
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic electrodes based on redox-active covalent organic frameworks for lithium batteries
Raquel
Dantas†
 a,
Catarina
Ribeiro†
a and
Manuel
Souto
*ab
a,
Catarina
Ribeiro†
a and
Manuel
Souto
*ab
aDepartment of Chemistry, CICECO-Aveiro Institute of Materials, University of Aveiro, Aveiro, 3810-393, Portugal. E-mail: manuel.souto@ua.pt
bCIQUS, Centro Singular de Investigación en Química Bioloxica e Materiais Moleculares, Departamento de Química-Física, Universidade de Santiago de Compostela, 15782, Santiago de Compostela, Spain
First published on 28th November 2023
Abstract
Electroactive organic materials have received much attention as alternative electrodes for metal–ion batteries due to their high theoretical capacity, resource availability, and environmental friendliness. In particular, redox-active covalent organic frameworks (COFs) have recently emerged as promising electrodes due to their tunable electrochemical properties, insolubility in electrolytes, and structural versatility. In this Highlight, we review some recent strategies to improve the energy density and power density of COF electrodes for lithium batteries from the perspective of molecular design and electrode optimisation. Some other aspects such as stability and scalability are also discussed. Finally, the main challenges to improve their performance and future prospects for COF-based organic batteries are highlighted.
 Raquel Dantas | Raquel Dantas (Viseu, 1996) is a PhD student at the Department of Chemistry of the University of Aveiro and CICECO-Aveiro Institute of Materials. She obtained her BSc and MSc (2022) in chemical engineering from the University of Aveiro. She completed her Master's thesis under the supervision of Manuel Souto. Her PhD work focuses on the design of redox-active COFs as electrode materials for metal–ion batteries. |
 Catarina Ribeiro | Catarina Ribeiro (Porto, 1995) obtained her BSc (2017) in Chemistry from the University of Porto, and her MSc (2019) in Medicinal Chemistry from the University of Minho. In 2022, she started her PhD at the University of Aveiro and CICECO – Aveiro Institute of Materials. Her research deals with the synthesis of novel electroactive organic building blocks and porous frameworks for energy storage applications. |
 Manuel Souto | Manuel Souto Salom (Valencia, 1988) is an Oportunius Research Professor and Principal Investigator at CIQUS (University of Santiago de Compostela). He is also a Guest Professor at the University of Aveiro. His main current research interest is in the design and synthesis of functional electroactive porous frameworks (e.g., COFs & MOFs) based on redox-active organic building blocks for energy storage applications. In 2021, he was awarded an ERC Starting Grant for the project ELECTROCOFS, which aims to design new redox-active COF-based electrodes for rechargeable batteries. |
Introduction
Electrical energy storage devices have proven to be key to the transition from conventional fossil fuels because they can efficiently store the electricity generated from renewable sources, helping to mitigate climate change. Lithium-ion batteries (LIBs) are among the most promising electrochemical energy storage devices due to their superior performance compared to other conventional rechargeable batteries, widespread use in portable electronics, and potential for the development of long-range electric vehicles.1,2 However, the current state-of-the-art of LIBs presents significant development bottlenecks in terms of efficiency and raw materials’ availability. For instance, transition metal oxide cathode materials have energy density limitations that are constrained by their specific capacities (e.g., ∼140 mA h g−1 for LiCoO2). In addition, the large-scale production of these materials is limited by their reduced resource availability and raises serious environmental concerns.3,4 Several critical elements used in LIBs, such as cobalt, would inevitably face supply shortages in the near future5 and raise some environmental and ethical concerns.6Electroactive organic materials have long been proposed as alternatives to inorganic electrodes due to their tuneable electrochemical performance, resource availability, environmental friendliness, and flexibility.4,7–11 They are composed of abundant elements (carbon, hydrogen, oxygen, nitrogen, sulphur, etc.) and may have higher specific capacities than conventional inorganic electrodes. In addition, some organic electrodes can be obtained from biomass and recycled.12 However, one of the main limitations of organic electrode materials (especially small organic molecules) is their high solubility in the electrolyte, resulting in rapid capacity fading.9 An efficient strategy to overcome such solubility issues is to covalently bind electroactive monomers to form organic polymers, which are normally insoluble in aqueous and organic electrolytes.13,14
After their discovery in the late 1970s,15 many conductive polymers were explored as electrodes in lithium metal–organic batteries, and even some companies (Bridgestone-Seiko and Varta/BASF) developed commercial batteries based on poly(pyrrole) and poly(aniline) as active electrode materials.14 Nevertheless, such polymer-based batteries were quickly discontinued due to self-discharge issues and poor performance compared to lithium-ion batteries, which were commercialised by Sony in 1991.16 After a period where organic electrodes received less attention, the discovery of organic radical batteries17,18 and the bright future for organic batteries vaticinated by Armand and Tarascon in 20083 revived the field with remarkable progress in the last 15 years. New materials, including electroactive porous organic polymers, and novel redox-active moieties beyond the well-known organic radicals, carbonyl and organosulfur compounds have been extensively investigated for different types of batteries in recent years.9 In addition, sophisticated operando and ex situ characterisation techniques19,20 have been developed to further investigate charge storage mechanisms in organic compounds, whereas machine learning methods are accelerating the identification and discovery of promising organic electrodes.21
Covalent organic frameworks (COFs) are a class of crystalline porous polymers composed of organic building blocks linked by strong covalent bonds with predictable 2D or 3D structures. The topology and pore size of the resulting frameworks can be predetermined by the appropriate choice of organic linkers. Their great chemical and structural versatility makes them potentially interesting for a wide range of applications, including gas sorption and separation, (photo)catalysis or sensing, among others.22–25 In particular, layered 2D COFs have received much attention towards electronics and energy applications, as their electrical, optical and magnetic properties can be finely tuned by the judicious selection of electroactive organic building blocks and conjugated linkers.26,27
In recent years, redox-active COFs have emerged as promising organic electrode materials for rechargeable batteries.28–33 Compared to conventional organic polymers, COFs offer the same advantages mentioned above, such as the abundant availability of raw materials, sustainability, high stability and insolubility in the electrolyte. In addition, COFs present ordered porous channels to facilitate ion diffusion and their high surface area allows better accessibility to multiple redox active sites. On the other hand, the possibility to introduce π-conjugation and improve electronic delocalisation through the framework can help to enhance the electronic conductivity of COFs.27 Another important difference with respect to amorphous linear and cross-linked polymers is their ordered crystalline nature which allows their structure to be predicted by computational modelling, being of great interest in predesigning COFs with specific topologies and properties. Like other conventional redox polymers, electroactive COFs can undergo redox processes in a reversible manner, thus being able to be reduced and oxidised during the charge and discharge of the battery. For example, in the case of LIBs where a n-type redox-active COF is used as a cathode, the electroactive centres from the COF are reduced during the discharge process, at the same time as Li+ cations are accommodated in the framework to balance the negative charge. In the charging process, the COF returns to the neutral state while the Li+ ions are released into the electrolyte.28
COFs have recently been explored as electrode materials in different metal–ion (including multivalent) batteries34 (Li+,35,36 Na+,37,38 K+,39,40 Mg2+,41 Ca2+,42 Zn2+,43 Al3+,44,45etc.) and lithium–sulphur (Li–S) batteries.46–49 The first reported COF electrodes for each type of battery are shown in Fig. 1. Note that we have also included the first reported covalent triazine frameworks (CTFs), which tend to be much more amorphous.
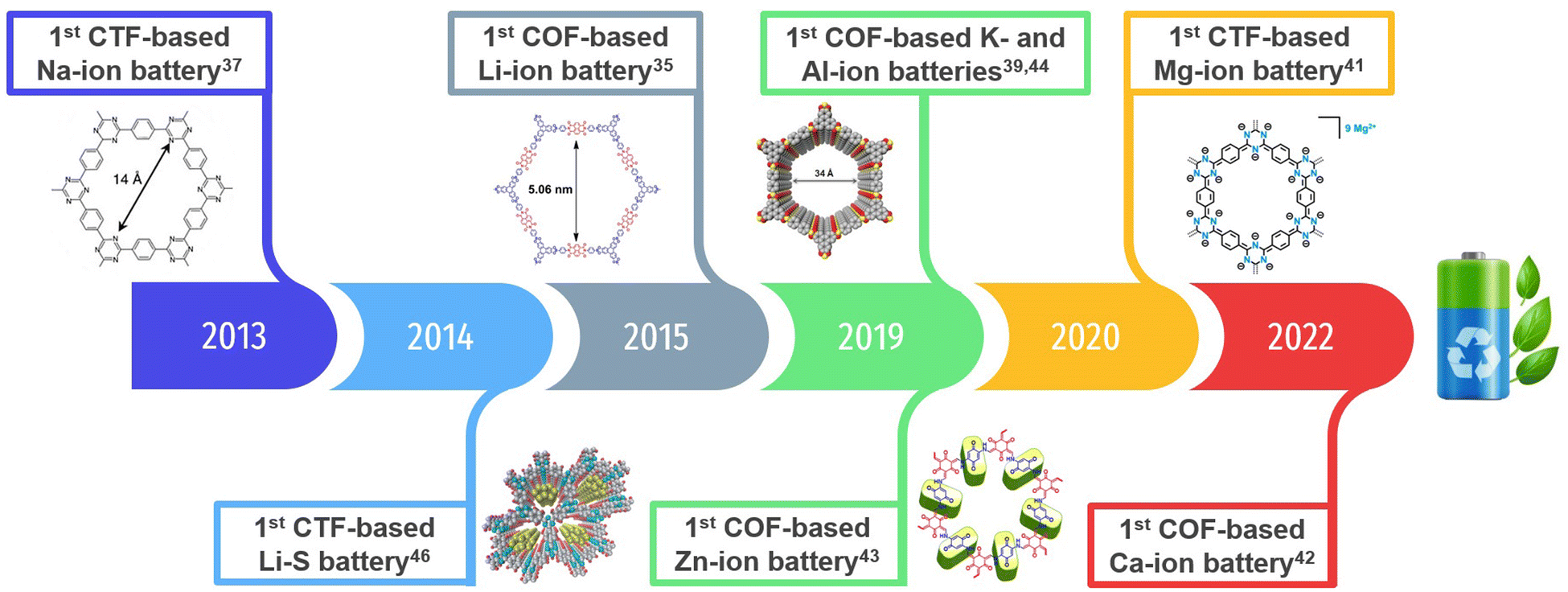 | ||
| Fig. 1 An overview of the development of COF-based electrodes (including CTFs) for batteries, highlighting the first reported examples for lithium-ion (LIBs), sodium-ion (SIBs), potassium-ion (PIBs), zinc-ion (ZIBs), magnesium-ion (MIBs), calcium-ion (CIBs), aluminium-ion (AIBs) and lithium–sulphur (Li–S) batteries. Reprinted in part with permission of ref. 35, 39, 41, 43 and 46. | ||
In this Highlight article, we provide an overview of recent design strategies to improve the electrochemical performance of COF electrode materials for metal–ion batteries within the context of the organic battery field. In particular, we discuss some principles for the molecular design of COF cathodes in lithium batteries to increase both the energy and power densities. We also highlight the need to optimise the COFs processing, and the electrode and electrolyte composition from a materials engineering point of view. Some other aspects such as cycling stability, scalability and recyclability are also considered. Finally, the main challenges and prospects for COF electrode materials for rechargeable batteries are highlighted.
