| PA4T | 0.084 (0.082 ± 0.003) | −5 | 104–105 | —e | 0.149 |
| PA4T-L | 0.062 (0.046 ± 0.008) | −14 | 102–103 | 97.1 | 0.149 |
| PA4T-C12-1 | 0.041 (0.023 ± 0.005) | −17 | 103–104 | 66.6 | 0.169 |
| PA4T-C12-2 | 0.059 (0.040 ± 0.006) | −16 | 104–105 | 79.6 | 0.143 |
| PA4T-Ra | 3.11 (2.39 ± 0.33) | −10 | 103–104 | 88.3 | 0.115 |
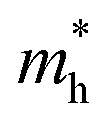
The use of chlorinated solvents during the fabrication of high mobility OFET devices has been a source of health and environmental concerns, which demands the substitution of eco-friendly non-chlorinated solvents, such as THF for ambient-processed OFETs. Given that non-chlorinated and non-aromatic tetrahydrofuran (THF) is structurally distinct from chlorobenzene (CB), we have selected eco-friendly THF as the solvent and systematically investigated the solubility, solution aggregation, film morphology and hole mobility of PA4T-Ra and its homologous polymers. As shown in Fig. S18a (ESI†), the solubilities of the PA4T, PA4T-L, PA4T-C12-1, PA4T-C12-2 and PA4T-Ra are 2.3, 4.5, 50.2, 48.9 and 10.2 mg mL−1 in THF, respectively, which are much lower than their respective solubility in CB because of the weaker dissolving capability of THF and follow the same trend of PA4T-C12-1 ≈ PA4T-C12-2 > PA4T-Ra > PA4T-L > PA4T. In addition, all five polymers processed in THF exhibited similar absorption features to those processed in CB, except that A0–0/A0–1 ratios in THF-deposited films were slightly lower than those obtained from CB, which indicated that solid-state interchain packing become less ordered when processed from THF (Fig. S18b and c, ESI†). The variable temperature UV-vis absorption spectra of all five polymers in THF (from 20 to 60 °C) are shown in Fig. S18d–h (ESI†), which demonstrates that the order of “relative aggregation strength” of the polymers roughly follows the trend of that in CB. Moreover, OFET devices based on these THF-processed polymers are also fabricated and measured in ambient air. As presented in Fig. S19 and summarized in Table S5 (ESI†), those OFET devices exhibited similar p-type transport behavior as those fabricated using chlorinated solvents. Impressively, PA4T-Ra delivers a hole mobility of up to 0.83 cm2 V−1 s−1, representing one of the first examples of quinoidal CP-based OFETs processed from a non-chlorinated solvent. Despite the lower mobility than that obtained from CB, the mobility of PA4T-Ra remains to rank the highest amongst the five polymers, underscoring the effectiveness of the skeletal randomization strategy in constructing high-performing non-chlorinated solvent-processed conjugated polymers. Thin film morphology and microstructure of PA4T-Ra deposited from THF are further studied to correlate with its hole transport properties (Fig. S20, ESI†). Sharp (h00) (h is up to 4) diffraction peaks, together with a weak (010) diffraction peak in the OOP direction and the lack of obvious IP patterns in annealed PA4T-Ra, indicate the presence of predominant edge-on oriented crystallites similar to those from CB. Moreover, the calculated CCL was as large as 356 Å while the g factor was only 9.2%, supporting the high crystallinity of the film.
2.7. OFET device stability
Operational stability under bias and storage stability in ambient air are essential for practical usage and future commercialization of OFETs, thus it is important to evaluate such characteristics of those AQM polymers. As depicted in Fig. 6a, after applying VG of −80 and 0 V at VDS of −80 V up to 9000 cycles (4000 s), PA4T and PA4T-L exhibited fluctuant on-current and off-current, showing a certain degree of operational instability. For PA4T-C12-1 and PA4T-C12-2 with low crystallinity, the operational instability became more pronounced, as characterized by larger fluctuations in on-current and off-current and obviously eroded on/off ratio. In contrast, impressive operational stability was observed for PA4T-Ra, of which the on/off currents and ratios remained nearly constant. The relative operational stability of the five polymers correlated well with their film crystallinity and structural disorder, i.e., polymers with higher crystallinity and less structural disorder delivered greater operational stability. Further repeatability of transfer characteristics and bias stress stability were also studied, again revealing trends that were consistent with the relative operational stability and the film crystallinity (Fig. 6b and c). Additionally, transfer characteristics of OFET devices based on those polymers were measured before and after storing in ambient air at a relative humidity of 56% for 30 days to reveal their storage stabilities (Fig. 6d). Among these results, PA4T-Ra-based devices displayed the slightest shift of its transfer characteristics, attesting to the best storage stability for PA4T-Ra. Overall, PA4T-Ra with the highest crystallinity and least structural disorder exhibits the best operational and environmental stabilities, a trend that is consistent with previous reports.36,83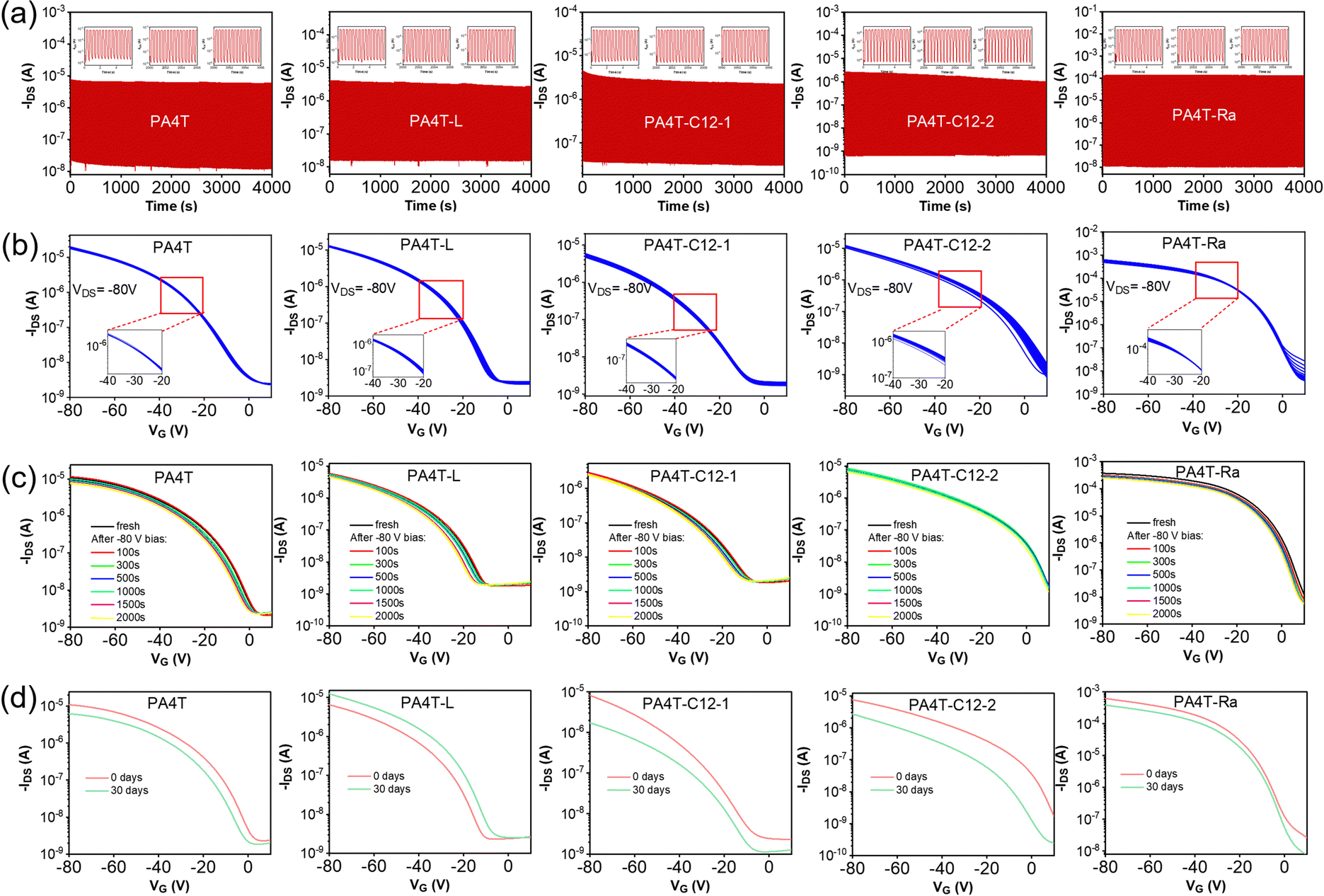 | ||
| Fig. 6 (a) On–off cycle tests (9000 cycles) on OFET devices by applying VG of −80 and 0 V at VDS of −80 V. (b) Repeatability of series of transfer curves (30 cycles) of OFET devices at VDS of −80 V. (c) Bias stress tests on OFET devices by applying prolonged bias voltage of −80 V for up to 2000 s. (d) Transfer curves of OFET devices before and after storing in ambient air for 30 days. | ||
3. Conclusion
In summary, a skeletal randomization strategy was introduced as a potent tool to tune the solubility and aggregation strength of quinoid-aromatic CPs for better charge transport in OFET devices. Various homologues of CPs with the same overall backbone composition were designed and synthesized according to three solubilization strategies, allowing for a comparative and systematic study to validate the effectiveness of this strategy. The influence of molecular structures on solubility, aggregation behavior, optoelectronic properties, film morphologies, microstructures and OFET performance was thoroughly investigated. Despite the disparity in the effectiveness of improving solubilities, more significant distinctions were observed in their interchain aggregation behavior in solution and thin film crystallinities. Remarkably, random polymer PA4T-Ra accessed using the facile skeletal randomization strategy exhibited modest aggregation strength, giving rise to the largest crystalline coherence length and lowest structural disorder in the film. Consequently, OFET devices based on PA4T-Ra displayed excellent hole mobilities of up to 3.11 cm2 V−1 s−1, amounting to more than 30 times improvement over the other PA4T homologues. Future investigation of the effect of temperature on polymer aggregation and film microstructures is desired for further optimization of charge transport properties. PA4T-Ra represents a rare and counterintuitive example where “disorder” introduced along the main chain of conjugated polymers has dramatically improved the film crystallinity and enhanced the charge carrier mobility, with the added benefit of improved solution processibility, and excellent operational and air stability of the resulting devices. Theoretical insights revealed that PA4T-Ra has the smallest effective hole mass among all the PA4T homologues, corroborating with efficient intrachain hole transport. This study, for the first time, offers a fresh perspective on the use of skeletal randomization to enhance the film crystallinity of CPs. We expect that the underutilized skeletal randomization strategy can be implemented more generally for developing practical, high-performing CPs, potentially accelerating the large-scale manufacturing of superior organic electronic devices.Author contributions
Quanfeng Zhou: conceptualization, data curation, formal analysis, methodology, and writing – original draft. Cheng Liu: data curation, formal analysis, methodology, software, and writing – original draft. Jinlun Li: data curation, formal analysis, visualization, and writing – original draft. Runze Xie: data curation, methodology, and software. Guoxiang Zhang: data curation, formal analysis, and software. Xiang Ge: data curation and formal analysis. Zesheng Zhang: data curation and formal analysis. Lianjie Zhang: data curation and resources. Junwu Chen: data curation, formal analysis, and resources. Xiu Gong, data curation, formal analysis, and resources. Chen Yang: resources and software. Yuanyu Wang: data curation, formal analysis, funding acquisition, resources, software, and writing – review & editing. Yi Liu: conceptualization, funding acquisition, methodology, supervision, and writing – review & editing. Xuncheng Liu: conceptualization, formal analysis, funding acquisition, project administration, supervision, and writing – original draft.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the Nature Science Foundation of Guizhou Provincial Science and Technology Department (QKHJC ZK [2021] general 247), the Natural Science Research Young Talents Project of Provincial Education Department (No. Guizhou Education & KY Word [2022] 105), and the National Natural Science Foundation of China (52363021, 22305050). Part of the work was performed as a user project at the Molecular Foundry, a national user facility supported by the Office of Science, Office of Basic Energy Sciences, the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors are thankful for the computing support of the State Key Laboratory of Public Big Data at Guizhou University.References
- M. Luo, L. Kong, J. Liang, Z. Zhang, S. Deng, L. Zhang, X. Qiao, D. Ma and J. Chen, Chem. Eng. J., 2023, 457, 141281 CrossRef CAS.
- R. Ma, K. Zhou, Y. Sun, T. Liu, Y. Kan, Y. Xiao, T. A. Dela Peña, Y. Li, X. Zou, Z. Xing, Z. Luo, K. S. Wong, X. Lu, L. Ye, H. Yan and K. Gao, Matter, 2022, 5, 725–734 CrossRef CAS.
- X. Liu, L. Nian, K. Gao, L. Zhang, L. Qing, Z. Wang, L. Ying, Z. Xie, Y. Ma, Y. Cao, F. Liu and J. Chen, J. Mater. Chem. A, 2017, 5, 17619–17631 RSC.
- X. Cao, H. Li, J. Hu, H. Tian, Y. Han, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2023, 62, e202212979 CrossRef CAS PubMed.
- J. Meng, J. Dou, Z. Zhou, P. Chen, N. Luo, Y. Li, L. Luo, F. He, H. Geng, X. Shao, H.-L. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2023, 62, e202301863 CrossRef CAS PubMed.
- J. Chen, W. Zhang, L. Wang and G. Yu, Adv. Mater., 2023, 35, 2210772 CrossRef CAS.
- Y. Shi, J. Li, H. Sun, Y. Li, Y. Wang, Z. Wu, S. Y. Jeong, H. Y. Woo, S. Fabiano and X. Guo, Angew. Chem., Int. Ed., 2022, 61, e202214192 CrossRef CAS PubMed.
- C. Gao, A. Shukla, H. Gao, Z. Miao, Y. Zhang, P. Wang, G. Luo, Y. Zeng, W. W. H. Wong, T. A. Smith, S.-C. Lo, W. Hu, E. B. Namdas and H. Dong, Adv. Mater., 2023, 35, 2208389 CrossRef CAS.
- C. Dong, S. Deng, B. Meng, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2021, 60, 16184–16190 CrossRef CAS PubMed.
- A. Nawaz, L. Merces, L. M. M. Ferro, P. Sonar and C. C. B. Bufon, Adv. Mater., 2023, 35, 2204804 CrossRef CAS PubMed.
- Q. Zhang, T. Jin, X. Ye, D. Geng, W. Chen and W. Hu, Adv. Funct. Mater., 2021, 31, 2106151 CrossRef CAS.
- Y. Woo, W. Hong, S. Y. Yang, H. J. Kim, J.-H. Cha, J. E. Lee, K. J. Lee, T. Kang and S.-Y. Choi, Adv. Electron. Mater., 2018, 4, 1800251 CrossRef.
- Q. Liu, Y. Wang, A. Kohara, H. Matsumoto, S. Manzhos, K. Feron, S. E. Bottle, J. Bell, T. Michinobu and P. Sonar, Adv. Funct. Mater., 2020, 30, 1907452 CrossRef CAS.
- C. Grand, W. Zajaczkowski, N. Deb, C. K. Lo, J. L. Hernandez, D. G. Bucknall, K. Müllen, W. Pisula and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 13357–13368 CrossRef CAS PubMed.
- I. Osaka, M. Saito, H. Mori, T. Koganezawa and K. Takimiya, Adv. Mater., 2012, 24, 425–430 CrossRef CAS.
- S. Lv, L. Li, Y. Mu and X. Wan, Polym. Rev., 2021, 61, 520–552 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- T. Lei, J.-Y. Wang and J. Pei, Chem. Mater., 2014, 26, 594–603 CrossRef CAS.
- Q. Chen, Y. H. Han, L. R. Franco, C. F. N. Marchiori, Z. Genene, C. M. Araujo, J.-W. Lee, T. N.-L. Phan, J. Wu, D. Yu, D. J. Kim, T.-S. Kim, L. Hou, B. J. Kim and E. Wang, Nano-Micro Lett., 2022, 14, 164 CrossRef CAS.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS.
- I. Osaka and K. Takimiya, Polymer, 2015, 59, A1–A15 CrossRef CAS.
- A. Zen, J. Pflaum, S. Hirschmann, W. Zhuang, F. Jaiser, U. Asawapirom, J. P. Rabe, U. Scherf and D. Neher, Adv. Funct. Mater., 2004, 14, 757–764 CrossRef CAS.
- Y. Ding, Y. Zhu, X. Wang, Y. Wang, S. Zhang, G. Zhang, X. Gu and L. Qiu, Chem. Mater., 2022, 34, 2696–2707 CrossRef CAS.
- Y. Ran, Q. Li, J. Li, Y. Sun, W. Shi, Y. Guo and Y. Liu, Adv. Electron. Mater., 2021, 7, 2100526 CrossRef CAS.
- K. Yang, Z. Chen, Y. Wang and X. Guo, Acc. Mater. Res., 2023, 4, 237–250 CrossRef CAS.
- M. Li, Z. Li, D. Yu, M. Wang, D. Wang and B. Wang, Chem. – Eur. J., 2023, 29, e202202930 CrossRef CAS PubMed.
- J. Huang and G. Yu, Mater. Chem. Front., 2021, 5, 76–96 RSC.
- X. Ji and L. Fang, Polym. Chem., 2021, 12, 1347–1361 RSC.
- Y. Choi, Y. Kim, Y. Moon, I.-B. Kim, H. Hwang and D.-Y. Kim, J. Mater. Chem. C, 2022, 10, 17874–17885 RSC.
- M. Yang, T. Du, X. Zhao, X. Huang, L. Pan, S. Pang, H. Tang, Z. Peng, L. Ye, Y. Deng, S. Mingliang, C. Duan, H. Fei and C. Yong, Sci. China: Chem., 2021, 64, 1219–1227 CrossRef CAS.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- Y. Kim, H. Hwang, N.-K. Kim, K. Hwang, J.-J. Park, G.-I. Shin and D.-Y. Kim, Adv. Mater., 2018, 30, 1706557 CrossRef.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 7725–7732 CrossRef CAS PubMed.
- X. Liu, C. L. Anderson and Y. Liu, Acc. Chem. Res., 2023, 56, 1669–1682 CrossRef CAS PubMed.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz, A. Navarro, Y. Lv and Y. Liu, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS.
- H. Liang, C. Liu, Z. Zhang, X. Liu, Q. Zhou, G. Zheng, X. Gong, L. Xie, C. Yang, L. Zhang, B. He, J. Chen and Y. Liu, Adv. Funct. Mater., 2022, 32, 2201903 CrossRef CAS.
- C. L. Anderson, H. Li, C. G. Jones, S. J. Teat, N. S. Settineri, E. A. Dailing, J. Liang, H. Mao, C. Yang, L. M. Klivansky, X. Li, J. A. Reimer, H. M. Nelson and Y. Liu, Nat. Commun., 2021, 12, 6818 CrossRef CAS PubMed.
- C. Liu, X. Liu, G. Zheng, X. Gong, C. Yang, H. Liu, L. Zhang, C. L. Anderson, B. He, L. Xie, R. Zheng, H. Liang, Q. Zhou, Z. Zhang, J. Chen and Y. Liu, J. Mater. Chem. A, 2021, 9, 23497–23505 RSC.
- C. L. Anderson, N. Dai, S. J. Teat, B. He, S. Wang and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 17978–17985 CrossRef CAS.
- X. Liu, B. He, C. L. Anderson, J. Kang, T. Chen, J. Chen, S. Feng, L. Zhang, M. A. Kolaczkowski, S. J. Teat, M. A. Brady, C. Zhu, L.-W. Wang, J. Chen and Y. Liu, J. Am. Chem. Soc., 2017, 139, 8355–8363 CrossRef CAS PubMed.
- X. Liu, B. He, A. Garzon-Ruiz, A. Navarro, T. L. Chen, M. A. Kolaczkowski, S. Feng, L. Zhang, C. A. Anderson, J. Chen and Y. Liu, Adv. Funct. Mater., 2018, 28, 201801874 Search PubMed.
- Y. Kim, H. Park, J. S. Park, J.-W. Lee, F. S. Kim, H. J. Kim and B. J. Kim, J. Mater. Chem. A, 2022, 10, 2672–2696 RSC.
- J.-W. Lee, C. Sun, S.-W. Lee, G.-U. Kim, S. Li, C. Wang, T.-S. Kim, Y.-H. Kim and B. J. Kim, Energy Environ. Sci., 2022, 15, 4672–4685 RSC.
- P. J. Leenaers, H. van Eersel, J. Li, M. M. Wienk and R. A. J. Janssen, Macromolecules, 2020, 53, 7749–7758 CrossRef CAS PubMed.
- S. W. Kim, H. Kim, J.-W. Lee, C. Lee, B. Lim, J. Lee, Y. Lee and B. J. Kim, Macromolecules, 2019, 52, 738–746 CrossRef CAS.
- E. L. Melenbrink, K. M. Hilby, M. A. Alkhadra, S. Samal, D. J. Lipomi and B. C. Thompson, ACS Appl. Mater. Interfaces, 2018, 10, 32426–32434 CrossRef CAS.
- J. B. Howard and B. C. Thompson, Macromol. Chem. Phys., 2017, 218, 1700255 CrossRef.
- H. H. Choi, J. Y. Baek, E. Song, B. Kang, K. Cho, S.-K. Kwon and Y.-H. Kim, Adv. Mater., 2015, 27, 3626–3631 CrossRef CAS.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS.
- J. Yuan, M. J. Ford, Y. Zhang, H. Dong, Z. Li, Y. Li, T.-Q. Nguyen, G. C. Bazan and W. Ma, Chem. Mater., 2017, 29, 1758–1768 CrossRef CAS.
- H. Zhong, C.-Z. Li, J. Carpenter, H. Ade and A. K. Y. Jen, J. Am. Chem. Soc., 2015, 137, 7616–7619 CrossRef CAS.
- J. Lee, S. H. Kang, S. M. Lee, K. C. Lee, H. Yang, Y. Cho, D. Han, Y. F. Li, B. H. Lee and C. Yang, Angew. Chem., Int. Ed., 2018, 57, 13629–13634 CrossRef CAS.
- K. H. Park, S. Y. Son, J. O. Kim, G. Kang, T. Park and D. Kim, J. Phys. Chem. Lett., 2018, 9, 3173–3180 CrossRef CAS PubMed.
- S. Y. Son, K. Choi, J. Lee, H. I. Kim, T. Park and M. Kim, Chem. Mater., 2022, 34, 4856–4864 CrossRef CAS.
- C. Zhou, Z. Chen, G. Zhang, C. McDowell, P. Luo, X. Jia, M. J. Ford, M. Wang, G. C. Bazan, F. Huang and Y. Cao, Adv. Energy Mater., 2018, 8, 1701668 CrossRef.
- S. Y. Son, J. W. Kim, J. Lee, G.-W. Kim, J. Hong, J. Y. Kim and T. Park, J. Mater. Chem. A, 2018, 6, 24580–24587 RSC.
- J. Ji, X. Wu, P. Deng, D. Zhou, D. Lai, H. Zhan and H. Chen, J. Mater. Chem. C, 2019, 7, 10860–10867 RSC.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. J. Janssen and W. Li, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- Z. Wang, M. Gao, C. He, W. Shi, Y. Deng, Y. Han, L. Ye and Y. Geng, Adv. Mater., 2022, 34, 2108255 CrossRef CAS.
- X. Yuan, Y. Zhao, T. Zhan, J. Oh, J. Zhou, J. Li, X. Wang, Z. Wang, S. Pang, P. Cai, C. Yang, Z. He, Z. Xie, C. Duan, F. Huang and Y. Cao, Energy Environ. Sci., 2021, 14, 5530–5540 RSC.
- Z. Wang, X. Song, Y. Jiang, J. Zhang, X. Yu, Y. Deng, Y. Han, W. Hu and Y. Geng, Adv. Sci., 2019, 6, 1902412 CrossRef CAS.
- L. Ye, W. Li, X. Guo, M. Zhang and H. Ade, Chem. Mater., 2019, 31, 6568–6577 CrossRef CAS.
- T. J. Fauvell, T. Zheng, N. E. Jackson, M. A. Ratner, L. Yu and L. X. Chen, Chem. Mater., 2016, 28, 2814–2822 CrossRef CAS.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS.
- H. Hu, P. C. Y. Chow, G. Zhang, T. Ma, J. Liu, G. Yang and H. Yan, Acc. Chem. Res., 2017, 50, 2519–2528 CrossRef CAS PubMed.
- J. Ma, Z. Liu, J. Yao, Z. Wang, G. Zhang, X. Zhang and D. Zhang, Macromolecules, 2018, 51, 6003–6010 CrossRef CAS.
- S. H. Yu, K. H. Park, Y.-H. Kim, D. S. Chung and S.-K. Kwon, Macromolecules, 2017, 50, 4227–4234 CrossRef CAS.
- M. Li, H. Bin, X. Jiao, M. M. Wienk, H. Yan and R. A. J. Janssen, Angew. Chem., Int. Ed., 2020, 59, 846–852 CrossRef CAS PubMed.
- Z.-F. Yao, Z.-Y. Wang, H.-T. Wu, Y. Lu, Q.-Y. Li, L. Zou, J.-Y. Wang and J. Pei, Angew. Chem., Int. Ed., 2020, 59, 17467–17471 CrossRef CAS PubMed.
- H.-L. Yi and C.-C. Hua, Macromolecules, 2019, 52, 332–340 CrossRef CAS.
- W. Zhang, K. Shi, J. Lai, Y. Zhou, X. Wei, Q. Che, J. Wei, L. Wang and G. Yu, Adv. Mater., 2023, 35, 2300145 CrossRef CAS.
- C. Cheng, H. Geng, Y. Yi and Z. Shuai, J. Mater. Chem. C, 2017, 5, 3247–3253 RSC.
- J. Yang, Z. Zhao, H. Geng, C. Cheng, J. Chen, Y. Sun, L. Shi, Y. Yi, Z. Shuai, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 201702115 Search PubMed.
- T. Mikie, M. Hayakawa, K. Okamoto, K. Iguchi, S. Yashiro, T. Koganezawa, M. Sumiya, H. Ishii, S. Yamaguchi, A. Fukazawa and I. Osaka, Chem. Mater., 2021, 33, 8183–8193 CrossRef CAS.
- Z.-F. Yao, J.-Y. Wang and J. Pei, Prog. Polym. Sci., 2023, 136, 101626 CrossRef CAS.
- J. Rivnay, S. C. B. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012, 112, 5488–5519 CrossRef CAS PubMed.
- J. Rivnay, R. Noriega, R. J. Kline, A. Salleo and M. F. Toney, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 045203 CrossRef.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313–5317 CrossRef CAS PubMed.
- R. Stalder, S. R. Puniredd, M. R. Hansen, U. Koldemir, C. Grand, W. Zajaczkowski, K. Müllen, W. Pisula and J. R. Reynolds, Chem. Mater., 2016, 28, 1286–1297 CrossRef CAS.
- Y. Xu, Y. Li, S. Li, F. Balestra, G. Ghibaudo, W. Li, Y.-F. Lin, H. Sun, J. Wan, X. Wang, Y. Guo, Y. Shi and Y.-Y. Noh, Adv. Funct. Mater., 2020, 30, 1904508 CrossRef CAS.
- H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2018, 17, 2–7 CrossRef CAS PubMed.
- W. H. Lee, H. H. Choi, D. H. Kim and K. Cho, Adv. Mater., 2014, 26, 1660–1680 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01143g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |