
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO2 cleavage by tantalum/M (M = iridium, osmium) heterobimetallic complexes†
Abdelhak
Lachguar
a,
Christopher Z.
Ye
 bc,
Sheridon N.
Kelly
bc,
Erwann
Jeanneau
d,
Iker
Del Rosal
e,
Laurent
Maron
e,
Laurent
Veyre
a,
Chloé
Thieuleux
a,
John
Arnold
*bc and
Clément
Camp
*a
bc,
Sheridon N.
Kelly
bc,
Erwann
Jeanneau
d,
Iker
Del Rosal
e,
Laurent
Maron
e,
Laurent
Veyre
a,
Chloé
Thieuleux
a,
John
Arnold
*bc and
Clément
Camp
*a
aLaboratory of Catalysis, Polymerization, Processes and Materials (CP2M UMR 5128) CNRS, Universite Claude Bernard Lyon 1, CPE-Lyon, Institut de Chimie de Lyon, 43 Bvd du 11 Novembre 1918, 69616 Villeurbanne, France. E-mail: clement.camp@univ-lyon1.fr
bDepartment of Chemistry, University of California, Berkeley, California 94720, USA. E-mail: arnold@berkeley.edu
cChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
dCentre de Diffractométrie Henri Longchambon, Universite Claude Bernard Lyon 1, 5 Rue de la Doua, 69100 Villeurbanne, France
eUniversité de Toulouse, CNRS, INSA, UPS, UMR5215, LCPNO, 135 Avenue de Rangueil, F-31077 Toulouse, France
First published on 8th July 2024
Abstract
A novel Ta/Os heterobimetallic complex, [Ta(CH2tBu)3(μ-H)3OsCp*], 2, is prepared by protonolysis of Ta(CHtBu)(CH2tBu)3 with Cp*OsH5. Treatment of 2 and its iridium analogue [Ta(CH2tBu)3(μ-H)2IrCp*], 1, with CO2 under mild conditions reveal the efficient cleavage of CO2, driven by the formation of a tantalum oxo species in conjunction with CO transfer to the osmium or iridium fragments, to form Cp*Ir(CO)H2 and Cp*Os(CO)H3, respectively. This bimetallic reactivity diverges from more classical CO2 insertion into metal–X (X = metal, hydride, alkyl) bonds.
The design of synthetic bimetallic complexes associating different metals with complementary Lewis acidic/Lewis basic behaviour has raised interest for cooperative reactivity,1–10 including CO2 activation.11–17 In many instances, these bifunctional complexes lead to CO2 adducts or insertion products, in which a bent CO2 fragment binds across the two metals.18–27 In contrast, only a few heterobimetallic complexes have clearly exhibited the capability to cleave the C–O bond within CO2. Thomas and colleagues reported oxidative CO2 cleavage across the early/late heterobimetallic complex Co(iPr2PNMes)3Zr(THF), yielding (OC)Co(iPr2PNMes)2(μ-O)Zr(iPr2PNMes) at ambient temperature (Scheme 1a).28 The Mazzanti group reported the potassium-assisted reductive cleavage of CO2 by a U(III) siloxide complex, resulting in CO evolution and the formation of a pentavalent uranium oxo complex (Scheme 1b). When the potassium cation is encapsulated in 18-crown-6, bimetallic cooperativity no longer occurs, and a carbonate complex is formed instead. Our group has developed an Ir/Al-based heterobimetallic complex proficient in CO2 deoxygenation, yielding Cp*Ir(CO)H2, Cp*IrH4, and [Al(Py)(OAr)(iBu)]2(μ-O) at room temperature (Scheme 1c).29 Recently, Campos and coworkers reported the use of Al(C6F5)3 for triggering the bimetallic cleavage of Fe-bound CO2 moiety, to form an oxo carbonyl complex (Scheme 1d). The choice of the Al-based Lewis acid partner plays a pivotal role in initiating this reaction, as boron, zinc, or gold Lewis acids did not exhibit activity in this transformation.22 Finding right bimetallic combinations therefore remains a major challenge for controlling reactivity.
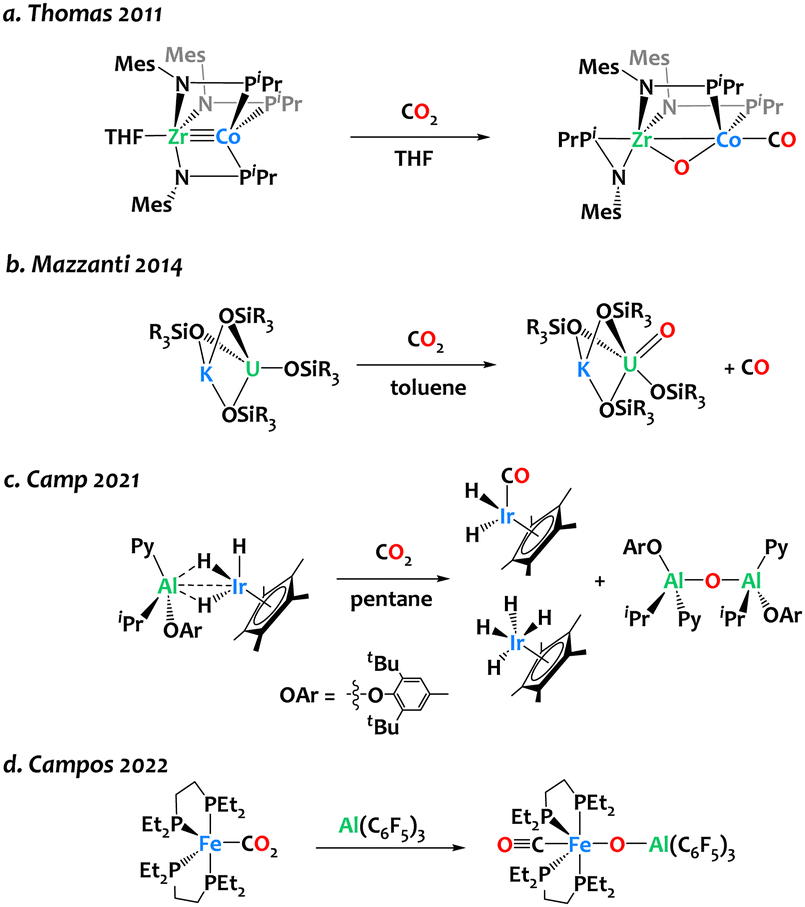 | ||
| Scheme 1 Reports of CO2 cleavage by heterobimetallic complexes relevant to the present study.22,28–30 | ||
The alkane elimination reaction between metal (poly)alkyls and (poly)hydride species has proven efficient for accessing heterobimetallic complexes.31–36 We used this strategy to synthesize compound [Ta(CH2tBu)3IrH2Cp*] 1 from the tantalum tris-neopentyl neopentylidene complex Ta(CHtBu)(CH2tBu)3 and the iridium tetrahydride complex Cp*IrH4 (Scheme 2-top).37 This prompted us to extend this chemistry by investigating the reactivity of Ta(CHtBu)(CH2tBu)3 towards related 6d metal polyhydrides. Treating Ta(CHtBu)(CH2tBu)3 with Cp*OsH538,39 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio in pentane at room temperature yields the heterobimetallic complex [Ta(CH2tBu)3(μ-H)3OsCp*] 2 in 98% isolated yield, accompanied by the elimination of one equivalent of neopentane (Scheme 2-middle). 1H NMR monitoring of the reaction of 2 with Cp*OsH5 (1 equiv.) suggests the slow formation of a trinuclear TaOs2 species (see Fig. S7, ESI†), analogous to the TaIr2 species previously reported.40 Surprisingly, Cp*ReH641 shows no reactivity towards Ta(CHtBu)(CH2tBu)3 either in pentane at room temperature or in C6D6 at 80 °C. DFT calculations indicate that the Ta/Re analogue should be thermodynamically stable: this observed lack of reactivity is thus surprising, and might be due to the lack of available coordination site at Re (see ESI† for discussion).
:1 stoichiometric ratio in pentane at room temperature yields the heterobimetallic complex [Ta(CH2tBu)3(μ-H)3OsCp*] 2 in 98% isolated yield, accompanied by the elimination of one equivalent of neopentane (Scheme 2-middle). 1H NMR monitoring of the reaction of 2 with Cp*OsH5 (1 equiv.) suggests the slow formation of a trinuclear TaOs2 species (see Fig. S7, ESI†), analogous to the TaIr2 species previously reported.40 Surprisingly, Cp*ReH641 shows no reactivity towards Ta(CHtBu)(CH2tBu)3 either in pentane at room temperature or in C6D6 at 80 °C. DFT calculations indicate that the Ta/Re analogue should be thermodynamically stable: this observed lack of reactivity is thus surprising, and might be due to the lack of available coordination site at Re (see ESI† for discussion).
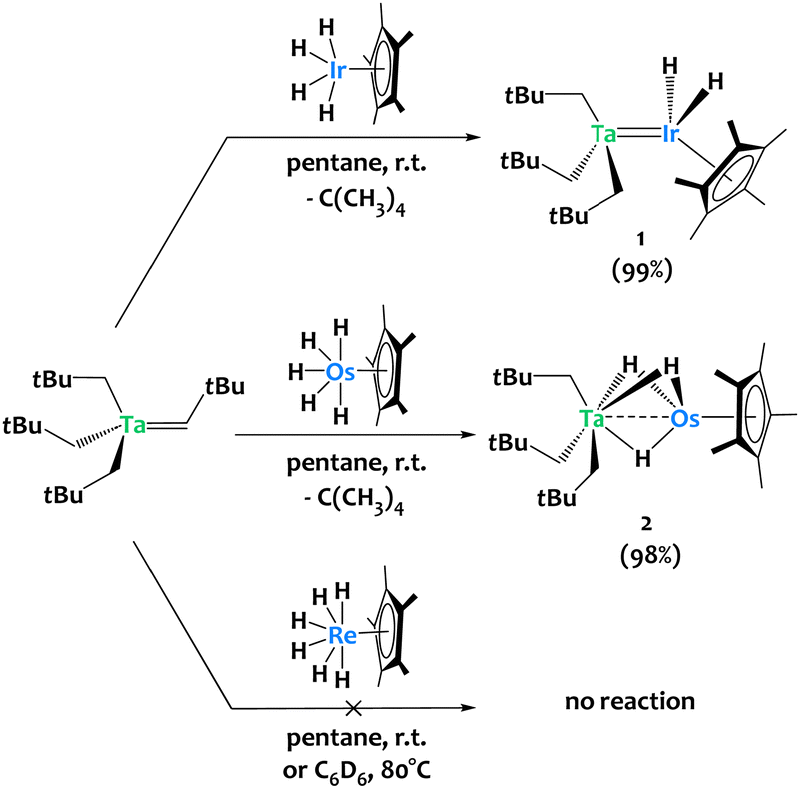 | ||
| Scheme 2 Reactivity of Ta(CHtBu)(CH2tBu)3 towards Cp*IrH4, Cp*OsH5 and Cp*ReH6. | ||
Identification of 2 is confirmed through a range of analytical methods including infrared (IR) and multinuclei (1H, 13C, 1H–1H COSY, 1H–13C HSQC and HMBC) solution NMR spectroscopy, elemental analysis, and X-ray diffraction studies. In the 1H-NMR spectrum of 2 obtained in a toluene-d8 solution, the hydride resonance appears as a high-field singlet at δ = −6.90 ppm, integrating for 3H. The hydride resonance in 2 exhibits a shift of Δδ = +4.3 ppm from Cp*OsH5 which is reminiscent of the observed shift from Cp*IrCH4 to 1, of approximately Δδ = +3.5 ppm.37 The IR spectrum of compound 2 displays a characteristic metal-hydride stretching vibration signal at 1961 cm−1, consistent with bridging hydrides. This value deviates significantly from that of complex [Ta(CH2tBu)3IrCp*(H)2], 1, featuring two terminal hydrides (νIr–H = 2061 cm−1, see Fig. S6, ESI†) and that of the Cp*OsH5 precursor, which exhibits a strong absorption at 2083 (s) cm−1 with a minor one at 2214 (w) cm−1.42 For comparison, the metal-hydride stretch is observed at 1982 cm−1 in [Hf(CH2tBu)3(μ-H)3IrCp*]34 and at 1952 and 1970 cm−1 for [Cp2Zr(X)(μ-H)3Os(PMe2Ph)3] (X = Cl or H respectively),43 where the two metal centres are bridged by three hydride ligands.
Single crystals of 2 suitable for X-ray diffraction were grown from a saturated pentane solution at −40 °C. The solid-state structure is depicted in Fig. 1. The nearly linear Ta–Os–Cp*centroid angle (178.1(1)°), indicates the presence of three bridging hydrides between the two metals, arranged in a tripod geometry around the {Cp*Os} core. This angle aligns well with values reported for systems featuring similar bridging hydride motifs, such as [CpRu(μ-H)4OsCp*] (179.2(9)°)44 and [Hf(CH2tBu)3(μ-H)3IrCp*] (179.2(3)°),34 but starkly contrasts with that found in complex 1, featuring two terminal Ir–H moieties (151.3(1)°). The Ta–CNp bond lengths (with an average value of 2.137(5) Å) are consistent with neopentyl groups.45–47 The Ta–Os distance in compound 2 is 2.4817(2) Å, which is 0.115 Å shorter than the sum of the metallic radii of tantalum (1.343 Å) and osmium (1.255 Å).48 This difference results in a formal shortness ratio (FSR) slightly below unity (FSR = 0.95),49 suggestive of some degree of metal–metal interaction, although the presence of bridging hydrides could also explain the proximity. This FSR value lies between those of complex [Hf(CH2tBu)3(μ-H)3IrCp*] (FSR = 0.99),34 where the close proximity between the Hf and Ir centres likely results from bridging hydrides, and the Ta/Ir complex 1 (FSR = 0.90),37 which exhibits clear double metal–metal bonding.
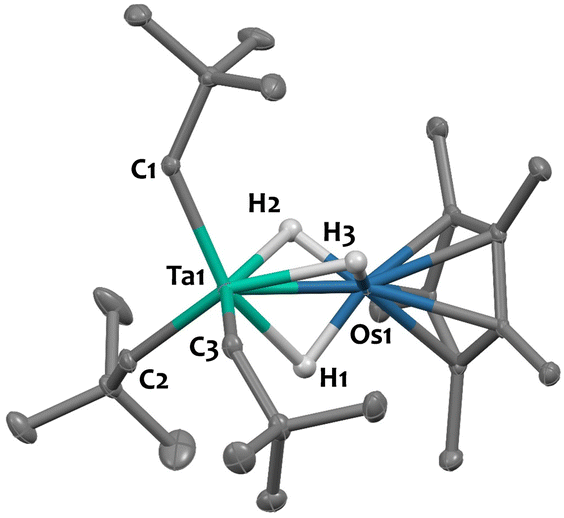 | ||
| Fig. 1 Solid-state molecular structure of 2 (30% probability ellipsoids). Hydrogen atoms from the hydrocarbon ligands are omitted for clarity. Selected bond distances (Å) and angles (°): Ta1–Os1 2.4817(2), Ta1–C1 2.132(3), Ta1–C2 2.136(3), Ta1–C3 2.143(3), Os1–H1 1.45(5), Os1–H2 1.56(5), Os1–H3 1.48(8), Ta1–H1 2.07(5), Ta1–H2 2.03(5), Ta1–H3 2.07(8), Ta1–Os1–Cp*centroid 178.1(1). | ||
To explore the potential of these heterobimetallic complexes in promoting cooperative reactivity, we investigated the reaction of 1 and 2 with CO2 (1 atm, ca. 6 equiv.). The reactions were carried out in tetrahydrofuran (THF) at ambient temperature, resulting in rapid discoloration of the reaction mixture within ca. 10 minutes in both cases. Analysis of the crude reaction mixtures by 1H NMR reveals the complete consumption of complexes 1 and 2, with clean and quantitative formation of compounds Cp*Os(CO)H33 and Cp*Ir(CO)H24, respectively derived from complexes 1 and 2, alongside the generation of a tantalum oxo complex, [Ta(O)(CH2tBu)3]x5 (refer to Fig. S9 and S10 in the ESI†). The insolubility of compound 5 in pentane facilitated its separation from the reaction mixtures by simple evaporation of THF followed by pentane extraction of 3 or 4. Compound 3 was isolated in 96% yield; 1H and 13C NMR data are in agreement with the literature (Scheme 3).29,50
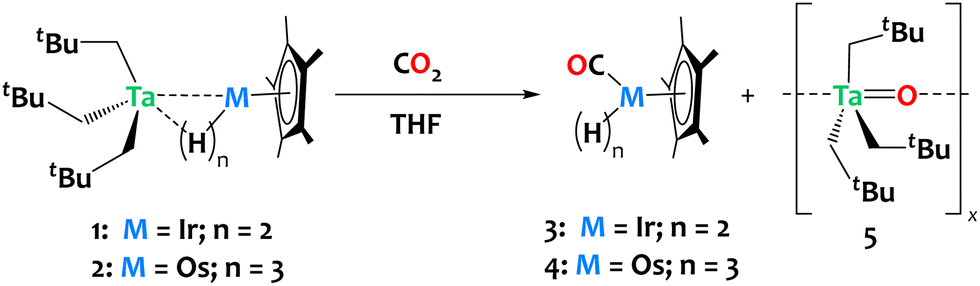 | ||
| Scheme 3 Reaction of compounds 1 and 2 with CO2, yielding Cp*Ir(CO)H2 and Cp*Os(CO)H3, respectively, together with the formation of Ta(O)(CH2tBu)3, 5. | ||
The 1H-NMR spectrum of 4, recorded in THF-d8, indicates that the three hydrides are not equivalent in solution, resulting in two signals at −10.48 ppm and −12.50 ppm integrating for 1H and 2H, respectively and coupling in the 1H–1H COSY NMR spectrum (Fig. S15, ESI†). These signals are assigned to hydrides in -trans and -cis positions relative to the CO group, respectively, which is consistent with literature data.51 The IR spectrum for 4 displays a broad terminal hydride stretching signal at 2075 cm−1, and νCO bands at 1932–1898 cm−1, as expected.51 Diluted THF solutions of complex 4 are stable at room temperature in the dark. Yet compound 4 is reported to be unstable in the solid-state,51 spontaneously eliminating H2 upon drying, which could explain the moderate 45% isolated yield. Regardless, single crystals suitable for X-ray diffraction were obtained by avoiding visible light and crystallisation from pentane at −40 °C. The solid-state structure of 4, determined for the first time in this study, is shown in Fig. 2. The Os1–C1 (1.851(3) Å) and O1–C1 (1.162(4) Å) distances are consistent with those observed in compound [Cp*Os(CO)(μ-H)]2, featuring Os–C bond lengths of 1.833(9) Å and C–O bond lengths of 1.18(1) Å.29
 | ||
| Fig. 2 Solid-state molecular structure of 4 (30% probability ellipsoids). Hydrogen atoms from the Cp* ligand are omitted for clarity. Selected bond distances (Å) and angles (°): Os1–H1 1.53(4), Os1–H2 1.58(4), Os1–H3 1.51(4), Os1–C1 1.851(3), O1–C1 1.162(4), Os1–Cp*centroid 1.916(1), C1–Os1–Cp*centroid 132.5(1). | ||
The 1H-NMR spectrum of 5 indicates that the three CH2tBu groups are equivalent in solution, resulting in two signals at 0.55 ppm and 1.12 ppm for the CH2 and tBu moieties, respectively. Analysis of the 13C{1H}-NMR spectrum of 5 reveals three distinct characteristic resonances at 104.3, 35.1 and 34.4 ppm assigned to the TaCH2, C(CH3)3 and C(CH3)3 moieties, respectively. These assignments are confirmed by the 2D 1H–1H COSY and 1H–13C HSQC and HMBC data (Fig. S19–S21, ESI†). Unfortunately, we were unable to determine the XRD structure of 5, which probably adopts oligomeric structures, given that terminal Ta-oxo species are rare in the literature.52–54 To confirm the identity of 5, we thus carried out a high resolution mass spectrometry analysis using an APCI source, which shows a clear signal for the ion [Ta(O)(CH2tBu)3 + H]+ at 411.2086 m/z (see Fig. S22, ESI†).
The computed reaction mechanism (DFT, B3PW91) is similar for 1 and 2. CO2 undergoes first a kinetically accessible (13 kcal mol−1 for 1, 11 kcal mol−1 for 2) nucleophilic attack by the Ir (or Os) center, which is assisted by oxygen-coordination to Ta. This results in 4-member metallacyclic intermediates shown on Fig. 3. The next step is a C–O bond breaking TS (barrier of 9 kcal mol−1 for 1 and 14 kcal mol−1 for 2) to yield to products 3 (or 4) and 5, which formation is strongly exothermic (see ESI† for reaction profiles).
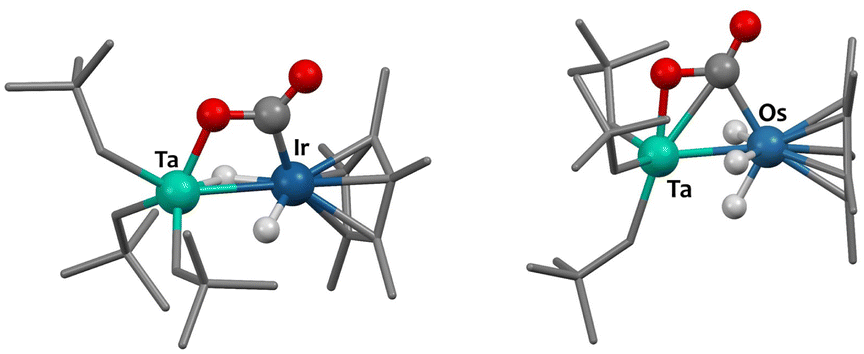 | ||
| Fig. 3 Computed (DFT) structures of the metallacyclic reaction intermediates. | ||
In summary, the reaction between Cp*OsH5 and Ta(CHtBu)(CH2tBu)3 affords a heterobimetallic Ta–Os complex, 2, in high yields via alkane elimination. Complex 2, along with its Ta–Ir analogue, 1, exhibit clean CO2 cleavage reactivity, driven by the formation of a tantalum oxo species in conjunction with late metal carbonyls. Given the propensity of related transition metal alkyls and hydrides for CO2 insertion,55–59 the selective, divergent bimetallic reactivity observed herein is notable. These results clearly further demonstrate how the synergistic action of early/late metal assemblies – particularly those based on tantalum – can facilitate the deoxygenation of CO2. This understanding contributes to advancing knowledge in CO2 activation and could lead to future applications in deoxygenative chemistry.
This work was funded by the European Union (ERC, DUO, 101041762) and the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences Heavy Element Chemistry Program of the U. S. Department of Energy (DOE) at LBNL under contract DE-AC02-05CH11231. Views and opinions expressed are however those of the authors only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. The authors acknowledge the HPCs CALcul en Midi-Pyrenees (CALMIP-EOS grant 0833). LM is a senior member of the Institut Universitaire de France.
Data availability
The data supporting this article have been included as part of the ESI.† CCDC 2351192 and 2351850 contain supplementary crystallographic data for this article; they can be obtained free of charge from The Cambridge Crystallographic Data Centre.Conflicts of interest
There are no conflicts to declare.References
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28–126 CrossRef CAS PubMed.
- E. K. Van Den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985–13011 CrossRef CAS.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- A. Lachguar, A. V. Pichugov, T. Neumann, Z. Dubrawski and C. Camp, Dalt. Trans., 2023, 53, 1393–1409 RSC.
- J. Campos, Nat. Rev. Chem., 2020, 4, 696–702 CrossRef CAS PubMed.
- N. P. Mankad, Chem. Commun., 2018, 54, 1291–1302 RSC.
- B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1–40 CrossRef CAS.
- B. Chatterjee, W. C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024–14055 CrossRef CAS.
- T. S. Hollingsworth, R. L. Hollingsworth, R. L. Lord and S. Groysman, Dalt. Trans., 2018, 47, 10017–10024 RSC.
- C. Z. Ye, I. Del Rosal, S. N. Kelly, I. J. Brackbill, L. Maron, C. Camp and J. Arnold, Chem. Sci., 2024, 15, 9784–9792 RSC.
- C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2023, 62, e202214665 CrossRef CAS PubMed.
- D. Ghosh, S. Sinhababu, B. D. Santarsiero and N. P. Mankad, J. Am. Chem. Soc., 2020, 142, 12635–12642 CrossRef CAS PubMed.
- M. Pérez-Jiménez, H. Corona, F. de la Cruz-Martínez and J. Campos, Chem. – A Eur. J., 2023, 29, e202301428 CrossRef PubMed.
- Z. B. G. Fickenscher, P. Lönnecke, A. K. Müller, O. Hollóczki, B. Kirchner and E. Hey-Hawkins, Molecules, 2023, 28, 2574 CrossRef CAS PubMed.
- J. Ye, R. C. Cammarota, J. Xie, M. V. Vollmer, D. G. Truhlar, C. J. Cramer, C. C. Lu and L. Gagliardi, ACS Catal., 2018, 8, 4955–4968 CrossRef CAS.
- J. R. Prat, C. A. Gaggioli, R. C. Cammarota, E. Bill, L. Gagliardi and C. C. Lu, Inorg. Chem., 2020, 59, 14251–14262 CrossRef CAS PubMed.
- A. Lachguar, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2024 DOI:10.1021/jacs.4c02172.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- M. Devillard, R. Declercq, E. Nicolas, A. W. Ehlers, J. Backs, N. Saffon-Merceron, G. Bouhadir, J. C. Slootweg, W. Uhl and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 4917–4926 CrossRef CAS PubMed.
- G. Fachinetti, C. Floriani and P. F. Zanazzi, J. Am. Chem. Soc., 1978, 100, 7405–7407 CrossRef CAS.
- H. Corona, M. Pérez-Jiménez, F. de la Cruz-Martínez, I. Fernández and J. Campos, Angew. Chem., Int. Ed., 2022, 61, e202207581 CrossRef CAS PubMed.
- C. Yoo and Y. Lee, Chem. Sci., 2016, 8, 600–605 RSC.
- E. G. Lundquist, J. C. Huffman and K. G. Caulton, J. Am. Chem. Soc., 1986, 108, 8309–8310 CrossRef CAS.
- T. A. Hanna, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 11363–11364 CrossRef CAS.
- J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21–23 CrossRef CAS.
- N. J. Hartmann, G. Wu and T. W. Hayton, Chem. Sci., 2018, 9, 6580–6588 RSC.
- J. P. Krogman, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2011, 133, 14582–14585 CrossRef CAS PubMed.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- M. V. Butovskii, C. Döring, V. Bezugly, F. R. Wagner, Y. Grin and R. Kempe, Nat. Chem., 2010, 2, 741–744 CrossRef CAS PubMed.
- C. J. Isaac, F. M. Miloserdov, A. F. Pécharman, J. P. Lowe, C. L. McMullin and M. K. Whittlesey, Organometallics, 2022, 41, 2716–2730 CrossRef CAS PubMed.
- L. Escomel, E. Jeanneau, C. Thieuleux and C. Camp, Inorganics, 2024, 12, 72 CrossRef CAS.
- S. Lassalle, J. Petit, R. L. Falconer, V. Hérault, E. Jeanneau, C. Thieuleux and C. Camp, Organometallics, 2022, 41, 1675–1687 CrossRef CAS.
- C. Z. Ye, I. Del Rosal, M. A. Boreen, E. T. Ouellette, D. R. Russo, L. Maron, J. Arnold and C. Camp, Chem. Sci., 2022, 14, 861–868 RSC.
- L. Escomel, N. Soulé, E. Robin, I. Del Rosal, L. Maron, E. Jeanneau, C. Thieuleux and C. Camp, Inorg. Chem., 2022, 61, 5715–5730 CrossRef CAS PubMed.
- S. Lassalle, R. Jabbour, P. Schiltz, P. Berruyer, T. K. Todorova, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2019, 141, 19321–19335 CrossRef CAS PubMed.
- C. L. Gross and G. S. Girolami, Organometallics, 2007, 26, 160–166 CrossRef CAS.
- C. L. Gross, S. R. Wilson and G. S. Girolami, J. Am. Chem. Soc., 1994, 116, 10294–10295 CrossRef CAS.
- S. Lassalle, R. Jabbour, I. Del Rosal, L. Maron, E. Fonda, L. Veyre, D. Gajan, A. Lesage, C. Thieuleux and C. Camp, J. Catal., 2020, 392, 287–301 CrossRef CAS.
- W. A. Herrmann, H. G. Theiler, P. Kiprof, J. Tremmel and R. Blom, J. Organomet. Chem., 1990, 395, 69–84 CrossRef CAS.
- C. L. Gross and G. S. Girolami, Organometallics, 2007, 26, 160–166 CrossRef CAS.
- J. W. Bruno, J. C. Huffman, M. A. Green and K. G. Caulton, J. Am. Chem. Soc., 1984, 106, 8310–8312 CrossRef CAS.
- T. Shima and H. Suzuki, Organometallics, 2005, 24, 3939–3945 CrossRef CAS.
- L. J. Guggenberger and R. R. Schrock, J. Am. Chem. Soc., 1975, 97, 2935 CrossRef CAS.
- R. Srivastava, E. A. Quadrelli and C. Camp, Dalt. Trans., 2020, 49, 3120–3128 RSC.
- R. Srivastava, R. Moneuse, J. Petit, P.-A. A. Pavard, V. Dardun, M. Rivat, P. Schiltz, M. Solari, E. Jeanneau, L. Veyre, C. Thieuleux, E. A. Quadrelli and C. Camp, Chem. – Eur. J., 2018, 24, 4361–4370 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- F. A. Cotton, Acc. Chem. Res., 1978, 11, 225–232 CrossRef CAS.
- D. M. Heinekey, D. A. Fine, T. G. P. Harper and S. T. Michel, Can. J. Chem., 1995, 73, 1116–1125 CrossRef CAS.
- J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3722–3723 CrossRef CAS.
- J. I. Fostvedt, M. A. Boreen, R. G. Bergman and J. Arnold, Inorg. Chem., 2021, 60, 9912–9931 CrossRef CAS PubMed.
- S. M. Mullins, R. G. Bergman and J. Arnold, Organometallics, 1999, 18, 4465–4467 CrossRef CAS.
- P. Horrillo-Martinez, B. O. Patrick, L. L. Schafer and M. D. Fryzuk, Dalt. Trans., 2012, 41, 1609–1616 RSC.
- M. K. Whittlesey, R. N. Perutz and M. H. Moore, Organometallics, 1996, 15, 5166–5169 CrossRef CAS.
- O. R. Allen, S. J. Dalgarno, L. D. Field, P. Jensen, A. J. Turnbull and A. C. Willis, Organometallics, 2008, 27, 2092–2098 CrossRef CAS.
- J. Sánchez-Nieves and P. Royo, J. Organomet. Chem., 2001, 621, 299–303 CrossRef.
- M. A. Rankin and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 10021–10023 CrossRef CAS PubMed.
- J. M. Mörsdorf and J. Ballmann, Inorg. Chem., 2021, 60, 18291–18295 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and additional characterization data. CCDC 2351192 and 2351850. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02207f |
| This journal is © The Royal Society of Chemistry 2024 |