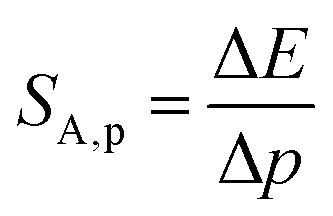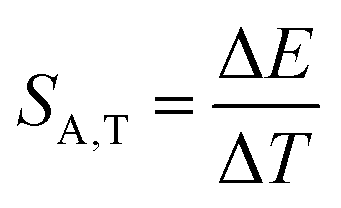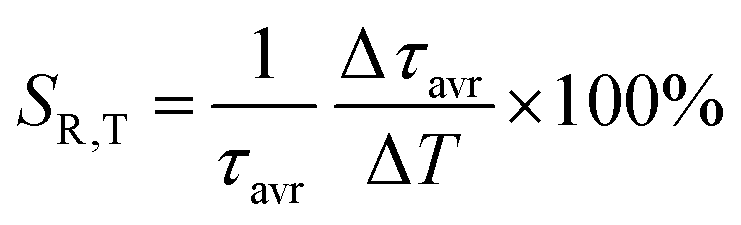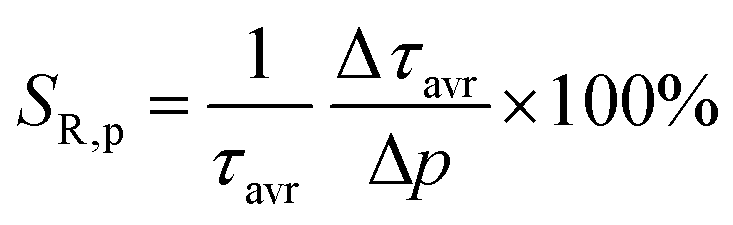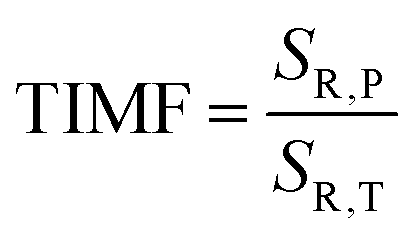DOI:
10.1039/D3TC00911D
(Paper)
J. Mater. Chem. C, 2023,
11, 11353-11360
Temperature invariant lifetime based luminescent manometer on Mn4+ ions†
Received
13th March 2023
, Accepted 24th July 2023
First published on 26th July 2023
Abstract
Luminescent manometry enables remote pressure readout with unprecedented spatial and pressure resolution. The need to image pressure in a temperature-invariant manner imposes the search for new solutions that offer such capabilities. In this study, we present an approach that enables remote pressure sensing using the luminescence kinetics of Mn4+ ions in SrGdAlO4. The uniqueness of this solution is related to the pressure induced prolongation of the lifetime of the 2E level of Mn4+ ions resulting from a change in the covalency of the Mn4+–O2− bond. Taking the advantage of this effect, the luminescence decay time was increased from 1.44 ms to 2.14 ms when the pressure was changed from ambient to 7.6 GPa. This allowed the development of a luminescent manometer with a maximum relative sensitivity of 7.85%/GPa at 3 GPa and sensitivities above SR > 5%/GPa in the pressure range of 1–7.6 GPa. Moreover, in the temperature range of 260–365 K, the lifetime value was independent of temperature changes, enabling a temperature invariant manometric factor of 134.
Introduction
Apart from the lighting market, luminescent materials are increasingly finding new applications, among which the sensing market has recently been one of the most developed owing to their ability to readout and even image physical and chemical quantities of objects under study, which can be done remotely by analyzing the spectroscopic parameters associated with the emitted light.1–6 In addition to the most popular luminescence thermometry, the possibility of remote pressure monitoring has also been intensively explored recently.7–11 Among the luminescent manometers described so far, the most common way to readout the pressure values in a given system is through spectral band shift analysis.11–26 This strategy applies both to luminophores with a narrow emission band headed by ruby and also to broadband luminophores. In this case, the pressure reading involves either an analysis of the position of the emission band centroid or a ratiometric reading of the ratio of luminescence intensities occurring in the two spectral bands.9,11,19–22 However, in the case of applications of luminescent manometers in environments where the medium is characterized by a dispersive dependence of the extinction coefficient, a reading based on analysis of the spectral response, including ratiometric approach and the band shift of the luminophore can lead to reduced reliability owing to the interactions of the emitted light with the medium in the form of absorption and/or scattering.23,24 Hence, in such applications, luminescent manometers using pressure-induced changes in luminescence kinetics are advantageous. Importantly and often overlooked, a luminescent pressure gauge should be characterized by insensitivity to temperature changes so that the pressure reading offered is unambiguous, even under extreme conditions of pressure and temperature. Although this type of manometer is rarely described in the literature, the vast majority of them are based on the luminescence of Ln3+ ions, where the dominant processes affecting the shortening of a lifetime are multiphonon and cross-relaxations,25 and their probabilities are significant for most Ln3+ ions at room temperature. This makes an unambiguous pressure reading difficult. On the other hand, due to the shielding of the 4f subshell by external orbitals, an increase in pressure does not significantly change the probability of radiative processes, hence little change in luminescence kinetics under increasing pressure is observed. Therefore, herein, we propose a new approach, in which we take the advantage of the fact that the spectroscopic properties of transition metal ions significantly depend on the strength of the crystal field, and therefore, on the applied pressure to lifetime-based manometers. Mn4+ ions, which are known for their red emission associated with the 2E → 4A2 electron transition, were used as model ions in this case.26–28 For Mn4+ ions, the rate of depopulation of the excited 2E level depends on the relative energy distance between the 2E level and the intersection point with the 4T2 state. The energy of the 4T2 state increases with increasing crystal field strength, hence it is expected that as the applied pressure increases the probability of the non-radiative processes will decrease, leading to the prolongation of the luminescence lifetime. In addition, a change in the applied pressure will affect the covalency of the Mn4+–O2− bond by changing the probability of non-radiative processes. The energy of the 4T2 level of Mn4+ ions significantly depends on the strength of the crystal field acting on it and thus depends on the applied pressure.
Experimental
SrGdAlO4 powders doped with different concentrations of Mn4+ ions were synthesized by a modified Pechini method.29 The materials used for synthesis: strontium nitrate (Sr(NO3)2 99.9965% purity from AlfaAesar), aluminum nitrate hydrate (Al(NO3)3·9H2O 99.999% purity from AlfaAesar), gadolinium oxide (Gd2O3 99.995% purity from Stanford Materials Corporation), manganese(II) chloride tetrahydrate (MnCl2·4H2O 99% purity from Sigma Aldrich), nitric acid (HNO3 65%; pure p.a.; from POCH), citric acid (≥99.5%, Sigma–Aldrich) and polyethylene glycol PEG 200 (H(OCH2CH2)nOH, n = 200, Alfa Aesar) were used without further purification. First, a stoichiometric amount of gadolinium oxide was dissolved in nitric acid to obtain gadolinium nitrate. Then, a three-fold recrystallization was carried out to evaporate the excess nitric acid. Appropriate amounts of strontium nitrate, aluminum nitrate, and manganese chloride were added to the gadolinium precursor and dissolved in 50 ml of distilled water. Next, citric acid was added in an amount six times the number of moles of the metal ions used in the synthesis. Once a clear solution was obtained, PEG was added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to the citric acid, and stirring continued for 2 h at room temperature. After this, the mixture was transferred to a porcelain crucible and dried at 360 K for 7 days. The obtained resin was then quenched at 873 K for 3 h, followed by grating in a mortar under hexane. This was placed in a crucible and annealed at 1323 K for 5 h in air.
:1 ratio to the citric acid, and stirring continued for 2 h at room temperature. After this, the mixture was transferred to a porcelain crucible and dried at 360 K for 7 days. The obtained resin was then quenched at 873 K for 3 h, followed by grating in a mortar under hexane. This was placed in a crucible and annealed at 1323 K for 5 h in air.
The obtained materials were then examined by X-ray powder diffraction (XRD) using a PANalytical X’Pert Pro diffractometer in Bragg–Brentano geometry equipped with an Anton Paar TCU1000 N temperature control unit using Ni-filtered Cu Kα radiation (V = 40 kV, I = 30 mA). Measurements were made in the range of 10–90°, and the acquisition time was approximately 30 min. Transmission electron microscopy (TEM) images were taken using a Philips CM-20 SuperTwin TEM microscope. The samples were dispersed in methanol, and a droplet of such suspension was put on a microscope copper grid. Next, the samples were dried and purified in a plasma cleaner. Studies were performed in a conventional TEM procedure with 160 kV parallel beam electron energy. The sizes were determined manually using ImageJ software by measuring the longest linear size (Feret diameter) of each particle.
Excitation and emission spectra were measured on a FLS1000 spectrometer from Edinburgh Instruments supplied with a 450 W xenon lamp as an excitation source, and a R928P side window photomultiplier tube from Hamamatsu as a detector. The temperature dependent emission spectra and luminescence decay profile were measured using the same system with a 445 nm laser diode from CNI Lasers used as the excitation source and a THMS 600 heating–cooling stage from Linkam to control the temperature with a precision of 0.1 K. Luminescence spectra and luminescence decay profile as a function of pressure were measured using the same excitation source. Pressure was applied to the sample owing to a gas membrane that transmitted it to the Diacell μScopeDAC-RT(G) diamond anvil cell (DAC) with 0.4 mm diamond culets. The gas used in the system is compressed nitrogen, which was supplied to the membrane using a Druck PACE 5000 which allowed for a relatively controlled change in pressure. Since the emission band of the Cr3+ ions of Al2O3:Cr3+ spectrally overlaps with the emission band of the analyzed SrGdAlO4:Mn4+, the spectral shift of the SrB4O7:Sm2+ 5D0 → 7F0 emission line was used as a pressure indicator in the DAC.30 The sample was placed in a ≈140 μm size hole drilled in a 250 μm thick stainless steel gasket. The methanol–ethanol solution (4:1 ratio) was used as a pressure transmission medium. The Raman spectra were measured in the pressure range from ≈0 to 9 GPa, in a backscattering geometry using a Renishaw InVia confocal micro-Raman system with a power-controlled 100 mW 532 nm laser diode. The laser beam was focused using an Olympus x20 SLMPlan N long working distance objective. The Raman spectra of the sample compressed in a methanol/ethanol/water – 16/3/1 (pressure transmitting medium) were measured in a DAC equipped with ultra-low fluorescence (IIas) diamond anvils.
Results and discussion
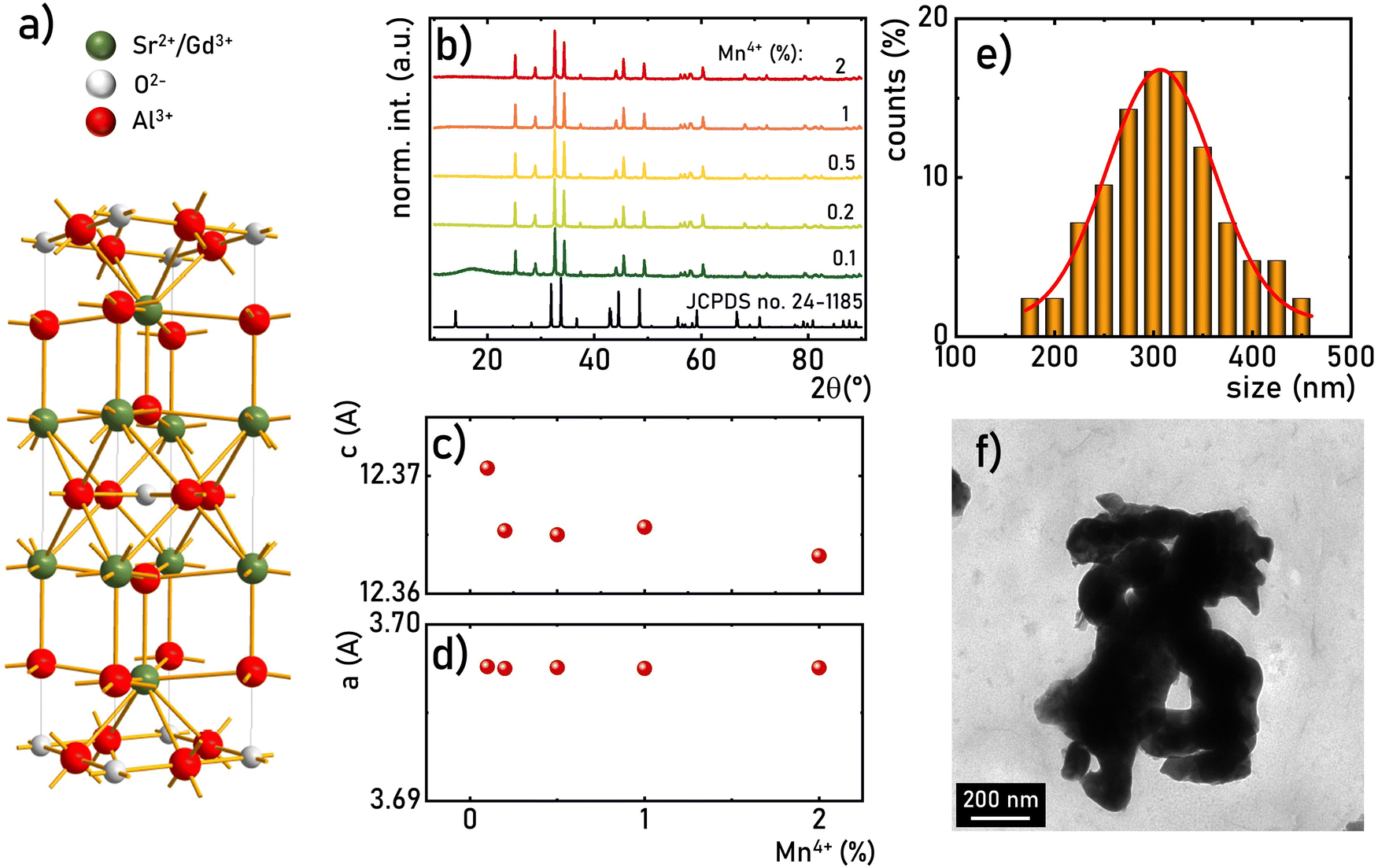
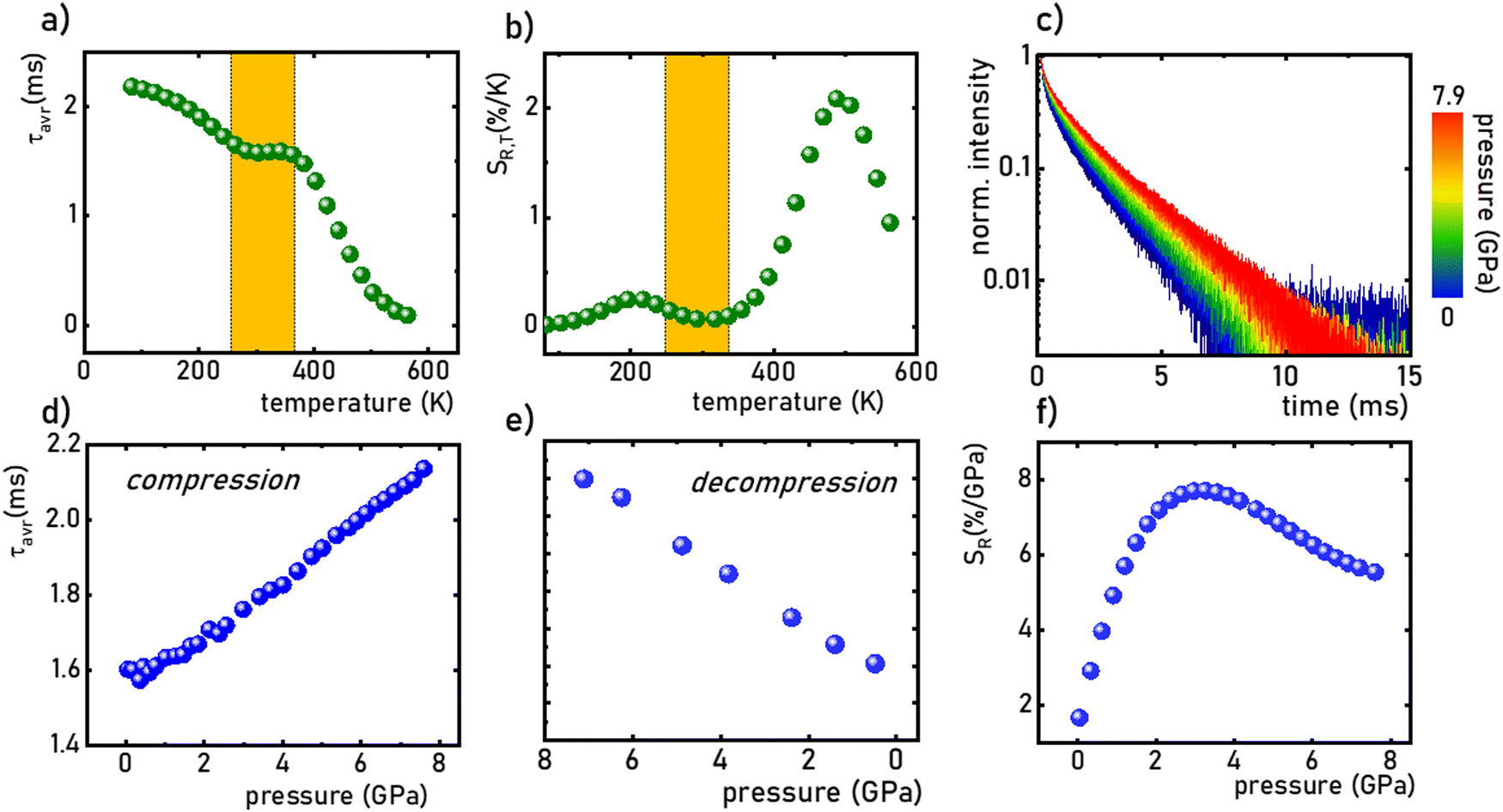
The SrGdAlO4 compound crystallizes in a tetragonal structure with the space group I4/mmm (a = b = 0.3663 nm, c = 1.1998 nm, α = β = γ = 90°).31–37 In this host material, Sr2+ and Gd3+ are nine-fold coordinated, whereas Al3+ ions are surrounded by six oxygen atoms forming (AlO6)9− octahedrons (Fig. 1a). Mn4+ ions, due to the ionic radius (rMn4+ = 0.530 Å) and preferred coordination number (CN = 6), effectively substitute the Al3+ site (rAl3+ = 0.535 Å CN = 6) of the C4v point symmetry in the SrGdAlO4 structure. Hence, no significant changes in the XRD patterns are observed for different concentrations of Mn4+ ions (Fig. 1b). All the diffraction reflexes correlate with the reference pattern for the bulk SrNdAlO4 (JCPDS no. 24-1185) confirming the lack of additional phases in the sample. The Rietveld analysis of the obtained diffractograms reveals that the c parameters slightly decrease with the increase of the Mn4+ concentration (Fig. 1c), while the a parameter remains independent of dopant concentration (Fig. 1d). The slight shrinkage of the c parameter results from the ionic difference between Mn4+ and the Al3+ ions. The morphological studies of the SrGdAlO4:Mn4+ material performed based on the TEM images, indicate that the obtained powder samples consist of well-crystalized particles of around 300 nm in diameter, (Fig. 1e, see also Fig. S1, ESI†) with a high degree of aggregation (Fig. 1f).
 |
| | Fig. 1 Visualization of the crystal structure of the SrGdAlO4 material (a); XRD patterns of the SrGdAlO4:Mn4+ for different concentrations of dopant ions (b); calculated c (c) and a (d) parameters for the SrGdAlO4:Mn4+ material as a function of Mn4+ concentration; particle size distribution (davr = 306 nm, FWHM = 98 nm) (e) and representative TEM image (f) of the SrGdAlO4:0.2%Mn4+. | |
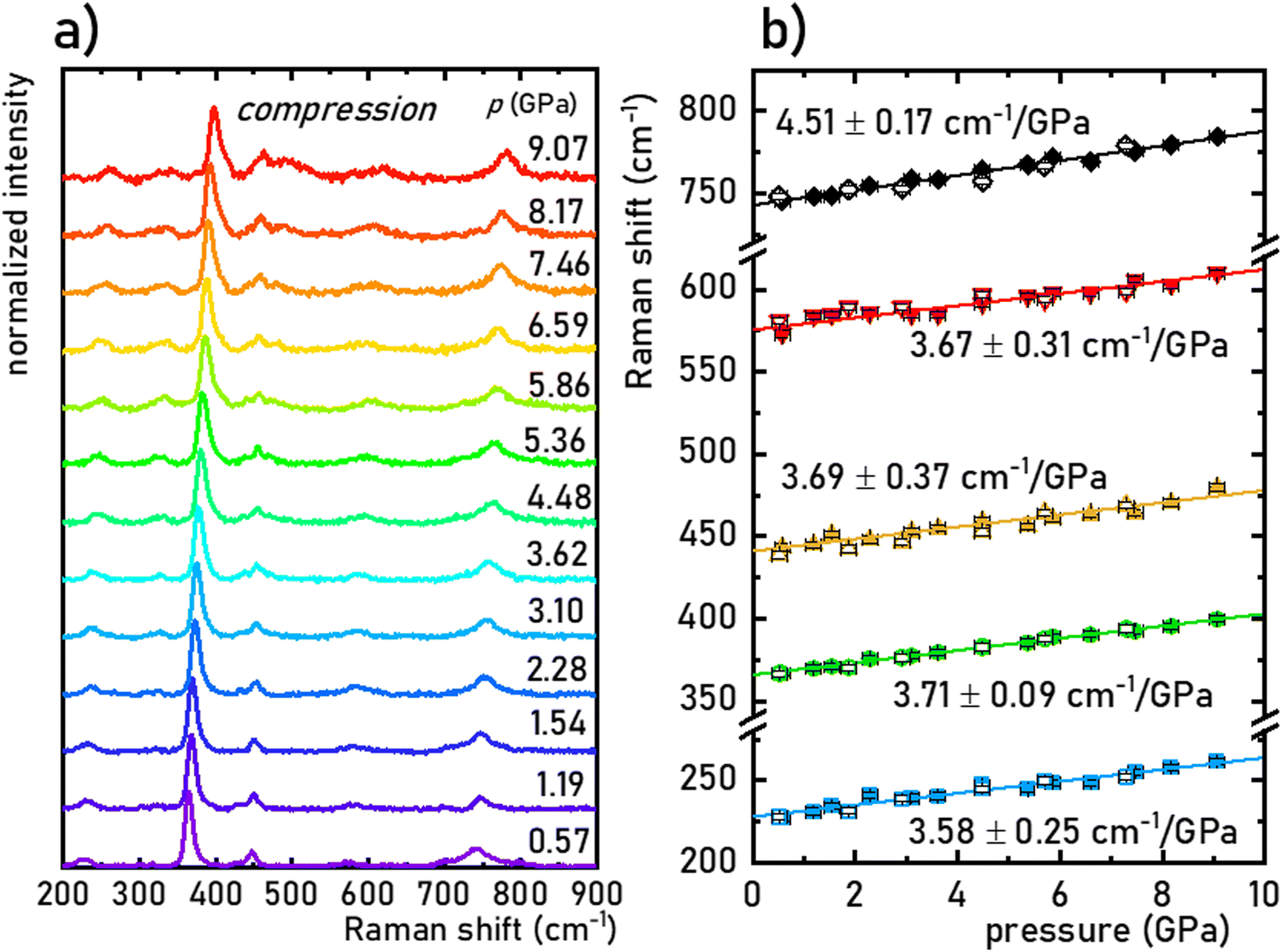
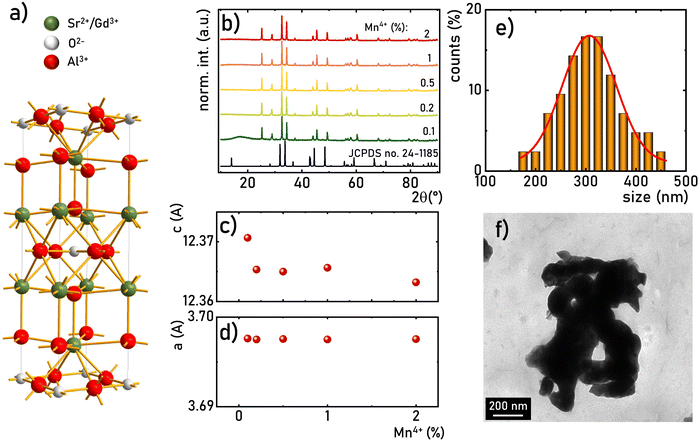
The structural stability of the SrGdAlO4 material was investigated by measuring the Raman spectra for the undoped compound, to avoid luminescence from the activator ions, which often obscure and superimpose with Raman peaks. The measured Raman spectrum for the SrGdAlO4 material at low pressure has five bands initially located around ≈230, 370, 440, 575, and 740 cm−1, as shown in Fig. 2a and b. With increasing pressure (material compression) the energies of the phonon modes increase, and the corresponding Raman bands shift towards higher wavenumbers, as shown in Fig. 2b. The observed effect originates from the bonds shortening, i.e., the decrease of the interatomic distances in the compressed structures. It is worth noting that the determined spectral shifts are monotonous and linear, which assure good structural stability of the investigated materials. The calculated shift rates (cm−1/GPa) for the observed Raman peaks are given in Table S1 in the ESI† data. Note that the observed deterioration of the Raman signal with pressure (lowering signal-to-noise ratio) is associated with increasing strains and crystal defects in the compressed crystal structures, which is typically observed for Raman measurements. Importantly, on releasing the pressure (decompression), the spectral positions and the shapes of all Raman bands return to the initial state (see Fig. 2b and Fig. S2, ESI†). The good convergence of the compression-decompression data indicates the reversibility of the high-pressure experiments. Such behavior is very important for optical pressure monitoring and implying the possibility of utilizing the synthesized SrGdAlO4 material as an optical pressure gauge-manometer.
 |
| | Fig. 2 Normalized Raman spectra for the SrGdAlO4 material measured for different pressure values, during the compression cycle (a); determined energies (peak centroids) of the most intense phonon modes as a function of pressure (b); filled symbols represent compression data and empty ones represent decompression data; the continuous lines are the linear fits applied for determination of the pressure shift rates of the corresponding Raman modes. | |
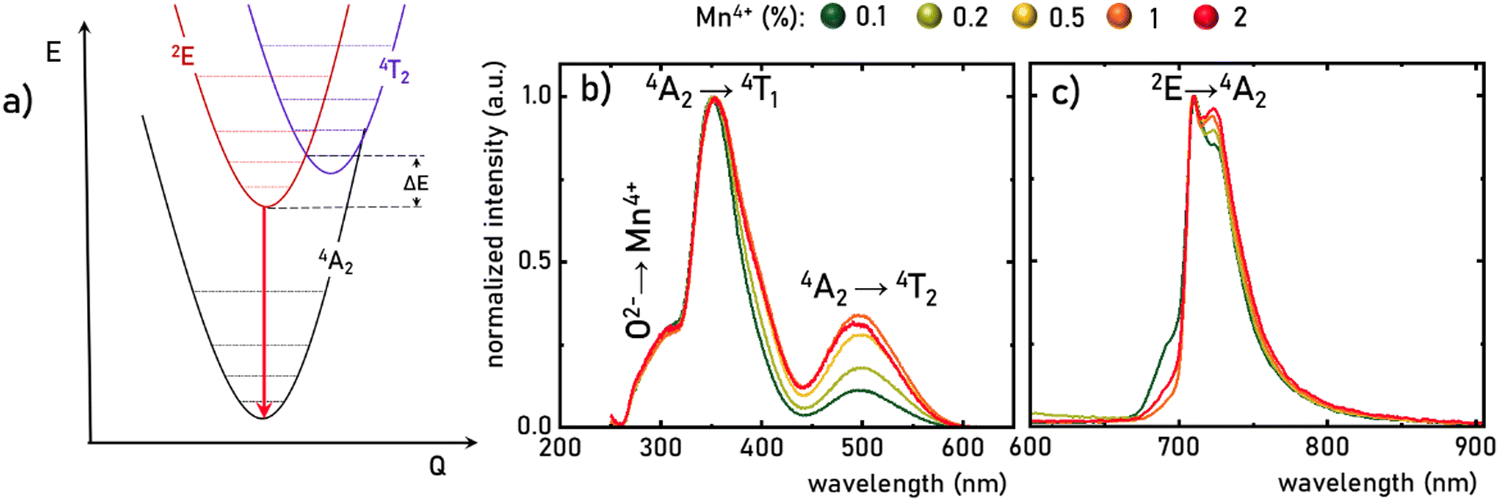
The luminescence properties of materials doped with Mn4+ ions are related to the electron transition from the excited 2E state to the ground 4A2 state (Fig. 3a). The parabola of the 2E level intersects with the 4T2 level at an energy called the activation energy (ΔE) and its value determines the thermal stability of the luminescence of Mn4+ ions, affecting the probability of non-radiative depopulation processes of the 2E level. Since the energy of the 4T2 level strongly depends on the strength of the crystal field interacting with the Mn4+ ions, the activation energy can be modified by appropriate selection of host material. To determine the strength of the crystal field for the SrGdAlO4:Mn4+ structure, a well-known procedure based on the excitation band energies of Mn4+ ions is used (eqn S1–S4, ESI†).28,38 As can be seen in Fig. 3b, the excitation spectra of the SrGdAlO4:Mn4+ have three bands with maxima at around 300, 350, and 500 nm, corresponding to the Mn4+ → O2−, 4A2 → 4T1, and 4A2 → 4T2 electronic transitions, respectively. It is clear that the spectral positions of the excitation bands do not significantly depend on the concentration of Mn4+ ions, and the determined strength of the crystal field is Dq/B = 2.36. However, with the increase in the dopant ion concentration, the intensity of the 4A2 → 4T2 band clearly increases compared to the other bands. The increase in the intensity of this band may explain the apparent change in the half-width of the 4A2 → 4T1 band, with increasing Mn4+ concentration. On the other hand, in the emission spectra, it can be seen that an increase in the concentration of Mn4+ ions causes a decrease in the intensity of the most intense component of the 2E → 4A2 band, located at around 710 nm, in respect to the entire band, which is most likely related to the reabsorption process, the probability of which increases with a shortening of the average distance between adjacent Mn4+ ions (Fig. 3c). Since no significant changes in the spectroscopic properties of SrGdAlO4:Mn4+ were found for different concentrations of Mn4+, the SrGdAlO4:0.2%Mn4+ sample was used as a representative material for further studies.
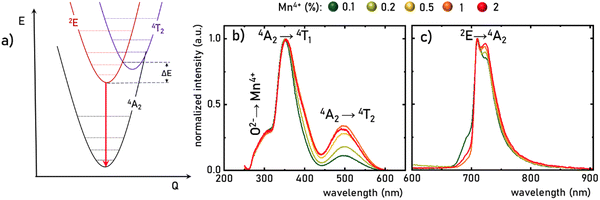 |
| | Fig. 3 Simplified configurational coordination diagram of Mn4+ ions (a), the comparison of the normalized room temperature excitation spectra (λem= 710 nm) (b), and emission spectra (λexc = 400 nm) (c) of SrGdAlO4:Mn4+ with different concentration of Mn4+ ions. | |
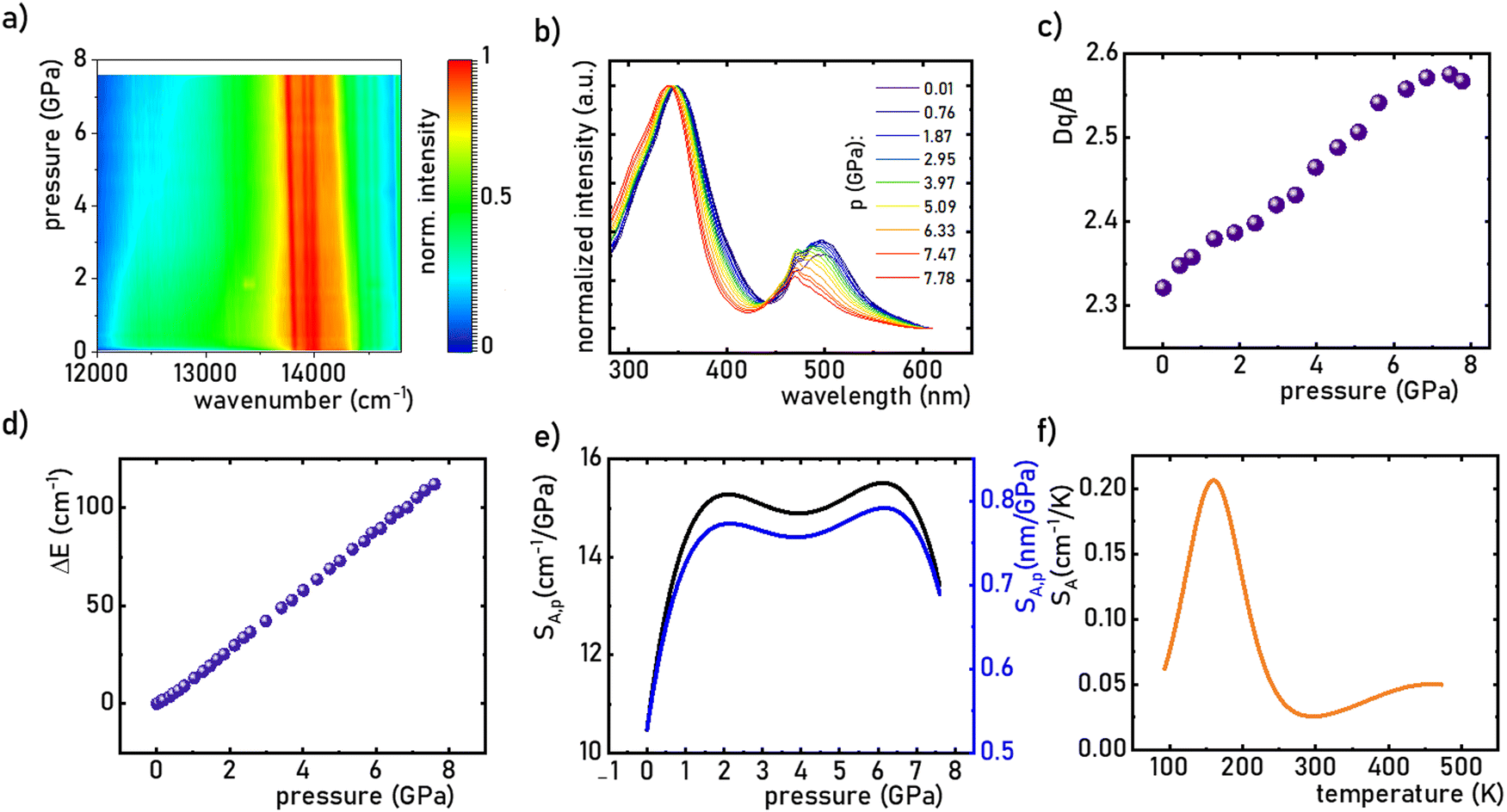
Investigating the effect of applied pressure on the luminescence properties of the SrGdAlO4:0.2%Mn4+ material, the in situ luminescence spectra during the compression-decompression cycle were measured. An increase in pressure from ambient to ≈7.9 GPa leads to the spectral shift of the band maximum from 13976 cm−1 (715.66 nm) to 13861 cm−1 (721.44 nm) (Fig. 4a, see also Fig. S3, ESI†). The change in the position of this band can be discussed in terms of increasing covalency of the Mn4+–O2− bond, leading to a nephelauxetic red-shift,39 which is an effect analogous to that used in the luminescence manometry of shifting the R1 line of the 2E → 4A2 band of Cr3+ ions in ruby.11 The analysis of the excitation spectra of SrGdAlO4:0.2%Mn4+ measured as a function of pressure indicates a blue shift of the Mn4+ absorption bands, which is associated with a change in the crystal field strength (Fig. 4b and Fig. S4, ESI†). Calculation of the Dq/B parameter (where B is the Racah parameter) reveals the gradual increase of its value from 2.36 at ambient pressure up to 2.59 at 7.8 GPa (Fig. 4c). As can be seen, the band energy changes monotonically and varies by approx. 115 cm−1 (6.4 nm) in the analyzed pressure range (Fig. 4d), resulting in the absolute sensitivity determined as follows:
| | 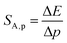 | (1) |
| | 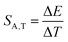 | (2) |
where Δ
E is the change in the band energy corresponding to a change in pressure (temperature) by Δ
p (Δ
T). The resulting
SA,p value is ≈15.5 cm
−1 GPa
−1 at around 6 GPa that corresponds to d
λ/d
p = 0.79 nm GPa
−1 and decreases with pressure (
Fig. 4e). On the other hand, in the case of temperature measurements, the emission intensity decreases and the band position shifts slightly with temperature (Fig. S3a, ESI
†). The maximum observed shift exceeds 25 cm
−1 (Fig. S2b, ESI
†). Making a pressure reading based on the spectral position of the
2E →
4A
2 band of Mn
4+ ions is strongly affected by temperature change. Consequently, SrGdAlO
4:
0.2%Mn
4+ exhibits an absolute temperature sensitivity of about
SA,T = 0.2 cm
−1 K
−1 at 100 K, which decreases with increasing temperature (
Fig. 4f). The corresponding d
λ/d
T = 0.0097 nm K
−1 for SrGdAlO
4:
0.2%Mn
4+ is only slightly higher compared to 0.007 nm K
−1 for ruby.
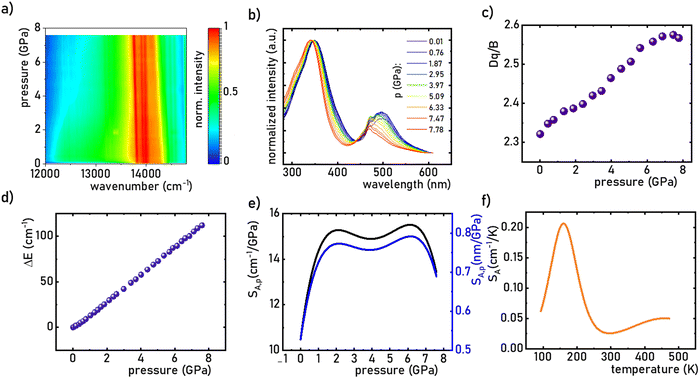 |
| | Fig. 4 The influence of the applied pressure on the normalized room temperature emission spectra of the SrGdAlO4:0.2%Mn4+ (a); the excitation spectra of SrGdAlO4:0.2%Mn4+ measured as a function of pressure (b); the change in the Dq/B as a function of applied pressure (c) determined change in the wavenumber of the emission band – ΔE (cm−1) (d) and corresponding SA,p (e) as a function of pressure; SA,T as a function of temperature f). | |
As already shown, the significant change in the SrGdAlO4:0.2%Mn4+ luminescence intensity with temperature may suggest that the emission lifetime of the 2E-excited level is strongly thermally shortened. In order to experimentally verify this hypothesis, the luminescence decay profiles for the SrGdAlO4:0.2%Mn4+ sample were measured in the range of 77–580 K (Fig. 5a and Fig. S5, ESI†). Due to the slight deviation of their shape from single-exponential profile, the average lifetimes (τavr) were determined according to the procedure described in ESI† (eqn S5 and S6, ESI†). At 77 K, the τavr equaled 2.1 ms and with increasing temperature it gradually shortened until 260 K, where it reached 1.61 ms. In the temperature range of 260–365 K, the τavr value remained almost constant (plateau), and above that threshold temperature a sharp reduction in the values was observed. A similar relationship for Mn4+ lifetimes was reported by Senden et al40 for the K2TiF6:Mn4+ compound. According to the interpretation presented in the literature, this behavior can be explained in terms of a thermal change in the radiative lifetime of the 2E state in the 77–365 K range, followed by the shortening associated with the nonradiative depopulation of the 2E state at higher temperature values. The shortening of the radiative lifetime observed in the initial temperature range is due to the thermal population of odd-parity vibrational modes at elevated temperatures. The quantitative analysis of the obtained results was carried out by determining the relative sensitivity as follows:
| | 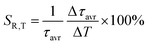 | (3) |
| | 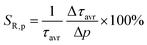 | (4) |
where Δ
τavr represents the change in
τavr corresponding to a change in temperature by Δ
T (pressure by Δ
p). The obtained results indicate that the maximum sensitivity of
SR,T = 2.1% K
−1 is observed at 490 K (
Fig. 5b). However, in the 260–365 K
T-range,
SR,T < 0.09% K
−1. This is direct evidence that the luminescence properties of the SrGdAlO
4:
0.2%Mn
4+ compound in this T-range are temperature-independent. As the pressure increases, a significant elongation of the luminescence decay profiles was observed (
Fig. 5c). In addition, it was noted that above 1.5 GPa the lifetime begins to approach a single-exponential profile with increasing pressure, and
τavr monotonically increases with pressure from 1.44 ms up to 2.14 ms at 7.6 GPa (
Fig. 5d and e). Importantly, its value shortens again during the decompression cycle, confirming that the observed changes are fully reversible, as shown previously in the Raman spectra. The observed effect can be explained by (i) a change in the probability of radiative processes resulting from a change in the covalency of the bond and depopulation of the odd-parity vibrational modes and (ii) a decrease in the probability of non-radiative processes through an increase in the activation energy resulting from an increase in the energy of the
4T
2 level. The analysis of the relative sensitivity of the lifetime-based manometer indicates that in the 1–7.6 GPa pressure range
SR,P > 5% GPa
−1, and the maximum value of
SR = 7.85% GPa
−1 is reached at 3 GPa (
Fig. 5f). This is a very high value comparable to the most sensitive luminescence manometers based on the spectral analysis of materials doped with Ln
3+ ions.
41,42 However, it should be borne in mind that luminescence kinetics is hardly affected by the dispersive dependence of light absorption and scattering by the medium, and therefore allows for a more reliable measurement as proven for luminescence thermometry.
43 In the case of luminescence manometers, it is essential to provide a temperature-invariant pressure readout. For this purpose, the temperature invariability manometric factor (TIMF) defined as follows was determined for the SrGdAlO
4:
0.2%Mn
4+ sensor material:
44| | 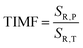 | (5) |
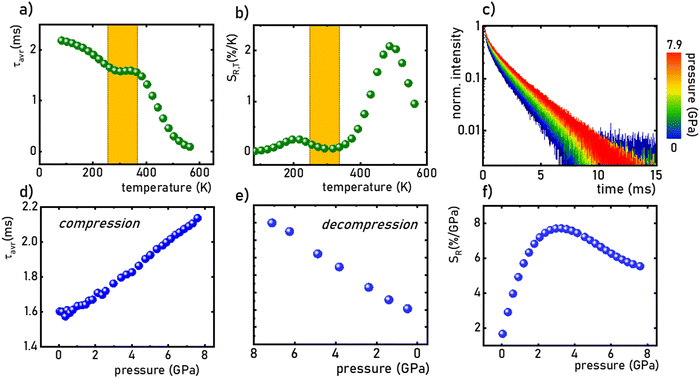 |
| | Fig. 5 The τavr of the SrGdAlO4:0.2%Mn4+ measured as a function of temperature (a) the corresponding SR,T (b); luminescence decay profile of SrGdAlO4:0.2%Mn4+ measured as a function of pressure (c); the τavr measured at room temperature as a function of pressure during compression (d) and decompression (e) and corresponding SR,p (f). | |
TIMF expresses the temperature change necessary to harness the change in spectroscopic properties corresponding to a 1 GPa change in pressure. The larger the TIMF value, the more temperature-independent the pressure reading. In the case of the SrGdAlO4:0.2%Mn4+ compound compressed to 2.1 GPa at room temperature, the TIMF is 134, which is the final confirmation of the high application potential of this material. Additional advantage of this manometer is the fact that τavr shows very high repeatability within the compression and decompression cycles (Fig. S6, ESI†). The calculation of the pressure determination uncertainty (eqn S7, ESI†) for lifetime-based luminescence manometry on SrGdAlO4:0.2%Mn4+ was as low as 0.07 GPa at 3 GPa, which confirms a high manometric performance of this luminescence manometer.
Conclusions
In this study, the spectroscopic properties of the SrGdAlO4:0.2%Mn4+ material were studied as a function of pressure and temperature to develop a lifetime-based luminescent manometer. It was shown that the 2E → 4A2 emission spectral band shifts with applied pressure, with the maximum absolute sensitivity SA,p ≈ 15.5 cm−1 GPa−1 at around 6 GPa (dλ/dp = 0.79 nm GPa−1) and temperature with SA,T = 0.2 cm−1 K−1 at 200 K. The luminescence intensity of the Mn4+ emission is rapidly quenched with temperature, indicating that the activation energy, and thus the energy of the 4T2 band is probably relatively low. Measurements of lifetimes as a function of temperature reveal that in the range of 77–360 K the change in lifetime values result from a change in the probability of radiative processes. Whereas, the rapid shortening of the τavr values observed above 360 K originates from an increase in the probability of non-radiative processes. Importantly, in the range of 260–360 K the τavr remains insensitive to temperature changes. The pressure-dependence analysis indicates that the τavr values significantly and monotonically prolong with pressure, at least up to ≈7.9 GPa, and the maximum sensitivity reaches SR,p = 7.85% GPa−1 at 3 GPa with the pressure determination uncertainty as low as 0.07 GPa at 3 GPa. Importantly, the observed changes in lifetime values are fully reversible. The high value of the TIMF parameter (≈134) confirms the high application potential of the SrGdAlO4:0.2%Mn4+ material for remote pressure readings based on the lifetime of the 2E excited state. We believe this study can trigger research interest in the use of Mn4+ doped phosphors to develop a new class of highly reliable luminescent manometers.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Science Center (NCN) Poland under project no. DEC-UMO-2020/37/B/ST5/00164. M. R. acknowledges support from Fondo Social Europeo and Agencia Estatal de Investigación (RYC2020-028778-I/AEI/10.13039/501100011033).
References
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- O. S. Wolfbeis, Anal. Chem., 2006, 78, 3859–3874 CrossRef CAS PubMed.
- M. D. Dramićanin, J. Appl. Phys., 2020, 128, 40902 CrossRef.
- L. Marciniak, K. Kniec, K. Elżbieciak-Piecka, K. Trejgis, J. Stefanska and M. Dramićanin, Coord. Chem. Rev., 2022, 469, 214671 CrossRef CAS.
- X. D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- B. R. Jovanić and J. P. Andreeta, Phys. Scr., 1999, 59, 274–276 CrossRef.
- T. Zheng, L. Luo, P. Du, S. Lis, U. R. Rodríguez-Mendoza, V. Lavín, I. R. Martín and M. Runowski, Chem. Eng. J., 2022, 443, 136414 CrossRef CAS.
- M. Szymczak, P. Woźny, M. Runowski, M. Pieprz, V. Lavín and L. Marciniak, Chem. Eng. J., 2023, 453, 139632 CrossRef CAS.
- R. R. Petit, S. E. Michels, A. Feng and P. F. Smet, Light: Sci. Appl., 2019, 8, 124 CrossRef CAS PubMed.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673 CrossRef CAS.
- M. Tian, Y. Gao, P. Zhou, K. Chi, Y. Zhang and B. Liu, Phys. Chem. Chem. Phys., 2021, 23, 20567–20573 RSC.
- Y. Masubuchi, S. Nishitani, S. Miyazaki, H. Hua, J. Ueda, M. Higuchi and S. Tanabe, Appl. Phys. Express, 2020, 13, 42009 CrossRef CAS.
- J. Liu and Y. K. Vohra, Appl. Phys. Lett., 1994, 64, 3386–3388 CrossRef CAS.
- Y. R. Shen, T. Gregorian and W. B. Holzapfel, High Pressure Res., 1991, 7, 73–75 CrossRef.
- B. Lorenz, Y. R. Shen and W. B. Holzapfel, High Pressure Res., 1994, 12, 91–99 CrossRef.
- F. H. Su, Z. L. Fang, B. S. Ma, K. Ding, G. H. Li and W. Chen, J. Phys. Chem. B, 2003, 107, 6991–6996 CrossRef CAS.
- N. J. Hess and G. J. Exarhos, High Pressure Res., 1989, 2, 57–64 CrossRef.
- J. Barzowska, T. Lesniewski, S. Mahlik, H. J. Seo and M. Grinberg, Opt. Mater., 2018, 84, 99–102 CrossRef CAS.
- U. R. Rodríguez-Mendoza, S. F. León-Luis, J. E. Muñoz-Santiuste, D. Jaque and V. Lavín, J. Appl. Phys., 2013, 113, 213517 CrossRef.
- L. M. M. Szymczak, M. Runowski and V. Lavín, Laser Photonics Rev., 2023, 17, 2200801 CrossRef.
- M. Pieprz, M. Runowski, K. Ledwa, J. J. Carvajal, A. Bednarkiewicz and L. Marciniak, ACS Appl. Opt. Mater., 2023, 1, 1080–1087 CrossRef CAS.
- L. Labrador-Páez, M. Pedroni, A. Speghini, J. García-Solé, P. Haro-González and D. Jaque, Nanoscale, 2018, 10, 22319–22328 RSC.
- A. Bednarkiewicz, L. Marciniak, L. D. Carlos and D. Jaque, Nanoscale, 2020, 12, 14405–14421 RSC.
- M. Runowski, J. Marciniak, T. Grzyb, D. Przybylska, A. Shyichuk, B. Barszcz, A. Katrusiak and S. Lis, Nanoscale, 2017, 9, 16030–16037 RSC.
- Q. Zhou, L. Dolgov, A. M. Srivastava, L. Zhou, Z. Wang, J. Shi, M. D. Dramićanin, M. G. Brik and M. Wu, J. Mater. Chem. C, 2018, 6, 2652–2671 RSC.
- M. G. Brik and A. M. Srivastava, J. Lumin., 2013, 133, 69–72 CrossRef CAS.
- M. G. Brik, S. J. Camardello and A. M. Srivastava, ECS J. Solid State Sci. Technol., 2015, 4, R39 CrossRef CAS.
-
M. Pechini, US Pat., US3330697A, 1967.
- T. Zheng, M. Sójka, P. Woźny, I. R. Martín, V. Lavín, E. Zych, S. Lis, P. Du, L. Luo and M. Runowski, Adv. Opt. Mater., 2022, 10, 2201055 CrossRef CAS.
- W. Xie, J. Feng, X. Liu, L. Yan, H. Guo, Y. Dai, Y. Xie, H. S. Jang and J. Lin, J. Alloys Compd., 2018, 753, 781–790 CrossRef CAS.
- A. Hooda, S. P. Khatkar, A. Khatkar, S. Chahar, S. Devi, J. Dalal and V. B. Taxak, Curr. Appl. Phys., 2019, 19, 621–628 CrossRef.
- P. Sehrawat, A. Khatkar, A. Hooda, M. Kumar, R. Kumar, R. K. Malik, S. P. Khatkar and V. B. Taxak, Ceram. Int., 2019, 45, 24104–24114 CrossRef.
- A. Hooda, S. P. Khatkar, A. Khatkar, R. K. Malik, J. Dalal, S. Devi and V. B. Taxak, Mater. Chem. Phys., 2019, 232, 39–48 CrossRef CAS.
- F. Zou, M. Li, Z. Du, Y. Song, L. Li and G. Li, Opt. Mater., 2022, 133, 112904 CrossRef CAS.
- A. Hooda, S. P. Khatkar, A. Khatkar, M. Kumar, M. Dalal and V. B. Taxak, J. Mater. Sci.: Mater. Electron., 2019, 30, 1297–1309 CrossRef CAS.
- Q. Sun, S. Wang, B. Devakumar, B. Li, L. Sun, J. Liang, D. Chen and X. Huang, RSC Adv., 2018, 8, 39307–39313 RSC.
- M. G. Brik, S. J. Camardello, A. M. Srivastava, N. M. Avram and A. Suchocki, ECS J. Solid State Sci. Technol., 2016, 5, R3067 CrossRef CAS.
- T. Zheng, L. Luo, P. Du, S. Lis, U. R. Rodríguez-Mendoza, V. Lavín and M. Runowski, Chem. Eng. J., 2022, 446, 136839 CrossRef CAS.
- T. Senden, R. J. A. Van DIjk-Moes and A. Meijerink, Light: Sci. Appl., 2018, 7, 8 CrossRef PubMed.
- M. Sójka, M. Runowski, P. Woźny, L. D. Carlos, E. Zych and S. Lis, J. Mater. Chem. C, 2021, 9, 13818–13831 RSC.
- M. A. Antoniak, S. J. Zelewski, R. Oliva, A. Żak, R. Kudrawiec and M. Nyk, ACS Appl. Nano Mater., 2020, 3, 4209–4217 CrossRef CAS.
- M. Runowski, S. Goderski, D. Przybylska, T. Grzyb, S. Lis and I. R. Martín, ACS Appl. Nano Mater., 2020, 3, 6406–6415 CrossRef CAS.
- M. Szymczak, M. Runowski, M. G. Brik and L. Marciniak, Chem. Eng. J., 2023, 143130 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |

 bc,
P.
Woźny
bc,
P.
Woźny