
Efficient lignin depolymerization by continuous flow microreactor-assisted electrochemical advanced oxidation in water/co-solvent system†
Lalida
Waura-angkura‡
ab,
Babasaheb M.
Matsagar‡
 a,
Kevin
Lee
b,
Varong
Pavarajarn
*b and
Kevin C.-W.
Wu
*ac
a,
Kevin
Lee
b,
Varong
Pavarajarn
*b and
Kevin C.-W.
Wu
*ac
aDepartment of Chemical Engineering, National Taiwan University, Taipei 10617, Taiwan. E-mail: kevinwu@ntu.edu.tw
bCenter of Excellence in Particle and Materials Processing Technology, Department of Chemical Engineering, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: Varong.P@chula.ac.th
cDepartment of Chemical Engineering and Materials Science, Yuan Ze University, Chung-Li, Taoyuan, Taiwan
First published on 10th October 2023
Abstract
This study presented for the first time a continuous flow microreactor system for efficient dealkaline lignin depolymerization applying an electrochemical advanced oxidation process (EAOP). The insolubility of dealkaline lignin in water poses a challenge for its chemical depolymerization via EAOP. To address this limitation, several co-solvent systems were developed and investigated to achieve efficient dealkaline lignin depolymerization. Five water-miscible organic solvents, namely methanol (MeOH), ethanol (EtOH), acetonitrile (MeCN), tetrahydrofuran (THF), and N,N-dimethylformamide (DMF), were used as co-solvents. With the co-solvent fraction of 1% in water, the solubility of dealkaline lignin followed the order of THF > DMF > MeCN > MeOH > EtOH. The electron spin resonance (EPR) of various co-solvents revealed that different co-solvents affected the concentration of in situ OH radical generation, leading to the difference in dealkaline lignin conversions. For dealkaline lignin in water–MeCN system, 72% conversion was observed within 100 s of residence time under ambient conditions with a 1 mA current. Moreover, the two main lignin model compounds (2-phenoxy-1-phenyl ethanol (PPE) containing β-O-4 and benzyl phenyl ether (BPE) containing α-O-4) were used to understand the kinetic analysis and degradation of dealkaline lignin. High conversions of 75% and 92% were achieved for the β-O-4 (PPE) and α-O-4 (BPE) lignin model compounds, respectively. All the products were confirmed using GC-MS analysis. Additionally, mechanistic insights for lignin depolymerization were provided using lignin model compounds.
1. Introduction
Lignin is an organic polymer found in lignocellulosic biomass, typically generated as a by-product of the paper and pulp industries, rendering it a form of bio-waste.1–4 Owing to the massive production of cellulose-based products, it is estimated that approximately 50 to 70 million tons of lignin are produced annually.5 However, in the paper and pulp industries, lignin is typically incinerated to generate heat in the pulping process.6 As lignin is composed of crosslinked phenolic polymer, it is abundantly available as a renewable feedstock for fuels and aromatic compounds.4 So far, only ca. 2% of lignin is commercially utilized due to its limited solubility and extremely complex molecular structure.7,8 Therefore, the valorization of lignin into high-value-added chemicals has garnered much attention in recent years.9–11Multiple techniques that have been well-established for lignin valorization include thermal conversion,12–14 biological depolymerization,15,16 enzymatic conversions,17,18 and inorganic catalysis.19–21 Although these techniques have been reported to be able to convert lignin into value-added chemicals, they have some major drawbacks. For instance, thermal conversion requires high energy, while biological depolymerization is limited to a narrow pH and temperature. Meanwhile, lignin conversion via catalysis is limited by the high catalyst cost, tedious catalyst synthesis and catalyst separation from the reaction solution. Therefore, a new technique for efficient depolymerization of lignin is keenly desired.
Recently, electrochemical conversion as a class of advanced oxidation processes (AOPs) has been reported as a potential method for polymer depolymerization under ambient conditions, thus reducing the total energy and cost required for the process.22 In electrochemical advanced oxidation processes (EAOPs), reactive oxygen species (ROS) are generated as an oxidizing agent for the depolymerization of polymers at the anode or cathode.23 In this case, the catalyst addition is unnecessary as the generated radical species actively initiate reactions. Du et al. reported that, via EAOP, lignin can be chemically oxidized by the OH radical (OH˙) that is produced on the anode via water dissociation.23 However, small mass transfer and the requirement of supporting electrolytes limit the efficiency of the process.24 To improve the efficiency of EAOPs, some researchers have applied microreactors in their work. Microreactors are microscale reactors that, compared to typical reactors, possess a high surface-to-volume ratio, low mass and heat resistances, simplified control over residence time, and greater control of reaction parameters.24–27
Continuous flow is the preferred system over batch processing in this context due to its highly efficient mass and heat transfer capabilities, all of which contribute to improved performance. Previously, our group reported the degradation of diuron, a chemically persistent herbicide, via EAOP in a microreactor system.28 It was demonstrated that the system can effectively convert diuron into CO2 and H2O with 90% conversion, under an applied current density of 0.157 mA cm−2 with a residence time of 100 s.28 Scialdone et al. also reported that more than 90% of tetrachloroethane can be degraded under 3 mA cm−2 within 20.25 s in the microreactor.29 Although the depolymerization of some compounds via EAOP in a microreactor has been reported, there is no report on the depolymerization of dealkaline lignin via EAOP in a microreactor so far. The EAOP of alkali lignin is relatively easier due to its solubility in water. However, EAOP for dealkaline lignin has not been explored and is challenging because of its limited solubility in water, which is essential for EAOP. Therefore, the investigation and utilization of various co-solvents to overcome the solubility issue of dealkaline lignin are proposed. Additionally, the evaluation of the role of co-solvent in the conversion of dealkaline lignin and lignin model compounds, i.e., 2-phenoxy-1-phenyl ethanol (PPE) and benzyl phenyl ether (BPE), representing β-O-4 and α-O-4 linkages, respectively, is systematically investigated. It is noted that these two linkages are the two major linkages in lignin structure, e.g., 50–65% and 6–8% for β-O-4 and α-O-4, respectively.5 For the first time, this work reports the efficient dealkaline lignin conversion under ambient conditions using EAOP. This study of continuous flow microreactor-assisted EAOP shows promising potential for economically feasible lignin valorization into value-added chemicals.
2. Experimental
2.1. Materials
All materials were of high purity and used without further purification. Tetrahydrofuran (THF; 99%) was purchased from Acros, while ethanol (EtOH; 99.8%), and hydrogen peroxide (H2O2; 30%) were obtained from Honeywell Fluka. Methanol (MeOH; anhydrous, ≥99.8%) was procured from Macron. N,N-Dimethylformamide (DMF; 99.9%) was obtained from Duksan. Acetonitrile (MeCN; ≥99.9%) and dealkaline and alkali lignin were purchased from TCI and Sigma-Aldrich, respectively. For these lignins, earlier research documented the detection of Na and S through ICP-OES and SEM-EDAX characterizations, meaning these lignins were likely isolated using the Kraft method, which commonly involves the Na2S and NaOH reagents use.30 While 2-phenoxy-1-phenyl ethanol (PPE; 97%) was procured from Combi-Blocks. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO; >97%) and benzyl phenyl ether (BPE; >98%) were purchased from Tokyo Chemical Industry (TCI). Pure graphite plate with the thickness of 1 mm was purchased from LNJ Bhilwara group, India. Ultrapure water with a resistance of 18.20 MΩ resistance was used for the experiment and was purified with a Milli-Q system (Millipore, Bedford, M, USA).2.2. Solubility test
The solubility of dealkaline lignin in various water/co-solvent systems was investigated to select a suitable co-solvent for lignin depolymerization. In typical sample preparation, 4 mg of dealkaline lignin was dissolved in a solution comprising 20 mL of co-solvent and water with 1, 5, 10, and 30% volumetric ratios. The samples were then sonicated for 5 min, followed by filtration. The co-solvents employed for initial screening were EtOH, MeOH, MeCN, DMF, and THF. The concentration of dealkaline lignin that dissolved was measured using a UV–visible spectrophotometer (Jasco, V-670), with the maximum absorbance (λmax) at 280 nm.31,32 In the experiment, Beer's law was used in conjunction with calibration curve using lignin standard with known concentrations to determine the concentration of lignin in the solution. We specifically evaluated the absorbance (A) directly related to the lignin concentration (c). A calibration curve generated for dealkaline lignin concentrations between 1–50 ppm in a 30% THF and other solvent system was used to convert absorbance to lignin concentration.2.3. Radical scavenging analysis
The efficiency of the EAOP largely depends on production of OH radicals, a highly reactive oxidant, at the anode and its concentration. However, the organic solvents used in this study may exhibit scavenging activity toward OH radicals. Therefore, the effect of the co-solvent on the generation of OH radicals was analyzed qualitatively using electron paramagnetic resonance (EPR) spectroscopy (Bruker, Bruker EPR-plus). The EPR analysis was carried out for six solutions containing 30% co-solvent in water, including EtOH, MeOH, MeCN, DMF, and THF. The solutions contained 500 ppm of H2O2, and 500 ppm of DMPO. Before analysis the solutions were irradiated with UV light for 20 min. The EPR spectra were then recorded in the 3400–3600 G range at 10 dB.2.4. Degradation of dealkaline lignin and alkali lignin
The microchannel was fabricated by sandwiching two conducting electrodes between a 250 μm thick Teflon sheet with a pre-determined gap, resulting in a microchannel with dimensions of 27 mm in length and 10 mm in width, as shown in Scheme 1. The anode and cathode were composed of graphite and stainless-steel plates, respectively. For lignin conversion, the reactant solution was prepared using 10–250 ppm of dealkaline lignin in 30% co-solvent. The flow rate was controlled by a syringe pump from 1 to 3.5 mL h−1, corresponding to a mean residence time of 243 to 69 s, respectively. The electrochemical reaction was started by applying direct current (DC) across the microchannel with an applied current varied from 0.5 to 3 mA. The lignin solution passed through the microchannel underwent lignin depolymerization. The products generated and the unreacted reactant were collected at the outlet of the microreactor. The amount of dealkaline lignin converted at steady state was quantified by measuring the reduction in dealkaline lignin concentration after degradation using UV–visible spectrophotometry (UV–Vis). For the depolymerization of alkali lignin, as it is soluble in water, the reactions of alkali lignin were carried out in an aqueous medium. The reaction solution was prepared by dissolving 130 ppm of alkali lignin in water, and a current of 1 mA was applied during the reaction.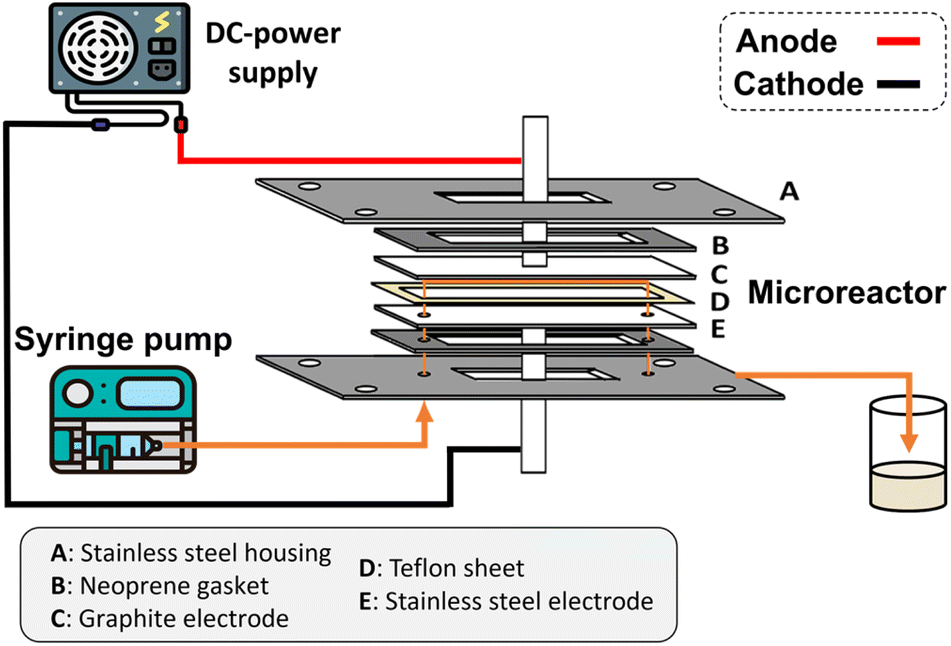 | ||
| Scheme 1 Schematic illustration of the complete setup of the microreactor. | ||
2.5. Degradation of the lignin model compound
The degradation of lignin model compounds (PPE and BPE) was performed using the identical reaction setup employed for the degradation of dealkaline and alkaline lignin. Reactions were performed with 100 ppm of lignin model compound in 30% co-solvent with the optimum applied current at 1 mA. The flow rate of the sample was varied in the range of 2.4 to 9 mL h−1, corresponding to a mean residence time of 100 to 27 s, respectively. To identify the chemical intermediates and products formed during the depolymerization of PPE and BPE model compounds, Gas Chromatography-Mass Spectrometry (GC-MS) analysis using an Agilent 6890 instrument was performed. For GC-MS analysis, an HP-5 ms capillary column (30 m × 0.2 μm) was utilized with helium gas (1.0 mL min−1) was used as the carrier gas. A 2 μL sample was injected in splitless mode, and both the injector and detector operated at 300 °C during analysis. The column oven temperature was initially held at 100 °C for 3 min, followed by an increase from 100 to 300 °C at a heating rate of 6 °C min−1, and finally held at 300 °C for 5 min. The products and intermediates were ionized and separated by a single quadrupole within the 20 to 500 m/z range. Meanwhile the mass spectra were analyzed by comparing them with the spectra of pure compounds from the NIST11 commercial library.3. Results and discussion
3.1. Effect of co-solvent on the solubility of dealkaline lignin
Although EAOP is an effective technique in the degradation of organic compounds, depolymerization of lignin via EAOP has been limited by low solubility of dealkaline lignin in water. Consequently, the addition of an organic co-solvent is indispensable. In this work, to gain insights into the efficient co-solvent for dissolving dealkaline lignin in water, the effect of different water/co-solvent systems on the solubility of the dealkaline lignin was investigated. The investigation was carried out by changing the volume fraction of organic solvent from 1 to 30% (v/v) in water with fixed content of dealkaline lignin corresponding to the concentration of 200 ppm. The solubility of dealkaline lignin was determined from the lignin concentration in the supernatant. Without co-solvent, the solubility of dealkaline lignin in pure water is only ca. 10 ppm. As expected, the solubility of dealkaline lignin in all water/co-solvent systems increased with increasing co-solvent content from 1 to 30% indicating that organic solvents can improve the solubility. Since the solubility of dealkaline lignin at a high co-solvent ratio was high in all co-solvents, the trend in the solubility can only be compared at a low fraction, i.e., 1% co-solvent fraction. As shown in Fig. 1, at 1% co-solvent fraction, the solubility of dealkaline lignin was found in order of THF > DMF > MeCN > MeOH > EtOH.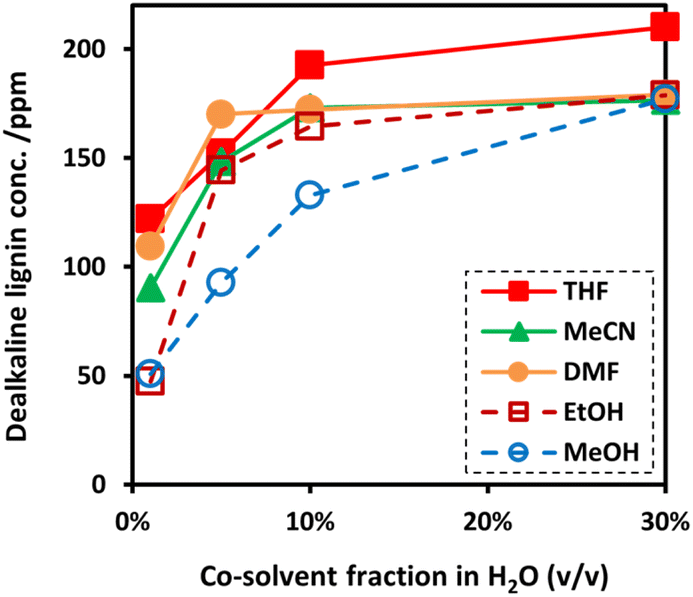 | ||
| Fig. 1 The soluble concentration of dealkaline lignin in co-solvent (conditions: concentration of dealkaline lignin 200 ppm; and sonication time 5 min). | ||
It is noted that polymers, such as lignin, are generally soluble in solvents with Hildebrand and Hansen solubility parameters (δt) values similar to their own.8,33 Also, with an optimal δt value, the lignin-dissolving ability increases with the increase in the hydrogen-bonding potential of the solvent.34,35 Therefore, the solubility of dealkaline lignin in co-solvent systems can be theoretically predicted by comparing how close the δt value of dealkaline lignin to co-solvents, i.e., the closer the value, the higher the solubility. It was reported that the δt value of lignin is ca. 12.87 (cal cm−3)½,35 while THF, MeCN, DMF, EtOH, and MeOH are 9.1, 11.9, 12.1, 12.7, and 14.5 (cal cm−3)½, respectively.36 From these reports, the δt value of EtOH is the closest to lignin, followed by DMF, MeCN, MeOH, and THF. It is evident that, apart from EtOH and THF, the order is similar to the result of the solubility test in this work. Although δt value of EtOH is the closest to the lignin, the solubility is very low. Shukry et al. reported that EtOH is not an efficient solvent for the dissolution of lignin owing to a very high hydrogen bonding parameter (δh) value (9.5 (cal cm−3)½).37 Therefore, for THF, it is plausible that the high solubility of lignin might be caused by a relatively low δh value, ca. 3.9 (cal cm−3)½.
3.2. Effect of co-solvent on the generated OH radicals
To investigate the impact of co-solvent on the in situ generation of OH radicals by water, the peak of OH radicals were compared at 3400–3600 G in pure water to that of different water/co-solvent systems using EPR analysis. As shown in Fig. 2, the presence of co-solvent significantly decreased the signals corresponding to OH radicals. The trend of the scavenging ability was observed as: EtOH > MeOH > DMF > MeCN > THF. MeOH, EtOH, DMF, and THF possessed a strong scavenging ability to OH radicals. These co-solvents have at least one O atom, which can form various active chemical bonds (e.g., C–O and H–O) or functional groups such as OH, ester, amide, and cyclic ether groups, which strongly scavenge OH radicals.38 Meanwhile, the scavenging ability of MeCN is primarily caused by the non-conjugated unsaturated bonds in the MeCN molecule (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).38
N).38
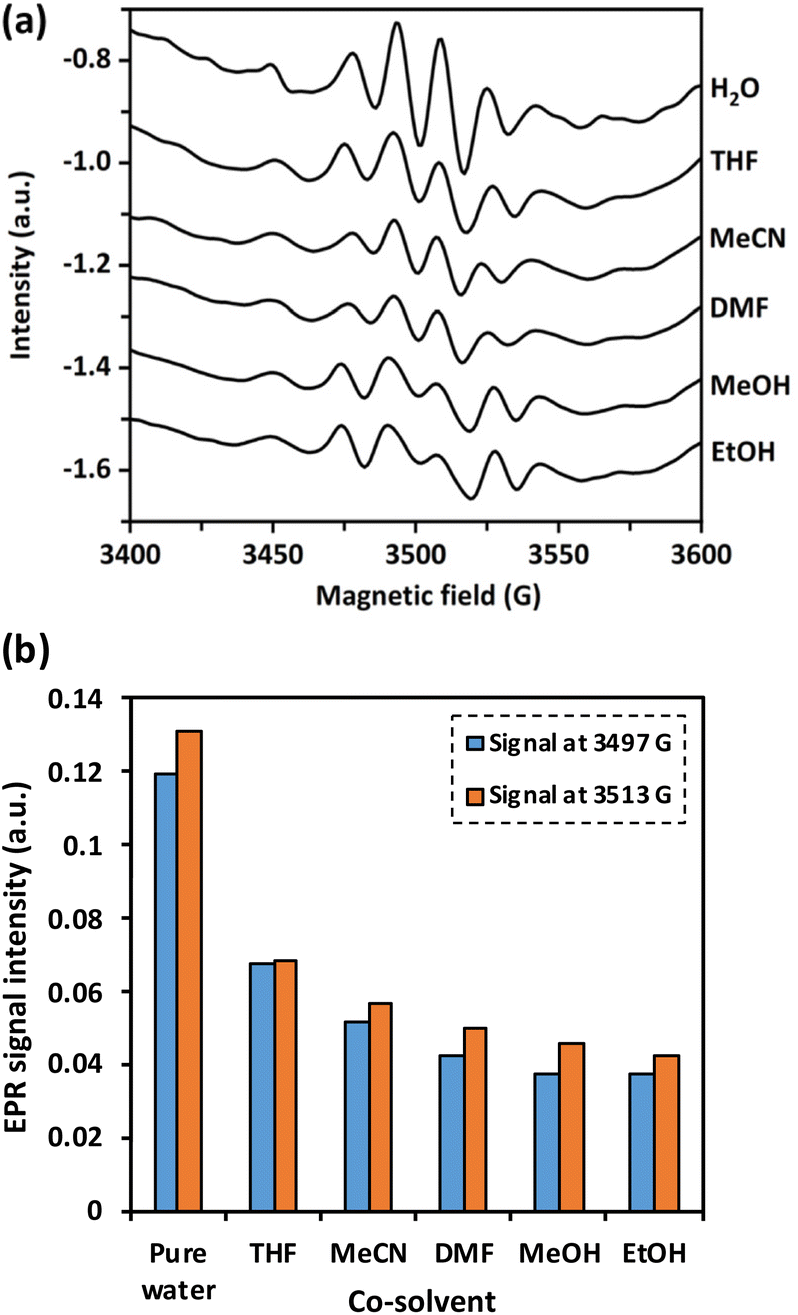 | ||
| Fig. 2 Effect of OH radical scavenger in different co-solvent systems (reaction conditions: co-solvent, 30% vol. in water; H2O2, 500 ppm; DMPO, 500 ppm; and UV light irradiation for 20 min); (a) EPR spectra, (b) intensities of EPR signal corresponding to OH radicals. | ||
To understand the effect of the scavenging ability of co-solvent systems on the degradation of dealkaline lignin, DMF and EtOH were chosen to represent co-solvents with strong scavenging ability, whereas MeCN was selected to represent a co-solvent system with low scavenging ability for the reaction. Although THF is the most common solvent for dissolving lignin, it affects the control of the reactant concentration due to its high vapor pressure (216 h Pa at 25 °C).39 As shown in Fig. 3, dealkaline lignin demonstrates the highest conversion in the MeCN co-solvent system, followed by DMF and EtOH under ambient conditions. This trend aligns generally well with the trend observed in the scavenging of OH radicals from the EPR analysis, suggesting that the concentration of OH radicals play a significant role in lignin conversion.
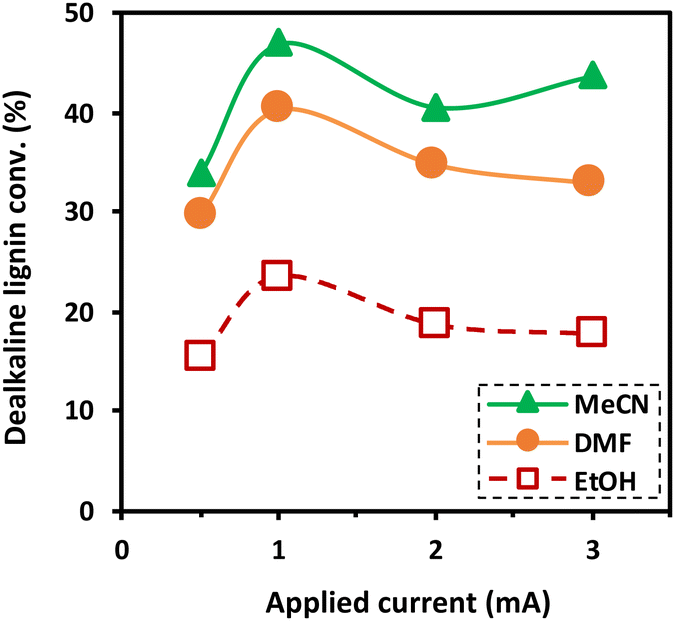 | ||
| Fig. 3 Effect of co-solvents and current density on degradation of dealkaline lignin (reaction conditions: co-solvent, 30% vol. in water; concentration of dealkaline lignin, 50 ppm; and residence time, 100 s). | ||
3.3. Effect of applied current on the degradation of dealkaline lignin
The effect of the applied current on the degradation of dealkaline lignin using different co-solvents was investigated and the results are shown in Fig. 3. The degradation of dealkaline lignin was initially increased when the applied current was raised from 0.5 to 1 mA in all co-solvents. In contrast, it subsequently decreased with further increasing the current from 2 to 3 mA. This clearly indicated that 1 mA is the optimal current for the reaction. The initial increase in lignin degradation is due to the increase in the OH radicals generated in the system, which allows for a higher degradation rate. The decrease in lignin degradation with a further increase in applied current from 1 to 3 mA is plausibly due to water splitting during the reaction. It was noted that bubbles were generated for the reactions performed at 2 and 3 mA, suggesting the generation of hydrogen and oxygen from the water-splitting reaction (eqn (2)).40 The generated gases decreases the contact between liquid and electrodes in the microreactor. Also, the excess current density initiates parasitic reactions such as the OH radicals recombination to form H2O2 (eqn (3)), and the combination of H2O2 with OH radical to H2O (eqn (4)).41,42| H2O → H˙ + OH˙ | (1) |
| H2O → H2 + ½O2 | (2) |
| HO˙ + OH˙ → H2O2 | (3) |
| H2O2 + OH˙ → HO2˙ + H2O | (4) |
The trend in the degradation of dealkaline lignin among the three water/co-solvent systems is consistent and inversely proportional to their scavenging ability. The scavenging ability of EtOH > DMF > MeCN suggests that a higher concentration of OH radical is responsible for efficient lignin degradation.
3.4. Kinetic study
In a microreactor system, the OH radicals are continuously generated at the surface of the anode, leading to full monolayer coverage during the reaction. Concomitantly, the reactants compete to react with the generated OH radicals. Therefore, the reaction rate in a continuous flow microreactor system is limited by the reactant concentration instead of the concentration of OH radicals.24 As a result, the degradation behaviour of dealkaline lignin was expected to follow the pseudo-first-order kinetic model.43,44 To confirm the hypothesis, the effect of the residence time of dealkaline lignin in different water/co-solvent systems was fitted to pseudo-first-order kinetic model (eqn (5)).| ln(Cin/Cout) = k(t) | (5) |
As shown in Fig. 4a, it is evident that the degradation of dealkaline lignin in all water/co-solvent systems followed the pseudo-first-order kinetic model with the correlation coefficient value (R2) of 0.9946, 0.9992, and 0.9956 for MeCN, DMF, and EtOH, respectively. The rate constant of dealkaline lignin degradation was found to be the highest in MeCN (3.3 × 10−3 s−1), followed by DMF (1.2 × 10−3 s−1), and EtOH (9 × 10−4 s−1), which generally follows the scavenging ability toward OH radical in order of EtOH > DMF > MeCN (Fig. 2). Since OH radicals are the key active species for the degradation of dealkaline lignin, the stronger the scavenging ability of a co-solvent, the lower the degradation rate, and vice versa. Therefore, due to higher in situ OH radicals in MeCN, high degradation rate of dealkaline lignin was observed in MeCN than in other co-solvent systems.
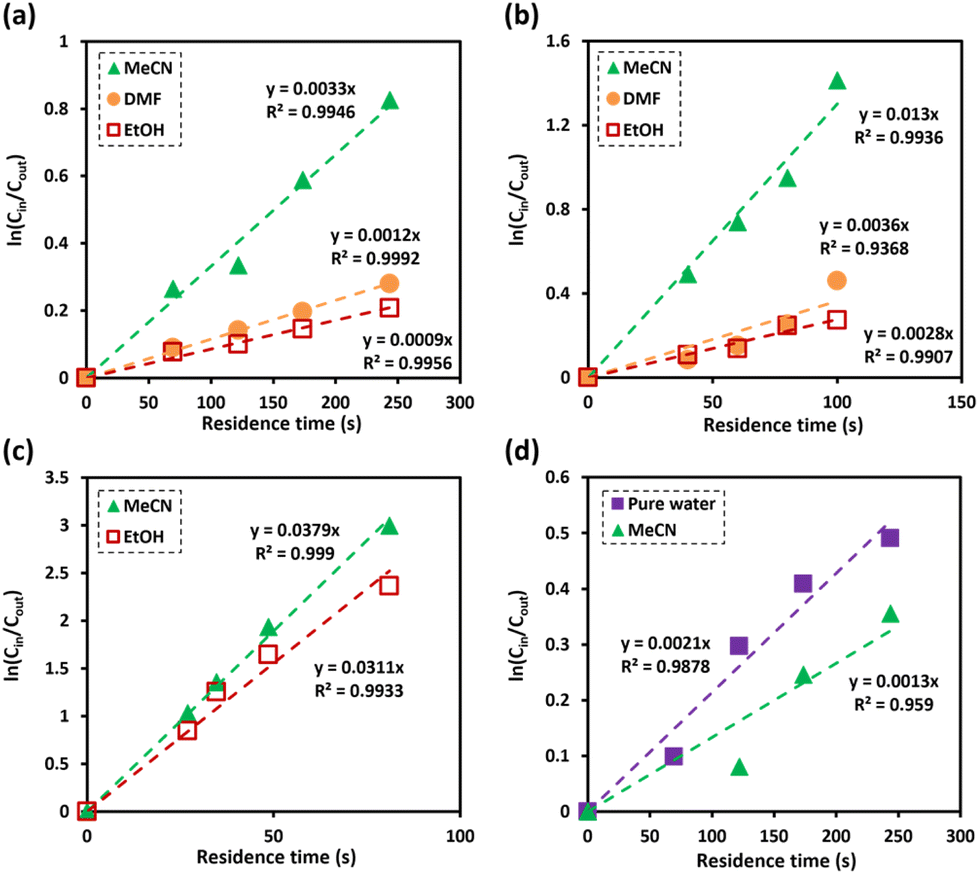 | ||
| Fig. 4 Kinetic analysis of degradation of (a) dealkaline lignin (reaction conditions: co-solvent, 30% vol. in water; dealkaline lignin, 130 ppm; and applied current, 1 mA), (b) PPE (reaction conditions: co-solvent, 30% vol. in water; PPE, 100 ppm; and applied current, 1 mA), (c) BPE (reaction conditions: co-solvent, 30% vol. in water; BPE, 100 ppm; and applied current, 1.25 mA), and (d) alkali lignin (reaction conditions: co-solvent, 30% vol. in water; alkalin lignin, 130 ppm; and applied current, 1 mA). | ||
For further investigation, lignin model compounds, i.e., PPE and BPE representing β-O-4 and α-O-4 linkages in lignin, respectively, were used for electrochemical oxidation. The degradation of both model compounds follows the pseudo-first-order kinetic model quite well (Fig. 4b and c), further confirming that these compounds could represent lignin. The rate constant for PPE degradation was found to be the highest in MeCN (1.3 × 10−2 s−1), followed by DMF (3.6 × 10−3 s−1) and EtOH (2.8 × 10−3 s−1), all of which are higher than the value previously reported45 even though no catalyst was employed in this work. In addition, the solvent used for the lignin depolymerization, and lignin model compounds conversion was recovered by rotary evaporator.
Based on the obtained reaction rate constants, the reaction in water/MeCN and in water/DMF is 4.64 and 1.29 times faster than that in water/EtOH, respectively. Interestingly, the EPR signals representing OH radicals (Fig. 2b) in water/MeCN and in water/DMF are only ca. 1.3 and 1.2 times higher than that in water/EtOH. Hence, the radical scavenging effect is not the only factor that the co-solvent imposes on the EAOP. One of the common reactions taking place during the degradation via OH radical is hydrogen atom abstraction from Cα–OH of PPE. It has been reported that the stabilization of the transition state (TS) formed by the abstraction of the H atom and the promotion of the reaction are driven by hydrogen bonding interactions among the OH radical and surrounding water molecules.46
The ability of the co-solvent to form hydrogen bond and participate in the polarized TS follows the order of EtOH > DMF > MeCN. EtOH has polarized H and O, which can participate in the TS along with water (Fig. S1†). However, the participation of EtOH weakens the TS, because EtOH is not as strongly polarized as water. DMF co-solvent has only polarized O, which can interfere with TS less significantly than EtOH. In contrast, MeCN is not involved in TS due to the absence of polarized H and O; so mainly water participates in TS, resulting in the most stable TS.46 The water participation for polarized TS stabilization for these water/co-solvent systems in the order of EtOH < DMF < MeCN further imposes on degradation rate in the EAOP, in addition to the scavenging ability of the co-solvent.
In contrast, the rate constants for the degradation of BPE in MeCN (3.7 × 10−2 s−1) and EtOH (3.1 × 10−2 s−1) were not significantly different, as shown in Table 1. Unlike PPE, BPE lacks highly polarized hydrogen atom in the –OH functional group to form hydrogen bonding interactions with solvent in TS. As a result, BPE degradation is unaffected by the participating ability of the co-solvents. The effect of the co-solvent in BPE degradation is mainly the result from radical scavenging ability, as supported by the ratio of rate constant in water/MeCN system to that in water/EtOH system (kMeCN/kEtOH = 1.2) that is similar to the ratio of EPR signals in water/MeCN to that in water/EtOH, i.e., 1.3.
| Lignin model compounds | Residence time (s) | Conversion (%) | ||
|---|---|---|---|---|
| EtOH | MeCN | DMF | ||
| All co-solvents used were 30% in H2O; the applied current used for PPE was 1 mA, and for BPE was 1.25 mA (n.a. stands for not attended). | ||||
| PPE | 100 | 23.8 | 75.6 | 36.8 |
| 80 | 21.9 | 61.3 | 22.3 | |
| 60 | 12.8 | 52.1 | 14 | |
| 40 | 10.1 | 38.8 | 7.9 | |
| k/s−1 | 2.8 × 10−3 | 1.3 × 10−2 | 3.6 × 10−3 | |
| BPE | 81 | 90.6 | 94.9 | n.a. |
| 49 | 80.7 | 85.5 | n.a. | |
| 35 | 71.4 | 74.2 | n.a. | |
| 27 | 57.1 | 64.3 | n.a. | |
| k/s−1 | 3.1 × 10−2 | 3.7 × 10−2 | n.a. | |
The degradation of alkaline lignin in the absence of co-solvents exhibited a similar rate constant as dealkaline lignin in MeCN co-solvent system (Fig. 4d). It is worth noting that only water participates in the TS stabilization in the water/MeCN co-solvent system, a similar mechanism as when OH radicals attack alkaline lignin in pure water. When MeCN was presented, the degradation rate of alkaline lignin was decreased by scavenging effect (Fig. 4d). It should be noted that lignin is a complex and heterogeneous polymer, and its degradation involves a wide range of reactions, the degradations of alkaline and dealkaline lignin could not be directly compared (Tables 1, 2 and 3).
| RT/min | MW | Monomer structure |
|---|---|---|
| All the products were confirmed using GC-MS analysis. | ||
| 14.91 | 184 |
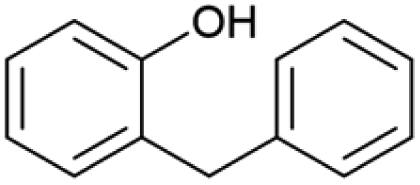
|
| 15.61 | 196 |
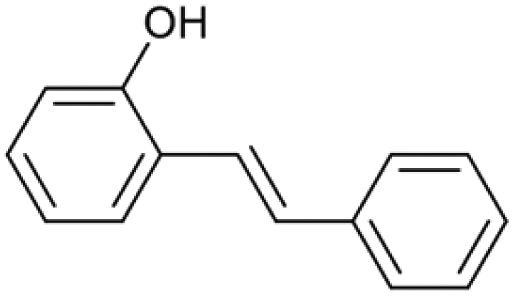
|
| 15.68 | 196 |
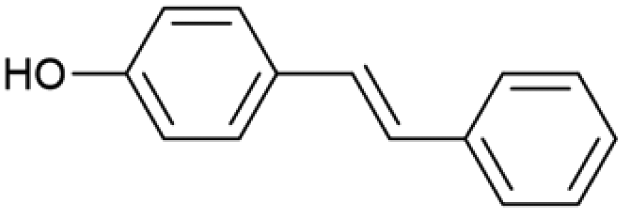
|
| 17.42 | 212 |
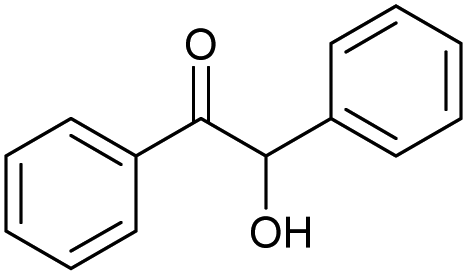
|
| 18.49 | 214 |
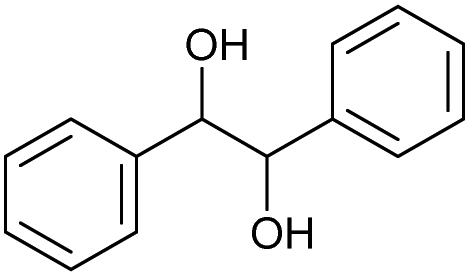
|
| 18.86 | 94 |
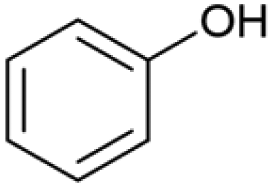
|
| 19.32 | 162 |
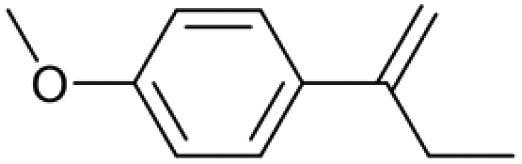
|
| 20.13 | 208 |
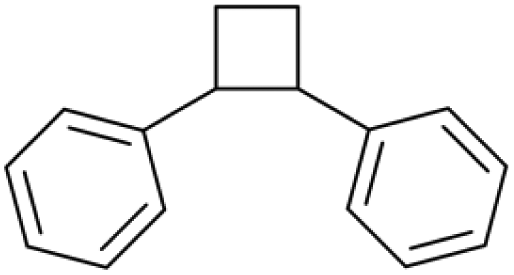
|
| 20.24 | 134 |
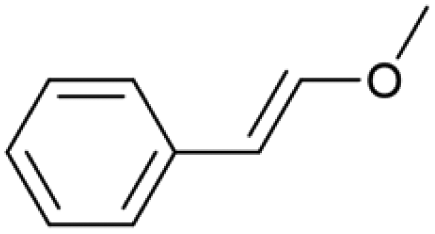
|
| 20.38 | 167 |
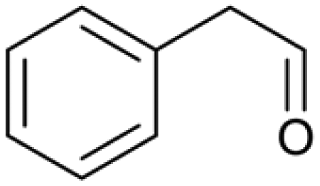
|
| 20.66 | 238 |
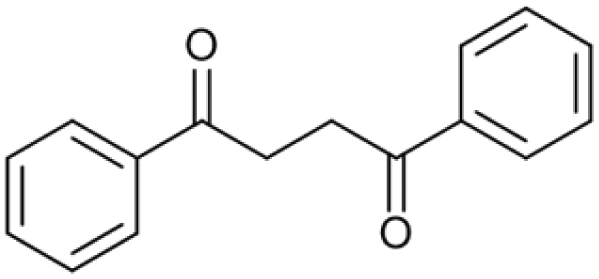
|
| 21.03 | 106 |
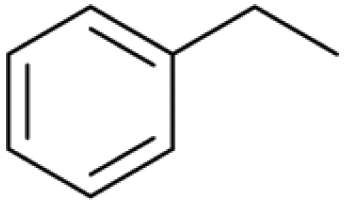
|
| 21.48 | 104 |
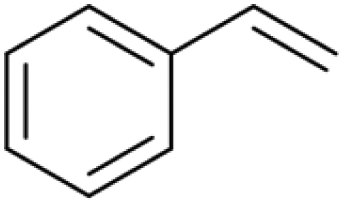
|
| 21.75 | 138 |
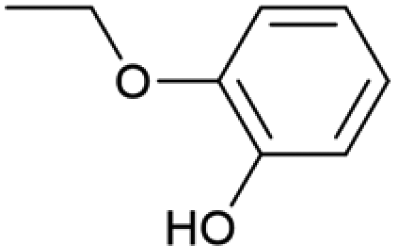
|
| 22.07 | 122 |
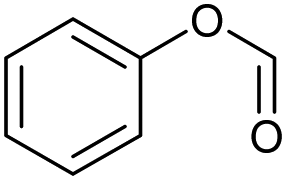
|
| 23.03 | 138 |
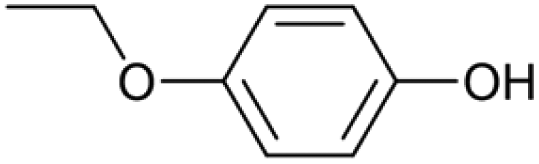
|
| RT/min | MW | Monomer structure |
|---|---|---|
| All the products were confirmed using GC-MS analysis. | ||
| 5.45 | 106 |
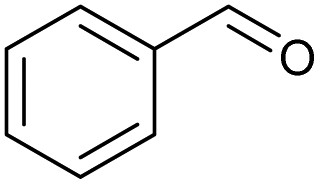
|
| 5.72 | 108 |
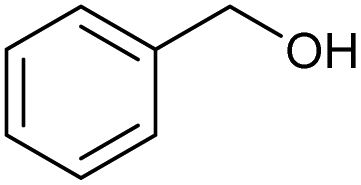
|
| 6.07 | 142 |
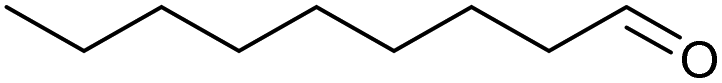
|
| 6.44 | 122 |
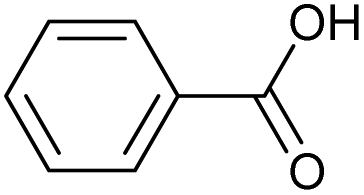
|
| 6.57 | 172 |
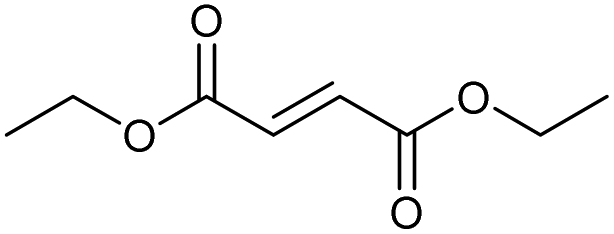
|
| 6.65 | 148 |
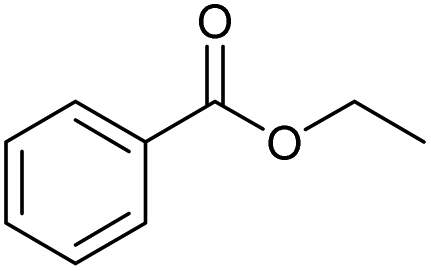
|
| 8.36 | 138 |
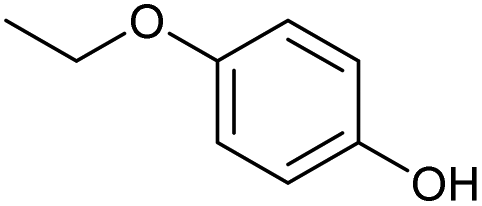
|
Overall, the EAOP of dealkaline lignin is achievable with the aid of co-solvent. The results clearly indicate that the degradation of dealkaline lignin, represented by BPE and PPE model compounds in the continuous flow microreactor systems, follows a pseudo-first-order kinetic. The co-solvent not only scavenges OH radicals, but also affects stabilization of transition state formed between the OH radicals and polarized functional group of lignin. The extent of the stabilization effect depends upon type of linkages in lignin structure.
3.5. Mechanistic insights
The oxidative cleavage of β-O-4 and α-O-4 linkages in lignin has several advantages compared to acid and alkaline hydrolysis, including higher activity under mild reaction conditions, higher yields of low molecular weight products, and higher product solubility in organic solvents.47,48 These advantages make oxidative cleavage a promising method for producing value-added products from lignin. As shown in the previous section, the EAOP of PPE and BPE, which represent β-O-4 and α-O-4 linkages in lignin, achieved high conversion under ambient condition within relatively short residence time. The GC-MS analysis confirmed the production of various aromatic products from PPE and BPE, as presented in Fig. S2 and Fig. S3,† respectively.The β-O-4 linkages are the most prevalent in lignin structure, and breaking them is critical for the efficient depolymerization of lignin into valuable chemicals.49 The cleavage of β-O-4 linkage is more challenging than α-O-4 linkage due to the higher bond dissociation energy (BDE) of β-O-4 linkage. In addition, the β-O-4 linkage has higher steric hindrance due to –OH functional group than the α-O-4 linkage further making it less reactive towards ether bond cleavage. Hence, the present study provides a plausible reaction pathway for the degradation of the PPE model compound.
Electrochemical conversion of PPE involves the transfer of electrons between the electrode, solvent, and PPE reactant, which takes place under an applied electric field. These transferred electrons can directly impact particular chemical bonds within the reaction substrates, leading to their cleavage. The formation of OH radical from the H2O of the water/co-solvent system on the surface of the electrode was confirmed using EPR analysis as active species within the system. The OH radicals then react with PPE, leading to various degradation product formations, as well as convert reactants into specific free radical intermediates, facilitating the continuous conversion of the intermediates. In addition, H2O2 is formed within the system viaeqn (3), which was confirmed by an iodometric technique similar to the one reported by Joshi et al.50 In this case, both OH radicals and H2O2 act as oxidizing agents.51
Scheme 2 presents the mechanism for PPE conversion in the EtOH co-solvent system. The mechanism involved in the Cα–Cβ and Cβ–O–Ar bond cleavage is discussed by understanding the degradation products of PPE. Fig. 5 shows the percentage of the peak area analysed from GC-MS, while Table 2 is the detailed information of all detected products (retention time, molecular weight, and chemical structure). Various co-solvent studies indicated a positive correlation between the effectiveness of bond cleavage and the concentration of in situ generated OH radicals. At relatively low concentration of OH radicals, such as in water/EtOH system, selective attack of the active sites occurred (Table 2). However, at high OH radical concentration, non-selective over-oxidation of the products was observed.
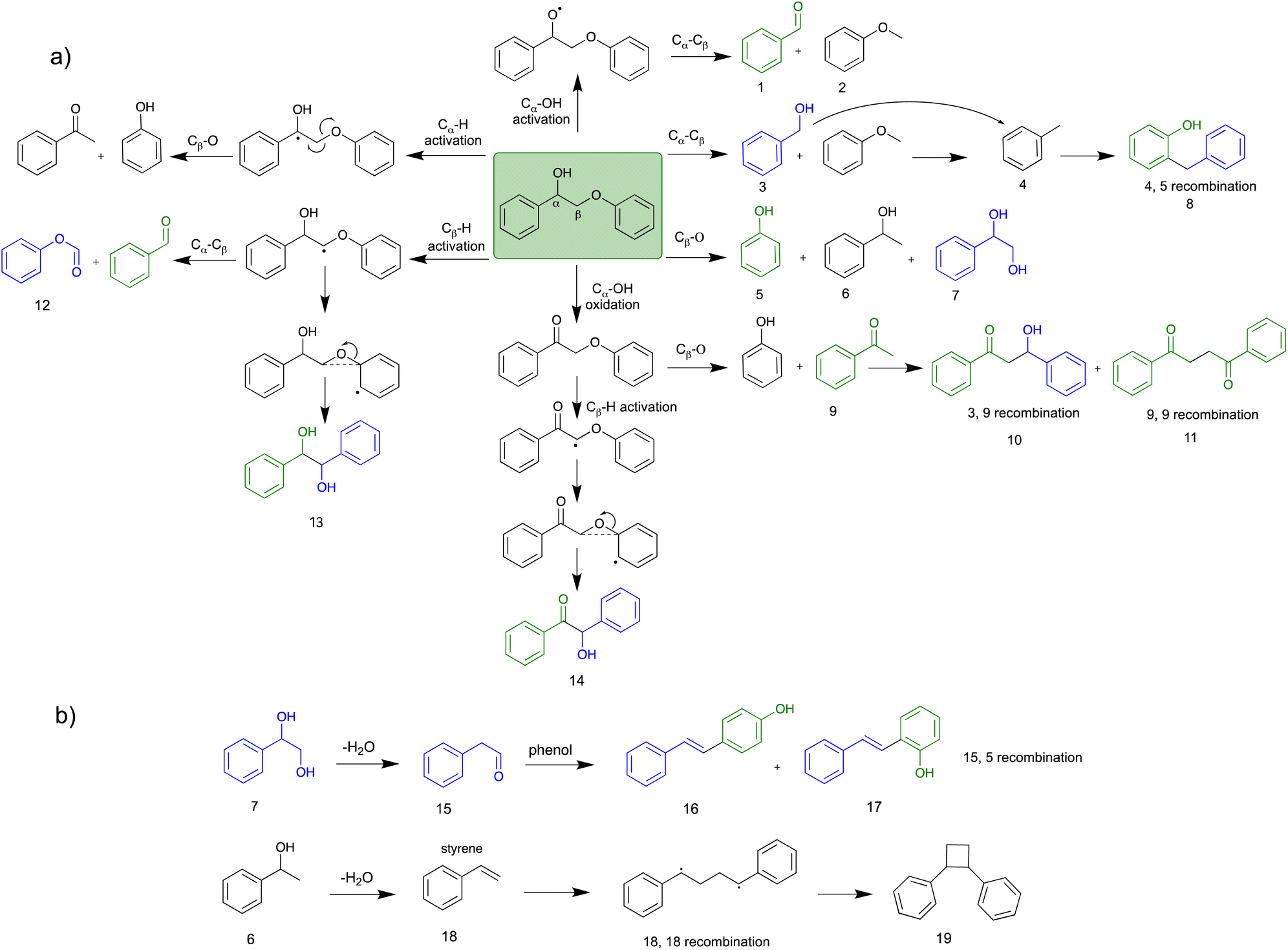 | ||
| Scheme 2 The proposed mechanism for the conversion of PPE using electrochemical oxidation under a continuous flow microreactor in EtOH-water co-solvent system. (a) PPE conversion mechanism and (b) conversion of products 6 and 7. | ||
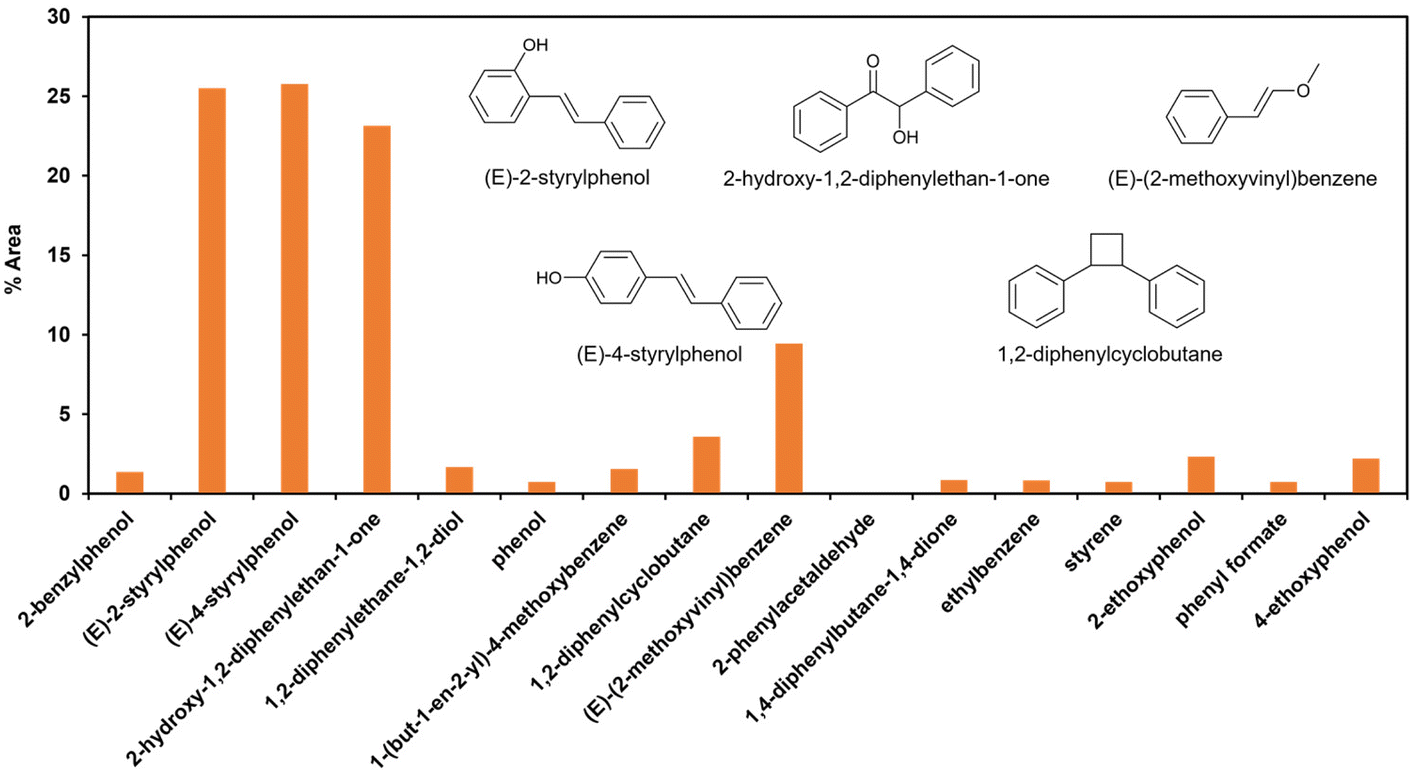 | ||
| Fig. 5 Percentage of peak area of the produced aromatic compound from GC-MS analysis for PPE model compound depolymerization in 30% co-solvent system. | ||
Recent studies have revealed two major pathways for PPE conversion. The first pathway (I), called Cα–OH activation, involves the oxidation of Cα–OH to form Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O via the hydrogen atom abstraction from hydroxyl group, which decreases the BDE of the remaining Cβ–O from 55 to 47 kcal mol−1.52 Benzaldehyde and anisole are formed by Cα–OH activation after further Cα–Cβ cleavage. The second pathway (II), called Cα–H activation, involves the hydrogen atom abstraction from Cα–H to form Cα radicals, which decreases the BDE of Cβ–O to 7.8 kcal mol−1.53 Subsequent Cβ–O homolytic cleavage yields acetophenone (CαO) and phenol, as shown in Scheme 2.
O via the hydrogen atom abstraction from hydroxyl group, which decreases the BDE of the remaining Cβ–O from 55 to 47 kcal mol−1.52 Benzaldehyde and anisole are formed by Cα–OH activation after further Cα–Cβ cleavage. The second pathway (II), called Cα–H activation, involves the hydrogen atom abstraction from Cα–H to form Cα radicals, which decreases the BDE of Cβ–O to 7.8 kcal mol−1.53 Subsequent Cβ–O homolytic cleavage yields acetophenone (CαO) and phenol, as shown in Scheme 2.
Although the reaction pathway (I) has a higher BDE compared to the pathway (II), hydroxyl hydrogen atom of PPE reacts more easily with OH radicals than the H atoms of CH2 and CH of PPE.46 Additionally, the co-solvent system plays a crucial role in the stabilization of the polarized intermediate step during the hydroxyl hydrogen abstraction of Cα–OH by OH radicals. The stabilization of the polarized TS of hydroxyl hydrogen abstraction by water molecules is shown in Fig. S4a.† The hydrogen bonding interaction for the TS is crucial for H abstraction; water can stabilize the developing negative charge on the oxygen of OH radical during TS by acting as a hydrogen donor.46 When EtOH is used as a co-solvent, the hydrogen bonding interaction for polarized TS of the pathway (I) is weaker than with pure water, making the Cα–OH hydrogen abstraction TS less stable, as shown in Fig. S4b.† On the other hand, DMF is unable to form a hydrogen bond with the oxygen of the OH radical in the TS because it lacks polarized hydrogen. DMF can only interact with the hydrogen of OH radical via oxygen of CO. Therefore, with DMF co-solvent, more water molecules participate in stabilizing the polarized TS, promoting Cα–OH activation (Fig. S4c†). Conversely, MeCN does not participate in stabilizing the TS due to the absence of polarized oxygen and hydrogen, resulting in the stabilization of polarized TS only by water, which decreases the free energy of the TS (Fig. S4a†). As a result, the use of MeCN as a co-solvent promotes pathway (I) due to the efficient stabilization of the polarized TS by water than the EtOH co-solvent system. All these results are consistent with kinetic study results for PPE conversion in different co-solvents.
In addition to Cα–OH activation, the Cα–OH can also be oxidized. The resulting oxidized product can lead to subsequent Cβ–H activation and rearrangement to form product 14. The high concentration of product 14 suggests that this is the major pathway (Fig. S2† and Scheme 2). On the other hand, if the Cα–OH oxidation is followed by Cβ–O cleavage, it results in the production of phenol and acetophenone. Acetophenone self-recombine and combine with benzyl alcohol to produce products 11 and 10 dimers, respectively.
The homolytic cleavage of Cα–Cβ of PPE results in the production of benzyl alcohol and anisole; benzyl alcohol further converted to toluene radical, which subsequently recombined with phenol intermediates to yield product 8, as shown in Scheme 2 and Table 2. However, cleavage of Cα–Cβ is not prominent due to the higher BDE of Cα–Cβ bond.54 Therefore, the lower concentration of product 8 was observed, as shown in Fig. S2.† The Cβ–O homolytic cleavage of PPE leads to the formation of phenol, 1-phenylethanol (6), and 1-phenyl-1,2-ethanediol (7). The Cβ–O homolytic cleavage is easier under oxidation conditions.55 Lastly, the Cβ–H activation of PPE results in the production of product 13 after molecular rearrangement and leads to the formation of benzaldehyde and phenyl-formate (12) after Cα–Cβ cleavage.
The degradation products listed in Table 2 indicate a complex reaction network in the EAOP of PPE. It is important to note that additional reactions can occur; the degraded intermediates interact with each other to form products mentioned in Scheme 2b. Product number 7, upon dehydration, followed by keto–enol tautomerism converts into 2-phenylacetaldehyde (15), which reacts with phenol to form product number 16 and 17. Furthermore, product 6 undergoes dehydration-producing styrene (18); self-recombination of styrene results in the formation of product 19.56 The products 16, 17, and 19 mentioned in Scheme 2(b) and Table 2 were confirmed using GC-MS analysis (Fig. S2†). The detection of radical intermediates using GC-MS analysis is limited due to the recombination of products and intermediates containing radicals forming dimeric products, as the radical intermediates are highly unstable. Although more research is needed to fully understand the reaction mechanisms involved in the lignin depolymerization using EAOP, the employment of EAOP shows a promising approach for lignin depolymerization into high-value products. The products formed with the BPE lignin model compound are presented in Table 3.
The final role of the co-solvent was observed from the degradation intermediates. Although it was not apparent when EtOH was used, N-containing products were detected from water/MeCN and water/DMF systems (Tables S3 and S4†). Since the N-containing solvents, such as MeCN and DMF, were unstable under an applied electric field during electrochemical conversion, radicals could be formed from the co-solvent. Mitroka et al. reported that the OH radical reacts with MeCN forming ˙CH2CN.46 These radicals are not as strong oxidizing agents as OH radicals; hence they should not significantly alter the degradation pathways. Nevertheless, they could recombine with the radicals formed from PPE degradation.
To understand the lignin depolymerization, the dealkaline lignin and depolymerized products of dealkaline lignin were characterized using two-dimensional heteronuclear single quantum coherence (2D-HSQC) NMR spectroscopy (Fig. 6). The HSQC NMR analysis was performed using DMSO-d6 solvent. For dealkaaline lignin, the signals of the aliphatic carbon chains were observed at (δC/δH = 15–25/0.5–2.0 ppm; Fig. 6a). In addition, the –OMe commonly due to coniferyl (G) and sinapyl alcohols (S), were observed at δC/δH = 55/3.73 ppm.57 After lignin depolymerization, the –OMe signal disappeared may be due to the demethoxylation reaction by OH radical led to products without –OMe functional groups.54 The dealkaline lignin showed the signals for Aγ (Cγ–Hγ) in β-O-4′ substructures (A) (δC/δH = 60/3.38 ppm),58 Cβ (Cβ–Hβ–C) in β-5′ phynylcoumaran (C) (δC/δH = 54/3.44 ppm), Bγ (Cγ–Hγ–B) in β–β′ resinol substructures (B) (δC/δH = 71.7/3.7 ppm) and Cγ (Cγ–Hγ–C) in β-5′ phenylcoumaran (δC/δH = 63/3.6 ppm),59 while similar signals were absent after depolymerization confirming the cleavage of β-O-4′ and resinol substructures.
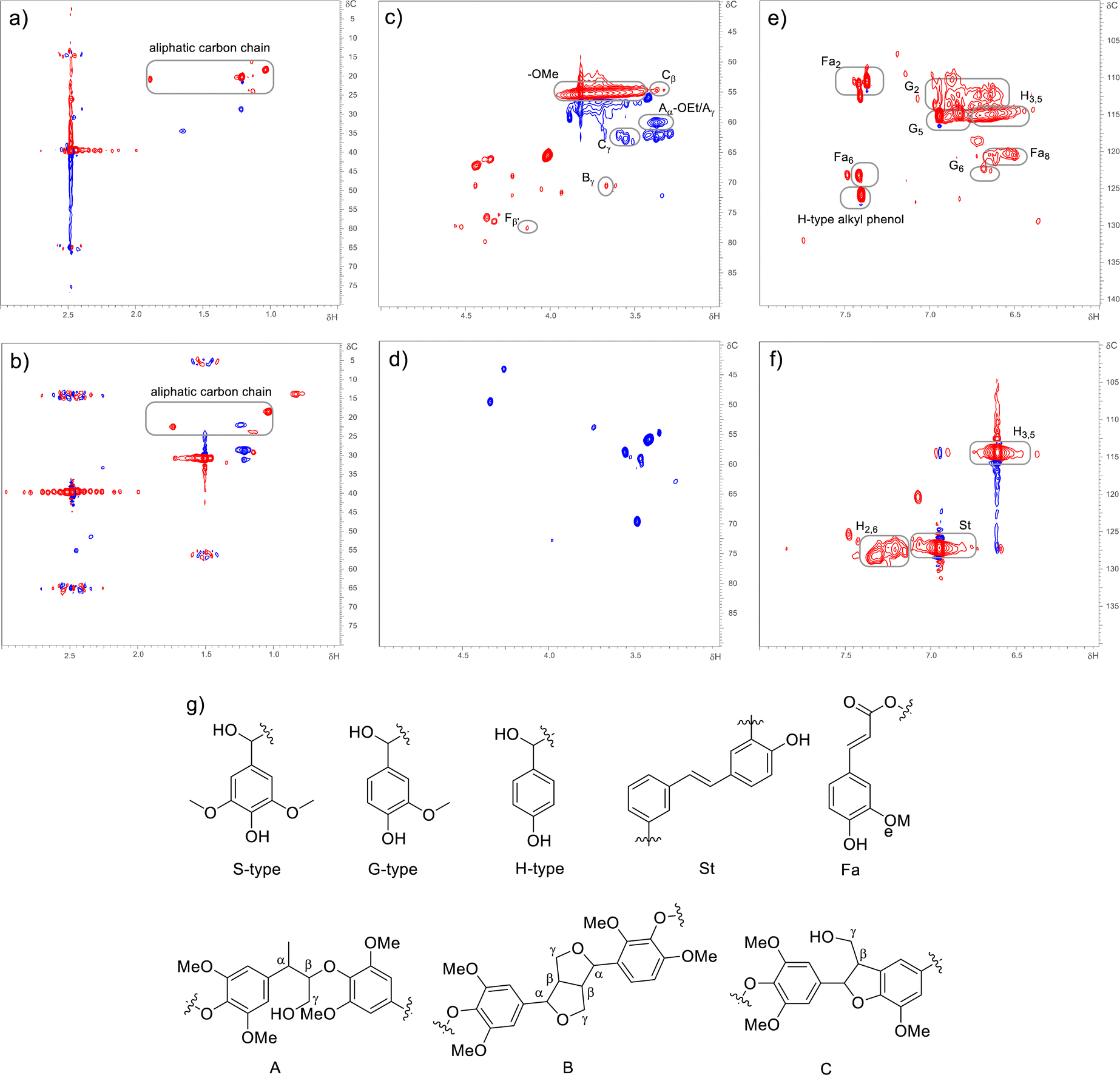 | ||
| Fig. 6 2D-HSQC NMR (800 MHz) analysis of dealkaline lignin (a, c, and e) and depolymerization products of dealkaline lignin (b, d, and f). Reaction conditions: MeCN, 30% v in water; dealkaline lignin, 130 ppm; and applied current, 1 mA. Sub-aromatic units of lignin structure (g). | ||
In the aromatic region of dealkaline lignin (δC/δH = 100–130/6–8 ppm (Fig. 6e)) signals for Fa2, Fa6, Fa8 (ferulate moieties), alkyl phenol; G2 (C2–H2), G5 (C5–H5) and G6 (C6–H6) in guaiacyl units (G) were observed. On the other hand, after depolymerization, these signals disappeared (Fig. 6f), which confirms the depolymerization of dealkaline lignin by EAOP. The aromatic compounds after depolymerization (δC/δH = 100–130/6–8 ppm; Fig. 6f) suggest that substitute groups on the aromatic ring were removed by bond cleavage. The presence of H3,5 (C3–H3 and C5–H5) and H2,6 (C2–H2 and C6–H6) in p-hydroxyphenyl units (H) signals for depolymerized products suggests the products contain p-hydroxyphenyl units.60 The β-O-4 model compound conversion showed the formation of stilbene (St) type of products (Scheme 2; product no. 16 and 17), which corroborated by 2D-HSQC NMR analysis where the signal due to St substructure was observed (δC/δH = 126.5/6.9 ppm) for depolymerized products. However, the St substructure signal was not present for dealakaline lignin. Overall, 2D-HSQC NMR analysis confirmed the depolymerization of dealkaline lignin by EAOP.
The 2D-HSQC NMR results also indicated the lignin depolymerization by the cleavage of the β-O-4 bond. The mechanism for the β-O-4 lignin model compound presented in Scheme 2 revealed that few products were obtained by the cleavage of the β-O-4 bond (product numbers 5, 6, 7, and 9 in Scheme 2). However, the concentrations of these products were lower because these products were involved in the recombination reactions. The direct β-O-4 cleavage is more common for acid-catalyzed lignin depolymerization reactions than OH radical-catalyzed lignin depolymerization reactions.
4. Conclusions
This work demonstrated an efficient EAOP depolymerizing alkaline and dealkaline lignin and lignin model compounds (BPE and PPE) under ambient conditions in a continuous flow microreactor system. As dealkaline lignin is insoluble in water, the effect of solubility in various co-solvent systems, including MeOH, EtOH, MeCN, THF, and DMF, on lignin depolymerization was investigated. The effect of co-solvent on the formation of OH radicals showed higher lignin conversion with MeCN co-solvent system due to the low OH radical scavenging ability of MeCN. The kinetic study revealed a higher rate constant for PPE and dealkaline lignin in the MeCN co-solvent. However, for BPE, nearly a similar rate constant was observed in EtOH and MeCN co-solvent systems. The detailed mechanism for OH radicals involved in the Cα–Cβ and Cβ–O–Ar bond cleavage was discussed by understanding the degradation products of PPE using GC-MS analysis. In addition, 2D-HSQC NMR analysis of dealkaline lignin and depolymerized products confirm the efficient lignin depolymerization by EAOP. The lignin conversion using continuous flow EAOP under ambient conditions represents a promising approach for lignin valorization offering advantages over conventional methods and enabling the efficient production of value-added chemicals from lignin. Furthermore, the degradation reaction kinetics and mechanism were also elucidated using BPE and PPE model compounds.Author contributions
Conceptualization: LWA, BMM, VP, KCWW; formal analysis: LWA, BMM; funding acquisition: KCWW, VP; investigation: LWA, BMM, KL; methodology: BMM, LWA, VP, KCWW; project administration: KCWW, VP; supervision: BMM, VP, KCWW; validation: LWA, BMM, KL; visualization: LWA, BMM, KL; writing-original draft: LWA, BMM; writing-review & editing: BMM, LWA, VP, KCWW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by Thailand Science Research and Innovation Fund Chulalongkorn University (CU_FRB65_bcg (21)_135_21_01). We acknowledge the Ministry of Science and Technology (MOST), Taiwan for research funding (108-2638-E-002-003-MY2).References
- L. Serrano, R. Luque, B. F. Sels and I. Agirre, Lignin Chemistry, Springer, 2020 Search PubMed.
- B. M. Matsagar and P. L. Dhepe, New J. Chem., 2017, 41, 6137–6144 RSC.
- B. M. Matsagar, R.-X. Yang, S. Dutta, Y. S. Ok and K. C. W. Wu, J. Mater. Chem. A, 2021, 9, 3703–3728 RSC.
- L. A. Z. Torres, A. L. Woiciechowski, V. O. de Andrade Tanobe, S. G. Karp, L. C. G. Lorenci, C. Faulds and C. R. Soccol, J. Cleaner Prod., 2020, 263, 121499 CrossRef.
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef CAS PubMed.
- S. Van den Bosch, S.-F. Koelewijn, T. Renders, G. Van den Bossche, T. Vangeel, W. Schutyser and B. Sels, Lignin Chem., 2020, 129–168 Search PubMed.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS.
- B. M. Matsagar, Z.-Y. Wang, C. Sakdaronnarong, S. S. Chen, D. C. W. Tsang and K. C.-W. Wu, ChemCatChem, 2019, 11, 4604–4616 CrossRef CAS.
- Y.-T. Liao, B. M. Matsagar and K. C. W. Wu, ACS Sustainable Chem. Eng., 2018, 6, 13628–13643 CrossRef CAS.
- Y. Cao, S. S. Chen, S. Zhang, Y. S. Ok, B. M. Matsagar, K. C. W. Wu and D. C. W. Tsang, Bioresour. Technol., 2019, 291, 121878 CrossRef CAS PubMed.
- S. Sethupathy, G. Murillo Morales, L. Gao, H. Wang, B. Yang, J. Jiang, J. Sun and D. Zhu, Bioresour. Technol., 2022, 347, 126696 CrossRef CAS.
- R. K. Sharma, J. B. Wooten, V. L. Baliga, X. Lin, W. G. Chan and M. R. Hajaligol, Fuel, 2004, 83, 1469–1482 CrossRef CAS.
- Y. Han, F. Chen, T. Ma, H. Gong, K. W. Al-Shwafy, W. Li, J. Zhang and M. Zhang, Ind. Eng. Chem. Res., 2019, 58, 23014–23024 CrossRef CAS.
- M. Brebu and C. Vasile, Cellul. Chem. Technol., 2010, 44, 353 CAS.
- Z. Chen and C. Wan, Renewable Sustainable Energy Rev., 2017, 73, 610–621 CrossRef CAS.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS PubMed.
- C. Huang, X. Jiang, X. Shen, J. Hu, W. Tang, X. Wu, A. Ragauskas, H. Jameel, X. Meng and Q. Yong, Renewable Sustainable Energy Rev., 2022, 154, 111822 CrossRef CAS.
- T. K. Kirk and R. L. Farrell, Annu. Rev. Microbiol., 1987, 41, 465–501 CrossRef CAS PubMed.
- T. M. Budnyak, A. Slabon and M. H. Sipponen, ChemSusChem, 2020, 13, 4344–4355 CrossRef CAS.
- C. A. Gasser, G. Hommes, A. Schäffer and P. F.-X. Corvini, Appl. Microbiol. Biotechnol., 2012, 95, 1115–1134 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- D. Mukhopadhyay, C. Chang, M. Kulsreshtha and P. Gupta, Int. J. Biol. Macromol., 2023, 227, 307–315 CrossRef CAS PubMed.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13, 4318–4343 CrossRef CAS.
- F. H. Adnan, M. N. Pons and E. Mousset, Electrochem. Sci. Adv., 2022, e2100210 Search PubMed.
- O. Scialdone, E. Corrado, A. Galia and I. Sirés, Electrochim. Acta, 2014, 132, 15–24 CrossRef CAS.
- O. Scialdone, A. Galia and S. Sabatino, Appl. Catal., B, 2014, 148, 473–483 CrossRef.
- S. Sabatino, A. Galia and O. Scialdone, ChemElectroChem, 2016, 3, 83–90 CrossRef CAS.
- W. Khongthon, G. Jovanovic, A. Yokochi, P. Sangvanich and V. Pavarajarn, Chem. Eng. J., 2016, 292, 298–307 CrossRef CAS.
- O. Scialdone, A. Galia, C. Guarisco and S. La Mantia, Chem. Eng. J., 2012, 189, 229–236 CrossRef.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2015, 5, 365–379 CrossRef CAS.
- O. Y. Abdelaziz and C. P. Hulteberg, Waste Biomass Valorization, 2017, 8, 859–869 CrossRef CAS.
- I. Morrison, J. Sci. Food Agric., 1972, 23, 455–463 CrossRef CAS.
- J. Howell, M. Roesing and D. Boucher, J. Phys. Chem. B, 2017, 121, 4191–4201 CrossRef CAS PubMed.
- L. P. Novo and A. A. S. Curvelo, Ind. Eng. Chem. Res., 2019, 58, 14520–14527 CrossRef CAS.
- J. Sameni, S. Krigstin and M. Sain, BioResources, 2017, 12, 1548–1565 CrossRef CAS.
- A. F. Barton, Chem. Rev., 1975, 75, 731–753 CrossRef CAS.
- N. Shukry, S. Fadel, F. Agblevor and S. El-Kalyoubi, J. Appl. Polym. Sci., 2008, 109, 434–444 CrossRef CAS.
- X. Li, Food Chem., 2013, 141, 2083–2088 CrossRef CAS.
- D. W. Scott, J. Chem. Thermodyn., 1970, 2, 833–837 CrossRef CAS.
- B. You, M. T. Tang, C. Tsai, F. Abild-Pedersen, X. Zheng and H. Li, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- H. Krause, B. Schweiger, E. Prinz, J. Kim and U. Steinfeld, J. Electrost., 2011, 69, 333–338 CrossRef CAS.
- N. Borras, R. Oliver, C. Arias and E. Brillas, J. Phys. Chem. A, 2010, 114, 6613–6621 CrossRef CAS.
- R. Tolba, M. Tian, J. Wen, Z.-H. Jiang and A. Chen, J. Electroanal. Chem., 2010, 649, 9–15 CrossRef CAS.
- K. Pan, M. Tian, Z.-H. Jiang, B. Kjartanson and A. Chen, Electrochim. Acta, 2012, 60, 147–153 CrossRef CAS.
- L. Jing, M. Xie, Y. Xu, C. Tong, H. Zhao, N. Zhong, H. Li, I. D. Gates and J. Hu, Appl. Catal., B, 2022, 318, 121814 CrossRef CAS.
- S. Mitroka, S. Zimmeck, D. Troya and J. M. Tanko, J. Am. Chem. Soc., 2010, 132, 2907–2913 CrossRef CAS PubMed.
- R. Panyadee, P. Posoknistakul, W. Jonglertjunya, P. Kim-Lohsoontorn, N. Laosiripojana, B. M. Matsagar, K. C. W. Wu and C. Sakdaronnarong, ACS Sustainable Chem. Eng., 2018, 6, 16896–16906 CrossRef CAS.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, C. A. E. Costa, A. E. Rodrigues, C. P. Hulteberg and A. Riisager, ChemSusChem, 2022, 15, e202201232 CrossRef CAS PubMed.
- M. Garedew, F. Lin, B. Song, T. M. DeWinter, J. E. Jackson, C. M. Saffron, C. H. Lam and P. T. Anastas, ChemSusChem, 2020, 13, 4214–4237 CrossRef CAS.
- A. A. Joshi, B. R. Locke, P. Arce and W. C. Finney, J. Hazard. Mater., 1995, 41, 3–30 CrossRef CAS.
- I. Janik, D. M. Bartels and C. D. Jonah, J. Phys. Chem. A, 2007, 111, 1835–1843 CrossRef CAS.
- J. Lin, X. Wu, S. Xie, L. Chen, Q. Zhang, W. Deng and Y. Wang, ChemSusChem, 2019, 12, 5023–5031 CrossRef CAS PubMed.
- J. Xu, F. Lin, J. Wang and Y. Wang, Chem. Phys. Lett., 2022, 805, 139981 CrossRef CAS.
- K. Wu, M. Cao, Q. Zeng and X. Li, Green Energy Environ., 2023, 8, 383–405 CrossRef CAS.
- S. Kim, S. C. Chmely, M. R. Nimlos, Y. J. Bomble, T. D. Foust, R. S. Paton and G. T. Beckham, J. Phys. Chem. Lett., 2011, 2, 2846–2852 CrossRef CAS.
- K. S. Khuong, W. H. Jones, W. A. Pryor and K. N. Houk, J. Am. Chem. Soc., 2005, 127, 1265–1277 CrossRef CAS.
- B. M. Matsagar, Z. Y. Wang, C. Sakdaronnarong, S. S. Chen, D. C. Tsang and K. C. W. Wu, ChemCatChem, 2019, 11, 4604–4616 CrossRef CAS.
- J.-P. Guo, F.-J. Liu, L.-L. Bie, X.-G. Si, Y.-H. Li, P. Song, N. Liu, Y.-P. Zhao, Z.-X. Huang, J.-P. Cao and X.-Y. Wei, Fuel, 2022, 316, 123338 CrossRef CAS.
- T.-Q. Yuan, S.-N. Sun, F. Xu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 10604–10614 CrossRef CAS PubMed.
- Z. Wang and P. J. Deuss, ChemSusChem, 2021, 14, 5186–5198 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03066k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |