
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the electrocatalytic prowess of a synergistic 1T-MoS2-metallic Ni composite towards alkaline hydrogen evolution†
Avishek
Roy
a,
Ayan
Mondal‡
 a,
Harish Reddy
Inta‡
ab,
Sourav
Ghosh
a,
Khushboo S
Paliwal
a,
Soumalya
Debnath
a,
Ajith Ambattuparambil
Valsan
a and
Venkataramanan
Mahalingam
*a
a,
Harish Reddy
Inta‡
ab,
Sourav
Ghosh
a,
Khushboo S
Paliwal
a,
Soumalya
Debnath
a,
Ajith Ambattuparambil
Valsan
a and
Venkataramanan
Mahalingam
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Kolkata, Mohanpur, 741246, Nadia, West Bengal, India. E-mail: mvenkataramanan@yahoo.com
bDepartment of Energy Science & Engineering, Daegu Gyeongbuk Institute of Science & Technology (DGIST), Daegu 42988, South Korea
First published on 8th February 2024
Abstract
Designing of suitable electrocatalysts for efficient alkaline hydrogen evolution reaction (HER) is a challenging task owing to the additional energy consumption to disintegrate the H–OH bond in the Volmer step. Under these circumstances, strong synergistic interactions between Ni/Co derivatives with 1T-MoS2 can frequently accelerate the alkaline HER. This study addresses the challenge of designing efficient electrocatalysts for the alkaline HER, focusing on minimizing additional energy consumption during the Volmer step. A composite structure, 1T-MoS2–Ni(18), was synthesized using 1T-MoS2 and metallic Ni for effective alkaline HER catalysis. Rigorous physical characterization confirmed the formation of an interfacial structure between 1T-MoS2 and metallic Ni. The resulting composite exhibited very good alkaline HER performance, requiring only a 120 mV overpotential for a standard 10 mA cm−2geo current density. The improved performance was attributed to feasible water dissociation over the metallic Ni promoter, facile electron migration kinetics through the interfacial structure, and enhanced per-site activity. In addition, for the practical execution of an alkaline electrolyzer, NiS nanoparticles were synthesized as an OER catalyst, exhibiting only a 310 mV overpotential (1.54 V vs. RHE) to attain a 10 mA cm−2geo current density. The alkaline electrolyzer, (NiS (+)||1T-MoS2–Ni(18) (−)), delivered a 1.68 V cell potential to sustain a 10 mA cm−2geo current density with excellent stability for up to 48 h.
Introduction
To accomplish the carbon-neutral fuel economy, the utilization of green H2 (hydrogen) as modern fuel is the most efficient way out owing to its impressive specific energy (∼142 MJ Kg−1) and emission of zero carbon derivatives.1–3 In this context, acidic (polymer electrolyte membrane) and alkaline water electrolyzers are the most sustainable sources for clean H2 production.4 Water electrolysis involves two individual half-cell reactions in which cathodic and anodic half-cells consist of hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), respectively.5 Despite the Pt group of metals being at the forefront for efficient HER electrocatalysts in acidic pH, their performances remarkably decline in alkaline conditions.6,7 The additional energy consumption in the water dissociation step (H–OH bond disintegration) creates substantial difficulty in alkaline HER kinetics.8,9 Hence, the development of a competent alkaline HER catalyst is particularly in demand. Further, it is essential to consider the following limiting factors while designing the HER catalyst: (i) optimal free energy of hydrogen adsorption (H(ad)), (ii) minimal energy consumption for water disintegration, and (iii) limited affinity of active sites towards developed hydroxyl centers (OH(ad)) to mitigate catalyst poisoning.9Recently, the design of interfacial structures through composite formation using transition metal-based materials has emerged as an effective strategy to achieve improved alkaline HER. In this context, synergistic coupling between water dissociation promoters (Ni/Co derivatives) and Pt is an efficient approach to developing catalysts for the alkaline HER. The former facilitates the water dissociation step, and subsequently, the latter drives the HER process.10 For instance, Subbaraman et al. reported the Ni(OH)2–Pt interfacial structure towards enhanced alkaline HER activity. This study reveals that the Ni(OH)2 surface aids in the water-dissociation Volmer step (H2O + e− → H(ad) + OH−) to produce H(ad). Subsequently, the H(ad) follows the Heyrovsky (H(ad) + H2O+ e− → H2 + OH−) or Tafel (H(ad) + H(ad) → H2) pathway to produce H2 on the Pt surface.11 Similarly, Chen and coworkers prepared a Ni(OH)2–PtO2 hybrid structure on Ti mesh, which delivered excellent catalytic activity towards the alkaline HER. The theoretical studies confirmed that the Ni(OH)2–PtO2 interfacial structure displays an effectively low energy barrier for water dissociation.12 In another report, Sun et al., showed a superior alkaline HER via NiCo hydrogen carbonate (NiCo(HCO3)2)-supported Pt nanoparticles. The NiCo(HCO3)2 facilitates the water dissociation process and thereby, strong electronic interaction with Pt results in enhanced HER activity.13 Pt, being an expensive metal, necessitates the search for suitable alternative catalyst materials that are less expensive yet efficient for catalyzing the HER.
2D layered transition metal dichalcogenides (TMDs) are emerging as potential candidates for catalyzing the HER in acidic pH due to optimum hydrogen binding free energy (ΔGH).14,15 In this context, MoX2 (X = S, Se) with exposed edges is highly effective towards HER catalysis.16,17 Among the different phases of MoS2, the 1H and 2H-phases, in particular, are semiconducting, thermodynamically stable, and have a hexagonal lattice array. In these phases, the six S atoms are rearranged to form a triangular prism-like structure where Mo is placed at the center and covalently connected to the S atoms. In addition, the 1H phase consists of a monolayer of MoS2 whereas in the 2H phase repeating patterns of hexagonal geometry are observed.18,19 On the other hand, the 1T-phase is metallic, thermodynamically meta-stable, and has an octahedral coordination. The Mo atoms are positioned at the center of the octahedral hole and sulfur atoms are rearranged above and below the octahedral planes.18,20 Especially, the metallic 1T-MoS2 displays promising catalytic performance as compared to 2H phases in an acidic medium.16,21,22 However, the catalyzing efficiency of 1T-MoS2 is highly compromised in alkaline conditions due to the poor water dissociation kinetics.23,24 Interestingly, the doping of foreign substances into 1T-MoS2 can significantly modulate the alkaline HER performance. For instance, 5% V doping into 1T-MoS2 nanosheet enhances the alkaline HER activity. The V aids in generating abundant active sites as well as leads to almost zero ΔGH value. The material required 173 mV overpotential to reach the standard 10 mA cm−2 current density.25 Combining 1T-MoS2 with (Ni/Co)-based materials also displayed efficient HER activity in an alkaline medium. For instance, Liu et al. reported the improved alkaline HER activity of Co-doped 1T-MoS2 through a synergistic ligand modulation strategy. The developed material required only 118 mV overpotential to attain a benchmark 10 mA cm−2 current density.26 Similarly, the electrochemical performance of 1T-MoS2 was enhanced through interfacial engineering with Co(OH)2. The developed Co(OH)2/1T-MoS2 heterostructure delivered 10 mA cm−2 current density at a 151 mV over potential.27 Along with Co, Ni-based materials also serve as water dissociation promoters to enhance the HER performance of 1T-MoS2. Particularly, nickel sulfide/hydroxide-based materials have been examined to improve the catalyzing power of 1T-MoS2 towards the alkaline HER. For example, Yu and co-workers have developed Ni(OH)2 nanoparticle-decorated 1T-MoS2 nanowires that display excellent alkaline HER activity.28 In another report, 1T/2H-MoS2–Ni3S2 nanorods were reported for improved alkaline HER activity. The material displayed a 73 mV overpotential to attain a current density of 10 mA cm−2.29 Moreover, theoretical studies show that Ni-based materials enhance the catalyzing power of 1T-MoS2 by significantly reducing the water decomposition energy barrier.24
However, reports on the use of metallic Ni with MoS2 for alkaline HER are scarce. The use of metallic Ni is advantageous due to its low cost, high electrical conductivity, and facile synthesis routes.30,31 Both theoretical and experimental studies revealed that metallic nickel has significantly higher HER activity as compared to other non-noble metals.30,32 This has encouraged researchers to integrate electrocatalysts with metallic Ni to improve their alkaline HER activity. For example, Pattengale et al. recently studied the alkaline HER performance of Ni single-atom-modified 1T-MoS2. In alkaline conditions, the Ni single atom is initially converted into the NiSxOy species, which reversibly transforms into catalytically active Ni0 at the basal planes of 1T-MoS2 and thereby activates the basal planes towards improved HER. The material shows excellent HER activity of 76 mV to reach the standard 10 mA cm−2 current density.33 In another report, Xu et al. showed the improved HER performance of 1T-2H-MoS2 by utilizing Ni metal clusters. Subsequently, a composite was prepared by combing the as-synthesized 1T-2H-MoS2@Ni cluster with RGO. In this case, the electronic energy states of the Ni clusters could supply additional electrons for better adsorption of hydrogen ions. Further, the combination with RGO resulted in the effective lowering of the hydrogen adsorption energy barrier and facilitated better alkaline HER. The material achieved the benchmark of 10 mA cm−2 current density with an overpotential of only 92 mV.34 Our idea is to use 1T-MoS2 as a template for the growth of metallic Ni. MoS2, being a layered structure, is expected to provide more surface for the growth of metallic Ni. Such growth is envisioned to result in compact Ni/MoS2 interfaces for an efficient alkaline electrocatalytic HER process.
In this work, first ammonia-intercalated 1T-MoS2 was prepared via a one-step solvothermal route, where thiourea was used as both a sulfurizing agent and an ammonia source. Subsequently, 1T-MoS2–Ni(X) (X = 9, 18, 27, 36) composite structures were developed using a facile wet chemical route. X denotes the amounts (mg) of NiCl2·6H2O used for the synthesis of 1T-MoS2–Ni(X) composite structures. During the composite structure formation, 1T-MoS2 acts as a support matrix for the uniform growth of metallic Ni. We emphasize that in the absence of 1T-MoS2, the synthesis results in the formation of Ni nanowires.35 The as-prepared composite materials showed better alkaline HER performance as compared to individual 1T-MoS2 and metallic Ni nanowires. The 1T-MoS2–Ni(18) composite exhibited the lowest overpotential of 120 mV to attain the standard 10 mA cm−2geo current density. The enhanced catalytic performance of the 1T-MoS2–Ni(18) composite could be attributed to (i) easy water disintegration step on the metallic Ni surface, (ii) facile electron transfer kinetics, and (iii) improved per-site activity through appropriate synergistic coupling between 1T-MoS2 and metallic Ni. In addition, the as-prepared 1T-MoS2–Ni(18) composite showed faster kinetics (Tafel slope 89 mV dec−1) as well as superior electrochemical stability for 24 h in 1 M KOH. Further, for the demonstration of an alkaline water electrolyzer, NiS was synthesized through a one-step hydrothermal route and used as an OER catalyst. The as-prepared NiS consumed ∼310 mV overpotential to attain the standard 10 mA cm−2geo current density. Finally, an alkaline electrolyzer was constructed using IT-MoS2–Ni(18) as the cathode and NiS as the anode material, delivering a cell voltage of 1.68 V to sustain the benchmark current density of 10 mA cm−2geo with 48 h stability in 1 M KOH. Post-chronopotentiometric physical characterization of the retained catalyst revealed the good mechanical robustness of the 1T-MoS2–Ni(18) composite structure and structural transformation of NiS into NiOOH after long catalysis.
Results and discussion
Phase and structural characterization
The synthetic protocol for the preparation of the composite material is schematically illustrated in Fig. 1. Initially, the one-step hydrothermal method was adopted to prepare 1T-MoS2 using ammonium heptamolybdate and thiourea as precursors. On the other hand, metallic Ni nanowire was prepared via a hot wet chemical pathway using nickel chloride as a precursor, ethylene glycol as a solvent, and hydrazine hydrate as a reducing agent. For the synthesis of the 1T-MoS2–Ni(X) composite structure, the as-prepared 1T-MoS2 was initially dispersed in an ethylene glycol medium. Subsequently, different amounts of nickel chloride solutions (9, 18, 27, 36 mg) were introduced into the dispersed solution and stirred well. The resulting solution was subjected to a similar wet chemical pathway to that used for the Ni nanowire synthesis (see the details in ESI†). The derived products were named 1T-MoS2–Ni(9), 1T-MoS2–Ni(18), 1T-MoS2–Ni(27), and 1T-MoS2–Ni(36). Among the different composite structures, 1T-MoS2–Ni(18) displayed the best electrocatalytic alkaline HER performances (vide infra). Therefore, 1T-MoS2–Ni(18) was chosen for detailed physical characterization. The powder X-ray diffraction (PXRD) analysis was carried out to investigate the phases of the synthesized materials. The PXRD pattern of the as-synthesized Ni nanowire was well indexed to metallic nickel (JCPDS # 00-004-0850) with a cubic crystal system (Fig. 2a). On the other hand, the PXRD pattern of only MoS2 matched the standard pattern of MoS2 as well as the literature reports.16,36 The broad nature of the peaks suggests the presence of amorphous regions in the material. Interestingly, the (002) plane was observed at 9.4° as compared to 14° for the bulk MoS2. In addition to the (002) plane, the corresponding second-order diffraction, i.e., (004) plane, was observed at 2θ value 18.6°. This implies an increase in the interlayer spacing as compared to the bulk (0.62), presumably caused by the intercalation of some foreign moieties, such as ammonia, formed through the dissociation of thiourea. Such intercalation and the associated expansion of interlayer distance resulted in the formation of the 1T phase of MoS2 together with the 2H phase (vide infra). The asymmetrical nature of the (110) and (100) planes signifies the presence of limited turbostatic disorder.17,37 On the other hand, the diffraction pattern of the 1T-MoS2–Ni (18) composite is similar to that observed for 1T-MoS2. However, the observed (002) plane of the composite further shifted to a lower 2θ value (8.9°). We believe that the observed shift is associated with the additional intercalation of NH4+/NH3 into MoS2 layers caused by the dissociation of hydrazine hydrate at high temperatures. However, hardly any peaks for metallic Ni were noted in the composite (Fig. 2a). Similar PXRD patterns were also observed for composite structures up to 27 mg of Ni content (Fig. S1, ESI†). Interestingly, when the metal content reached 36 mg, all the diffraction peaks associated with individual metallic Ni and 1T-MoS2 became visible in the 1T-MoS2–Ni(36) composite (Fig. S1, ESI†). This result supports the formation of the composite structure.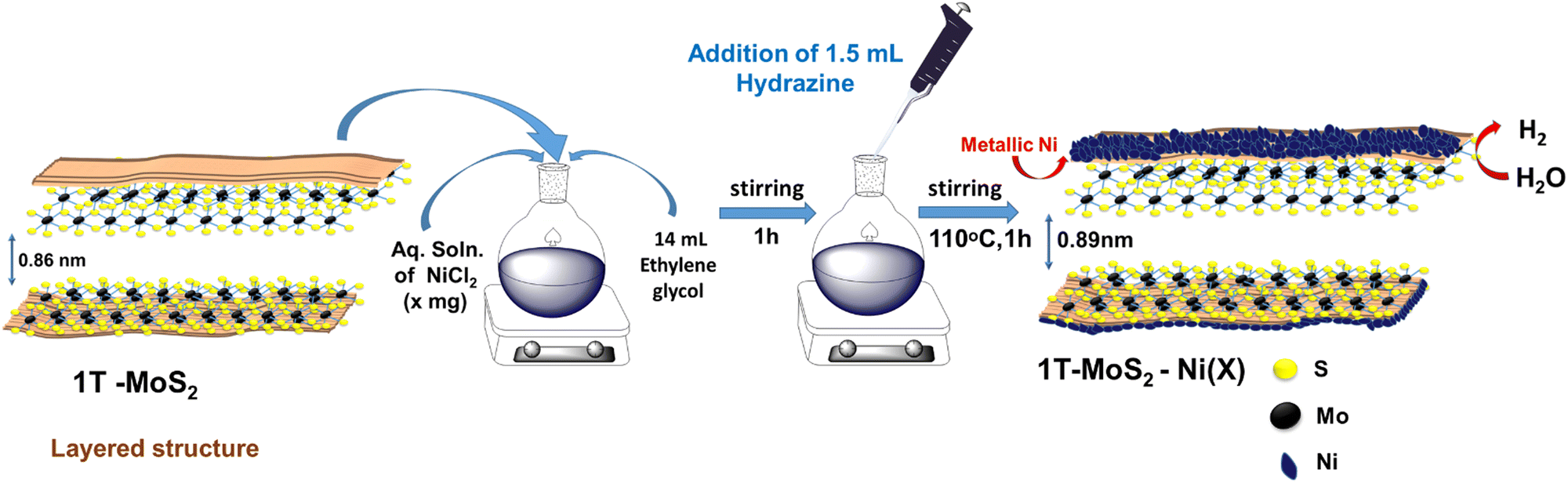 | ||
| Fig. 1 Schematic representation of the synthetic protocol to prepare the 1T-MoS2–Ni(X) composite structure. | ||
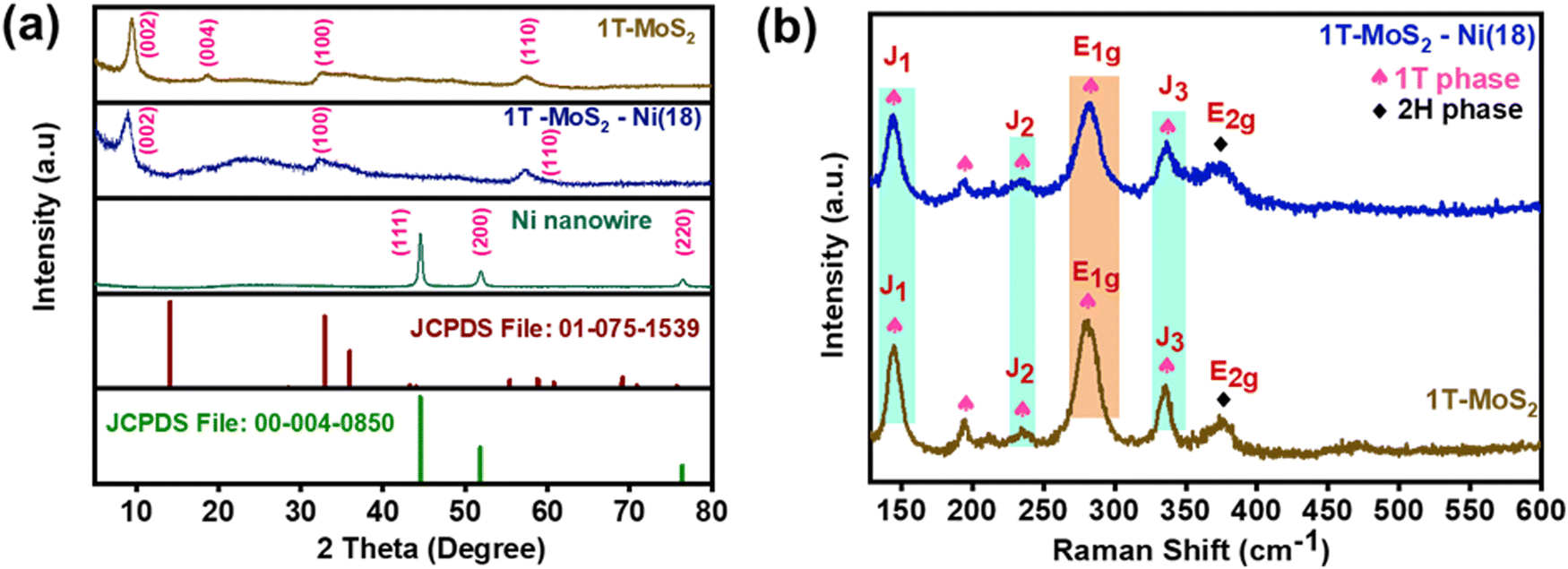 | ||
| Fig. 2 (a) PXRD patterns of the Ni nanowire, 1T-MoS2 and 1T-MoS2–Ni(18) as compared with the standard patterns of MoS2 (JCPDS File No. 01-075-1539) and metallic Ni (JCPDS File No. 00-004-0850). (b) Raman spectra of the as-synthesized 1T-MoS2 and 1T-MoS2–Ni(18) composite material. | ||
FTIR and Raman analyses
The presence of ammonia in the 1T-MoS2 structure was confirmed through FTIR analysis. The peaks near 1624 cm−1, 1402 cm−1, and 3136 cm−1 were assigned to the N–H symmetrical bending and stretching modes in NH3, respectively (Fig. S2, ESI†).38,39 This result confirms the presence of NH3 presumably intercalated between the MoS2 layers. A similar result was noted for 1T-MoS2–Ni(18), indicating the retention of NH3 intercalation in the composite structure. To validate the formation of the 1T phase of MoS2, Raman studies were performed. The existence of the three major peaks, J1 (144.8 cm−1), J2 (234.6 cm−1), and J3 (335.7 cm−1) confirmed the formation of metallic 1T-MoS2 (Fig. 2b).40,41 The presence of the E1g mode at 280.3 cm−1 along with the 194.7 cm−1 peak in the Raman spectrum further supported the existence of the 1T phase in the as-synthesized MoS2.42 The appearance of the weak-intensity E2g mode at 375.4 cm−1 in the spectrum implies the existence of the semiconducting 2H-MoS2 as the minor phase43,44 Further, the observed identical Raman active modes for the 1T-MoS2–Ni(18) composite structure suggests the retention of the 1T phase of MoS2 in the as-synthesized composite network (Fig. 2b).Morphological characterization
To gain insight into the surface textural properties of the as-synthesized materials, FESEM (field emission scanning electron microscopy) and TEM (transmission electron microscopy) analyses were performed. The FESEM images of 1T-MoS2 display hierarchical agglomerated nanoflakes (Fig. S3a and b, ESI†). On the other hand, in the case of metallic Ni, the particles are assembled to form a nanowire-like architecture with long-range connectivity (Fig. S3c and d, ESI†). However, the 1T-MoS2–Ni(18) composite network acquired a floret-like assembly due to the agglomeration of thin nanoflakes (Fig. S3e and f, ESI†). Similar textural patterns were observed for other composite structures as well (Fig. S4, ESI†). The formation of the floret assembly is presumably due to different kinds of inter-layer stacking interactions in the MoS2.17 Close inspection of TEM images showed the flake-like microstructure of 1T-MoS2 (Fig. 3a). HRTEM analysis indicated the presence of 002 crystal planes of MoS2 with a higher interlayer distance (0.86 nm) (Fig. 3b). This is consistent with PXRD analysis and further supports an increase in interlayer spacing, which is likely due to the intercalation of NH3 along the (002) planes. On the other hand, the presence of circular rings in the corresponding FFT pattern indicates the poor crystalline nature of the as-prepared 1T-MoS2. The TEM and STEM (scanning transmission electron microscopy) images of metallic Ni show a nanowire-like texture with some thin spikes sticking out from the surface (Fig. S5, ESI†). Similar textural patterns were also observed in our earlier report.35 The HRTEM analysis additionally supports the formation of metallic Ni (Fig. S6, ESI†). TEM images of the 1T-MoS2–Ni(18) composite structure display an isotropic flake-like microstructure (Fig. 3c and d). Close inspection of the HRTEM analysis indicated the distinguishable interfaces shared by metallic Ni and 1T-MoS2. Particularly, the presence of the (111) and (002) planes of metallic Ni and 1T-MoS2 confirmed the formation of the interfacial structure (Fig. 3e). The calculated interlayer spacing along the (002) plane (d002) is 0.89 nm, indicating additional NH4+/NH3 intercalation between MoS2 layers. This observation is in good accordance with the PXRD result. Further, the EDS study and the elemental mapping analyses confirmed the presence of Mo, Ni, and S and their uniform distribution in the 1T-MoS2–Ni(18) composite material, respectively (Fig. 3f, (g–j)). | ||
| Fig. 3 (a) and (b) TEM and HRTEM images of 1T-MoS2, respectively (the FFT patterns of the corresponding HRTEM images are shown in the inset of figure b). (c) and (d) TEM and (e) HRTEM images of the 1T-MoS2–Ni(18) composite material, (f) EDS analysis, and (g)–(j) elemental mapping study of as-synthesized 1T-MoS-Ni(18) composite material. | ||
Chemical state analysis
The chemical state information of the as-synthesized materials was studied using X-ray photoelectron spectroscopy (XPS). The XPS survey scan of 1T-MoS2 confirmed the presence of Mo, S, and N elements in the material (Fig. S7, ESI†). Further, the deconvolution of the high-resolution Mo 3d spectrum displayed four major peaks. The lower binding energy (BE) peaks located at 228.6 eV (Mo 3d5/2) and 231.8 eV (Mo 3d3/2) correspond to Mo+4 formations as well as support the presence of the metallic or 1T phase of MoS2.45,46 In addition, two weak peaks were observed at relatively high BE 229.5 eV (Mo 3d5/2) and 233.1 eV (Mo 3d3/2). This observation corroborates the presence of the semi-conducting 2H-MoS2 along with the 1T-MoS2 phase (Fig. 4a).47 Also, two comparatively weak peaks were noted at 236.2 eV and 226.1 eV. This indicates the presence of Mo+6 (partial oxidation of Mo+4) and S 2s in the as-prepared 1T-MoS2.48,49 Similarly, the peak-fitted S 2p spectrum displayed two strong peaks at lower BE, positioned at 161.6 eV (2p3/2) and 163 eV (2p1/2). The observed BE values indicated the formation of 1T-MoS2 (Fig. 4b).47,50 Two additional weak peaks were observed at 162.6 eV (2p 3/2) and 164 eV (2p 1/2), ascribed to the S 2p of 2H-MoS2.47,51 The aforementioned results suggest the predominant formation of the metallic 1T phase of MoS2 as compared to the semiconducting 2H phase, which is in agreement with Raman analysis. In addition, the deconvoluted N1s spectrum displayed two peaks at 401.8 eV and 398.7 eV, which are ascribed to the existence of NH4+ and NH3, respectively, in the as-synthesized material (Fig. S8, ESI†).52,53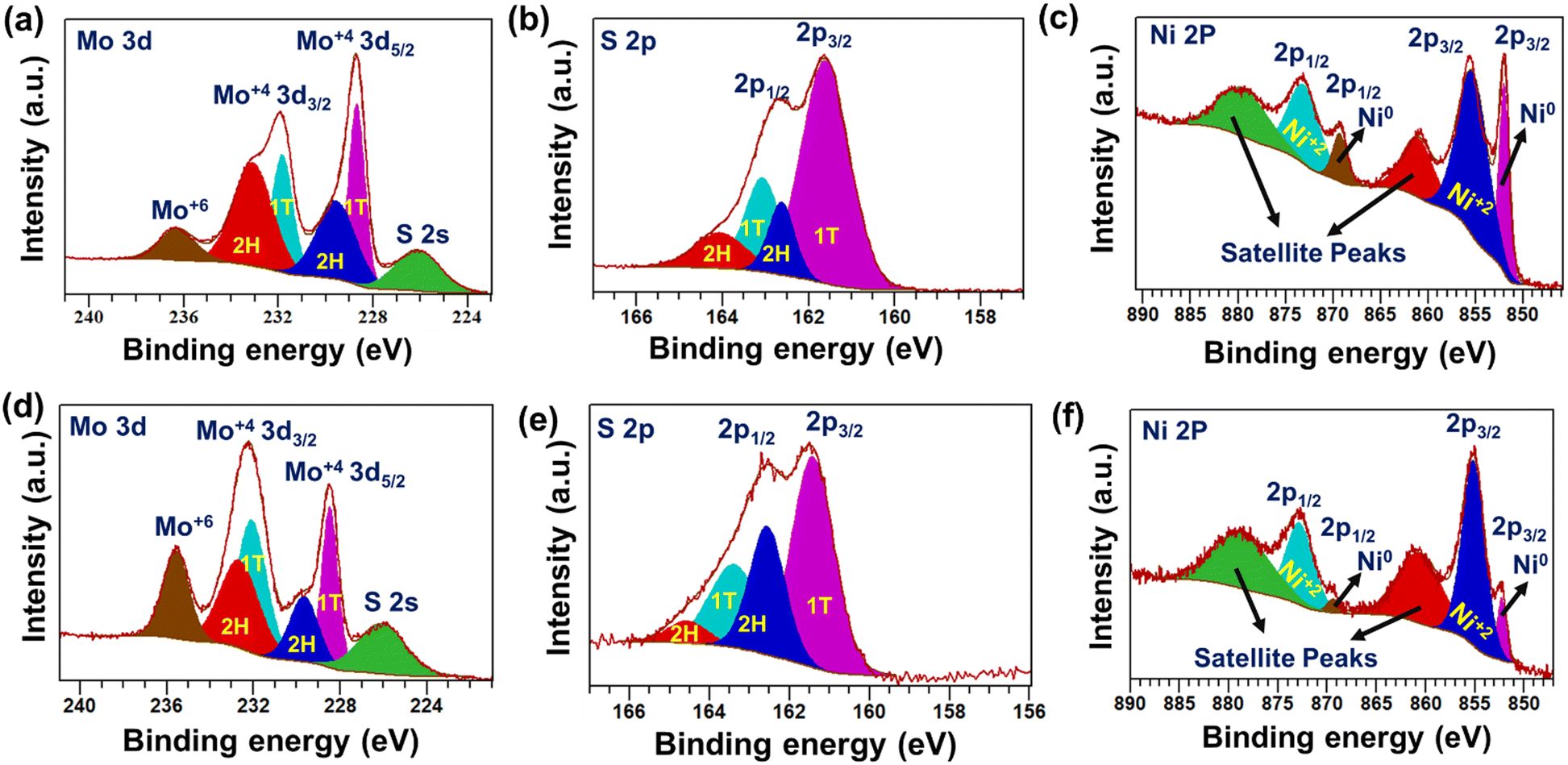 | ||
| Fig. 4 High-resolution deconvoluted XPS scan of (a) Mo 3d, (b) S 2p for 1T-MoS2, (c) Ni 2p for metallic Ni nanowire, (d) Mo 3d, (e) S 2p, and (f) Ni 2p for as-prepared 1T-MoS2–Ni(18) composite material. | ||
The peaks at 395.6 eV and 394.5 eV indicated the formation of the Mo–N bond and the existence of Mo3p3/2, respectively, in the as-prepared 1T-MoS2 (Fig. S8, ESI†).54,55 These results support ammonia intercalation, preferably between the MoS2 layers, during the solvothermal growth process. Further, the surface composition of metallic Ni nanowires was identified through XPS analysis (Fig. S9, ESI†). The deconvoluted Ni 2p XPS spectrum indicates the presence of both Ni0 and Ni+2 (Fig. 4c). The corresponding peak positions are shown in Table S1 (ESI†).35,56–59 The presence of Ni+2 on the surface of the Ni nanowire is most likely due to the partial oxidation of metallic Ni0. Further, the detailed chemical composition of the as-prepared 1T-MoS2–Ni(18) composite structure was determined through XPS. The survey scan confirmed the presence of Mo, Ni, S, and O in the as-prepared composite material (Fig. S10, ESI†). The deconvoluted Mo 3d scan revealed four peaks, as observed for pristine 1T-MoS2. The peaks positioned at 228.4 eV (3d5/2) and 232.1 eV (3d3/2) are ascribed to the presence of Mo+4 as well as the 1T phase in the composite network (Fig. 4d).60–62 Two weak intensity peaks were noted at 229.6 eV. (3d5/2) and 232.6 eV (3d3/2), indicating the presence of 2H-MoS2.51,60,62,63 The presence of Mo+6 (peak at 235.5 eV) in the composite is most likely due to the surface oxidation of Mo+4.64 Deconvolution of the S 2p spectrum resulted in two strong peaks at 161.4 eV (2p 3/2) and 163.3 eV (2p 1/2) (Fig. 4e). These peaks were assigned to S associated with the 1T phase of MoS2 in the as-prepared 1T-MoS2–Ni(18) composite structure.47 In addition, two weak intensity peaks were noted at 162.5 eV (2p3/2) and 164.5 eV (2p1/2), corresponding to the presence of the 2H phase of MoS2 in the composite.65 Further, the high-resolution N 1s scan of the composite structure displayed peaks at 398.1 eV and 399.5 eV, which are ascribed to the intercalated NH3 and NH4+, respectively (Fig. S11, ESI†).53,66 This observation manifests the excellent stability of the ammonia-intercalated 1T phase of MoS2 in the composite structure. Further, a peak-fitted high-resolution Ni 2p XPS scan indicated the formation of metallic Ni0. The noted peak positions are 852.2 eV and 869.3 eV, assigned to 2p3/2 and 2p1/2, respectively, and the BE values ascribed the formation of metallic Ni0 in the as-prepared 1T-MoS2–Ni(18) composite network (Fig. 4f).56 Along with this, two extra peaks were also noted at 855.1 eV and 872.7 eV (BE difference of 17.6 eV), attributed to the presence of Ni+2 due to the partial oxidation of metallic Ni0.57 The above-mentioned experimental evidence supports that the composite structure (1T-MoS2–Ni(18)) was formed between 1T-MoS2 and metallic Ni0.
Electrochemical HER performance
To evaluate the electrochemical HER activity, all the catalyst materials were initially preconditioned for 20 CV (cyclic voltammetry) cycles in freshly prepared 1 M KOH solution (see the details in the ESI†). Subsequently, the backward cathodic polarization curve (LSV curve) was considered to estimate the required overpotential to reach the benchmark 10 mA cm−2geo current density.67 The backward LSV curve (100% manually iR-corrected) shows that the as-prepared 1T-MoS2–Ni(18) composite structure has better alkaline HER activity as compared to individual 1T-MoS2 and metallic Ni nanowires (Fig. 5a). The measured overpotential value for the 1T-MoS2–Ni(18) composite network is ∼120 mV at −10 mA cm−2geo current density, which is much less as compared to individual 1T-MoS2 (243 mV), as well as Ni nanowires (190 mV), but higher than standard Pt/C (25 mV) at the same current density (Fig. 5b). The calculated overpotential values (@ −10 mA cm−2geo) for other composites like 1T-MoS2–Ni(36), 1T-MoS2–Ni(27), and 1T-MoS2–Ni(9) are 144 mV, 158 mV, and 138 mV, respectively (Fig. S12a and b, ESI†). Thus, the 1T-MoS2–Ni(18) composite material displays better catalytic performances among all 1T-MoS2–Ni(X) composite materials, and the measured overpotential value is quite cognate with the recently reported composite structures towards the alkaline HER (Table S6, ESI†). Hence, the superior catalytic activity of all 1T-MoS2–Ni(X) composite structures draws attention to the strong interfacial coupling present in the composite structures between 1T-MoS2 and metallic Ni. On the other hand, the excellent alkaline HER performances of the 1T-MoS2–Ni(18) composite structure encourage us to evaluate the catalytic competency at higher current densities. The close inspection of the LSV curve of 1T-MoS2–Ni(18) revealed that the composite material can attain current densities of −25 mA cm−2geo, −50 mA cm−2geo, and −100 mA cm−2geo with overpotentials of only 160, 199 and 246 mV, respectively (Fig. S13, ESI†).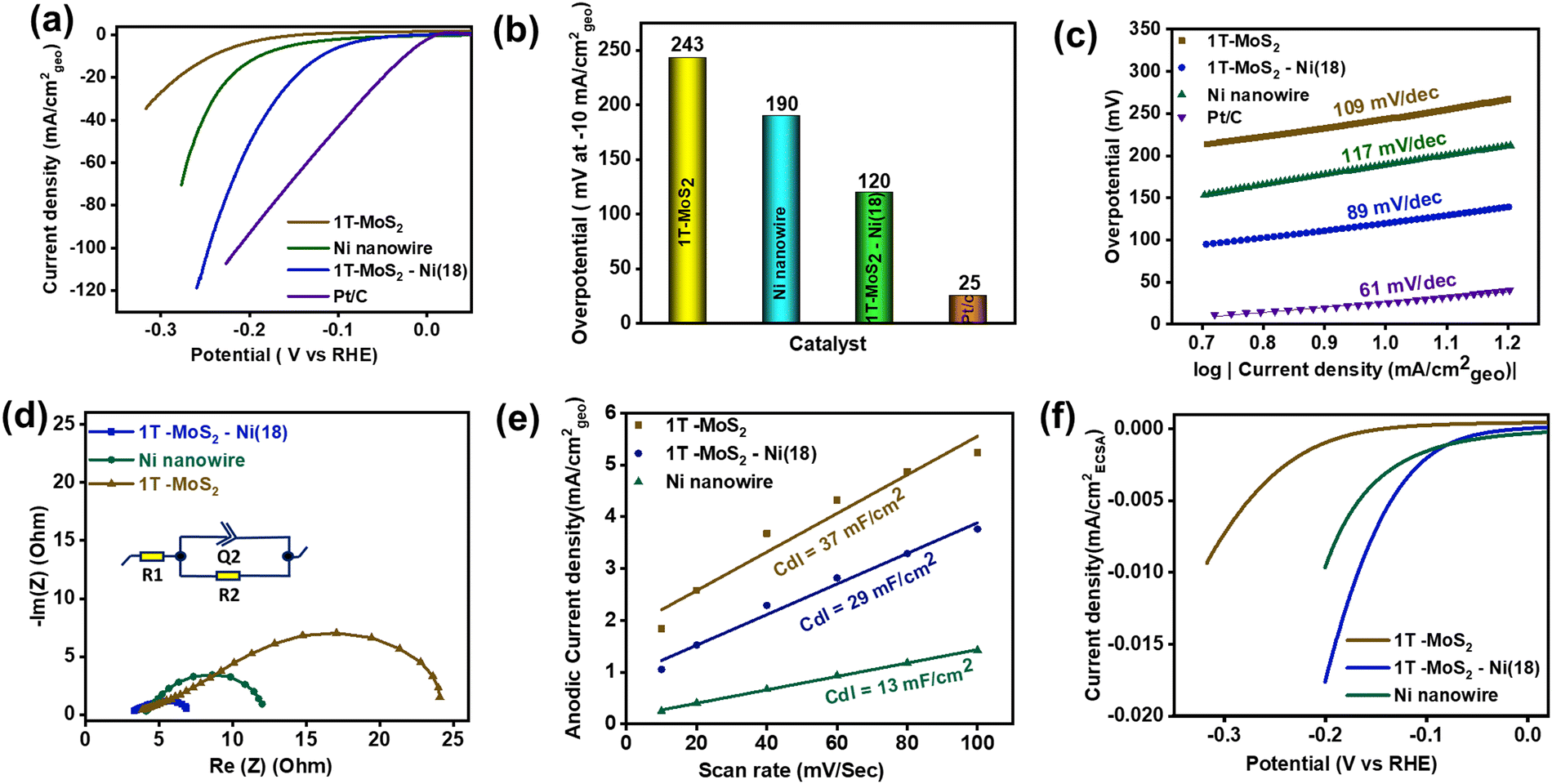 | ||
| Fig. 5 (a) Cathodic backward polarization curves (100% iR corrected) of the materials measured at a scan rate of 5 mV s−1. (b) The bar diagram corresponds to the overpotential value of the materials at a current density of −10 mA cm−2geo. (c) Tafel slope analysis. (d) Nyquist plots of the materials estimated at 300 mV overpotential. (e) Linear fitting of the current (anodic) vs. scan rate plot for Cdl estimation. (f) ECSA-normalized LSV of the material to unveil the intrinsic activity. | ||
Mechanism
The Tafel slope analysis was performed to unveil the alkaline HER kinetics as well as the reaction mechanism. The Tafel slopes of the materials were evaluated from the manually 100% iR-corrected backward cathodic LSV curves. The measured Tafel slope values for 1T-MoS2 and Ni nanowires were 109 and 117 mV dec−1, respectively (Fig. 5c). This implies the poor water-breaking kinetics (H2O + M + e− → H* + OH−) of individual 1T-MoS2 and metallic Ni nanowires. Interestingly, the corresponding value for 1T-MoS2–Ni(18) is 89 mV dec−1, suggesting enhanced interfacial HER kinetics in the composite network. Further, literature surveys revealed that the HER proceeds through a three-step pathway in alkaline conditions. In the first step (Volmer step), H2O dissociation reveals adsorbed hydrogen species (H*). The Tafel slope associated with this step (H2O + M (metal) + e− → H* + OH−) is ∼120 mV dec−1. Eventually, the H2 generation takes place through two different pathways. Another possible pathway is the Heyrovsky pathway (H* + H2O + e− → H2 + OH−, Tafel slope value ∼40 mV dec−1) where H* is recombined with another H2O molecule with the aid of an electron. The other pathway is known as the Tafel step where the adsorbed hydrogens are combined (H* + H* → H2, Tafel slope value ∼30 mV dec−1) and release the H2. Hence, the observed Tafel value (89 mV dec−1) of 1T-MoS2–Ni(18) indicates that the reaction is most likely to follow the Volmer–Heyrovsky mechanism.68 It is, therefore, reasonable to suggest that the integration of metallic Ni with 1T-MoS2 facilitates water disintegration (Volmer step) kinetics in the composite structure, resulting in enhanced HER performance. This observation is a good analogy with earlier literature where metallic Ni serves as a good HER catalyst.69–71 As such, it is relevant to propose that in the composite structure, water dissociation happens over metallic Ni under an alkaline environment, which ultimately results in adsorbed hydrogen species (H*). Subsequently, the resulting H* is expected to drift through the interface of metallic Ni and 1T-MoS2. Finally, the close proximity to neighbouring catalytically efficient edge sites of 1T-MoS2 produces H2 under the Heyrovsky mechanism (Fig. S14, ESI†).This validates the significance of synergistic coupling between 1T-MoS2 and metallic Ni towards improved HER performance. Further, the interfacial charge transfer kinetics of the as-synthesized materials were investigated through electrochemical impedance spectroscopy (EIS) at an overpotential of 300 mV (Fig. 5d). The semicircle diameter of the Nyquist plot reveals the requisite charge transfer resistance (Rct) between the interface of the electrode and electrolytic solution. The obtained Rct values for the as-prepared 1T-MoS2 and metallic Ni nanowire are 20.1 and 8.4 Ω, respectively. These results indicate that electron migration is facile in metallic Ni as compared to 1T-MoS2. Interestingly, with an adequate quantity of Ni incorporation into 1T-MoS2, the as-prepared 1T-MoS2–Ni(18) composite displayed an effectively low Rct value (4.4 Ω).
The above results further proved that the metallic Ni facilitates easier interfacial electron transfer kinetics between the catalytic material and electrolyte and, thereby, promotes the improved alkaline HER activity of the 1T-MoS2–Ni(18) composite structure. The Nyquist plot and the corresponding Rct values of other composite materials are shown in Fig. S15 and Table S2 (ESI†). Further, to understand the electrochemical active surface area (ECSA) of the materials, the double layer capacitances (Cdl) of 1T-MoS2, metallic Ni, and 1T-MoS2–Ni(18) composite material were calculated. The double layer is a constructive electrical parameter used to estimate ECSA (see the details in the ESI†). Initially, CVs of the materials were recorded by varying the scan rates within the potential region of −0.05 V to 0.05 V (at the non-faradaic region) at open circuit potential (EOCP) (Fig. S16, ESI†). Subsequently, the plot of anodic current density (ja) at EOCPVs. scan rate (ν) was made, where the corresponding slope values provide the desired Cdl values for the catalytic materials (Fig. 5e). The measured Cdl values for individual 1T-MoS2 and Ni nanowires are 37, and 13 mF cm−2, respectively. The above results suggest that metallic Ni has a significantly lower ECSA as compared to 1T-MoS2. Interestingly, the 1T-MoS2–Ni(18) composite network displayed a Cdl value of 29 mF cm−2, which was slightly lower as compared to 1T-MoS2 but higher than metallic Ni. These results motivated us to probe the intrinsic catalytic proficiency of the as-synthesized materials through the ECSA-normalized backward cathodic LSV scan (Fig. 5f). The Ni nanowire displayed improved catalytic activity as compared to 1T-MoS2, indicating the higher per-site activity of Ni nanowires. On the other hand, for the 1T-MoS2–Ni(18) composite structure, superior catalytic competency was observed, indicating improved per-site activity of 1T-MoS2 due to the optimal synergistic interaction with metallic Ni nanowires. Therefore, it is reasonable to propose that the metallic Ni simultaneously modulates the total number of electrochemically active sites, as well as improves per-site efficiency in the 1T-MoS2–Ni(18) composite structure. Therefore, the enhanced HER performance of 1T-MoS2–Ni(18) could be ascribed to the following: (1) better water disintegration over the metallic Ni surface, (2) facilitating interfacial kinetics due to optimal synergistic coupling between metallic Ni and 1T-MoS2, (3) improvement in the potential of individual active sites of the 1T-MoS2–Ni(18) composite as compared to separate 1T-MoS2 and Ni nanowires, and (4) reduced electron migration resistance between the composite material and the electrolyte solution.
To examine the robustness of the catalyst, a long-term durability test of the 1T-MoS2–Ni(18) composite material was performed using steady-state chronopotentiometry (CP) and accelerated degradation studies (ADS) (cyclic stability). The long-term stability experiment was performed using Hg/HgO as the reference electrode and graphite rod as the counter electrode. The steady-state CP study of 1T-MoS2–Ni(18) was performed for 24 h at the current density of −10 mA cm−2geo (Fig. 6a). The LSV curves before and after the 24 h of the CP test revealed that only 23 mV of extra potential was required to maintain the same −10 mA cm−2geo current density (Fig. 6b). This observation further authenticates the superior mechanical robustness of the as-synthesized 1T-MoS2–Ni(18) composite structure. In addition, we performed the stability test of the as-prepared 1T-MoS2 and observed that although it has poor catalytic activity, it is quite stable in the alkaline medium for a long period (Fig. S17, ESI†). In addition, the ADS (cyclic stability test) experiment of 1T-MoS2–Ni(18) was also carried out by the continuous execution of 2000 CV cycles in the voltage window of 0 to −0.2 V (vs. RHE). The comparison of LSV curves between the 1st and 2001st cycle showed that the 1T-MoS2–Ni(18) composite demands only an additional 16 mV overpotential to sustain the −10 mA cm−2geo current density (Fig. 6c). The above-mentioned results further support the superior mechanical stability of the 1T-MoS2–Ni(18) composite even after long-term electrocatalysis performed in strongly alkaline conditions.
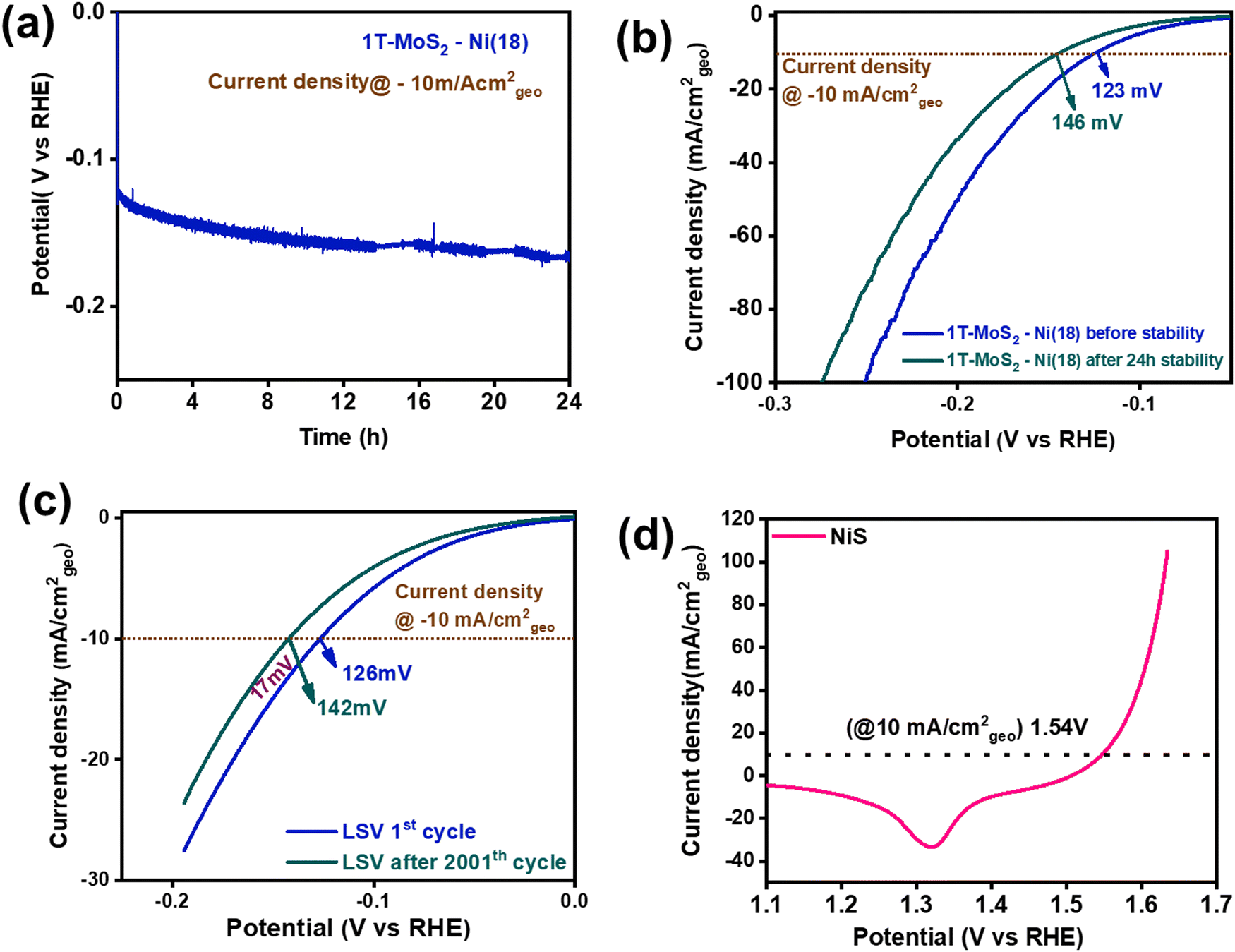 | ||
| Fig. 6 (a) A 24-h-long durability study (steady state CP test) at a current density of −10 mA cm−2geo, (b) comparison of the cathodic polarization curves before and after the 24-h stability test. (c) ADS experiment (cyclic stability test) of 1T-MoS2–Ni(18) towards the alkaline HER. (d) Backward anodic polarization curve (100% iR corrected) for NiS towards the alkaline OER. | ||
To fabricate an efficient alkaline electrolyzer, a good OER electrocatalyst is also required to display impressive two-electrode performance. In this context, we have synthesized NiS nanoparticles by reacting NiCl2·6H2O solution (1 ml) with thiourea and hydrazine in an ethylene glycol medium using a solvothermal process. (See details in the Experimental section of the ESI.†) The PXRD analysis revealed that the as-synthesized material matched well with the standard pattern of NiS (JCPDS File: 01-086-2281.) This confirms the phase purity of the material (Fig. S18, ESI†). In addition, FTIR spectroscopy was performed to understand the chemical functional groups present in the NiS material. The sharp vibrational band near 1098 cm−1 corresponds to the asymmetrical stretching mode of NiS. Two additional bands were observed at 768 cm−1 and 620 cm−1, which were attributed to the symmetrical stretching mode of NiS (Fig. S19, ESI†).72,73 The XPS study was performed to understand the chemical state information of the synthesized NiS material. The XPS survey scan confirmed the presence of Ni, S, and O in the material (Fig. S20, ESI†). The presence of oxygen (O) in the survey scan may be attributed to adsorbed water molecules on the surface of NiS. Moreover, the high-resolution deconvoluted Ni 2p spectrum indicated the co-existence of Ni+2 and Ni+3 (Fig. S21a, ESI†). The formation of Ni+3 is most likely due to the partial surface oxidation of the exposed Ni centers on the surface of the catalyst. For instance, Wu et al. recently observed similar Ni+3 formation in β NiS.74 Further, the deconvoluted S 2p confirmed the formation of S2− (Fig. S21b, ESI†).75 The respective peak positions of the corresponding high-resolution XPS spectra are shown in Table S3 (ESI†). The above-mentioned experimental results prove the formation of NiS. Further, the surface textural properties of the as-synthesized NiS were investigated by the FESEM study. The FESEM images divulged a random particle-like morphology with an average particle size of ∼30–50 nm (Fig. S22, ESI†). The elemental mapping analysis displays a uniform distribution of Ni and S in the material (Fig. S23a and b, ESI†). The EDS study showed that the atomic composition of Ni and S is 50.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49.3, which further authenticates the formation of NiS (Fig. S23c and d, ESI†).
:49.3, which further authenticates the formation of NiS (Fig. S23c and d, ESI†).
Electrochemical OER activity
The electrochemical proficiency of the as-synthesized NiS as an OER catalyst was investigated in 1 M KOH solution at room temperature by drop-casting it over the carbon paper substrate (0.5 cm × 0.5 cm). The details of the catalyst loading and reaction conditions are provided in the ESI.† Prior to determining the catalytic efficiency, the NiS electrode material was preconditioned within the potential range of 1 to 1.7 V vs. RHE with a high scan rate of 50 mV s−1. Subsequently, the CV was recorded at a low scan rate of 5 mV s−1, which displayed a distinct redox peak located at 1.48 V (without iR correction) and 1.43 V (with 100% iR correction) corresponding to Ni+2/Ni+3 redox behaviour (Fig. S24, ESI†).76 Further, the (iR-corrected) backward anodic polarization curve of NiS revealed that an overpotential of about 310 mV (1.54 V vs. RHE) was required to attain the benchmark current density of 10 mA cm−2geo (Fig. 6d). NiS also displayed overpotentials of 340 mV, 370 mV, 390 mV and 400 mV at current densities of 25, 50, 75, and 100 mA cm−2geo (Fig. S25, ESI†). However, the 1T-MoS2–Ni(18) sample displayed very poor OER activity (Fig. S26, ESI†). These results indicate the suitability of NiS as an OER catalyst. To evaluate the OER kinetics, Tafel slope analysis was performed using a 100% iR-corrected backward LSV curve. The as-prepared NiS displayed a Tafel slope value of 74 mV dec−1, suggesting good OER kinetics (Fig. S27, ESI†). To study the charge transfer resistance, an EIS study was conducted for NiS (Fig. S28, ESI†). The Nyquist plot and the corresponding experimental parameters are shown in detail in Table S4 (ESI†). The above-mentioned results suggest that NiS nanoparticles are good OER electrocatalysts in an alkaline medium.Overall water splitting activity in alkaline medium
An alkaline electrolyzer was demonstrated using 1T-MoS2–Ni(18) as a cathode and NiS as an anode material in 1 M KOH at room temperature (Fig. S29, ESI†). The individual NiS and 1T-MoS2–Ni(18) composite material required 1.54 V and −0.120 V vs. RHE to deliver 10 mA cm−2geo current density towards OER (anodic) and HER (cathodic) half-cells, respectively (Fig. 7a). The overall CV of the alkaline electrolyzer (two-electrode configuration) is shown in Fig. S30 (ESI†). Further, the backward LSV scan (recorded at 5 mV s−1 scan rate) for NiS(+)||1T-MoS2–Ni(18)(−) electrolyzer indicated that an overpotential of about 450 mV (1.68 V cell voltage) is required to attain a current density of 10 mA cm−2geo in 1 M KOH (Fig. 7b). The calculated cell potential value was slightly higher than the commercially available RuO2(+)||Pt/C(−) electrolyzer, up to a current density of 25 mA cm−2geo. However, at a relatively higher current density (50 mA cm−2geo), the NiS(+)||1T-MoS2–Ni(18)(−) electrolyzer displayed better catalytic proficiency (cell voltage 1.87 V) as compared to the standard RuO2(+)||Pt/C(−) electrolyzer (cell voltage 1.93 V) (Fig. 7c). Further, for practical viability, the long-term durability of the alkaline electrolyzer was monitored through a steady-state CP test at the current density of 10 mA cm−2geo. The voltage response was almost constant, even after a 48-h-long catalytic study, indicating excellent stability (Fig. 7d). In addition, the LSV curves before and after the durability study revealed a slight rise in the overpotential value of 10 mV to maintain the same current density (Fig. 7e). This observation further authenticates the mechanical robustness of both NiS and the 1T-MoS2–Ni(18) composite material towards total water-splitting reactions. The alkaline electrolyzer with NiS and 1T-MoS2–Ni(18) as the anode and cathode materials, respectively, is demonstrated in Fig. 7f.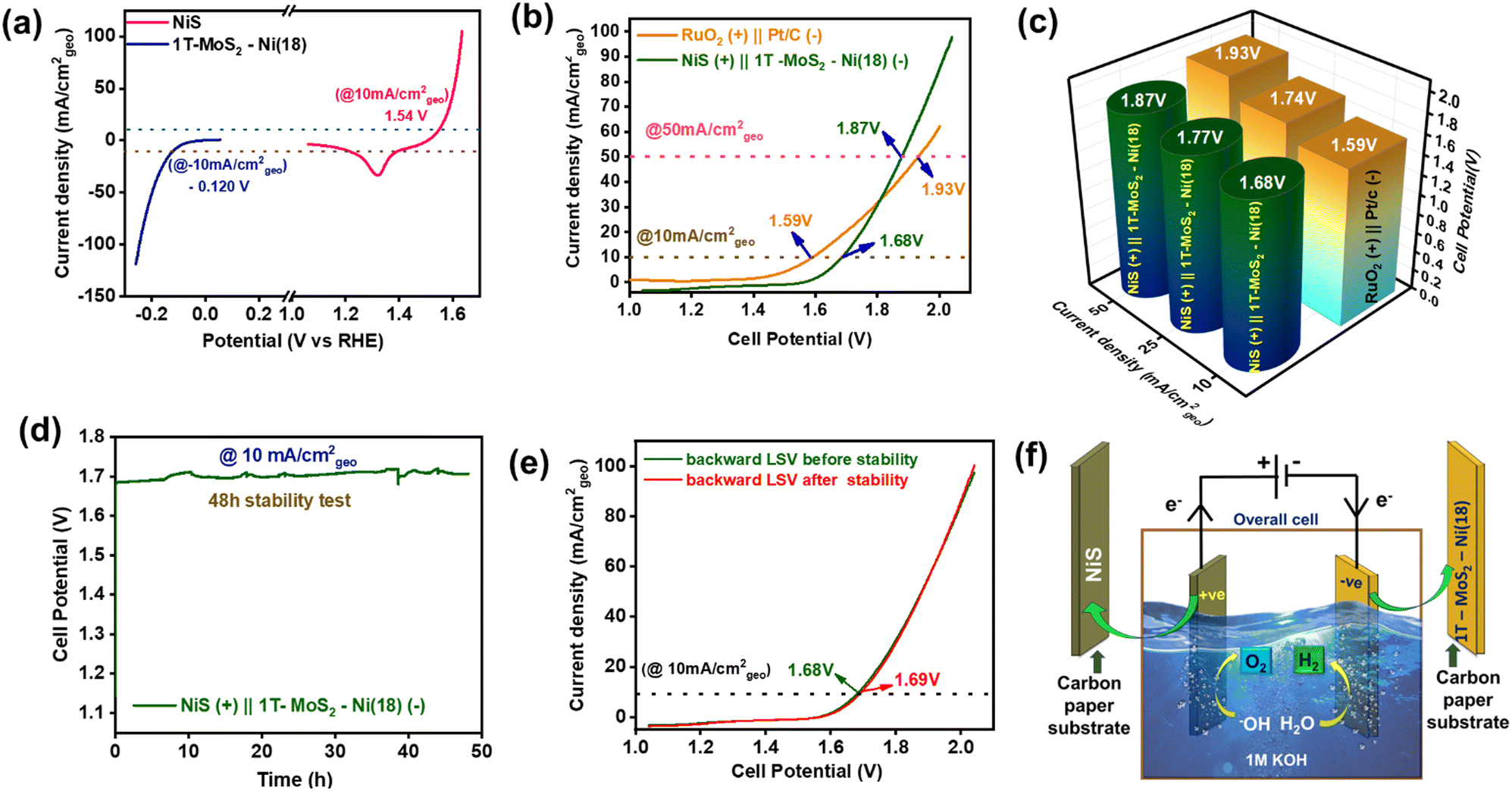 | ||
| Fig. 7 (a) Backward polarization curves of 1T-MoS2–Ni(18) and NiS toward the HER and OER. (b) Backward polarization curves of total alkaline water splitting in a two-electrode set-up (alkaline electrolyzer), (c) 3D bar diagram for the comparison of the overall cell potential at different current densities (mA cm−2geo) between NiS (+)||1T-MoS2–Ni(18) (−) and RuO2(+)||Pt/C (−) electrolyzers. (d) Long durability (48 h) study of the alkaline electrolyzer. (e) Comparison of the overall backward LSV curve before and after 48 h of stability. (f) Schematic demonstration of total water splitting using 1T-MoS2–Ni(18) as the cathode and NiS as the anode material. | ||
Post electrochemical characterization
Different post-physical characterizations were performed to recognize any structural alteration in the 1T-MoS2–Ni(18) and NiS after the course of long electrocatalysis. In this context, PXRD analysis was performed on the catalyst-coated carbon paper substrate to examine possible alterations in the phase of the 1T-MoS2–Ni(18) composite structure. The PXRD pattern of the 1T-MoS2–Ni(18) composite showed hardly any change (Fig. S31, ESI†). This observation indicated the retention of intercalation in the composite structure, even after an extended stability experiment. The post-FTIR study of the 1T-MoS2–Ni(18) composite material showed the retention of the N–H bending mode. This indicates the existence of intercalated ammonia even after long catalysis (Fig. S32, ESI†). The XPS study on the catalyst after the extended durability test revealed no significant change in the chemical state. The deconvoluted Mo 3d and S 2p spectra indicated the presence of the metallic 1T phase of MoS2 along with 2H-MoS2 in the composite material (Fig. S33a and b, ESI†). This observation indicates the excellent mechanical stability of 1T-MoS2 in the composite even after a prolonged electrocatalytic alkaline HER. The deconvoluted Ni 2p spectrum also manifested the existence of metallic Ni (Ni0) (Fig. S33c, ESI†). The high-resolution N 1s XPS scan indicated the presence of NH3/NH4+ in the post-catalyst material, which additionally supports the stability of ammonia intercalation in the 1T-MoS2–Ni(18) composite (Fig. S33d, ESI†). The microstructural change after the catalysis was investigated by FESEM and TEM analyses. The post-catalytic FESEM study of the 1T-MoS2–Ni(18) composite structure revealed hardly any alteration in the surface textural patterns (Fig. S34, ESI†). In addition, the post-stability TEM images also displayed an isotropic flake-like architecture, indicating the microstructural robustness of the material (Fig. S35, ESI†). These results indicate the robustness of the 1T-MoS2–Ni(18) composite catalyst for the long-term electrocatalytic alkaline HER process. On the other hand, the post-catalytic FESEM images of NiS hardly showed any observable changes in the morphology (Fig. S36, ESI†). However, the post-XPS analyses of NiS displayed significant catalytic transformations. For instance, the high-resolution peak-fitted Ni 2p XPS spectrum displays the presence of Ni+3. However, hardly any Ni+2 signals were observed, suggesting that during anodic oxidation, all Ni+2 ions were transformed into Ni+3 (Fig. S37a, ESI†). Further, the post-catalytic O1s spectra revealed the existence of a hydroxyl group (OH) at 530.8 eV (Fig. S37b, ESI†).77 This observation indicates the (oxy)hydroxide (MOOH (M = Ni)) formation after the OER catalysis. On the other hand, the XPS spectrum of S 2p showed no observable signal, indicating etching from the as-prepared NiS material in 1 M KOH during the anodic oxidation process (Fig. S37c, ESI†). A similar observation was reported in earlier studies.78 Fig. S37d (ESI†) displays a comparison of S intensity before and after the stability test. Hence, the results mentioned above indicate the transformation of NiS into NiOOH during the water oxidation process. The peak positions of the respective elements are shown in Table S5 (ESI†). This catalytic transformation is analogous to previous nickel chalcogenide literature where catalytic materials are usually converted into active NiOOH species during the OER.76,79–81 The post-stability EDS study of NiS revealed the presence of O and the absence of S, which additionally supports the etching of sulfur moieties during OER and validates the XPS results (Fig. S38, ESI†). The results suggest that the surface of NiS is catalytically transformed into active NiOOH species during the anodic oxidation process, which displays excellent alkaline OER performance.Conclusions
This work presents a two-step synthetic approach to preparing the 1T-MoS2 composite with metallic Ni for improved alkaline HER performance. Initially, different quantities of aqueous NiCl2·6H2O were introduced into the dispersed 1T-MoS2 (in ethylene glycol medium). Subsequent reaction with hydrazine under a wet chemical synthesis route resulted in the formation of the 1T-MoS2–Ni(X) composite structure. The detailed structural and morphological analyses of the 1T-MoS2–Ni(18) composite proved the formation of an interfacial structure between metallic Ni and 1T-MoS2. The as-prepared 1T-MoS2–Ni(18) composite structure displayed excellent alkaline HER activity as compared to individual 1T-MoS2 and metallic Ni nanowires. The feasible water-disintegration kinetics, facile electron migration through the interfacial structure, and the improvement in the per-site activity are the main reasons for the noted enhanced alkaline HER performances of the composite structure. For the practical demonstration of an alkaline electrolyzer, NiS nanoparticles were synthesized and used as an OER catalyst. Finally, an alkaline electrolyzer was constructed using the as-prepared NiS as the anode and 1T-MoS2–Ni(18) as the cathode material. The electrolyzer exhibited a 1.68 V cell potential to attain the standard 10 mA cm−2geo current density and was sustained for 48 h, demonstrating long stability without any major performance drop. The superior stability of the materials indicates their suitability for real applications. To the best of our knowledge, this is the first detailed study on the interfacial structure shared between metallic Ni and 1T-MoS2 for alkaline HER applications through composite formation. This synthetic scheme could be extended to make composites with other metals for electrocatalytic applications.Author contributions
A. R., A. M., H. R. I., and V. M. made the entire idea of the work. A. R. performed all the material synthesis, the entire physical characterization, and the total electrochemical study. A. M., H. R. I., S. G., K. S. P., and V. M., continuously helped A. R. during the manuscript writing. S. D. and A. A. V. aid A. R. in making the schematic figure of work. V. M. supervised the entire work. Prior to the submission, all authors have approved the ultimate version of the manuscript.Conflicts of interest
There are no such competing financial interests to announce.Acknowledgements
A. R. sincerely acknowledges DST-Inspire for the Fellowship. H. R. I. and A. M. thank UGC and CSIR, India, for their fellowship. S. G. acknowledges DST, India, for providing N-PDF. KSP, SD acknowledge IISER Kolkata for their Fellowships. The author V. M., thanks DST, India, (DST/TMD/MES/2K17/70) for project funding. The authors thank IISER, Kolkata, for providing different Instrumental facilities, and HRTEM supported by DIST-FIST-TEM (SR/FST/CS11-029/2014). A. R. also sincerely acknowledges Antarip Mitra, Viplove Mishra, K. V. S. H. R. Murthy, Gouri Tudu, Athma E. P., Jaydeep Kumar, Diya Raveendran, Koustav Banerjee, and Saikat Mondal for their valuable suggestions and mental support throughout the work.Notes and references
- W. Liu, Y. Wan, Y. Xiong and P. Gao, Int. J. Hydrogen Energy, 2022, 47, 24584–24591 CrossRef CAS.
- P. Aggarwal, D. Sarkar, P. W. Menezes and K. Awasthi, Int. J. Hydrogen Energy, 2022, 47, 41795–41805 CrossRef CAS.
- L. Sharma, T. Botari, C. S. Tiwary and A. Halder, ACS Appl. Energy Mater., 2020, 3, 5333–5342 CrossRef CAS.
- J. D. Benck, T. R. Hellstern, J. Kibsgaard, P. Chakthranont and T. F. Jaramillo, ACS Catal., 2014, 4, 3957–3971 CrossRef CAS.
- Z. Wu, Z. Zou, J. Huang and F. Gao, J. Catal., 2018, 358, 243–252 CrossRef CAS.
- S. Anantharaj, Curr. Opin. Electrochem., 2022, 33 Search PubMed.
- K. A. Stoerzinger, M. Favaro, P. N. Ross, J. Yano, Z. Liu, Z. Hussain and E. J. Crumlin, J. Phys. Chem. B, 2018, 122, 864–870 CrossRef CAS PubMed.
- Z. Li, W. Niu, Z. Yang, A. Kara, Q. Wang, M. Wang, M. Gu, Z. Feng, Y. Du and Y. Yang, Energy Environ. Sci., 2020, 13, 3110–3118 RSC.
- N. Mahmood, Y. Yao, J. W. Zhang, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- P. Wang, X. Zhang, J. Zhang, S. Wan, S. Guo, G. Lu, J. Yao and X. Huang, Nat. Commun., 2017, 8, 14580 CrossRef CAS PubMed.
- K. C. Lee, M. R. Sprague, B. J. Sussman, J. Nunn, N. K. Langford, X. M. Jin, T. Champion, P. Michelberger, K. F. Reim, D. England, D. Jaksch and I. A. Walmsley, Science, 2011, 334, 1253–1256 CrossRef CAS PubMed.
- L. Xie, X. Ren, Q. Liu, G. Cui, R. Ge, A. M. Asiri, X. Sun, Q. Zhang and L. Chen, J. Mater. Chem. A, 2018, 6, 1967–1970 RSC.
- J. Sun, D. Zhu, Y. Sun, L. Ma, J. Guo, Q. Liu and X. Zhang, J. Alloys Compd., 2020, 833, 155131 CrossRef CAS.
- K. Guruprasad, T. Maiyalagan and S. Shanmugam, ACS Appl. Energy Mater., 2019, 2, 6184–6194 CrossRef CAS.
- A. Kagkoura, M. Pelaez-Fernandez, R. Arenal and N. Tagmatarchis, Nanoscale Adv., 2019, 1, 1489–1496 RSC.
- H. Reddy Inta, T. Biswas, S. Ghosh, R. Kumar, S. Kanti Jana and V. Mahalingam, ChemNanoMat, 2020, 6, 685–695 CrossRef CAS.
- A. Mondal, H. R. Inta, V. Bheemireddy, S. Ghosh and V. Mahalingam, ChemNanoMat, 2021, 7, 1063–1071 CrossRef CAS.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Adv. Energy Mater., 2018, 8, 1703482 CrossRef.
- S. Mansingh, K. K. Das and K. Parida, Sustainable Energy Fuels, 2021, 5, 1952–1987 RSC.
- J. Tang, J. Huang, D. Ding, S. Zhang and X. Deng, Int. J. Hydrogen Energy, 2022, 47, 39771–39795 CrossRef CAS.
- M. Li, B. Cai, R. Tian, X. Yu, M. B. H. Breese, X. Chu, Z. Han, S. Li, R. Joshi, A. Vinu, T. Wan, Z. Ao, J. Yi and D. Chu, Chem. Eng. J., 2021, 409, 128158 CrossRef CAS.
- J. Xiong, J. Li, J. Shi, X. Zhang, W. Cai, Z. Yang and H. Cheng, Appl. Catal., B, 2019, 243, 614–620 CrossRef CAS.
- Y. Huang, Y. Sun, X. Zheng, T. Aoki, B. Pattengale, J. Huang, X. He, W. Bian, S. Younan, N. Williams, J. Hu, J. Ge, N. Pu, X. Yan, X. Pan, L. Zhang, Y. Wei and J. Gu, Nat. Commun., 2019, 10, 982 CrossRef CAS PubMed.
- W. Chen, J. Gu, Y. Du, F. Song, F. Bu, J. Li, Y. Yuan, R. Luo, Q. Liu and D. Zhang, Adv. Funct. Mater., 2020, 30, 2000551 CrossRef CAS.
- M. Li, B. Cai, R. Tian, X. Yu, M. B. H. Breese, X. Chu, Z. Han, S. Li, R. Joshi, A. Vinu, T. Wan, Z. Ao, J. Yi and D. Chu, Chem. Eng. J., 2020, 409, 128158 CrossRef.
- H. J. Liu, S. Zhang, Y. M. Chai and B. Dong, Angew. Chem. Int. Ed., 2023, 13845 Search PubMed.
- N. P. Dileep, P. V. Sarma, R. Prasannachandran, V. Surendran and M. M. Shaijumon, ACS Appl. Nano Mater., 2021, 4, 7206–7212 CrossRef CAS.
- K. Wang, Z. Liu, Q. Gao, N. Li and K. Yu, Appl. Surf. Sci., 2022, 593, 153408 CrossRef CAS.
- Y. Zhao, S. Wei, F. Wang, L. Xu, Y. Liu, J. Lin, K. Pan and H. Pang, Chem. – Eur. J., 2020, 26, 2034–2040 CrossRef CAS PubMed.
- H. Sun, Z. Yan, C. Tian, C. Li, X. Feng, R. Huang, Y. Lan, J. Chen, C. P. Li, Z. Zhang and M. Du, Nat. Commun., 2022, 13, 3857 CrossRef CAS PubMed.
- X. He, W. Zhong, C. T. Au and Y. Du, Nanoscale Res. Lett., 2013, 8, 446 CrossRef PubMed.
- M. Wang, H. Yang, J. Shi, Y. Chen, Y. Zhou, L. Wang, S. Di, X. Zhao, J. Zhong, T. Cheng, W. Zhou and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 5771–5777 CrossRef CAS PubMed.
- B. Pattengale, Y. Huang, X. Yan, S. Yang, S. Younan, W. Hu, Z. Li, S. Lee, X. Pan, J. Gu and J. Huang, Nat. Commun., 2020, 11, 4114 CrossRef CAS PubMed.
- H. Xu, N. Liang, Z. Bai, B. Yang, D. Chen and H. Tang, Molecules, 2023, 28, 6658 CrossRef CAS PubMed.
- A. E. Praveen, S. Ganguli and V. Mahalingam, Nanoscale Adv., 2020, 2, 1927–1938 RSC.
- M. Chatti, T. Gengenbach, R. King, L. Spiccia and A. N. Simonov, Chem. Mater., 2017, 29, 3092–3099 CrossRef CAS.
- A. S. Goloveshkin, N. D. Lenenko, V. I. Zaikovskii, A. S. Golub, A. A. Korlyukov and I. S. Bushmarinov, RSC Adv., 2015, 5, 19206–19212 RSC.
- W. Hu, H. Liu, W. Dong, H. Akif Munir, X. Fan, X. Tian and L. Pang, J. Electroanal. Chem., 2023, 949, 117882 CrossRef CAS.
- Y. Xu, L. Wang, X. Liu, S. Zhang, C. Liu, D. Yan, Y. Zeng, Y. Pei, Y. Liu and S. Luo, J. Mater. Chem. A, 2016, 4, 16524–16530 RSC.
- X. Zhao, X. Ma, Q. Lu, Q. Li, C. Han, Z. Xing and X. Yang, Electrochim. Acta, 2017, 249, 72–78 CrossRef CAS.
- Z. Lei, J. Zhan, L. Tang, Y. Zhang and Y. Wang, Adv. Energy Mater., 2018, 8 Search PubMed.
- Y. Yin, P. Miao, Y. Zhang, J. Han, X. Zhang, Y. Gong, L. Gu, C. Xu, T. Yao, P. Xu, Y. Wang, B. Song and S. Jin, Adv. Funct. Mater., 2017, 27, 1606694 CrossRef.
- Y. Yan, B. Xia, X. Ge, Z. Liu, J. Y. Wang and X. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 12794–12798 CrossRef CAS PubMed.
- M. A. Bissett, I. A. Kinloch and R. A. W. Dryfe, ACS Appl. Mater. Interfaces, 2015, 7, 17388–17398 CrossRef CAS PubMed.
- S. Venkateshwaran and S. M. Senthil Kumar, ACS Sustainable Chem. Eng., 2019, 7, 2008–2017 CrossRef CAS.
- H. Xia, L. Zan, P. Yuan, G. Qu, H. Dong, Y. Wei, Y. Yu, Z. Wei, W. Yan, J. S. Hu, D. Deng and J. N. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202218282 CrossRef CAS PubMed.
- S. Das, G. Swain, B. P. Mishra and K. Parida, New J. Chem., 2022, 46, 14922–14932 RSC.
- X. Yin, Y. Li, H. Meng and W. Wu, Appl. Surf. Sci., 2019, 486, 362–370 CrossRef CAS.
- W. Hou, Y. Sun, Y. Zhang, T. Wang, L. Wu, Y. Du and W. Zhong, J. Alloys Compd., 2021, 859 Search PubMed.
- X. Li, X. Lv, X. Sun, C. Yang, Y. Z. Zheng, L. Yang, S. Li and X. Tao, Appl. Catal., B, 2021, 284 CAS.
- S. Das, L. Acharya, L. Biswal, B. P. Mishra and K. Parida, Catal. Sci. Technol., 2023, 13, 2827–2840 RSC.
- Z. Wu, C. Tang, P. Zhou, Z. Liu, Y. Xu, D. Wang and B. Fang, J. Mater. Chem. A, 2015, 3, 13050–13056 RSC.
- F. Z. Wang, M. J. Zheng, B. Zhang, C. Q. Zhu, Q. Li, L. Ma and W. Z. Shen, Sci. Rep., 2016, 6, 31092 CrossRef CAS PubMed.
- L. Pang, A. Barras, Y. Zhang, M. A. Amin, A. Addad, S. Szunerits and R. Boukherroub, ACS Appl. Mater. Interfaces, 2019, 11, 31889–31898 CrossRef CAS PubMed.
- X. Zhang, J. Xu, L. Chai, T. He, F. Yu and P. Wang, Appl. Surf. Sci., 2017, 406, 30–38 CrossRef CAS.
- X. Li, Y. C. Chang, J. Y. Chen, K. W. Lin, R. D. Desautels, J. van Lierop and P. W. T. Pong, Phys. Lett. A, 2018, 382, 2886–2893 CrossRef CAS.
- X. Zhang, X. Gao, D. Li, C. Duanmu, J. Jiang, J. Chen, X. Yu and P. Dong, J. Colloid Interface Sci., 2020, 563, 354–362 CrossRef CAS PubMed.
- Y. Li and C. Zhao, Chem. Mater., 2016, 28, 5659–5666 CrossRef CAS.
- Y. W. Dong, X. Y. Zhang, J. C. Zhou, F. L. Wang, R. N. Luan, X. Liu, B. Liu, Y. M. Chai and B. Dong, Int. J. Hydrogen Energy, 2022, 47, 31656–31664 CrossRef CAS.
- D. Tang, J. Li, Z. Yang, X. Jiang, L. Huang, X. Guo, Y. Li, J. Zhu and X. Sun, Chem. Eng. J., 2021, 130954 Search PubMed.
- N. Joseph and C. B. Arumugam, J. Electroanal. Chem., 2020, 874, 114461 CrossRef.
- A. T. Borgogoi, D. J. Borah and A. T. T. Mostako, Phys. B, 2023, 660, 414896 CrossRef CAS.
- A. Mondal, H. R. Inta, A. Roy, A. Kumar Mahato and V. Mahalingam, ACS Appl. Nano Mater., 2023, 6, 12040–12049 CrossRef CAS.
- X. Zhao, W. Gao, Q. Liu, C. Cui, W. Zhou, X. Wang, X. L. Zhang, L. Zhao, Y. Sang and H. Liu, Chem. Eng. J., 2021, 404, 126972 CrossRef CAS.
- J. Prasad, A. K. Singh, A. N. Yadav, A. Kumar, M. Tomar, A. Srivastava, P. Kumar, V. Gupta and K. Singh, ACS Appl. Mater. Interfaces, 2020, 12, 40828–40837 CrossRef CAS PubMed.
- C. Nethravathi, J. Prabhu, S. Lakshmipriya and M. Rajamathi, ACS Omega, 2017, 2, 5891–5897 CrossRef CAS PubMed.
- S. Anantharaj, S. R. Ede, K. Karthick, S. Sam Sankar, K. Sangeetha, P. E. Karthik and S. Kundu, Energy Environ. Sci., 2018, 11, 744–771 RSC.
- W. Wang, L. Yang, F. Qu, Z. Liu, G. Du, A. M. Asiri, Y. Yao, L. Chen and X. Sun, J. Mater. Chem. A, 2017, 5, 16585–16589 RSC.
- T. Wang, Q. Zhou, X. Wang, J. Zheng and X. Li, J. Mater. Chem. A, 2015, 3, 16435–16439 RSC.
- S. Wang, J. Wang, M. Zhu, X. Bao, B. Xiao, D. Su, H. Li and Y. Wang, J. Am. Chem. Soc., 2015, 137, 15753–15759 CrossRef CAS PubMed.
- L. Dai, Z. N. Chen, L. Li, P. Yin, Z. Liu and H. Zhang, Adv. Mater., 2010, 32, 1906915 CrossRef PubMed.
- S. Nachimuthu, K. Kannan, S. Thangavel and K. Gurushankar, Appl. Surf. Sci. Adv., 2022, 6, 100209 CrossRef.
- M. A. AlMalki, Z. A. Khan and W. A. El-Said, Appl. Surf. Sci. Adv., 2021, 7, 100187 CrossRef.
- T. H. Wu, Y. C. Lin, B. W. Hou and W. Y. Liang, Catalysts, 2020, 10, 1–11 Search PubMed.
- Q. Li, D. Wang, C. Han, X. Ma, Q. Lu, Z. Xing and X. Yang, J. Mater. Chem. A, 2018, 6, 8233–8237 RSC.
- H. R. Inta, S. Ghosh, A. Mondal, G. Tudu, H. V. S. R. M. Koppisetti and V. Mahalingam, ACS Appl. Energy Mater., 2021, 4, 2828–2837 CrossRef CAS.
- K. Andersson, A. Nikitin, L. G. M. Pettersson, A. Nilsson and H. Ogasawara, Phys. Rev. Lett., 2004, 93, 196101 CrossRef CAS PubMed.
- C. Yan, J. Huang, C. Wu, Y. Li, Y. Tan, L. Zhang, Y. Sun, X. Huang and J. Xiong, J. Mater. Sci. Technol., 2020, 42, 10–16 CrossRef CAS.
- C. Jin, P. Zhai, Y. Wei, Q. Chen, X. Wang, W. Yang, J. Xiao, Q. He, Q. Liu and Y. Gong, Small, 2021, 17, 2102097 CrossRef CAS PubMed.
- G. Tudu, S. Ghosh, H. R. Inta, H. V. S. R. M. Koppisetti, S. Ganguli, A. K. Mahato and V. Mahalingam, ChemNanoMat, 2022, 8, e202200199 CrossRef CAS.
- X. Ding, W. Li, H. Kuang, M. Qu, M. Cui, C. Zhao, D. C. Qi, F. E. Oropeza and K. H. L. Zhang, Nanoscale, 2019, 11, 23217–23225 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma01077e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |