
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparison of the impact of cation chemistry in ionic liquid-based lithium battery electrolytes†
Faezeh
Makhlooghiazad
 *a,
Colin S. M.
Kang
a,
Mojtaba
Eftekharnia
a,
Patrick C.
Howlett
a,
Oliver
Hutt
b,
Maria
Forsyth
a,
Luke A.
O’Dell
a and
Jennifer M.
Pringle
*a
*a,
Colin S. M.
Kang
a,
Mojtaba
Eftekharnia
a,
Patrick C.
Howlett
a,
Oliver
Hutt
b,
Maria
Forsyth
a,
Luke A.
O’Dell
a and
Jennifer M.
Pringle
*a
aDeakin University, Melbourne, Institute for Frontier Materials, Victoria 3125, Australia. E-mail: f.makhlooghiazad@deakin.edu.au; jenny.pringle@deakin.edu.au
bBoron Molecular, 500 Princes Hwy, Noble Park, VIC 3174, Australia
First published on 14th September 2023
Abstract
There is an increasing interest in ionic liquid electrolytes for battery applications because they are potentially safer alternatives to conventional liquid electrolytes. As the properties of ionic liquid electrolytes strongly depend on the chemistry of the constituent cations and anions, with phosphonium cations often being more favourable, here-in, we compare electrolytes based on methyl-substituted phosphonium and ammonium cations, namely a new tris(amino)-based phosphonium cation with the bis(fluorosulfonyl)imide anion (P1(DMA)3][FSI]) and the previously reported hexamethylguanidinium [HMG][FSI]. Highly concentrated electrolytes containing 50 mol% LiFSI are investigated for their thermal properties and dynamic behaviour as well as in lithium cells. Glass transition temperature, viscosity, ionic conductivity, and the ion diffusion coefficients show dependency on the nature of the cation. The electrochemical performance of the two electrolytes in Li metal symmetrical cells was compared under different cycling conditions, and the interfacial resistance monitored by EIS. Under milder cycling rates (0.5 mA cm−2), the [HMG][FSI] based electrolyte shows lower overpotentials compared to the phosphonium analogue, but this is reversed when Li metal is cycled at high current (1.5 mA cm−2). A higher interfacial resistance during the first 5 cycles was observed in the cell with 50 mol% LiFSI in [HMG][FSI] when cycled at 1.5 mA cm−2 which was consistent with the SEI analysis performed using ex situ X-ray photoelectron spectroscopy that showed the presence of higher amount of carbonate species at the Li metal surface. Finally, both electrolytes supported full cell cycling with LiFePO4 cathodes (2 mA h cm−2) with good rate performance and stability, although P1(DMA)3][FSI]/50 mol% LiFSI gave higher areal capacity values than the HMG analogue when cycled at C/5 (1.6 vs. 1.53 mA h cm−2).
Introduction
Advanced energy storage technologies are imperative to meet the increasing demand of electronic devices and electric vehicles.1–3 Li-ion batteries with graphite anodes have been extensively used in portable electronic devices due to their high energy density compared to other battery technologies.4,5 However, a higher energy density than what Li-ion batteries can presently offer is needed for future technologies and transport. The Li metal anode with its high theoretical specific capacity (3861 mA h g−1, more than 10 times higher than that of a graphite anode with 327 mA h g−1 capacity), and the lowest reductive potential of all metallic anode materials (3.04 V vs. standard hydrogen), is an ideal alternative anode material.6–8 However, the practical application of lithium metal batteries is hampered due to some fundamental challenges including Li dendrite growth and the formation of an unstable solid electrolyte interphase (SEI). These challenges are due to the incompatibility of the reactive Li metal with organic solvent electrolytes that have commonly been used in Li-ion batteries, leading to low cycling efficiency and battery failure.9–11 Therefore, developing new solvent chemistries for the electrolyte that are non-flammable liquids such as deep eutectic solvents (DESs) made from lithium salts and a hydrogen bond donor such as an amide,12 or ionic liquids (ILs),13 could provide potential solutions to the abovementioned problems.In the past few years, there has been an increasing interest in the use of ionic liquids (ILs) (water-free organic salts that exhibit melting points below their decomposition temperatures),14 and their solid-state analogues, organic ionic plastic crystals (OIPCs), in electrochemical devices due to their unique properties such as high ionic conductivity, high thermal and electrochemical stability, nonflammability, negligible volatility and ability to form stable and conductive SEIs.15–24 The physicochemical behaviour and SEI formation properties of these salts can be tailored by altering the nature of the anions and cations as well as the electrolyte composition. It has been shown that high concentrations of alkali salts (50 mol% salt or more) added to ILs or OIPCs can form electrolytes with high target ion transport numbers that can support high charge/discharge rates in Li and Na devices.23,25–31 It is important to note that for OIPCs, depending on the binary phase diagram of the OIPC/salt combination, the resultant electrolytes can be solid, quasi-solid state or liquid at the chosen composition and temperature.
Previous reports have indicated the importance of the IL cation chemistry in improving Li and Na metal cycling.32,33 For example, Li metal cells were cycled with high efficiency of >99.2% by using a high concentration trimethylisobutylphosphonium bis(fluorosulfonyl)imide (P111i4FSI) ionic liquid of 3.8 m LiFSI in a Li|LiNi0.33Mn0.33Co0.33O2 (NMC111, ∼0.3 mA h cm−2) cell with capacities of 130 mA h g−1 for 200 cycles at room temperature.34–36 Stable Li stripping/plating behaviour was also achieved using 50 mol% LiFSI in triethylmethylphosphonium FSI ([P1222][FSI]) or N,N-diethylpyrrolidinium FSI, both of which form liquid electrolytes at room temperature despite the neat materials being solid.37,38
While alkyl substituted quaternary amines and phosphines have been the dominant cation chemistry in Li and Na based electrolytes, recently we have been exploring the influence of changing the shape and charge delocalisation of the IL cation. For example, the hexamethylguanidinium FSI, [HMG][FSI], OIPC was recently used as an electrolyte and presented interesting properties when mixed with Na and Li salts.39–42 Biernacka et al. investigated the effect of different anions and salt concentrations on the physicochemical properties of [HMG][FSI] by creating mixtures of [HMG][FSI]/LiFSI and [HMG][FSI]/LiTFSI. The results showed that 50 mol% LiFSI or LiTFSI in [HMG][FSI] is liquid with glass transition temperatures at −58 °C and −50 °C, respectively, whereas, [HMG][FSI] with 10 mol% and 90 mol% of LiFSI or LiTFSI acts as quasi-solid electrolytes with melting points at 89 °C for 90 mol% LiFSI and 120 °C for 90 mol% LiTFSI. Overall, the [HMG][FSI]/LiFSI mixtures showed higher ionic conductivity and greater Li ion diffusivity compared to the LiTFSI mixtures.40 The mixture of [HMG][FSI] with NaFSI showed a eutectic composition at 25 mol% NaFSI and a eutectic temperature of 44 °C. It exhibited a high ionic conductivity of 5.9 × 10−5 S cm−1 at 40 °C and a high sodium transference number of 0.36 at 50 °C. Stable stripping/plating of Na in a symmetrical coin cell was demonstrated with this electrolyte at 0.5 mA cm−2 current density with 1 h polarisation time with stable and low polarisation potential of 45 mV.
In a very recent work, we reported a new family of OIPCs with the tris(amino)-based phosphonium cation in combination with FSI or TFSI anions, where the FSI based OIPCs showed higher ionic conductivity.43 The structure of this new cation is of particular interest as it is similar to that of the HMG, which has shown very promising properties, but incorporates a phosphorus heteroatom instead of the central carbon. Phosphorus-based ILs and OIPCs have previously been shown to be very effective electrolytes for Li batteries, as discussed above. The electrochemical performance of the new phosphorus cation with ethyl-substituents, [P1(DEA)3][FSI], in combination with 50 mol% NaFSI, for Na metal battery application was demonstrated.43 However, the properties and performance of the new phosphorus-based salts with Li is yet to be studied. Further, there has been no direct comparison of the properties and electrochemical performance of the new phosphorus-based cation with that of the analogous HMG-based cation.
In this work, we selected the phosphonium cation with the smaller methyl chain [P1(DMA)3][FSI], chosen to exhibit closer structural resemblance to the HMG cation. After an initial study of both low and high concentrations of LiFSI on the phase behaviour, we then focused on the high salt content IL electrolyte (50 mol% LiFSI in [P1(DMA)3][FSI]). Our study aims to explore the thermal and transport properties of this bulk electrolyte in comparison to the 50 mol% LiFSI in [HMG][FSI]. This exhibits a wide liquid range and high ionic conductivities similar to those exhibited by DESs made from lithium salts and hydrogen bond donors such as an amide.12 Prior studies have revealed the importance of anion and cation chemistries on interphase properties and electrochemical performance in IL and OIPC electrolytes.32,44–46 Thus, here, we used X-ray photoelectron spectroscopy (XPS) analysis of the SEI layer to study the interphase properties of the two electrolytes. Finally, successful cycling of a Li/50 mol% LiFSI in [P1(DMA)3][FSI]/LiFePO4 cell at C/5 and C/2 at 50 °C is demonstrated, paving the way for application of these new electrolytes in future high energy density Li-metal batteries.
Experimental methods
Electrolyte preparation
The detailed synthesis of the OIPCs ([P1(DMA)3][FSI] and [HMG][FSI]) are provided in ref. 39 and 43. Briefly, the [P1(DMA)3]+ cation was synthesised by the quarternisation of tris(dimethylamino)phosphine with methyl iodide followed by filtration and drying under vacuum. Then, ion-exchange took place with [P1(DMA)3][I] and LiFSI (/KFSI) in water, and was extracted five times with dichloromethane. A white sticky solid was formed after drying in vacuo. The [HMG]+ cation was synthesised via the quarternisation of dimethylammonium chloride and dimethylamine. After removing the solvent and recrystallisation, further drying afforded [HMG][Cl] as a white solid. After ion-exchange with KFSI, the product was extracted with chloroform five times. A white sticky solid was formed after drying in vacuo.The neat OIPCs were dried under vacuum on a Schlenk-line at 60 °C for 48 hours before use. Lithium bis(fluorosulfonyl)imide (LiFSI) (99.5% purity) salt was purchased from Nippon Shokubai (Osaka, Japan). The 50 mol% LiFSI electrolytes were prepared by mixing the calculated amount of LiFSI with the OIPCs under magnetic stirring at 50 °C to form a homogeneous solution. The IL was then dried under vacuum on a Schlenk-line at 50 °C for 48 hours until the moisture content was less than 50 ppm as measured by Karl–Fischer analysis. For making 10 mol% LiFSI in [P1(DMA)3][FSI], after adding LiFSI to the OIPC, a few droplets of anhydrous acetonitrile (Sigma) were added to achieve a clear solution. Then the acetonitrile was evaporated, and the electrolyte was dried under vacuum on a Schlenk-line at 50 °C for 48 hours. The chemical structure of [P1(DMA)3], [HMG], and FSI are presented in Fig. 1.
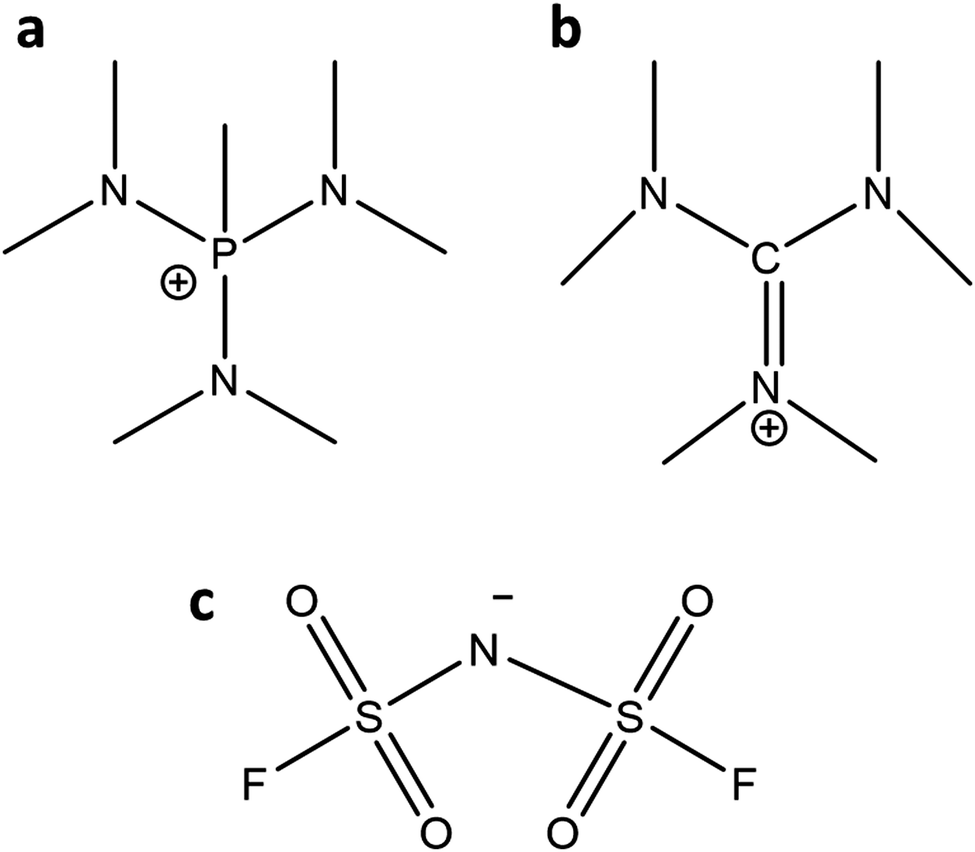 | ||
| Fig. 1 Molecular structure of (a) [P1(DMA)3]+, (b) [HMG]+ cations and (c) [FSI]− anion. | ||
Differential scanning calorimetry (DSC)
DSC was measured on a Netzch 214 Polyma DSC with liquid N2 cooling, driven by Proteus 80 software with a scan rate of 10 °C min−1. 5–10 mg of sample were sealed into an Al DSC pan in an Ar-filled glove box. The melting point of 10 mol% LiFSI in [P1(DMA)3][FSI] was also visually determined using a Gallenkamp melting point apparatus.Density and viscosity
The density of the ionic liquids (50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI]) were measured by a density meter from Anton Paar (DMA4500M) in the temperature range between 30 to 90 °C with an error of ±0.001 g cm−3. Their viscosity was measured by a rolling-ball viscometer from Anton Paar (Lovis 2000 M/ME) within the same temperature range of 30 to 90 °C. A 10 mm long capillary tube (+1.8 mm diameter) with automatic tilted angle was used to measure viscosity.Electrochemical impedance spectroscopy (EIS).
A Biologic MTZ-35 impedance analyzer (Bio-Logic Science Instruments, France), equipped with a Eurotherm 2204e temperature controller was used to measure electrochemical impedance spectroscopy (EIS). A custom-made conductivity dip-cell with two platinum electrodes was used for conductivity measurements of the liquid electrolytes. 15 min equilibration time was used per sample and three heating and cooling cycles were performed for each sample. The cell assembly was done inside an Ar-filled glovebox in the temperature range 30 to 80 °C for the mixed electrolytes and to 90 °C for the neat [P1(DMA)3][FSI]. The cell constant was determined by measuring EIS of a standard reference of 10 mM KCl solution at 30 °C and the data were analysed on MT-Lab software.Pulsed-field gradient nuclear magnetic resonance (PFGNMR)
The diffusivities of the [P1(DMA)3], [HMG], Li cations and [FSI] anion for the 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI] electrolytes were measured by 1H, 7Li and 19F pulse-field gradient NMR at 60 °C. The measurements were performed on a 7 T Bruker Avance III spectrometer equipped with a Bruker Diff50 probe using a double stimulated echo pulse sequence, gradient pulses of 2 ms, a diffusion time of 20 ms, and 16 gradient steps up to a maximum value of 3000 G cm−1. The data were analysed with TopSpin.Surface characterization
X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific Nexsa surface analysis system equipped with a hemispherical analyzer using an Al Kα X-rays source at a power of 72 W. High-resolution region spectra were acquired at 0.1 eV energy step. The pressure in the analysis chamber was lower than 5.0 × 10−9 mbar. CASA XPS software (v. 2.3.22PR1.0) was used. The XPS data processing and curve fitting were used to determine the chemical composition on the surface of the plated Li metal after 5 and 100 cycles at 1.5 mA cm−2|0.5 mA h cm−2. The spectra were calibrated based on C–C peak at 284.8 eV. To prepare the Li samples for XPS characterization, the cycled coin cells were disassembled in an Ar-filled glovebox, cleaned and dried under vacuum for 1 hour prior to being carried to the XPS instrument using a sealed chamber.Symmetric cell fabrication
Li|Li symmetrical cells were assembled in coin cells, (CR2032, Hohsen Co) inside an Ar-filled glovebox. A 100 μm thickness Li metal was placed on a 1 mm thick, 16 mm diameter stainless-steel spacer. To provide a constant internal pressure (1–2 kg cm−2) and good contact between the cell components a stainless-steel spring was used. A Celgard 3501 separator saturated with 80 μL electrolyte was sandwich between two Li metal disks. The cells were rested for 24 h at 50 °C before being cycled using a VMP3 (BioLogic) multichannel potentiostat. The cell interfacial resistance was measured before cycling and after each 10 cycles at 50 °C, with spectra recorded from 1 to 50 MHz with an amplitude of 10 mV.Lithium transference number (tLi+).
Li|Li symmetrical cells were prepared as previously mentioned in the symmetric cell fabrication section. The Li+ transference number measurements were done at 50 °C based on the Vincent-Bruce technique47 by applying a constant potential of 10 mV and measuring the initial (I0) and steady state (Iss) currents. Moreover, the cell impedance before (R0) and after (Rss) polarization was measured to calculate tLi+ using the following equation: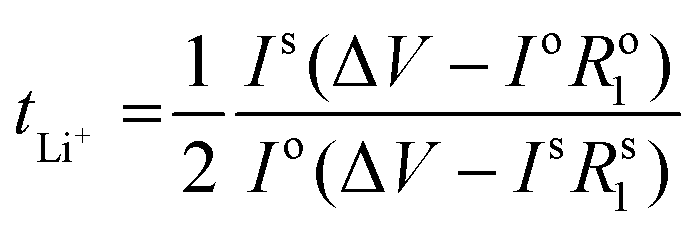
Full cell fabrication and cycling
Li|LiFePO4 (LFP) cells were assembled in coin cells (CR2032, Hohsen) in an Ar-filled glovebox using 8 mm cathode disks, 16 mm Celgard separator, 10 mm lithium metal disks with 100 μm thickness, and 80 μL of electrolyte. The coin cells were stored at 50 °C for 24 h before cycling to ensure high absorption of the electrolytes into the LFP electrodes. The cells were cycled within the potential range of 2.8 V and 3.8 V vs. Li/Li+ using a VMP3 (BioLogic) multichannel potentiostat battery cycler at 50 °C.Rate capability tests were conducted at 50 °C and at C-rates of C/20, C/10, C/5, C/2 1C, 2C and 5C using the 50 mol% LiFSI in [P1(DMA)3][FSI] electrolyte and LFP electrodes (Aleees) with areal capacity of 0.4 mA h cm−2 (mass loading of 2.25 mg cm−2). Long term cycling test was performed at 50 °C and at C/5 using 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI] electrolytes and LFP electrodes with areal capacity of 2 mA h cm−2.
To prepare low capacity (0.4 mA h cm−2) LFP electrodes, LFP powder and carbon black were first dry mixed in FlackTek SpeedMixer (USA), and polyvinylidene fluoride (PVDF) dissolved in N-methyl-2-pyrrolidone (NMP) was added to the dry mixture. The weight ratio of active material, carbon black and PVDF was 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10. The slurry was then cast on an Al foil using a doctor blade. The electrodes were first dried at 80 °C overnight before they were cut and dried again at 110 °C. The higher capacity (2 mA h cm−2) LFP electrodes were obtained from Customcells, Germany.
:10:10. The slurry was then cast on an Al foil using a doctor blade. The electrodes were first dried at 80 °C overnight before they were cut and dried again at 110 °C. The higher capacity (2 mA h cm−2) LFP electrodes were obtained from Customcells, Germany.
Results and discussion
Electrolyte properties: thermal phase behaviour, viscosity, and ionic conductivity
The performance of electrolytes strongly depends on their phase behaviour (melting transition, solid–solid phase transitions as well as glass transition temperature) and transport properties. The phase behaviours of the neat [P1(DMA)3][FSI] and its mixtures with LiFSI (10 and 50 mol%) were determined by DSC and are presented in Fig. 2a. The properties of the neat OIPC [P1(DMA)3[FSI] were also described in a recent paper by Sun et al.43 This cation was explored to further examine prospective electrolytes, due to the high performance of [HMG][FSI]-NaFSI electrolytes observed in other work.41,48 [P1(DMA)3][FSI] also features the presence of three dimethylamino functional groups stemming from the central atom. However, the [P1(DMA)3]+ cation differs by its central phosphorus atom, and that the charge is localised on the phosphorus atom whereas it can exist on any of the three nitrogen atoms for the [HMG]+ cation.39 Furthermore, the additional methyl group on the [P1(DMA)3]+ cation can lead to different structural interactions than with the [HMG]+ cation. [P1(DMA)3[FSI] exhibits a higher melting point by over 150 °C and displays wider phase II and phase I temperature ranges indicating significantly different phase behaviour (of 5 solid–solid phase transitions) to the [HMG][FSI] analogue.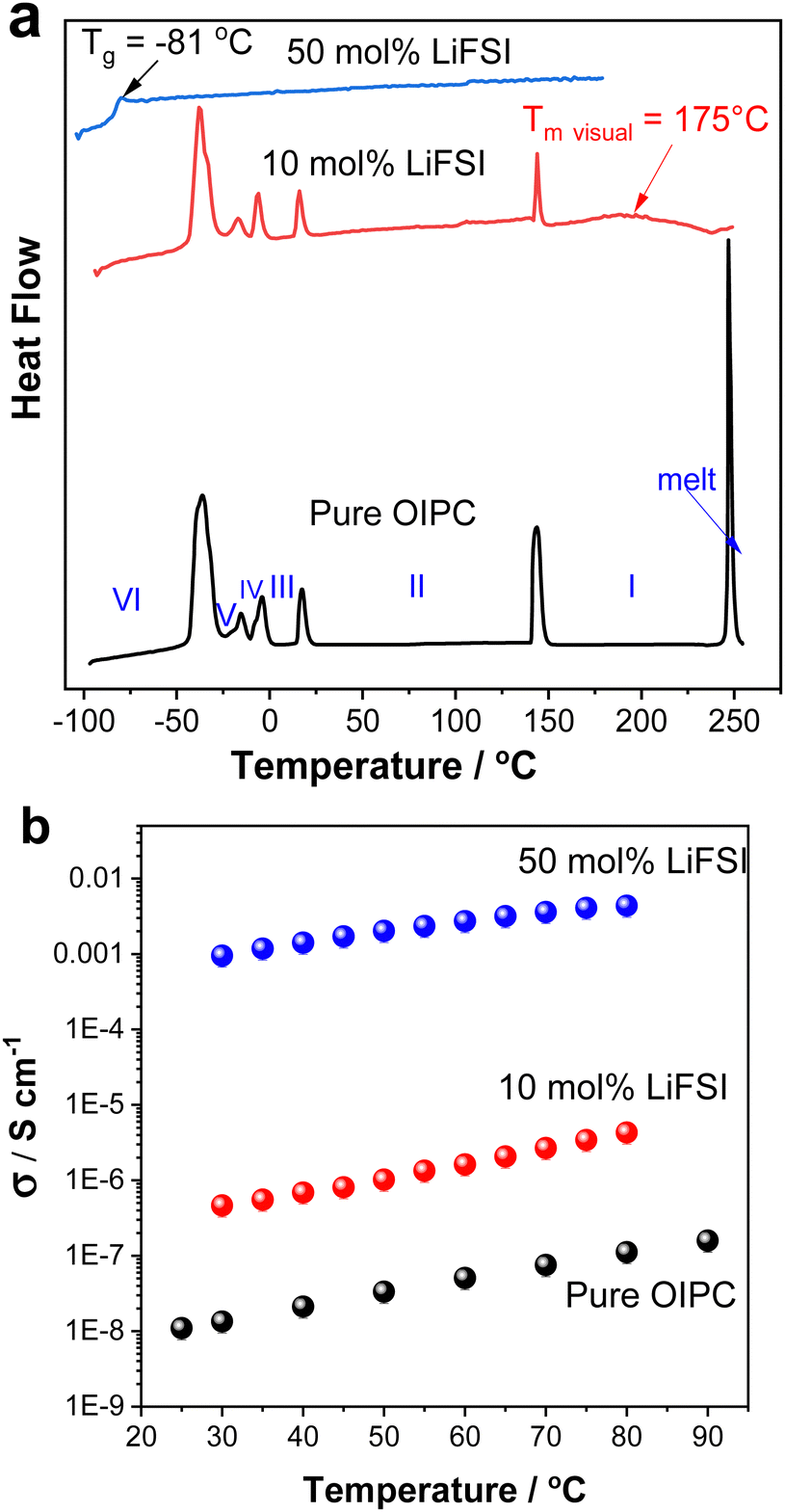 | ||
| Fig. 2 (a) DSC heating traces (b) ionic conductivity, of the pure OIPC ([P1(DMA)3][FSI]), 10 mol% and 50 mol% LiFSI in the OIPC. | ||
Adding 10 mol% LiFSI did not alter the enthalpies or onset temperatures of the phase VI–V, V–IV, IV–III and III–II transitions, however, the enthalpy of the phase II–I transition reduced from 8 J K−1 mol−1 for the pure OIPC to 3.5 J K−1 molOIPC−1 for the 10 mol% LiFSI mixture. This indicates that upon addition of 10 mol% LiFSI, the OIPC becomes more disordered in phase I (above 144 °C). In addition, the melting transition was significantly broadened and shifted to a lower temperature in the 10 mol% LiFSI electrolytes, (the melting temperature at 175 °C was confirmed visually, compared to the neat OIPC at 246 °C).
Upon increasing the LiFSI content to 50 mol%, a transparent and colourless liquid with Tg at −81 °C was formed. Suppression of the crystalline phase is common in lithium/sodium-salt mixtures with OIPCs and was reported previously in several electrolytes.40,49,50
The effect of Li-salt concentration on the ionic conductivity was studied by comparing the salt with 10 mol% LiFSI and 50 mol% LiFSI to that of the neat [P1(DMA)3][FSI] (Fig. 2b). It can be seen that the addition of LiFSI increased the ionic conductivity in all three samples across the temperature range, as was previously demonstrated in other plastic crystal systems.51–54 However, a more significant conductivity enhancement occurred when LiFSI was increased from 10 mol% to 50 mol% LiFSI (4.6 × 10−7 to 9.5 × 10−4 S cm−1 at 30 °C and 4.3 × 10−6 to 4.3 × 10−3 S cm−1 at 80 °C). As discussed above, adding 50 mol% LiFSI to the OIPC totally interrupted the long-range order of the neat OIPC, resulting in an ionic liquid phase. The high salt content ionic liquid system ([P1(DMA)3][FSI]–50 mol% LiFSI) was selected for further analysis and its properties were compared with 50 mol% LiFSI in [HMG][FSI]. As previously reported, the [HMG][FSI] based OIPC has been identified as having promising transport properties in combination with Na and Li salts.40,41
The physicochemical properties including thermal behaviour, ionic conductivity, density, dynamic viscosity of the [P1(DMA)3][FSI]–50 mol% LiFSI and the [HMG][FSI]–50 mol% LiFSI, have been studied and are presented in Fig. 3 and Fig. S1 (ESI†). With the addition of 50 mol% LiFSI to the [HMG][FSI] OIPC, the crystallisation of the OIPC is also suppressed and the solution behaves as an ionic liquid with a low glass transition, −58 °C, which is higher than [P1(DMA)3][FSI]–50 mol% LiFSI electrolyte (−83 °C) (Fig. 3a). This reflects significantly different interactions and slower dynamics in the [HMG]-based electrolyte, particularly at low temperature, with a more rapidly increasing viscosity leading to a glassy state at higher temperatures (−58 °C) compared to that of the P1(DMA)3-based electrolyte. Previous work in [HMG][FSI]–NaFSI suggested that the HMG cation may order in the IL electrolyte, which could explain the higher Tg, although more evidence is needed to support this assertion.41 The temperature dependences of dynamic viscosity and ionic conductivity are presented in Fig. 3b and c. The viscosity of the [HMG][FSI]–50 mol% LiFSI sample is higher at room temperature and is slightly lower at temperatures above 40 °C (97 mPa s at 60 °C) than for the [P1(DMA)3][FSI]–50 mol% LiFSI (113 mPa s at 60 °C). The ionic conductivity of the [P1(DMA)3][FSI]–50 mol% LiFSI is slightly higher from 35 °C, with a larger difference at higher temperatures. Comparing the ionic conductivity of these electrolytes with the well-studied 50 mol% LiFSI in [P111i4][FSI] IL, the latter has a higher ionic conductivity (1.4 × 10−3 30 °C) compared to 1.0 × 10−3 S cm−1 for the 50 mol% LiFSI in [P1(DMA)3][FSI] at 30 °C. However, their viscosity values are comparable to the 50 mol% LiFSI in [P111i4][FSI] electrolyte (362 mPa s at 30 °C in the [HMG][FSI]–50 mol% LiFSI, 264 mPa s at 30 °C in [P1(DMA)3][FSI]–50 mol% LiFSI and 323 mPa s at 25 °C in [P111i4][FSI]–50 mol% LiFSI).34
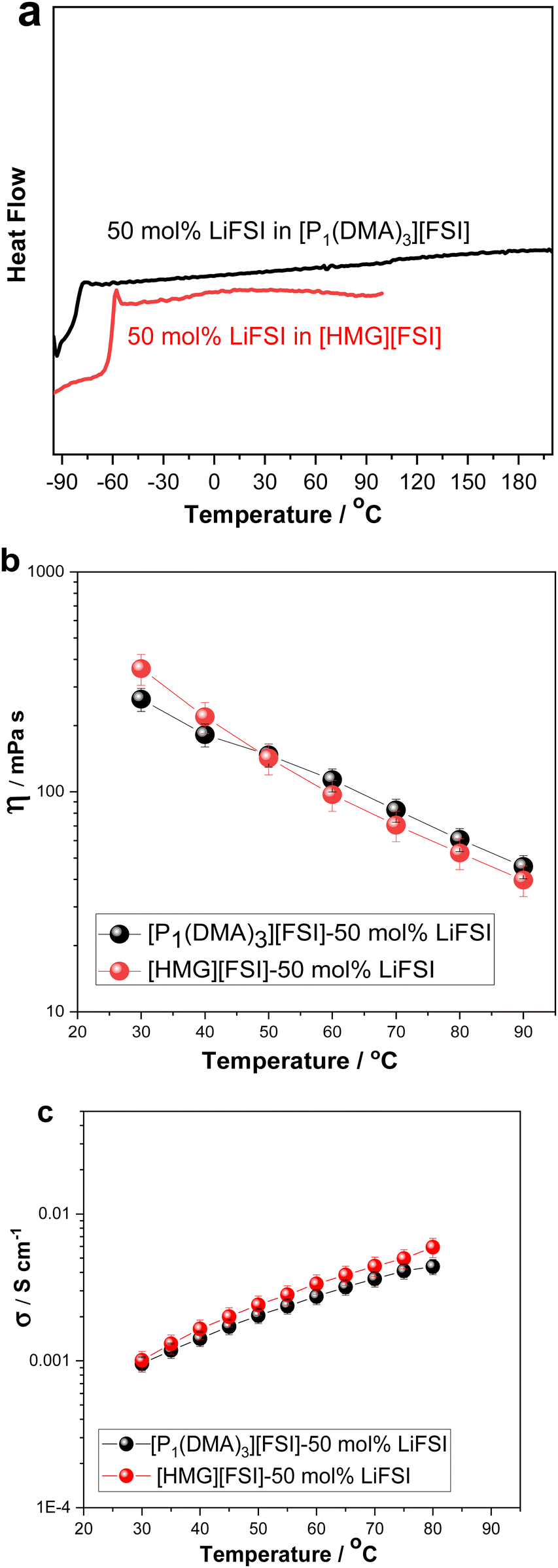 | ||
| Fig. 3 (a) DSC heating traces, (the feature at −90 °C in the [P1(DMA)3][FSI] system is an instrumental artefact as a result of the initial temperature conditions on the DSC instrument) (b) dynamic viscosity, (c) ionic conductivity of 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI]. | ||
Fig. S2 (ESI†) shows Arrhenius plot of temperature-dependent conductivities and viscosity for the [HMG][FSI]–50 mol% LiFSI, and [P1(DMA)3][FSI]–50 mol% LiFSI. Neither plot shows Arrhenius like behaviour, instead they show a curvature. Thus, the data was fit using the Vogel–Fulcher–Tammann (VFT) equation based on eqn (1).55 The VFT plots for conductivity and viscosity are presented in Fig. S2 (ESI†) and fit VFT parameters are summarized in Tables S1 and S2 (ESI†).
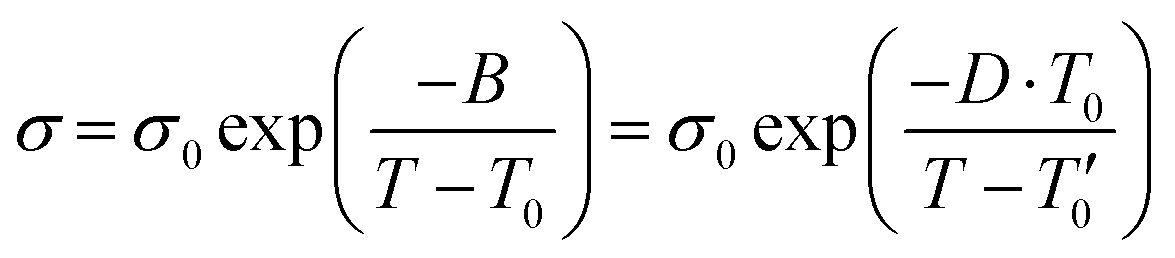 | (1) |
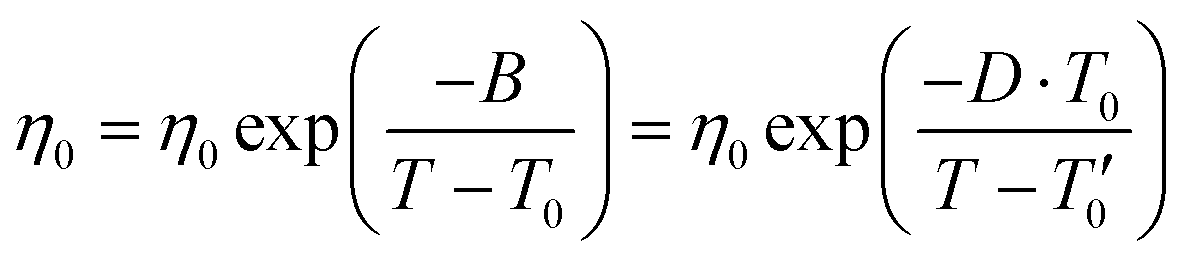 | (2) |
σ 0 and η0 are pre-exponential factors, related to the ionic conductivity and viscosity at infinite temperature. T is the temperature, T0 is the temperature at which ions start to show mobility. B is pseudo-activation energy for mobility. D is the fragility factor, (small D values are typical for fragile glasses (D < 30) while strong glasses show large D values (D > 30).
The pseudo-activation energy B for both conductivity and viscosity appears higher for [HMG][FSI]–50 mol% LiFSI (575), in contrast to [P1(DMA)3][FSI]–50 mol% LiFSI (560), indicating that the latter requires less energy for ion mobility. Additionally, the value of T0 is lower in the [P1(DMA)3][FSI]-system, suggesting that [P1(DMA)3][FSI] IL electrolyte is more prone to forming fragile glasses. The parameter Tg − T0 is relatively low in both electrolytes, and smaller (0.4) in the [P1(DMA)3][FSI]–50 mol% LiFSI system. This observation points towards a greater ion dissociation in this electrolyte compared to the [HMG][FSI] electrolyte. It is worth noting that both electrolytes exhibit relatively small fragility factor, D, consistent with the behaviour of liquids that form fragile glass structures. A comparison of the VFT parameters with other IL-salt electrolytes,55,56 indicates that the pseudo activation energies (B) in both electrolytes are either in the same range or lower than the other electrolytes. A comparison of the T0 values also shows that they are lower in comparison to some ILs reported in the literatures. To illustrate, the T0 values are lower than the value observed in 0.5 M LiTFSI in [N2(2O2O1)3][TFSI] (210 K),55 2.7 M LiFSI in [N2(2O2O1)3][TFSI] (207 K),55 or in 10 mol% LiFSI in [C1O1mmor][FSI] (202 K).56
High ionic conductivity is an indicator of a promising electrolyte in terms of the applicability of the electrolytes in a device. However, all ions in the high salt content IL contribute to the measured ionic conductivity; high Li ion conductivity is also important to achieve good device performance. To investigate the impact of the cation on this parameter, the lithium transference numbers of the two electrolytes was measured at 50 °C using the Bruce–Vincent method, based on CV polarisation and EIS measurements,47 and is presented in Fig. S3 (ESI†). This showed that the transference numbers are very close in the two electrolytes, at 0.44 ± 0.02 in the [HMG][FSI]–50 mol% LiFSI and 0.41 ± 0.03 in the [P1(DMA)3][FSI]–50 mol% LiFSI. It is worth noting that the transference number is also comparable to the [P111i4][FSI]–50 mol% LiFSI electrolyte, which was previously reported as 0.4 ± 0.02 at 25 °C.21
Ion diffusivity and ionicity analysis
To further study the mobility of the Li cations compared to the other ions in the electrolytes, the diffusion coefficients of 7Li, 1H (representative of the IL cations) and 19F (representative of the FSI anion) in 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI] were measured by pulsed-field gradient (PFG) NMR at 60 °C, and are shown in Fig. 4. While the diffusivity values of all ions are quite similar in the two electrolyte systems, they are slightly higher in the [HMG][FSI] system, consistent with higher ionic conductivity and slightly lower viscosity of this electrolyte at 60 °C. However, the 7Li is the most diffusive species in 50 mol% LiFSI in [P1(DMA)3][FSI], but not in the 50 mol% LiFSI in [HMG][FSI] electrolyte. In both Li-doped electrolyte systems, the diffusion coefficient of Li ions (DLi+) are faster than the diffusion coefficient of the cations (DH+), where the ratio of DLi+/DH+ equals 1.4 for 50 mol% LiFSI in [P1(DMA)3][FSI], and 1.2 for 50 mol% LiFSI in [HMG][FSI]. In other words, the Li ions are more decoupled from the other ions in the former system. This faster diffusivity of Li ions compared to the IL cations (DLi+ > DH+) in high salt content IL electrolytes is consistent with prior reports,25,34 where it was hypothesised that the higher diffusivity of the Li ions is due to structural rearrangement of the Li coordination environment, which leads to Li+ transport through an interconnected network. Marginally higher DLi+/DH+ ratio in the [P1(DMA)3]+ system may reflect more Li ions decoupled from the IL and less hindered by ion-association and is consistent with the better electrochemical performance using [P1(DMA)3][FSI] electrolyte discussed below. This is probed further below using Walden plot analysis. Moreover, the observed lower diffusion coefficient of the FSI anion, relative to the diffusion coefficient of Li ions (DLi+/DFSI− = 1.2), reinforces the likelihood of a significant structural rearrangement mechanism within this system.57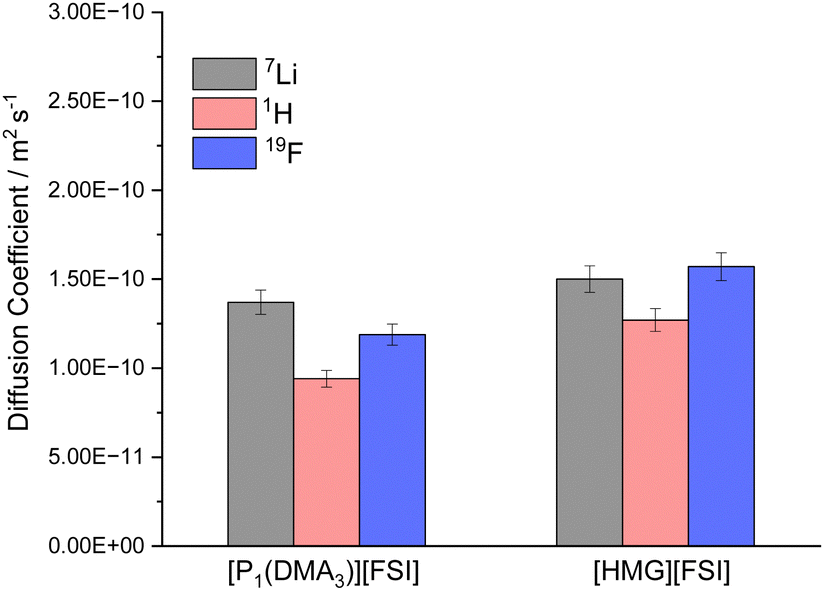 | ||
| Fig. 4 Diffusivity of the ions in 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI] at 60 °C. | ||
The extent of ionic interactions in an ionic liquid can be qualitatively characterized using a Walden plot, which is calculated based on the relationship between viscosity (fluidity (1/η)) and molar conductivity.58 The Walden plot of the two IL systems is presented in Fig. 5. Data for a dilute salt solution (0.01 M aqueous KCl solution,58 is also shown in a straight line in Fig. 6 as a reference to an ideal system in which the ions are fully dissociated and have equal mobility. Ideal systems have also been reported with 1 M KCl,59 or 0.1 M KCl.60 Deviation from the ideal line indicates the degree of ion association in the IL system. The dashed line in Fig. 5 indicates where the 10% of the molar conductivity for a given viscosity exists. The Walden plot of both electrolyte systems, 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI], show that they lie between the region of being ionic and sub-ionic according to Angell et al.,59 which likely indicate ionic interactions that prevent independent ion dissociation. However, this is common to Li-salt electrolyte systems as similarly observed in other Li-based electrolyte systems.61,62 However, the [P1(DMA)3][FSI]–50 mol% LiFSI solution shows a larger degree of dissociation at temperatures above 50 °C compared to the [HMG][FSI]–50 mol% LiFSI IL. This is in agreement with the trends in the ion diffusivity diffusion coefficient data where self-diffusion of ions in the [HMG][FSI]–50 mol% LiFSI IL is higher than in the [P1(DMA)3][FSI]–50 mol% LiFSI IL, but the ratio of Li diffusivity (DLi+) to IL cation diffusivity (DH+) is higher in the P1(DMA)3][FSI]–50 mol% LiFSI electrolyte. In comparison, Walden plot data for [P111i4][FSI]–50 mol% LiFSI IL lies fairly close to the ILs reported here, thus, in all three ionic liquids with 50 mol% LiFSI, the ion dissociation is good and of a similar order.34 According to the viscosity, conductivity, ionicity and Li transference data both electrolytes are relatively high conductivity with close viscosity and Li transference values, indicating potential applicability of both electrolytes in Li battery applications.
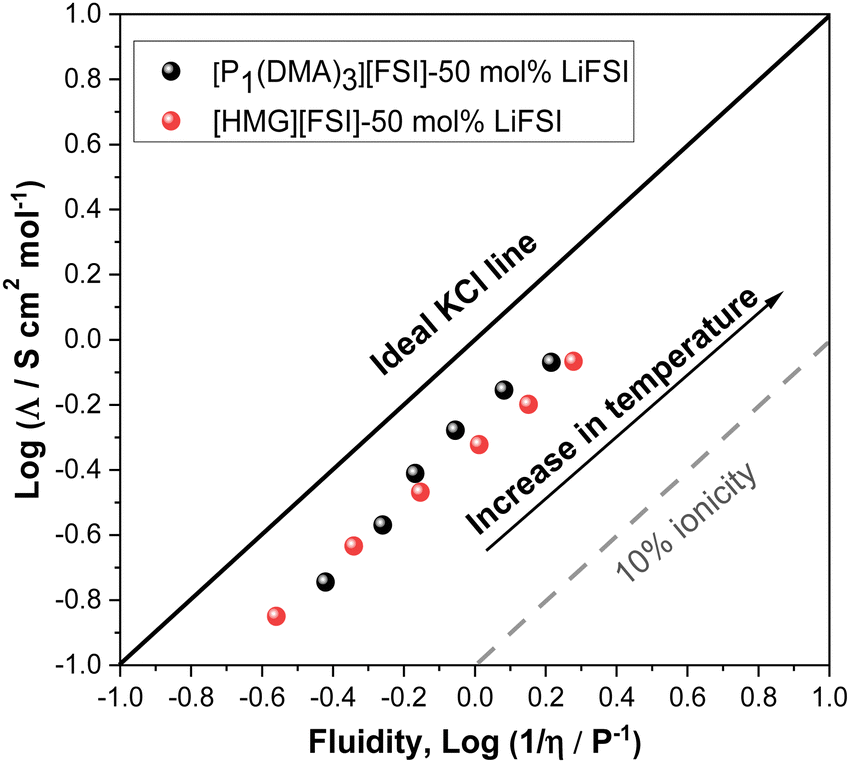 | ||
| Fig. 5 Walden plot showing relationship between inverse viscosity and molar conductivity for 50 mol% LiFSI in [P1(DMA)3][FSI] and 50 mol% LiFSI in [HMG][FSI]. | ||
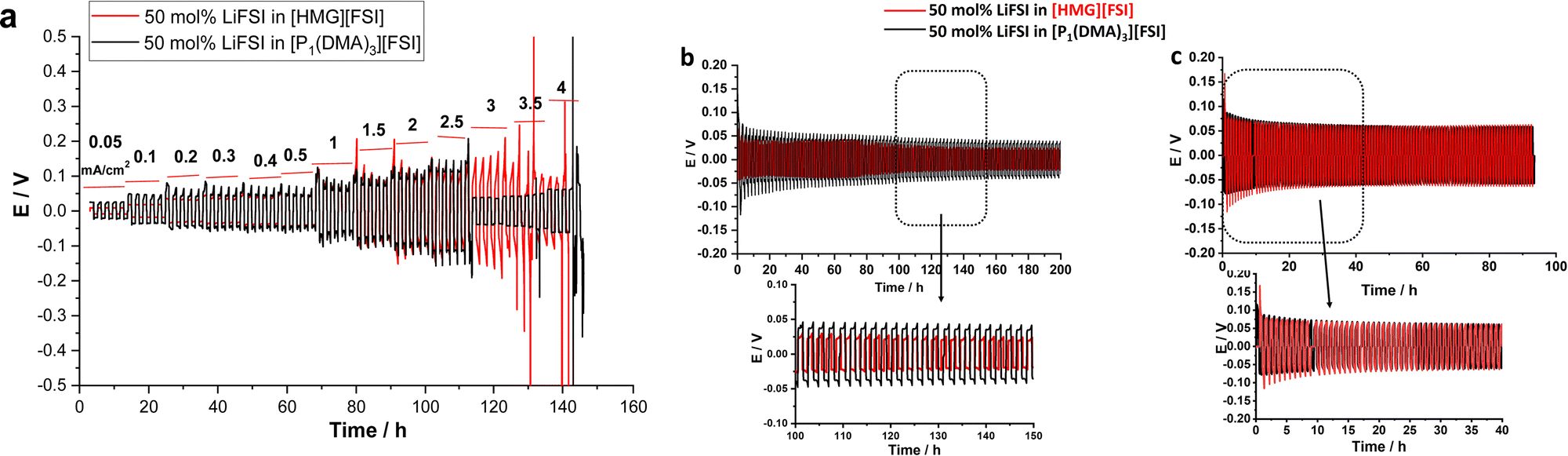 | ||
| Fig. 6 (a) Rate capability of the 50 mol% LiFSI in [P1(DMA)3][FSI] and [HMG][FSI] in Li symmetrical cell, 1 hour polarization time at 50 °C, (b) long term cycling of the 50 mol% LiFSI in [P1(DMA)3][FSI] and [HMG][FSI] in Li symmetrical cell at 0.5 mA cm−2 (c) 1.5 mA cm−2, 0.5 mA h cm−2 at 50 °C. | ||
Cycling stability of lithium symmetrical cells
To investigate the compatibility of the electrolytes with lithium metal electrodes and investigate the effect of the cation on the interfacial reactivity, the reversibility of Li plating and stripping in Li symmetrical cells was studied. Fig. 7a shows the galvanostatic cycling of Li symmetrical cells with [P1(DMA)3][FSI]–50 mol% LiFSI and [HMG][FSI]–50 mol% LiFSI electrolytes at 50 °C by applying various current densities ranging from 0.05 mA cm−2 to 4 mA cm−2 and a polarization interval of 1 hour. As can be seen in Fig. 6a, a low and stable polarization potential is observed in both electrolytes up to 2.5 mA cm−2 (2.5 mA h cm−2) indicating that both electrolytes have excellent compatibility with the Li electrodes and can support the electrochemistry of Li as well as Li ion transport for charges up to 2.5 mA h cm−2. Interestingly, the polarization potential is lower at lower current densities, from 0.05 up to 0.4 mA cm−2, in the [HMG][FSI]–50 mol% LiFSI electrolyte (9 mV at 0.05 mA cm−2 and 43 mV at 0.4 mA cm−2) compared to the [P1(DMA)3][FSI]–50 mol% LiFSI electrolyte (25 mV at 0.05 mA cm−2 and 45 mV at 0.4 mA cm−2). The polarization potential is similar at currents higher than 0.4 mA cm−2.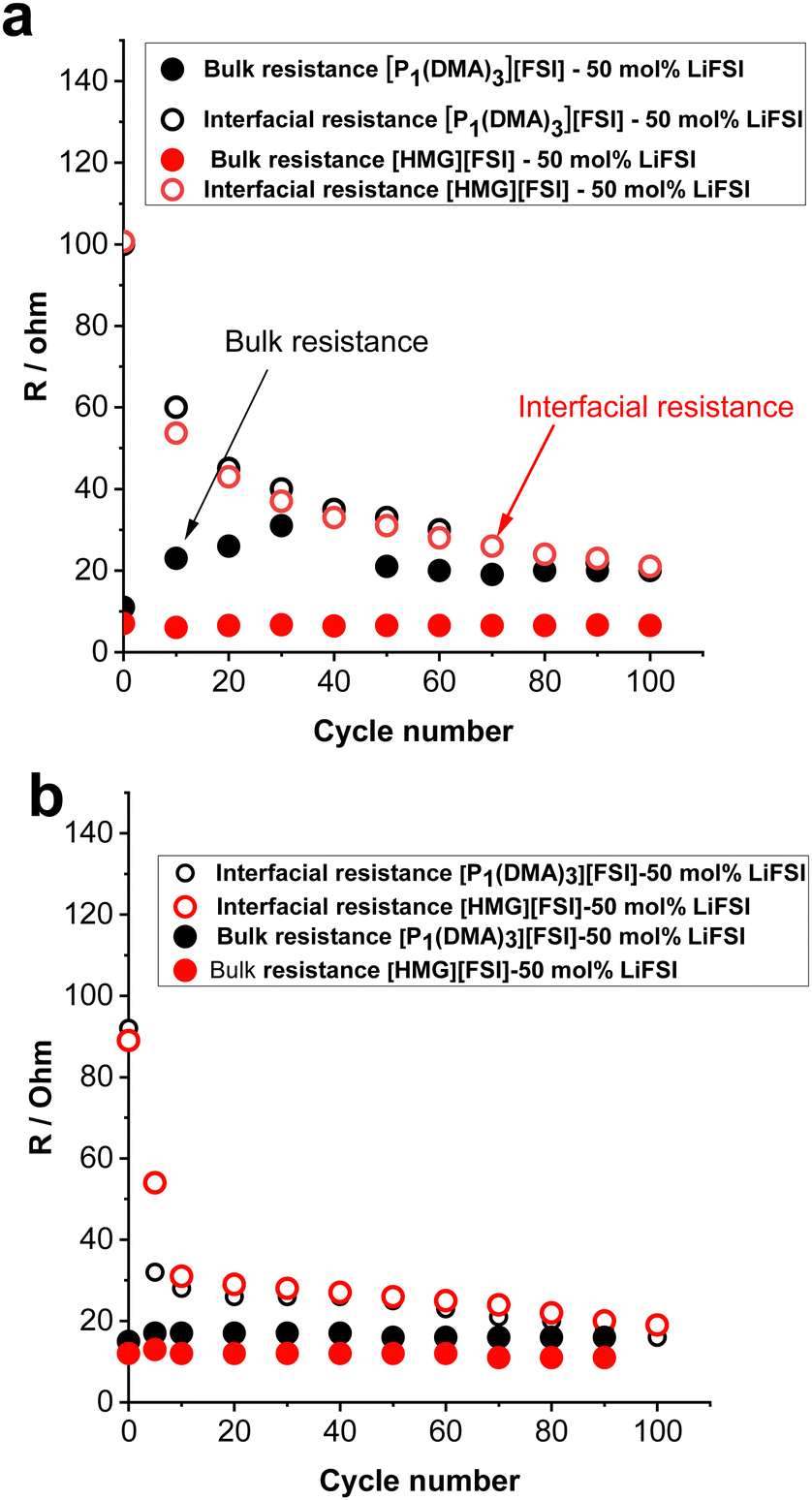 | ||
| Fig. 7 Bulk and interfacial resistance of the Li symmetrical cells obtained before polarisation and after every 10 cycles for [P1(DMA)3][FSI]–50 mol% LiFSI and [HMG][FSI]–50 mol% LiFSI, (a) 0.5 mA cm−2 (0.5 mA h cm−2), (b) 1.5 mA cm−2 (0.5 mA h cm−2). | ||
Longer-term symmetrical cell cycling was conducted at 50 °C for both electrolytes, after resting at 50 °C for 24 h. Since the [HMG]-based electrolyte showed lower polarization potential at lower current densities, in the long-term cycling study, the amount of charge cycled was set to a constant 0.5 mA h cm−2, but two different current densities, 0.5 and 1.5 mA cm−2, were applied. Fig. 6b shows the cycling potential profile of the Li|Li cells with the different electrolytes over 100 cycles (each electrode was plated/stripped for 1 h at 0.5 mA cm−2). In general, the voltage profiles of the Li|Li cells cycled in both highly concentrated IL electrolytes show impressive Li stripping and plating stability, with a low overpotential for 100 cycles without any breakdown. However, the polarisation potential for the cell with the [HMG][FSI] electrolyte is lower over the whole 100 cycles, which is consistent with the rate capability data where at lower current density the polarisation potential for the HMG-based electrolyte was lower. When a higher current density of 1.5 mA cm−2 was applied (Fig. 6c), initially the overpotential for the HMG system was higher, which then decreased upon continued cycling, stabilising at values only slightly higher than those of the [P1(DMA)3] system.
The cycling results indicate that the cell overpotential and cycling stability depends on the cation chemistry and the applied current densities and is influenced by multiple factors including conductivity and Li ion transport, as well as interfacial processes and the SEI. This is consistent with the recent report on Na metal cells at moderate current densities (1 mA cm−2) with 50 mol% NaFSI in [C3mpyr][FSI] IL which showed lower overpotentials compared to the [P111i4][FSI] IL system.44 However, upon applying higher rate cycling of (1 mA cm−2 and 4.0 mA h cm−2) the system with the [C3mpyr][FSI] IL exhibited a shorting failure while the phosphonium IL sustained 800 h (100 cycles) at this current density. The Na surface characterization showed different compositions formed at the surface of the Na metals when different cation chemistry and different current densities were used.
Interfacial properties at a Li metal surface
To gain further understanding about the interfacial layer, electrochemical impedance (EIS) measurements were conducted before cycling and after every 10 cycles, for the Li symmetrical cells containing each electrolyte (Fig. 7). The impedance spectra are fitted based on the equivalent electric circuit shown in Fig. S4 (ESI†). The interfacial resistance values for the [HMG][FSI] and [P1(DMA)3][FSI] systems were similar at 100 ± 5 Ω before cycling. However, when applying a low current density of 0.5 mA cm−2, the interfacial resistance decreased in both systems after 10 cycles, attributed to the formation of a new SEI layer on the lithium metal. The resistance value for the [HMG][FSI] electrolyte after 10 cycles at 0.5 mA cm−2 decreased to 53 ± 5 Ω, while for the [P1(DMA)3][FSI] electrolyte the resistance decreased to 60 ± 5 Ω. This more rapid decrease in resistance for the [HMG][FSI] electrolyte is reflected in the cycling profile as a lower overpotential (Fig. 7). Interestingly, the bulk resistance of the [HMG][FSI]–50 mol% LiFSI is stable at 7 ± 5 Ω before and after cycling, whereas for the [P1(DMA)3][FSI] electrolyte the bulk resistance increased from 11 ± 1 to 31 ± 2 Ω after 30 cycles and stabilised at 21 ± 2 Ω after 50 cycles. Battery charging/discharging may lead to concentration gradients in the [P1(DMA)3][FSI]–50 mol% LiFSI and induce crystallization of the LiFSI salt rich phase. It is worth noting that neat [P1(DMA)3][FSI] is a higher melting point OIPC compared to the [HMG][FSI] OIPC. Thus it is possible that the compositions around 50 mol% LiFSI (i.e., slightly higher or lower concentrations) in the [P1(DMA)3][FSI] OIPC appeared in the solid or quasi-solid phase. In general, the bulk resistance of 50 mol% LiFSI in the [HMG][FSI] is slightly lower than [P1(DMA)3][FSI], which is consistent with the ionic conductivity results presented in Fig. 3c. When applying a higher current density of 1.5 mA cm−2, the bulk resistances for both electrolytes are stable and again slightly higher for the [P1(DMA)3][FSI]–50 mol% LiFSI (17 ± 2 Ω) than [HMG][FSI]–50 mol% LiFSI (12 ± 1 Ω). The interfacial resistances for both electrolytes decreased more sharply in the [P1(DMA)3][FSI]–50 mol% LiFSI electrolytes after 5 cycles to reach 32 ± 2 Ω compared to 54 ± 2 Ω in the [HMG][FSI]–50 mol% LiFSI. The higher interfacial resistance in the first 5 cycles with the [HMG][FSI]–50 mol% LiFSI is reflected in the slightly higher polarisation potential with this electrolyte presented in Fig. 6c. This reveals the evolution of the stable interphase during the first stripping/plating cycles in the [HMG][FSI]–50 mol% LiFSI electrolyte. The interfacial resistances become close in both electrolytes after 5 cycles.In order to understand the reason for the higher initial polarisation potential in the cell with the 50 mol% LiFSI in [HMG][FSI] electrolyte at higher current densities (1.5 mA cm−2), the interphases formed on the Li metal surface from the different ionic liquid electrolytes were studied by XPS analysis after 5 and 100 Li stripping/plating cycles. Fig. 8a provides a graphical representation of XPS survey spectra, illustrating the relative proportion of various chemical elements of various chemical elements (F, O, N, C, S, P, and Li) on plated Li surfaces. This figure immediately shows some significant differences between the two electrolyte systems. Firstly, whereas the top surface of the metal cycled in the [HMG][FSI] electrolyte system changes significantly upon increasing the cycling from 5 to 100 cycles, in contrast, the [P1(DMA)3][FSI] IL electrolyte sustains a more stable SEI composition as observed from all the XPS spectra. This behaviour has been noted before for Li metal cycled in [C3mpyr][FSI] under different cycling rates; when high rates were used to deposit Li metal the subsequent SEI remained stable throughout the cycling duration and, moreover, this correlated with better cycling efficiencies.63,64 Similarly for Na metal electrodes cycled in ammonium and phosphonium IL electrolytes, improved electrochemical performance for the phosphonium electrolyte appeared to coincide with a less changeable SEI composition.44 Notably, the data from both electrolyte systems demonstrate a consistent pattern of increasing the relative amount of F from 5 cycles to 100 cycles. In [HMG][FSI], the proportion of F rises from 1.51% after 5 cycles to 3.18% after 100 cycles, while in [P1(DMA)3][FSI], it increases from 2.01% after 5 cycles to 7.31% after 100 cycles. This observation indicates the formation of an interphase layer on the Li surface during cycling. Furthermore, the percentage of S, which likely represents FSI-reduction components, demonstrates an upward trend with successive cycling. This finding further supports the growth of the SEI layer over the cycling period. Notably, the percentage of these two components are higher on the Li surface with [P1(DMA)3][FSI] IL.
Taking a closer look at Li 1s, F 1s, C 1s, O 1s, and N 1s spectra shown in Fig. 8b–f, the nature of the chemical species and their relative amounts can be ascertained as follows. The Li 1s spectra of all the samples have a peak at ∼54 eV that shows the presence of lithium fluoride (LiF) and a peak at ∼55 eV related to lithium carbonate (Li2CO3) species.65,66 68% and 86% of Li is LiF in the 50 mol% LiFSI in [P1(DMA)3][FSI] IL after 5 and 100 cycles respectively, and 43% and 68% of Li is LiF, after 5 and 100 cycles respectively, when 50 mol% LiFSI in [HMG][FSI] electrolyte was used. Table S3 (ESI†) summarizes the composition of the SEI on the Li surface after 5 and 100 cycles. This shows that the LiF content on the Li surface from the [HMG][FSI] IL increased significantly upon cycling, and is consistently lower than in the 50 mol% LiFSI in [P1(DMA)3][FSI] IL.
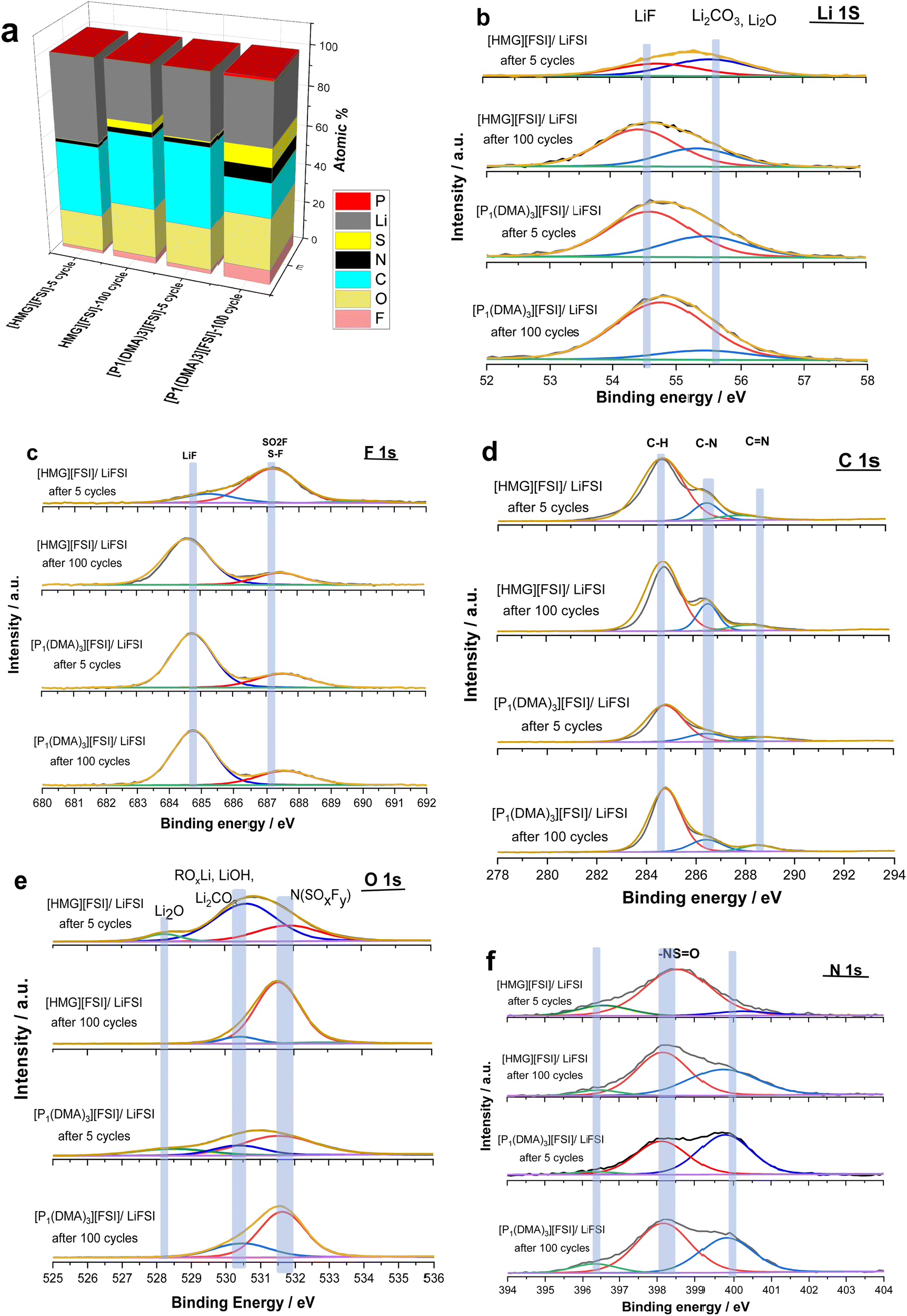 | ||
| Fig. 8 (a) Chemical composition (atomic%) summary determined from the survey spectra (b) Li 1s spectra (c) F 1s spectra (d) C 1s spectra (e) O 1s spectra and f) N 1s spectra of the Li surface after 5 and 100 cycles using 50 mol% LiFSI in [P1(DMA)3][FSI] and [HMG][FSI]. | ||
It has been reported that bulk LiF has high chemical and mechanical stability and allows high Li diffusion, which stabilizes the SEI layer and allows homogeneous Li ion diffusion, hence better cycling performance.6,67 In all cases, the F 1s spectra also show a peak at 685 eV consistent with the presence of a layer of LiF on the surface,68 as indicated by the Li 1s spectra.
Quantification of the amount of LiF in the F 1s spectra indicates that it has rapidly formed on the Li surface when [P1(DMA)3][FSI] IL was used (75% after 5 cycles) and remained stable up to 100 cycles (75%). In contrast, it appears to take more time to build up on the surface of Li metal when the [HMG][FSI] electrolyte was used (21% after 5 cycles and 79% after 100 cycles). On all the surfaces another peak was observed at 687 eV, which is related to a covalent –F species like an –SO2F group, presumably resulting from decomposition of the FSI anion.69,70 The C 1s spectra all show a broad peak at 285 eV corresponding to the presence of aliphatic C species (C–C and/or C–H) and a C–N peak at 286 eV. There is also a small peak in the [HMG][FSI] electrolyte spectra at 288 eV attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, and a peak at 289 eV assigned to C–P in the [P1(DMA)3][FSI] IL based electrolyte, both of which are presumably related to the decomposition of the IL cation or residual salt on the surface.70,71
N, and a peak at 289 eV assigned to C–P in the [P1(DMA)3][FSI] IL based electrolyte, both of which are presumably related to the decomposition of the IL cation or residual salt on the surface.70,71
In the N 1s spectra from all the electrolytes, there are two peaks corresponding to the N-species which are attributed to degradation of FSI anions, appearing at 400 and 398 eV, in agreement with previous reports.72 A small quantity of nitride species is also observed at 396 eV, most likely related to species existing at the surface of pristine Li discs.65 All of the O 1s spectra have peaks indicating the formation of Li2CO3 at around 530 eV, consistent with the appearance of this species in the Li 1s spectra, and N(SOxFy) species (532 eV) presumably from decomposition of the FSI anions at the Li surface of all the electrolytes.73 A peak at 528 eV was also observed in the [HMG][FSI] and [P1(DMA)3][FSI] IL after 5 cycles that is associated with Li2O. Interestingly, the Li2O peak is no longer evident in the spectra of the surfaces after 100 cycles in either electrolyte. Thus, the Li2O species possibly exists on the surface of the Li metal in the native SEI or it was formed initially. The intensity of the Li2CO3 peak in the O 1s spectra decreased after 5 cycles from 65% to 8% after 100 cycles in the [HMG][FSI] electrolyte and the intensity of the peak associated with the FSI decomposition at 532 eV (–SO2–, –SO3–2) becomes more evident (28% after 5 cycles to 89% after 100 cycles). However, the intensity of these compositions did not significantly change in the [P1(DMA)3][FSI] electrolyte. Fig. S5 (ESI†) summarizes the SEI composition on the Li surface in the presence of 50 mol% LiFSI within [P1(DMA)3][FSI] and [HMG][FSI] electrolytes, examined after 5 and 100 cycles. Notably, the formation of LiF is observed after 5 cycles, exhibiting a gradual augmentation up to 100 cycles. This trend underscores the favourable stability of the SEI layer over extended cycling periods.
As a tentative conclusion, the differences in the XPS spectra between the two Li-metal electrodes cycled in two different electrolytes is most distinct after 5 cycles. Specifically, the [P1(DMA)3][FSI] IL facilitates a more rapid formation and conversion of the SEI into a LiF-rich film. This transformation results in a lower resistance and a reduced polarisation potential, as previously discussed. These effects highlight the potential advantages of the [P1(DMA)3][FSI] IL as compared to the alternative electrolyte system. While further investigations are warranted to fully understand the underlying mechanisms, the initial findings suggest that the [P1(DMA)3][FSI] IL promotes the development of a more favourable SEI composition, leading to enhanced electrochemical performance.
Li-metal battery performance
To demonstrate the application of the new electrolytes, [P1(DMA)3][FSI]–50 mol% LiFSI electrolyte was selected to be cycled with low mass loading (2.25 mg cm−2 = 0.38 mA h cm−2) LFP cathode. The rate capability of this cell was investigated at different current rates between C/20 and 5C, with C/20 performed for two cycles and the other current rates (C/10 to 5C) performed for five cycles, as presented in Fig. 9a. The cell shows an initial discharge capacity of 169 mA h g−1 at C/20 and C/10, which means all the theoretical capacity (169 mA h g−1) was delivered. When the current density was increased to 5C, a reversible capacity of 143 mA h g−1 was achieved. Importantly, when a high rate of C/2 was applied at the end of the rate capability cycles, the cell retained a capacity of 164 mA h g−1 (Fig. 9a).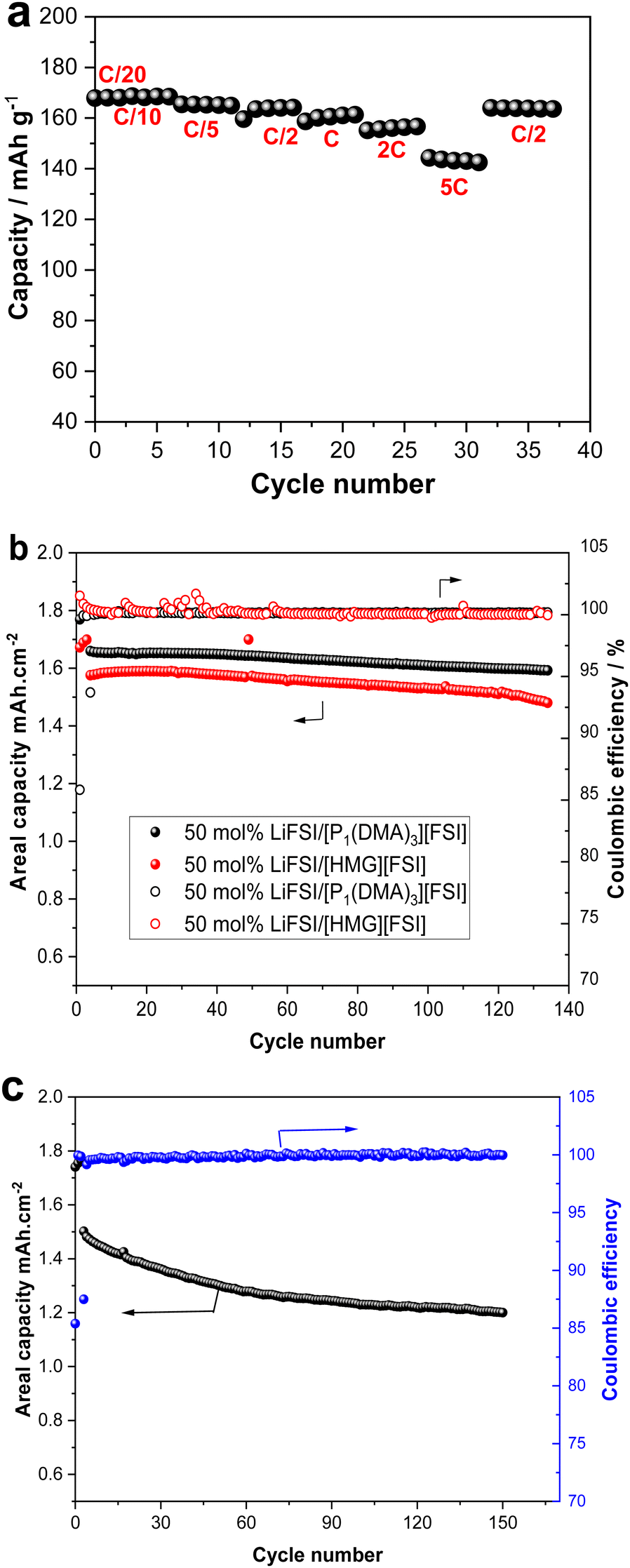 | ||
| Fig. 9 (a) Rate capability of Li|50 mol% LiFSI/[P1(DMA)3][FSI]|LFP (b) Cycling performance of Li|50 mol% LiFSI/[P1(DMA)3][FSI]|LFP and Li|50 mol% LiFSI/[HMG][FSI]|LFP at C/5 and (c) Cycling performance of Li|50 mol% LiFSI/[P1(DMA)3][FSI]|LFP at C/2 rate at 50°. | ||
As the cells exhibited good rate capability with a low-loading mass cathode electrode at 50 °C, long-term cycling of the cells fabricated with higher mass loading LFP electrodes (2 mA h cm−2) was performed with both [P1(DMA)3][FSI]–50 mol% LiFSI and [HMG][FSI]–50 mol% LiFSI electrolytes. The cells exhibited stable, long-term cycling at C/5 at 50 °C, as presented in Fig. 9b. The areal capacity of 1.6 mA h cm−2 with a capacity retention of 97% with ∼99.9% coulombic efficiency was achieved over 100 cycles in a cell with [P1(DMA)3][FSI]–50 mol% LiFSI and 1.53 mA h cm−2 with ∼99.9% coulombic efficiency was attained when [HMG][FSI]–50 mol% LiFSI was used. The excellent cycling stability is also confirmed by the corresponding charge–discharge plots in Fig. S6 (ESI†).
As the [P1(DMA)3][FSI]–50 mol% LiFSI showed higher areal capacity, this electrolyte was subjected to higher current rates, C/2, at 50 °C. The cell showed stable, long-term cycling at C/2 with a first-discharge reversible capacity of 1.5 mA h cm−2 and a reversible discharge capacity value of 1.2 mA h cm−2 after 150 cycles (80% capacity retention and 99.9% Coulombic efficiency, (Fig. 9c). Table S4 (ESI†) provides a summary of the Li battery performance of the reported high salt content IL-based electrolytes.
Conclusions
This work presents a comprehensive study and comparison of the phase behaviour, viscosity, ionic conductivity, diffusion and electrochemical properties of high LiFSI content ILs developed from two novel OIPCs where the cations had some structural similarities; one having a phosphorous centre and the other carbon based. Compared with the [P1(DMA)3][FSI]–50 mol% LiFSI, the [HMG][FSI]–50 mol% LiFSI has slightly higher viscosity at room temperature and marginally lower viscosity at temperatures above 40 °C, together with slightly higher ionic conductivity above 35 °C. Despite the slightly lower ionic conductivity in the [P1(DMA)3][FSI]–50 mol% LiFSI, the Li ions appeared more decoupled from the other ions in this electrolyte; NMR analysis showed that the ratio of Li diffusivity (DLi+) to IL cation diffusivity (DH+) is slightly higher, while a Walden plot showed that the degree of dissociation at temperatures above 50 °C is larger in the [P1(DMA)3][FSI]–50 mol% LiFSI.The cycling of Li symmetrical cells indicated that Li can be cycled consistently and reversibly in both electrolytes at 50 °C up to current densities of 2.5 mA cm−2, with a lower polarization potential at lower current densities (up to 0.4 mA cm−2) in the [HMG][FSI]–50 mol% LiFSI. Both electrolytes showed very good long-term cycling stability with current densities of 0.5 and 1.5 mA cm−2 and excellent reversibility in long-term Li plating/stripping tests. Slightly higher polarization potential was observed for the [HMG][FSI]–50 mol% LiFSI at higher current density of 1.5 mA cm−2, and correspondingly higher interfacial resistance was observed in the EIS data with cycling. Finally, full Li-metal batteries with LiFePO4 cathodes demonstrated stable cycling with very good charge/discharge reversibility (CE = 99.9%) at C/5 after more than 100 cycles at 50 °C with both electrolytes. The best capacity retention was achieved using the cells with [P1(DMA)3][FSI]–50 mol% LiFSI. Thus, this work introduces two new Li metal battery electrolytes to the field, both of which show excellent device performance, and insights into the effect of the unique cation structures on the bulk and interfacial properties. Future investigations will involve conducting XPS depth profiling to compare the thickness of the solid electrolyte interphase (SEI) in both electrolyte systems. Additionally, molecular dynamic (MD) simulations will be employed to gain further insights into the structure of the different cations present at the metal surface.
Author contributions
Faezeh Makhlooghiazad: conceptualization, experimental design, methodology, investigation, data analysis, writing original draft. Colin Kang: materials synthesis, analysis. Mojtaba Eftekharnia: full cell assembly and cell cycling, analysis. Luke A. O’Dell: NMR supervision and NMR data interpretation. Oliver Hutt: material synthesis supervision. Jennifer M. Pringle: conceptualization, supervising, scientific discussion and understanding of concept. Patrick C. Howlett: scientific discussion and understanding of concepts, Maria Forsyth: scientific discussion and understanding of concepts, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Australian Research Council Training Centre for Future Energy Storage Technologies (StorEnergy) for funding through IC180100049. The authors acknowledge use of facilities at the Battery Research and Innovation Hub.References
- W. P. Wang, J. Zhang, J. Chou, Y. X. Yin, Y. You, S. Xin and Y. G. Guo, Adv. Energy Mater., 2021, 11, 2000791 CrossRef CAS.
- F. Yang, W. Li and B. Tang, Chem. Eng. J., 2018, 334, 2021–2029 CrossRef CAS.
- J. Wan, W.-P. Chen, G.-X. Liu, Y. Shi, S. Xin, Y.-G. Guo, R. Wen and L.-J. Wan, J. Energy Chem., 2022, 67, 780–786 CrossRef CAS.
- Y. Liang, C. Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J. Q. Huang, D. Yu, Y. Liu, M. M. Titirici and Y. L. Chueh, InfoMat, 2019, 1, 6–32 CrossRef CAS.
- A. Manthiram, ACS Cent. Sci., 2017, 3, 1063–1069 CrossRef CAS PubMed.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- E. Cha, J. H. Yun, R. Ponraj and D. K. Kim, Mater. Chem. Front., 2021, 5, 6294–6314 RSC.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu and A. Manthiram, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- Q. Yang and C. Li, Energy Storage Mater., 2018, 14, 100–117 CrossRef.
- D. Aurbach, E. Zinigrad, Y. Cohen and H. Teller, Solid State Ionics, 2002, 148, 405–416 CrossRef CAS.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- M. E. Di Pietro and A. Mele, J. Mol. Liq., 2021, 338, 116597 CrossRef CAS.
- G. Kaur, H. Kumar and M. Singla, J. Mol. Liq., 2022, 351, 118556 CrossRef CAS.
- A. Mariani, M. Bonomo, X. Gao, B. Centrella, A. Nucara, R. Buscaino, A. Barge, N. Barbero, L. Gontrani and S. Passerini, J. Mol. Liq., 2021, 324, 115069 CrossRef CAS.
- J. Sun, L. A. O’Dell, M. Armand, P. C. Howlett and M. Forsyth, ACS Energy Lett., 2021, 6, 2481–2490 CrossRef CAS.
- T. C. Mendes, X. Zhang, Y. Wu, P. C. Howlett, M. Forsyth and D. R. Macfarlane, ACS Sustainable Chem. Eng., 2019, 7, 3722–3726 CrossRef CAS.
- M. Eftekharnia, M. Hasanpoor, M. Forsyth, R. Kerr and P. C. Howlett, ACS Appl. Energy Mater., 2019, 2, 6655–6663 CrossRef CAS.
- F. Makhlooghiazad, M. Sharma, Z. Zhang, P. C. Howlett, M. Forsyth and L. F. Nazar, J. Phys. Chem. Lett., 2020, 11, 2092–2100 CrossRef CAS PubMed.
- A. Basile, M. Hilder, F. Makhlooghiazad, C. Pozo-Gonzalo, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Adv. Energy Mater., 2018, 8 Search PubMed.
- S. A. Ferdousi, M. Hilder, A. Basile, H. Zhu, L. A. O'Dell, D. Saurel, T. Rojo, M. Armand, M. Forsyth and P. C. Howlett, ChemSusChem, 2019, 12, 1700–1711 CrossRef CAS PubMed.
- M. Forsyth, G. M. Girard, A. Basile, M. Hilder, D. MacFarlane, F. Chen and P. Howlett, Electrochim. Acta, 2016, 220, 609–617 CrossRef CAS.
- R. Hagiwara, K. Matsumoto, J. Hwang and T. Nohira, Chem. Rec., 2019, 19, 758–770 CrossRef CAS PubMed.
- K. Matsumoto, Y. Okamoto, T. Nohira and R. Hagiwara, J. Phys. Chem. C, 2015, 119, 7648–7655 CrossRef CAS.
- H. Yang, J. Hwang, Y. Wang, K. Matsumoto and R. Hagiwara, J. Phys. Chem. C, 2019, 123, 22018–22026 CrossRef CAS.
- K. Araño, D. Mazouzi, R. Kerr, B. Lestriez, J. Le Bideau, P. C. Howlett, N. Dupre, M. Forsyth and D. Guyomard, J. Electrochem. Soc., 2020, 167, 120520 CrossRef.
- M. Forsyth, G. M. A. Girard, A. Basile, M. Hilder, D. R. MacFarlane, F. Chen and P. C. Howlett, Electrochim. Acta, 2016, 220, 609–617 CrossRef CAS.
- H. Zhang, W. Qu, N. Chen, Y. Huang, L. Li, F. Wu and R. Chen, Electrochim. Acta, 2018, 285, 78–85 CrossRef CAS.
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406 CrossRef CAS.
- D. A. Rakov, F. Chen, S. A. Ferdousi, H. Li, T. Pathirana, A. N. Simonov, P. C. Howlett, R. Atkin and M. Forsyth, Nat. Mater., 2020, 19, 1096–1101 CrossRef CAS PubMed.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1–9 Search PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS PubMed.
- F. Makhlooghiazad, J. Guazzagaloppa, L. A. O’Dell, R. Yunis, A. Basile, P. C. Howlett and M. Forsyth, Phys. Chem. Chem. Phys., 2018, 20, 4721–4731 RSC.
- G. A. Giffin, J. Mater. Chem. A, 2016, 4, 13378–13389 RSC.
- G. M. Girard, M. Hilder, H. Zhu, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. Macfarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 8706–8713 RSC.
- G. M. A. Girard, M. Hilder, N. Dupre, D. Guyomard, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D. R. MacFarlane and P. C. Howlett, ACS Appl. Mater. Interfaces, 2018, 10, 6719–6729 CrossRef CAS PubMed.
- G. Girard, Deakin University, 2016.
- R. Kerr, N. Singh, T. S. Arthur, T. Pathirana, F. Mizuno, K. Takechi, M. Forsyth and P. C. Howlett, Sustainable Energy Fuels, 2018, 2, 2276–2283 RSC.
- D. Al-Masri, R. Yunis, H. Zhu, L. Jin, P. Bruce, A. F. Hollenkamp and J. M. Pringle, J. Mater. Chem. A, 2019, 7, 25389–25398 RSC.
- R. Yunis, A. F. Hollenkamp, C. Forsyth, C. M. Doherty, D. Al-Masri and J. M. Pringle, Phys. Chem. Chem. Phys., 2019, 21, 12288–12300 RSC.
- K. Biernacka, D. Al-Masri, R. Yunis, H. Zhu, A. F. Hollenkamp and J. M. Pringle, Electrochim. Acta, 2020, 357, 136863 CrossRef CAS.
- K. Biernacka, F. Makhlooghiazad, I. Popov, H. Zhu, J.-N. Chotard, C. M. Forsyth, R. Yunis, L. A. O’Dell, A. P. Sokolov and J. M. Pringle, J. Phys. Chem. C, 2021, 125, 12518–12530 CrossRef CAS.
- K. Biernacka, J. Sun, F. Makhlooghiazad, A. Balkis, I. E. Gunathilaka, L. A. O’Dell, M. G. Mestres, P. C. Howlett, J. M. Pringle and M. Forsyth, J. Phys.: Energy, 2022, 5, 014006 Search PubMed.
- J. Sun, C. S. M. Kang, G. Huang, L. A. O’Dell, M. G. Mestres, O. Hutt, P. C. Howlett, M. Forsyth and J. M. Pringle, J. Mater. Chem. A, 2023 Search PubMed.
- S. A. Ferdousi, L. A. O’Dell, J. Sun, Y. Hora, M. Forsyth and P. C. Howlett, ACS Appl. Mater. Interfaces, 2022, 14, 15784–15798 CrossRef CAS PubMed.
- M. Hilder, P. C. Howlett, D. Saurel, E. Gonzalo, A. Basile, M. Armand, T. Rojo, M. Kar, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2018, 268, 94–100 CrossRef CAS.
- A. Bagno, C. Butts, C. Chiappe, F. D'Amico, J. C. Lord, D. Pieraccini and F. Rastrelli, Org. Biomol. Chem., 2005, 3, 1624–1630 RSC.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- K. Biernacka, D. Al-Masri, R. Yunis, H. Zhu, A. F. Hollenkamp and J. M. Pringle, Electrochim. Acta, 2020, 357, 136863 CrossRef CAS.
- F. Makhlooghiazad, P. C. Howlett, X. Wang, M. Hilder, D. R. MacFarlane, M. Armand and M. Forsyth, J. Mater. Chem. A, 2017, 5, 5770–5780 RSC.
- Q. Zhou, W. A. Henderson, G. B. Appetecchi and S. Passerini, J. Phys. Chem. C, 2010, 114, 6201–6204 CrossRef CAS.
- E. Cooper and C. Angell, Solid State Ionics, 1986, 18, 570–576 CrossRef.
- W. A. Henderson, D. M. Seo, Q. Zhou, P. D. Boyle, J. H. Shin, H. C. De Long, P. C. Trulove and S. Passerini, Adv. Energy Mater., 2012, 2, 1343–1350 CrossRef CAS.
- M. Moriya, D. Kato, W. Sakamoto and T. Yogo, Chem. Commun., 2011, 47, 6311–6313 RSC.
- Z.-B. Zhou and H. Matsumoto, Electrochem. Commun., 2007, 9, 1017–1022 CrossRef CAS.
- M. Hilder, M. Gras, C. R. Pope, M. Kar, D. R. MacFarlane, M. Forsyth and L. A. O’Dell, Phys. Chem. Chem. Phys., 2017, 19, 17461–17468 RSC.
- A. Warrington, L. A. O’Dell, O. E. Hutt, M. Forsyth and J. M. Pringle, Energy Adv., 2023, 2, 530–546 RSC.
- M. Brinkkötter, A. Mariani, S. Jeong, S. Passerini and M. Schönhoff, Adv. Energy Sustainability Res., 2021, 2, 2000078 CrossRef.
- K. Ueno, H. Tokuda and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 1649–1658 RSC.
- C. A. Angell, N. Byrne and J.-P. Belieres, Acc. Chem. Res., 2007, 40, 1228–1236 CrossRef CAS PubMed.
- J. Stoimenovski, E. I. Izgorodina and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2010, 12, 10341–10347 RSC.
- H. Yoon, A. S. Best, M. Forsyth, D. R. MacFarlane and P. C. Howlett, Phys. Chem. Chem. Phys., 2015, 17, 4656–4663 RSC.
- X. Gao, F. Wu, A. Mariani and S. Passerini, ChemSusChem, 2019, 12, 4185–4193 CrossRef CAS PubMed.
- K. Periyapperuma, E. Arca, S. Harvey, T. Pathirana, C. Ban, A. Burrell, C. Pozo-Gonzalo and P. C. Howlett, ACS Appl. Mater. Interfaces, 2020, 12, 42236–42247 CrossRef CAS PubMed.
- K. Periyapperuma, E. Arca, S. Harvey, C. Ban, A. Burrell, D. R. MacFarlane, C. Pozo-Gonzalo, M. Forsyth and P. C. Howlett, J. Mater. Chem. A, 2020, 8, 3574–3579 RSC.
- P. C. Howlett, N. Brack, A. F. Hollenkamp, M. Forsyth and D. R. Macfarlane, J. Electrochem. Soc., 2006, 153, A595 CrossRef CAS.
- A. Budi, A. Basile, G. Opletal, A. F. Hollenkamp, A. S. Best, R. J. Rees, A. I. Bhatt, A. P. O’Mullane and S. P. Russo, J. Phys. Chem. C, 2012, 116, 19789–19797 CrossRef CAS.
- W. Xue, Z. Shi, M. Huang, S. Feng, C. Wang, F. Wang, J. Lopez, B. Qiao, G. Xu and W. Zhang, Energy Environ. Sci., 2020, 13, 212–220 RSC.
- K. Edstroem, T. Gustafsson and J. O. Thomas, Electrochim. Acta, 2004, 50, 397–403 CrossRef CAS.
- U. Pal, D. Rakov, B. Lu, B. Sayahpour, F. Chen, B. Roy, D. R. MacFarlane, M. Armand, P. C. Howlett and Y. S. Meng, Energy Environ. Sci., 2022, 15, 1907–1919 RSC.
- A. Basile, A. Bhatt and A. O’Mullane, Nat. Commun., 2016, 7, 1–11 Search PubMed.
- D. Rakov, M. Hasanpoor, A. Baskin, J. W. Lawson, F. Chen, P. V. Cherepanov, A. N. Simonov, P. C. Howlett and M. Forsyth, Chem. Mater., 2021, 34, 165–177 CrossRef.
- B. Philippe, R. M. Dedryvere, M. Gorgoi, H. K. Rensmo, D. Gonbeau and K. Edstrom, J. Am. Chem. Soc., 2013, 135, 9829–9842 CrossRef CAS PubMed.
- N. Schulz, R. Hausbrand, C. Wittich, L. Dimesso and W. Jaegermann, J. Electrochem. Soc., 2018, 165, A833 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00336a |
| This journal is © The Royal Society of Chemistry 2023 |