
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-efficiency unbiased water splitting with photoanodes harnessing polycarbazole hole transport layers†
Jin Wook
Yang
a,
Su Geun
Ji
a,
Chang-Seop
Jeong
b,
Jaehyun
Kim
a,
Hee Ryeong
Kwon
a,
Tae Hyung
Lee
a,
Sol A
Lee
a,
Woo Seok
Cheon
a,
Seokju
Lee
a,
Hyungsoo
Lee
b,
Min Sang
Kwon
 a,
Jooho
Moon
*b,
Jin Young
Kim
*a and
Ho Won
Jang
*ac
a,
Jooho
Moon
*b,
Jin Young
Kim
*a and
Ho Won
Jang
*ac
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea. E-mail: jykim.mse@snu.ac.kr; hwjang@snu.ac.kr
bDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: jmoon@yonsei.ac.kr
cAdvanced Institute of Convergence Technology, Seoul National University, Suwon 16229, Republic of Korea
First published on 22nd February 2024
Abstract
The construction of uniform heterojunctions for effective hole transport in a nanoporous BiVO4 photoanode is highly challenging, despite its promise for unbiased photoelectrochemical (PEC) water splitting. Herein, we grew a nanoscale conjugated polycarbazole framework (CPF-TCB) on nanoporous Mo:BiVO4 and exhaustively assessed its hole extraction capability. Type II band alignment in the CPF-TCB/Mo:BiVO4 heterostructure enabled effective hole transport by suppressing charge recombination, enhancing both the fill factor and stability of the photoanode after loading a cocatalyst. The NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode generates a superlative water oxidation photocurrent density of 6.66 mA cm−2 at 1.23 V versus the reversible hydrogen electrode. By combining the photoanode with a perovskite photocathode and a perovskite/Si solar cell, two types of PEC tandem devices exhibit solar-to-hydrogen conversion efficiencies of 6.75% and 9.02%, which are the topmost records under tandem illumination mode. This work provides a significant step for designing high-performance organic–inorganic hybrid photoelectrodes for solar hydrogen production.
Broader contextPhotoelectrochemical (PEC) water splitting is a promising technology to produce green hydrogen and reduce the carbon footprint. High solar-to-hydrogen (STH) conversion efficiency and long-term stability of tandem devices must be achieved for the commercialization of the PEC approach. Bismuth vanadate (BiVO4), providing a high photovoltage, has been considered an excellent photoanode material for tandem devices, but nanostructuring and hole transport layers (HTL) are necessary to overcome its charge recombination issue. However, it is very challenging not only to explore HTL materials with suitable band structures for BiVO4, but also to uniformly coat it on the nanostructure. Herein, we demonstrate a conjugated polycarbazole framework (CPF-TCB) as a new hole extraction layer for a nanoporous molybdenum-doped BiVO4 photoanode for unassisted solar hydrogen production. The intimately contacted CPF-TCB/Mo:BiVO4 heterostructure forms a type-II band alignment, reinforcing charge separation. After loading a cocatalyst, the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode combined with a perovskite photocathode and a perovskite/Si photovoltaic cell affords remarkable STH conversion efficiencies of 6.75% and 9.02%, respectively. The high-performance tandem devices demonstrate the prospective potential of organic–inorganic hybrid photoanodes for unassisted solar hydrogen production. |
Introduction
Photoelectrocatalysis, which is more efficient than photovoltaic–electrocatalysis in the manufacturing process and photocatalysis in yield, is highly promising for solar hydrogen production.1–3 The trade-off between photovoltage and photocurrent based on the band gap of a single photoelectrode requires an electrical bias to exceed the water splitting voltage, which is a challenge in the photoelectrochemical (PEC) approach.4 To overcome this challenge, unassisted water splitting using tandem devices comprising dual light absorbers has been attempted to maximize photovoltage via light harvesting.5 In terms of solar-to-hydrogen (STH) conversion efficiency for tandem devices, bismuth vanadate (BiVO4) is an essential photoanode material owing to its suitable band gap, enabling both high photovoltage and absorption spectrum separation with posterior absorber materials.6 Nevertheless, a short carrier diffusion length (∼70 nm) incurs charge recombination,7 lowering its photocurrent density far below the theoretical value of 7.5 mA cm−2.In addition to the nanostructuring of BiVO4 to reduce the diffusion path, it is necessary to introduce charge transport layers to reinforce charge separation.8 Specifically, since hole transport is more susceptible to being blocked by potential barriers along the carrier passage, it is necessary to develop a competent hole transport layer (HTL) to improve the fill factor (FF) of BiVO4 in order to secure high operating points of tandem devices. Unlike the various metal oxides, such as SnO2,9 WO3,10 TiO2,11 and In2O3,12 that plainly construct type II band alignments favorable to electron transport under BiVO4, the inorganic material pool for HTL is highly limited. Owing to its stringent requirements such as a low band gap, high durability, and more negative conduction band edge than that of BiVO4, only few types of research on HTL have been reported. Quantum dot synthesis from carbon13 and black phosphorous14 or elemental doping for carbon nitride15 has been attempted to form staggered band edges onto BiVO4. Although these 2-step approaches of coating after phase formation are advantageous for ensuring conductivity, possible degeneration in PEC performances by clogging the pores of nanostructured BiVO4 or blocking light also exists.
Conjugated polymers have been widely investigated in organic photovoltaics16 and perovskite solar cells.17 Their fascinating nature of tailored band structures stems from the diversity of functional groups.18 In addition, high electrical conductivity is retained without post-processing since the π-electrons delocalized by alternating single bonds and double bonds facilitate charge flow.19 These delocalized electrons in the π-conjugated backbone also contribute to electrochemical stability by preventing charge accumulation. Despite these favorable features for HTL, few studies have been applied to nanostructured photoelectrodes owing to the difficulty of their direct growth without reducing the specific surface area. Forming a heterojunction between nanoporous BiVO4 and conjugated polymers with optimal band structures through conformal growth provides a breakthrough to the chronic issues in the efficiency and durability of photoanodes.
Here, we demonstrate the hole extraction of a conjugated polycarbazole framework (CPF-TCB) layer on the nanoporous molybdenum-doped BiVO4 (Mo:BiVO4) photoanode. Electropolymerization is utilized to conformally deposit CPF-TCB on Mo:BiVO4 by elaborately controlling the growth via sweep cycle, and their uniform thickness is identified by transmission electron microscopy (TEM). After coating a tannic acid-based NiFeCoOx cocatalyst, the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode exhibits exceptional PEC water oxidation with a high photocurrent density of 6.66 mA cm−2 at 1.23 V versus a reversible hydrogen electrode (VRHE). X-ray spectroscopic analyses reveal the electronic environment of CPF-TCB/Mo:BiVO4, and density functional theory (DFT) calculations suggest a theoretical basis for the charge transport within the heterojunction. Systematic studies on the charge carrier dynamics verify the hole transport capability of CPF-TCB, and confirm that the enhanced PEC performances originate from the type II band structures. For unbiased solar hydrogen production, two types of PEC tandem devices using the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode with a perovskite photocathode and a perovskite/Si solar cell are fabricated. These tandem devices measured under tandem illumination mode (mode T) attain excellent STH conversion efficiencies of 6.75% and 9.02%, respectively, which are the foremost records among not only BiVO4-based, but also mode T tandem devices. This study reveals that CPF-TCB is a key material in photoanodes for high-efficiency and high-durability PEC tandem devices.
Results and discussion
The fabrication processes of photoanodes are schematically illustrated in Fig. S1 (ESI†). All designs of the photoanode were carried out with a focus on efficiently separating and transferring charges generated in BiVO4. Firstly, BiVO4 was synthesized from electrodeposited BiOI nanoplates as a precursor to possess nanopores, providing a large specific surface area and short diffusion path.6 During the annealing process, the removal of surface trap sites in BiVO4 was induced by molybdenum doping.15 For maximization of the surface area and electron extraction, vertical nanorod-type SnO2 was introduced as an electron transport layer (ETL) under the photoanode.9 The thin CPF-TCB layer was directly synthesized onto the surface of Mo:BiVO4 using electropolymerization, and the CPF-TCB/Mo:BiVO4 photoanode was dip-coated with tannic acid-based NiFeCoOx acting as a cocatalyst. Conformal coating of conductive polymers on BiVO4 without filling its nanopores is highly challenging. As illustrated in Fig. 1(a), the electropolymerization we adopted is sophisticated in that the monomers can intimately contact all surfaces of the photoanode. As a building block to construct the conjugated polycarbazole framework, we employed 1,3,5-tris(N-carbazolyl)benzene (TCB) which contains carbazole. Among various aromatic units, such as benzene, thiophene, and pyridine, carbazole applied in the conjugated polymer backbone contributes to the electrochemical stability because of its electrophilic substitution in the nitrogen atom and the aromatic ring.20,21 The CPF-TCB films were electropolymerized on the Mo:BiVO4 photoanode by linear sweep voltammetry, and their thickness was controlled by the sweep cycle. In the current density–potential (J–V) curves (Fig. S2, ESI†), increases in the current density were observed around 1.2 V as the sweep number increased. This indicates that as the monomers polymerized, the formed larger frameworks increased the number of sites where additional monomers could attach.22 We observed that the bright yellowish Mo:BiVO4 surfaces became cloudy after polymerization, and also confirmed a color change to yellow when coated on the fluorine-doped tin oxide (FTO), as shown in Fig. S3 (ESI†).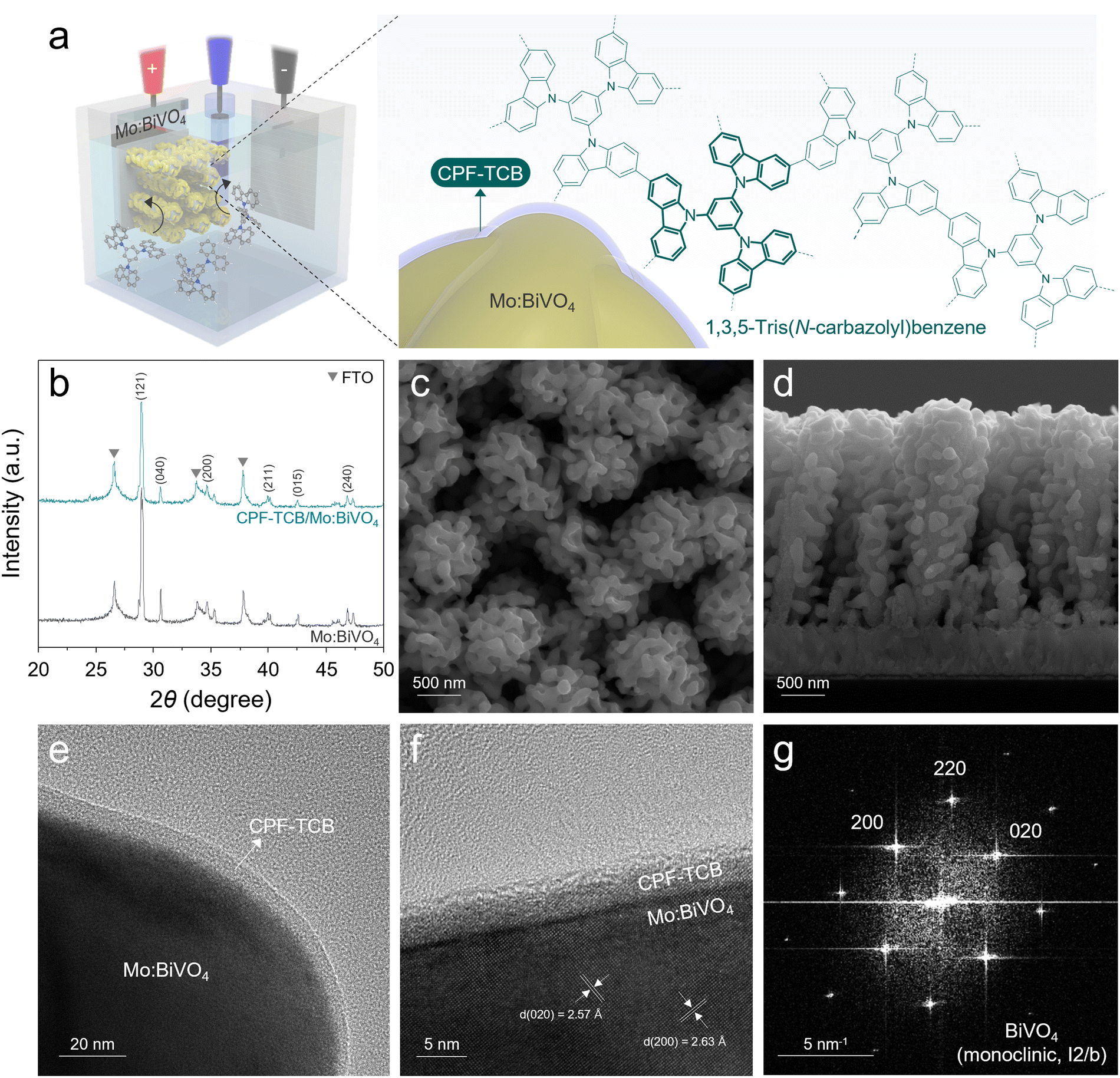 | ||
| Fig. 1 Structural characterization. (a) Schematic illustration of the electropolymerization of CPF-TCB on Mo:BiVO4. (b) XRD of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (c) Top-view and (d) cross-sectional SEM images of CPF-TCB/Mo:BiVO4. (e) TEM and (f) HRTEM images of CPF-TCB/Mo:BiVO4. (g) FFT pattern of monoclinic Mo:BiVO4. | ||
The X-ray diffraction (XRD) patterns in Fig. 1(b) were indexed to monoclinic BiVO4 (PDF# 04-010-5173), showing that the crystal structure of BiVO4 was unchanged after deposition of CPF-TCB. In the top field-emission scanning electron microscopy (FESEM) image of CPF-TCB/Mo:BiVO4 (Fig. 1(c)), the coexistence of nanopores and ETL-derived mesopores was observed, leading to the maximized surface area of the photoanode. The overall morphology of the 2.5 μm-thick vertical photoanode suitable for light absorption was confirmed through the cross-sectional FESEM image (Fig. 1(d)). Since the CPF-TCB thin layer with optimal sweep cycle was indistinguishable, we prepared Mo:BiVO4 with 40 time-swept CPF-TCB. As exhibited in Fig. S4 (ESI†), even at high cycle numbers, the CPF-TCB layer was uniformly coated on Mo:BiVO4 without agglomeration and clogging of nanopores. The corresponding energy dispersive X-ray (EDX) mapping indicated the uniform distribution of C and N elements from CPF-TCB on the BiVO4 surface. The CPF-TCB film with the optimal sweep cycle was revealed in the TEM image. As shown in Fig. 1(e), the CPF-TCB thin layer conformally covered Mo:BiVO4. The high-resolution TEM (HRTEM) image (Fig. 1(f)) showed that under the optimal sweep cycle, CPF-TCB grew uniformly onto Mo:BiVO4 with a thickness of 5 nm. The amorphous nature of CPF-TCB was consistent with the characteristics of electropolymerized polycarbazole frameworks in previous studies,22,23 and also corresponded to the XRD results showing no difference before and after deposition (Fig. 1(b)). The lattice fringes with interplanar spacings of 2.57 and 2.63 Å in two directions correspond to the (020) and (200) planes, respectively. The fast Fourier transformation (FFT) patterns in Fig. 1(g) show the diffraction spots for the (200) and (020) planes, indicating the crystalline monoclinic phase (I2/b group). These results matched well with the XRD analysis, identifying the suitable phase for water oxidation.24
The photoelectrochemical water oxidation of the fabricated photoanodes was evaluated in a three-electrode cell with 1 M potassium borate (K–Bi) buffer (pH 9.5) under AM1.5G solar illumination. To investigate the optimal thickness of the CPF-TCB layer for Mo:BiVO4, the electropolymerization of CPF-TCB was executed with different sweep cycles (5, 10, and 20 cycles). In the J–V curves of the CPF-TCB/Mo:BiVO4 photoanodes (Fig. S5, ESI†), they exhibited similar photocurrent densities at 1.23 VRHE, whereas CPF-TCB with 10 cycles exhibited the largest polarization. This indicated effective charge separation, and it was confirmed to grow to a thickness of 5 nm under 10 cycles (Fig. 1(f)). The PEC characteristics of Mo:BiVO4 before and after the 10-cycle deposition of the CPF-TCB layer were evaluated with and without the NiFeCoOx cocatalyst. The J–V curves for four types of photoanodes are presented in Fig. 2(a). The photocurrent density of CPF-TCB/Mo:BiVO4 was measured to 3.82 mA cm−2 at 1.23 VRHE, which is higher than that of Mo:BiVO4 (3.30 mA cm−2). The improvement was more pronounced with the cocatalyst, with NiFeCoOx/CPF-TCB/Mo:BiVO4 reaching a remarkable photocurrent density of 6.66 mA cm−2 at 1.23 VRHE, which is higher than that of NiFeCoOx/Mo:BiVO4 (6.09 mA cm−2). This means that CPF-TCB contributes to effective hole transport from Mo:BiVO4 to the cocatalyst. To verify the exceptional photocurrent density, linear sweep voltammetry (LSV) measurements of five NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes fabricated by the same process were conducted. Fig. S6 (ESI†) shows a minor variation in PEC performances between samples, indicating that the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode has good reproducibility. As shown in the SEM images of NiFeCoOx/CPF-TCB/Mo:BiVO4 (Fig. S7, ESI†), no structural changes were observed in the photoanode before and after the LSV measurement, indicating that CPF-TCB and NiFeCoOx effectively protect Mo:BiVO4 and promote water oxidation. The resulting significant enhancement in 0.4 ∼ 0.8 VRHE is advantageous to provide high power at the general range of the operating point in PEC tandem devices.25 The contribution of CPF-TCB to the power of the photoanode was quantitatively evaluated by the fill factor, as shown in Fig. 2(b). It is noteworthy that the maximum power point with the CPF-TCB layer was achieved at a more cathodic potential of 0.65 VRHE compared to that without the layer (0.75 VRHE), indicating that CPF-TCB served to elevate both photocurrent and photovoltage. Based on the rectangularity at the maximum power point, we calculated the FF of the photoanodes using the following equation:
| FF (%) = [JMP(1.23 − VMP)]/[JSC(1.23 − VON)] |
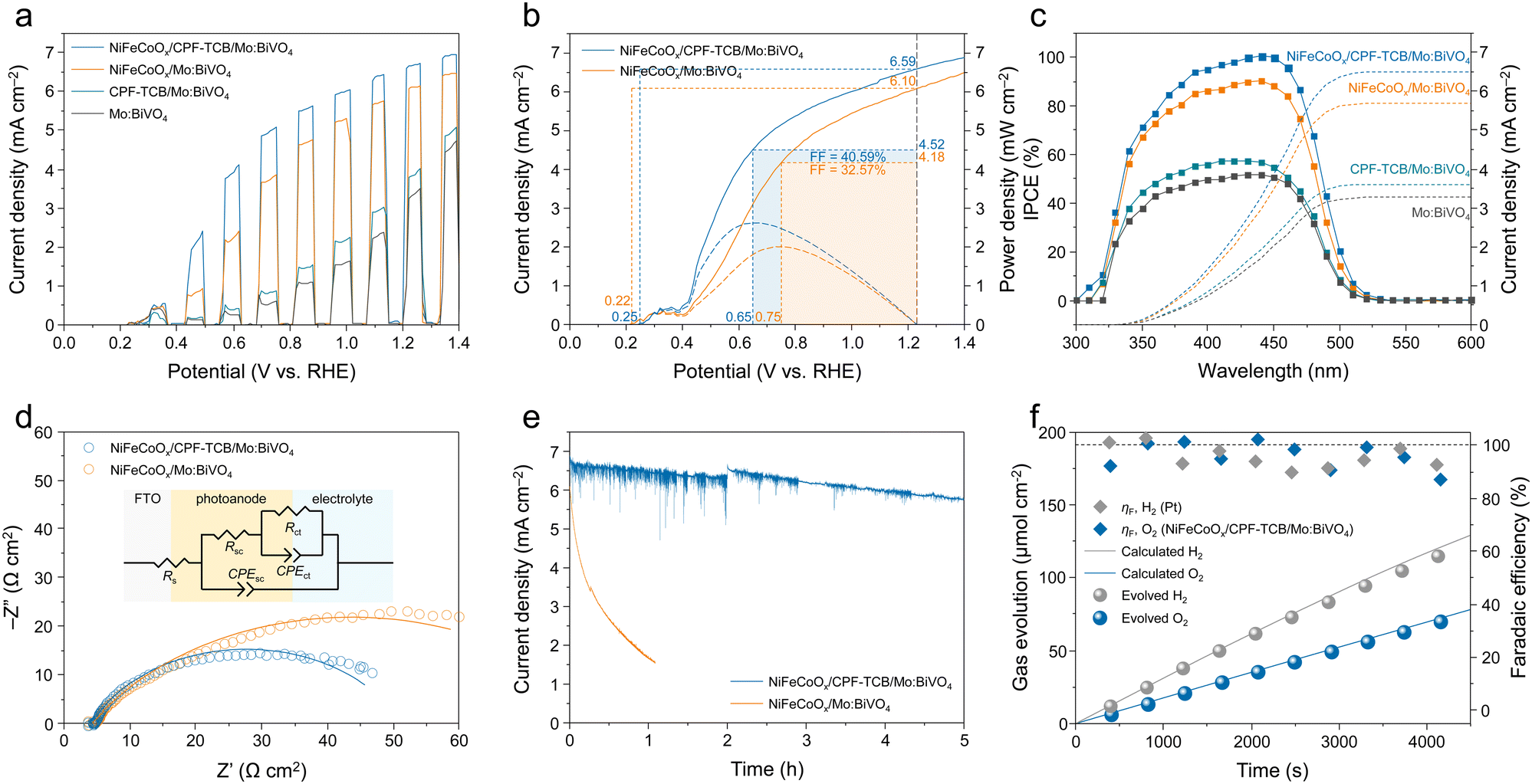 | ||
| Fig. 2 Photoelectrochemical water oxidation. (a) J–V curves of Mo:BiVO4, CPF-TCB/Mo:BiVO4, NiFeCoOx/Mo:BiVO4, and NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes. (b) Fill factors of NiFeCoOx/Mo:BiVO4 and NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes. (c) IPCE at 1.23 VRHE and integrated current density of the Mo:BiVO4, CPF-TCB/Mo:BiVO4, NiFeCoOx/Mo:BiVO4, and NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes. (d) EIS Nyquist plots at 0.5 VRHE and (e) J–t curves at 1.23 VRHE of the NiFeCoOx/Mo:BiVO4 and NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes. (f) Faradaic efficiency and gas evolution at 1.23 VRHE of the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanodes. All measurements were conducted in K–Bi buffer (pH 9.5) under AM1.5G 1 sun illumination. | ||
The hole extraction ability of CPF-TCB was further proven by electrochemical impedance spectroscopy (EIS) measurements conducted at 0.5 VRHE, where large photoactivity differences appeared. The Nyquist plots were fitted with an equivalent circuit as indicated in Fig. 2(d), and their values are tabulated in Table S1 (ESI†). The charge transfer resistance (Rct) of NiFeCoOx/CPF-TCB/Mo:BiVO4 (41.45 Ω cm2) was similar to that of NiFeCoOx/Mo:BiVO4 (42.65 Ω cm2), showing the same charge injection of the cocatalyst. For the charge transport resistance (Rsc) in the photoanode, N iFeCoOx/CPF-TCB/Mo:BiVO4 had a lower value of 5.525 Ω cm2 compared to NiFeCoOx/Mo:BiVO4 (36.13 Ω cm2). The significant decrease in Rsc indicates that the enhancement of the PEC performance sprang from the hole extraction behavior of CPF-TCB. The durability of the photoanodes was examined by chronoamperometry at 1.23 VRHE. As shown in the current density–time (J–t) curves (Fig. 2(e)), the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode ran stably for 5 h, whereas the NiFeCoOx/Mo:BiVO4 photoanode exhibited a sharp degradation within 1 h. This means that CPF-TCB plays a role as the protective layer as well as the HTL for Mo:BiVO4. The improved stability is also indirect evidence of the efficient hole extraction of CPF-TCB. The instability of the photoanode is derived from the hole accumulation causing self-oxidation of BiVO4. The expedited charge separation by CPF-TCB prevents the hole accumulation, enhancing the PEC stability. Furthermore, gas chromatography was conducted to collect the evolved gas and measure the faradaic efficiency. As shown in Fig. 2(f), faradaic efficiencies of nearly 100% were acquired in both the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode and Pt cathode, indicating that generated charges were fully consumed for oxygen and hydrogen evolution. It was also confirmed that the actual gas evolution rates were close to the calculated values.
The formation of the CPF-TCB layer was further proved by Fourier transform-infrared (FT-IR) spectroscopy, as shown in Fig. 3(a). Within the wavenumber range of 1000 to 1800 cm−1 where typical polymer bonding is observed, no peaks were observed for Mo:BiVO4. On the other hand, several peaks were identified in CPF-TCB/Mo:BiVO4, among which peaks at 1223, 1329, 1438, 1462, and 1591 cm−1 were found to originate from the carbazole functional group in CPF-TCB. These spectra most directly verify that the amorphous CPF-TCB thin film was well formed on Mo:BiVO4. To further demonstrate the charge transport behavior of CPF-TCB and the resulting changes in the electronic states of Mo:BiVO4, X-ray absorption spectroscopy (XAS) was employed for the V K-edge of Mo:BiVO4 with and without CPF-TCB. In the Fourier-transformed extended X-ray absorption fine structure (EXAFS) spectra (Fig. S8, ESI†), the peak corresponding to V–O bonding was observed around 1.3 Å for both samples, confirming that CPF-TCB had no influence on the interatomic structure of Mo:BiVO4. X-ray absorption near edge structure (XANES) spectra for the V K-edge in Fig. 3(b) show the electronic state changes. After forming the CPF-TCB layer on Mo:BiVO4, the pre-edge peak shifted from 5469.4 to 5469.8 eV, and the edge peak shifted from 5501.4 to 5502.6 eV. The prominent shifts indicate the relative electron transport towards Mo:BiVO4 due to charge separation caused by the hole extraction of CPF-TCB.26
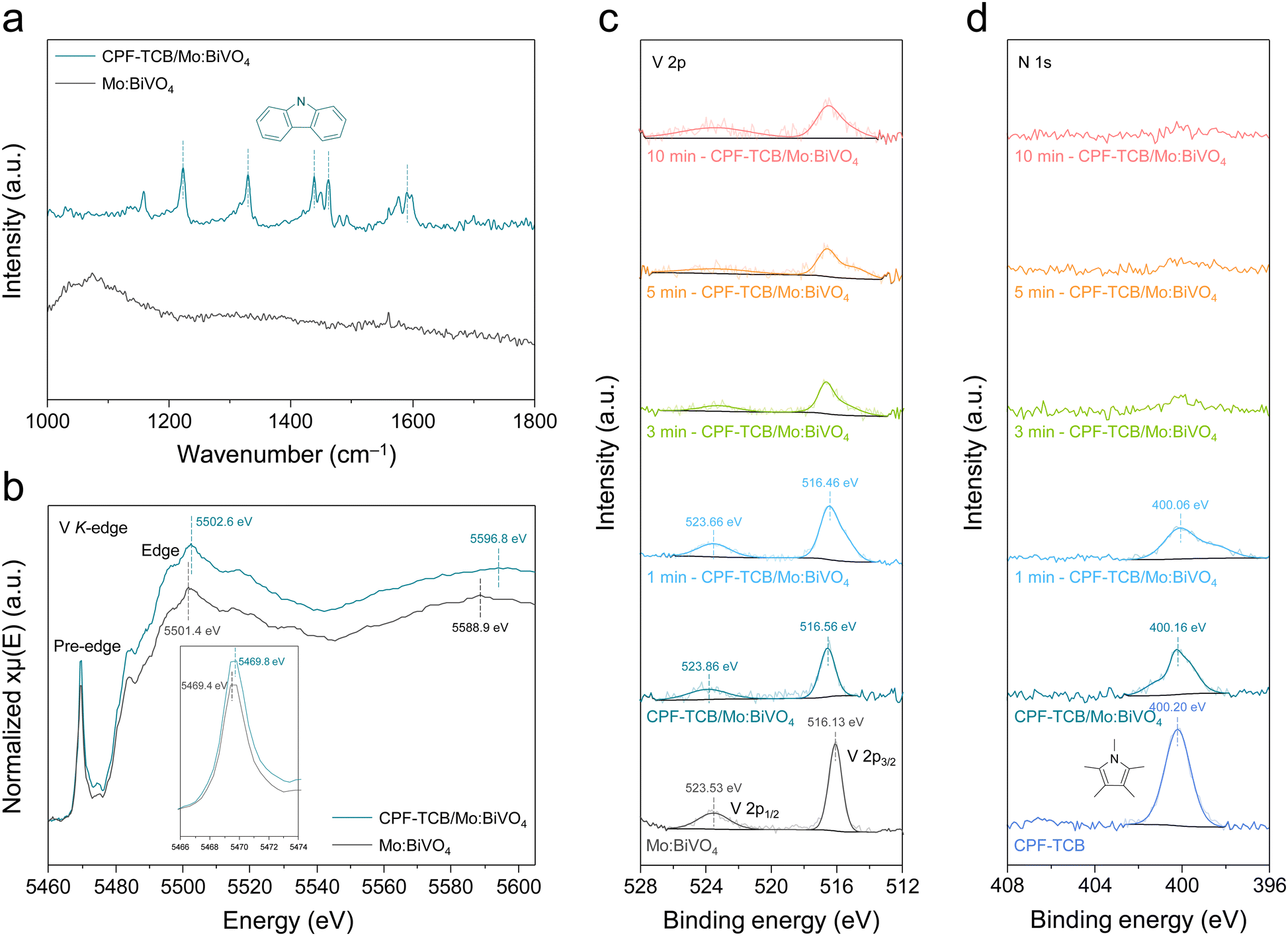 | ||
| Fig. 3 Spectroscopic analyses of the conjugated polycarbazole frameworks. (a) FT-IR spectra of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (b) V K-edge XANES spectra of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (c) V 2p XPS spectra of Mo:BiVO4 and CPF-TCB/Mo:BiVO4 with different Ar plasma etching times. (d) N 1s XPS spectra of CPF-TCB and CPF-TCB/Mo:BiVO4 with different Ar plasma etching times. | ||
X-ray photoelectron spectroscopy (XPS) analysis of Mo:BiVO4, CPF-TCB, and their heterojunction was carried out to scrutinize the electronic structures, and the wide spectra are presented in Fig. S9 (ESI†). As shown in Fig. 3(c), after the deposition of CPF-TCB, the V 2p1/2 and 2p3/2 peaks shifted from 523.53 and 516.13 eV to the higher binding energies of 523.86 and 516.56 eV, respectively. The Bi 4f5/2 and 4f7/2 peaks also shifted from 163.83 and 158.53 eV to the higher binding energies of 164.16 and 158.86 eV, respectively (Fig. S10, ESI†). In contrast, the N 1s peak (400.20 eV) in carbazole slightly moved to the lower binding energy of 400.16 eV, as exhibited in Fig. 3(d). These tendencies in the binding energy are attributed to the strong interfacial interactions between CPF-TCB and Mo:BiVO4.27,28 Depth profiling was performed while etching the surface of CPF-TCB/Mo:BiVO4 by irradiating Ar plasma with time variation. The N 1s peak intensity gradually decreased over time and completely disappeared after 10 min plasma, indicating that the 5 nm-thick CPF-TCB layer was etched in 10 min. On the other hand, the V 2p peak intensity remained constant after a slight decrease over time, and the Bi 4f peaks maintained high intensity regardless of time. The transition of peak positions during etching was ascribed to the change in the interaction between CPF-TCB and Mo:BiVO4. Meanwhile, the final NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode was also analyzed by XPS, as shown in Fig. S11 (ESI†). The compositional elements of the cocatalysts were revealed in the Ni 2p (873.3 and 855.6 eV for 2p1/2 and 2p3/2), Fe 2p (724.3 and 710.9 eV for 2p1/2 and 2p3/2), and Co 2p (796.5 and 781.8 eV for 2p1/2 and 2p3/2) spectra. The C 1s peak was deconvoluted into three peaks representing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.6 eV), C–O (285.5 eV), and C–C (284.5 eV) bondings, which were derived from the tannic acid chelating metal ions.29 Metal–(hydro)oxide bondings (M–O and M–OH) that appeared in the deconvoluted O 1s peaks were also attributed to the cocatalyst.30
O (288.6 eV), C–O (285.5 eV), and C–C (284.5 eV) bondings, which were derived from the tannic acid chelating metal ions.29 Metal–(hydro)oxide bondings (M–O and M–OH) that appeared in the deconvoluted O 1s peaks were also attributed to the cocatalyst.30
To theoretically scrutinize the charge transport behavior within the CPF-TCB/Mo:BiVO4 heterostructure, DFT calculations were utilized. As suggested in Fig. S12 (ESI†), a single layer of CPF-TCB positioned on the (010) surface of BiVO4 was established as the DFT calculation model to elucidate the spatial distribution of charge density differences. Fig. 4(a) shows the 3-dimensional charge density difference across the CPF-TCB/Mo:BiVO4 interface. The charge redistribution mainly occurred at the interface of CPF-TCB/Mo:BiVO4, and electrons accumulated around the surface of Mo:BiVO4. It is one piece of evidence indicating favorable charge transport at the interface of the heterostructure, which is consistent with the aforementioned results of XAS and XPS analysis.23,31 Additionally, the planar-averaged charge density difference along the z-direction normalized to the CPF-TCB/Mo:BiVO4 heterostructure was calculated to investigate quantitative changes (Fig. 4(b)). Strong charge accumulation on the surface of Mo:BiVO4 indicates that electrons are mainly transported from CPF-TCB to Mo:BiVO4 at their interface. According to Bader charge analysis, about 0.05 electrons per supercell are transported from the CPF-TCB layer to Mo:BiVO4 (010). After reaching the equilibrium state, a built-in electric field is developed by the net charge accumulation across the CPF-TCB/BiVO4 heterostructure, promoting charge separation.32 Thereby, these calculations provide support for the crucial charge transport role of CPF-TCB shown in PEC measurements.
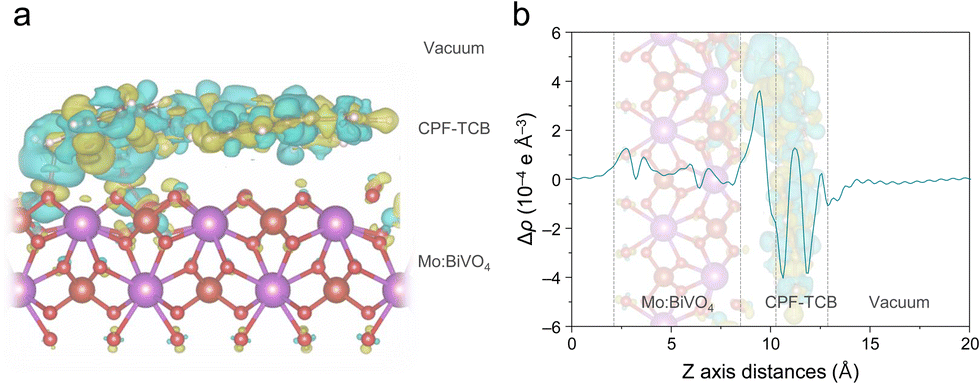 | ||
| Fig. 4 DFT calculations. (a) Charge density difference across CPF-TCB/Mo:BiVO4. (b) Planar-averaged charge density difference Δρ (Δρ = ρjunction − ρBiVO4 − ρCPF-TCB) along the z-direction for CPF-TCB/BiVO4. | ||
The hole extraction capability of CPF-TCB was further demonstrated by Mott–Schottky (M–S) analysis under light-off. Comparing the M–S plots shown in Fig. 5(a), the flat band potentials of TCB/Mo:BiVO4 and Mo:BiVO4 were almost identical. It was in line with the J–V curves in Fig. 2(a), where there was no difference in the onset potential of the two photoanodes. However, the slope of CPF-TCB/Mo:BiVO4 was lower than that of Mo:BiVO4, indicating that the donor density (ND) of Mo:BiVO4 increases when it forms a heterojunction with CPF-TCB. The donor density was calculated by the following equation and tabulated in Table S2 (ESI†):
| C−2 = (2/εε0A2eND)[V − EFB − (kBT)/e] |
| τn = −[(kBT)/e](dOCP/dt)–1 |
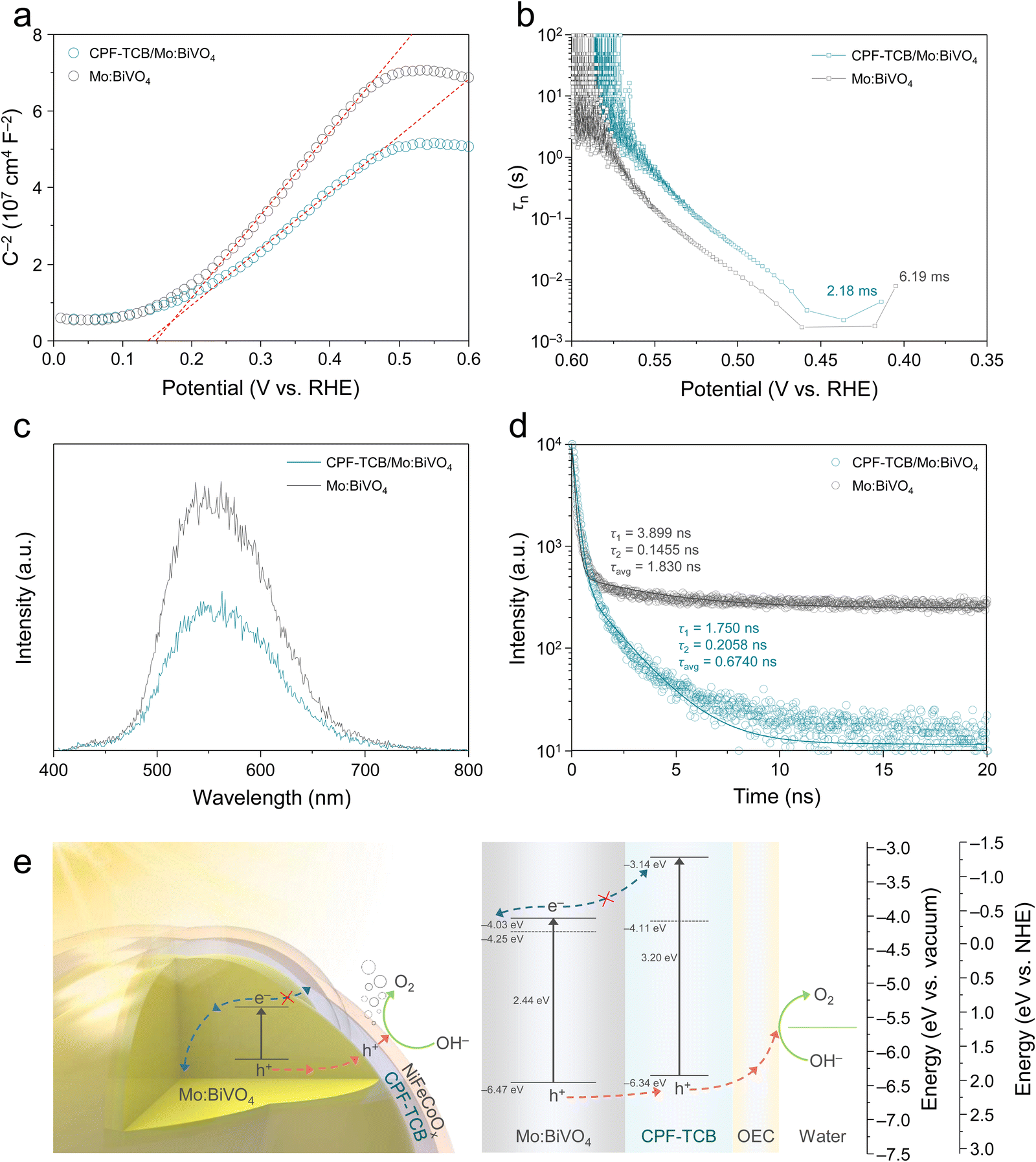 | ||
| Fig. 5 Charge carrier dynamics. (a) M–S plots of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (b) OCP-derived carrier lifetimes of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (c) Steady-state and (d) time-resolved PL of Mo:BiVO4 and CPF-TCB/Mo:BiVO4. (e) Energy band diagram of CPF-TCB/Mo:BiVO4. | ||
To make a comparison of the charge recombination behavior in the two BiVO4-based photoanodes, photoluminescence (PL) analysis was executed. Fig. 5(c) shows the steady-state PL spectra of Mo:BiVO4 with and without CPF-TCB, displaying peaks at 550 nm originating from radiative charge recombination. The PL emission was intensively quenched in forming a CPF-TCB/Mo:BiVO4 heterojunction, meaning that the HTL suppressed the intrinsic radiative recombination of charges in Mo:BiVO4.36 The benefit of the heterojunction was further supported by time-resolved photoluminescence (TRPL), elucidating the charge carrier kinetics (Fig. 5(d)). The decay obtained at the PL peak of 550 nm was fitted by the biexponential decay model as shown below, offering two carrier lifetimes (τ1 and τ2).
| I (t) = A1e−t/τ1 + A2e−t/τ2 |
The fundamental cause of effective charge separation in CPF-TCB/Mo:BiVO4 was disclosed in the band structure analysis. Ultraviolet-visible (UV-vis) spectroscopy was measured, and the absorption spectra calculated by the measured transmittance and reflectance were converted to Tauc plots, as shown in Fig. S14 (ESI†). The optical band gaps of Mo:BiVO4 and CPF-TCB were 2.44 and 3.20 eV, respectively. Ultraviolet photoelectron spectroscopy (UPS) was utilized to precisely determine the work functions and band edge positions, as suggested in Fig. S15 (ESI†). The secondary electron emission (SEE) spectra revealed the work function of Mo:BiVO4 (4.25 eV) and CPF-TCB (4.11 eV) from the difference with the source energy. From the valence band (VB) spectra in the low binding energy region, the energy differences between the Fermi level and VB maximum were verified. The band structure of CPF-TCB/Mo:BiVO4 was constructed by combining UV-vis spectra and UPS, as illustrated in Fig. 5(e). The CPF-TCB formed a type II heterojunction with Mo:BiVO4. This band alignment is energetically favorable for not only the efficient extraction of photogenerated holes, but also for blocking photogenerated electrons in Mo:BiVO4, reinforcing the charge separation. As a result, the aforementioned characteristics of CPF-TCB/Mo:BiVO4, such as inhibited charge recombination, rapid hole extraction, and resulting increased carrier concentration, all stem from the band structure.
To implement unbiased solar hydrogen production, the ultimate goal of PEC water splitting, we fabricated two types of tandem devices using the final NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode. The key requirement for the bottom cell is to avoid overlapping absorption of the photoanode to generate high photocurrent through light harvesting, while being able to generate sufficient photovoltage.38 Thus, for optimal band gap matching with 2.44 eV of Mo:BiVO4, halide perovskite-based bottom cells were prepared. Firstly, a photoanode–photocathode (PA–PC) tandem device was fabricated by combining the NiFeCoOx/CPF-TCB/Mo:BiVO4 PA and the perovskite PC. The photocathode illustrated in Fig. S16 (ESI†) exhibited prominent PEC performances (photocurrent density of 19.0 mA cm−2 at 0 VRHE and onset potential of 1.03 VRHE in potassium phosphate (K–Pi) buffer). As shown in Fig. 6(a), the PA–PC tandem device was measured in the K–Bi buffer under tandem illumination mode. The operating current density (JOP) determined by the intersection of 3 electrode J–V curves of the PA and PC behind it was 5.49 mA cm−2 at 0.81 VRHE., which corresponded to a STH conversion efficiency of 6.75%. Secondly, a photovoltaic–photoanode (PV–PA) tandem device was fabricated by combining the PA and the perovskite/Si solar cell. As displayed in Fig. S17 (ESI†), the PV cell made a similar current density (short-circuit current density of 19.2 mA cm−2) but higher voltages (open-circuit voltage of 1.81 V) compared to photocathodes, since Si generated additional photovoltage by absorbing light transmitted from perovskite.39 The higher photovoltage allowed the operating point of the PV–PA tandem device to be formed at a higher voltage, delivering a higher JOP. Fig. 6(b) shows the 2-electrode J–V curves of the PEC cell comprising a Pt cathode and the NiFeCoOx/CPF-TCB/Mo:BiVO4 PA, and the PV cell behind it under mode T. The JOP of the PV–PA tandem device was 7.33 mA cm−2 at 1.30 V, which corresponded to a STH conversion efficiency of 9.02%.
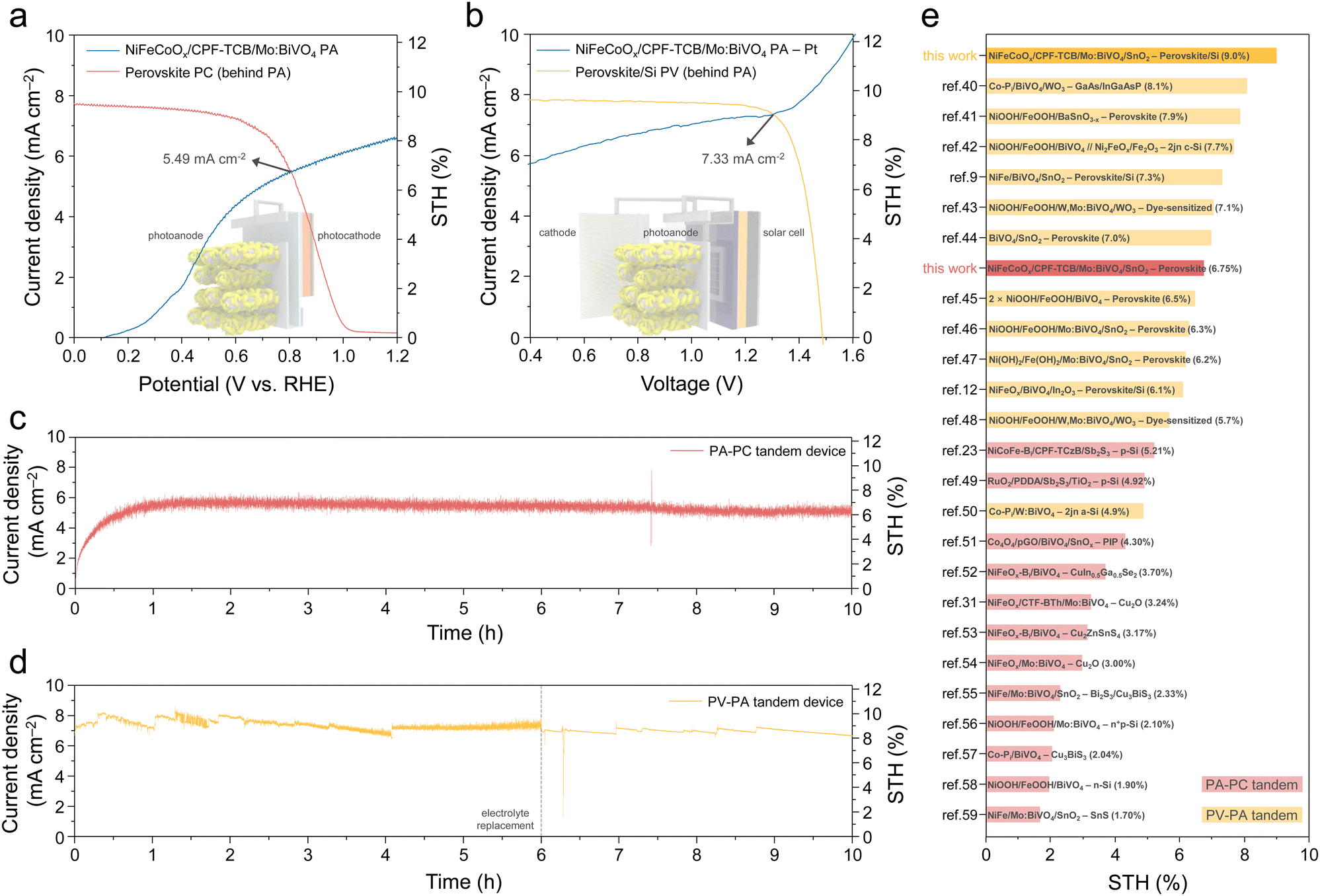 | ||
| Fig. 6 Unbiased solar hydrogen production. (a) Three-electrode J–V curves of the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode and perovskite-based photocathode (behind the photoanode). (b) Two-electrode J–V curves of the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode–Pt cathode and perovskite/Si solar cell (behind the photoanode). J–t curves of the (c) unbiased PA–PC and (d) PV–PA tandem devices. (e) STH conversion efficiency benchmarks of the PA–PC and PV–PA tandem devices in mode T. All measurements were conducted in K–Bi buffer (pH 9.5) under AM1.5G 1 sun illumination. | ||
Finally, two fabricated tandem devices were measured by chronoamperometry without an external bias under mode T (Fig. S18, ESI†). As shown in the J–t curve (Fig. 6(c)), the PA–PC tandem device recorded a current density that was consistent with the JOP and the corresponding STH conversion efficiency, durably operating for 10 h. Also, the PV–PA tandem device was stably operated for 10 h, generating a current density in accordance with JOP and STH conversion efficiency of 9% (Fig. 6(d)). The PA–PC and PV–PA tandem device we implemented shows the best STH conversion efficiency among each type of tandem device with BiVO4-based photoanodes, as shown in Fig. 6(e). Moreover, it is noteworthy that both types recorded the highest STH conversion efficiency among the mode T tandem devices, which require systematic engineering for the separation of the absorption wavelength (Tables S4 and S5 (ESI†)).40–59 These results emerge from the high-efficiency tandem device-tailored photoanode, in which a remarkable HTL, CPF-TCB, simultaneously increases the photocurrent and photovoltage of Mo:BiVO4.
Conclusion
We have successfully expedited the hole transport of the nanoporous Mo:BiVO4 photoanode by harnessing a CPF-TCB layer via electropolymerization, where the sophisticated control enabled the direct and uniform growth of CPF-TCB without plugging nanopores. Effective charge transport in the CPF-TCB/Mo:BiVO4 heterojunction was disclosed by XANES and XPS analyses, and depth profiling reaffirmed the heterostructure. The planar-averaged charge density difference by the DFT calculations substantiated strong charge separation in CPF-TCB/Mo:BiVO4. Based on the investigation of the charge carrier dynamics, we proved that enhanced hole transport through the CPF-TCB layer suppressed charge recombination and elevated the charge density of the photoanode. Consequently, the NiFeCoOx/CPF-TCB/Mo:BiVO4 photoanode generated an outstanding water oxidation photocurrent density of 6.66 mA cm−2 at 1.23 VRHE. Also, its high fill factor played a key role in both types of tandem devices for unassisted solar hydrogen production. The PA–PC tandem device combined with the perovskite photocathode and PV–PA tandem device assembled with perovskite/Si solar cell recorded STH conversion efficiencies of 6.75% and 9.02%, respectively, which are the topmost records among not only BiVO4-based, but also mode T tandem devices. This work broadens the material pool and provides novel strategies for designing high-performance organic–inorganic hybrid photoelectrodes for solar hydrogen production.Experimental section
Synthesis of nanoporous Mo:BiVO4 photoanodes
SnO2 NRs as an electron transport layer were fabricated by e-beam evaporation on the FTO glasses with a size of 1 × 1.5 cm2. After depositing a 50 nm-thick SnO2 film as an adhesive layer, 2.5 μm-thick SnO2 NRs were formed with a glancing angle of 85° and a rotation of 80 rpm. Nanoporous Mo:BiVO4 was prepared by the modified two-step electrodeposition on the SnO2 NRs annealed at 550 °C for 2 h in the air.9,15 The 50-mL aqueous solution with pH 1.8 contained 0.4 M KI (Daejung, 99.5%), 15 mM Bi(NO3)3·5H2O (Junsei, 98%), and 30 mM lactic acid (Sigma-Aldrich, 85%). The 20 mL of ethanol (Daejung, 99.9%)-dissolved 46 mM p-benzoquinone (Junsei, 98%) was slowly mixed with the as-prepared solution. After cooling the solution temperature to 22 °C, BiOI was electrodeposited in the three-electrode cell with a Ag/AgCl electrode and Pt mesh. Nucleation of BiOI with a potential of −0.35 VAg/AgCl for 20 s was followed by its growth with a potential of −0.1 VAg/AgCl for 4 min. The 10 μL aqueous solution with 0.1 M Na2MoO4 (Sigma-Aldrich, 98%) was added into a 5-mL dimethyl sulfoxide (DMSO) (Kanto, 98%)-dissolved 0.2 M VO(acac)2 (Sigma-Aldrich, 98%) solution. The as-deposited BiOI was converted to Mo:BiVO4 by annealing at 450 °C for 2 h in the air after impregnating the solution. The excess V2O5 was removed from the Mo:BiVO4 photoanode by immersing it in 1 M NaOH solution (Daejung) for 10 min.Surface modification with CPF-TCB hole transport layers and NiFeCoOx cocatalysts
The CPF-TCB films were fabricated by electropolymerization on Mo:BiVO4 photoanodes. The mixed solvent with 28 mL acetonitrile (Daejung, 99.7%) and 42 mL dichloromethane (Daejung, 99.5%) attained conductivity by dissolving 0.1 M tetrabutylammonium perchlorate (Sigma-Aldrich, 95%). 1,3,5-Tris(N-carbazolyl)benzene (4 mM, Sigma-Aldrich, 97%) was dissolved in the solution for 10 min. The potential of the Mo:BiVO4 photoanode in the three-electrode cell with Ag/AgCl and Pt plate was linearly swept in the range of −0.8 to 1.4 VAg/AgCl with a scan rate of 20 mV s−1. After electropolymerization, the photoanode was rinsed with acetonitrile and ethanol, followed by drying with N2 flow. The NiFeCoOx cocatalysts were introduced by dip coating, as modified from previous methods.12 In a vessel, 2 mL of 10 mM FeCl3·6H2O (Daejung, 98%), 7 mL of 10 mM NiCl2·6H2O (Daejung, 96%), and 1 mL of 10 mM CoCl2·6H2O (Daejung, 97%) solutions were combined and mixed. After immersing the CPF-TCB/Mo:BiVO4 photoanode in the vessel for 15 min, 10 mL of 9 mg L−1 tannic acid (Sigma-Aldrich) solution and 75 μL of 2 M NaOH (Daejung, 97%) solution were sequentially added. After 1 h, the photoanode was rinsed with deionized water and dried with N2 gas flow.Fabrication of the perovskite photocathodes
The FTO glass was patterned by a laser-etching instrument (Korthem Science), followed by UV treatment for 15 min. As an ETL, the SnO2 colloidal dispersion (Alfa Aesar) with deionized water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4; volume ratio) was spin-coated at 3000 rpm for 60 s and heated at 170 °C for 1 h. The perovskite solution was prepared by dissolving 240.8 mg of FAI (Greatcell Solar, 99.99%), 8.25 mg of MABr (Greatcell Solar, 99.99%), 33.76 mg of MACl (Greatcell Solar, 99.99%), 18.19 mg of CsI (Sigma-Aldrich, 99.999%), 705.3 mg of PbI2 (Sigma-Aldrich, 99.999%), and 27.04 mg of PbBr2 (Sigma-Aldrich, 99.999%) in 0.96 mL of DMF (Sigma-Aldrich, 99.8%) and 0.24 mL DMSO (Sigma-Aldrich, 99.9%) mixed solvent. Subsequently, the solution was stirred for 1 h in an ambient air atmosphere. The perovskite solution was spin-coated at 1200 rpm for 12 s and 5800 rpm for 20 s, followed by annealing at 100 °C for 1 h. An anti-solvent, ethyl acetate (0.8 mL), was dripped onto the substrate for 10 s before the end of the spin-coating process. As an HTL, the spiro-OMeTAD solution comprising 72 mg of spiro-OMeTAD (Sigma-Aldrich, 99%), 8.8 μL of Li-TFSI (Sigma-Aldrich, 99.99%) solution (520 mg of Li-TFSI in 1 mL of acetonitrile), and 4.4 μL of 4-tert-butylpyridin (Sigma-Aldrich, 98%) was dissolved in 1 mL of chlorobenzene and spin-coated on the perovskite at 3000 rpm for 60 s. An 80 nm-thick Au top electrode was deposited on the HTL by thermal evaporation. Cu tape and wire were attached to the Au electrode, followed by sealing with an epoxy. Finally, the Pt cocatalyst was deposited on the active area by using a sputter coater (Cressington 108 Auto, Ted Pella).
:4; volume ratio) was spin-coated at 3000 rpm for 60 s and heated at 170 °C for 1 h. The perovskite solution was prepared by dissolving 240.8 mg of FAI (Greatcell Solar, 99.99%), 8.25 mg of MABr (Greatcell Solar, 99.99%), 33.76 mg of MACl (Greatcell Solar, 99.99%), 18.19 mg of CsI (Sigma-Aldrich, 99.999%), 705.3 mg of PbI2 (Sigma-Aldrich, 99.999%), and 27.04 mg of PbBr2 (Sigma-Aldrich, 99.999%) in 0.96 mL of DMF (Sigma-Aldrich, 99.8%) and 0.24 mL DMSO (Sigma-Aldrich, 99.9%) mixed solvent. Subsequently, the solution was stirred for 1 h in an ambient air atmosphere. The perovskite solution was spin-coated at 1200 rpm for 12 s and 5800 rpm for 20 s, followed by annealing at 100 °C for 1 h. An anti-solvent, ethyl acetate (0.8 mL), was dripped onto the substrate for 10 s before the end of the spin-coating process. As an HTL, the spiro-OMeTAD solution comprising 72 mg of spiro-OMeTAD (Sigma-Aldrich, 99%), 8.8 μL of Li-TFSI (Sigma-Aldrich, 99.99%) solution (520 mg of Li-TFSI in 1 mL of acetonitrile), and 4.4 μL of 4-tert-butylpyridin (Sigma-Aldrich, 98%) was dissolved in 1 mL of chlorobenzene and spin-coated on the perovskite at 3000 rpm for 60 s. An 80 nm-thick Au top electrode was deposited on the HTL by thermal evaporation. Cu tape and wire were attached to the Au electrode, followed by sealing with an epoxy. Finally, the Pt cocatalyst was deposited on the active area by using a sputter coater (Cressington 108 Auto, Ted Pella).
Fabrication of the perovskite/Si solar cells
The perovskite/Si tandem solar cells were fabricated by the previously reported method.39 Briefly, on the top of the Si bottom cell with 20 nm-thick of indium tin oxide (ITO) recombination layer, 7 mg mL−1 poly(triarylamine) (PTAA, Sigma-Aldrich) solution in toluene was spin-coated at 6000 rpm for 30 s and annealed at 100 °C for 10 min. The 1.3 M perovskite (Cs0.3DMA0.2MA0.5PbI3) solution with 10 mol% of MACl (Lumtec, 99.5%), 1.5 mol% of PEASCN (Greatcell Solar), and 2 mol% of Pb(SCN)2 additives in dimethylformamide (DMF, Sigma-Aldrich, 99.8%) and N-methyl-2-pyrroldone (Sigma-Aldrich, 99%) mixed solvent (4:1; volume ratio) was spin-coated on PTAA at 3000 rpm for 24 s, and 50 μL of methyl acetate (Sigma-Aldrich, 99%) was dripped at 20 s after the start of spinning, followed by annealing at 100 °C for 10 min. On the perovskite layer, a 15 nm-thick C60 (Nano-C) layer was deposited by thermal evaporation, and 0.2 wt% polyethyleneimine (Sigma-Aldrich, 80% ethoxylated) solution in methanol was dynamically spin-coated at 6000 rpm for 30 s. A 40 nm-thick ITO layer was deposited by radio frequency sputtering at room temperature (base pressure: 2 × 10−6 Torr, working pressure: 2 × 10−3 Torr, power: 50 W), and a 500 nm-thick, 50 μm-width Ag grid was deposited by thermal evaporation. Finally, a 105 nm-thick MgF2 (Sigma-Aldrich, 99.99%) layer was deposited as an anti-reflection layer.
Characterizations
The crystal structures of the samples were identified using XRD (D8 Advance, Bruker) with Cu Kα (1.5406 Å) radiation. The surface morphology and EDX of the samples were investigated by FESEM (MERLIN Compact, ZEISS) with acceleration voltages of 5 and 15 kV, respectively. The HRTEM images and FFT patterns of CPF-TCB/Mo:BiVO4 were analyzed by TEM (JEM-2100F, JEOL) with an acceleration voltage of 200 kV. FT-IR spectra were recorded by FT-IR spectrometer (Nicolet iS50, Thermo Fisher Scientific). The XAS data at V K-edge was obtained on the 7D beamline at the Pohang Light Source (PLS) in the Pohang Accelerator Laboratory (PAL), Republic of Korea. The data were acquired in the fluorescence mode by a solid-state detector and fitted using Athena software (Demeter). The electronic states and chemical compositions were analyzed by XPS (AXIS HSi, KRATOS) with monochromatic Al Kα (1486.6 eV) radiation. Depth profiling was conducted with time variation of Ar ion etching, and the data were fitted using CasaXPS software (CasaXPS). PL spectra were measured at room temperature on the fluorescence spectrometer (FlouTime 300, PicoQuant) with an excitation laser (405 nm). The TRPL data were obtained at the wavelength of 550 nm and fitted with FlouFit software (PicoQuant). Tauc plots were acquired from the absorption spectra calculated by the transmittance and reflectance spectra from the UV-vis spectrometer (V-770, JASCO). The UPS spectra were measured using XPS (AXIS Nova, KRATOS) with He I (21.22 eV) source.Photoelectrochemical measurements
A Xe arc lamp (LS 150, Abet Technologies) was used as a light source. Its illumination and intensity were calibrated with AM1.5G filter and Si photodiode (S300, McScience), respectively. The PEC measurements were carried out using a potentiostat (Ivium-n-Stat, Ivium Technologies) in the three-electrode cell with the Ag/AgCl electrode and Pt mesh. Water oxidation of the photoanodes was measured in 1 M K–Bi buffer (pH 9.5), and proton reduction of the perovskite photocathodes was measured in 0.5 M K–Pi buffer (pH 7.0). All of the measured potentials were converted into RHE by the Nernst equation:| ERHE = EAg/AgCl + E0Ag/AgCl + 0.059 × pH |
| STH (%) = JOP × 1.23 × ηF/Pin |
Computational details
The spin-polarized DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP) with the projector-augmented wave method for the core region and a plane-wave kinetic energy cutoff of 400 eV. The generalized gradient approximation (GGA) in the form of Perdew–Burke–Ernzerhof (PBE) for the exchange–correlation potentials was used. The DFT+U calculations were performed with Hubbard–U correction of U = 3.25 eV to the d-electrons of Bi to account for the on-site correlation effects. A 7 × 5 × 7 gamma-centered Monkhorst–pack sampled k-point grid was employed to sample the reciprocal space for the bulk BiVO4. Monkhorst–pack k-point meshes of 2 × 2 × 1 were used for structure optimization and property calculation of the BiVO4 (010) slab, CPF-TCB, and the heterojunction CPF-TCB/BiVO4 (010) heterostructure. The large vacuum layers of these slab models were set at least 15 Å in the z-direction for the isolation of the surface to prevent the interaction between two periodic units. The optimized lattice constants of BiVO4 were a = 7.338 Å, b = 11.727 Å, c = 5.188 Å, α = γ = 90.00°, and β = 135.01°, respectively. The surface of BiVO4 (010) was carried out using the slab model composed of p(4 × 3) supercells with four metal atomic monolayers. The bottom two atomic monolayers of BiVO4 (010) were fixed at their bulk positions, while the remaining atomic layers and 2-dimensional CPF-TCB layer were free to move in all directions until the convergence of energy and residual force on each atom were less than 1 × 10−4 eV and 0.05 eV Å−1, respectively. To describe the charge transfer process in the CPF-TCB/BiVO4 heterostructure, the dipole moment on the z-direction was calculated, and the planar-averaged charge density was acquired by averaging the charge density on the xy plane toward the z-direction.Data availability
All data supporting the findings of this study are available within the main text and the ESI.† All relevant data are available from the corresponding authors upon reasonable request.Author contributions
H. W. J., J. Y. K., J. M., and M. S. K. supervised the project. H. W. J. and J. W. Y. conceived the project and designed the experiments. J. W. Y. fabricated and measured the devices, and analyzed the experimental results. S. G. J. fabricated the perovskite/Si solar cells. C. S. J. fabricated the perovskite photocathodes. J. K. performed the density functional theory calculations and helped to analyze the X-ray absorption spectra. H. R. K. helped to analyze the carrier dynamics. T. H. L. carried out the transmission electron microscopic characterizations. S. A. L. helped to analyze the experimental results. W. S. C. conducted gas chromatography measurements. S. L. helped to synthesize conjugated polymers. H. L. helped to measure the photocathode. J. W. Y. and H. W. J. mainly wrote the manuscript. All authors discussed the results and commented on the manuscript at all stages.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government Ministry of Science and ICT (MSIT) (2021R1A2B5B03001851, 2021M3H4A1A03057403). This work was also supported by the KRISS (Korea Research Institute of Standards and Science) MPI Lab. program. H. W. J. gratefully acknowledges the KRISS MPI Lab. program. J. W. Y. acknowledges the NRF grant funded by the Korea government MSIT (RS-2023-00213786). The Inter-University Semiconductor Research Center and Institute of Engineering Research at Seoul National University provided research facilities for this work. We thank the Pohang Accelerator Laboratory for providing the synchrotron radiation source at the 7D beamline.References
- M. Grätzel, Nature, 2001, 414, 338–344 CrossRef.
- V. Andrei, G. M. Ucoski, C. Pornrungroj, C. Uswachoke, Q. Wang, D. S. Achilleos, H. Kasap, K. P. Sokol, R. A. Jagt, H. Lu, T. Lawson, A. Wagner, S. D. Pike, D. S. Wright, R. L. Z. Hoye, J. L. MacManus-Driscoll, H. J. Joyce, R. H. Friend and E. Reisner, Nature, 2022, 608, 518–522 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- K. Sivula and R. Van De Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- T. W. Kim and K. S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- D. K. Lee and K. S. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- S. Corby, R. R. Rao, L. Steier and J. R. Durrant, Nat. Rev. Mater., 2021, 6, 1136–1155 CrossRef CAS.
- J. W. Yang, I. J. Park, S. A. Lee, M. G. Lee, T. H. Lee, H. Park, C. Kim, J. Park, J. Moon, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2021, 293, 120217 CrossRef CAS.
- M. J. Choi, T. L. Kim, K. S. Choi, W. Sohn, T. H. Lee, S. A. Lee, H. Park, S. Y. Jeong, J. W. Yang, S. Lee and H. W. Jang, ACS Appl. Mater. Interfaces, 2022, 14, 7788–7795 CrossRef CAS PubMed.
- M. G. Lee, J. W. Yang, H. Park, C. W. Moon, D. M. Andoshe, J. Park, C. K. Moon, T. H. Lee, K. S. Choi, W. S. Cheon, J. J. Kim and H. W. Jang, Nano-Micro Lett., 2022, 14, 48 CrossRef CAS PubMed.
- M. G. Lee, J. W. Yang, I. J. Park, T. H. Lee, H. Park, W. S. Cheon, S. A. Lee, H. Lee, S. G. Ji, J. M. Suh, J. Moon, J. Y. Kim and H. W. Jang, Carbon Energy, 2023, 5, e321 CrossRef CAS.
- K. H. Ye, Z. Wang, J. Gu, S. Xiao, Y. Yuan, Y. Zhu, Y. Zhang, W. Mai and S. Yang, Energy Environ. Sci., 2017, 10, 772–779 RSC.
- B. Jin, Y. Cho, C. Park, J. Jeong, S. Kim, J. Jin, W. Kim, L. Wang, S. Lu, S. Zhang, S. H. Oh, K. Zhang and J. H. Park, Energy Environ. Sci., 2022, 15, 672–679 RSC.
- K. H. Ye, H. Li, D. Huang, S. Xiao, W. Qiu, M. Li, Y. Hu, W. Mai, H. Ji and S. Yang, Nat. Commun., 2019, 10, 3687 CrossRef PubMed.
- S. Gu, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- D. Xu, Z. Gong, Y. Jiang, Y. Feng, Z. Wang, X. Gao, X. Lu, G. Zhou, J. M. Liu and J. Gao, Nat. Commun., 2022, 13, 7020 CrossRef CAS PubMed.
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- Q. Chen, M. Luo, P. Hammershøj, D. Zhou, Y. Han, B. W. Laursen, C. G. Yan and B. H. Han, J. Am. Chem. Soc., 2012, 134, 6084–6087 CrossRef CAS PubMed.
- K. Takagi, H. Takao and T. Nakagawa, Polym. J., 2013, 45, 396–400 CrossRef CAS.
- Z. Zhou, X. Li, D. Guo, D. B. Shinde, D. Lu, L. Chen, X. Liu, L. Cao, A. M. Aboalsaud, Y. Hu and Z. Lai, Nat. Commun., 2020, 11, 5323 CrossRef CAS PubMed.
- L. Wang, W. Lian, B. Liu, H. Lv, Y. Zhang, X. Wu, T. Wang, J. Gong, T. Chen and H. Xu, Adv. Mater., 2022, 34, 2200723 CrossRef CAS PubMed.
- M. G. Lee, J. W. Yang, H. R. Kwon and H. W. Jang, CrystEngComm, 2022, 24, 5838–5864 RSC.
- B. Liu, S. Wang, G. Zhang, Z. Gong, B. Wu, T. Wang and J. Gong, Chem. Soc. Rev., 2023, 52, 4644–4671 RSC.
- J. Chen, C. L. Dong, D. Zhao, Y. C. Huang, X. Wang, L. Samad, L. Dang, M. Shearer, S. Shen and L. Guo, Adv. Mater., 2017, 29, 1606198 CrossRef PubMed.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 2001 CrossRef PubMed.
- M. Zhu, Z. Sun, M. Fujitsuka and T. Majima, Angew. Chem., Int. Ed., 2018, 130, 2182–2186 CrossRef.
- M. Huang, Z. Huang and H. Zhu, Nano Energy, 2020, 70, 104487 CrossRef CAS.
- Y. Shi, Y. Yu, Y. Liang, Y. Du and B. Zhang, Angew. Chem., Int. Ed., 2019, 131, 3809–3813 CrossRef.
- Y. Zhang, H. Lv, Z. Zhang, L. Wang, X. Wu and H. Xu, Adv. Mater., 2021, 33, 2008264 CrossRef CAS PubMed.
- J. Li, L. Cai, J. Shang, Y. Yu and L. Zhang, Adv. Mater., 2016, 28, 4059–4064 CrossRef CAS PubMed.
- H. Zhang, D. Li, W. J. Byun, X. Wang, T. J. Shin, H. Y. Jeong, H. Han, C. Li and J. S. Lee, Nat. Commun., 2020, 11, 4622 CrossRef PubMed.
- M. Zhong, T. Hisatomi, Y. Kuang, J. Zhao, M. Liu, A. Iwase, Q. Jia, H. Nishiyama, T. Minegishi, M. Nakabayashi, N. Shibata, R. Niishiro, C. Katayama, H. Shibano, M. Katayama, A. Kudo, T. Yamada and K. Domen, J. Am. Chem. Soc., 2015, 137, 5053–5060 CrossRef CAS PubMed.
- Y. Lin, Y. Xu, M. T. Mayer, Z. I. Simpson, G. McMahon, S. Zhou and D. Wang, J. Am. Chem. Soc., 2012, 134, 5508–5511 CrossRef CAS PubMed.
- B. Liu, X. Wang, Y. Zhang, L. Xu, T. Wang, X. Xiao, S. Wang, L. Wang and W. Huang, Angew. Chem., Int. Ed., 2023, 135, e202217346 CrossRef.
- S. Wang, T. He, P. Chen, A. Du, K. Ostrikov, W. Huang and L. Wang, Adv. Mater., 2020, 32, 2001385 CrossRef CAS PubMed.
- J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- S. G. Ji, I. J. Park, H. Chang, J. H. Park, G. P. Hong, B. K. Choi, J. H. Jang, Y. J. Choi, H. W. Lim, Y. J. Ahn, S. J. Park, K. T. Nam, T. Hyeon, J. Park, D. H. Kim and J. Y. Kim, Joule, 2022, 6, 2390–2405 CrossRef CAS.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef.
- M. Kim, B. Lee, H. Ju, J. Y. Kim, J. Kim and S. W. Lee, Adv. Mater., 2019, 31, 1903316 CrossRef.
- J. H. Kim, J. W. Jang, Y. H. Jo, F. F. Abdi, Y. H. Lee, R. Van De Krol and J. S. Lee, Nat. Commun., 2016, 7, 13380 CrossRef CAS.
- X. Shi, H. Jeong, S. J. Oh, M. Ma, K. Zhang, J. Kwon, I. T. Choi, I. Y. Choi, H. K. Kim, J. K. Kim and J. H. Park, Nat. Commun., 2016, 7, 11943 CrossRef CAS PubMed.
- Z. Xu, L. Chen, C. J. Brabec and F. Guo, Small Methods, 2023, 7, 2300619 CrossRef CAS PubMed.
- S. Wang, P. Chen, Y. Bai, J. H. Yun, G. Liu and L. Wang, Adv. Mater., 2018, 30, 1800486 CrossRef.
- S. Xiao, C. Hu, H. Lin, X. Meng, Y. Bai, T. Zhang, Y. Yang, Y. Qu, K. Yan, J. Xu, Y. Qiu and S. Yang, J. Mater. Chem. A, 2017, 5, 19091–19097 RSC.
- Y. Qiu, W. Liu, W. Chen, G. Zhou, P. C. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, Sci. Adv., 2016, 2, 1501764 CrossRef PubMed.
- X. Shi, K. Zhang, K. Shin, M. Ma, J. Kwon, I. T. Choi, J. K. Kim, H. K. Kim, D. H. Wang and J. H. Park, Nano Energy, 2015, 13, 182–191 CrossRef CAS.
- Y. S. Park, X. Jin, J. Tan, H. Lee, J. Yun, S. Ma, G. Jang, T. Kim, S. G. Shim, K. Kim, J. Lee, C. U. Lee, S. J. Hwang and J. Moon, Energy Environ. Sci., 2022, 15, 4725–4737 RSC.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. Van De Krol, Nat. Commun., 2013, 4, 2195 CrossRef PubMed.
- S. Ye, W. Shi, Y. Liu, D. Li, H. Yin, H. Chi, Y. Luo, N. Ta, F. Fan, X. Wang and C. Li, J. Am. Chem. Soc., 2021, 143, 12499–12508 CrossRef CAS PubMed.
- H. Kobayashi, N. Sato, M. Orita, Y. Kuang, H. Kaneko, T. Minegishi, T. Yamada and K. Domen, Energy Environ. Sci., 2018, 11, 3003–3009 RSC.
- D. Huang, K. Wang, L. Li, K. Feng, N. An, S. Ikeda, Y. Kuang, Y. Ng and F. Jiang, Energy Environ. Sci., 2021, 14, 1480–1489 RSC.
- L. Pan, J. H. Kim, M. T. Mayer, M. K. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- S. Moon, J. Park, H. Lee, J. W. Yang, J. Yun, Y. S. Park, J. Lee, H. Im, H. W. Jang, W. Yang and J. Moon, Adv. Sci., 2023, 10, 2206286 CrossRef CAS PubMed.
- W. Vijselaar, P. Westerik, J. Veerbeek, R. M. Tiggelaar, E. Berenschot, N. R. Tas, H. Gardeniers and J. Huskens, Nat. Energy, 2018, 3, 185–192 CrossRef CAS.
- D. Huang, L. Li, K. Wang, Y. Li, K. Feng and F. Jiang, Nat. Commun., 2021, 12, 3795 CrossRef CAS PubMed.
- B. Liu, S. Feng, L. Yang, C. Li, Z. Luo, T. Wang and J. Gong, Energy Environ. Sci., 2020, 13, 221–228 RSC.
- H. Lee, J. W. Yang, J. Tan, J. Park, S. G. Shim, Y. S. Park, J. Yun, K. Kim, H. W. Jang and J. Moon, Adv. Sci., 2021, 8, 2102458 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ee03353h |
| This journal is © The Royal Society of Chemistry 2024 |