
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnlocking the potential of metal ligand cooperation for enantioselective transformations
Tizian-Frank
Ramspoth†
 ,
Johanan
Kootstra†
and
Syuzanna R.
Harutyunyan
*
,
Johanan
Kootstra†
and
Syuzanna R.
Harutyunyan
*
Institute for Chemistry, University of Groningen Institution Nijenborgh 4, 9747 AG, Groningen, The Netherlands. E-mail: s.harutyunyan@rug.nl
First published on 21st February 2024
Abstract
Metal–ligand cooperation, in which both the metal and the ligand of a transition metal complex actively participate in chemical transformations leading to enhanced reactivity or selectivity in chemical reactions, has emerged as a powerful and versatile concept in catalysis. This Viewpoint discusses the development trajectory of transition metal-based complexes as catalysts in (de)hydrogenative processes, in particular those cases where metal–ligand cooperation has been invoked to rationalise the observed high reactivities and excellent selectivities. The historical context, mechanistic aspects and current applications are discussed with the suggestion to explore the potential of the MLC mode of action of such catalysts in enantioselective transformations beyond (de)hydrogenative processes.
 Tizian-Frank Ramspoth | Tizian-Frank Ramspoth obtained his BSc and MSc in Chemistry from the University of Bielefeld. He conducted his masters research at the Helmholtz Institute for Pharmaceutical Research (HIPS) in the group of Professor A. K. H. Hirsch. Afterwards he did a research internship at Bayer AG Wuppertal. Currently he is pursuing a PhD in Chemistry on sustainable cooperative catalysis for crosslinking reactions in the group of Professor S.R. Harutyunyan, University of Groningen. |
 Johanan Kootstra | Johanan Kootstra obtained his BSc and MSc in Chemistry from the University of Groningen. During these studies, he performed research in the group of Prof. Syuzanna R. Harutyunyan, working on copper-catalysed asymmetric conjugate addition reactions. Currently, he is pursuing his PhD in the same group, working on aerobic oxidations. |
 Syuzanna R. Harutyunyan | Syuzanna R. Harutyunyan received her MSc in Chemistry from Yerevan State University, after which she moved to Moscow, to undertake her PhD under the supervision of Prof. Belokon. Following her PhD, she joined the research group of Prof. Feringa at the University of Groningen as a postdoctoral Research Fellow. Syuzanna started her independent research career in 2010 and is currently a full professor at the University of Groningen. Her research activities include homogeneous catalysis, out-of-equilibrium systems and mechanistic studies. |
Introduction
The selection of a suitable ligand is a critical factor in the design of an effective homogeneous catalyst, as ligands can affect the reactivity, selectivity and the stability of a catalyst. Usually they remain chemically inert while all the key reaction steps take place at the metal centre.1,2 Over the last few decades, however, an increasing number of systems have been developed in which the ligands in metal complexes have been proposed to play a more active co-operative role, leading to improved reactivities and selectivities.1,2 These developments led to the introduction of the term ‘metal–ligand cooperativity’ (MLC), which can manifest itself in a variety of ways. In its most common use in catalysis, MLC refers to reactions catalysed by transition metal complexes that involve active participation of both ligand and metal in at least one mechanistically relevant bond-forming or bond-breaking step of the catalytic reaction.3 MLC catalysis with metal complexes bearing functional ligands, a design element not unknown to nature,4 allowed for significant developments in the field of catalytic hydrogenation including asymmetric versions.5The earliest example of bond activation through MLC can be traced back to the work reported by Fujiwara and Moritani in 1967,6 describing a method to access stilbenes via aromatic C–H bond cleavage, that years later was shown to operate through a concerted metalation-deprotonation pathway.7–10 In the following years, Shvo and co-workers reported a ruthenium cyclopentadienone complex as a hydrogenation catalyst, capable of heterolytic cleavage of hydrogen–hydrogen bonds through MLC.11 This catalyst class has been proven effective for (de)hydrogenations of C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N/CN bonds and was mechanistically a forerunner of many follow-up methodologies involving (transfer-) hydrogenation (including asymmetric versions) that proposed the concept of MLC as a mechanistic rational.12–14
O and C–N/CN bonds and was mechanistically a forerunner of many follow-up methodologies involving (transfer-) hydrogenation (including asymmetric versions) that proposed the concept of MLC as a mechanistic rational.12–14
Despite the success of this MLC concept in catalytic hydrogenations and the demonstration of its potential beyond dihydrogen activation (e.g. Si–H, B–H, and C–H),15,16 its application in asymmetric catalysis remains largely limited to (de)hydrogenative transformations. Moreover, the reported mechanistic aspects of hydrogenative transformations have been revisited several times and the proposed ‘classical’ MLC pathway has been challenged following new computational and experimental data.17–21 How catalytic mechanisms extend to systems beyond hydrogenation processes requires further investigation and it presents a great opportunity not only to develop new synthetic methodologies but also to test mechanistic hypotheses.
In order to assess the potential of already developed MLC-type hydrogenation catalysts and their structural analogues for applications in asymmetric methodologies beyond hydrogenative synthesis, here we review the development of relevant (de)hydrogenation catalysts and mechanistic proposals as well as recently developed non-hydrogenative transformations.
Before proceeding, it is worth noting that different literature reports use the terms ‘MLC catalysis’ and ‘metal–ligand bifunctional catalysis’ to describe a similar concept with subtle differences in the role of the ligand, with MLC catalysis being narrowed down to cases involving participation of both the metal and the ligand in the bond cleavage/formation events through their chemical modification, meaning the ligand becomes chemically non-innocent.14,18,22–24 In this work, we will use these terms synonymously, implying that both terms cover cases with active ligand participation, including ligand deprotonation/protonation as well as ligand-driven substrate polarization or outer-sphere stabilization of the product intermediate via hydrogen bonding, thus involving both chemically innocent and non-innocent ligands. When discussing different mechanistic aspects, we will use these definitions interchangeably, trying to adhere to the terms described in the original reports.
Finally, the viewpoint does not aim to be comprehensive but rather focuses on the examples relevant for its conclusions.
MLC in asymmetric (transfer) hydrogenation
In 1995, Noyori and co-workers described a ruthenium-based complex, Cat-1, bound to both a chiral diphosphine and a diamine ligand, which turned out to be a highly efficient catalyst for the enantioselective hydrogenation of arylketones with excellent stereoselectivity and favouring hydrogenation of carbonyl groups over olefin groups with remarkable chemoselectivity (Scheme 1a).25,26 Later, Noyori and Ikariya developed another ruthenium catalyst that contains a similar diamine functionality together with an η6-coordinated arene, Cat-2, which shows exceptional performance in asymmetric transfer hydrogenation (ATH) reactions (Scheme 1b).27 Catalysts based on these original designs have found numerous applications in the pharmaceutical industry due to their high reactivity, chemo- and stereoselectivity.28 Because of this remarkable contribution to the field of asymmetric catalysis, Noyori was one of the Nobel prize laureates in 2001.28–30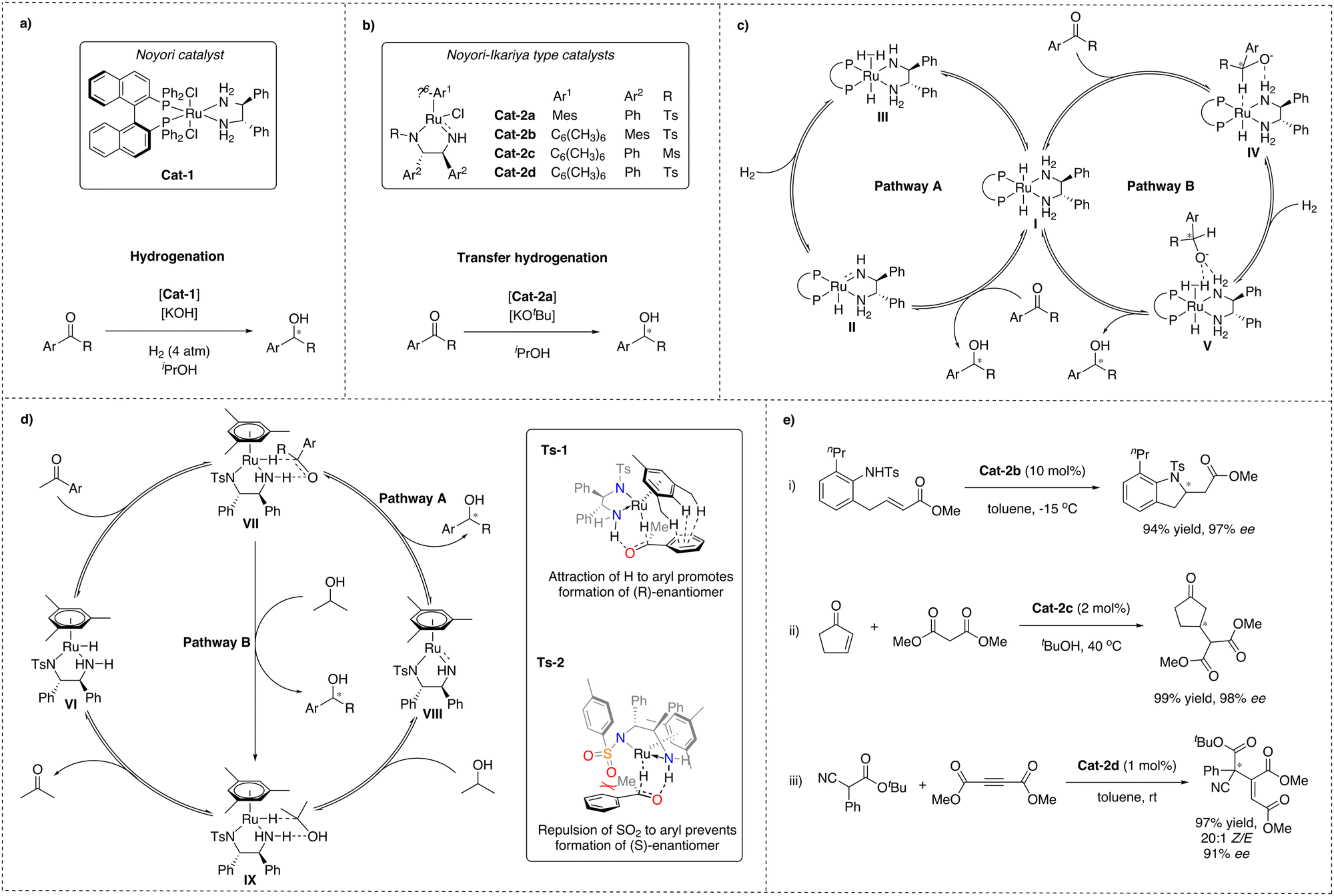 | ||
| Scheme 1 (a) Asymmetric hydrogenation using Noyori catalyst; (b) asymmetric transfer hydrogenation using Noyori–Ikariya type catalysts; (c) simplified mechanisms for asymmetric hydrogenation using Noyori's catalyst; (d) simplified mechanism for asymmetric transfer hydrogenation using Noyori–Ikariya type catalysts with stereodetermining catalyst–substrate interactions TS-1 and TS-2; (e) selected examples of CH and NH bond activation by Noyori–Ikariya type catalysts. | ||
For both catalyst types, the effect of the NH-functionality was found to be crucial for the catalytic performance in hydrogenations and transfer hydrogenations, as tertiary amines were found to be inactive, pointing at an MLC mode of action and chemical non-innocence of the ligand. Extensive effort was made to understand the mechanistic details responsible for the high (up to 99%) CO/CC chemo- and enantioselectivities, exhibited by Noyori and Ikariya catalysts and the role of the NH-moiety of the ligand. This has led to the identification of two main scenarios (Scheme 1c and d).27,31,32
In the first scenario (pathway A), based on early studies by Noyori and Ikariya, the NH motif of the diamine ligand undergoes protonation and deprotonation during the course of the reaction. It was proposed that in the case of hydrogenation, the H2 cleavage would occur via cooperative action of the metal and a deprotonated amide ligand formed in the presence of a base, leading to amine-hydride complex I.33 In the case of transfer hydrogenation, the Noyori–Ikariya catalyst (Scheme 1b) would transform into the active hydride species VI in the presence of a sacrificial hydrogen donor, e.g. isopropanol, often introduced as a solvent, and a catalytic amount of an alkoxide base. Subsequent concerted hydride and proton transfer to the substrate was proposed to occur next. Finally, the active form of the catalysts is regenerated from the resulting Ru-amido complex II and VIII upon reaction with H2 for the former and isopropanol for the latter.34,35 This scenario implies that hydrogenation and transfer hydrogenation take place through the outer-sphere of the metal catalysts via concerted transfer of both the metal RuH and the protic NH from the ligand to the carbonyl group via a six-membered transition state. This is opposite to the at the time more commonly accepted inner-sphere mechanism of transition metal-based hydrogenation catalysts.36
Later studies of these metal–ligand bifunctional catalytic systems revealed a second scenario, constituting an alternative, more complex pathway (pathway B) for both Noyori hydrogenation and Noyori–Ikariya transfer hydrogenation catalysts. According to this revised mechanism, the ketone substrate is in both cases predominantly reduced via consecutive outer-sphere hydride transfer from the catalyst's metal center to the unbound substrate, followed by proton transfer from an η2-H2 ligand and/or protic solvent.14,32 This also suggests that the NH functionality of the ligands is involved in the catalytic reaction via stabilization of the determining transition states through N–H⋯O hydrogen-bonding interactions, rather than via the originally proposed reversible proton transfer, and therefore, the ligand in this case would be chemically innocent.18
Both scenarios have been studied experimentally and in silico, leading to the conclusion that a stepwise process is energetically more favorable in solution.14,17,18,31,33,34,37,38 Furthermore, it was suggested that the source of protonation of the product intermediate in the revised mechanism of Noyori–Ikariya transfer hydrogenation is dependent on the reaction conditions. Solvents such as water or formic acid mixtures favor proton transfer from the protic solvent (pathway B), while use of isopropanol as solvent or increased acidity of the NH group of the ligand favor pathway A, although not in a concerted, but stepwise manner. It cannot be ruled out that both protonation pathways contribute to the overall reaction profile.17,32,37
Finally, to explain the experimentally observed acceleration of the reaction rate in the presence of high loadings of an inorganic base, for Noyori's catalyst an additional variation of pathway B has been proposed to contribute as well, in which the NH hydrogen atom is replaced by a metal cation, able to stabilise the alkoxylate via electrostatic forces, thereby replacing the hydrogen bond interaction.17,38,39
In both scenarios, hydride transfer from the ruthenium center to the ketone is considered to be the stereodetermining step.33,37,40 For the Noyori–Ikariya catalyst enantiodiscrimination has been mainly attributed to attractive π-CH interactions between the aryl moiety of the arylketone and the η6-arene moiety of the ruthenium complex (Scheme 1d, TS-1) as well as to repulsive interactions between the π-electrons of the arylketone and the lone pair on the SO2 moiety of the diamine ligand (Scheme 1d, TS-2).40,41
C–H acidic heterolytic bond activation for C–C and C–N couplings
Importantly, in addition to hydrogenative transformations, Noyori–Ikariya type catalysts have also been reported to be effective for enantioselective C–C and C–N bond formations via activation of CH and NH acidic substrates (Scheme 1e). Specific examples are the intramolecular aza-Michael reaction (i),42 conjugate addition of 1,3-dicarbonyl substrates to Michael acceptors (ii)43,44 and addition of α-cyanoacetates to acetylenic esters (iii)45 and dimethylazodicarboxylates.46 A common mechanistic characteristic of these examples are substrate activation by deprotonation via the amido ligand. Interestingly, the reaction with acetylenic esters shows a high Z-selectivity, which has been rationalised by a stereospecific protonation of the coordinated reaction intermediate via the NH functionality of the catalyst.47 This indicates that the NH moiety might have two functions: facilitating stereoselective coordination of the substrate to the metal and serving as a donor for stereospecific protonation. Nevertheless, the scope of the reported non-hydrogenative asymmetric transformations catalysed by Noyori–Ikariya type catalysts is mainly limited to these few examples.Pincer complexes
Noyori's discovery has also influenced the development of another class of catalyst structures that contain a tridentate, predominantly meridionally coordinated ligand and also an acidic functionality capable of engaging in MLC upon base activation.24,48 This class of catalysts, referred to as ‘pincer’ complexes, a terminology introduced by van Koten et al., exhibits unique properties.49 The general structure of pincer complexes consists of a central donor atom (e.g. NH, NPyridine, C) connected to two flanking donor atoms (P, N, S,O, C) (Scheme 2a).50 This blueprint allows for facile tuning of ligand properties. Where the flanking donor atom can be varied to stabilise different metal ions, tune electron density at the metal center and introduce steric hindrance, the nature of the central donor ligand is pivotal for the reactivity of the corresponding complex.48 These remarkable characteristics of pincer complexes have resulted in the development of numerous methodologies, including (de)hydrogenations and dehydrogenative couplings relevant for industry and sustainable synthesis efforts.51–54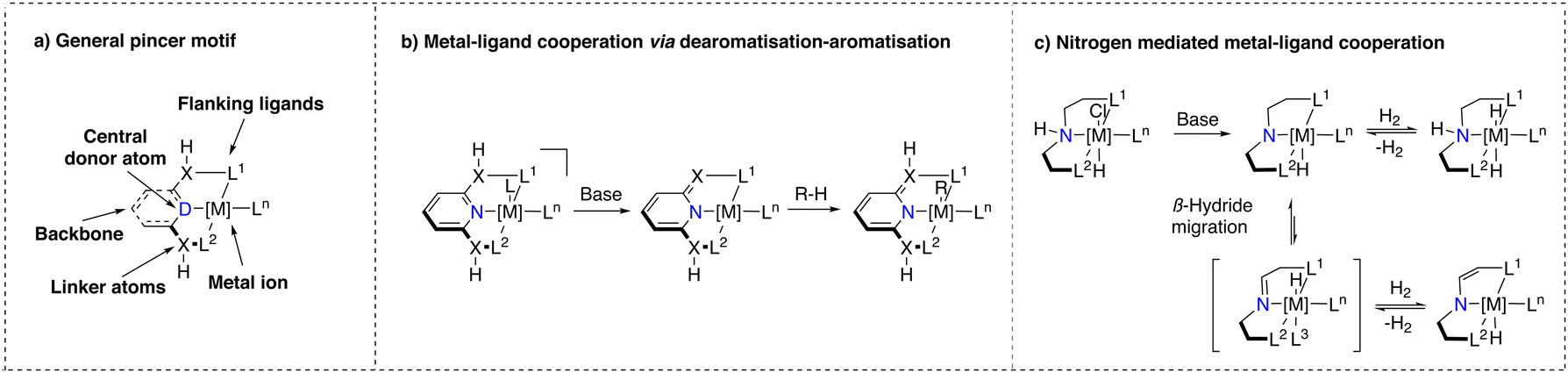 | ||
| Scheme 2 (a) General pincer complex structure; (b) base induced dearomatisation–aromatisation MLC activation mode of Milstein pyridyl type pincer complexes; (c) base induced MLC activation modes of NH-only type pincer complexes. | ||
Bond activation with pincer complexes through MLC
Pincer complexes (Scheme 2a) that were suggested to engage in MLC can be divided into two major classes: (i) ones that bear ligands consisting of a pyridine core with either a CH254 or an NH55 linker, which we will refer to as ‘Milstein pyridyl’ type pincer complexes (Scheme 2b) and (ii) ones that bear a central secondary amine donor atom,24,50 which we will refer to as ‘NH-only’ type pincer complexes (Scheme 2c). In the presence of a base, the former class undergoes a dearomatisation process initiated by deprotonation of the linker, whereas the latter creates a basic amido moiety. Upon deprotonation, the corresponding complexes can activate reactants through MLC.In 2005, Milstein and co-workers were the first to report on pincer complexes that can undergo reversible dearomatisation.56 These complexes were found to be effective for activating various bonds and it is proposed that the linker of the dearomatised ligand plays a crucial role as nucleophilic acceptor to activate an electrophilic substrate followed by re-aromatisation.3,54 This MLC action mode has attracted much interest from both experimentalists and theoreticians who have extensively studied the mechanism involving the aromatisation/dearomatisation process during the catalytic cycle of the corresponding reactions. However, several recent studies have also pointed out that the originally proposed mechanism of hydrogenation/dehydrogenation reactions with Milstein type catalyst may not necessarily follow the ‘classical’ MLC pathway observed under stoichiometric conditions due to likely differences in speciation/conditions when comparing stoichiometric and catalytic reactions.17,57–61
In case of the NH-only type pincer complexes (Scheme 2c), their ability to engage in MLC bond activation was first proposed by Fryzuk in the 1980s for the hydrogenation of olefins.62–64 The proposed reactivity pattern of these complexes is strikingly similar to that of the Noyori–Ikariya catalysts.
Deprotonation of the secondary amine results in a basic amido moiety, capable of stabilizing a coordinatively unsaturated metal species and activating H–H bonds. Depending on the nature of the metal and ligands, a β-hydride migration step, followed by subsequent dihydrogen elimination (or deprotonation) can lead to an enamine modification of the pincer ligand, which influences the geometry and hence the ligand field distortion (Scheme 2c).65 This structure shows similarities to the dearomatised Milstein-pyridyl complexes described earlier. NH-only type pincer complexes have been shown to exhibit high catalytic activity and stability in numerous hydrogen-based transformations.66–69 However, further heterolytic HX bond activations are scarce.70
MLC pincer complexes in enantioselective reactions
As the field progressed, asymmetric transformations using chiral MLC pincer complexes were explored as well. For the Milstein pyridyl type complexes, chirality was introduced primarily at the linker moiety and the flanking donor ligand (Scheme 3a). In 2015, Castillón and Díaz reported a ruthenium-based complex bearing P-stereogenic flanking donors for the hydrogenation of arylketones.71 Good results with enantioselectivities up to 95% were obtained only at low temperature (−40 °C). It is suggested that conformational equilibria of the catalyst may account for this temperature effect.72 In addition, Mezzetti and co-workers reported in 2018 an earth abundant variant using iron which was subjected to enantioselective hydrogenation of acetophenone.73 In contrast to the work of Castillón and Díaz, rapid conformational interconversion could not be prevented, resulting in mediocre enantiomeric excesses for this transformation.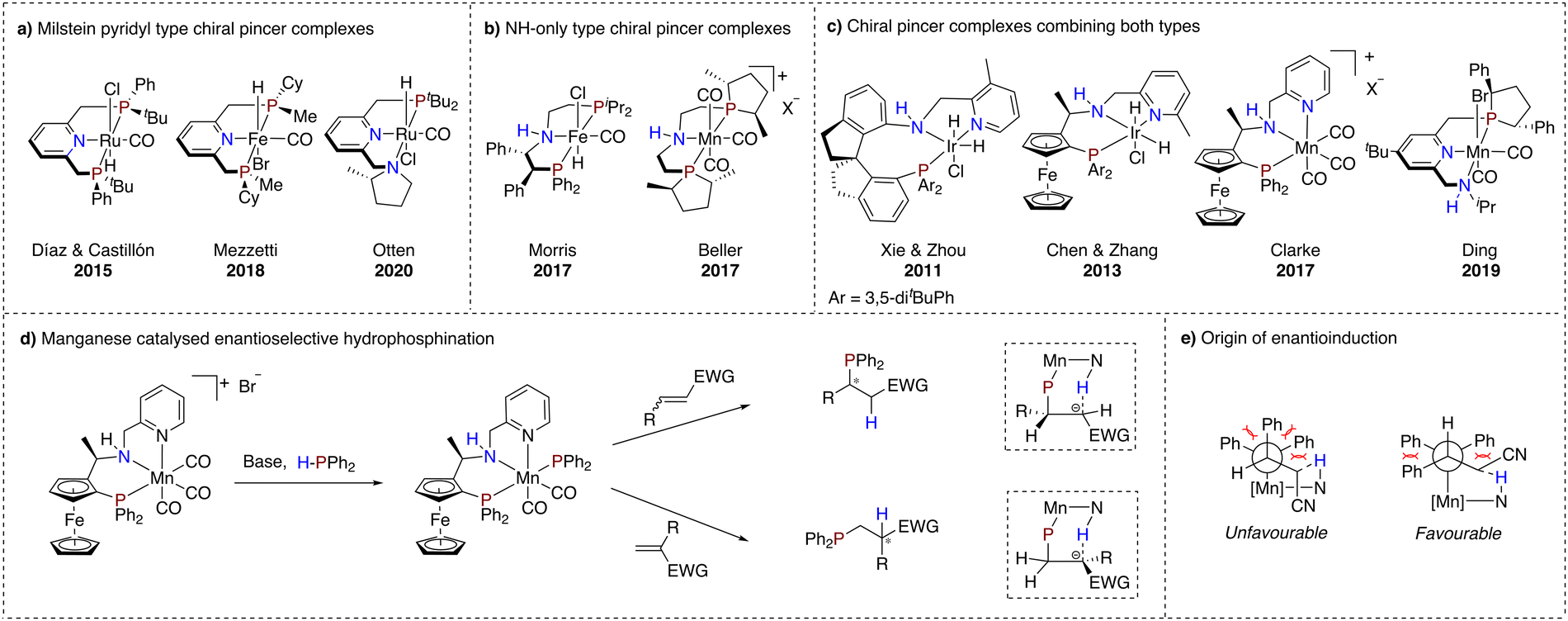 | ||
| Scheme 3 (a) Chiral Milstein pyridyl type pincer complexes; (b) chiral NH-only type pincer complexes; (c) chiral pincer complexes with merged structural features of both MLC active pincer types; (d) enantioselective hydrophosphination of α,β-unsaturated compounds catalysed by Clarke's manganese catalyst; (e) proposed origin of enantioinduction for the manganese catalysed enantioselective hydrophosphination. | ||
The conformational problems of these types of complexes were further described by Otten and co-workers, showcasing two challenges for stereoretention of dearomatised asymmetric ruthenium hydride PNN pincer complexes with a chiral pyrrolidine flanking donor (Scheme 3a).74 The issues are associated with inversion of stereochemistry on the chiral amine and epimerisation of a metal-hydride bond facilitated by the dearomatised linker moiety.
On the other hand, NH-only type pincer complexes have successfully been employed in asymmetric (transfer) hydrogenation of ketones and imines. Remarkably, earth abundant metal complexes have not fallen short within this development trajectory (Scheme 3b). In fact, chirality introduced in an iron complex via substitution on the ethyl linker,75,76 as well as in a manganese complex through chiral phospholane flanking donor substituents, has led to chiral alcohol and amine products with enantiomeric excesses exceeding 80%.77
The two types of reviewed pincer motifs display almost complementary advantages. The Milstein pyridyl type complexes have been suggested to engage in cooperative activation of various bonds, but are less developed for asymmetric transformations. On the other hand, NH-only type pincer complexes are known to catalyse asymmetric (transfer) hydrogenations, but are underexplored in terms of further HX bond activation. Thus, combining these two different ligand motifs in one pincer complex potentially offers more opportunities in asymmetric catalysis (Scheme 3c).
In this context, a very effective iridium complex for the hydrogenation of arylketones with both a dearomatisable pyridine ring and a central NH moiety was reported by Xie and Zhou in 2011.78 MLC was suggested to operate via the NH moiety, while the pyridine ring was proposed to simply add stability to the catalyst. Following up on this work, in 2013, Chen and Zhang reported an enantioselective hydrogenation reaction with a similar iridium complex, containing planar chirality as a chiral element in addition to a carbon stereocenter.79 In 2017, Clarke and co-workers reported the first example of a manganese catalysed enantioselective hydrogenation using a similar ligand structure.80 Interestingly, a facial coordination was found for this complex. Another example of a manganese-based catalyst that merges structural features of both types of MLC-capable pincer complexes was reported by Ding and co-workers.81 Mechanistic studies in this case suggest an outer sphere type mechanism, mediated by the NH functionality of the flanking donor.
Surprisingly, to date only the manganese complex reported by Clarke has been demonstrated to catalyse different asymmetric transformations. In 2021, the group of Harutyunyan reported the first enantioselective hydrophosphination of unsaturated conjugated nitriles using this catalyst.82 It is noteworthy that both internal and terminal unsaturated nitriles yielded enantiomerically pure products, where the former results from enantioselective phosphination and the latter from stereospecific protonation, which was attributed to an MLC mode of action of the catalyst through the NH functionality of the ligand (Scheme 3d).
In addition to the hydrophosphination of nitriles, the catalyst system was also successfully applied for the enantioselective hydrophosphination of α,β-unsaturated esters, ketones, carboxamides, phosphine oxides and trifluoromethyl substituted substrates.83–85 Based on initial experimental and theoretical studies, it was proposed that activation of the H–P bond occurs through MLC, initiated by treatment of the manganese catalyst with a base (Scheme 3d). The transfer of the resulting phosphide from the manganese atom to the alkene was put forward as the stereodetermining step of this transformation. Steric interactions between the alkene substrate and the phenyl substituents at the phosphorus atom, as well as by hydrogen bonding between the NH functionality of the catalyst and the α-carbon of the alkene, are the main processes that control the stereochemistry (Scheme 3e). The observed enantioselectivity for terminal alkenes was rationalised by the subsequent same face stereospecific intramolecular protonation step, taking place after the transfer of the diphenylphosphide from the manganese center to the β-carbon of the alkene.82 Crucial for the successful H–P bond activation and subsequent enantioselective hydrophosphination are the interplay between the metal and suggested non-innocence of NH and pyridine functionalities of the ligand.
Conclusions and outlook
Since its discovery, MLC has become a versatile tool for catalysis, especially in the field of (transfer) hydrogenation. Noyori's first reports described a bifunctional ruthenium catalyst with a diamine ligand bearing an NH functionality for the enantioselective (transfer) hydrogenation of aryl ketones. This work foreshadowed the development of MLC-active tridentate pincer complexes, which subsequently found widespread application in catalysis. The two most investigated MLC activation modes either operate via dearomatisation of a pyridyl moiety with subsequent re-aromatisation upon bond activation or contain a central non-innocent NH functionality that can act as a hydrogen bond donor or proton relay. The former activation mode has been shown to activate various bonds, such as NH, CN and CH, but is underdeveloped for enantioselective transformations. In contrast, pincer scaffolds with a central NH functionality have been successfully used in various enantioselective (transfer) hydrogenation methodologies by introducing chirality to the linker and flanking donor framework, but their ability to activate other chemical bonds has not been studied in depth.Both types of ligand motifs (a dearomatisable pyridyl flanking donor and a secondary NH functionality) have also been combined, leading to, among others, the highly efficient facially coordinated manganese pincer complex reported by Clarke. Apart from catalysing enantioselective (transfer) hydrogenations, this complex enabled the development of a general catalytic enantioselective hydrophosphination method for a wide range of activated terminal and internal alkenes. The interplay between the two modes of activation of the MLC present within the single pincer complex was suggested to be a crucial feature for the observed broad substrate scope in this transformation. The various reactivity patterns underline the potential of MLC in pincer complexes for versatile enantioselective bond activation.
Neither ligand nor coordinated metal exploration has been exhausted so far. New MLC chiral pincer complexes with earth abundant metal ions and ligand variations could further expand the type of bonds that can be activated with these complexes, particularly with respect to heterolytic HX bond activation.86 Opportunities lie, for instance, in exploration of the reactivity of unsaturation in the ligand backbone in conjunction with NH-only functionalities. Furthermore, the tunable charge density of an activated amido functionality could provide control over electrostatic interactions and substrate polarisation, potentially relevant for nucleophilic substitution reactions. Finally, the reversibility of the discussed MLC activation modes could result in proton relay, facilitating isomerisation reactions and the stabilisation of reactive species.87
Nevertheless, the mechanistic picture of operating base-activated MLC pincer complexes remains intricate. Mechanistic details on the role of the NH functionality in (transfer-) hydrogenations, for example, are still under investigation, and different mechanistic routes are possible depending on the reaction conditions. Especially for chiral pincer ligands, formation of several isomers of the resulting complexes, including facial and meridional coordination must be taken into account.70,75 Despite this ambiguity, the initially proposed MLC concept has resulted in the development of a large number of methodologies, used in many useful applications. However, opportunities arising from this mechanistic framework go hand in hand with low predictability of reactivity profiles. Hence, more work is required on rational design and mechanistic understanding of chiral pincer complexes, especially for structures combining different MLC activation modes. This development direction may allow the promising potential of MLC pincer complexes to be fully exploited in various asymmetric transformations, including for instance heterolytic bond activation of phosphines, amines, alcohols and thiols, as well as activation of CH acids, nucleophilic substitution reactions and isomerisations.88
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. C. Chadwick, R. Duchateau, Z. Freixa and P. W. N. M. van Leeuwen, Homogeneous Catalysts: Activity–Stability–Deactivation, Wiley-VCH, Weinheim, 2011 Search PubMed.
- Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, ed. B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Wiley-VCH, Weinheim, 3rd edn, 2017 Search PubMed.
- Metal-Ligand Co-operativity, ed. G. van Koten, K. Kirchner and M.-E. More, Springer, Cham, 2020, vol. 68 Search PubMed.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2018, 2, 0099 CrossRef CAS.
- Asymmetric Hydrogenation and Transfer Hydrogenation, ed. V. Ratovelomanana-Vidal and P. Phansavath, Wiley-VCH, Weinheim, 2021 Search PubMed.
- I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119–1122 CrossRef.
- M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed.
- M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496–16497 CrossRef CAS PubMed.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754–13755 CrossRef CAS PubMed.
- D. García-Cuadrado, A. A. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066–1067 CrossRef PubMed.
- Y. Shvo, D. Czarkie, Y. Rahamin and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- R. Karvembu, R. Prabhakaran and N. Natarajan, Coord. Chem. Rev., 2005, 249, 911–918 CrossRef CAS.
- P. A. Dub and J. C. Gordon, Nat. Rev. Chem., 2018, 2, 396–408 CrossRef CAS.
- C. Gunanathan and D. Milstein, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed.
- T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756–6781 RSC.
- D. G. Gusev, Organometallics, 2020, 39, 258–270 CrossRef CAS.
- D. G. Gusev and D. M. Spasyk, ACS Catal., 2018, 8, 6851–6861 CrossRef CAS.
- P. A. Dub, B. L. Scott and J. C. Gordon, J. Am. Chem. Soc., 2017, 139, 1245–1260 CrossRef CAS PubMed.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- H. Gruetzmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- T. Ohkuma, H. Ooka, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 10417–10418 CrossRef CAS.
- T. Ohkuma, H. Ikehira, T. Ikariya and R. Noyori, Synlett, 1997, 467–468 CrossRef CAS.
- S. Hashiguchi, A. Fuji, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS PubMed.
- W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998–2007 CrossRef CAS.
- K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2024–2032 CrossRef CAS PubMed.
- T. Ikariyah and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef PubMed.
- N. V. Tkachenko, P. Rublev and P. A. Dub, ACS Catal., 2022, 12, 13149–13157 CrossRef CAS.
- C. A. Sandoval, T. Ohkuma, K. Muñiz and R. Noyori, J. Am. Chem. Soc., 2003, 125, 13490–13503 CrossRef CAS PubMed.
- K.-J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285–288 CrossRef CAS.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466–1478 CrossRef CAS.
- A. Comas-Vives, G. Ujaque and A. Lledós, Adv. Inorg. Chem., 2010, 62, 231–260 CrossRef CAS.
- P. A. Dub and T. Ikariya, J. Am. Chem. Soc., 2013, 135, 2604–2619 CrossRef CAS PubMed.
- P. A. Dub, N. J. Henson, R. L. Martin and J. C. Gordon, J. Am. Chem. Soc., 2014, 136, 3505–3521 CrossRef CAS PubMed.
- Y. Wang, S. Liu, H. Yang, H. Li, Y. Lan and Q. Liu, Nat. Chem., 2022, 14, 1233–1241 CrossRef CAS PubMed.
- M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef CAS PubMed.
- P. A. Dub, N. V. Tkachenko, V. K. Vyas, M. Wills, J. S. Smith and S. Tretiak, Organometallics, 2021, 40, 1402–1410 CrossRef CAS.
- K. Muñiz, A. Lishchynskyi, J. Streuff, M. Nieger, E. C. Escudero-Adán and M. Martínez Belmonte, Chem. Commun., 2011, 47, 4911–4913 RSC.
- M. Watanabe, K. Murata and T. Ikariya, J. Am. Chem. Soc., 2003, 125, 7508–7509 CrossRef CAS PubMed.
- M. Watanabe, A. Ikagawa, H. Wang, K. Murata and T. Ikariya, J. Am. Chem. Soc., 2004, 126, 11148–11149 CrossRef CAS PubMed.
- Y. Hasegawa, I. D. Gridnev and T. Ikariya, Angew. Chem., Int. Ed., 2010, 49, 8157–8160 CrossRef CAS PubMed.
- Y. Hasegawa, M. Watanabe, I. D. Gridnev and T. Ikariya, J. Am. Chem. Soc., 2008, 130, 2158–2159 CrossRef CAS PubMed.
- Y. Hasegawa, I. D. Gridnev and T. Ikariya, Bull. Chem. Soc. Jpn., 2012, 85, 316–334 CrossRef CAS.
- E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC.
- G. van Koten, Pure Appl. Chem., 1989, 61, 1681–1694 CrossRef CAS.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- S. Elangovan, C. Topf, S. Fischer, H. Jiao, A. Spannenberg, W. Baumann, R. Ludwig, K. Junge and M. Beller, J. Am. Chem. Soc., 2016, 138, 8809–8814 CrossRef CAS PubMed.
- P. Nad and A. Mukherjee, Asian J. Org. Chem., 2021, 10, 1958–1985 CrossRef CAS.
- C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- H. Li, T. P. Gonçalves, D. Lupp and K.-W. Huang, ACS Catal., 2019, 9, 1619–1629 CrossRef CAS.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, J. Am. Chem. Soc., 2005, 127, 10840–10841 CrossRef CAS PubMed.
- X. Yang and M. B. Hall, J. Am. Chem. Soc., 2010, 132, 120–130 CrossRef CAS PubMed.
- H. Li and M. B. Hall, J. Am. Chem. Soc., 2014, 136, 383–395 CrossRef CAS PubMed.
- H. Li and M. B. Hall, ACS Catal., 2015, 5, 1895–1913 CrossRef CAS.
- D. G. Gusev, Organometallics, 2020, 39, 258–270 CrossRef CAS.
- T. He, J. C. Buttner, E. F. Reynolds, J. Pham, J. C. Malek, J. M. Keith and A. R. Chianese, J. Am. Chem. Soc., 2019, 141, 17404–17413 CrossRef CAS PubMed.
- M. D. Fryzuk and P. A. MacNeil, Organometallics, 1983, 2, 682–684 CrossRef CAS.
- M. D. Fryzuk, P. A. MacNeil and S. J. Rettig, Organometallics, 1985, 4, 1145–1147 CrossRef CAS.
- M. D. Fryzuk, P. A. MacNeil and S. J. Rettig, J. Am. Chem. Soc., 1987, 109, 2803–2812 CrossRef CAS.
- S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412–429 CrossRef CAS.
- C. M. Hert, J. B. Curley, S. P. Kelley, N. Hazari and W. H. Bernskoetter, Organometallics, 2022, 41, 3332–3340 CrossRef CAS.
- D. H. Nguyen, X. Trivelli, F. Capet, J.-F. Paul, F. Dumeignil and R. M. Gauvin, ACS Catal., 2017, 7, 2022–2032 CrossRef CAS.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- W. Kuriyama, T. Matsumoto, O. Ogata, Y. Ino, K. Aoki, S. Tanaka, K. Ishida, T. Kobayashi, N. Sayo and T. Saito, Org. Process Res. Dev., 2012, 16, 166–171 CrossRef CAS.
- P. Hermosilla, P. Lopéz, P. García-Orduña, F. J. Lahoz, V. Polo and M. A. Casado, Organometallics, 2018, 37, 2618–2629 CrossRef CAS.
- I. Arenas, O. Boutureira, M. I. Matheu, Y. Díaz and S. Castillón, Eur. J. Org. Chem., 2015, 3666–3669 CrossRef CAS.
- R. Huber, A. Passera and A. Mezzetti, Chem. Commun., 2019, 55, 9251–9266 RSC.
- R. Huber, A. Passera and A. Mezzetti, Organometallics, 2018, 37, 396–405 CrossRef CAS.
- J. Bootsma, B. Guo, J. G. de Vries and E. Otten, Organometallics, 2020, 39, 544–555 CrossRef CAS.
- P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367–1380 CrossRef CAS PubMed.
- S. A. M. Smith, P. O. Lagaditis, A. Luepke, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2017, 23, 7212–7216 CrossRef CAS PubMed.
- M. Garbe, K. Junge, S. Walker, Z. Wie, H. Jiao, A. Spannenberg, S. Bachmann, M. Scalone and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 11237–11241 CrossRef CAS PubMed.
- J.-H. Xie, X.-Y. Liu, J.-B. Xie, L.-X. Wang and L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 7329–7332 CrossRef CAS PubMed.
- H. Nie, G. Zhou, Q. Wang, W. Chen and S. Zhang, Tetrahedron: Asymmetry, 2013, 24, 1567–1571 CrossRef CAS.
- M. B. Widegren, G. J. Harkness, A. M. Z. Slawin, D. B. Cordes and M. L. Clarke, Angew. Chem., Int. Ed., 2017, 56, 5825–5828 CrossRef CAS PubMed.
- L. Zhang, Y. Tang, Z. Han and K. Ding, Angew. Chem., Int. Ed., 2019, 58, 4973–4977 CrossRef CAS PubMed.
- J. M. Pérez, R. Postolache, M. Castiñeira Reis, E. G. Sinnema, D. Vargová, F. de Vries, E. Otten, L. Ge and S. R. Harutyunyan, J. Am. Chem. Soc., 2021, 143, 20071–20076 CrossRef PubMed.
- R. Postolache, J. M. Pérez, M. Castiñeira Reis, L. Ge, E. G. Sinnema and S. R. Harutyunyan, Org. Lett., 2023, 25, 1611–1615 CrossRef CAS PubMed.
- L. Ge and S. R. Harutyunyan, Chem. Sci., 2022, 13, 1307–1312 RSC.
- L. Ge, E. G. Sinnema, J. M. Pérez, R. Postolache, M. Castiñeira Reis and S. R. Harutyunyan, Sci. Adv., 2023, 9, eadf8742 CrossRef PubMed.
- Y. Liang, U. K. Das, J. Luo, Y. Diskin-Posner, L. Avram and D. Milstein, J. Am. Chem. Soc., 2022, 144, 19115–19126 CrossRef CAS PubMed.
- H. Liu, D. Leow, K.-W. Huang and C.-H. Tan, J. Am. Chem. Soc., 2009, 131, 7212–7213 CrossRef CAS PubMed.
- S. Perdriau, D. S. Zijlstra, H. J. Heeres, J. G. de Vries and E. Otten, Angew. Chem., Int. Ed., 2015, 54, 4236–4240 CrossRef CAS PubMed.
Footnote |
| † Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |