
Quantitative separation of polystyrene nanoparticles in environmental matrices with picogram detection limits using capillary electrophoresis†
Michael A.
Caprise
a,
Ana C.
Quevedo
b and
Kathryn R.
Riley
 *a
*a
aDepartment of Chemistry & Biochemistry, Swarthmore College, 500 College Ave, Swarthmore, PA 19081, USA. E-mail: kriley1@swarthmore.edu; Tel: +1 610-690-3904
bDepartment of Chemical Engineering, McGill University, Montreal, Quebec H3A0C5, Canada
First published on 27th November 2023
Abstract
We developed a capillary electrophoresis method to separate polystyrene particles (PSPs) with different sizes or different surface functionalities. Separations were performed in buffer and 100 mg L−1 clay or 100 mg L−1 Suwanee River humic acid. In all solutions, PSPs were baseline or near-baseline resolved in less than 15 minutes.
Globally, about 320 million metric tons of plastic are produced each year and will continue to rise.1 Of concern, macroscale plastics released into the environment undergo biotic and abiotic degradation to produce nano- and micro-scale plastic particles.1–3 The small size of nanoplastics (1–1000 nm) alters their physical and chemical properties relative to their macroscale counterpart and influences their environmental fate and toxicity.3,4 For example, nanoplastics can aggregate with natural particles or other nanoplastic particles to form micro-scale clusters that alter their sedimentation properties and bioavailability.4–10 Due to their small size and unique surface chemistry, environmental molecules adsorb onto nanoplastic surfaces to form an eco-corona, which strongly alters their aggregation properties, bioavailability, and toxicity.5–7,11–16 However, our understanding of nanoplastic fate and transport mechanisms is still limited by our ability to detect, quantify, and separate nanoplastics in environmental matrices.
Several analytical methods have been proposed for the separation and detection of nanoplastics, including filtration, centrifugation, fluorescence microscopy, Raman spectroscopy, and pyrolysis gas chromatography mass spectrometry.17,18 However, these techniques have several limitations, including the particle size range, separation efficiency, detection sensitivity, and ability to detect particles in complex matrices. Asymmetric flow field-flow fractionation (AF4) has emerged as a promising technique because it can distinguish nanoplastics with different sizes and can be coupled to several different detectors for nanoplastic detection (e.g., UV, fluorescence, light scattering, and total organic carbon).8,19 However, AF4 suffers from long analysis times (≈1 h) that prevent rapid data acquisition and lead to peak broadening and poor resolution of analyte bands. Depending on the choice of detector, AF4 detection sensitivity may also be reduced for PSPs in environmental samples.8 Thus, further improvements are needed for the separation and sensitive detection of PSPs in the environment.
Capillary electrophoresis (CE) is a separation technique that separates analytes based on their electrophoretic mobilities. Previously, CE has been applied for the separation of noble metal and polymer-based nanomaterials ranging from 5 nm to >1 μm in diameter.20–23 While some separations suffer from poor peak resolution and band broadening, CE focusing techniques can improve the separation quality.24,25 Analytes are often detected using an online UV detector and most commercial instruments can be integrated with fluorescence detectors or mass spectrometers. Recently, researchers have coupled CE with inductively coupled plasma mass spectrometry for the analysis of metal nanoparticles in complex matrices26,27 and with AF4 for the analysis of plastic particles.19,28
We describe the development of CE for the highly efficient separation and sensitive detection of polystyrene particles (PSPs) in environmental matrices. We optimized CE conditions for the separation of PSPs differing by size (50, 100, or 200 nm) and surface functionality (non-functionalized, amino, or carboxyl). Detection was performed with an integrated UV-vis detector and quantitative analysis of each PSP was performed in bicarbonate buffer and in environmental matrices (clay colloids and humic acid). To the best of our knowledge this is the first demonstration of capillary electrophoresis as a standalone technique for the quantitative detection of PSPs in environmental matrices. We demonstrate near baseline resolution for the separation of PSPs based on size and surface functionality, an improvement on current techniques that are primarily limited to size-based separations. Further, the limits of quantitation (LOQs) afforded by the CE method we describe are orders of magnitude better than techniques like AF4, allowing for the detection of picogram quantities of PSPs.
Experimental methods are fully described in the ESI† (Sections S1–S9). Briefly, PSPs were prepared in bicarbonate buffer, which we chose as a model environmental buffer. We used a 10 mM bicarbonate buffer (pH 8.5) for the size-based separation and a 15 mM bicarbonate buffer (pH 8.5) for the separation based on PSP surface functionality unless otherwise stated. Polystyrene microspheres were purchased from Polysciences, Inc. (Warrington, PA) and are abbreviated as follows: 50 nm non-functionalized (PS-50), 100 nm non-functionalized (PS-100), 200 nm non-functionalized (PS-200), 100 nm amino-functionalized (PS-NH2), and 100 nm carboxyl-functionalized (PS-COOH). Since commercial PSPs are often dialyzed to remove residual surfactants or preservatives from the synthesis, we dialyzed our particles prior to analysis.29,30 We confirmed removal of the preservative using inductively coupled plasma-optical emission spectrometry, ICP-OES (ESI,† Section S3) and determined the concentration of each PSP following dialysis using nanoparticle tracking analysis, NTA (ESI,† Section S4 and Table S1). All PSPs were characterized using dynamic light scattering (DLS) to determine hydrodynamic diameters and polydispersity indices (PDIs), zeta potential measurements to determine the surface charge, and attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) and X-ray photoelectron spectroscopy (XPS) to confirm the particle surface chemistry (ESI,† Sections S5–S7). Finally, all CE analyses were performed using an AB SCIEX P/ACE MDQ Plus instrument and an uncoated fused-silica capillary with UV detection at 214 nm (ESI,† Sections S8 and S9).
Triplicate samples of PS-50, PS-100, and PS-200 were prepared in 10 mM bicarbonate buffer (pH 8.5) and their hydrodynamic diameters and zeta potentials were measured. All particle diameters were close to their nominal size and were determined as 61.6 ± 0.6 nm, 102 ± 1 nm, and 226 ± 2 nm, respectively (Fig. 1A and Table S2, ESI†). All particles were colloidally stable in the bicarbonate buffer, as demonstrated by their narrow particle size distributions and low PDIs (Fig. S1 and Table S2, ESI†). The zeta potentials of the particles increased with increasing particle diameter and were determined as −51 ± 1 mV, −60 ± 5 mV, and −82 ± 7 mV, respectively (Fig. 1B and Table S2, ESI†). Typically, PS is expected to be non-ionic, however, the manufacturer of the PSPs used in this study states that all functionalized polystyrene particles have residual surface sulfate ester groups that remain after the synthesis and impart a negative surface charge that increases with increasing PSP surface area. Negative zeta potentials have also been observed in previous studies of non-functionalized PSPs obtained from other manufacturers.8 Finally, the composition of the PSPs was confirmed using ATR-FTIR and XPS. Identified peaks were consistent with the composition of polystyrene (Fig. S2A, S3A and Tables S3, S4, ESI†).
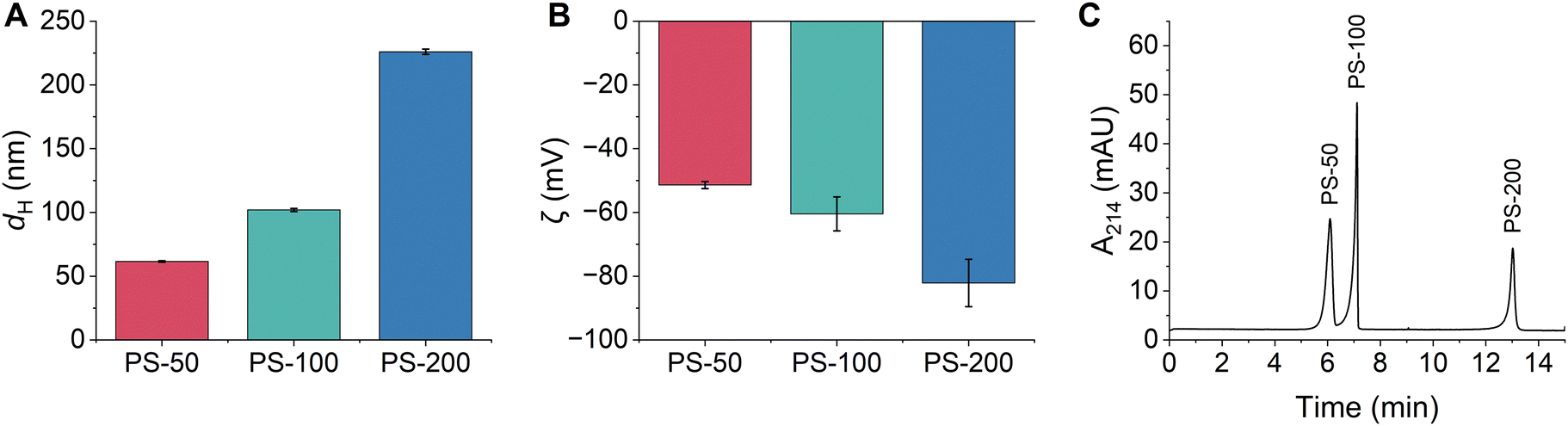 | ||
Fig. 1 (A) Hydrodynamic diameters and (B) zeta potentials of PSPs with different sizes measured using DLS. Error bars represent the standard deviation of triplicate samples. (C) Representative CE electropherogram demonstrating the separation of PSPs based on their size. All PSP stock solutions (Table S1, ESI†) were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in 10 mM bicarbonate buffer (pH 8.5) from the stock solution (Table S1, ESI†). :100 in 10 mM bicarbonate buffer (pH 8.5) from the stock solution (Table S1, ESI†). | ||
CE separation conditions were optimized to separate and detect PS-50, PS-100, and PS-200 based on differences in their size and charge. Optimal conditions were selected based on a balance of several factors including peak efficiency and resolution, reproducibility of peak migration times, and overall analysis time. The bicarbonate buffer concentration (5–50 mM) and pH (7.0–8.8) were systematically evaluated, and the ideal conditions were 10 mM bicarbonate with a pH of 8.5 (Fig. S4 and S5, ESI†). The separation voltage was also optimized between 25–30 kV. While a separation voltage of 30 kV offered a slight reduction in analysis time, a separation voltage of 25 kV was ultimately chosen since it led to the best reproducibility in peak migration times (RSD ≤ 1.3% for all PSPs; Fig. S6 and Table S5, ESI†). Under the optimal separation conditions, PS-50, PS-100, and PS-200 were separated in less than 14 minutes, PS-50 and PS-100 were nearly baseline resolved, and PS-100 and PS-200 were completely baseline resolved (Fig. 1C). Peak identities were confirmed by comparing migration times between the mixture and individual injections of each PSP and by spiking experiments (Fig. S7, ESI†). The PSP migration order followed the expected trend for negatively charged particles under normal polarity mode (anode at the inlet end of the capillary and cathode at the outlet). In this case, differences in charge dominated the separation, such that PS-50 (the least negatively charged) migrated first and PS-200 (the most negatively charged PSP) migrated last. Notably, the optimized separation represents a significant improvement over AF4 and CE-AF4 separations of similar size particles.8,19,28 The analysis time is approximately 4 times shorter, and sharp, well-resolved peaks are detected instead of poorly resolved, broad bands.
Triplicate samples of PS-NH2, PS-100, and PS-COOH were prepared in 15 mM bicarbonate buffer (pH 8.5) and their hydrodynamic diameters and zeta potentials were measured. Particle diameters were determined as 114 ± 1 nm, 102 ± 1 nm, and 99 ± 1 nm, respectively (Fig. 2A and Table S6, ESI†). All particles were colloidally stable in the bicarbonate buffer, exhibiting narrow particle size distributions and low PDIs (Fig. S8 and Table S6, ESI†). We anticipated that the different PSP surface chemistries would result in distinct surface charges. Instead, the zeta potentials of the PSPs were quite similar and were determined as −49 ± 2 mV, −56 ± 8 mV, and −54 ± 5 mV, respectively (Fig. 2B and Table S6, ESI†). The similar surface charges are likely due to residual surface sulfate ester groups that remain after the synthesis and impart a negative charge on all PSPs. Finally, the composition of the PSPs was confirmed using ATR-FTIR and XPS. Identified peaks were consistent with polystyrene and the expected surface chemistry of each PSP (Fig. S2B, S3B and Tables S3, S4, ESI†).
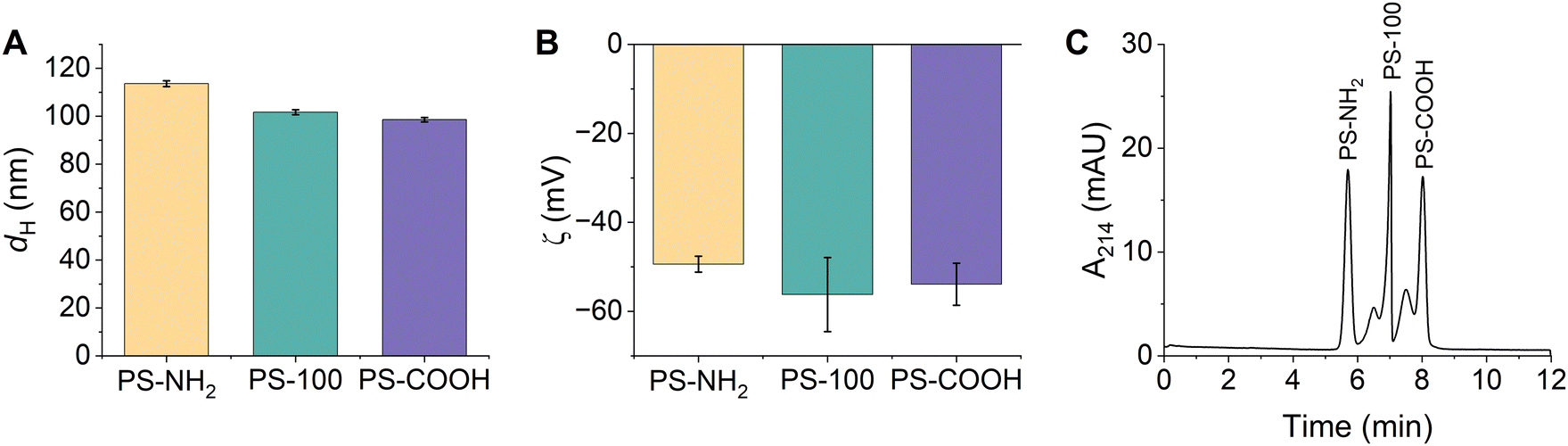 | ||
| Fig. 2 (A) Hydrodynamic diameters and (B) zeta potentials of PSPs with different surface functionalities measured using DLS. Error bars represent the standard deviation of triplicate samples. (C) Representative CE electropherogram demonstrating the separation of PSPs based on their surface functionality. All PSP stock solutions (Table S1, ESI†) were diluted 1:100 in 15 mM bicarbonate buffer (pH 8.5). | ||
CE conditions were optimized to separate and detect PS-NH2, PS-100, and PS-COOH. The ideal separation conditions included a 15 mM bicarbonate separation buffer (pH 8.5) and a 25 kV separation voltage. Despite the subtle differences in their size and charge, PS-NH2, PS-100, and PS-COOH were separated in under 10 minutes and all peaks were nearly baseline resolved (Fig. 2C). Individual injections of each PSP and spiking experiments were used to confirm peak identities (Fig. S9, ESI†). PSP charge and size both played a role in the migration order. PS-NH2 were the least negatively charged and had the largest diameter, so they migrated first. Within error, PS-100 and PS-COOH had the same charge, but PS COOH was slightly smaller, so PS-COOH migrated last. Markedly, PS-100 and PS COOH have a small shoulder peak at earlier migration time. This peak is observed when PS-COOH is injected individually. Since the particle size distribution is monomodal and the PDI is low (Table S6, ESI†), the peak is likely due to heterogeneity in the sample. For PS-100, the shoulder peak is not observed when PS-100 is injected individually, suggesting that PS-100 may interact with the other PSPs in the mixture, altering its migration.
Using the optimized separation conditions for each mixture (PS-50, PS-100, and PS-200 or PS-NH2, PS-100, and PS-COOH), we performed a calibration experiment to determine the CE quantitation limits for each PSP. The preparation and analysis of calibration standards is described in Sections S8 and S9 of the ESI.† In bicarbonate buffer, the LOQs for all particles were within the range of 1.2 × 108–18 × 108 particles mL−1 (Table 1 and Fig. S10, S11, ESI†). Given the small CE injection volume (18 nL), this corresponds to a quantitation limit of ≈2.1 × 103–3.2 × 104 particles or ≈1.2–12 pg. In comparison, AF4 analysis of similar PSP samples yielded LOQs between 0.14–0.28 μg.8 Thus, the optimized CE method offers orders of magnitude (≈105) better sensitivity compared to AF4.
| Separation | PSP | Bicarbonate buffer | 100 mg L−1 clay | 100 mg L−1 SRHA | |||
|---|---|---|---|---|---|---|---|
| LOQ (×108 particles mL−1) | Estimated mass (pg) | LOQ (×108 particles mL−1) | Estimated mass (pg) | LOQ (×108 particles mL−1) | Estimated mass (pg) | ||
| Size | PS-50 | 18 ± 7 | 4 ± 1 | 35 ± 4 | 8 ± 5 | 14 ± 8 | 3 ± 1 |
| PS-100 | 12 ± 3 | 12 ± 3 | 8 ± 5 | 8 ± 4 | 4.73 ± 0.5 | 4.73 ± 0.5 | |
| PS-200 | 6 ± 3 | 1.2 ± 0.8 | 8 ± 5 | 1.7 ± 1 | 6 ± 1 | 1.4 ± 0.3 | |
| Surface functionality | PS-NH2 | 7 ± 2 | 7 ± 2 | 0.5 ± 0.2 | 0.5 ± 0.2 | 1.2 ± 0.6 | 1.2 ± 0.6 |
| PS-100 | 1.2 ± 0.3 | 1.7 ± 0.4 | 0.81 ± 0.08 | 1.1 ± 0.1 | 1.5 ± 0.6 | 20 ± 8 | |
| PS-COOH | 1.1 ± 0.5 | 1.1 ± 0.5 | 0.6 ± 0.2 | 0.6 ± 0.2 | 8 ± 2 | 8 ± 2 | |
Given the superior resolution and detection sensitivity afforded by CE, we next evaluated whether CE could be used for the quantitative analysis of PSPs in environmental matrices. Kaolin clay and Suwanee River humic acid (SRHA) were used as model environmental matrices. DLS was used to characterize the clay and SRHA in both 10 mM and 15 mM bicarbonate buffer (pH 8.5). In both buffers, the clay and SRHA had measured diameters around 400 nm and zeta potentials around −50 mV and −35 mV, respectively (Table S7 and Fig. S12, ESI†). Calibration standards of each PSP mixture were prepared in bicarbonate buffer that contained either 100 mg L−1 clay or 100 mg L−1 SRHA and analyzed using CE (Fig. S10 and S11, ESI†). The presence of the clay and SRHA did not significantly impact the separation quality or the LOQs for either mixture (Fig. 3 and Table 1). The only exception is that the peak corresponding to PS-200 was much broader in the SRHA solution. Notably, the shoulder peak observed for PS-COOH persists in both environmental matrices (Fig. 3B), further suggesting that this small peak is due to inherent sample heterogeneity. The PS-100 shoulder peak is not present in either environmental matrix (Fig. 3B), which may suggest that the clay and SRHA reduce interactions between PS-100 and other particles in the mixture.
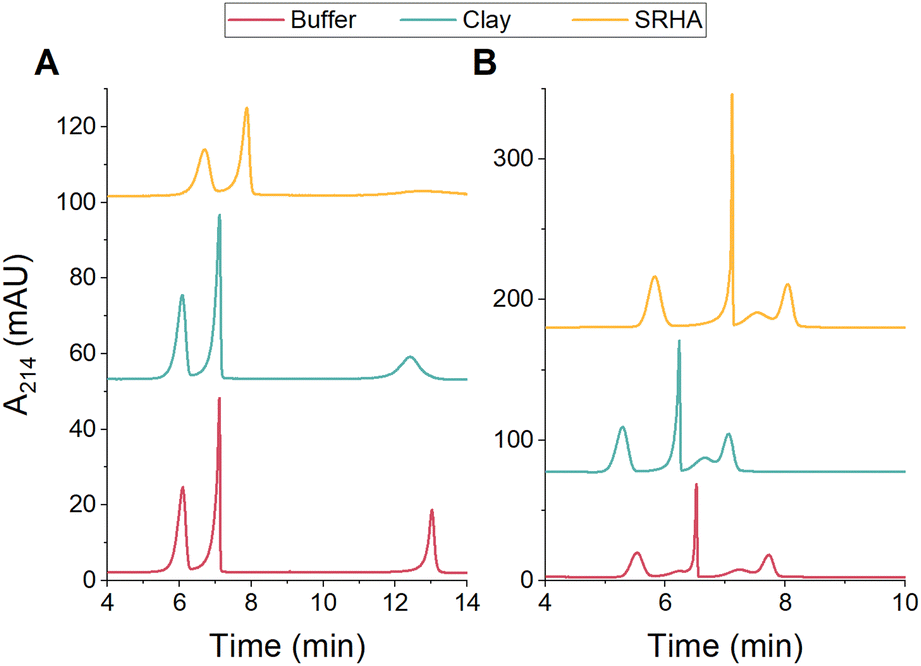 | ||
| Fig. 3 Representative CE electropherograms for the separation of PSPs with (A) different sizes (PS-50, PS-100, and PS-200) or (B) different surface functionalities (PS-NH2, PS-100, and PS-COOH) in environmental matrices. All PSP stock solutions (Table S1, ESI†) were diluted 1:100 in 10 mM (A) or 15 mM (B) bicarbonate buffer (pH 8.5) or in buffer containing 100 mg L−1 clay or 100 mg L−1 SRHA. | ||
In conclusion, this study demonstrates the superior resolving power and detection sensitivity of CE for the analysis of PSPs in environmental matrices. CE offers improved resolution of PSPs based on size and surface functionality, reduced analysis times, and orders of magnitude lower quantitation limits compared to AF4. PSPs can be quantitatively determined in environmental samples, including clay suspensions and SRHA solutions, without significant impacts on the separation quality. Future work should explore further optimization of separation conditions and the application of other CE detectors (i.e., fluorescence or MS) to improve resolution of PSPs and detection and quantitation sensitivity.
M. Caprise – experiments (DLS, zeta potential, ATR-FTIR, CE), data analysis, writing; A. Quevedo – experiments (ICP-OES, NTA, XPS), data analysis, writing; K. Riley – project conception, data analysis, writing.
We would like to thank Stacey Louie and Marfua Mowla for helpful discussions and initial materials to begin this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. P. H. Arp, D. Kühnel, C. Rummel, M. MacLeod, A. Potthoff, S. Reichelt, E. Rojo-Nieto, M. Schmitt-Jansen, J. Sonnenberg, E. Toorman and A. Jahnke, Environ. Sci. Technol., 2021, 55, 7246–7255 CrossRef CAS PubMed.
- J. Gigault, A. ter Halle, M. Baudrimont, P.-Y. Pascal, F. Gauffre, T.-L. Phi, H. El Hadri, B. Grassl and S. Reynaud, Environ. Pollut., 2018, 235, 1030–1034 CrossRef CAS PubMed.
- J. Gigault, H. El Hadri, B. Nguyen, B. Grassl, L. Rowenczyk, N. Tufenkji, S. Feng and M. Wiesner, Nat. Nanotechnol., 2021, 16, 501–507 CrossRef CAS PubMed.
- A. ter Halle and J. F. Ghiglione, Environ. Sci. Technol., 2021, 55, 14466–14469 CrossRef CAS PubMed.
- C. Veclin, C. Desmet, A. Pradel, A. Valsesia, J. Ponti, H. El Hadri, T. Maupas, V. Pellerin, J. Gigault, B. Grassl and S. Reynaud, ACS EST Water, 2022, 2, 88–95 CrossRef CAS.
- A. Pradel, S. Ferreres, C. Veclin, H. El Hadri, M. Gautier, B. Grassl and J. Gigault, ACS EST Water, 2021, 1, 1198–1208 CrossRef CAS.
- O. S. Alimi, J. M. Farner, L. Rowenczyk, A. R. Petosa, D. Claveau-Mallet, L. M. Hernandez, K. J. Wilkinson and N. Tufenkji, J. Hazardous Mater. Adv., 2022, 7, 100115 CrossRef CAS.
- M. Mowla, S. Shakiba and S. M. Louie, Chem. Commun., 2021, 57, 12940–12943 RSC.
- D. Shaniv, I. Dror and B. Berkowitz, Chemosphere, 2021, 262, 127854 CrossRef CAS PubMed.
- A. Brewer, I. Dror and B. Berkowitz, ACS EST Water, 2021, 1, 48–57 CrossRef CAS.
- C. L. Schultz, S. Bart, E. Lahive and D. J. Spurgeon, Environ. Sci. Technol., 2021, 55, 6065–6075 CrossRef CAS.
- I. Hansjosten, M. Takamiya, J. Rapp, L. Reiner, S. Fritsch-Decker, D. Mattern, S. Andraschko, C. Anders, G. Pace, T. Dickmeis, R. Peravali, S. Rastegar, U. Strähle, I.-L. Hsiao, D. Gilliland, I. Ojea-Jimenez, S. V. Y. Ambrose, M.-F. A. Belinga-Desaunay-Nault, A. O. Khan, I. Lynch, E. Valsami-Jones, S. Diabaté and C. Weiss, Environ. Sci.: Nano, 2022, 9, 375–392 RSC.
- B. Huang, Z.-B. Wei, L.-Y. Yang, K. Pan and A.-J. Miao, Environ. Sci. Technol., 2019, 53, 3871–3879 CrossRef CAS PubMed.
- S. Ducoli, S. Federici, R. Nicsanu, A. Zendrini, C. Marchesi, L. Paolini, A. Radeghieri, P. Bergese and L. E. Depero, Environ. Sci.: Nano, 2022, 9, 1414–1426 RSC.
- X. Li, E. He, B. Xia, Y. Liu, P. Zhang, X. Cao, L. Zhao, X. Xu and H. Qiu, Environ. Sci.: Nano, 2021, 8, 1560–1570 RSC.
- Y. Zhang, F. Cheng, T. Zhang, C. Li, J. Qu, J. Chen and W. J. G. M. Peijnenburg, Environ. Sci. Technol., 2022, 56(5), 3085–3095 CrossRef CAS PubMed.
- H. Cai, E. G. Xu, F. Du, R. Li, J. Liu and H. Shi, Chem. Eng. J., 2021, 410, 128208 CrossRef CAS.
- B. Nguyen, D. Claveau-Mallet, L. M. Hernandez, E. G. Xu, J. M. Farner and N. Tufenkji, Acc. Chem. Res., 2019, 52, 858–866 CrossRef CAS PubMed.
- M. Jing, W. Gao and P. Hutchins, Anal. Chem., 2023, 95, 3840–3847 CrossRef CAS PubMed.
- F.-K. Liu, Y.-Y. Lin and C.-H. Wu, Anal. Chim. Acta, 2005, 528, 249–254 CrossRef CAS.
- N. G. Vanifatova, B. Y. Spivakov, J. Mattusch and R. Wennrich, J. Chromatogr. A, 2000, 898, 257–263 CrossRef CAS PubMed.
- H. K. Jones and N. E. Ballou, Anal. Chem., 1990, 62, 2484–2490 CrossRef CAS.
- K. R. Riley, S. Liu, G. Yu, K. Libby, R. Cubicciotti and C. L. Colyer, J. Chromatogr. A, 2016, 1463, 169–175 CrossRef CAS PubMed.
- K. R. Riley, H. El Hadri, J. Tan, V. A. Hackley and W. A. MacCrehan, J. Chromatogr. A, 2019, 1598, 216–222 CrossRef CAS PubMed.
- K.-H. Lin, T.-C. Chu and F.-K. Liu, J. Chromatogr. A, 2007, 1161, 314–321 CrossRef CAS.
- H. Qu, T. K. Mudalige and S. W. Linder, Anal. Chem., 2014, 86, 11620–11627 CrossRef CAS PubMed.
- H. Qu, S. W. Linder and T. K. Mudalige, Anal. Bioanal. Chem., 2017, 409, 979–988 CrossRef CAS PubMed.
- Z. You, N. Nirmalananthan-Budau, U. Resch-Genger, U. Panne and S. M. Weidner, J. Chromatogr. A, 2020, 1626, 461392 CrossRef CAS PubMed.
- O. Pikuda, E. G. Xu, D. Berk and N. Tufenkji, Environ. Sci. Technol. Lett., 2019, 6, 21–25 CrossRef CAS.
- E. J. Petersen, A. C. Barrios, T. B. Henry, M. E. Johnson, A. A. Koelmans, A. R. Montoro Bustos, J. Matheson, M. Roesslein, J. Zhao and B. Xing, Environ. Sci. Technol., 2022, 56(22), 15192–15206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, nanoparticle characterization data, CE optimization data, and CE quantitative data. See DOI: https://doi.org/10.1039/d3cc04588a |
| This journal is © The Royal Society of Chemistry 2024 |