Molecular design of redox-active COFs to enhance the electrochemical performance
Energy density
Rechargeable batteries are mainly evaluated in terms of energy density, power density, and cycling stability.8 The energy density (Ed) is directly related to the specific capacity (Q) and output voltage (E) (eqn (1)), so both parameters should be increased to improve the battery performance through molecular design, electrode composition, electrolyte optimisation, etc.8,9| Ed = E × Q | (1) |
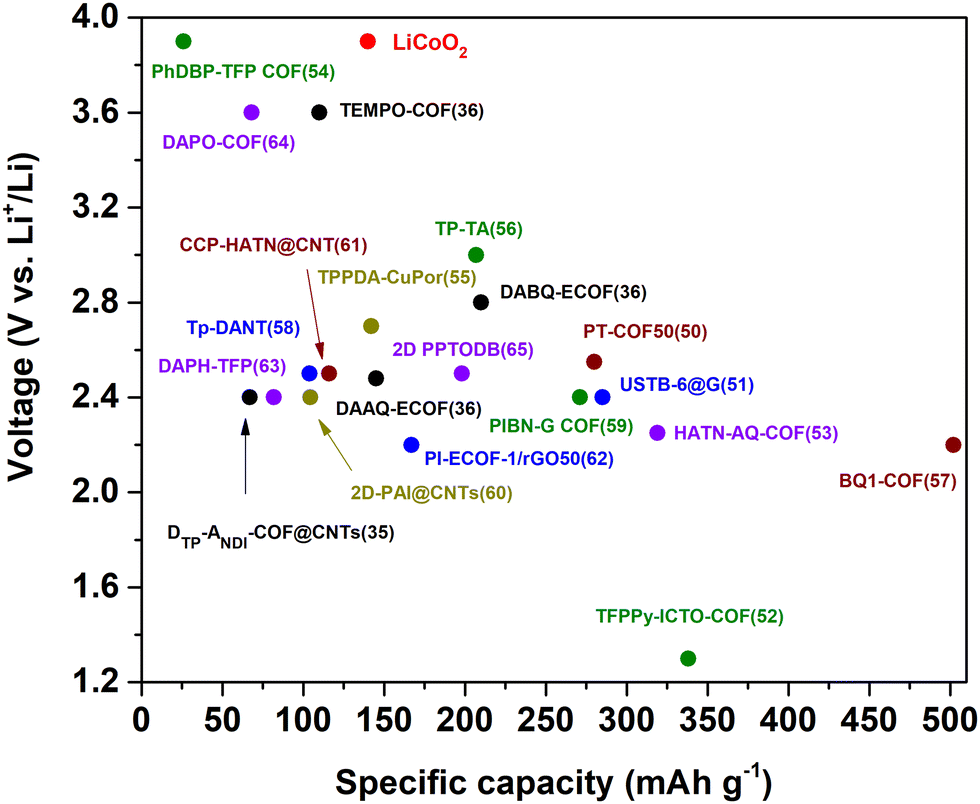 | ||
| Fig. 2 Average discharge voltage and practical specific capacity of some representative COF-based cathode materials for rechargeable lithium batteries. The specific capacity refers to the maximum reported reversible capacity (irrespective of the rate used). | ||
Capacity
According to eqn (1), high specific capacity is required to achieve high energy density. The specific capacity is defined as the electric charge that can be stored in a cell per unit mass of the active material.13 The theoretical specific capacity (Qtheo) of an electrode material can be calculated using the following equation: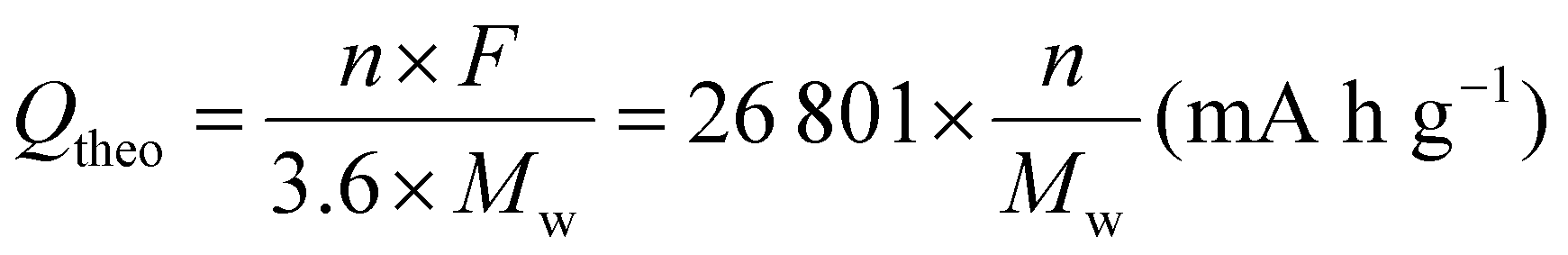 | (2) |
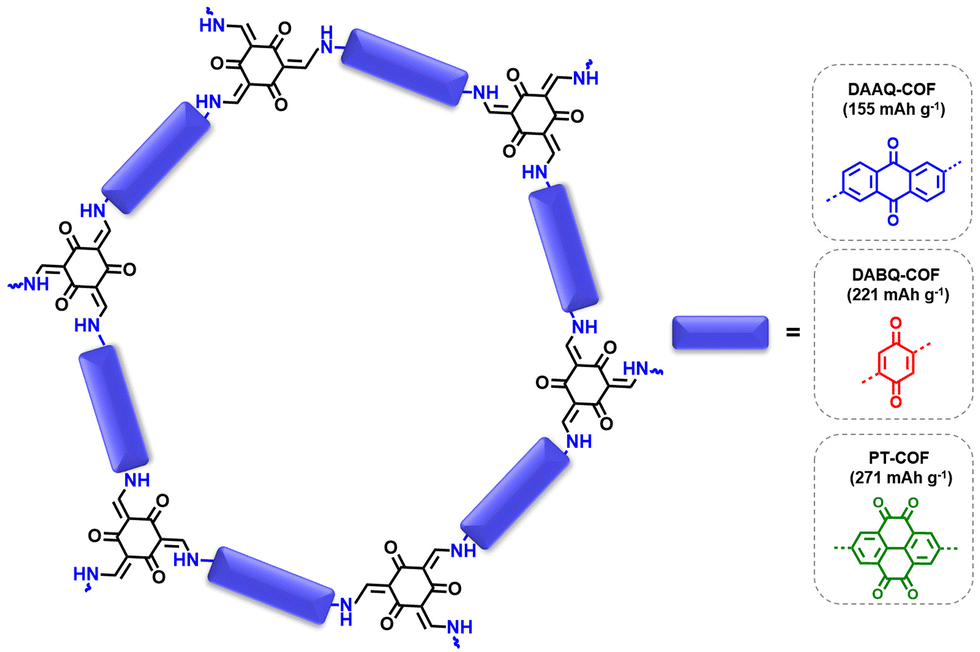 | ||
| Fig. 3 Schematic representation of the DAAQ-COF, DABQ-COF and PT-COF and their corresponding theoretical capacities. | ||
Reducing the number or molecular weight of inactive moieties in the framework is also critical to maximise the theoretical capacity of COF electrode materials. An example is the replacement of 1,3,5-triphenylbenzene (Mw = 306 g mol−1) by triphenylamine (Mw = 245 g mol−1) in PI-COFs, increasing the theoretical capacity of PI-COF-1 up to 142 mA h g−1 (Fig. 4).62 Therefore, the connection of linear redox-active building blocks by using electroactive linkers/linkages (quinoxalines,51 tetraphenyl-p-phenylenediamine,56 triazines,45 polyimides,53 phenazines,66etc.) (Fig. 5) is an efficient strategy to increase the active site density. Moreover, the geometry and length of the linkers are determinants for the topology and pore size of the frameworks, which can influence the electrolyte diffusion and thus the practical capacity. However, COFs with small pore size can lead to more rigid structures and be more efficient for charge transport, so the pore diameter needs to be optimised.33 Other factors affecting the practical capacity, such as the electronic conductivity or particle size, are discussed below.
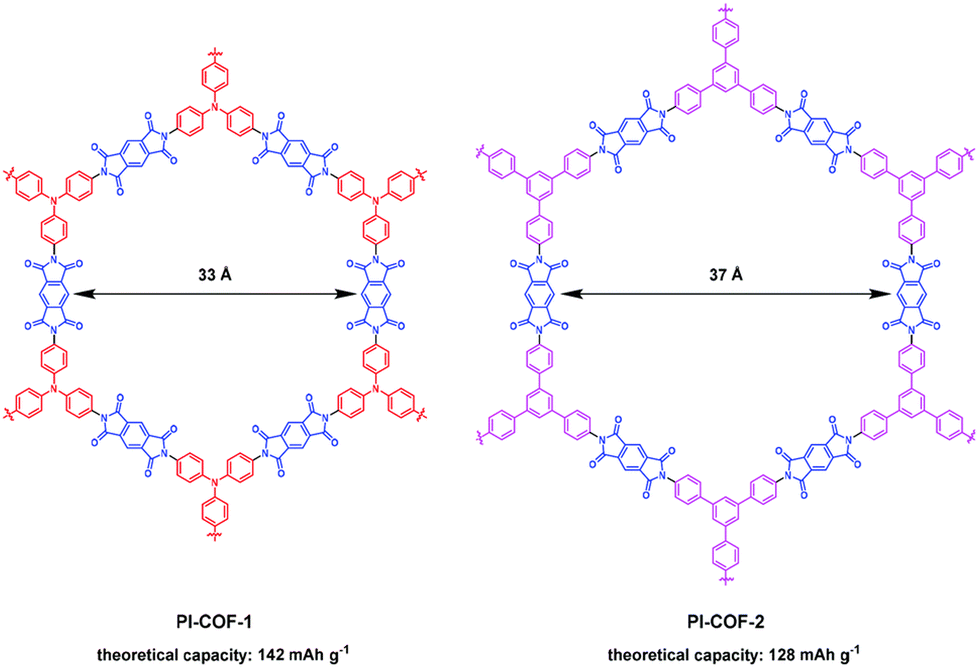 | ||
| Fig. 4 Chemical structures and theoretical capacities of PI-COF-1 and PI-COF-2. Reproduced with permission from ref. 62. | ||
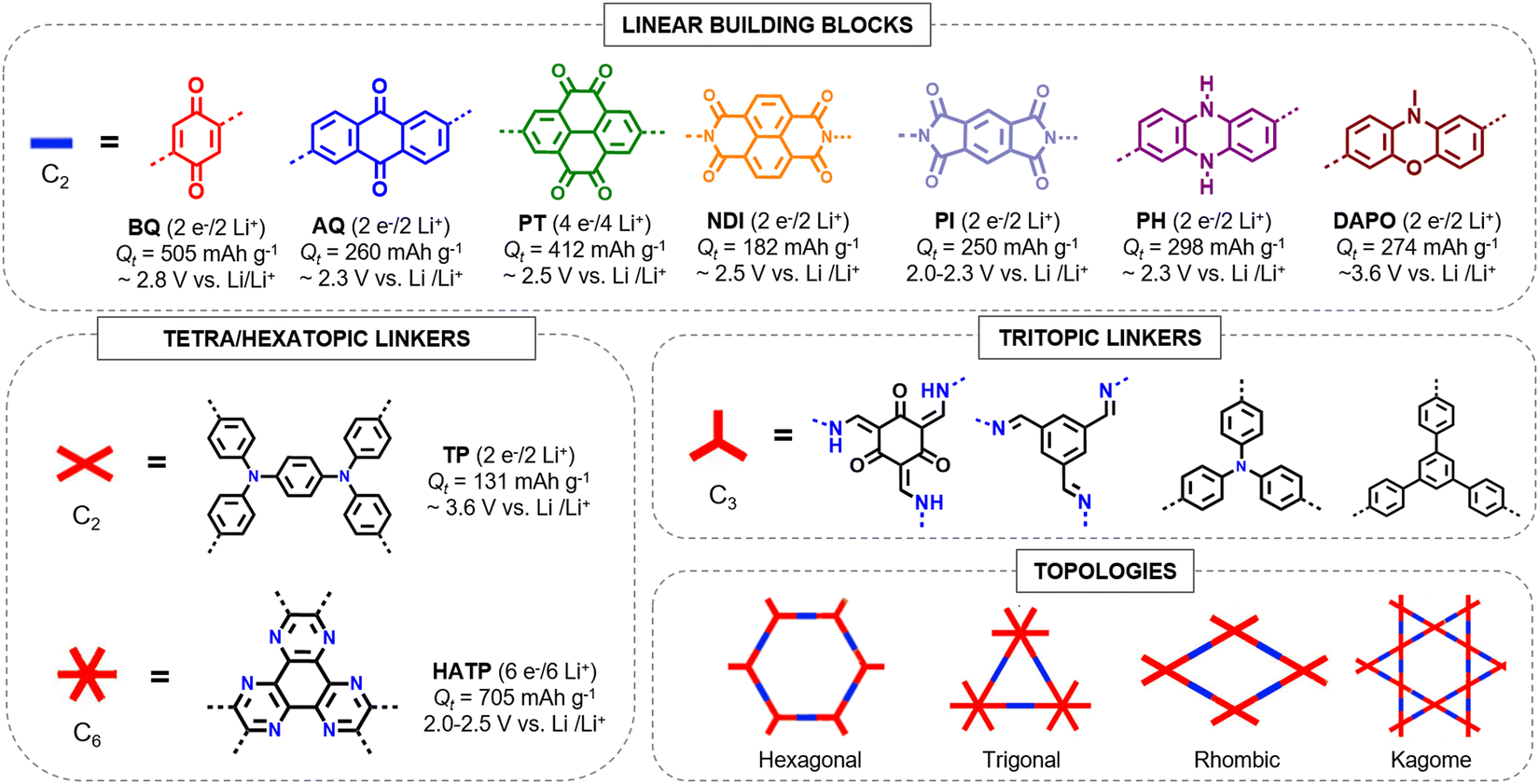 | ||
| Fig. 5 Library of some representative electroactive organic building blocks (theoretical capacities and average discharge voltages), linkers and topologies used for the synthesis of redox-active COF cathodes in LIBs. BQ (benzoquinone), AQ (anthraquinone), PT (pyrene-tetraone), NDI (naphthalene diimide), PI (polyimide), PH (phenazine), DAPO (phenoxazine), TP (tetraphenyl-p-phenylenediamine), and HATP (hexaazatriphenylene). | ||
The highest practical capacity among COF-based cathode materials in LIBs reported to date is 502 mA h g−1 (0.05C) for the BQ1-COF (theoretical capacity of 773 mA h g−1) (Fig. 2).57 Notably, the estimated energy density for this COF electrode at high current density (20C) was ∼350 W h kg−1, being superior to that of commercial transition metal oxide cathodes such as LiCoO2 or LiFePO4 (100–140 W h kg−1).67 Some key features of this material are its high density of active C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN groups and its high electrical conductivity, although its structural model remains unclear.68 In general, the specific capacities refer only to the COF active material, so they can vary significantly when the mass of the whole electrode (conductive additives, binders, etc.) is taken into account. In any case, the specific capacities of some reported redox-active COFs are higher than those of conventional transition metal oxides and among the highest values for any type of organic cathode for lithium batteries.14,69,70
O and CN groups and its high electrical conductivity, although its structural model remains unclear.68 In general, the specific capacities refer only to the COF active material, so they can vary significantly when the mass of the whole electrode (conductive additives, binders, etc.) is taken into account. In any case, the specific capacities of some reported redox-active COFs are higher than those of conventional transition metal oxides and among the highest values for any type of organic cathode for lithium batteries.14,69,70
Output voltage
Besides capacity, another way to maximise the energy density is to increase the output voltage. The theoretical voltage of a battery can be estimated from the operating potentials of the cathode and anode as shown in eqn (3).8,13 Therefore, in order to achieve a high voltage, it is essential to use cathode and anode materials with high and low operating potentials, respectively.| E = cathode potential – anode potential | (3) |
Working potentials of organic electrode materials depend mainly on the molecular structure, which can be finely tuned, for example, by incorporating electron withdrawing or donating groups.8,72 In general, n-type organic compounds can store charges in the moderate voltage range (<3 V vs. Li/Li+), whereas p-type organic electrodes exhibit high working potentials (>3 V vs. Li/Li+). For this reason, p-type compounds are mainly used as cathodes, while n-type compounds can also be used as anodes if they have a relatively low potential. In addition, redox kinetics of p-type compounds usually exhibit faster rates.9,73 The redox potentials of representative redox-active groups used in COFs have recently been summarised.33 In the particular case of COF cathodes for LIBs, the highest voltages have been achieved with the TEMPO radical (∼3.6 vs. Li/Li+),36 phenoxazine- (∼3.6 vs. Li/Li+),64 and dibenzopentalene-based COFs (∼3.9 vs. Li/Li+)54 (Fig. 2). However, the capacity of such COFs is rather limited. In contrast, the voltage of COFs with the highest capacity (>250 mA h g−1), which are based on n-type building blocks, does not exceed 2.5 V vs. Li/Li+. Therefore, new strategies need to be designed to simultaneously maximise the voltage and specific capacity, as well as to explore new electroactive building blocks for the construction of redox-active COFs with improved electrochemical performance. Remarkably, some p-type organic compounds with high redox potential widely studied in the field of organic batteries, such as phenothiazine, remain unexplored for the design of COFs used as electrode materials for batteries.
Redox-bipolar COFs
As mentioned above, the design of COFs with both high specific capacity and high voltage is one of the current challenges to achieve high energy densities. In this sense, the construction of COFs combining n-type and p-type building blocks can be an efficient approach to increase both parameters. One of the prototypical examples is a bipolar porous organic electrode (BPOE) which was used as a cathode in sodium-organic energy storage devices.37 This material is a porous covalent triazine framework with a honeycomb structure which has a high specific power of 10 kW kg−1 and a specific energy of 500 W h kg−1 when used in sodium batteries. Due to its bipolar nature, the BPOE can exhibit n- and p-doping processes, achieving a high working potential window (4.1–1.3 V vs. Na/Na+). This concept was also demonstrated by the synthesis of an imine-linked 2D COF (TP-TA) with Kagome topology based on the condensation reaction between tetraphenyl-p-phenylenediamine (TP) and terephthaldehyde (TA).56 In this case, the CN linkages can be reduced acting as n-type moieties, while the p-type TP building block can undergo 2 e− oxidation and combine with two PF6− from the electrolyte. This bipolar-type COF showed a specific capacity of 207 mA h g−1 (at 200 mA g−1), an average redox voltage of 3.6 V vs. Li/Li+ and good cycling stability (93% retention after 1500 cycles). Another representative example of this design strategy is a 2D COF (TPPDA-CuP or COF) composed of tetratopic p-type TP and bipolar-type porphyrin building blocks (Fig. 6), which provides a high redox potential and a high number of redox-active centres.55 Indeed, the TPPDA-CuPor COF cathode exhibited an average voltage of 2.7 V vs. Li/Li+ and a specific capacity of 142 mA h g−1 at 60 mA g−1, resulting in a calculated energy density of 371 W h kg−1. In addition, the bipolar redox mechanism was further confirmed by DFT calculations and ex situ XPS.
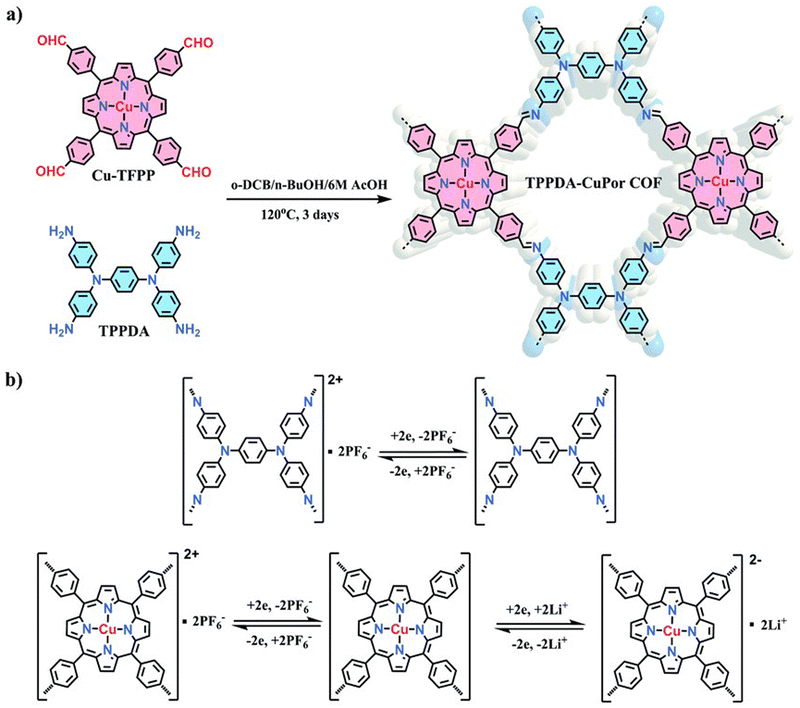 | ||
| Fig. 6 (a) Synthesis of TPPDA-CuPor COF. (b) Redox reaction of the p-type TP and bipolar-type Cu-TFPP building blocks. Reproduced with permission from ref. 55. | ||
More recently, Feng and co-workers have reported a redox-bipolar COF based on n-type imides and p-type triazine moieties that was investigated as a cathode for rechargeable aluminium batteries achieving a specific capacity of 132 mA h g−1 and excellent cycling stability (97% capacity retention after 4000 cycles).45 H. Chen and co-workers have also synthesised a porous organic polymer based on triazine and viologen building blocks which presents multiple co-storage modes, namely PF6−/Li+, OTF−/Mg2+ and OTF−/Zn2+ in the same host.74 The PF6−/Li+ co-storage in the porous polymer exhibited both high energy and power densities of 878 W h kg−1 and 28 kW kg−1, respectively, after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. One of the key features of this material is the Coulomb interaction between cationic and anionic carriers in the framework, promoting fast ion carrier migration. We also note that a similar approach has been used for the synthesis of another porous organic polymer based on the polyimide condensation of mellitic acid trianhydride (n-type) and diamino N-methyl phenothiazine (p-type).75 Such dual-redox porous organic polymer was used as an electrode in symmetric all-organic batteries displaying a specific capacity of 57 mA h g−1 (2C) and a capacity retention of 57% at high rates (60C). The design of bipolar-type COFs is still in its infancy and new combinations of building blocks as well as advanced in situ/ex situ mechanism studies are still needed to boost the performance of this promising type of COF electrodes.
000 cycles. One of the key features of this material is the Coulomb interaction between cationic and anionic carriers in the framework, promoting fast ion carrier migration. We also note that a similar approach has been used for the synthesis of another porous organic polymer based on the polyimide condensation of mellitic acid trianhydride (n-type) and diamino N-methyl phenothiazine (p-type).75 Such dual-redox porous organic polymer was used as an electrode in symmetric all-organic batteries displaying a specific capacity of 57 mA h g−1 (2C) and a capacity retention of 57% at high rates (60C). The design of bipolar-type COFs is still in its infancy and new combinations of building blocks as well as advanced in situ/ex situ mechanism studies are still needed to boost the performance of this promising type of COF electrodes.
Power density
The power density (Pd) is another important parameter to evaluate rechargeable batteries and is directly related to the charge–discharge ability of organic electrodes at different rates.8 Materials with high power density transfer a large amount of energy in a short time. The power density depends on the output voltage (E) and rate capability (I) as follows:| Pd = E × I | (4) |
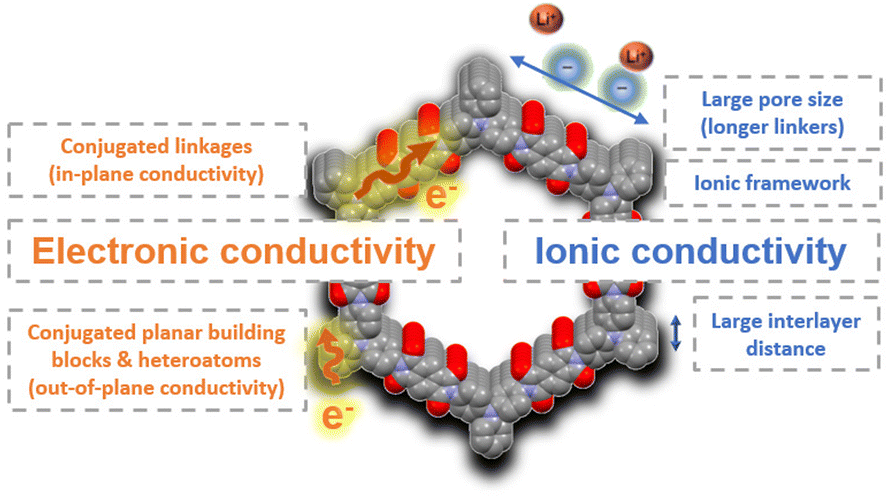 | ||
| Fig. 7 Summary of some molecular design strategies to improve electronic and ionic conductivity in COFs. | ||
Like most organic compounds, 2D COFs typically exhibit low intrinsic electronic conductivity although some strategies have recently been identified to improve the electronic transport in these materials.27 For example, conjugated planar building blocks are likely to exhibit a better π-orbital overlap between the layers which can facilitate the out-of-plane conductivity. On the other hand, the choice of conjugated linkages, such as vinylene or pyrazine, is critical to ensure efficient electron delocalisation throughout the framework, thereby increasing the in-plane conductivity. Band structure calculations are also a useful tool to predict important information about the charge transport through different directions and to extract structure–property relationships for the design of conductive COFs. One of the highest conductivities (10−3 S cm−1) among neutral (non-doped) COFs has very recently been reported for a fully conjugated vinylene-linked COF (TFPPy-ICTO-COF).52 This COF has been used as a cathode in LIBs, providing excellent rate capability without the addition of conductive additives (e.g., carbon nanotubes, graphene, etc.). Another conjugated 2D COF (BQ1-COF)57 showed a relatively high intrinsic electronic conductivity (10−6 S cm−1) for a neutral COF, which was responsible for the excellent performance as a cathode for lithium batteries. The incorporation of heteroatoms or electron-withdrawing groups in COFs is another effective approach to reduce their band gap and increase the conductivity. For example, the introduction of fluorine atoms into a covalent triazine framework (FCTF) significantly narrowed its band gap (from 2.35 to 1.45 eV), resulting in superior electron transport and rate capability.76 In addition, thiazole-linked COFs have also proved to exhibit high out-of-plane conductivity because of the efficient p orbital overlap of the azo groups across the layers,77 resulting in a superior rate performance compared to other similar COFs when used as electrodes in LIBs.
In addition to electronic conductivity, rate performance can be significantly enhanced by improving the ionic conductivity through chemical design.78 The simplest approach to modulate ionic conductivity is to modify the size and arrangement of the pores, as this can facilitate ion diffusion. In general, COFs with smaller pore size may present diffusion problems at high rates, especially for those metal ions and counterions with large ionic radii. For example, HATN-AQ-COF53 (pore size = 3.8 nm) shows a higher active sites utilisation efficiency of 63% at high current density (10C) than that of BQ1-COF57 (pore size = 1.4 nm, active site utilisation of 22% at 10C) when explored as cathode materials for LIBs. This reveals that the design of COFs with larger pore sizes and well-defined channels may be key to improving ion diffusion. The design of hierarchical COFs in the presence of both meso- and micropores has also proven to facilitate ion transport.56 The interlayer distance can also be modulated to improve the ionic conductivity. In general, COFs present stacked layers that often hinder the ion transport during electrochemical processes, so larger interlayer distances can favour better ion diffusion, higher utilisation of the redox-active sites, and higher rate performance. This approach was explored in a redox-active piperazine–terephthaldehyde COF based on nonplanar linkages exhibiting a chair-shaped conformation, resulting in a large interlayer distance of 6.2 Å.79 This interlayer distance facilitated ion transport, improving the electrochemical performance even at high rates (207 mA h g−1 at 5C) when used as an anode in LIBs. However, this strategy contrasts with a better interlayer overlap which is favourable for efficient out-of-plane electronic conductivity. Other strategies to increase ionic conductivity in COFs are based on the preparation of cationic frameworks,80 the incorporation of sulfonate groups,81 or post-functionalisation of the channels by anchoring, for example, oligo(ethylene oxide) moieties.82,83 However, these strategies significantly reduce the specific capacity and are therefore more focused on solid electrolytes’ applications.
Electrode and electrolyte optimisation
Influence of the electrolyte and binder on the electrochemical performance
The choice of electrolyte and binder has a direct impact on the practical capacity and stability of the electrode, as it can affect the solubility and efficiency of active sites’ utilisation. For example, optimising the nature and concentration of the electrolyte can lead to significant improvements in capacity and cycling stability.84 Very recently, we have reported the first comprehensive study of the influence of the electrolyte and binder on the performance of an anthraquinone-based COF (DAAQ-TFP-COF).85 Our results showed that the worst performance was obtained when using carbonate-based electrolytes (e.g., LiPF6 in ethylene carbonate/diethyl carbonate) (Fig. 8) which could be related to undesired side reactions. In contrast, the best capacity retention was obtained when using LiFTSI as the electrolyte in ether-based solvents (Fig. 8). The electrochemical performance was further improved by substituting polyvinylidene fluoride (PVdF) with poly(tetrafluoroethylene) (PTFE) as the binder, resulting in practical capacities close to the theoretical one and high capacity retention (99% after 100 cycles). One of the main reasons for the improved performance when using PTFE as a binder is its porous fibre-like structure, which can lead to better ion diffusion and accessibility to the active sites. Furthermore, the DAAQ-TFP-COF was also explored as a cathode in magnesium batteries using two electrolytes (MgCl2 and one containing weakly coordinating anions) that lead to different electrochemical performances. Therefore, in addition to the proper design and processing of the active material, the selection of the appropriate electrolyte and binder is critical to achieve optimal performances in COF-based batteries.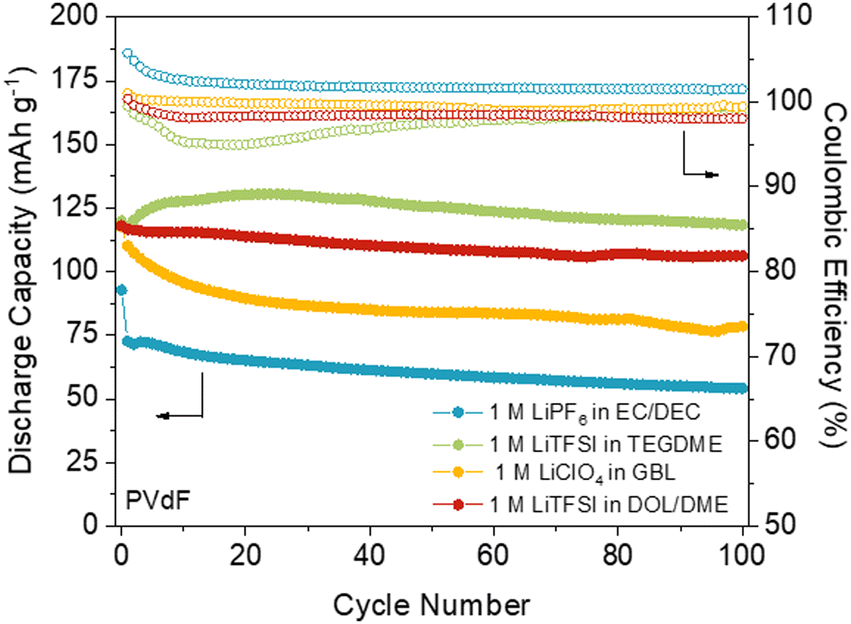 | ||
| Fig. 8 DAAQ-TFP-COF electrode performance with PVdF as a binder at 150 mA g−1 (1C) in four different electrolytes: 1 M LiTFSI in DOL/DME (1:1, vol%) (red), 1 M LiTFSI in TEGDME (green), 1 M LiPF6 in EC/DEC (1:1, vol%) (blue), and 1 M LiClO4 in GBL (yellow). Adapted with permission from ref. 85. | ||
Hybridisation with conductive additives
As discussed above, the rate capability depends on the electronic conductivity of organic electrodes. However, most organic compounds are insulators (σ < 10−9 S cm−1) and their low intrinsic electronic conductivity limits the performance of batteries based solely on organic active materials.9 For this reason, electroactive organic compounds are usually mixed with large amounts (typically from 30 to 70% in weight)86 of conductive additives (e.g., carbon black), significantly reducing the specific capacity and energy density of the whole organic electrode. It is therefore necessary to deduct capacity contributions from carbon black and other conductive additives to determine the real specific capacity of the active material as well as calculate the energy density taking into account the mass of the whole electrode.In the specific case of COF cathodes for LIBs, the amount of carbon black can vary from 10 to 70% in weight, depending on the intrinsic conductivity of the COF. In addition, a commonly used strategy to increase the electronic conductivity is the hybridisation of COFs with carbon nanotubes (CNTs),35,50,61,65,87 reduced graphene oxide,62,88 or conductive polymers.63,89 The COF/CNT composites are usually synthesised by in situ polymerisation, where COF layers are grown on the CNT surface (Fig. 9).90 In general, such COF/CNT composites result in a higher utilisation of active sites, high rate performance, and can help to improve cycling stability. For example, the preparation of PT-COF/CNTs composite cathodes for LIBs can increase the active site utilisation from 71% (PT-COF) to 98% (PT-COF composites with 50 wt% of CNTs).50 However, the PT-COF/CNTs composite shows a lower overall specific capacity (109 mA h g−1) than that of PT-COF (128 mA h g−1) when considering the mass of the active material and conductive additives. This proves that the addition of too many inactive conductive additives can significantly reduce the battery energy density. It is also worth noting that most COF composites still require a large amount of extra conductive additives such as carbon black to achieve a satisfactory performance. Thus, improving the intrinsic conductivity of COFs and developing new strategies to minimise the amount of conductive additives in the electrode remain the main challenges for the design of high performance COF electrodes.
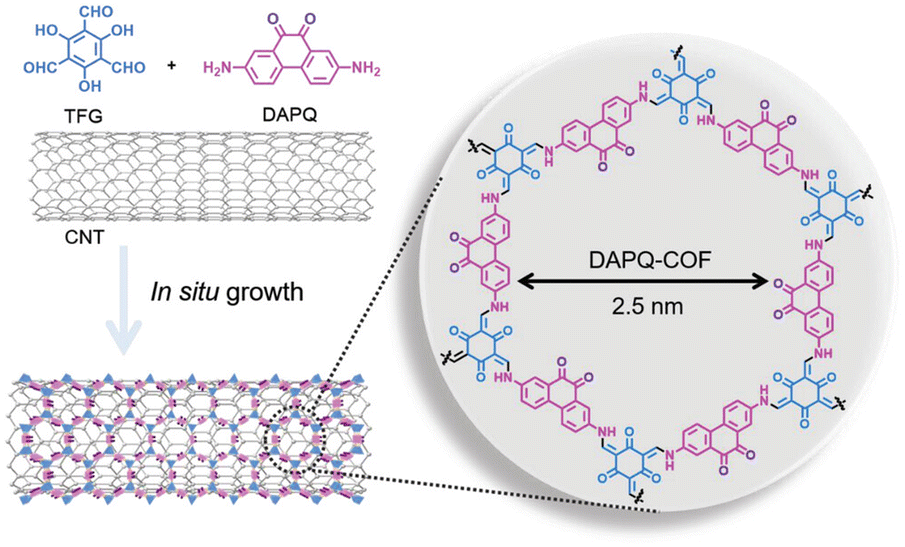 | ||
| Fig. 9 Representative example of COF/CNT composite synthesis via in situ growth. Reproduced with permission from ref. 90. | ||
Exfoliation of COFs into few-layered nanosheets
In general, bulk COFs are used directly as active materials and mixed with conductive carbon and the binder to prepare the electrode. However, the processing of electroactive organic materials to reduce the particle size or to increase the specific surface area is an effective way to improve the practical capacity.8 Most bulk COFs present strong π–π interactions between layers, which prevent ions from reaching the buried active sites and make them inaccessible, limiting the practical capacity. Exfoliation of 2D COFs into few layered nanosheets is a successful strategy to increase the number of accessible redox sites and assist ion diffusion, increasing the performance of COF-based batteries (Fig. 10).36,38,62 In general, there are several strategies for the preparation of COF nanosheets using top-down methods based on different types of exfoliation (including mechanical and chemical exfoliation) to bottom-up approaches based on the preparation of well-ordered nanostructures.29,91,92 In the particular case of COF nanosheets for batteries, different exfoliation techniques have been employed for their preparation: mechanical (ball milling,36,62,93 ultrasound-assisted exfoliation,38,94 grinding,38etc.), chemical exfoliation (using external agents such as maleic anhydride,95 MnO2,96 acids,97 thiol–ene reactions,98etc.) and self-exfoliation.99,100 Regarding bottom-up approaches, an example is the in situ growth of graphene-supported COF nanosheets that are uniformly dispersed on carbon layers.51 The relationship between thickness and practical capacity was systematically investigated using a redox-active COF (DAAQ-COF) as an anode in sodium batteries.38 In this case, three different exfoliation techniques were used to obtain different thicknesses: grinding (100–250 nm), ball milling (100–180 nm) and ultrasound-assisted exfoliation in methanesulfonic acid (4–12 nm). The thinnest (4–12 nm) and thickest (100–250 nm) samples showed capacities of 500 and 182 mA h g−1 (at 50 mA g−1), respectively. The resistance of the electrode also decreased from 428 Ω to 146 Ω after exfoliation. This study demonstrates that reducing the COF thickness can be an efficient approach to improve the practical capacity and rate capability. However, the preparation of COF nanosheets still presents some challenges that need to be addressed. First, the thicknesses of the nanosheets are always obtained in a wide size range, so precise thickness control remains challenging and can be problematic for reproducibility. On the other hand, the yields obtained for the preparation of nanosheets are usually relatively low and some exfoliation procedures can affect the crystallinity of the material.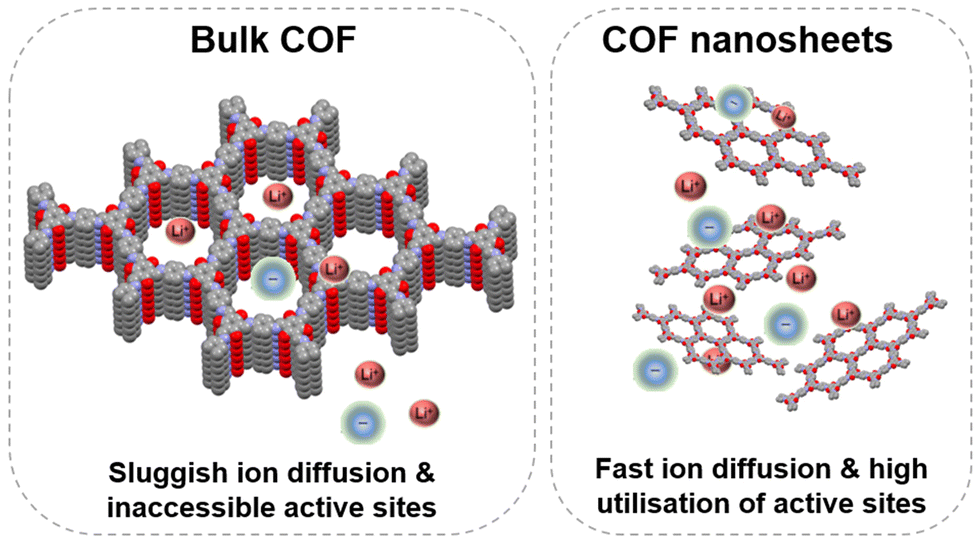 | ||
| Fig. 10 Schematic representation of the sluggish diffusion in bulk COFs and fast diffusion in exfoliated COF nanosheets. | ||
Other parameters
Cycling stability
Stability is one of the most important parameters for evaluating organic electrodes in batteries. In general, organic compounds may present solubility problems in the electrolyte (especially small molecules), leading to possible undesired side reactions, self-discharge issues and poor cycling stability. Other factors leading to low stability can be the instability of intermediates or volume change.8 Polymerisation is one of the main strategies to avoid solubility problems, along with salification and electrolyte optimisation (see above). When evaluating the stability of organic electrodes, we can take as a reference the capacity retention of some inorganic electrodes such as LiCoO2 or LiMn2O4 which exceeds 80% after more than 1000 cycles.9 Some COF-based electrodes show similar or even higher capacity retention after 1000 cycles, especially those based on β-ketoenamine,36,50 (cyano)vinylene,52,61 phenazine,66 imide45 and piperazine101,102 linkages (Fig. 11), as they lead to most stable frameworks.103 Normally COFs based on more reversible reactions such as boronate ester104 or imine condensations105 result in poorer stability. While increased reversibility results in higher crystallinity and may allow for error correction during linkage formation, reversible bond formation limits the stability of COFs. Therefore, stability and crystallinity are considered to be inversely related to each other.105 The strategy of pre-orientation, based on an initial reversible reaction followed by irreversible locking of the labile bond, can lead to very crystalline and highly stable COFs.106 Such a strategy has recently been explored to synthesise a thiazole-linked COF that was used as a cathode in lithium–sulphur batteries.107 We also note that in some cases, COFs are hybridised with insoluble conductive substrates such as CNTs, which can help to improve the stability.8 As mentioned above, electrolyte optimisation is also crucial to improve the cycling stability of COFs, as it may affect dissolution or lead to side reactions.85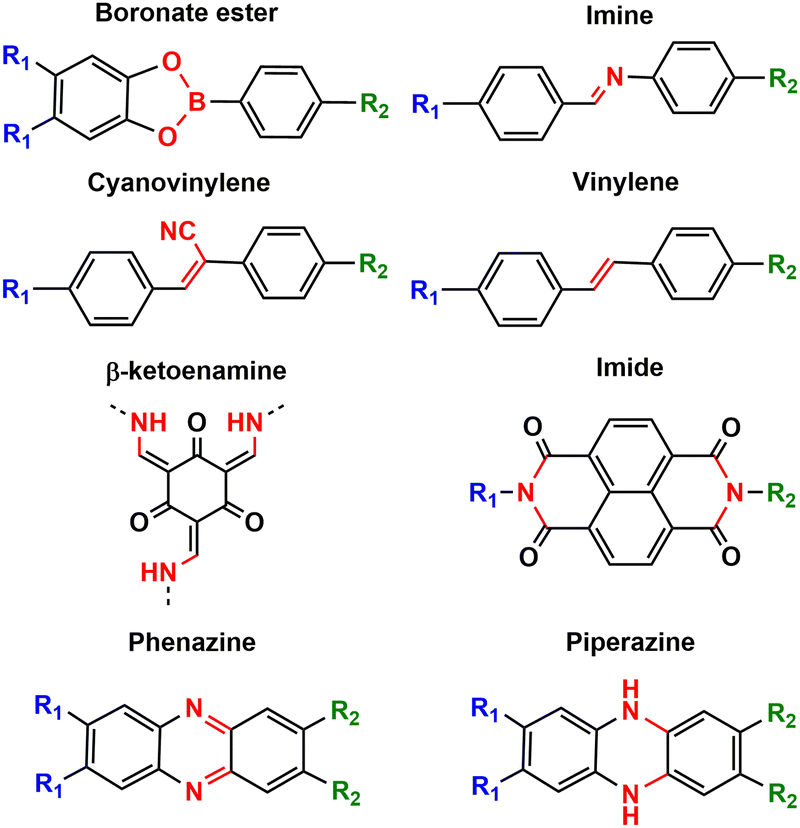 | ||
| Fig. 11 Some typical linkages employed for the synthesis of COFs used as electrodes in metal–ion batteries. | ||
Scalability
Although large-scale applications of organic electrodes for metal–ion batteries are still a long way to go, the possibility of scalability and production using simple low-cost methods needs to be evaluated.8 Some organic electrodes have already been scaled up at relatively low cost using high mass loading and pouch cells.108 Most COFs are synthesised using solvothermal conditions (typically between 120 and 150 °C), acid catalysts and high boiling point organic solvents such as mesitylene, 1,4-dioxane, or N,N-dimethylacetamide.105 More sustainable and environmentally friendly syntheses have already been proposed by replacing organic solvents with water,109,110 using supercritical CO2,111 or solvent-free methods.112 Microwave-assisted synthesis of COFs has also been extensively explored to considerably reduce reaction times.113 Recent studies have reported scalable methods to obtain COF nanosheets on a gram scale in a short time and under mild conditions.114 Although most of the COFs used as electrodes in batteries have been obtained via solvothermal reactions, alternative, greener and cost- and time-efficient syntheses should be considered towards large-scale applications in the long term. As for most organic electrodes, the cost of such materials is too difficult to estimate until large-scale commercial scale-up is feasible.9Degradation and recyclability
In addition to scalability, another important aspect to take into account in the near future is the end-of-life and recyclability of organic electrodes for the development of sustainable and greener batteries in a circular economy.86,115 In this direction, some naturally occurring and biodegradable organic electrode materials have been reported.116,117 A more recent example is based on metal-free polypeptide-based batteries, in which redox-active groups such as viologens and nitroxide radicals were incorporated along polypeptide backbones to be used as anode and cathode materials, respectively.118 The most important feature of such polypeptide-based battery is its high stability during battery operation and on-demand degradation under acidic conditions. The recycling of some organic electrodes has also been recently proposed by using a simple extraction method without the decomposition of the material and with high yields.108 As for COFs, although the biodegradability of some of them has been studied for other applications,119,120 future studies on the degradation and recycling of COF electrodes should be systematically carried out.Conclusions and outlook
The emergence of redox-active covalent organic frameworks as promising electrodes for rechargeable metal–ion batteries has revitalised the field of organic batteries in recent years. Some of their advantages over conventional electroactive organic polymers are the presence of ordered channels to improve ion diffusion and their crystalline nature, which allows their structures and properties to be predicted. In this Highlight article, we have discussed some of the recent strategies to improve their electrochemical performance (energy density, power density, cycling stability) either by molecular design or by electrode optimisation.In terms of energy density, some n-type COFs with high density of active sites show practical capacities of up to 500 mA g−1, being higher than those of conventional inorganic electrodes. Otherwise, high voltages (up to 3.9 vs. Li/Li+) can also be achieved by incorporating specific p-type moieities (TEMPO radical, phenoxazine, dibenzopentalenes, etc.) into the framework, although the specific capacity of the resulting COFs is limited. The design of COFs with both high capacity and high potential remains a major challenge to give rise to superior energy density. In this sense, redox-bipolar COFs have emerged as promising systems as they allow the combination of electroactive p- and n-type building blocks that can lead to high potential and high capacity at the same time.
Regarding power density, as for most organic compounds, low electronic conductivity is the main limitation in achieving high rate capabilities. To overcome this issue, some strategies at the molecular design level (planar building blocks, heteroatoms, conjugated linkages, etc.) can help to construct delocalised frameworks to improve the in-plane and out-of-plane electron transport. However, the intrinsic conductivities of COFs remain relatively low, and large amounts of conductive additives are still required, reducing the energy density of the battery. At the same time, ionic conductivity needs to be optimised, for example, by modulating porosity to facilitate ion diffusion. Therefore, a proper balance between the two conductivities is critical to maximise the rate performance. In addition, in situ/ex situ techniques combined with molecular dynamic simulations can provide important insights into the ion transport and charge storage in COFs.
Electrode optimisation is also key to improve the electrochemical performance of COFs. For example, the proper choice of an electrolyte and binder can significantly increase the practical capacity, whereas the preparation of COF composites with conductive carbon substrates can help to enhance rate capability. However, the energy density can be reduced if the amount of conductive additives is excessive. The exfoliation of bulk COFs into nanosheets is another approach to increase the accessibility of active sites. However, there is still a need for more straightforward synthetic approaches with improved yield and thickness control towards large-scale applications. Systematic studies for each of these strategies will be useful to optimise the composition and processing of COF electrodes.
Cycling stability is one of the key parameters for evaluating COF-based electrodes. Although the stability of COFs has been significantly improved in recent years, some design strategies can lead to new, much more robust linkages. The evaluation of capacity retention after several (>1000) cycles as well as post-mortem analysis of COF electrodes can contribute to a better understanding of the stability of these materials. Although there is still a long way to go, the scalability, cost and recycling of these materials should be considered in the near future for further applications.
In summary, in terms of molecular design, one of the main challenges will be to build frameworks that combine redox-active moieties that can give rise to both high voltage (p-type) and high capacity (n-type), with redox-bipolar COFs being the most promising systems. At the same time, inactive moieties should be minimised to maximise energy density. Particular attention should be paid to electroactive organic building blocks that have been extensively explored in the field of organic batteries,86 such as phenothiazines,73 thianthrenes,121 or specific organic radicals,122 which can lead to high voltages. In addition, it will be necessary to construct electron-delocalised frameworks using conjugated and robust linkages to ensure efficient electronic conductivity and stability to the framework. In this sense, vinylene-based linkages or even the direct C–C connection of electroactive building blocks could result in very stable frameworks, despite the synthetic challenge of obtaining crystalline materials. The pre-orientation strategy will also allow the discovery of new linkages to construct electroactive COFs showing both high crystallinity and stability. Furthermore, the pore size should be optimised to facilitate ion diffusion at high rates. Theoretical calculations supported by machine learning methods and molecular dynamic simulations will also be useful to design and identify the most promising materials with specific capacities and voltages, low bandgaps, efficient ion diffusion, etc.
In terms of COF processing and electrode optimisation, the exfoliation of bulk COFs has been shown to be one of the most promising techniques to increase the utilisation efficiency of redox active sites. Further comparative studies on different top-down (mechanical, chemical, and self-exfoliation) and bottom-up techniques should be carried out to identify the most appropriate COFs in each case and to optimise them to improve the yield and homogeneity of COF nanosheets. The preparation of composites by hybridising COFs with carbon substrates such as CNTs can result in substantial improvement in conductivity and rate performance, but the amount of such conductive additives should be minimised so as not to influence the energy density of the battery significantly. For this reason, the design of intrinsically conductive COFs with high performance or new strategies to minimise the amount of conductive additives in the electrode are some of the main challenges in the field. Finally, the proper selection of the electrolyte and binder has proven to be key to maximising the practical capability and stability of COF-based electrodes. In this respect, alternative binders such as PTFE can considerably improve the utilisation of active sites without the need for further COF processing. Therefore, it is expected that the optimisation of new electrolytes and binders can maximise the performance of COF-based electrodes.
The remarkable progress in the design of electroactive COFs and their use in numerous types of rechargeable (including multivalent)123,124 batteries has demonstrated their great potential as electrodes, revitalising the field of organic batteries. It is expected that this Highlight article will contribute to stimulate the design and optimisation of novel redox-active COF electrodes for energy storage devices in the coming years.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Research Council (ERC) under the European Union's Horizon Europe Framework Programme (ERC-2021-Starting Grant, grant agreement no. 101039748-ELECTROCOFS). This work was developed within the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 & LA/P/0006/2020, financed by national funds through the FCT/MEC (PIDDAC). This study was supported by the PRR – Plano de Recuperação e Resiliência and by the NextGenerationEU funds allocated at the University of Aveiro, as part of the Agenda for Business Innovation “New Generation Storage” (Project No. 58 with the application C644936001-00000045). We also thank FCT for funding the project with DOI: https://doi.org/10.54499/PTDC/QUI-ELT/2593/2021. This work has received financial support from the Xunta de Galicia (Centro singular de investigación de Galicia accreditation 2019–2022, ED431G 2019/03), the Oportunius program (Gain) and the European Union (European Regional Development Fund – ERDF).Notes and references
- J. B. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef.
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- A. Zeng, W. Chen, K. D. Rasmussen, X. Zhu, M. Lundhaug, D. B. Müller, J. Tan, J. K. Keiding, L. Liu, T. Dai, A. Wang and G. Liu, Nat. Commun., 2022, 13, 1341 CrossRef CAS PubMed.
- É. Lèbre, M. Stringer, K. Svobodova, J. R. Owen, D. Kemp, C. Côte, A. Arratia-Solar and R. K. Valenta, Nat. Commun., 2020, 11, 4823 CrossRef PubMed.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Y. Lu, Q. Zhang, L. Li, Z. Niu and J. Chen, Chem, 2018, 4, 2786–2813 CAS.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- A. Banerjee, N. Khossossi, W. Luo and R. Ahuja, J. Mater. Chem. A, 2022, 10, 15215–15234 RSC.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2022, 8, 54–70 CrossRef.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS PubMed.
- N. Goujon, N. Casado, N. Patil, R. Marcilla and D. Mecerreyes, Prog. Polym. Sci., 2021, 122, 101449 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- A. Yoshino, Angew. Chem., Int. Ed., 2012, 51, 5798–5800 CrossRef CAS PubMed.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359, 351–354 CrossRef CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- A. P. Black, A. Sorrentino, F. Fauth, I. Yousef, L. Simonelli, C. Frontera, A. Ponrouch, D. Tonti and M. R. Palacín, Chem. Sci., 2023, 14, 1641–1665 RSC.
- A. Vizintin, J. Bitenc, A. Kopač Lautar, K. Pirnat, J. Grdadolnik, J. Stare, A. Randon-Vitanova and R. Dominko, Nat. Commun., 2018, 9, 661 CrossRef PubMed.
- R. P. Carvalho, D. Brandell and C. M. Araujo, Energy Storage Mater., 2023, 102865 CrossRef.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 6328 CrossRef PubMed.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- M. Souto, K. Strutyński, M. Melle-Franco and J. Rocha, Chem. – Eur. J., 2020, 26, 10912–10935 CrossRef CAS PubMed.
- M. Souto and D. F. Perepichka, J. Mater. Chem. C, 2021, 9, 10668–10676 RSC.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1–23 Search PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Y. Lu, Y. Cai, Q. Zhang and J. Chen, J. Phys. Chem. Lett., 2021, 12, 8061–8071 CrossRef CAS PubMed.
- D. Zhu, G. Xu, M. Barnes, Y. Li, C. Tseng, Z. Zhang, J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef CAS.
- S. Kandambeth, V. S. Kale, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Adv. Energy Mater., 2022, 12, 2100177 CrossRef CAS.
- S. Haldar, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2023, 145, 13494–13513 CrossRef CAS PubMed.
- Z. Wang, J. Hu and Z. Lu, Batteries Supercaps, 2023, 6, e202200545 CrossRef CAS.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5, 8225 CrossRef CAS PubMed.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed.
- K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef PubMed.
- S. Gu, S. Wu, L. Cao, M. Li, N. Qin, J. Zhu, Z. Wang, Y. Li, Z. Li, J. Chen and Z. Lu, J. Am. Chem. Soc., 2019, 141, 9623–9628 CrossRef CAS PubMed.
- X. Chen, H. Zhang, C. Ci, W. Sun and Y. Wang, ACS Nano, 2019, 13, 3600–3607 CrossRef CAS PubMed.
- X. Luo, W. Li, H. Liang, H. Zhang, K. Du, X. Wang, X. Liu, J. Zhang and X. Wu, Angew. Chem., Int. Ed., 2022, 61, e202117661 CrossRef CAS PubMed.
- R. Sun, S. Hou, C. Luo, X. Ji, L. Wang, L. Mai and C. Wang, Nano Lett., 2020, 20, 3880–3888 CrossRef CAS PubMed.
- L. Li, G. Zhang, X. Deng, J. Hao, X. Zhao, H. Li, C. Han and B. Li, J. Mater. Chem. A, 2022, 10, 20827–20836 RSC.
- A. Khayum M, M. Ghosh, V. Vijayakumar, A. Halder, M. Nurhuda, S. Kumar, M. Addicoat, S. Kurungot and R. Banerjee, Chem. Sci., 2019, 10, 8889–8894 RSC.
- Q. Zhang, H. Wei, L. Wang, J. Wang, L. Fan, H. Ding, J. Lei, X. Yu and B. Lu, ACS Appl. Mater. Interfaces, 2019, 11, 44352–44359 CrossRef CAS PubMed.
- Y. Liu, Y. Lu, A. Hossain Khan, G. Wang, Y. Wang, A. Morag, Z. Wang, G. Chen, S. Huang, N. Chandrasekhar, D. Sabaghi, D. Li, P. Zhang, D. Ma, E. Brunner, M. Yu and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e202306091 CrossRef CAS PubMed.
- H. Liao, H. Ding, B. Li, X. Ai and C. Wang, J. Mater. Chem. A, 2014, 2, 8854–8858 RSC.
- S. Haldar, M. Wang, P. Bhauriyal, A. Hazra, A. H. Khan, V. Bon, M. A. Isaacs, A. De, L. Shupletsov, T. Boenke, J. Grothe, T. Heine, E. Brunner, X. Feng, R. Dong, A. Schneemann and S. Kaskel, J. Am. Chem. Soc., 2022, 144, 9101–9112 CrossRef CAS PubMed.
- W. Liu, L. Gong, Z. Liu, Y. Jin, H. Pan, X. Yang, B. Yu, N. Li, D. Qi, K. Wang, H. Wang and J. Jiang, J. Am. Chem. Soc., 2022, 144, 17209–17218 CrossRef CAS PubMed.
- B. Hu, J. Xu, Z. Fan, C. Xu, S. Han, J. Zhang, L. Ma, B. Ding, Z. Zhuang, Q. Kang and X. Zhang, Adv. Energy Mater., 2023, 13, 2203540 CrossRef CAS.
- H. Gao, A. R. Neale, Q. Zhu, M. Bahri, X. Wang, H. Yang, Y. Xu, R. Clowes, N. D. Browning, M. A. Little, L. J. Hardwick and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9434–9442 CrossRef CAS PubMed.
- X. Liu, Y. Jin, H. Wang, X. Yang, P. Zhang, K. Wang and J. Jiang, Adv. Mater., 2022, 34, 2203605 CrossRef CAS PubMed.
- X. Xu, S. Zhang, K. Xu, H. Chen, X. Fan and N. Huang, J. Am. Chem. Soc., 2023, 145, 1022–1030 CrossRef CAS PubMed.
- X. Yang, L. Gong, X. Liu, P. Zhang, B. Li, D. Qi, K. Wang, F. He and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202207043 CrossRef CAS PubMed.
- J. Sprachmann, T. Wachsmuth, M. Bhosale, D. Burmeister, G. J. Smales, M. Schmidt, Z. Kochovski, N. Grabicki, R. Wessling, E. J. W. List-Kratochvil, B. Esser and O. Dumele, J. Am. Chem. Soc., 2023, 145, 2840–2851 CrossRef CAS PubMed.
- L. Gong, X. Yang, Y. Gao, G. Yang, Z. Yu, X. Fu, Y. Wang, D. Qi, Y. Bian, K. Wang and J. Jiang, J. Mater. Chem. A, 2022, 10, 16595–16601 RSC.
- M. Wu, Y. Zhao, R. Zhao, J. Zhu, J. Liu, Y. Zhang, C. Li, Y. Ma, H. Zhang and Y. Chen, Adv. Funct. Mater., 2022, 32, 1–8 Search PubMed.
- M. Wu, Y. Zhao, B. Sun, Z. Sun, C. Li, Y. Han, L. Xu, Z. Ge, Y. Ren, M. Zhang, Q. Zhang, Y. Lu, W. Wang, Y. Ma and Y. Chen, Nano Energy, 2020, 70, 104498 CrossRef CAS.
- D. H. Yang, Z. Q. Yao, D. Wu, Y. H. Zhang, Z. Zhou and X. H. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- Z. Luo, L. Liu, J. Ning, K. Lei, Y. Lu, F. Li and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 9443–9446 CrossRef CAS PubMed.
- G. Wang, N. Chandrasekhar, B. P. Biswal, D. Becker, S. Paasch, E. Brunner, M. Addicoat, M. Yu, R. Berger and X. Feng, Adv. Mater., 2019, 31, 1–6 Search PubMed.
- S. Xu, G. Wang, B. P. Biswal, M. Addicoat, S. Paasch, W. Sheng, X. Zhuang, E. Brunner, T. Heine, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 849–853 CrossRef CAS PubMed.
- Z. Wang, Y. Li, P. Liu, Q. Qi, F. Zhang, G. Lu, X. Zhao and X. Huang, Nanoscale, 2019, 11, 5330–5335 RSC.
- E. Vitaku, C. N. Gannett, K. L. Carpenter, L. Shen, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2020, 142, 16–20 CrossRef CAS PubMed.
- Z. Meng, Y. Zhang, M. Dong, Y. Zhang, F. Cui, T.-P. Loh, Y. Jin, W. Zhang, H. Yang and Y. Du, J. Mater. Chem. A, 2021, 9, 10661–10665 RSC.
- C. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. J. Xu, D. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed.
- M. K. Shehab, K. S. Weeraratne, T. Huang, K. U. Lao and H. M. El-Kaderi, ACS Appl. Mater. Interfaces, 2021, 13, 15083–15091 CrossRef CAS PubMed.
- C. Peng, G.-H. Ning, J. Su, G. Zhong, W. Tang, B. Tian, C. Su, D. Yu, L. Zu, J. Yang, M.-F. Ng, Y.-S. Hu, Y. Yang, M. Armand and K. P. Loh, Nat. Energy, 2017, 2, 17074 CrossRef CAS.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Chen, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- H. Lyu, X.-G. Sun and S. Dai, Adv. Energy Sustainability Res., 2021, 2, 2000044 CrossRef CAS.
- M. E. Bhosale, S. Chae, J. M. Kim and J. Y. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC.
- S. Lee, J. Hong and K. Kang, Adv. Energy Mater., 2020, 10, 2001445 CrossRef CAS.
- W. Wang, V. S. Kale, Z. Cao, Y. Lei, S. Kandambeth, G. Zou, Y. Zhu, E. Abouhamad, O. Shekhah, L. Cavallo, M. Eddaoudi and H. N. Alshareef, Adv. Mater., 2021, 33, 2103617 CrossRef CAS PubMed.
- P. Acker, L. Rzesny, C. F. N. Marchiori, C. M. Araujo and B. Esser, Adv. Funct. Mater., 2019, 29, 1906436 CrossRef CAS.
- W. Sun, C. Zhou, Y. Fan, Y. He, H. Zhang, Z. Quan, H. Kong, F. Fu, J. Qin, Y. Shen and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300158 CrossRef CAS PubMed.
- N. Goujon, M. Lahnsteiner, D. A. Cerrón-Infantes, H. M. Moura, D. Mantione, M. M. Unterlass and D. Mecerreyes, Mater. Horiz., 2023, 10, 967–976 RSC.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 1–10 Search PubMed.
- Y. Cao, M. Wang, H. Wang, C. Han, F. Pan and J. Sun, Adv. Energy Mater., 2022, 12, 2200057 CrossRef CAS.
- M. Wu, Y. Zhao, H. Zhang, J. Zhu, Y. Ma, C. Li, Y. Zhang and Y. Chen, Nano Res., 2022, 15, 9779–9784 CrossRef CAS.
- H. Chen, H. Tu, C. Hu, Y. Liu, D. Dong, Y. Sun, Y. Dai, S. Wang, H. Qian, Z. Lin and L. Chen, J. Am. Chem. Soc., 2018, 140, 896–899 CrossRef CAS PubMed.
- K. Jeong, S. Park, G. Y. Jung, S. H. Kim, Y.-H. Lee, S. K. Kwak and S.-Y. Lee, J. Am. Chem. Soc., 2019, 141, 5880–5885 CrossRef CAS PubMed.
- Q. Xu, S. Tao, Q. Jiang and D. Jiang, J. Am. Chem. Soc., 2018, 140, 7429–7432 CrossRef CAS PubMed.
- G. Zhang, Y. Hong, Y. Nishiyama, S. Bai, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2019, 141, 1227–1234 CrossRef CAS PubMed.
- C. Guo, K. Zhang, Q. Zhao, L. Pei and J. Chen, Chem. Commun., 2015, 51, 10244–10247 RSC.
- O. Lužanin, R. Dantas, R. Dominko, J. Bitenc and M. Souto, J. Mater. Chem. A, 2023, 11, 21553–21560 RSC.
- B. Esser, F. Dolhem, M. Becuwe, P. Poizot, A. Vlad and D. Brandell, J. Power Sources, 2021, 482, 228814 CrossRef CAS.
- X. Li, H. Wang, Z. Chen, H. S. Xu, W. Yu, C. Liu, X. Wang, K. Zhang, K. Xie and K. P. Loh, Adv. Mater., 2019, 31, 1–9 Search PubMed.
- M. Ibrahim, H. N. Abdelhamid, A. M. Abuelftooh, S. G. Mohamed, Z. Wen and X. Sun, J. Energy Storage, 2022, 55, 105375 CrossRef.
- T. Günther, K. Oka, S. Olsson, M. Åhlén, N. Tohnai and R. Emanuelsson, J. Mater. Chem. A, 2023, 11, 13923–13931 RSC.
- H. Gao, Q. Zhu, A. R. Neale, M. Bahri, X. Wang, H. Yang, L. Liu, R. Clowes, N. D. Browning, R. S. Sprick, M. A. Little, L. J. Hardwick and A. I. Cooper, Adv. Energy Mater., 2021, 11, 2101880 CrossRef CAS.
- D. Rodríguez-San-Miguel, C. Montoro and F. Zamora, Chem. Soc. Rev., 2020, 49, 2291–2302 RSC.
- Y. Tao, W. Ji, X. Ding and B.-H. Han, J. Mater. Chem. A, 2021, 9, 7336–7365 RSC.
- G. Zhao, H. Li, Z. Gao, L. Xu, Z. Mei, S. Cai, T. Liu, X. Yang, H. Guo and X. Sun, Adv. Funct. Mater., 2021, 31, 1–9 Search PubMed.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed.
- S. Haldar, K. Roy, R. Kushwaha, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2019, 9, 1902428 CrossRef CAS.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31, 1901640 CrossRef PubMed.
- Y. Zhu, X. Chen, Y. Cao, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Chem. Commun., 2019, 55, 1434–1437 RSC.
- K. Wang, H. Zhang, Y. Xiao, S. Ren, Y. Wang and L. Li, Chem. Eng. J., 2023, 454, 140283 CrossRef CAS.
- S. Haldar, K. Roy, S. Nandi, D. Chakraborty, D. Puthusseri, Y. Gawli, S. Ogale and R. Vaidhyanathan, Adv. Energy Mater., 2018, 8, 1702170 CrossRef.
- Y. Cao, C. Liu, M. Wang, H. Yang, S. Liu, H. Wang, Z. Yang, F. Pan, Z. Jiang and J. Sun, Energy Storage Mater., 2020, 29, 207–215 CrossRef.
- R. Zhou, Y. Huang, Z. Li, S. Kang, X. Wang and S. Liu, Energy Storage Mater., 2021, 40, 124–138 CrossRef.
- Y. Yue, H. Li, H. Chen and N. Huang, J. Am. Chem. Soc., 2022, 144, 2873–2878 CrossRef CAS PubMed.
- X. Li, S. Cai, B. Sun, C. Yang, J. Zhang and Y. Liu, Matter, 2020, 3, 1507–1540 CrossRef.
- L. Frey, J. J. Jarju, L. M. Salonen and D. D. Medina, New J. Chem., 2021, 45, 14879–14907 RSC.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- R. Yan, B. Mishra, M. Traxler, J. Roeser, N. Chaoui, B. Kumbhakar, J. Schmidt, S. Li, A. Thomas and P. Pachfule, Angew. Chem., Int. Ed., 2023, 62, e202302276 CrossRef CAS PubMed.
- Y. Chen, H. Dai, K. Fan, G. Zhang, M. Tang, Y. Gao, C. Zhang, L. Guan, M. Mao, H. Liu, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202302539 CrossRef CAS PubMed.
- J. Á. Martín-Illán, D. Rodríguez-San-Miguel, C. Franco, I. Imaz, D. Maspoch, J. Puigmartí-Luis and F. Zamora, Chem. Commun., 2020, 56, 6704–6707 RSC.
- M. M. Unterlass, Angew. Chem., Int. Ed., 2018, 57, 2292–2294 CrossRef CAS PubMed.
- T. Xue, O. A. Syzgantseva, L. Peng, M. A. Syzgantseva, R. Li, G. Xu, D. T. Sun, R. Qiu, C. Liu, S. Zhang, T. Su, P. Su, S. Yang, J. Li and B. Han, Chem. Mater., 2022, 34, 10584–10593 CrossRef CAS.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- B. Díaz de Greñu, J. Torres, J. García-González, S. Muñoz-Pina, R. Reyes, A. M. Costero, P. Amorós and J. V. Ros-Lis, ChemSusChem, 2021, 14, 208–233 CrossRef PubMed.
- S.-X. Gan, C. Jia, Q.-Y. Qi and X. Zhao, Chem. Sci., 2022, 13, 1009–1015 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- T. Sun, Z. Li, H. Wang, D. Bao, F. Meng and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 10662–10666 CrossRef CAS PubMed.
- N. Delaporte, G. Lajoie, S. Collin-Martin and K. Zaghib, Sci. Rep., 2020, 10, 3812 CrossRef CAS PubMed.
- T. P. Nguyen, A. D. Easley, N. Kang, S. Khan, S.-M. Lim, Y. H. Rezenom, S. Wang, D. K. Tran, J. Fan, R. A. Letteri, X. He, L. Su, C.-H. Yu, J. L. Lutkenhaus and K. L. Wooley, Nature, 2021, 593, 61–66 CrossRef CAS PubMed.
- B. Sun, Z. Ye, M. Zhang, Q. Song, X. Chu, S. Gao, Q. Zhang, C. Jiang, N. Zhou, C. Yao and J. Shen, ACS Appl. Mater. Interfaces, 2021, 13, 42396–42410 CrossRef CAS PubMed.
- W.-Y. Li, J.-J. Wan, J.-L. Kan, B. Wang, T. Song, Q. Guan, L.-L. Zhou, Y.-A. Li and Y.-B. Dong, Chem. Sci., 2023, 14, 1453–1460 RSC.
- M. E. Speer, M. Kolek, J. J. Jassoy, J. Heine, M. Winter, P. M. Bieker and B. Esser, Chem. Commun., 2015, 51, 15261–15264 RSC.
- C. Friebe and U. S. Schubert, Top. Curr. Chem., 2017, 375, 19 CrossRef PubMed.
- L. Xie, K. Xu, W. Sun, Y. Fan, J. Zhang, Y. Zhang, H. Zhang, J. Chen, Y. Shen, F. Fu, H. Kong, G. Wu, J. Wu, L. Chen and H. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300372 CrossRef CAS PubMed.
- S. Zhang, Y.-L. Zhu, S. Ren, C. Li, X.-B. Chen, Z. Li, Y. Han, Z. Shi and S. Feng, J. Am. Chem. Soc., 2023, 145, 17309–17320 CrossRef CAS PubMed.
Footnote |
| † All authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |