
Expanding the horizons of covalent organic frameworks: sub-stoichiometric synthesis as an emerging toolkit for functional COFs
Samiyappan
Vijayakumar
 ab,
Ayyappanpillai
Ajayaghosh
*ab and
Sreejith
Shankar
*ab
ab,
Ayyappanpillai
Ajayaghosh
*ab and
Sreejith
Shankar
*ab
aChemical Sciences and Technology Division, CSIR-NIIST, Thiruvananthapuram, 695 019, India. E-mail: ajayaghosh@niist.res.in; sreejith.shankar@niist.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad, 201002, India
First published on 15th November 2023
Abstract
Covalent organic frameworks (COFs) are a class of porous materials generated from organic molecules resulting in two-dimensional (2D) or three-dimensional (3D) networks. Multifunctional molecules with defined symmetry react to give all-linked-COFs with pre-determined topologies. COFs and COF-based materials have continuously evolved via the exploration of new linkages, synthesis, and processing methodologies over nearly two decades. Several studies have contributed to the understanding of the formation mechanism and the influence of solvent(s), catalysts, and the reaction environment in the crystallization process. Apart from all-linked COFs with a “fully formed” network, sub-stoichiometric synthesis has been a recent development, where the change in stoichiometry of monomers results in generating unconventional topologies along with active functional groups within the pores of these materials. Thus, the generation and utilization of these “functional” COFs is an interesting challenge for reticular chemists, particularly in terms of the vast possibilities of applications that are not likely to emerge from conventional synthesis.
1. Introduction
The fundamental mechanistic understanding behind chemical transformations has evolved through a large realm of reagents, catalysis, and related synthetic strategies. The functionalization prospects of molecular materials have demonstrated the power of new synthetic methodologies in developing several intriguing chemical structures.1,2 Though the possibilities for selectively functionalizing a particular position in the presence of many similar ones offer a toolbox for diversifying the properties and utility of a molecule, developing such a reliable methodology in terms of reagents, catalysts, and reaction conditions has been a challenging task.3,4 The subtle differences in the intrinsic reactivity of similar functional groups in a molecule, under carefully controlled reaction conditions, could lead to site-selective and target-oriented syntheses, with a high degree of complementarity to conventional top-down and bottom-up approaches.5,6 Site-selective approaches are indeed utilized by nature and envisage a better redox economy with improved synthetic modalities in natural product chemistry, drug discovery, and materials science.7 Such strategies in biochemistry are aimed at understanding the mechanism behind biochemical processes, structure–activity relationship, and improved functions. For instance, non-covalent protection strategies using nucleic acid aptamers have been demonstrated for the selective functionalization of molecules.5With all the developments achieved in chemo- and stereo-selective syntheses over the past century, differentiating the reactivity among two or more reactive positions with identical or similar functional groups having negligible differences in their activation energy has been challenging.4,8–10 Such a synthetic methodology is yet to be demonstrated to provide a general solution for the functional modification of complex molecular architectures in high yields and selectivity. While the use of protecting groups has been a preferred strategy for the selective reaction of similar functional groups in a molecule, the need for protection–deprotection steps hampers the efficiency of chemical reactions.11–14 Therefore, modern synthetic chemistry advocates precise and selective recognition of minuscule differences in electronic and steric factors within potential reactive sites at the molecular level as the best approach towards achieving site-selective synthesis of complex chemical structures.4,9,10 Identification of reaction conditions, including reagents, solvents, and catalysts, that complement the electronic, steric, and stereo-electronic attributes of the target functional group among several similar functional groups is critical for achieving archetypical site-selective transformations in synthetic chemistry.8
Achieving site selectivity among similar functional groups in polymerisation reactions is not fully explored with regard to the synthesis and utilization of functional polymeric systems. Reticular chemistry that utilizes the austerity and rigor of both organic and inorganic synthetic strategies has led to the development of a large number of extended porous crystalline solids, such as metal–organic frameworks (MOFs)15–19 and covalent organic frameworks (COFs).20–25 The dynamic but directional bond formation with microscopic reversibility in most of these materials has resulted in several intriguing properties emanating from highly functional complexities.26–29 The structural matrices optimized through iso-reticular principles combined with the possibilities for post-synthetic modifications make these framework materials even more interesting.30–33 The synthesis of complex structures of covalent organic frameworks (COFs) from organic molecular building units linked through covalent bonds, in this context, is a formidable task to achieve via functional group selective transformations. Nevertheless, unlike organic molecules, topology-driven selectivity towards chemically equivalent functional groups in reticular synthesis can be achieved by controlling the stoichiometry of the reactants.34 While the dynamic covalent linkage stitches the atoms to the network backbone, unreacted functional groups decorate the pores, resulting in unprecedented properties and scope for post-synthetic modifications.34–37 Therefore, in COFs, where organic building units are linked through covalent bonds, the choice of monomeric units allows for creating the desired network with unreacted functional groups, in line with site-selective synthesis in organic chemistry.
2. Selective transformations in COF synthesis
Reticular synthesis of COFs has shown wide applicability and has witnessed tremendous growth over the past couple of decades.22,24,38 Connecting multifunctional molecules via preferentially dynamic covalent linkages to yield frameworks of various topologies has been accompanied by challenges related to crystallinity and structural elucidation.39 Nevertheless, the synergistic efforts from synthetic chemists, theoreticians, and materials scientists have brought systematic advancements in this field.40–43 Though we do not have a complete understanding of the formation of these networks, introduction of new synthetic approaches, crystallization methodologies, linkage chemistry, novel functional units, and processing strategies make this area an expanding and exciting field of research.32,44–48Reaction of multifunctional monomers to give a fully formed network of COFs has been an ideal proposal for a long time.41 The synthetic journey of COFs started with typically reversible linkages and later expanded to irreversible linkages and one-pot multicomponent reactions.49–52 The field has also witnessed the utilization of unreacted sites, the introduction of non-interfering functional groups, and post-synthetic modifications that functionalize the pores or transform the linkages.30,33,53 The functional versatility of such framework materials has led to various applications, including gas adsorption/separation,54–57 host–guest chemistry,58–61 sensing,62–68 ion capture/transport,69 catalysis,70–76 and electrochemical applications.77
Focussed research activities on COFs that started with the first report via the formation of dynamic boroxine and boronate ester bonds have now been extended to seeded growth of 2D COFs, graphene nanoribbon 2D COFs, and recently to water harvesting frameworks.78,79 One of the most investigated classes of COFs is imine-linked, which involves the reaction between aldehydes and amines.80–83 Several synthetic approaches,44,84–86 processing methodologies,47,87,88 studies in terms of imparting structural stability30,89–91 and mechanistic understanding of the formation under different conditions81,85,92–97 have emerged over time. Post-synthetic introduction of primary amine groups or the presence of unreacted amino or carbonyl groups in imine-linked COFs have been shown to result in functional properties that are otherwise difficult to attain in fully formed networks.98–101 In this context, several synthetic strategies, including functional group interconversions,102–105 transamination reactions,106,107 and site-selective108–112 and regioselective synthesis,113 have been reported to introduce primary amine groups in 2D COFs.
However, approaches like post-synthetic modification, transamination reactions, and site-selective and regioselective approaches are not devoid of limitations like complexity in the design of monomers, multiple steps, and challenges related to purification. These multistep synthetic approaches are still being utilized for incorporating active amine groups within imine-linked COFs.114,115 While site-selective strategy is limited to selected orthogonally reactive monomers, the sub-stoichiometric approach allows for the facile synthesis of designed network materials solely based on their symmetry. The bottleneck in this approach is to identify the right solvent combination that facilitates the formation of crystalline networks with frustrated functional groups, which, however, is a challenge in conventional COF synthesis as well. Thus, the sub-stoichiometric approach is advantageous as compared to the existing methodologies for constructing functional COFs with active amine or aldehyde sites within imine-linked COFs. While the sub-stoichiometric synthesis of COFs is still in its infancy, several significant contributions demonstrating applications in various fields have emerged over the last five years. This article attempts to comprehensively review the current developments and provide future perspectives on the sub-stoichiometric synthesis of COFs and their applications. We believe that this article would support detailed investigations on sub-stoichiometric synthesis of 2D and 3D COFs, in order to advance further developments in this exciting area of research.
3. Sub-stoichiometric synthesis of 2D and 3D COFs
The last few years have witnessed the emergence of sub-stoichiometric synthesis capable of yielding either free amine or free aldehyde groups in imine-linked COFs (Fig. 1). This approach has not only resulted in unconventional topologies but also revived our understanding of mechanisms behind COF formation.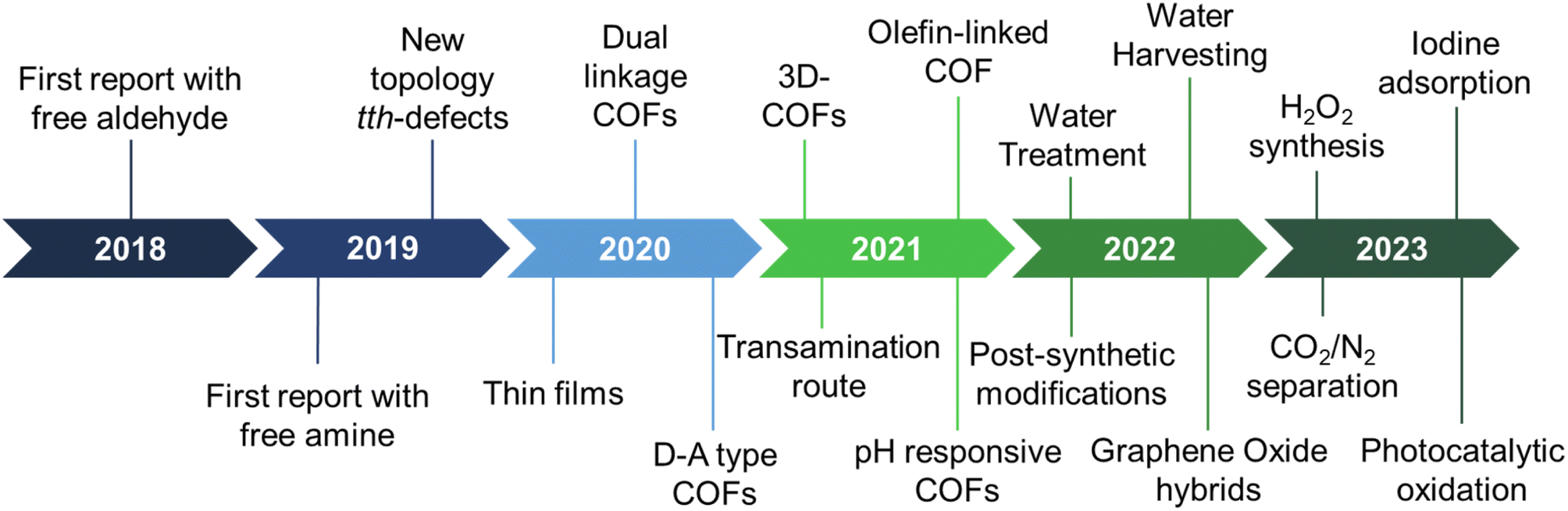 | ||
| Fig. 1 Evolution of sub-stoichiometric synthesis of COFs and related reports. | ||
Though strategies like different solvent combinations and variations in stoichiometry of reactants were used, stoichiometry control gained significance with time, thereby giving rise to the name sub-stoichiometric synthesis.34,116 With the first report in 2018,116 the sub-stoichiometric approach has so far yielded several 2D and 3D COFs via direct condensation of the monomeric units. Combined with functionalization prospects such as post-synthetic modification of frustrated groups, this approach has paved the way to utilize these networks in water harvesting, gas adsorption and separation, and extensively in photocatalysis. We anticipate this class of COFs, and the underlying synthetic challenges provide enough opportunities for reticular chemists to explore and comprehend. A brief timeline of the evolution of sub-stoichiometric approach in COF synthesis is given in Fig. 1.
While incomplete reactions may lead to unreacted functional groups, such COFs would possess low ordering and poor crystallinity, low stability, and non-uniform pore distributions within the network. Majority of the sub-stoichiometric (Type III) COFs are shown to be ordered, highly crystalline, stable, and possessing the expected porosity. The crystalline nature and ordering in sub-stoichiometric COFs are generally evident from XRD and porosity measurements. Theoretical calculation of the unit cell parameters further confirms the resulting structures. Moreover, the sub-stoichiometric approach generally yields unconventional topologies that are, in principle, not expected for all-linked COFs.34 And the sub-stoichiometric structures obtained from tetratopic linkers are always presented with all-linked COFs. These observations are in favour of the “sub-stoichiometric” composition of monomers in Type III COFs as compared to incomplete reactions in all-linked COFs.
3.1 Evolution of new topologies and mixed linkages
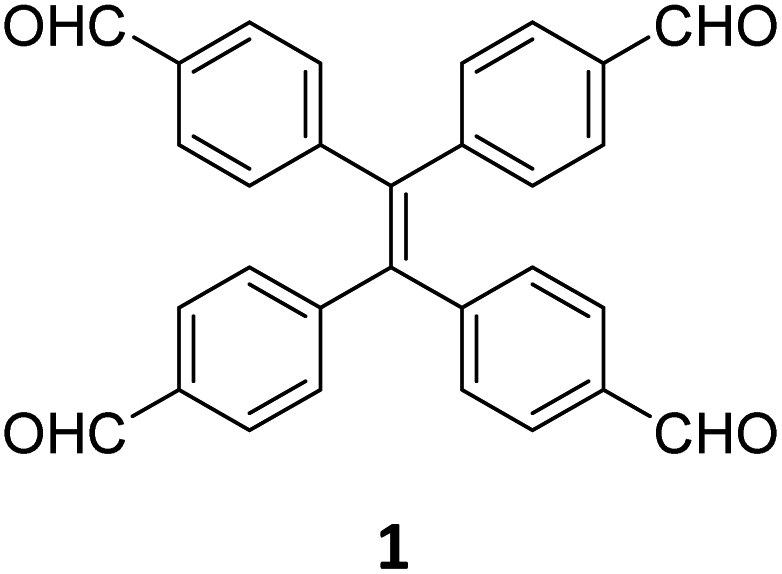
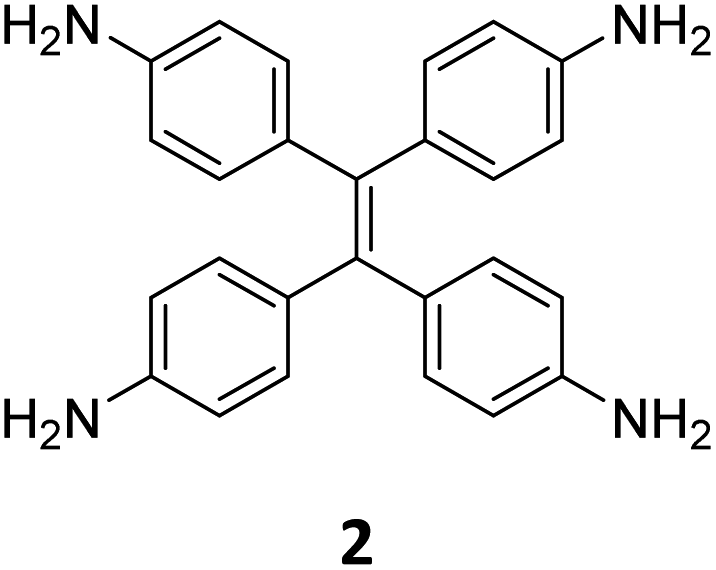
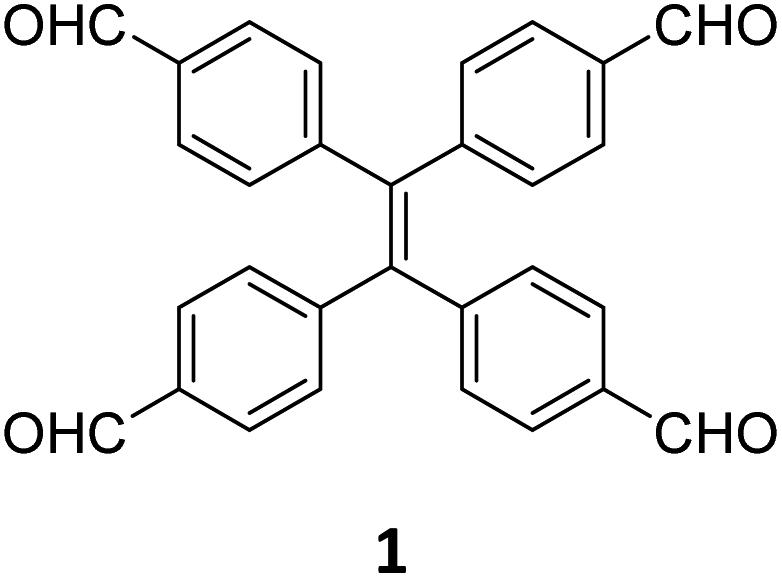
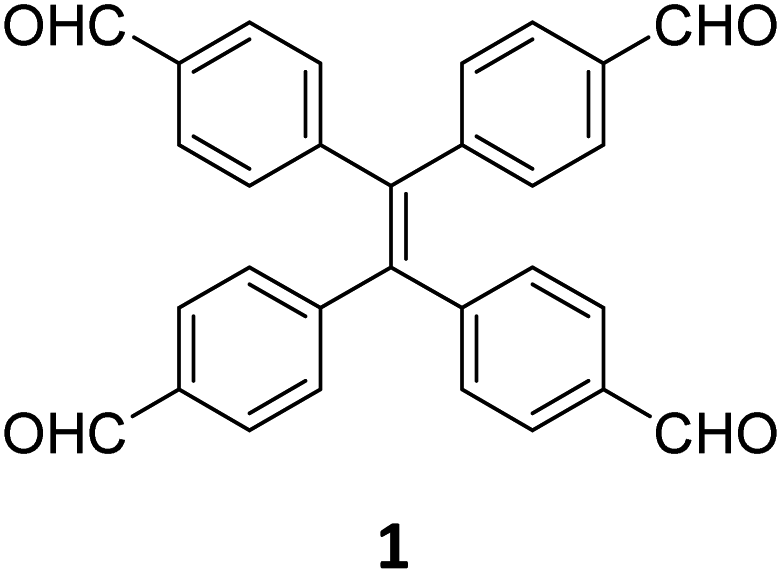
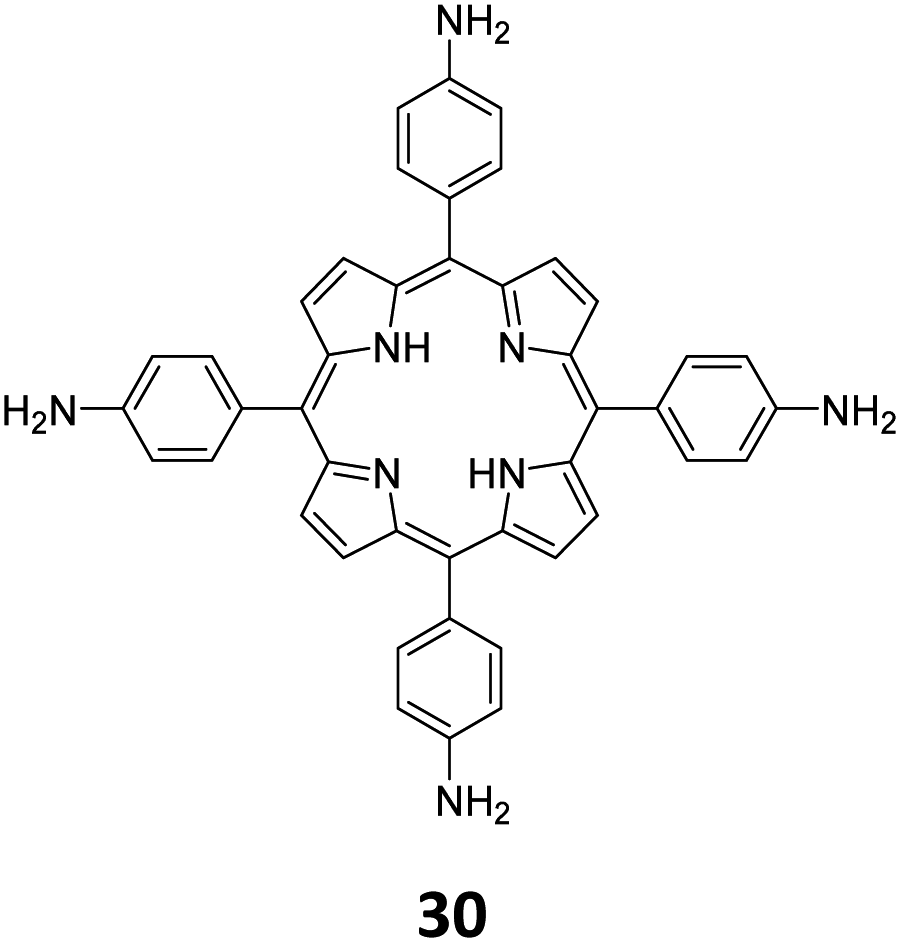
While sub-stoichiometric approach provides a powerful tool to introduce either free aldehyde or amine groups in imine-linked COFs, the first three reports described the emergence of new topologies in 2D COFs.34,116,117 Obtaining new topologies via sub-stoichiometric synthesis has aroused significant interest in terms of structure and applications of the resulting COFs, and subsequently, several groups have reported such COFs with multifarious applications.The first report on sub-stoichiometric synthesis of COFs appeared in 2018 when Loh and co-workers utilized structurally similar tetratopic aldehyde 1 and tetraphenylethylene (TPE) amine 2 to give fully condensed TPE-COF-I and partially condensed TPE-COF-II with frustrated carbonyls.116 Variations in solvent and stoichiometry of the precursors 1 and 2 were found to result in two topologies. A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the aldehyde 1 and amine 2 in o-dichlorobenzene/n-butanol mixture yielded TPE-COF-Ivia a [4 + 4] condensation pathway, and a 2:1 ratio in 1,4-dioxane, via an unusual [2 + 4] pathway, yielded TPE-COF-II (Fig. 2). Since the influence of solvent(s), steric factors and nucleation mechanism was critical to the formation of the COFs, the approach was termed solvent-directed divergent synthesis. The unreacted linker units within the pores of TPE-COF-II were shown to have a significant impact on its CO2 uptake capacity and a typically high value of 23.2 wt% (118.8 cm3 g−1) adsorption was observed at 1 atm and 273 K.
:1 ratio of the aldehyde 1 and amine 2 in o-dichlorobenzene/n-butanol mixture yielded TPE-COF-Ivia a [4 + 4] condensation pathway, and a 2:1 ratio in 1,4-dioxane, via an unusual [2 + 4] pathway, yielded TPE-COF-II (Fig. 2). Since the influence of solvent(s), steric factors and nucleation mechanism was critical to the formation of the COFs, the approach was termed solvent-directed divergent synthesis. The unreacted linker units within the pores of TPE-COF-II were shown to have a significant impact on its CO2 uptake capacity and a typically high value of 23.2 wt% (118.8 cm3 g−1) adsorption was observed at 1 atm and 273 K.
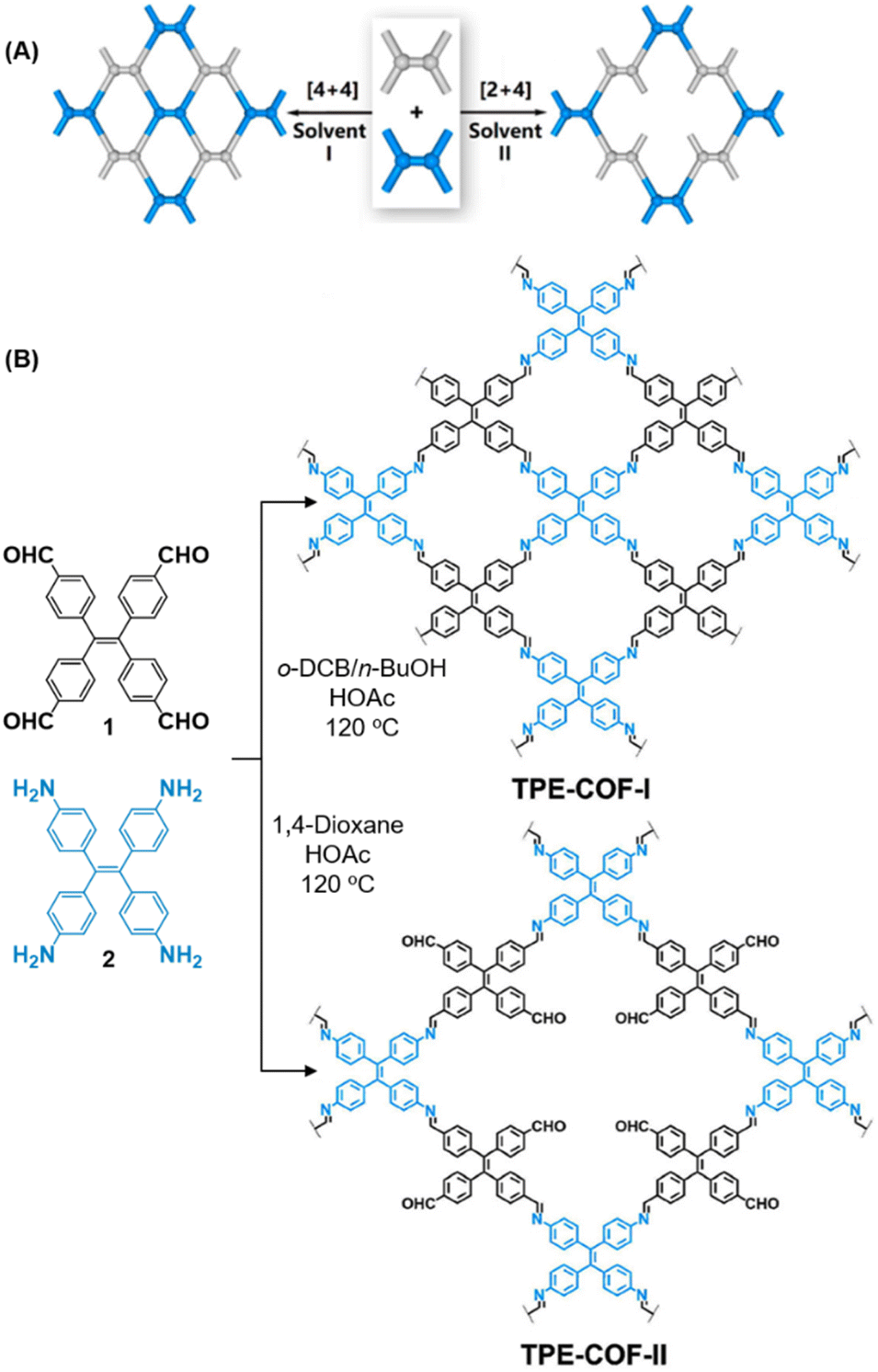 | ||
| Fig. 2 (A) Solvent-directed strategies used by Loh and co-workers for the construction of 2D-COFs with different topologies: fully formed network (TPE-COF-I, left) and partially formed frustrated-bonding structure (TPE-COF-II, right). (B) The corresponding synthetic scheme and the structures of the monomers (1, 2) and the COFs. Reproduced from ref. 116 with permission from American Chemical Society, copyright 2018. | ||
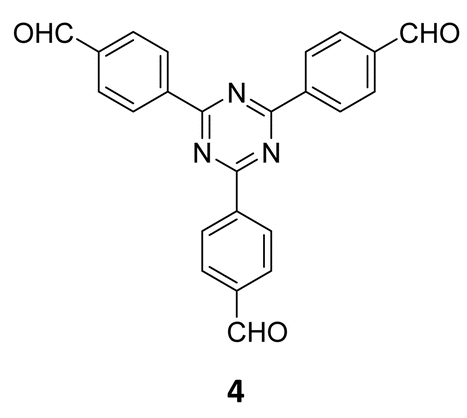
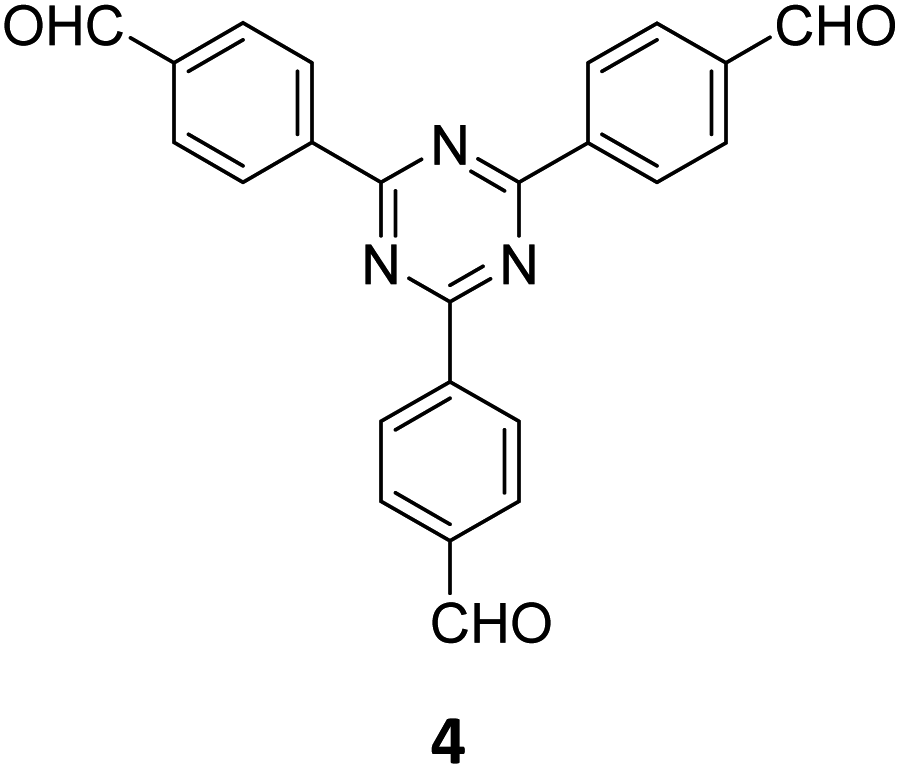
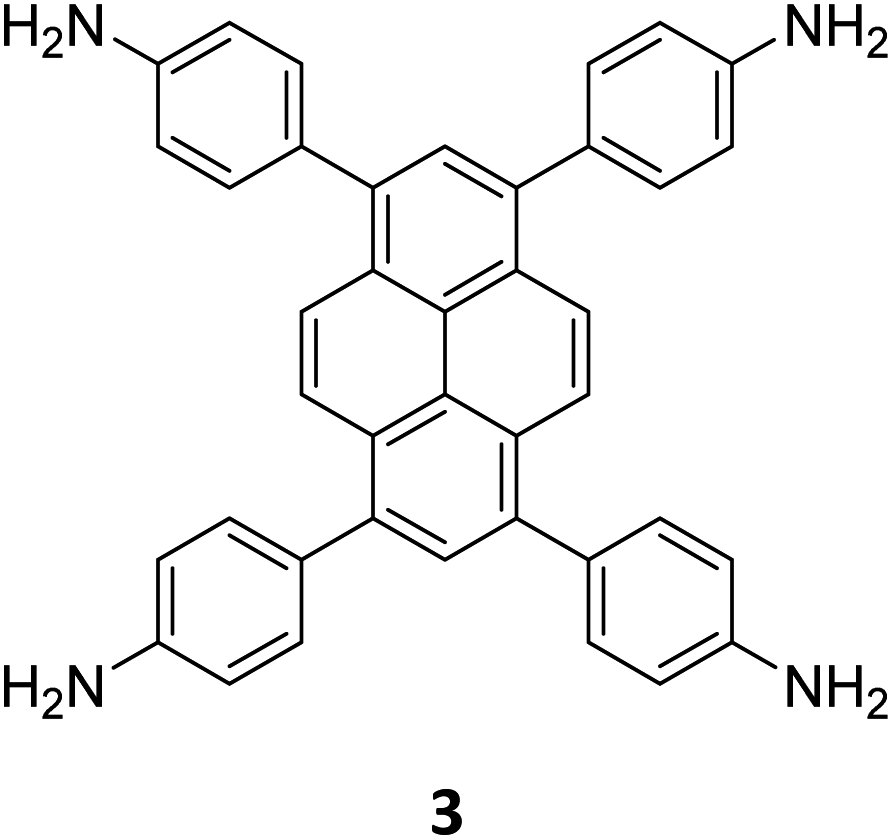
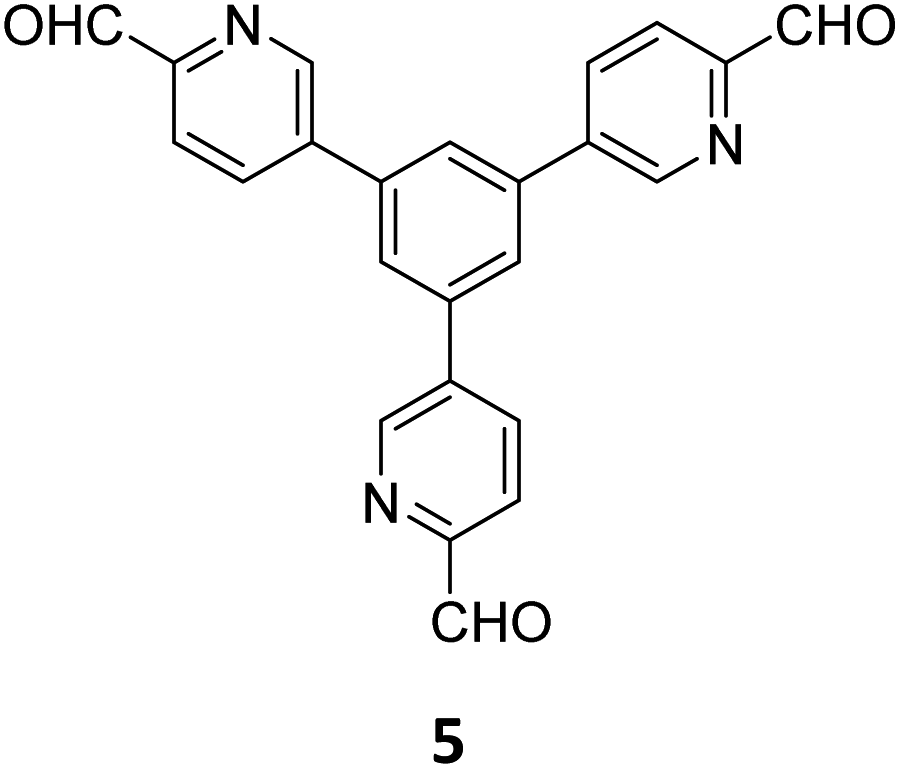
The term sub-stoichiometric synthesis was introduced in the very next year by Lotsch and co-workers and classified them as Type III COFs.34 Tetratopic pyrene tetraaniline 3 upon condensation with tritopic triazine tribenzaldehyde 4 or benzene tripicolinaldehyde 5, in an equimolar ratio in 1:1 mesitylene/dioxane under solvothermal conditions, yielded PT- and PY-COFs (Fig. 3). Sub-stoichiometric 2D COFs featuring imine linkages with an unexpected bex net topology and periodic unreacted amino groups were obtained from tri- and tetratopic linkers via a [4+3] mechanism. While fully formed networks and reasonable crystallinity cannot be expected from monomeric units with such symmetry, PT- and PY-COFs were obtained as crystalline solids. Contrary to the previous report from Loh and co-workers, variations in monomer feed ratio and solvent(s) did not yield distinctly different networks. In terms of functionalities, the free amino groups in PT- and PY-COFs led to better CO2 sorption properties, post-synthetic modifications and organocatalytic performance. A tricomponent approach was further demonstrated to form net-like topologies using a ditopic linker leading to [4 + 3 + 2] COFs.
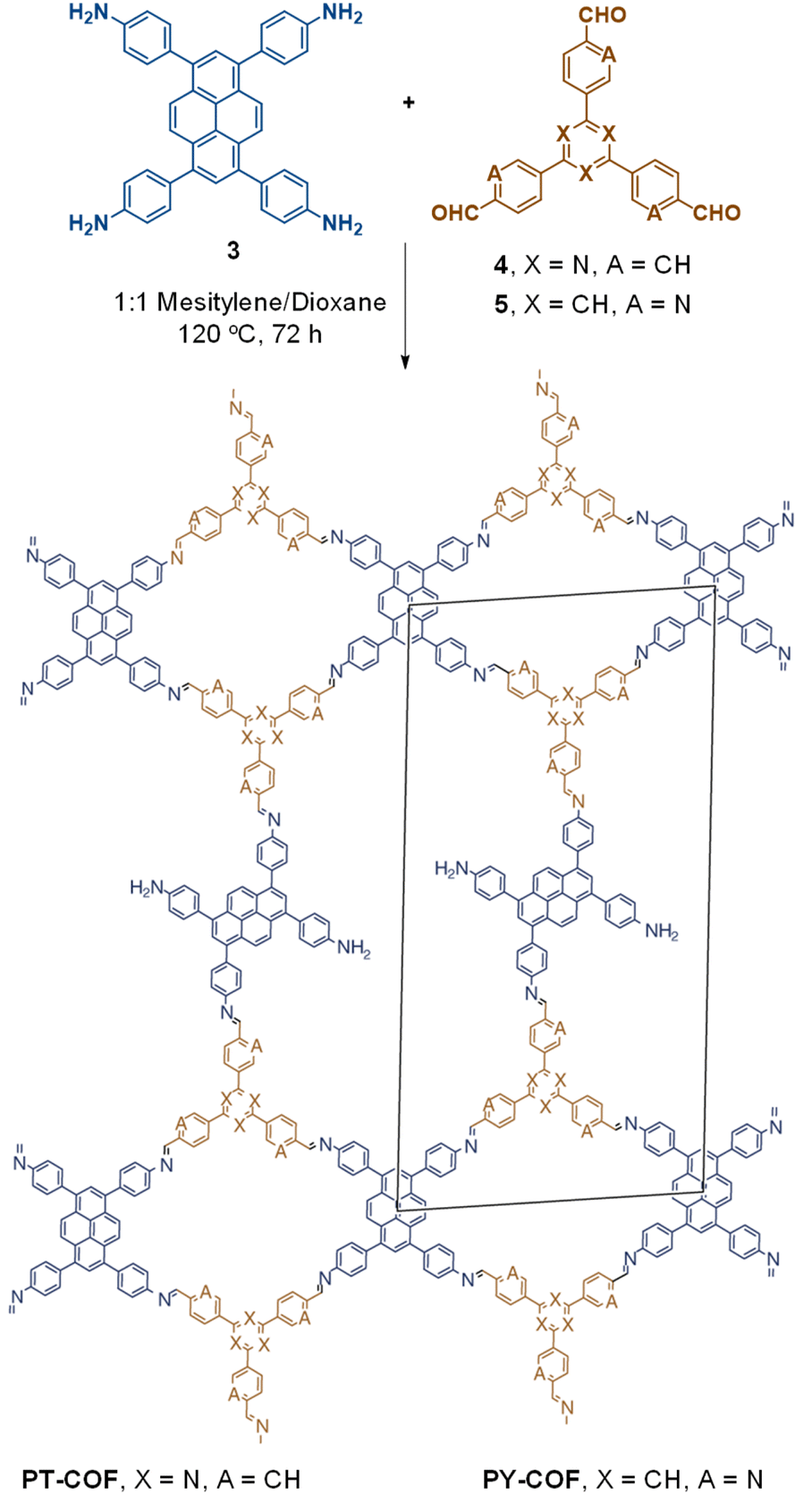 | ||
| Fig. 3 Sub-stoichiometric synthesis of the imine linked PT- and PY-COFs from tetratopic pyrene tetraaniline 3 and triangular tritopic triazine tribenzaldehyde 4 or benzene tripicolinaldehyde 5. Reproduced from ref. 34 with permission from Springer Nature, copyright 2019. | ||
Concurrent with Lotsch's report on sub-stoichiometric synthesis,34 Yaghi et al. published the synthesis of multinary COFs using hexa, tetra and trivalent linkers 6, 9, and 2 (Fig. 4).117 Condensation reaction involving hexaminophenyl benzene 6, tetrakis(4-aminophenyl) ethane 2, and 1,3,5-tris(p-formylphenyl)benzene 8 resulted in a complex network of COF-346 with unprecedented tth topology. In addition, crystalline, porous COF-360 with a rare kgd topology and COF-340 with a defective tth topology were also reported in the same study. However, variations in the ratio of the monomers were not found to afford COFs with different structural attributes or topologies.
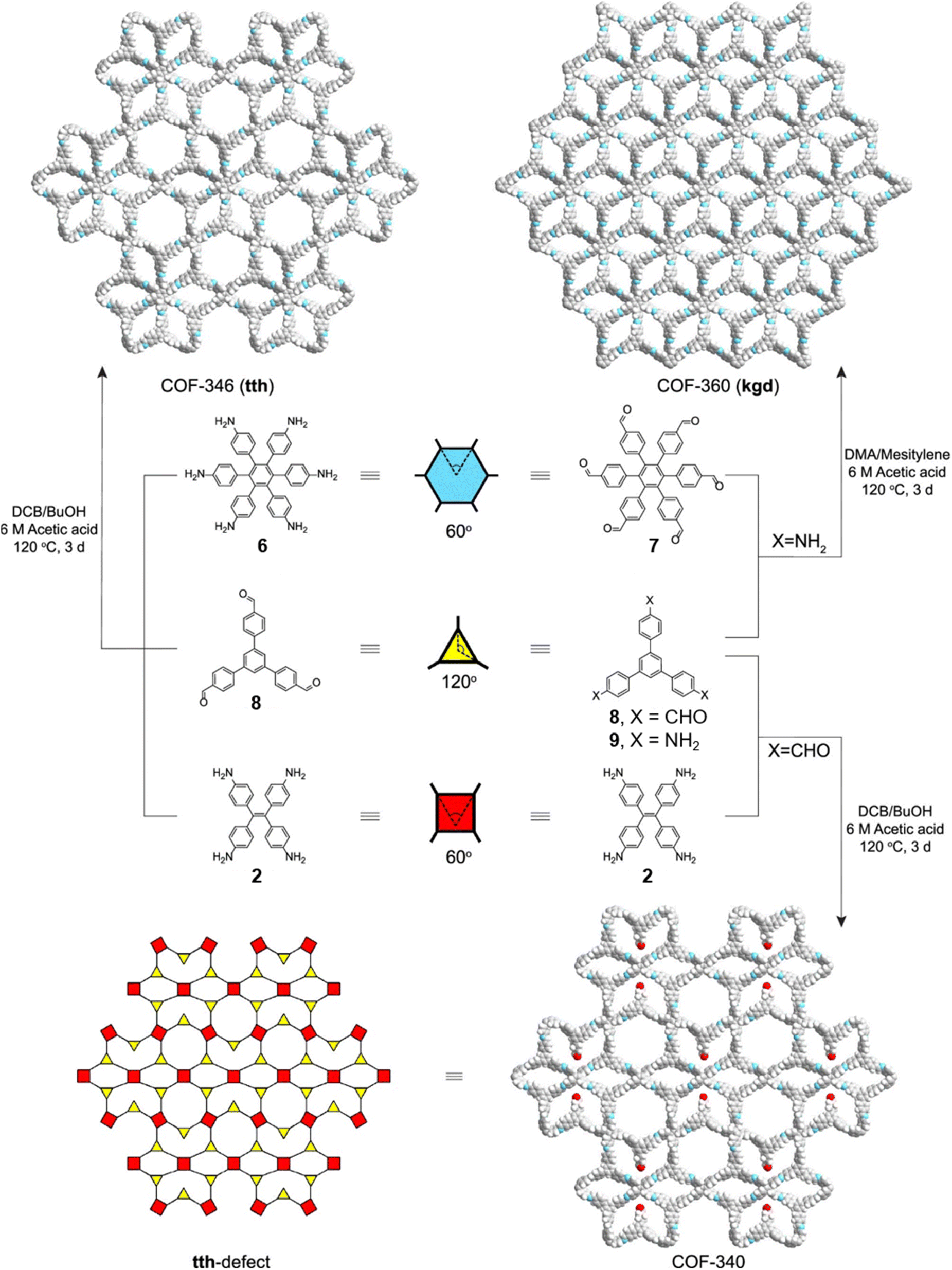 | ||
| Fig. 4 Synthetic scheme for COF-346, COF-360 and COF-340. The structures of the corresponding monomeric units are also shown along with the topologies of the COFs. Reproduced from ref. 117 with permission from American Chemical Society, copyright 2019. | ||
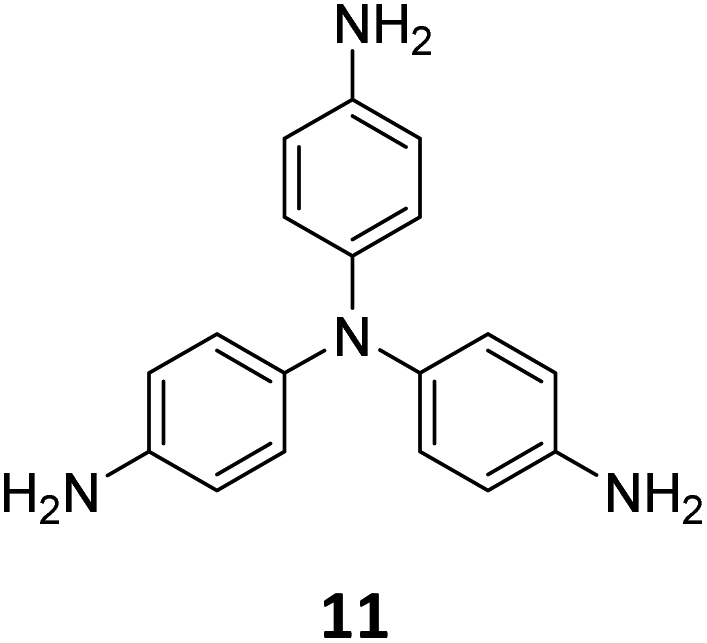
The sub-stoichiometric approach has also allowed the construction of 1D COFs with residual functionalities, which could further be converted to 2D COFs with mixed linkages. A year after the reports on COF-340, COF-346 and COF-360, Yaghi and co-workers constructed 1D ribbons of COF-76 with unreacted amino groups from 1,3,6,8-tetrakis(p-formylphenyl)pyrene 10 and tris(4-aminophenyl)amine 11. The free amines in these 1D ribbons were subsequently interlinked using aldehyde or anhydride to yield 2D frameworks of imine linked COF-77 or imide linked COF-78, respectively (Fig. 5).35 The 1D COF-76 was prepared via modulator (p-toluidine) assisted synthesis using C3 symmetric monomers 10 and 11 in a 1:2 sub-stoichiometric ratio, catalysed by trifluoroacetic acid. The structural stability and porosity of the 1D ribbons facilitated the crosslinking reaction involving free amino groups to yield the corresponding 2D networks. A one-pot synthesis for the construction of COF-77 and two step one-pot synthesis for the construction of COF-78 were also reported. This work further demonstrates the use of sub-stoichiometric synthesis for the construction of COFs with dual linkages.
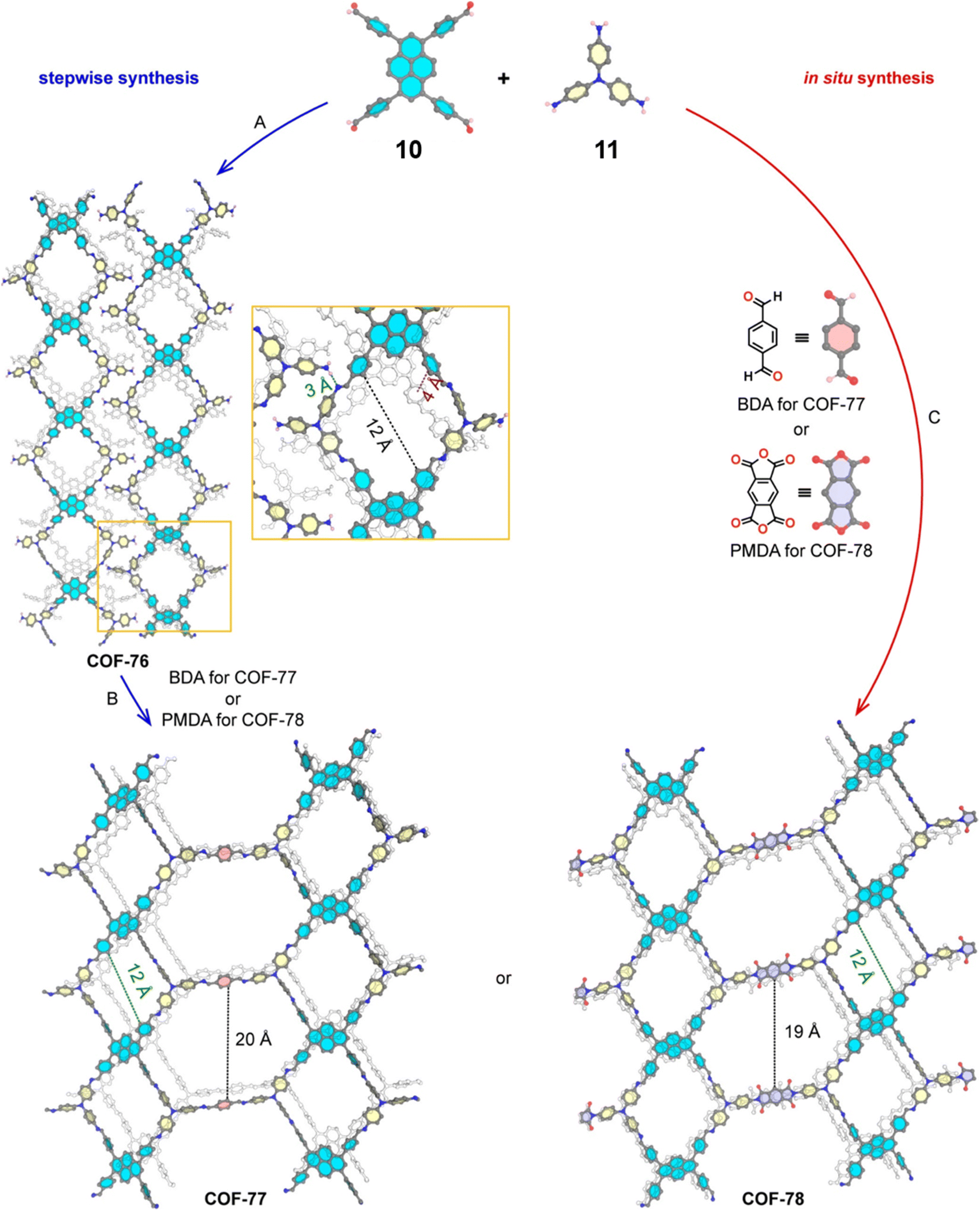 | ||
| Fig. 5 Scheme for the sub-stoichiometric imine linked 1D COF-76 with residual amines from the tetra-aldehyde 10 and tri-amine 11 in a 1:2 stoichiometric ratio and further conversion into 2D COF-77 and COF-78 using aldehyde and anhydride monomers via stepwise (A and B) and in situ methods (C). Reproduced from ref. 35 with permission from American Chemical Society, copyright 2020. | ||
3.2 Sub-stoichiometric synthesis of 3D COFs
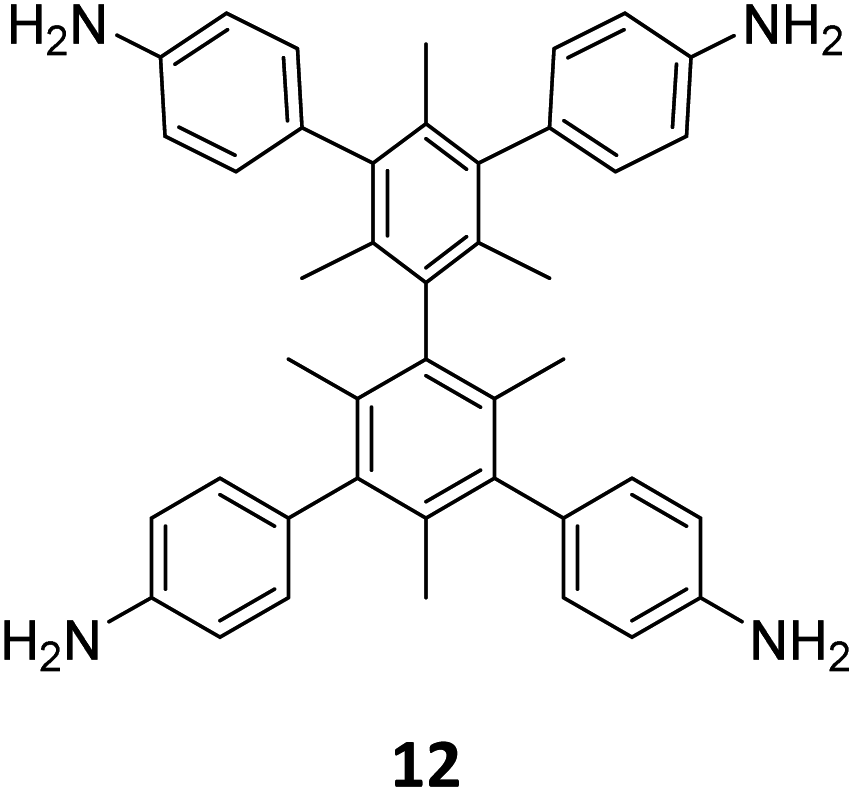
Extension of the sub-stoichiometric approach to construct 3D COFs was first demonstrated in 2021 by Peng and co-workers.118 During an attempt to construct a network of she/nts or het topologies, a frustrated 3D network with unreacted carbonyls was obtained. A 2D COF (2D sql COF) was also synthesized via the partial condensation of hexaldehyde phenyl benzene 7 and a tetramine derivative of tetraphenylethylene 2 (Fig. 6A and B). However, use of 3,3′,5,5′-tetra(p-aminophenyl)-bimesitylene 12 as the amino counterpart resulted in a 3D COF (3D pts COF) with pts topology (Fig. 6A and C).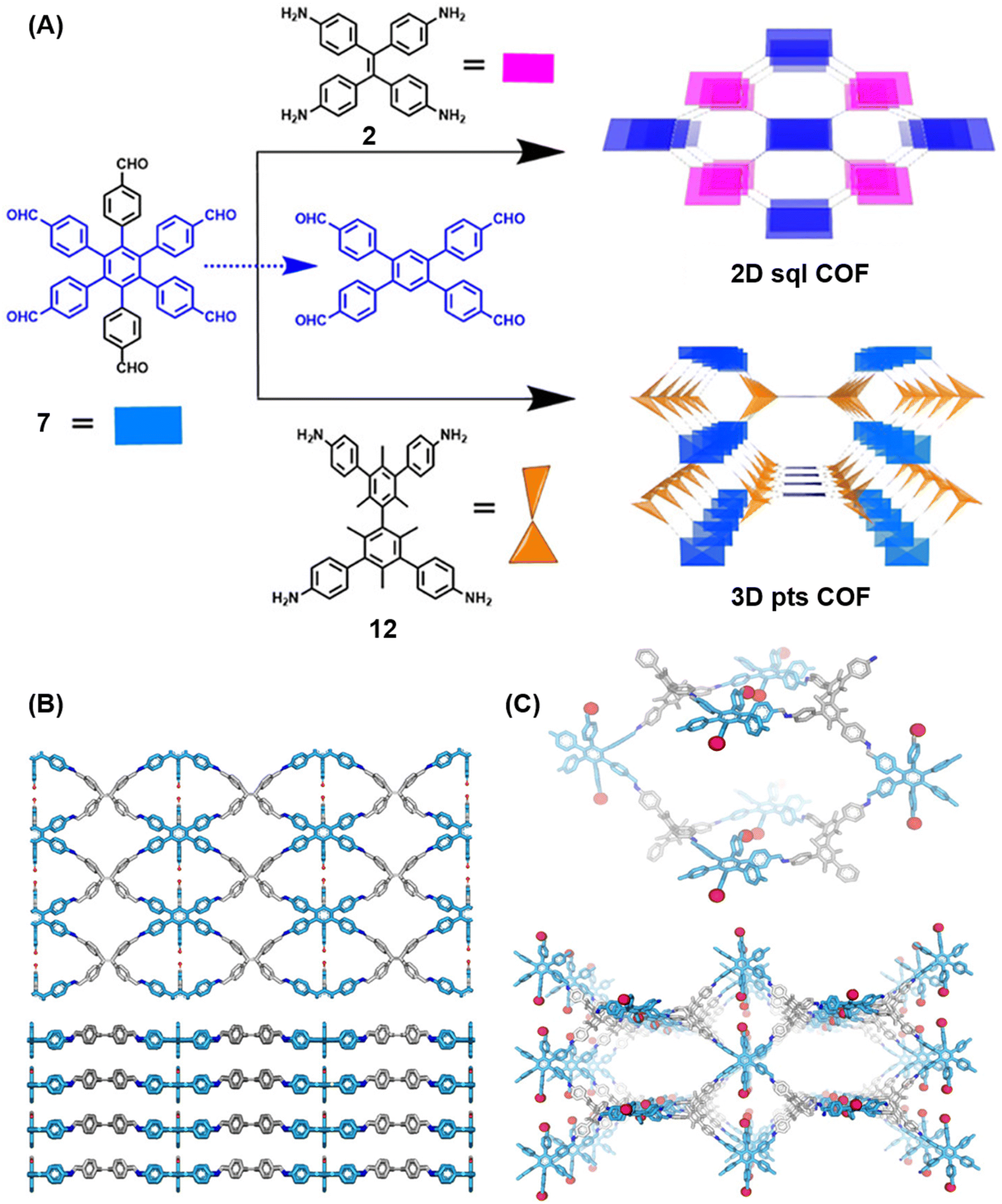 | ||
| Fig. 6 (A) Sub-stoichiometric synthesis of COFs with sql and pts topologies from the monomeric units 7, 2, and 12. (B) Top and side views of the ball-and-stick structural model for 2D sql COF. 7 is shown in blue, 2 is shown in grey, and O atoms are shown in red. (C) Cage type architecture and the ball-and-stick structural model for 3D pts COF. 7 is shown in blue, 12 is shown in grey, and O atoms are shown in red. Reproduced from ref. 118 with permission from American Chemical Society, copyright 2021. | ||
3D pts COF characterized by a surface area of 3478 m2 g−1, which is among the highest reported for 3D imine-linked COFs.118 The high surface area as well as the presence of unreacted aldehyde groups in the COFs further resulted in excellent gas storage properties. Both 2D sql COF and 3D pts COF exhibited preferential gas adsorption properties in the order C2H2 > CO2 > CH4 at ambient conditions. Though 3D pts COF was found to possess a higher surface area compared to 2D sql COF, the adsorption capacities of the former were found be inferior to those of the latter. It is proposed that the higher adsorption of C2H2 over CO2 was most likely due to the polarizability and quadruple moment of the gases, which interact with residual carbonyls in pores. Theoretical modelling corroborated that hydrogen bonding between the acetylene hydrogen and the residual carbonyls is the reason behind high adsorption capacities of the COFs.
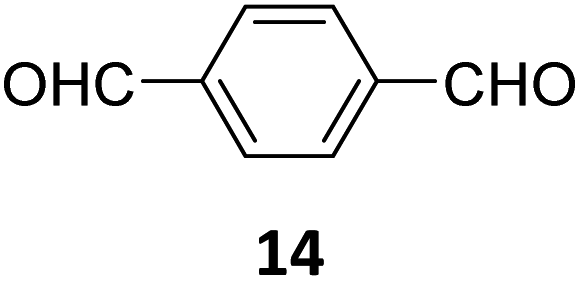
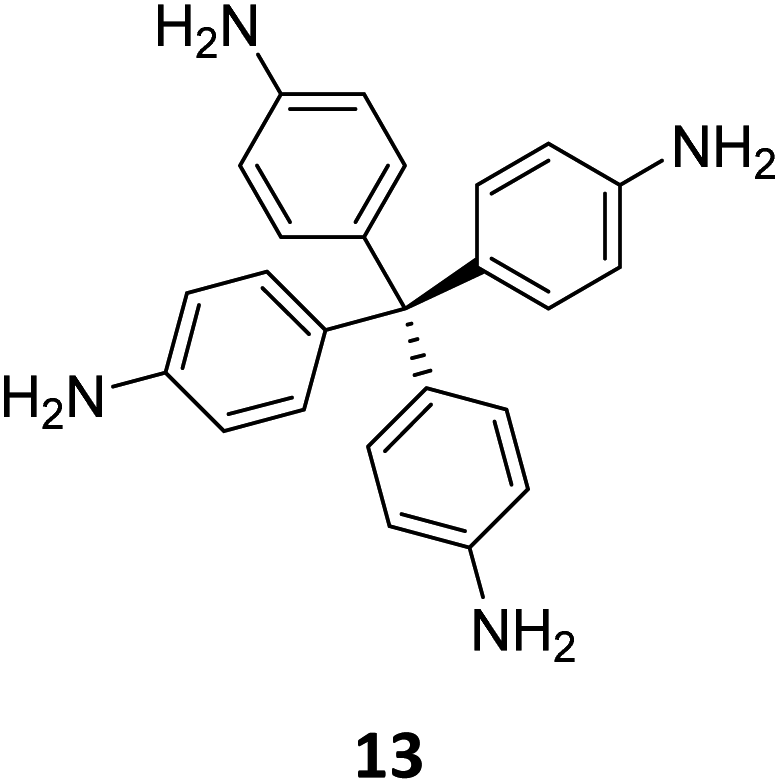
Creating a long-range microporous order in framework materials has been an interesting field of investigation over the past decade.119–127 Yue and co-workers have reported the synthesis of amine functionalised 3D COFs COF-300-0914 and COF-300-1114via a non-stoichiometric synthon (NSS) strategy by varying the ratio of tetrakis(4-anilyl) methane 13 and terephthaldehyde 14 (Fig. 7).36 Adjusting the ratio of the monomers 13:14 to 11:14 in a solvothermal condensation procedure led to COF-300-1114 with unreacted amino groups which allowed the pores to be impregnated with a larger amount of Pd(II) salts, that upon hydrogenation resulted in Pd nanoparticle decoration of the pores, while retaining the structure of the parent COF (Fig. 7). The presence of Pd NPs was confirmed through energy dispersive X-ray spectroscopy and ICP-MS techniques, resulting in a mass loading of 1–2%, while more than 90% of the Pd content was confirmed to be Pd(0) by XPS. While both COF-300-0914 and COF-300-1114 confined ultrasmall Pd nanoclusters within their pores, the presence of more unreacted amino groups in the latter restricted NP aggregation via Pd–N coordination. These Pd-decorated COFs were further used as catalysts for benzyl alcohol oxidation reactions with up to 97% conversion with 99% selectivity.
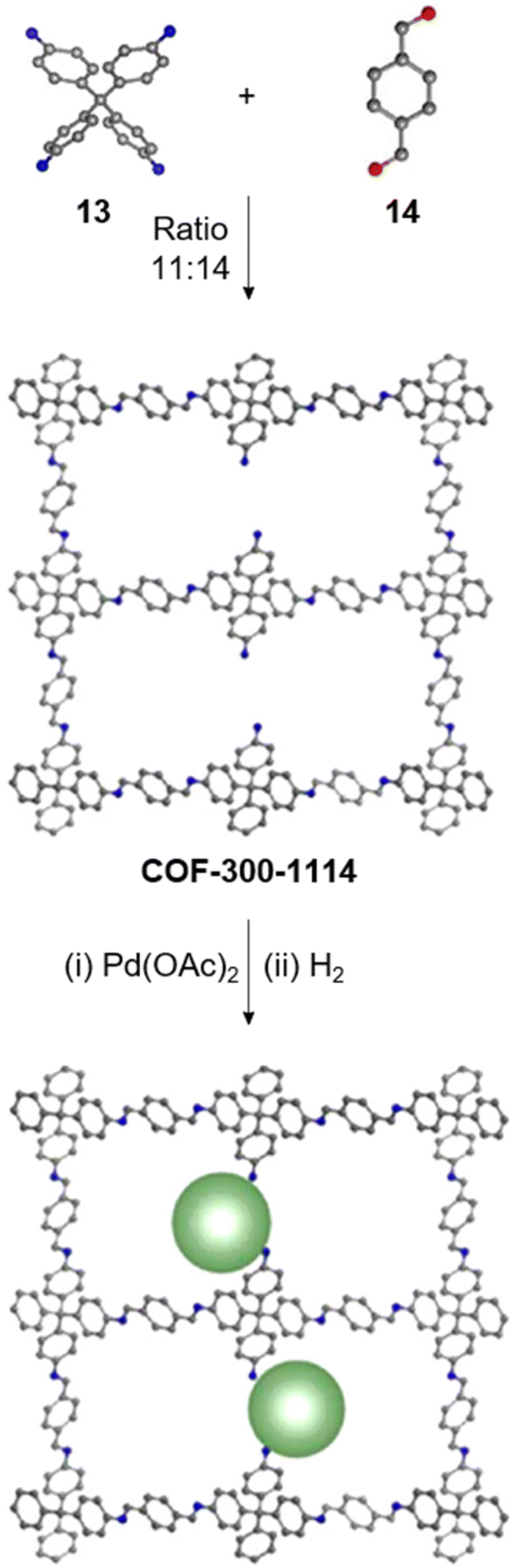 | ||
| Fig. 7 Non-stoichiometric synthesis of COF-300-1114 from the amine 13 and aldehyde 14 followed by Pd impregnation and subsequent reduction yielding the Pd/COF composite. Color code: grey, carbon; blue, nitrogen; red, oxygen. H-atoms are omitted for clarity. Reproduced from ref. 36 with permission from American Chemical Society, copyright 2020. | ||
3.3 Applications of COFs constructed via sub-stoichiometric synthesis
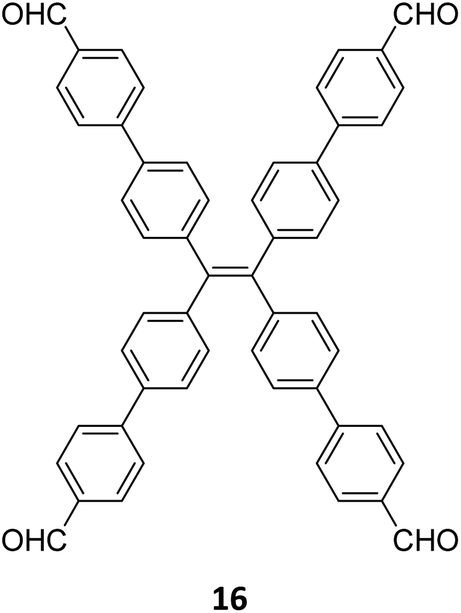
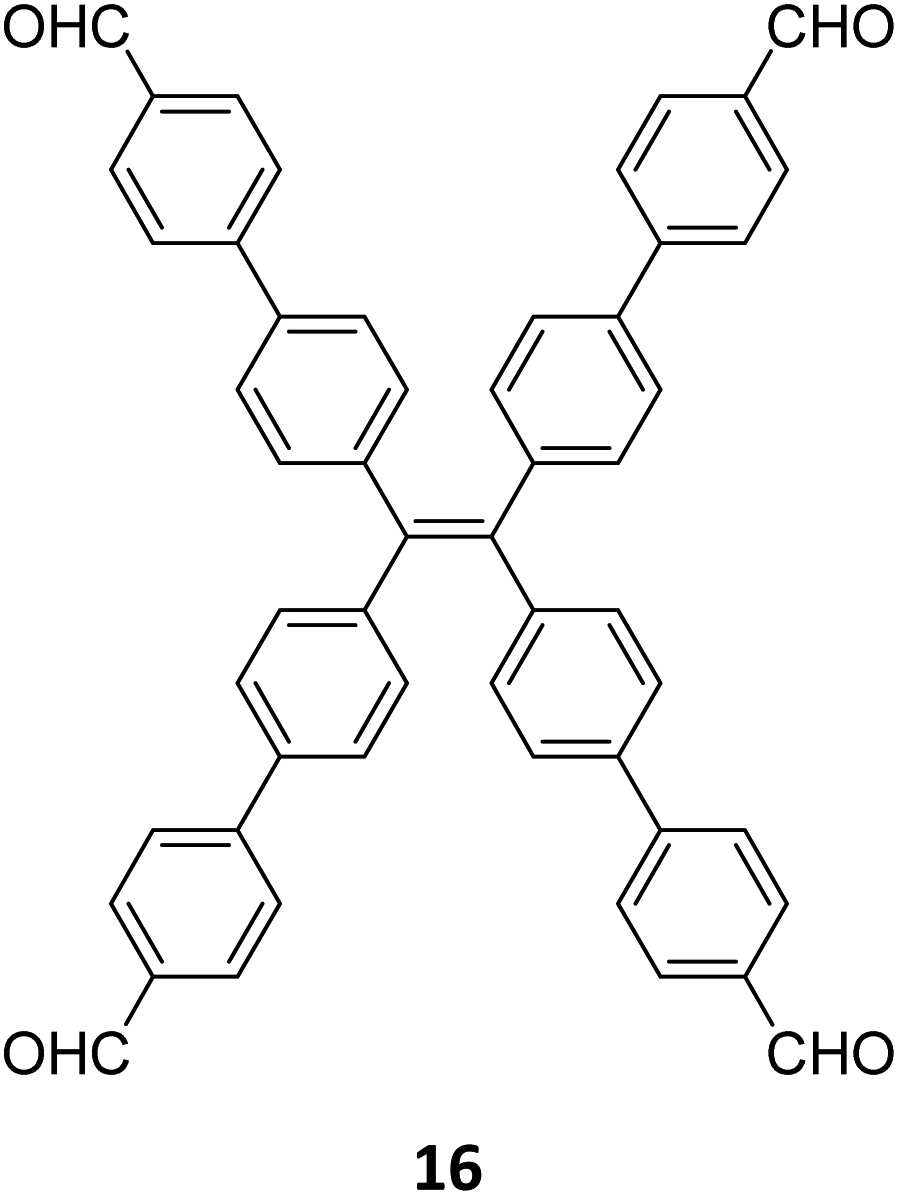
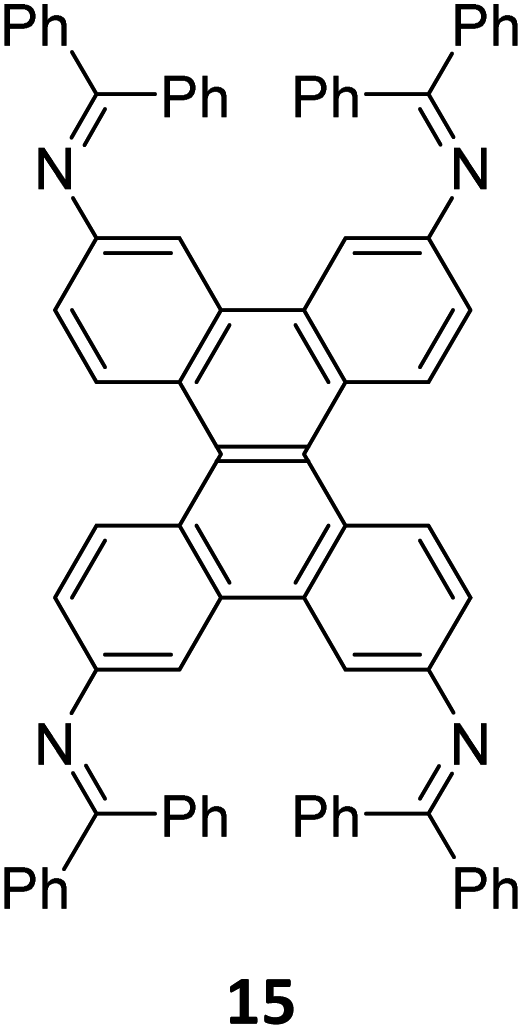
Robust morphology, high surface area, large pore volume, and high thermal and chemical stabilities make COFs suitable candidates for several applications including gas storage/separation, catalysis, energy storage, etc.23,38,128,129 While the formation of ideal crystals of framework materials is challenging, the defects or unreacted sites in such materials open up new avenues that are otherwise difficult to explore.98,99,130–132 Nevertheless, the introduction of free amine and carbonyl groups with periodic order is generally difficult to achieve in conventional COFs with an imine linkage. Therefore, several reports on the sub-stoichiometric synthesis of COFs are aimed at constructing network materials with a rational arrangement of either of the functional groups, for expanding their applications. This section of the article attempts to review the applications of COFs obtained via sub-stoichiometric approaches.Chen and co-workers have demonstrated a transamination approach to construct COFs with frustrated aldehyde and amino groups.138 This was the first instance of constructing a COF with free amine and aldehyde functionalities using a sub-stoichiometric approach from the same monomeric units by simple alteration in their ratios. Different molar ratios (1:2, 1:1 and 2:1) of dibenzo[g,p]chrysene 15 and tetraphenylethene tetraldehyde derivative 16 were used to construct three COFs, DBC-TPE-[CHO], DBC-TPE and DBC-TPE-[NH2], respectively (Fig. 8). Surface area exceeding 1000 m2 g−1 and thermal stability above 400 °C were observed for all three COFs. DBC-TPE-[NH2] with free amino groups exhibited the highest CO2 uptake of 3.16 mmol g−1, followed by DBC-TPE-[CHO] with free aldehyde groups (2.56 mmol g−1) and the fully formed network DBC-TPE (2.39 mmol g−1) at 273 K. The free functionalities in these COFs were also shown to adsorb water, most likely due to hydrogen bonding. Volumetric water vapor sorption experiments at 298 K revealed water uptake capacities of 0.42 g g−1, 0.40 g g−1, and 0.26 g g−1 for DBC-TPE-[NH2], DBC-TPE and DBCTPE-[CHO], respectively. The relatively higher water adsorption properties of DBC-TPE-[NH2] are attributed to the lowering of the inflexion point due to the presence of hydrophilic amines, suggesting better water adsorption properties under less humid conditions.
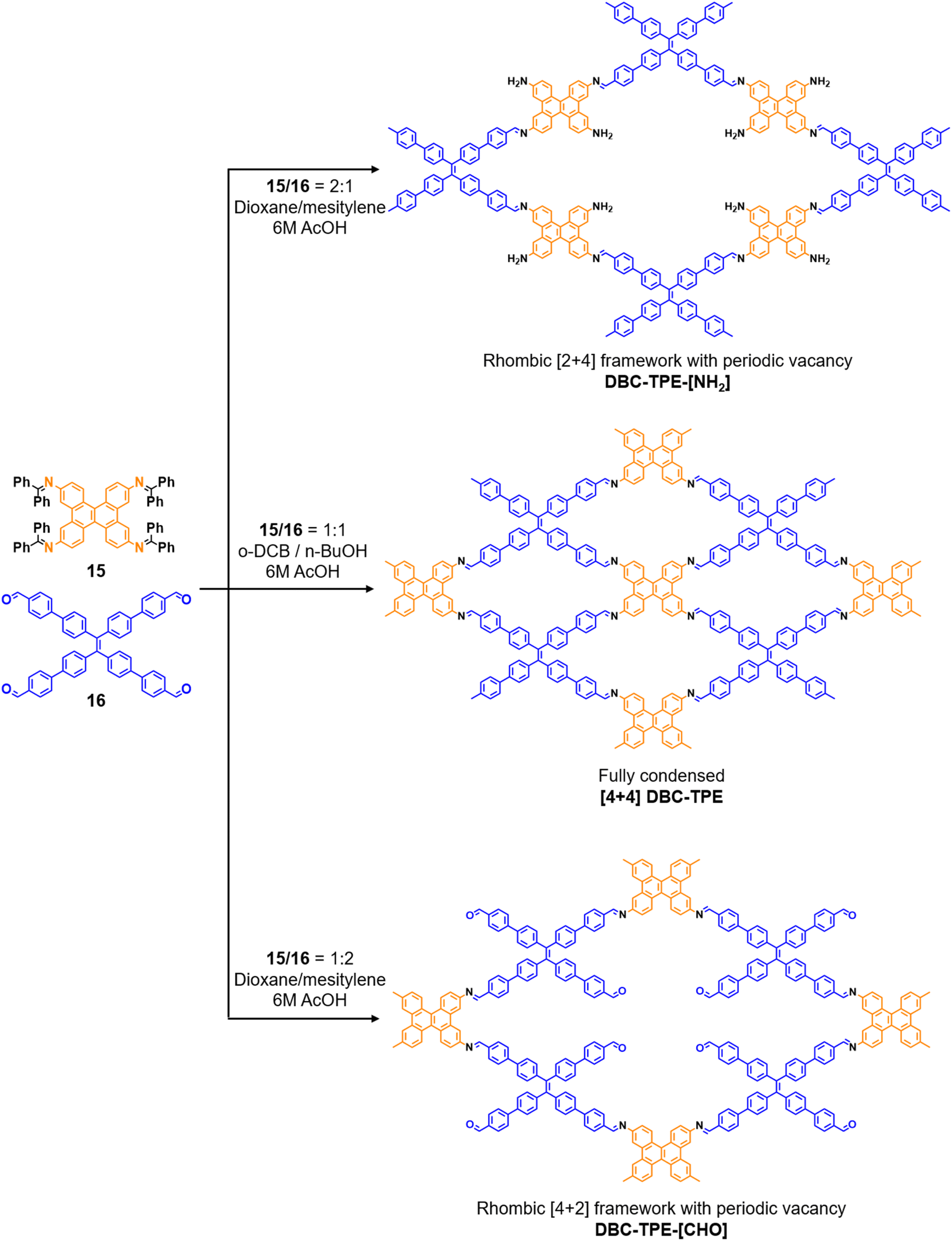 | ||
| Fig. 8 Sub-stoichiometric synthesis of imine linked 2D COFs DBC-TPE-[CHO], DBC-TPE and DBC-TPE-[NH2] with residual aldehydes and amines from the same monomers via a transamination approach. | ||
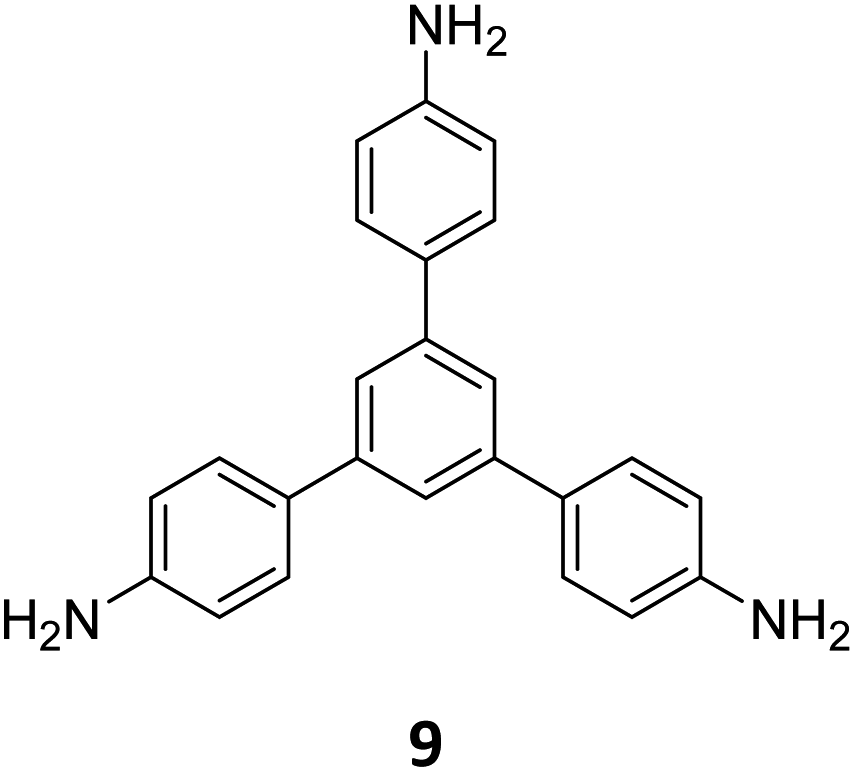
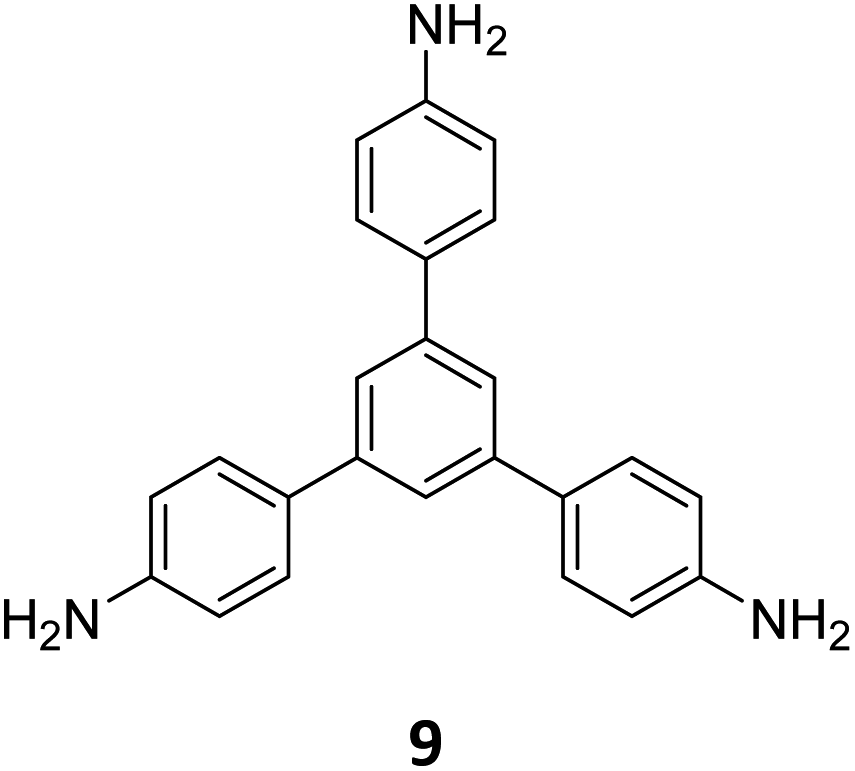
Ma and co-workers have recently reported COFs with residual carbonyls for selective adsorption of CO2.134 Three COFs, HFPB-Stb-D, HFPB-TAPB-D, and HFPB-ETTA-D, were constructed via sub-stoichiometric reactions involving C6-symmetric aldehyde monomers (hexa(4-formylphenyl)-benzene 7) and amine monomers of various symmetry (C2 – 4,4′-diaminostilbene 17; C3 – 1,3,5-tris(4-aminophenyl) benzene 9; and C4 – tetrakis(4-aminophenyl)ethene 2, Fig. 9A). This work tendered, for the first time, monomer symmetry regulated directional defects for in situ functionalization of COFs with a periodic distribution of aldehyde groups for selective interactions with CO2. Better structural stability, availability of aldehyde groups and good crystallinity of HFPB-Stb-D, HFPB-TAPB-D, and HFPB-ETTA-D resulted in high CO2 uptake values of 128.9 cm3 g−1 (25.3 wt%), 109.6 cm3 g−1 (21.5 wt%) and 44.9 cm3 g−1 (8.8 wt%), respectively at 1 bar pressure and 273 K. Their CO2 adsorption capacities at low pressure (0.1 bar) and at 298 K were also seen to follow the same order. The CO2 adsorption capacity of the defective COFs was found to depend preferentially on the number of homogeneously distributed aldehyde groups in the framework as compared to other factors such as specific surface area, pore volume, or nitrogen content. In fact, HFPB-Stb-D exhibited one of the best reported selective separation values (45.8 from Ideal Adsorbed Solution Theory (IAST)) for CO2/N2 among other COFs. This differential adsorption of gases is due to the reasonably lower quadrupole moment of N2 compared to residual aldehydes as validated by theoretical calculations (Fig. 9B).
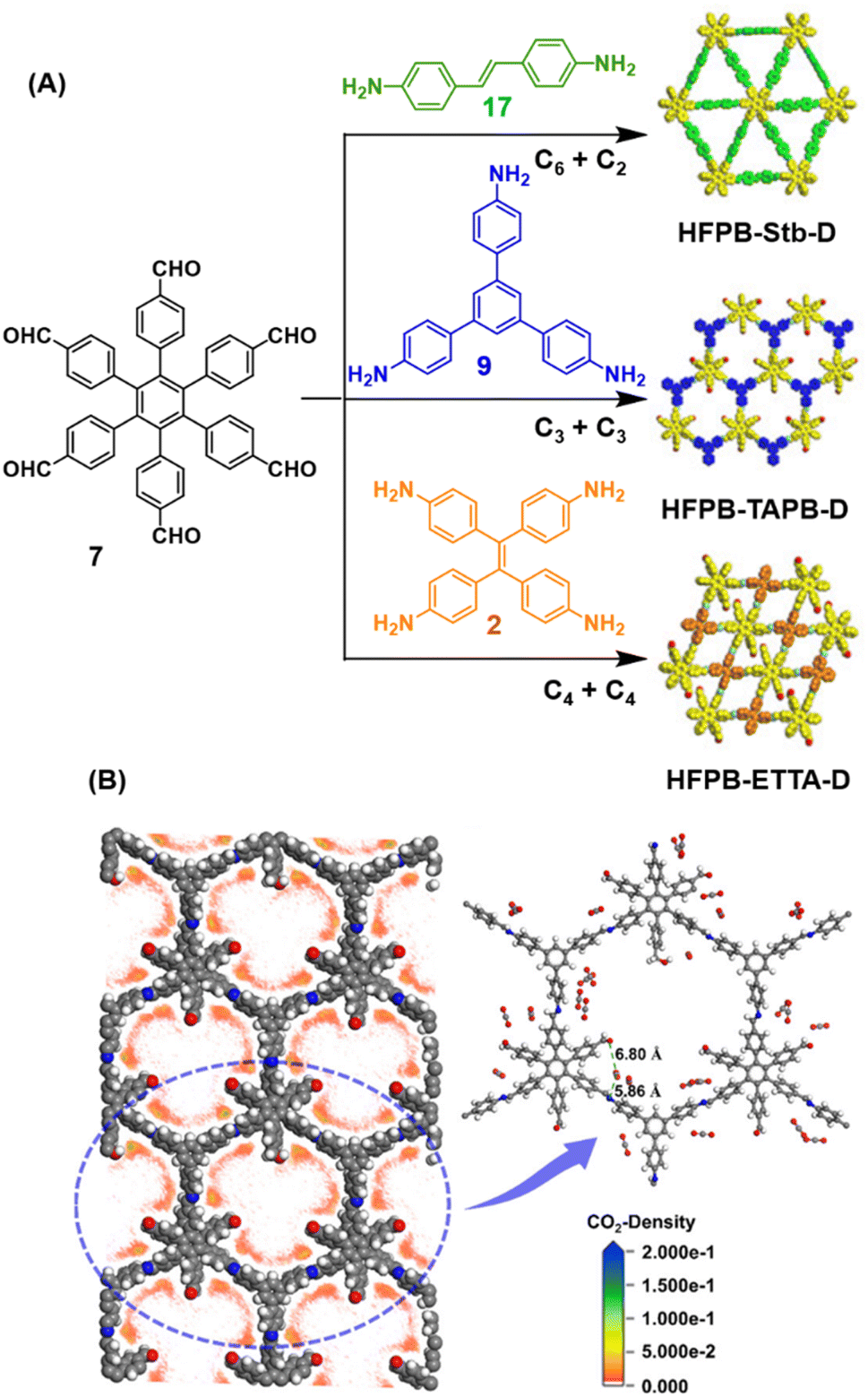 | ||
| Fig. 9 (A) Synthesis of the COFs HFPB-Stb-D, HFPB-TAPB-D, and HFPB-ETTA-D from the C6-symmetric aldehyde monomer (hexa(4-formylphenyl)-benzene 7) and amine monomers of various symmetry (C2 – 4,4′-diaminostilbene 17; C3 – 1,3,5-tris(4-aminophenyl) benzene 9; and C4 – tetrakis(4-aminophenyl)ethene 2). (B) CO2 distribution density in HFPB-TAPB-D (left) and the simplified stick model (right), where H, C, N, and O are marked in white, grey, blue, and red, respectively. Reproduced from ref. 134 with permission from American Chemical Society, copyright 2023. | ||
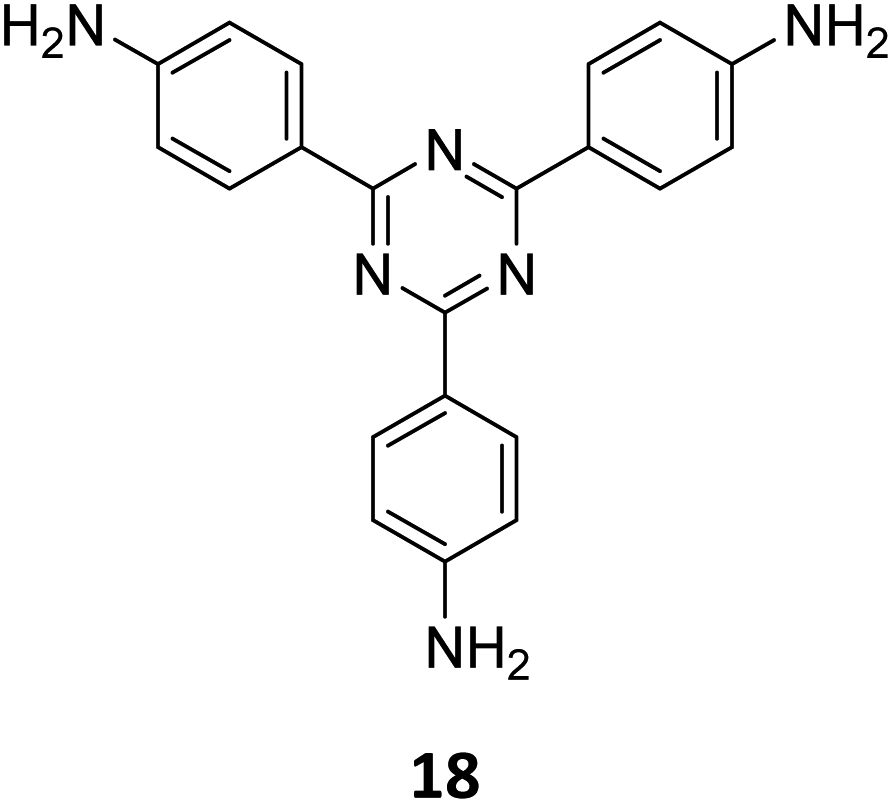
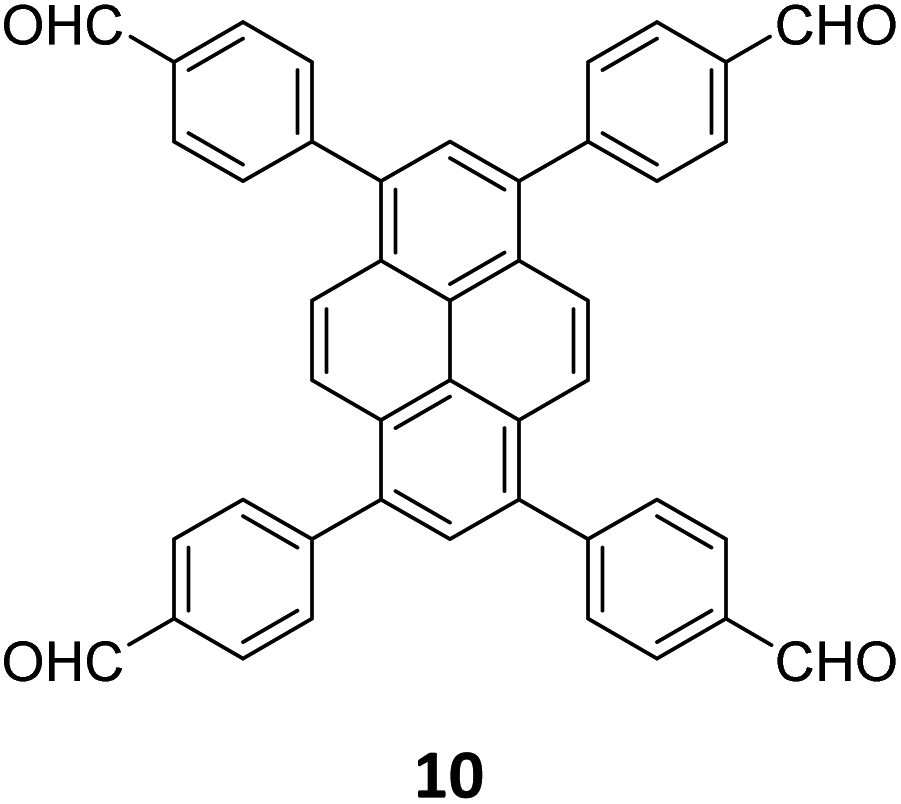
An emissive donor–acceptor type COF-SMU-1 having frustrated aldehyde groups, with a quantum yield of 6.8%, has recently been reported via sub-stoichiometric synthesis involving a triamine 18 and a tetra-aldehyde 10 by Das and co-workers (Fig. 10A).148COF-SMU-1 exhibited solvatochromic properties due to polarity dependent π–π interactions of pyrene units present in the neighbouring layers. The color of COF-SMU-1 was found to undergo a rapid change from yellow to reddish brown when exposed to water vapour (Fig. 10B). COF-SMU-1 when dispersed in acetonitrile, showed a slight bathochromic shift and a gradual decrease in emission intensity upon addition of water (3.6% v/v). Water that hydrogen bonds with triazine, imine and aldehyde impacts the D–A charge transfer (Fig. 10B, right). This interaction of water with residual carbonyls was established via spectroscopic techniques and a water-uptake value as high as 14.0 mmol g−1 (0.25 g g−1) was observed for COF-SMU-1 at 25 °C. Even in the presence of hydrophobic aromatic polycycles, high water adsorption properties are correlated to the abundance of imine and carbonyl groups in the COF structure. The absence of a significant reduction in porosity for prolonged exposure to moisture suggests that COF-SMU-1 may have application as a potential water harvester.
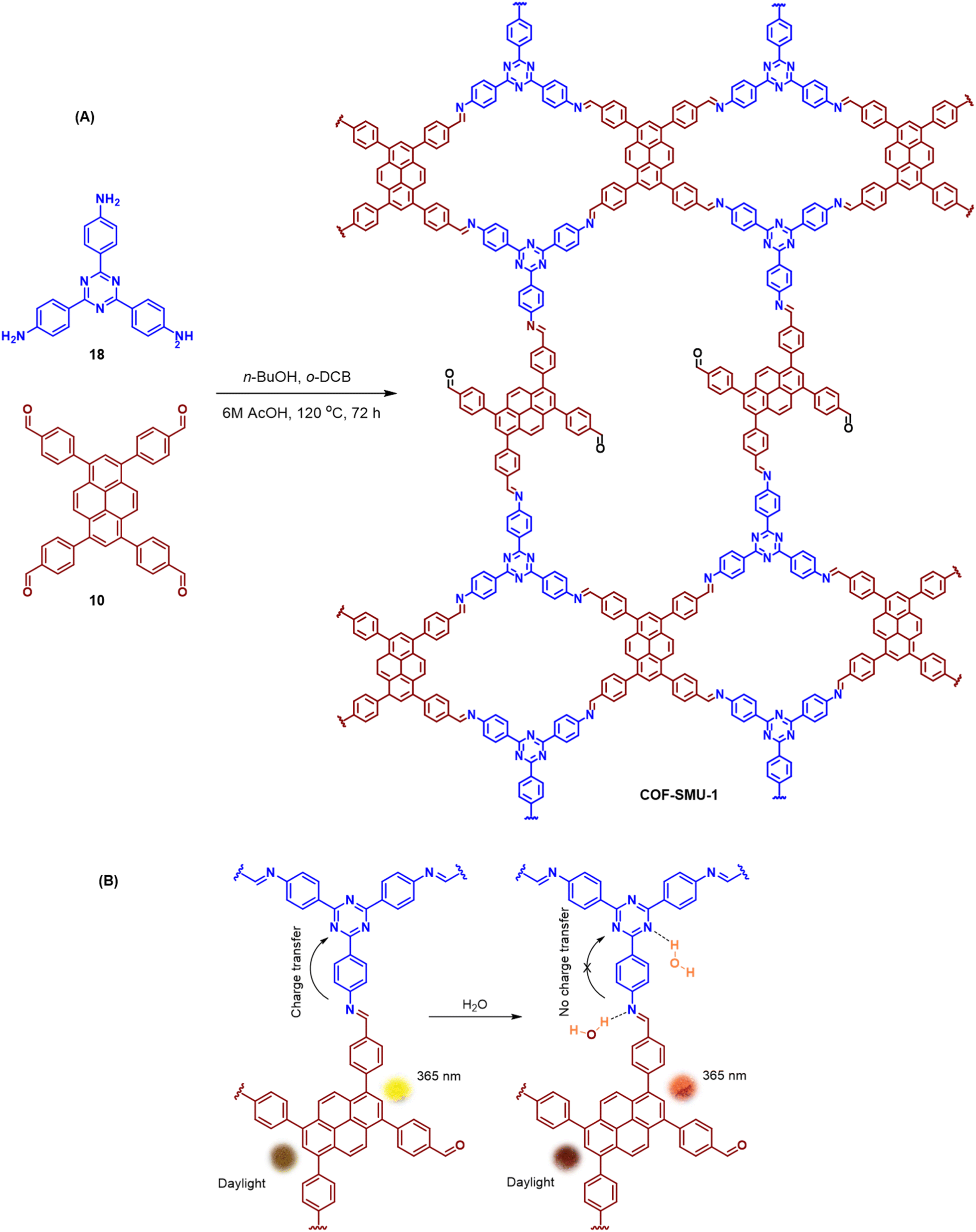 | ||
| Fig. 10 (A) Sub-stoichiometric synthesis of the COF COF-SMU-1 from a triamine 18 and a tetra-aldehyde 10 based monomeric unit. The unreacted aldehyde sites are shown in green. (B) Repeating units and water sensing mechanism of COF-SMU-1. Adapted from ref. 148 with permission from Wiley, copyright 2023. | ||
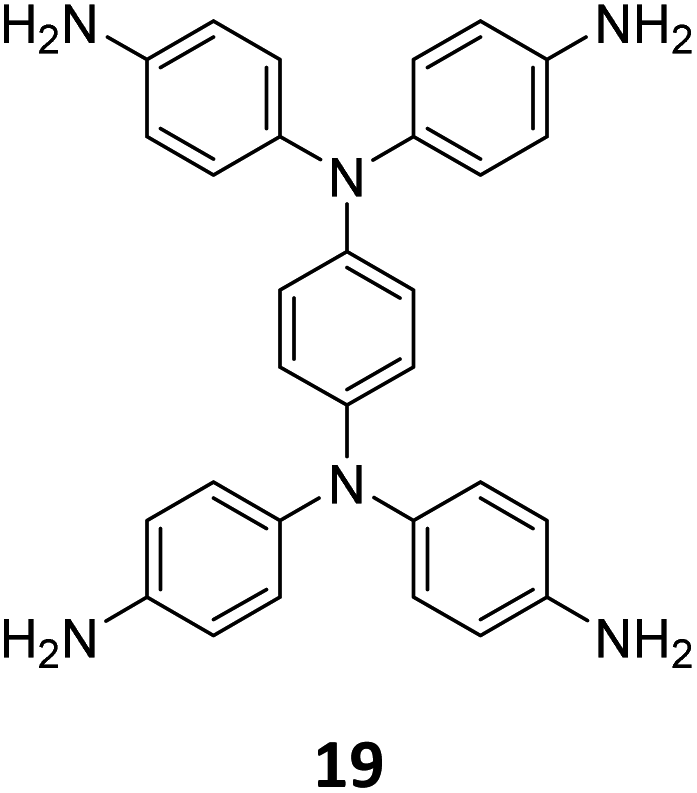
In 2023, Tian and co-workers reported the synthesis of [C4 + C4] COF PyT-2 and [C4 + C2] COF PyT-1 from tetrabenzaldehyde 19 and tetraamine 10via the variation in the choice of solvents alone (Fig. 11A).149 While both the COFs exhibited good thermal, solvent, acid–base stabilities, PyT-1 was found to have comparably higher BET surface area (1186.94 m2 g−1) and pore volume (0.89 cm3 g−1). Significant iodine vapour adsorption was observed for both PyT-1 and PyT-2 for the first 10 h and attained saturation around 90 h (>3.4 g g−1, Fig. 11B). Desorption was effected by rinsing the COFs in ethanol. PyT-1 was found to retain an adsorption efficiency >90% after five adsorption–desorption cycles, while PyT-2 showed a lower efficiency of 75–76% (Fig. 11C). Solution phase adsorption studies were conducted using n-hexane solutions of iodine and both COFs were found to follow pseudo second order kinetics.
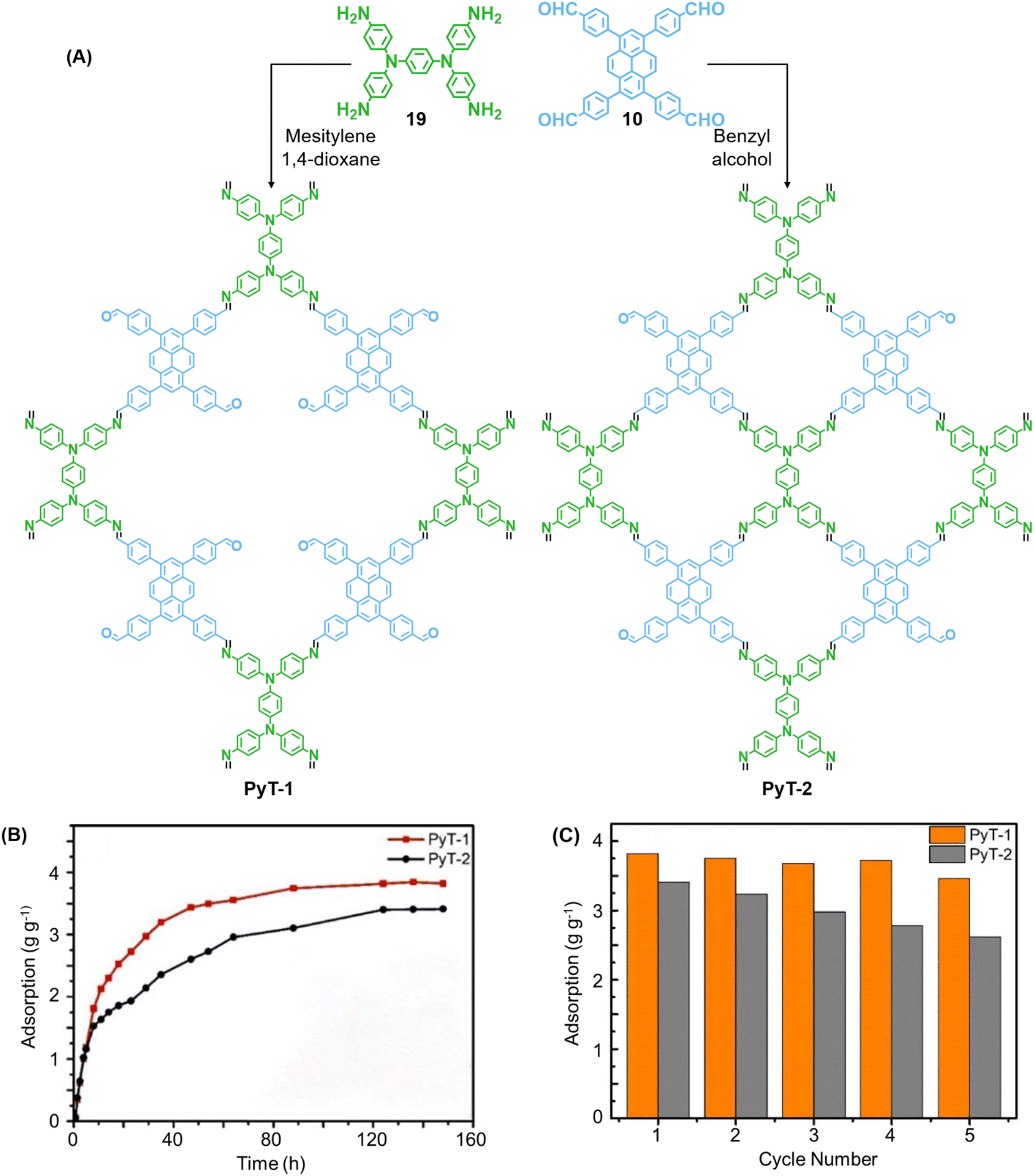 | ||
| Fig. 11 (A) Synthesis of the COFs PyT-1 and PyT-2 from tetratopic monomers 19 and 10. (B) Iodine vapor adsorption capacities of PyT-1 and PyT-2 at 75 °C and atmospheric pressure and (C) the corresponding cycling performance. Reproduced from ref. 149 with permission from MPDI, copyright 2023. | ||
3.3.2.1 Heterogeneous organocatalysis. Lotsch and co-workers, while introducing the term ‘sub-stoichiometric synthesis’ in COFs, further demonstrated free amines in PT- and PY-COFs (Fig. 3) capable of catalysing room temperature synthesis of substituted chromenes.34 The aromatic amines in the frameworks were found to catalyse the regioselective cyclization–substitution cascade reactions of 2-hydroxylcinnamaldehyde with trimethylsilyl enol ether, with yields of 22% and 8%, respectively, for PT- and PY-COFs. The proposed catalytic cycle begins with a Schiff base adduct formed between the residual amines and 2-hydroxylcinnamaldehyde. As previously mentioned, the Pd-decorated COF-300-0914 and COF-300-1114 (Fig. 7), reported by Yue et al.,36 were also used as catalysts for benzyl alcohol oxidation reactions with excellent yield and selectivity.
In 2020, Xie and co-workers demonstrated the first instance of using type III COFs as photocatalysts. Three D–A type COFs TTCOFs 1-3 were synthesized from C2 symmetric tetraphenylethylene based monomers 1, 5, and 2, and C3 symmetric triazine-based monomers 4 and 17, under acidic conditions over 5–9 days (Fig. 12A).150 While TTCOFs 1 and 2 featured a bex topology with optical bandgaps >2 eV, TTCOF-3 was characterized by a tth topology with a lower band gap of 1.73 eV. These COFs were used as photocatalysts in visible light driven selective α-functionalization of tertiary amines. TTCOF-2 was found to provide the best results in direct aerobic cross-dehydrogenative coupling between 2-phenyl-1,2,3,4-tetrahydroisoquinoline 19 and nitromethane or ketones via a radical mechanism (Fig. 12B). Furthermore, the same COF was also found to be active in photoconversion of aryl boronic acids into the corresponding phenols.
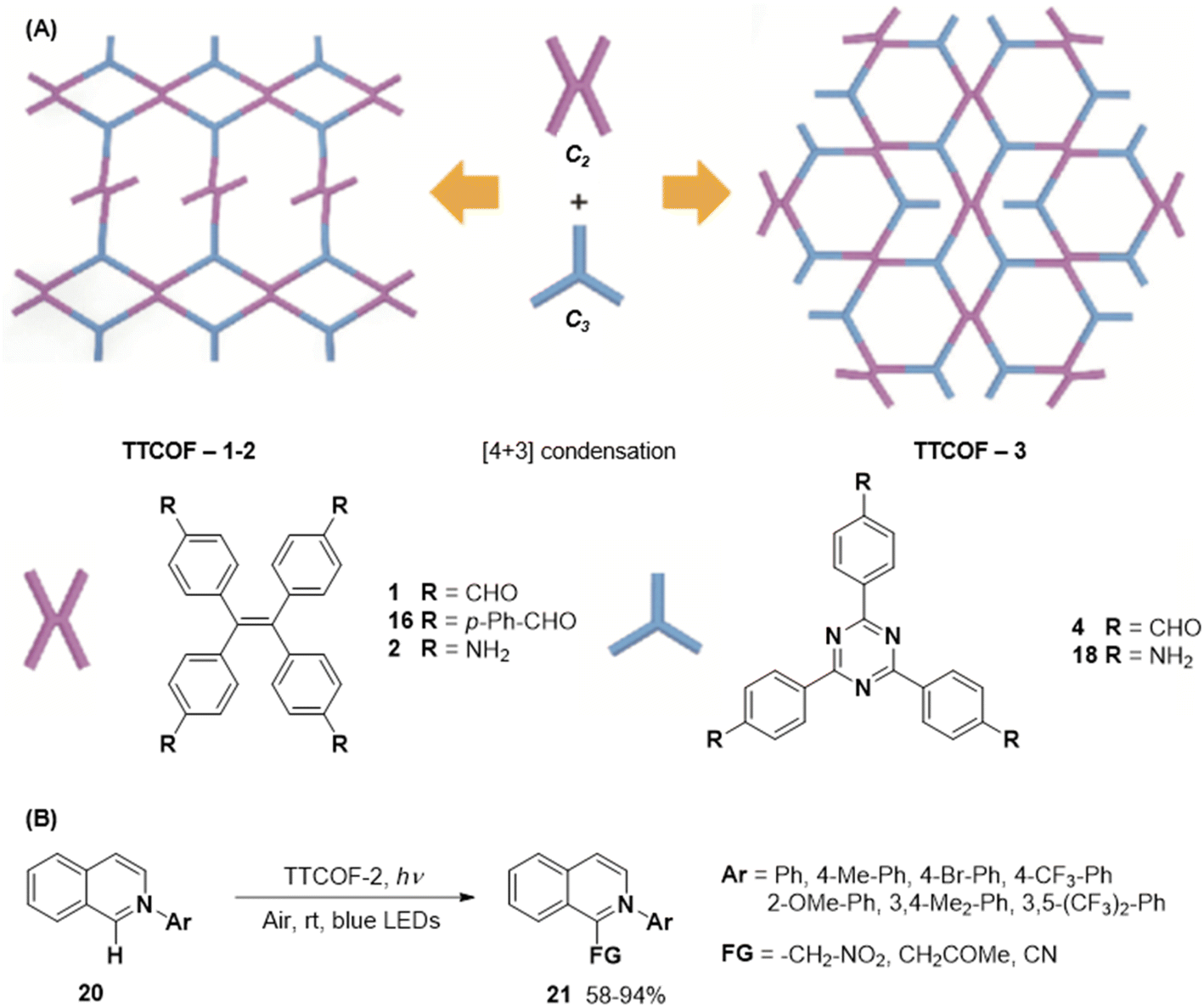 | ||
| Fig. 12 (A) Schematic representation of the synthesis of TTCOFs–1-3 from C2 symmetric tetraphenylethylene based monomers 1, 16, and 2, and C3 symmetric triazine-based monomers 4 and 18. (B) Photocatalyzed aerobic cross-dehydrogenative coupling between 2-phenyl-1,2,3,4-tetrahydroisoquinoline 20 and nitromethane or ketones. Reproduced from ref. 150 with permission from Science China Press, copyright 2020. | ||
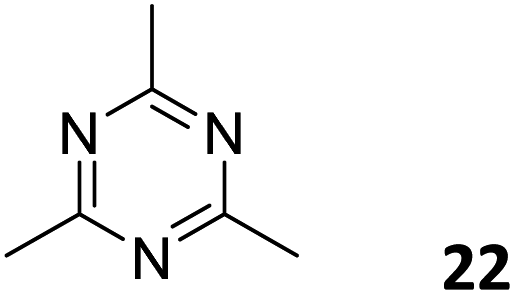
3.3.2.2 Photocatalysis. While the photoconversion of aryl boronic acids into the corresponding phenols catalysed by TTCOF-2 was reported by Xie and co-workers in 2020,150 the extension of the sub-stoichiometric approach to the synthesis of olefin linked COFs was first demonstrated by Cai and co-workers in 2021.151PTO-COF with a bex net topology was synthesized through sub-stoichiometric Knoevenagel condensation of 1,3,6,8-tetrakis(p-formylphenyl)-pyrene 10 and 2,4,6-trimethyl-1,3,5-triazine 22. The mechanochemical synthesis of β-keto enamine linked COF TpMaCOF from triformylphloroglucinol 23 and melamine 24 was integrated with the mechanochemical delamination of bulk PTO-COF to construct a heterojunction. Subsequently, 2D/2D TpMa/PTO-COFs heterojunctions were constructed by one-pot ball milling of the monomers 23 and 24 and PTO-COF in different ratios, vizTpMa
:PTO = 1:2, 1:1, 2:1, and 3:1 (Fig. 13) with bandgaps of ∼2.5 eV. These heterojunctions exhibited high photocurrents and electron transfer efficiency with a small interfacial resistance compared to the individual parent COFs. Photocatalytic performance of these materials towards the degradation of sulfamethazine, an antibiotic model, under the irradiation of visible light (λ > 420 nm) was further evaluated. The 2D/2D COF with a TpMa:PTO ratio of 2:1 was found to show a high degradation rate of 98.4% within 50 min of light irradiation. The rate constant of degradation (1.04 × 10−3 s−1) was indeed more than twice that of PTO-COF and approximately double that of TpMa-COF. The intense photocurrent response coupled with small charge transfer impedance is most likely the reason behind the better photocatalytic properties of the heterojunction, and the photocatalytic degradation was found to originate from the photogenerated holes and the superoxide radicals.
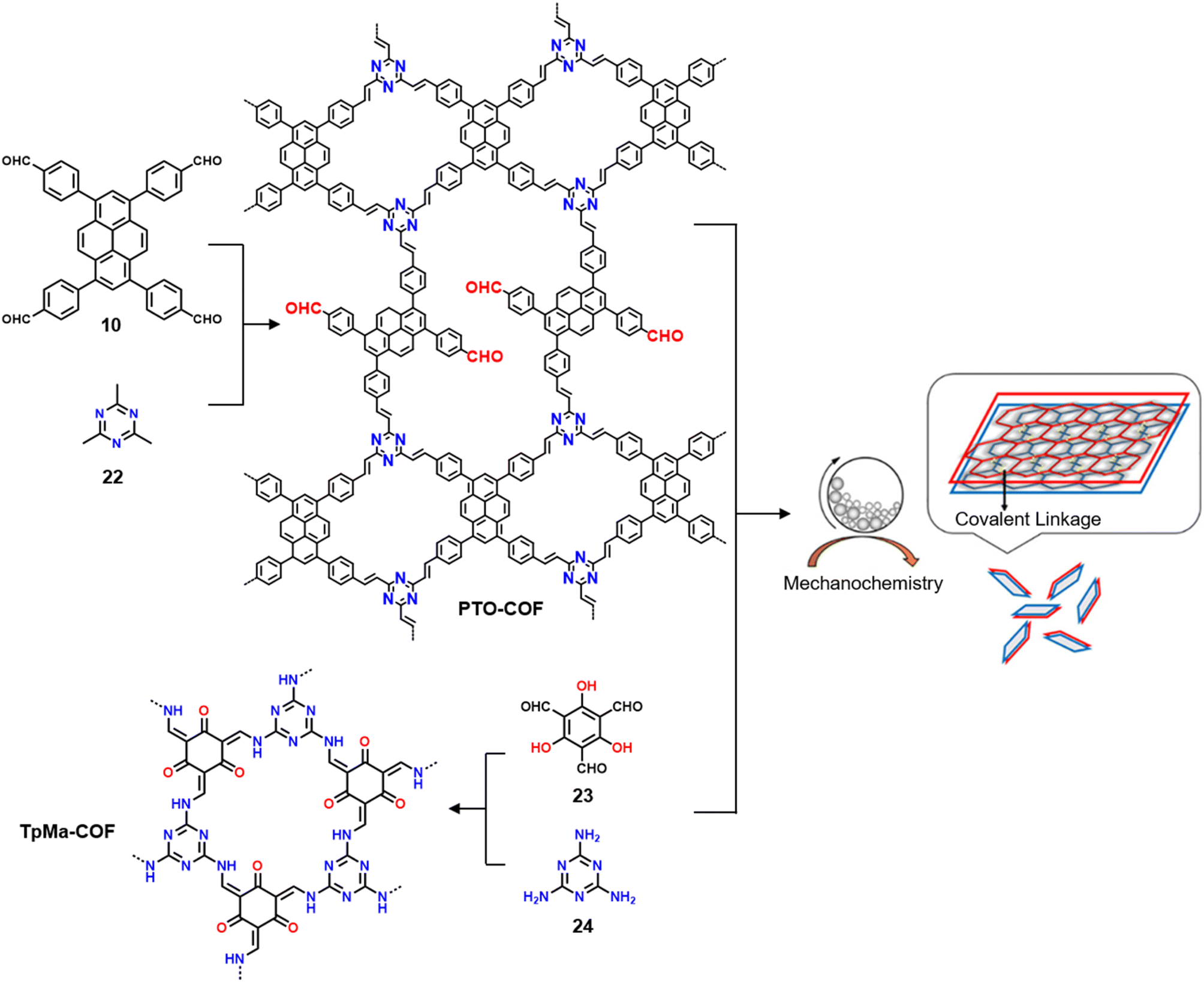 | ||
| Fig. 13 Schematic representation of the synthesis of the PTO-COF (from monomers 10 and 22), TpMa-COF (from monomers 23 and 24), and the 2D/2D TpMa/PTO COF heterojunctions. Reproduced from ref. 151 with permission from American Chemical Society, copyright 2021. | ||
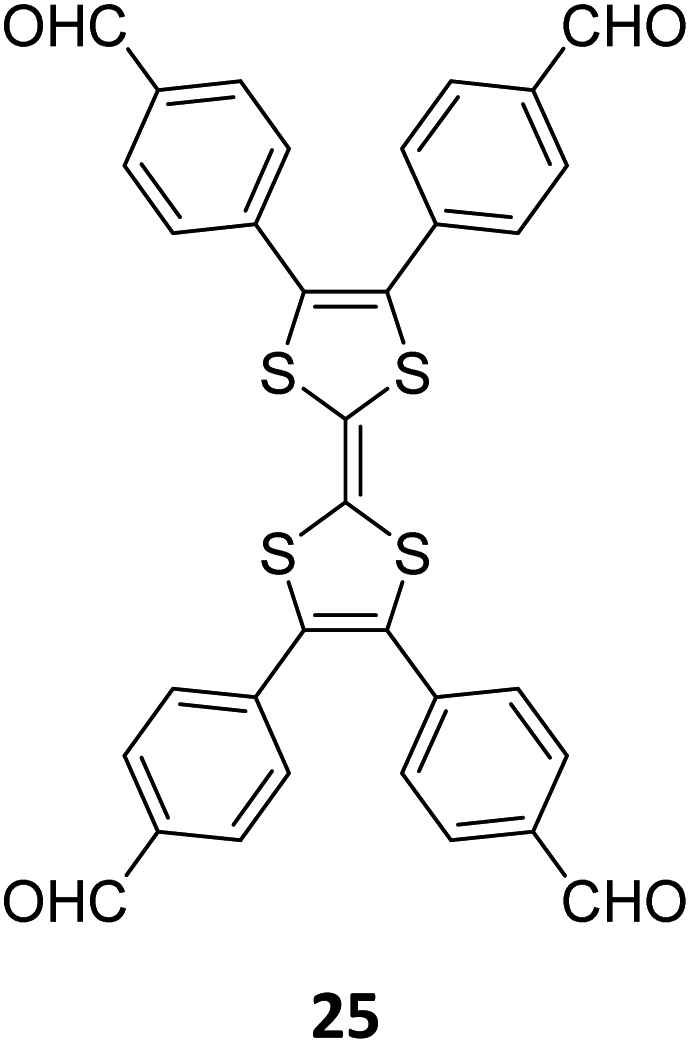
A year later, Lan and co-workers reported the sub-stoichiometric synthesis of imine-linked TCOF and ECOF with a bex net topology from tri- and tetra-topic linkers 25, 16, and 18.37 Addition of the amine functionalized polyoxometalate (POM) cluster of MnMo6–2NH2 resulted in its covalent attachment with uncondensed aldehydes in the pores of these COFs (Fig. 14A–C). Incorporation of POM within the pores was confirmed by FT-IR, XPS, elemental mapping, transmission electron microscopy and NMR. The amount of the POM in TCOF-MnMo6 was found to be 25.9 wt%.
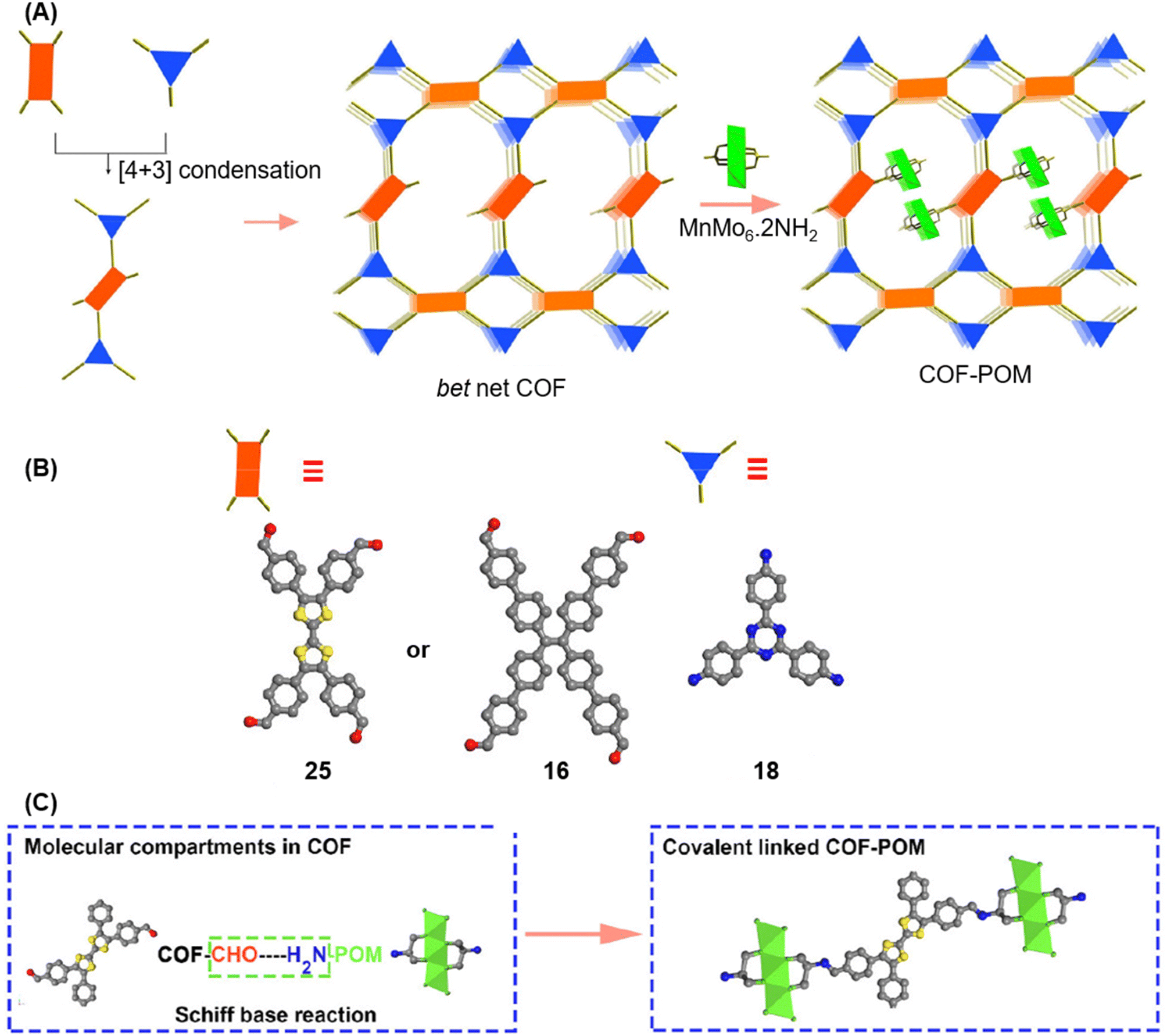 | ||
| Fig. 14 (A) Schematic representation of the synthesis of TCOF and ECOF and uniformly dispersed POM clusters within their pores via covalent linkages. (B) Ball and stick model of the monomers 25, 16, and 18. (C) Schematic representation of the molecular compartments in the COFs formed via Schiff Base reaction (left) and the formation of covalently linked COF-POM hybrids (right). Color code: grey, carbon; blue, nitrogen; red, oxygen. H-atoms are omitted for clarity. Reproduced from ref. 37 with permission from American Chemical Society, copyright 2022. | ||
The COF with its inherent light absorption combined with the catalytic properties of CO2 adsorbing POM crystals was proposed as an effective catalyst for the photoreduction of CO2 to CO. Reaction in a gas–solid reactor with CO2 and water vapour yielded more than two times conversion for TCOF-MnMo6 (796.6 μmol g−1) compared to ECOF-MnMo6 (365.3 μmol g−1) and exceeded that for previously reported POM- and COF-based CO2 photoreduction catalysis. The photocatalytic activity of individual COFs, POM alone or a physical mixture of both the materials was found to be rather low compared to the hybrid, which in turn confirmed the functional significance of incorporating a POM with a COF through a covalent linkage.
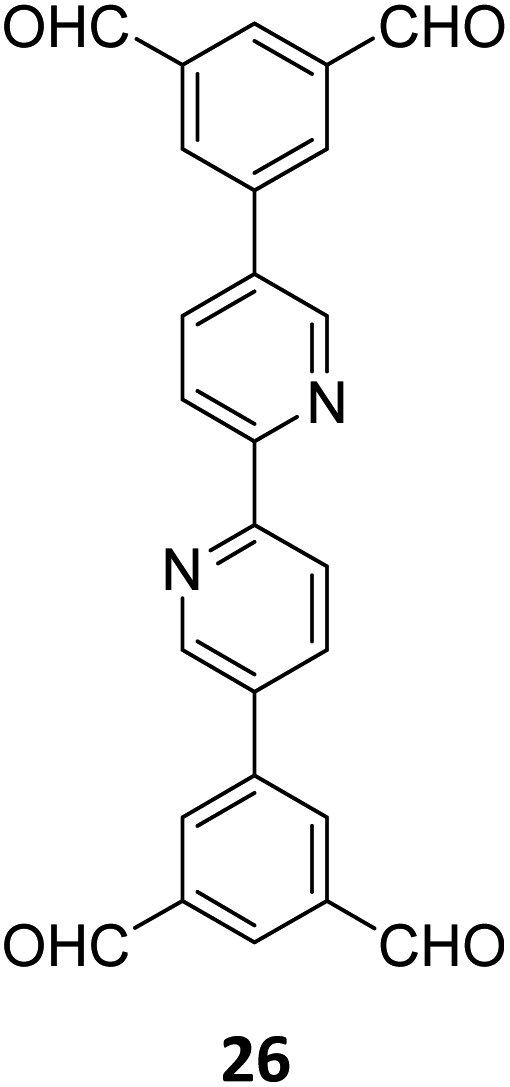
Gu and co-workers have recently reported a COF Bpy-TAPT with dual active sites and polar aldehyde groups, comprising of triazine and bipyridine units, from a tetratopic linker 26 and the tritopic linker 18, via a sub-stoichiometric approach (Fig. 15A).133 While other derivatives and analogues were also synthesized, a high photocatalytic generation of H2O2 from water (4038 μmol h−1 g−1) was observed for Bpy-TAPT. This rate of H2O2 generation in the absence of any sacrificial reagents and stabilizers was significantly higher compared to prior reports on COF-based photocatalysts (Fig. 15B). Theoretical calculations confirmed that the HOMO and LUMO of Bpy-TAPT were partly localized on the bipyridine and triazine units, suggesting them to be the oxidative and reductive sites, respectively. The dual active sites were found to improve the visible-light-response and enhanced charge generation, while the free aldehyde groups allowed for effective separation of carriers and adsorption of O2/H+ on the surface of Bpy-TAPT.
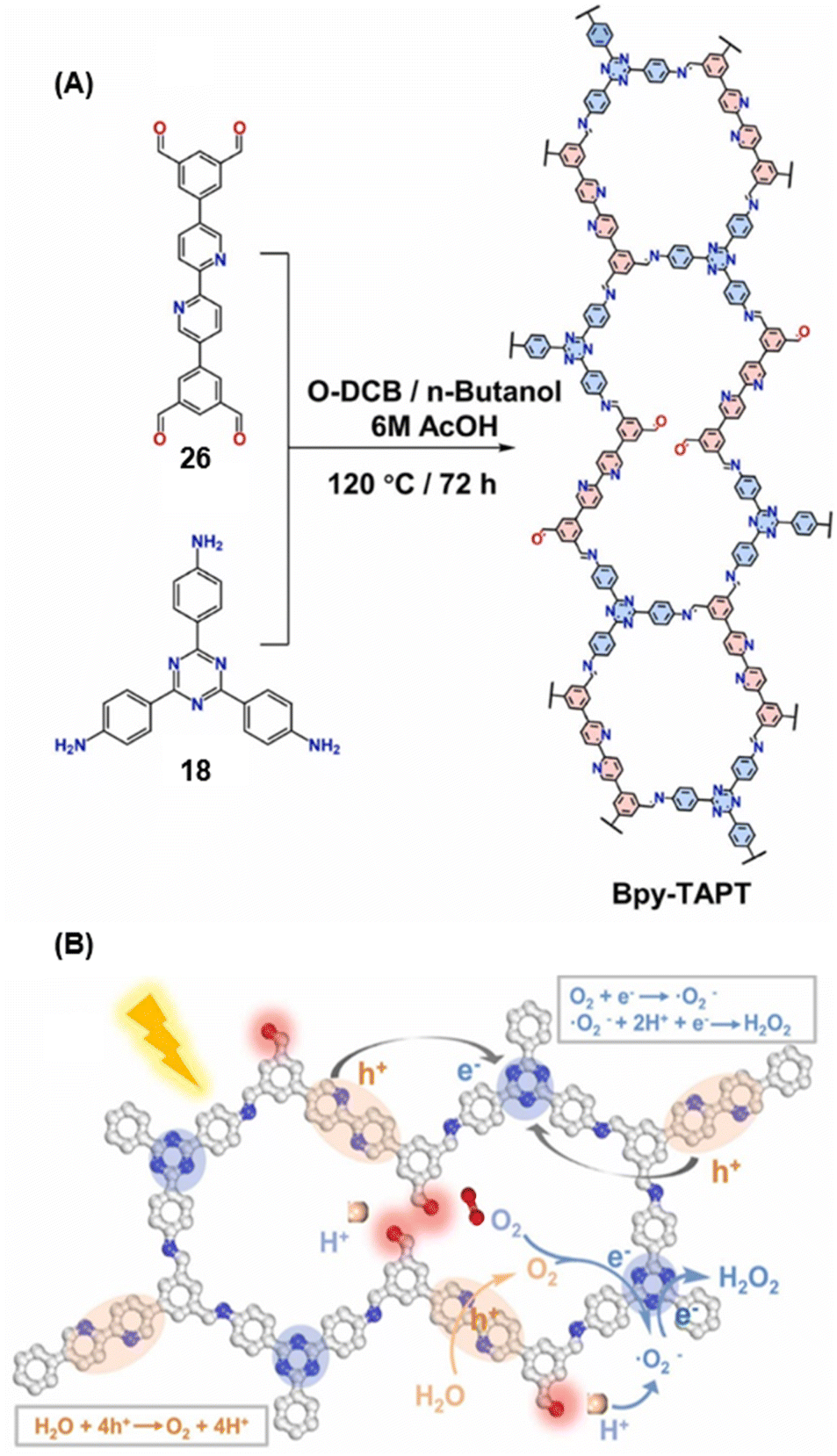 | ||
| Fig. 15 (A) Sub-stoichiometric synthesis of the COF Bpy-TAPT from the monomers 26 and 18, showing the presence of unreacted aldehyde sites. (B) Photocatalytic generation of H2O2 from water using Bpy-TAPT and its postulated mechanism. Reproduced from ref. 133 with permission from Elsevier, copyright 2023. | ||
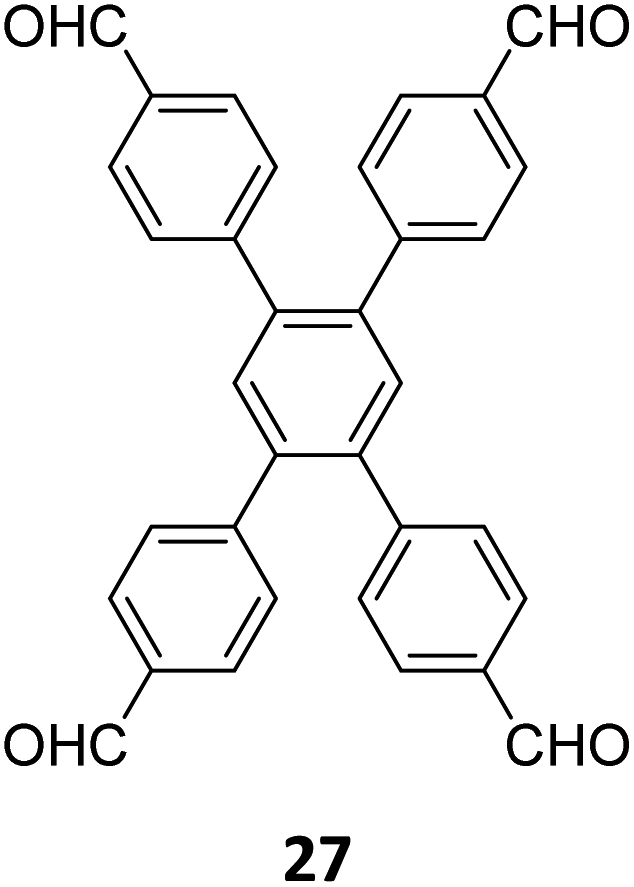
Hu and coworkers have reported two COFs TPE-TPB-A (Type-I) and TPE-TPB-B (Type-III) just by varying the solvent composition (n-butanol/mesitylene or n-butanol/dioxane), using a 1:1 molar ratio of the amine 2 and the aldehyde 27 in the presence of 6 M acetic acid for 5 days at 120 °C (Fig. 16A).152 Both the COFs exhibited high surface area (∼1000 m2 g−1), with similar water adsorption characteristics (∼450 cm3 g−1 at P/Po = 0.98). The stepwise water adsorption characteristics and comparably low water contact angle (78.9°) of TPE-TPB-B signifies the role of dangling formyl groups in making the pores hydrophilic. Experimental observations and theoretical calculations related to the energy and active sites for water adsorption confirmed that TPE-TPB-B is more hydrophilic than TPE-TPB-A due to the presence of carbonyl groups. The photocatalytic hydrogen evolution (PHE) from both the COFs was analysed using a platinum cocatalyst and ascorbic acid as a sacrificial agent. Irradiating TPE-TPB-A and TPE-TPB-B with visible light (>420 nm, 6 h) resulted in hydrogen generation at a rate of 43.53 and 510.2 μmol h−1 g−1, respectively (Fig. 16B). Both the COFs exhibited reasonably high photostability under continuous irradiation for 24 h. TPE-TPB-B displayed almost 15.3 times higher PHE rate than TPE-TPB-A (Fig. 16C), mediated by efficient electron transfer from the COF to the platinum cocatalyst.
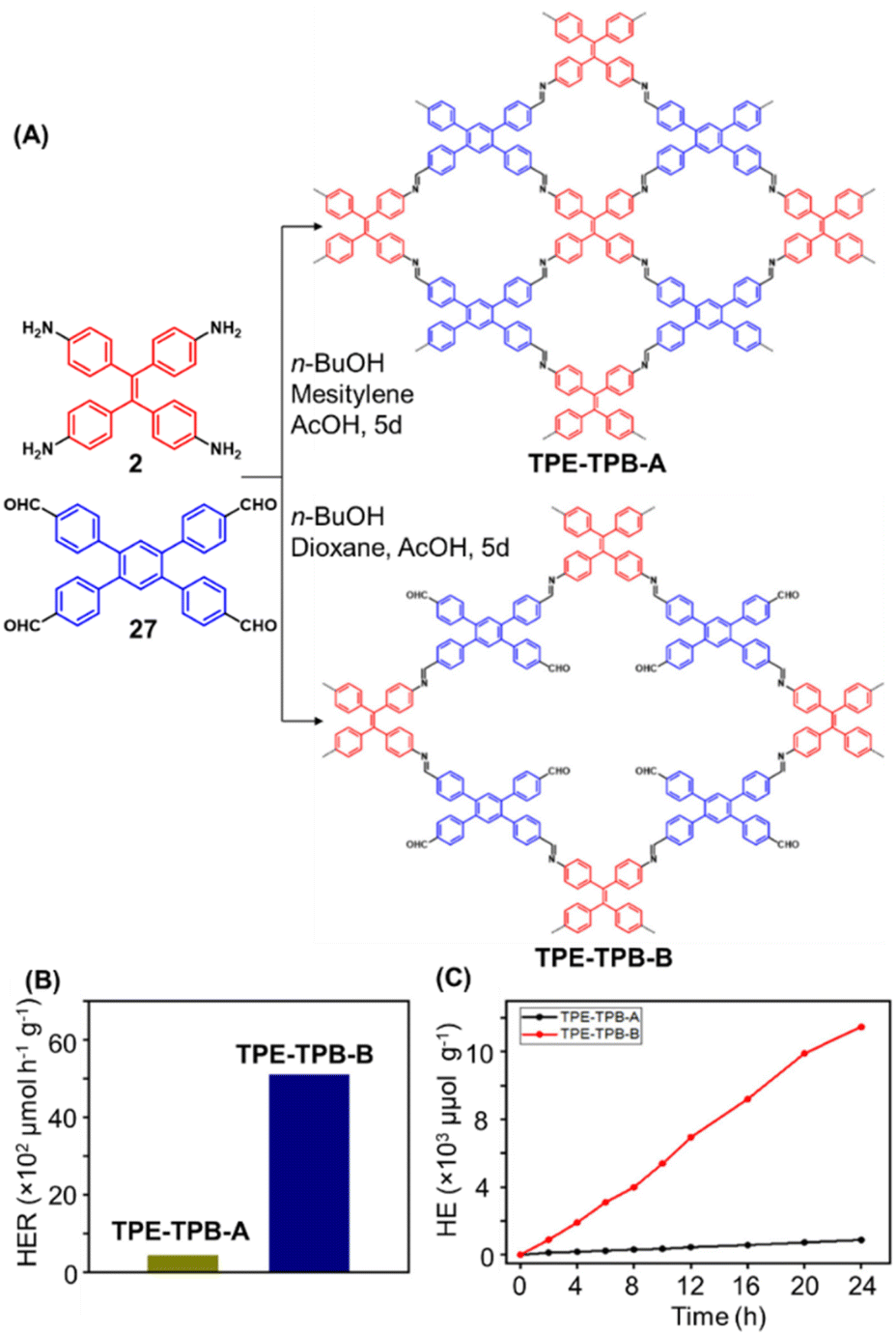 | ||
| Fig. 16 (A) Synthesis of the COFs TPE-TPB-A (type-I) and TPE-TPB-B (type-III) from the monomers 2 and 27. Unreacted aldehyde sites are shown in TPE-TPB-B. (B and C) Comparison of the photocatalytic hydrogen generation capabilities of TPE-TPB-A and TPE-TPB-B. Reproduced from ref. 152 with permission from American Chemical Society, copyright 2023. | ||
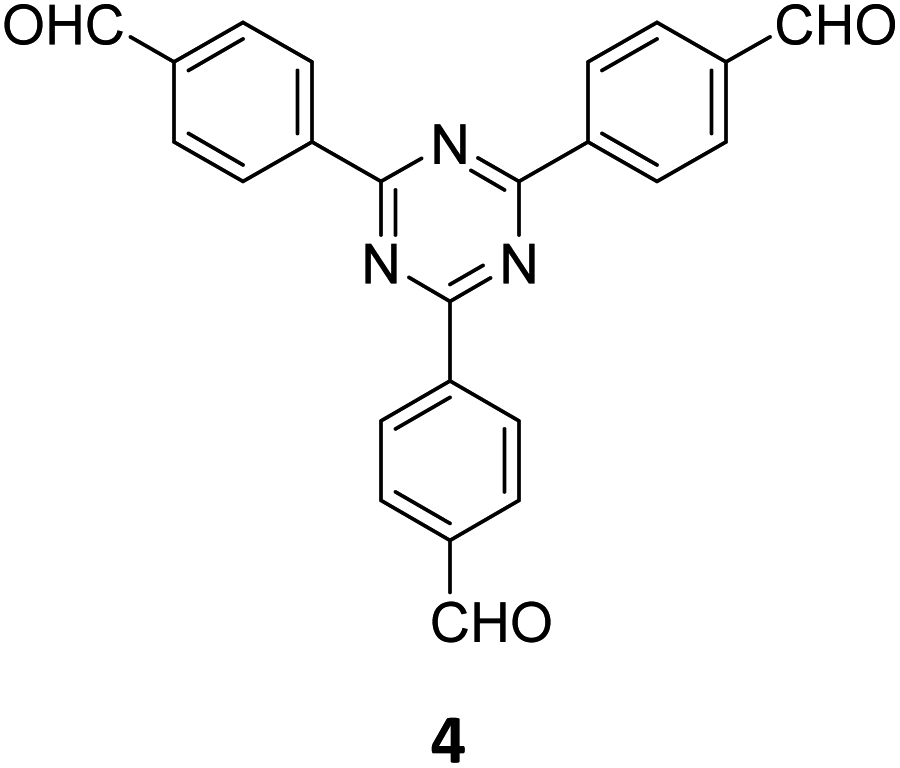
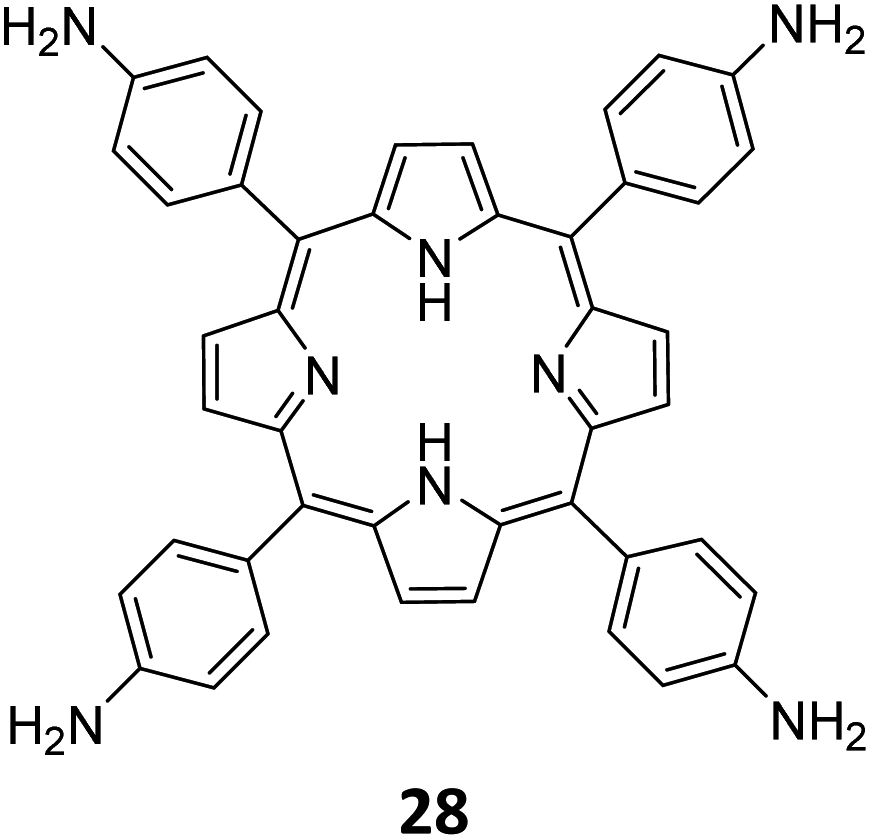
Qiu and co-workers have reported a sub-stoichiometric photoresponsive COF TAPP-TFPT using a porphyrin derivative 28 and the triazine trialdehyde 4 as monomers.153 Silver nanoparticles were then deposited over the COF via a post-synthetic modification to obtain the composite material Ag@TAPP-TFPT for the photocatalytic conversion of toxic bis(2-chloroethyl) sulfide (CEES) to non-toxic bis(2-chloroethyl) sulfoxide (Fig. 17A). The parent COF TAPP-TFPT and the composite Ag@TAPP-TFPT were evaluated for the degradation of CEES. While the oxidation of CEES was found to be complete within 15 min (t1/2 = 6.5 min), when using Ag@ TAPP-TFPT as a photocatalyst, TAPP-TFPT took 25 min for the reaction (t1/2 = 10.5 min, Fig. 17B).
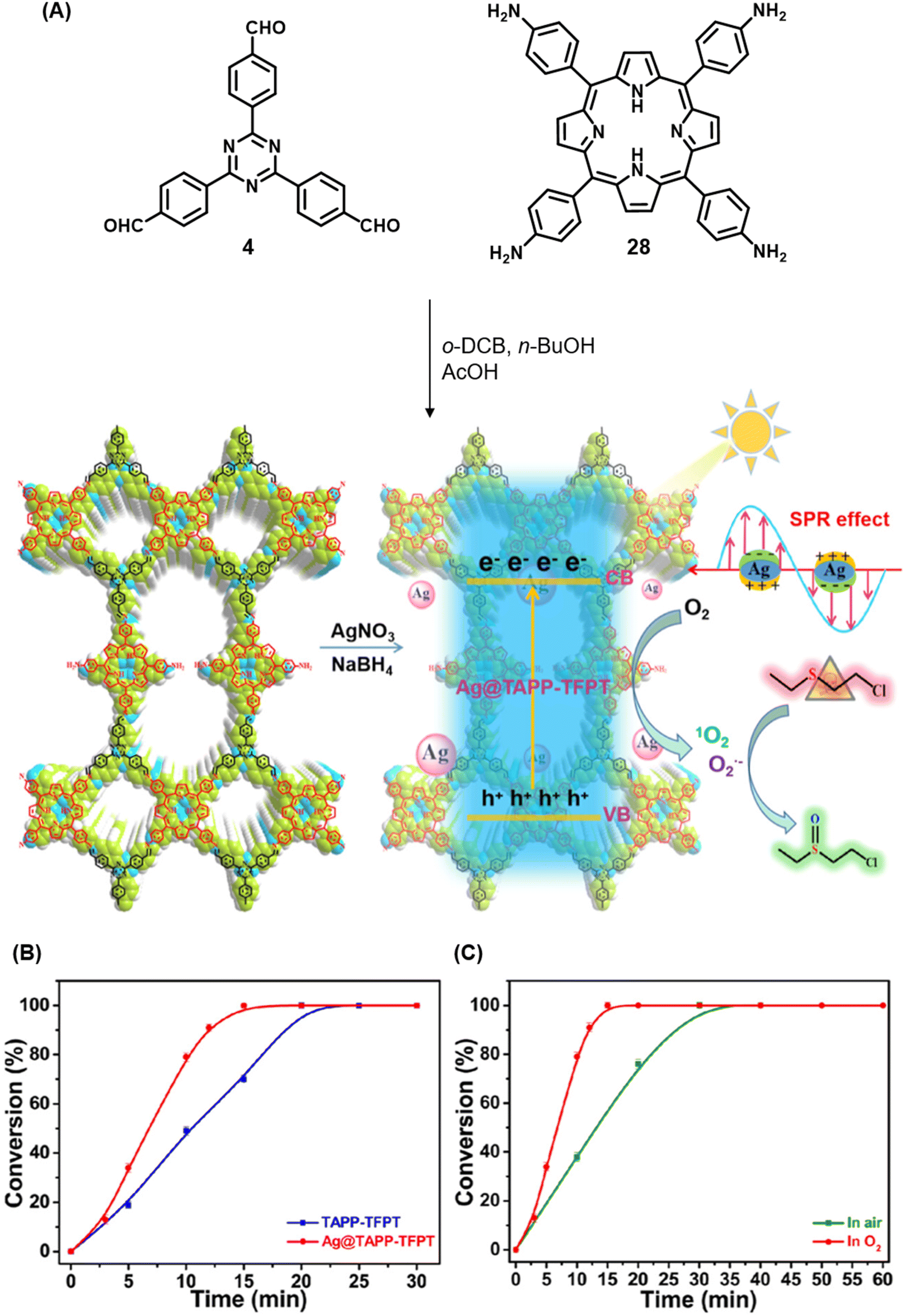 | ||
| Fig. 17 (A) Synthesis of the COFs TAPP-TFPT and Ag@TAPP-TFPT from the monomers 4 and 28. Silver nanoparticles were deposited over TAPP-TFPTvia a post-synthetic modification to obtain Ag@TAPP-TFPT. (B) Comparison of the photocatalytic efficiency of TAPP-TFPT and Ag@TAPP-TFPT in converting bis(2-chloroethyl) sulphide (CEES) to bis(2-chloroethyl) sulfoxide. (C) Comparison of the catalytic efficiency under ambient air and oxygen. Reproduced from ref. 153 with permission from American Chemical Society, copyright 2023. | ||
The catalytic efficiency was found to be two-fold higher under an oxygen atmosphere than in ambient air (Fig. 17C), with 98% conversion retained after five cycles. The strong surface plasmon resonance of silver nanoparticles along with efficient electron–hole separation in Ag@TAPP-TFPT resulted in increased absorption in the visible region with a concomitant decrease in the band gap, thereby fostering enhanced photocatalytic activity. EPR measurements confirmed that both 1O2 and O2˙− were involved in the oxidation process.
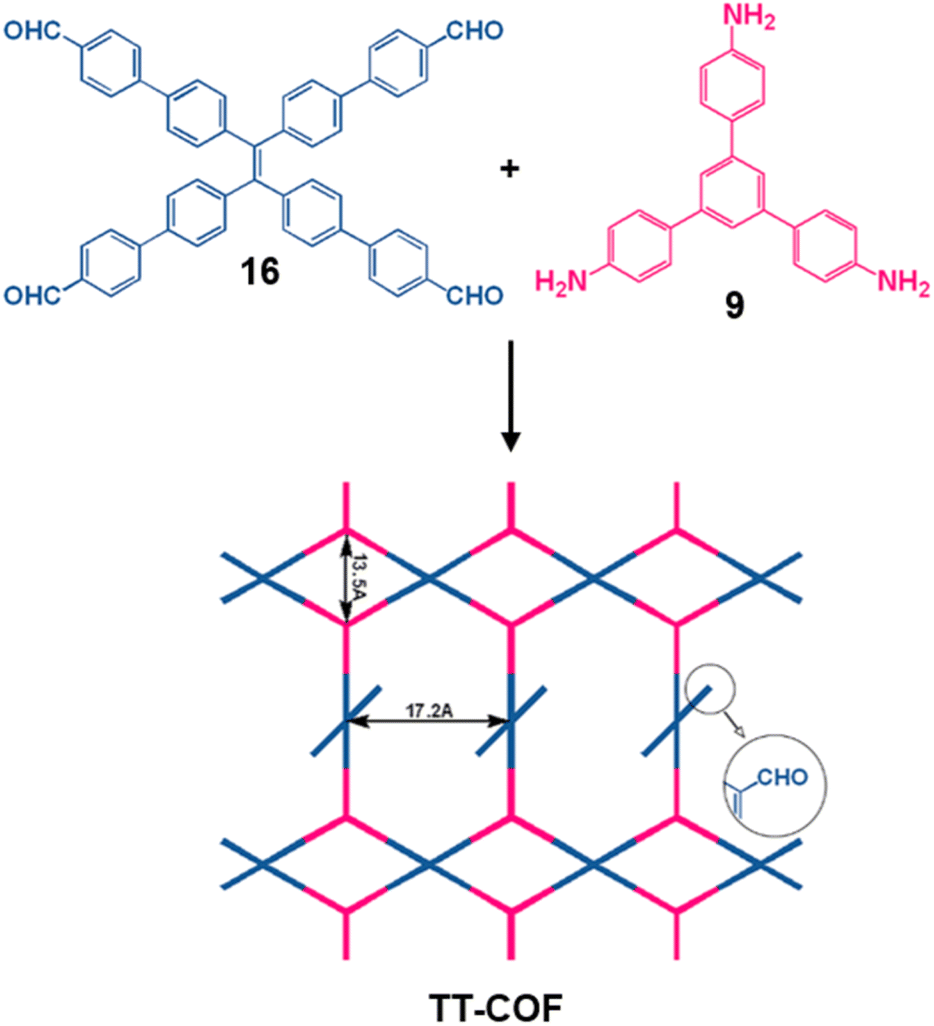 | ||
| Fig. 18 Sub-stoichiometric synthesis of the TT-COF from the monomers 16 and 9, showing the presence of unreacted aldehyde sites. Reproduced from ref. 166 with permission from Elsevier, copyright 2022. | ||
The hybrid material N6-GO@TT-COF-x (x = amount of COF, 0–8 mg) was prepared by spreading TT-COF over dispersed GO on a Nylon-66 substrate. Exposed formyl groups were found to facilitate the dispersion of the COF on GO, and large area defect-free continuous layers were fabricated via hot pressing. Owing to the presence of active sites and vertical channels stacked by regular pores, TT-COF exhibited a large flux of 309.99 L m−2 h−1 bar−1 and high rejection of 99.5% for cationic dyes with molecular weight above 500 Da. The membrane was unaffected by acidic or basic conditions and could be reused for many cycles without any detectable loss in crystallinity or activity.
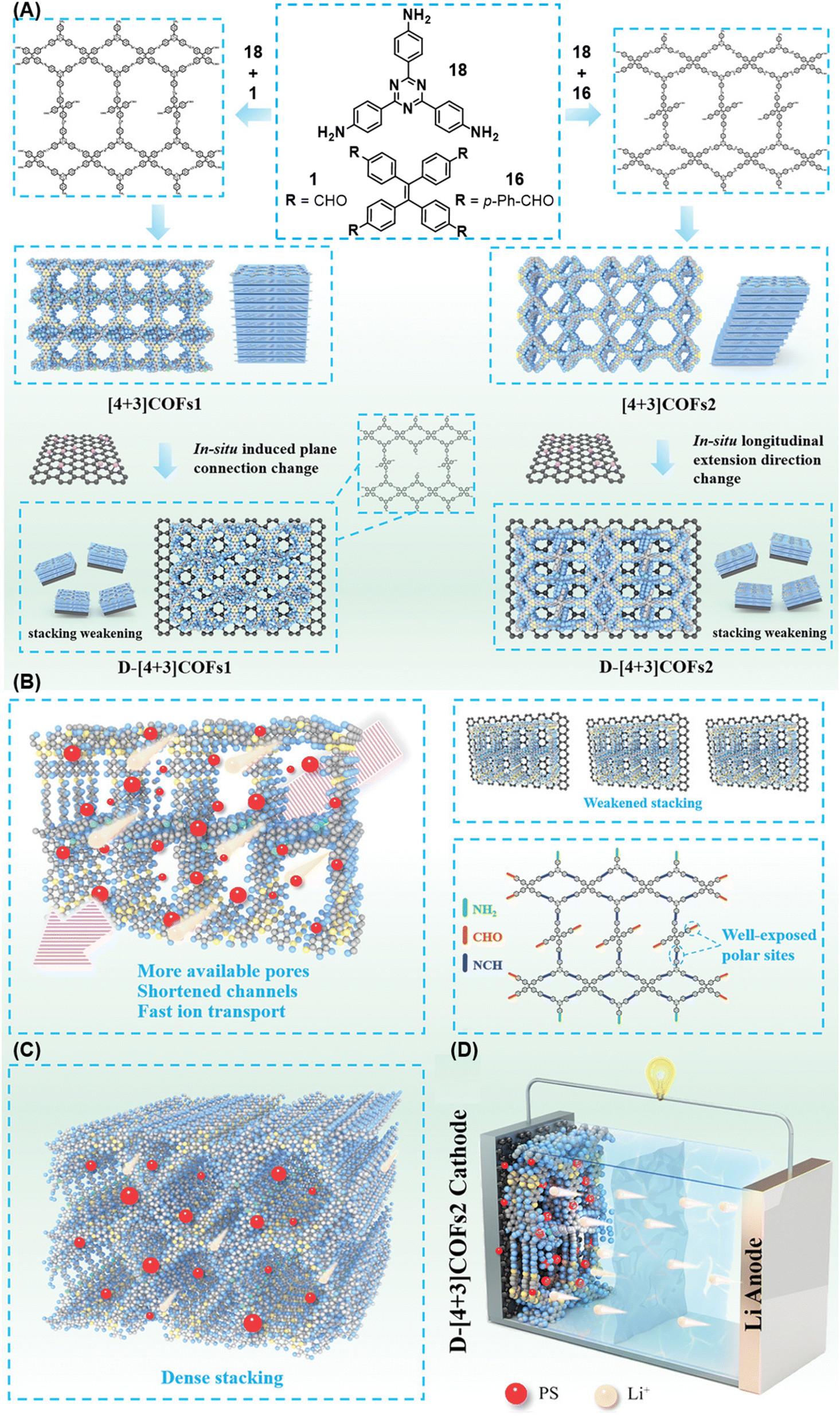 | ||
| Fig. 19 Schematic representation of (A) the sub-stoichiometric synthesis of [4+3] and D-[4+3] COFs and the topology deformation of D-[4+3] COFsvia the introduction of graphene oxide during synthesis, the (B) reduced and (C) dense stacking in [4+3] and D-[4+3] COFs, respectively, and (D) the Li–S battery assembled with D-[4+3] COFs. Reproduced from ref. 167 with permission from Wiley, copyright 2023. | ||
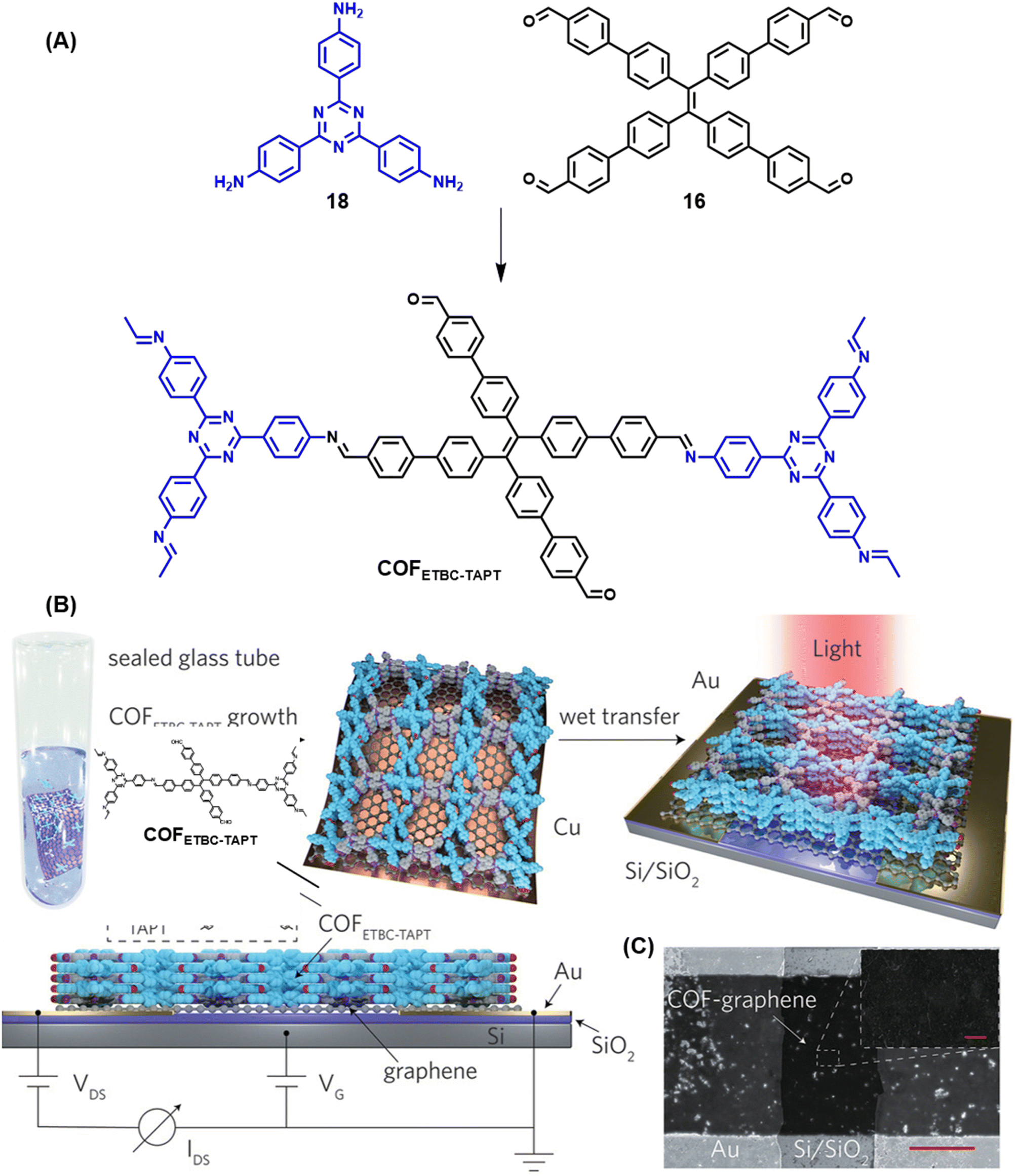 | ||
| Fig. 20 Schematic representations of (A) the sub-stoichiometric synthesis of COFETBC–TAPT from the monomers 18 and 16, and (B) oriented growth of COFETBC–TAPT on Cu-supported graphene and its wet transfer on to Au electrodes on a Si/SiO2 substrate (top) and the side view of the photodetector set-up (bottom). (C) The SEM images of the photodetector constructed using COFETBC–TAPT (scale bar = 20 μm) and the corresponding enlarged image (scale bar = 1 μm). Reproduced from ref. 168 with permission from Wiley, copyright 2020. | ||
A clean thin film structure of COFETBC–TAPT was confirmed by scanning electron microscopy (Fig. 20C). The COFETBC–TAPT–graphene heterostructure was characterized by a p-type architecture and a photocurrent of 1.79 μA was observed under a low illumination power of 0.67 μW cm−2. A photoresponsivity as high as 3.2 × 107 A W−1 for a light power of 0.1 pW (3.3 nW cm−2), with an external quantum efficiency of 8.5 × 109% and a fast response time of 1.14 ms, with cycling stabilities exceeding 800 on–off cycles, was observed for the COF hybrid. Furthermore, polar molecules like ammonia and ethanol were found to chemically interact with the free aldehyde groups in the pores of COFETBC–TAPT and had a corresponding impact on the photoelectric performance of the device, with a sensitivity of 16.2% and 5.8% for 1% NH3 and 1% ethanol vapour in air, respectively.
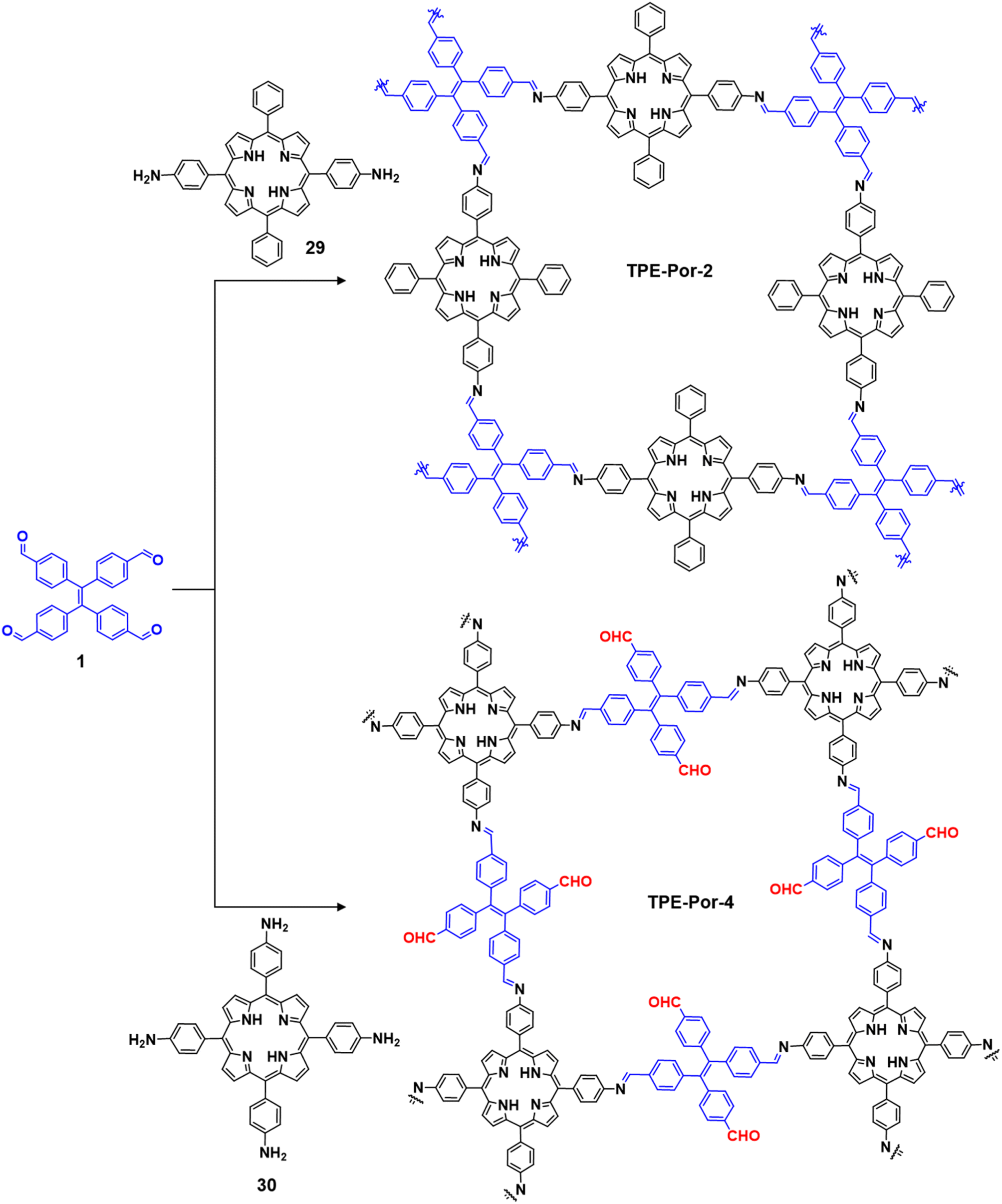 | ||
| Fig. 21 Synthesis of the porphyrin-based COFs TPE-Por-2 and TPRE-Por-4. | ||
4. Conclusions
Apart from tunable porosity and high physicochemical stability, their capability to form new topologies and accommodate diverse active functional groups within their pores makes these COFs interesting in terms of new properties and diverse applications. Selective transformations of seemingly similar functional groups during the synthesis of imine-linked COF were effected via protection, post synthetic modification, ligand exchange and site-selective synthesis. However, sub-stoichiometric synthesis of Type III COFs that shows a high degree of tolerance to chemically equivalent reactive sites and yields the products via orthogonal reactions is a recent development. The unusual selectivity observed during sub-stoichiometric synthesis of COFs not only widens their applications but assists in obtaining a better understanding of the general pathways behind COF formation. The literature reports on COFs synthesized through the sub-stoichiometric approach, including their topologies, linkages and applications are summarized in Table 1. Though several reports on the synthesis and applications of Type-III COFs have emerged over the last five years, extension to other linkages is still in its infancy. Therefore, we believe that this approach provides enough opportunities and challenges for focussed research in terms of new linkages, monomeric units, topologies, and applications.| Linker 1 | Linker 2 | Topology | Application | Ref. |
|---|---|---|---|---|
|
|
Rhombic | CO2 absorption | 116 |
|
tth defect | Photocatalysis | 150 | |
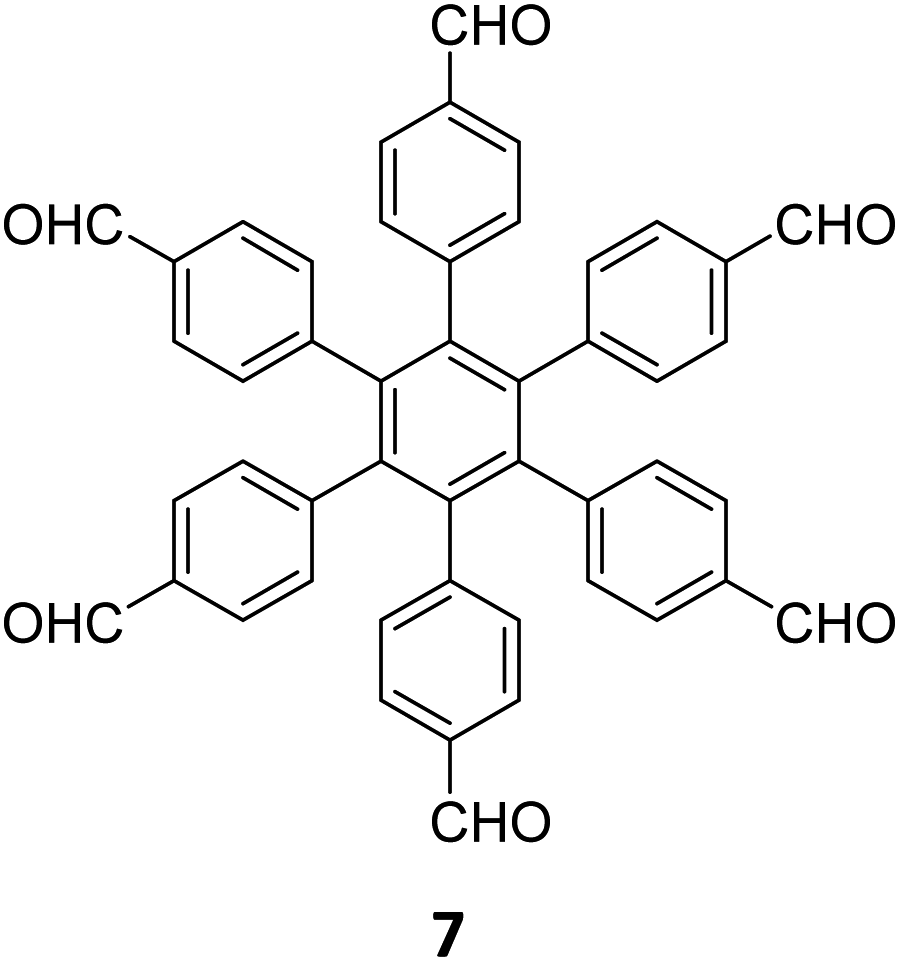
|
sql | 134 | ||
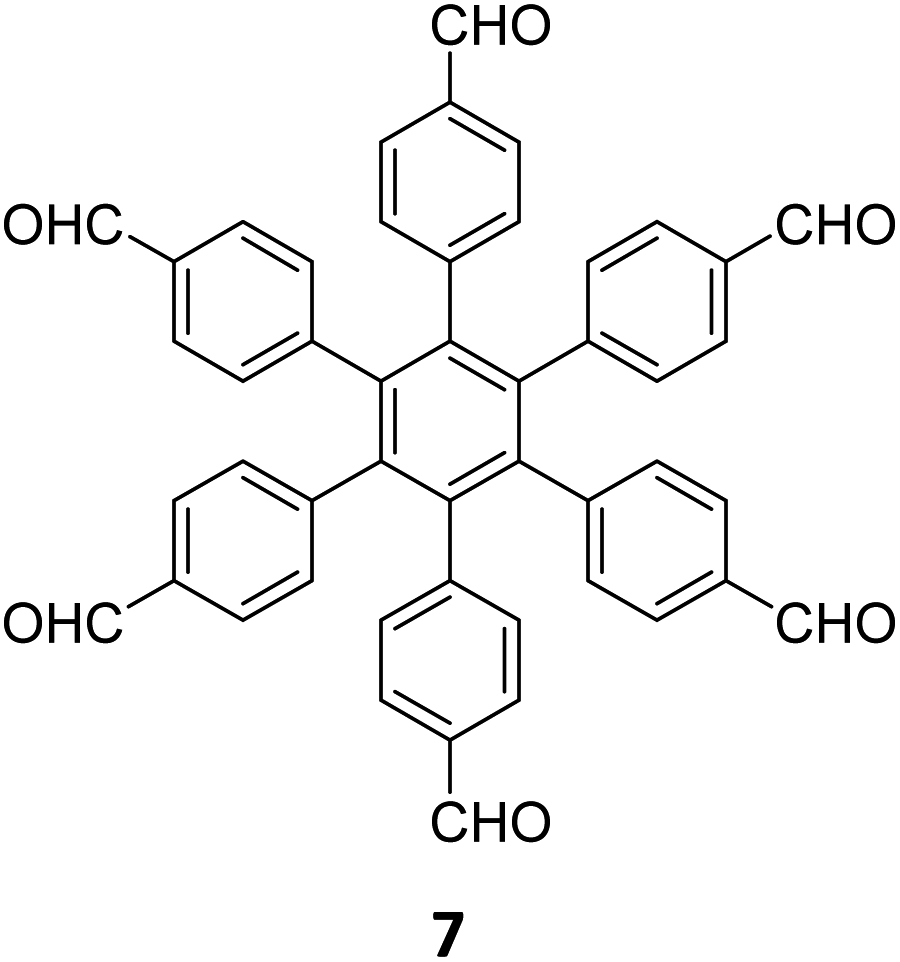
|
tth defect | 117 | ||
|
Rhombic | Photocatalysis | 152 | |
|
|
bex | Photocatalysis | 150 |
| Li–S battery | 167 | |||
|
bex | Water sensing and harvesting | 148 | |
|
bex | Photocatalysis | 150 | |
| Photodetector | 168 | |||
| Li–S battery | 167 | |||
|
bex | Photocatalysis | 37 | |
|
bex | Photocatalysis | 133 | |
|
|
bex | CO2 adsorption | 34 |
|
Catalysis | |||
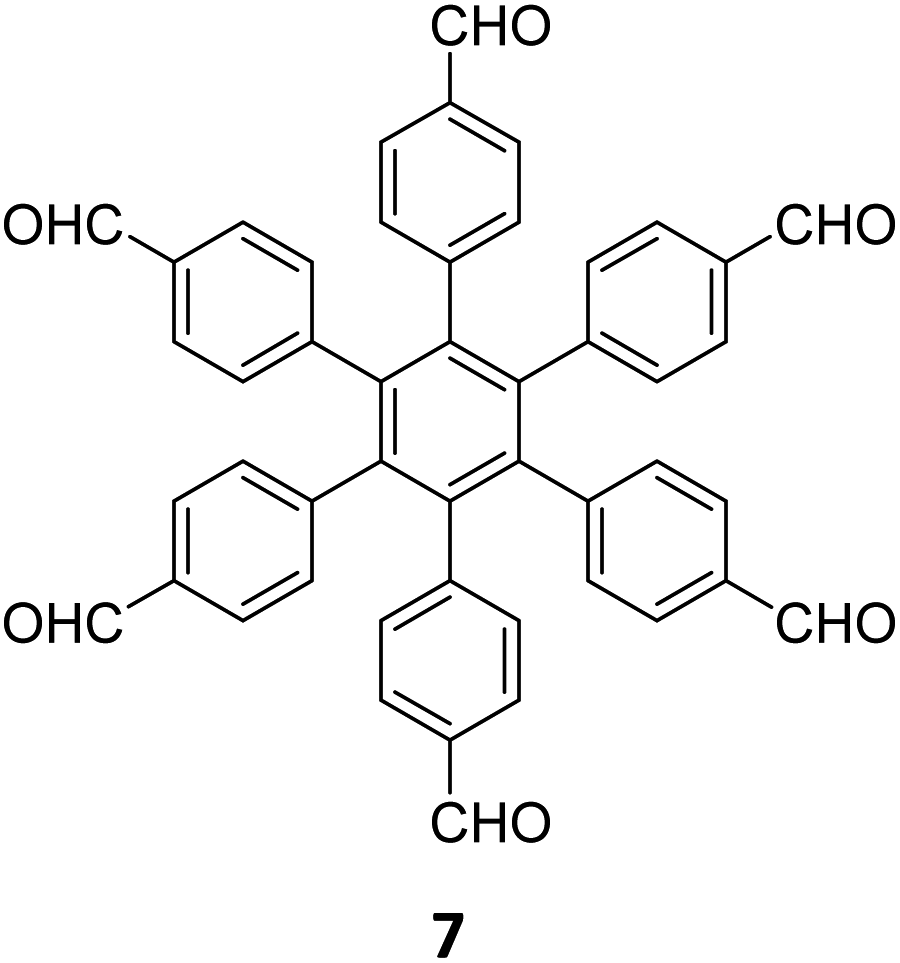
|
|
pts | Gas adsorption and separation | 118 |
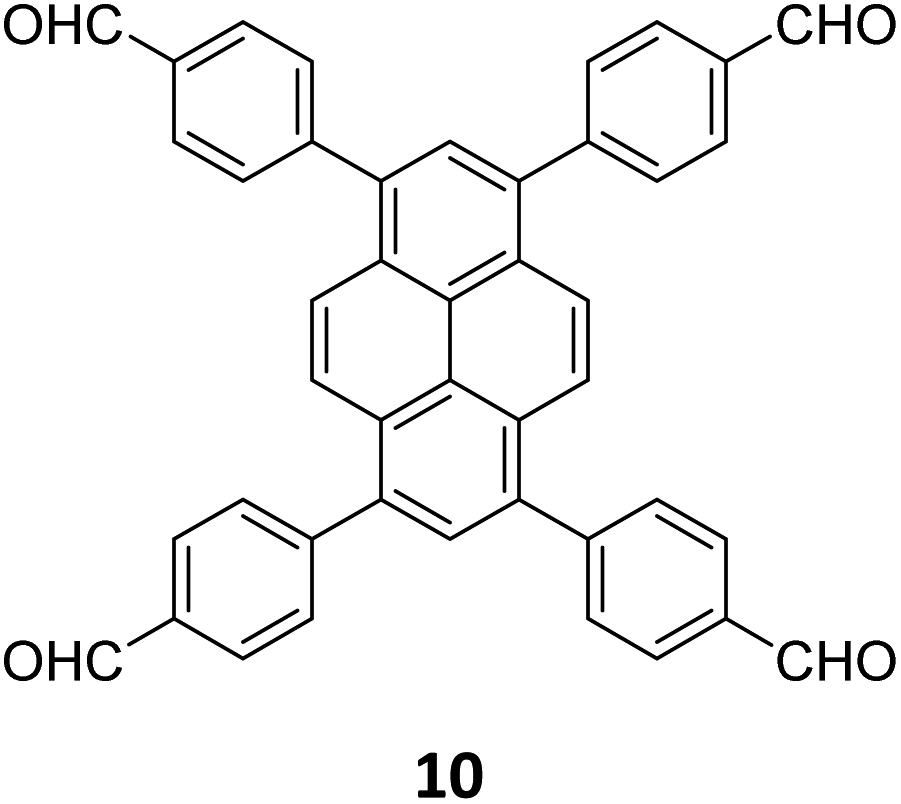
|
hcb | |||
|
|
bex | Photocatalysis | 151 |
|
1D COF | Dual linked 2D COFs | 35 | |
|
Rhombic | Iodine adsorption | 149 | |
|
|
dia | Heterogeneous | 36 |
| Catalysis | ||||
|
|
Rhombic | CO2 and H2O absorption | 138 |
|
bex | Dye removal | 166 | |
|
|
bex | Photocatalysis | 153 |
|
|
Rhombic | pH response | 169 |
5. Future prospects
While COFs, in general, and sub-stoichiometric COFs, in particular, have shown a wide range of applications emanating from their structural attributes, Type III COFs have the additional advantage of having the periodic presence of unreacted heteroatomic functional groups. Such active functional groups benefit from the possibilities for further chemical modifications, specific interactions with ions/molecules, catalysis, energy storage, etc., while retaining long-range order and crystallinity, robustness, and porosity. These unique prospects of type III COFs that retain the advantages of stoichiometric COFs lead to improved properties and applications that are otherwise difficult in conventional COFs.The sub-stoichiometric approach is a synthetic tool in terms of generating more functionalities in COFs aiming at unique properties and applications. Although Type III COFs present an interesting scenario for further exploration in terms of practical applicability, the field is still in its infancy.170 In this section, we discuss the possible ways to expand the use of monomers, linkages and topologies, multicomponent approaches, polymorphism, isomerism, modifications in physicochemical properties, practical applicability as a synthetic tool, and new avenues for applications.
5.1 Extending to new linkages and monomers
Type III COFs are generally stoichiometry-controlled products, and reaction conditions including solvents and temperature are as critical as the choice of monomers. While the majority of the reports are based on Schiff base chemistry leading to imine-linked Type III COFs, there is only a single report with an olefin linkage.151 This observation suggests that the sub-stoichiometric approach can be extended to new or existing linkages, resulting in COFs with periodic arrays of various functional groups. Covalent and non-covalent chemistry of the active functional groups in these COFs allows for further structural diversification and improvement in properties. This also calls for the identification of new monomeric units for the generation of COFs with new and stable linkages.5.2 Exploration of new topologies and multicomponent reactions
Though several reports hypothesized COF formation mechanisms, unknown aspects behind covalent bond crystallisation can be better revealed by exploring Type III COFs. The structural and functional complexities are enhanced in COFs synthesized via the sub-stoichiometric approach. While a good number of existing reports on sub-stoichiometric COFs are hypothesis-driven in terms of structural elucidation, attempts using combinations of monomers of various symmetry combined with the efforts on crystallography, theoretical modelling, and X-ray and electron diffraction techniques are expected to result in reliable predictions of the resulting unconventional topologies.171While the realization of new topologies and elucidating the exact structure of COFs may not be straightforward, bex topology alone has a lot to offer.34 As discussed above, this field has so far witnessed the use of the combination of only tetratopic and tritopic/tetratopic/hexatopic monomers to yield 2D and 3D COFs. Use of other tritopic or tetratopic monomers expected to yield only bex topology makes theoretical predictions and structural elucidations facile. Working with defined topologies also helps to understand the role of flexible cores and bifunctional group containing monomers in deciding the final structure of the COFs.
In addition, synthesis of 1D COFs via sub-stoichiometric approaches and subsequent crosslinking to the corresponding 2D architecture, yielding dual linked COFs, is yet another less explored but interesting area for investigation.35 These 1D chains can be effectively wrapped over nanotubes, or other nanostructures through covalent linkages, which could possibly yield new hybrid materials with interesting properties. Extending such approaches to 2D COFs results in diverse porous materials or hybrids with unprecedented structural and functional complexities.
Lotsch and co-workers have also demonstrated that replacing a tetratopic linker with a ditopic linker can generate all-linked analogues of sub-stoichiometric COFs.34 Such an approach is yet to be attempted in other sub-stoichiometric COFs. This approach could possibly be viewed as a strategy to construct all-linked bex topology with [4 + 3 + 2] linkages or sub-stoichiometric COFs with [4 + 3 + 4] linkages. In the latter case, one of the tetratopic linkers selectively behaves as a ditopic linker in generating sub-stoichiometric COFs with two different tetratopic linkers in the framework. Such attempts are envisioned to result in COFs with tuneable photochemical and electro-optical characteristics.
5.3 Isomerism and polymorphism
While reticular chemistry has led to significant progress in the development of framework materials, practical realization of isomeric COFs has not been explored extensively. Dimensional isomers are generally constructed from the same building blocks under different reaction conditions, but extend in different dimensions (2D or 3D).172 Different orientations of the molecular building blocks in these isomeric frameworks alter their physicochemical properties and functions. Isomerization in COFs is a recently realized phenomenon where the influence of solvent and/or temperature yielding different isomers and can also be extended to type-III COFs.173 Conversion of a kinetic product to a stable thermodynamic product suggests that transformation of sub-stoichiometric COFs to all-linked COFs (Type III to Type I) could also possibly be achieved particularly for rhombic topologies. This two-step process may yield better surface area and/or crystallinity, which is yet to be demonstrated. This could also provide a new synthetic methodology that attempts to impart kinetic/thermodynamic stability to COFs or to achieve both types of COFs in a continuous flow system174,175 where the isomers are obtained just by controlling the monomer feed ratio, solvent compositions or temperature. The sub-stoichiometric approach is probably the most practical strategy for the creation of such isomers of COFs using flexible monomers, with a multitude of active functionalities within the framework structure, though such reports are rarely found.Since the first report on sub-stoichiometric COFs, there are only three instances of polymorphism reported.116,149,152 These reports on Type III COFs involve the use of tetratopic monomers and rhombic topologies. In these attempts, COFs with unreacted carbonyls were realized just by changing the solvent(s) rather than the stoichiometry of the monomers. Unlike other topologies dealt with in sub-stoichiometric COFs, the rhombic topology allows the exclusive generation of polymorphic COFs using different solvent combinations, while the ratio of starting monomeric linkers remains unaltered. Though these are not strictly sub-stoichiometric syntheses where variations in stoichiometry of the monomers result in unreacted functional groups in the COF structure, we have deliberately included these reports in this review, since the resulting COFs are indeed Type III with active functional groups.
5.4 As a synthetic tool
The reversible nature of the linkages in COFs can be utilized for selective functional group interconversions. Unreacted functional groups of sub-stoichiometric COFs can undergo post synthetic modifications. The resulting COFs in principle can be promoted to undergo transamination reactions to yield an all-linked COF and diagonally functionalized aldehydes or amines. This approach, if realized, is envisioned to provide a useful synthetic tool to generate organic molecules which are difficult to be obtained via selective functionalization in their monomeric states. Moreover, reconstruction strategies, as reported by Cooper and co-workers, via “the pre-organization of monomers using a reversible and removable covalent tether, followed by confined polymerization”176 present a yet another promising avenue for sub-stoichiometric synthesis towards realizing stable and crystalline COFs with active functionalities for further modifications.5.5 Avenues for new applications
Broadening the scope and applicability of COFs demands unique structural diversity and pertinent functionalities that are exigent via conventional synthetic approaches. Though post-synthetic modification provides an option to achieve such diversification of COF architectures, the choices are limited by strictly orthogonal reaction requirements. We have elaborated the applications of Type III COFs in Section 3.3 of this article. While these are not an exhaustive list of applications of this class of COFs, we expect that concerted efforts from the research community working on these materials with emerging linkages, monomers and topologies combined with advancements in structural characterization tools would result in a large realm of applications in the near future.Author contributions
SV and SS wrote the manuscript. AA and SS conceptualized and supervised the project. AA and SS reviewed and edited the manuscript. All authors approved the submission of the reviewed draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SV thanks CSIR for a senior research fellowship. This work was supported by DST-SERB, Government of India (SS, Ramanujan Fellowship, SB/S2/RJN-058/2016 and AA, J. C. Bose National Fellowship, SB/S2/JCB-11/2014). The authors acknowledge the funding support from CSIR (MLP 75), KSCSTE, Government of Kerala (GAP 164839) and DST, Government of India (Nanomission – GAP 162939, IC-MAP – GAP 163939).Notes and references
- A. R. Brill, E. Koren and G. de Ruiter, J. Mater. Chem. C, 2021, 9, 11569–11587 RSC.
- M. Franz, S. Chandola, M. Koy, R. Zielinski, H. Aldahhak, M. Das, M. Freitag, U. Gerstmann, D. Liebig, A. K. Hoffmann, M. Rosin, W. G. Schmidt, C. Hogan, F. Glorius, N. Esser and M. Dähne, Nat. Chem., 2021, 13, 828–835 CrossRef CAS PubMed.
- Take aim (Editorial), Nat. Chem., 2012, 4, 955 Search PubMed.
- P. M. Tadross and E. N. Jacobsen, Nat. Chem., 2012, 4, 963–965 CrossRef CAS PubMed.
- A. A. Bastian, A. Marcozzi and A. Herrmann, Nat. Chem., 2012, 4, 789–793 CrossRef CAS PubMed.
- S. K. Silverman, Nat. Chem., 2012, 4, 774–775 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 1, 193–205 CrossRef CAS PubMed.
- Z. Huang and G. Dong, Acc. Chem. Res., 2017, 50, 465–471 CrossRef CAS PubMed.
- B. C. Wilcock, B. E. Uno, G. L. Bromann, M. J. Clark, T. M. Anderson and M. D. Burke, Nat. Chem., 2012, 412(4), 996–1003 CrossRef PubMed.
- P. A. Lichtor and S. J. Miller, Nat. Chem., 2012, 412(4), 990–995 CrossRef.
- C. Hui, F. Chen, F. Pu and J. Xu, Nat. Rev. Chem, 2019, 32(3), 85–107 CrossRef.
- T. Newhouse, P. S. Baran and R. W. Hoffmann, Chem. Soc. Rev., 2009, 38, 3010–3021 RSC.
- G. Sartori, R. Ballini, F. Bigi, G. Bosica, R. Maggi and P. Righi, Chem. Rev., 2004, 104, 199–250 CrossRef CAS PubMed.
- I. S. Young and P. S. Baran, Nat. Chem., 2009, 13(1), 193–205 CrossRef PubMed.
- W. Lu, Z. Wei, Z. Y. Gu, T. F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle, M. Bosch and H. C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, J. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 1–15 Search PubMed.
- N. Hanikel, M. S. Prévot and O. M. Yaghi, Nat. Nanotechnol., 2020, 15, 348–355 CrossRef CAS PubMed.
- Z. Ji, H. Wang, S. Canossa, S. Wuttke and O. M. Yaghi, Adv. Funct. Mater., 2020, 30, 2000238 CrossRef CAS.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, eaal1585 CrossRef PubMed.
- N. Huang, P. Wang and D. L. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- S. J. Lyle, P. J. Waller and O. M. Yaghi, Trends Chem., 2019, 1, 172–184 CrossRef CAS.
- H. Wang, Y. Yang, X. Yuan, W. L. Teo, Y. Wu, L. Tang and Y. Zhao, Mater. Today, 2022, 53, 106–133 CrossRef CAS.
- A. M. Evans, M. J. Strauss, A. R. Corcos, Z. Hirani, W. Ji, L. S. Hamachi, X. Aguilar-Enriquez, A. D. Chavez, B. J. Smith and W. R. Dichtel, Chem. Rev., 2022, 122, 442–564 CrossRef CAS PubMed.
- X. Guan, F. Chen, S. Qiu and Q. Fang, Angew. Chem., Int. Ed., 2023, 62, e202213203 CrossRef CAS PubMed.
- J. Jiang, Y. Zhao and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 3255–3265 CrossRef CAS PubMed.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS PubMed.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050–5091 CrossRef CAS PubMed.
- L. Deng, Z. Ding, X. Ye and D. Jiang, Acc. Mater. Res., 2022, 3, 879–893 CrossRef CAS.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- X. Huang, C. Sun and X. Feng, Sci. China: Chem., 2020, 63, 1367–1390 CrossRef CAS.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- H. Ding, A. Mal and C. Wang, Mater. Chem. Front., 2020, 4, 113–127 RSC.
- T. Banerjee, F. Haase, S. Trenker, B. P. Biswal, G. Savasci, V. Duppel, I. Moudrakovski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2019, 10, 2689 CrossRef PubMed.
- H. L. Nguyen, C. Gropp and O. M. Yaghi, J. Am. Chem. Soc., 2020, 142, 2771–2776 CrossRef CAS PubMed.
- C. Xu, J. Lin, D. Yan, Z. Guo, D. J. Austin, H. Zhan, A. Kent and Y. Yue, ACS Appl. Nano Mater., 2020, 3, 6416–6422 CrossRef CAS.
- M. Lu, M. Zhang, J. Liu, T.-Y. Yu, J.-N. Chang, L.-J. Shang, S.-L. Li and Y.-Q. Lan, J. Am. Chem. Soc., 2022, 144, 1861–1871 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- L. Bourda, C. Krishnaraj, P. Van Der Voort and K. Van Hecke, Mater. Adv., 2021, 2, 2811–2845 RSC.
- C. Gropp, S. Canossa, S. Wuttke, F. Gándara, Q. Li, L. Gagliardi and O. M. Yaghi, ACS Cent. Sci., 2020, 6, 1255–1273 CrossRef CAS PubMed.
- K. T. Tan, S. Ghosh, Z. Wang, F. Wen, D. Rodríguez-San-Miguel, J. Feng, N. Huang, W. Wang, F. Zamora, X. Feng, A. Thomas and D. Jiang, Nat. Rev. Methods Primers, 2023, 3, 1 CrossRef CAS.
- H. Lyu, Z. Ji, S. Wuttke and O. M. Yaghi, Chem, 2020, 6, 2219–2241 CAS.
- F. Haase and B. V. Lotsch, Chem. Soc. Rev., 2020, 49, 8469–8500 RSC.
- J. Hu, Z. Huang and Y. Liu, Angew. Chem., Int. Ed., 2023, 62, e202306999 CrossRef CAS PubMed.
- L. Bourda, C. Krishnaraj, P. Van Der Voort and K. Van Hecke, Mater. Adv., 2021, 2, 2811–2845 RSC.
- A. R. Bagheri and N. Aramesh, J. Mater. Sci., 2021, 56, 1116–1132 CrossRef CAS.
- H. S. Sasmal, A. Kumar Mahato, P. Majumder and R. Banerjee, J. Am. Chem. Soc., 2022, 144, 11482–11498 CrossRef CAS PubMed.
- S. Karak, K. Dey and R. Banerjee, Adv. Mater., 2022, 34, 2202751 CrossRef CAS PubMed.
- S. Xu, M. Richter and X. Feng, Acc. Mater. Res., 2021, 2, 252–265 CrossRef CAS.
- F. Guo, W. Zhang, S. Yang, L. Wang and G. Yu, Small, 2023, 2207876 CrossRef CAS PubMed.
- Q. Guan, L. Zhou and Y. Dong, J. Am. Chem. Soc., 2023, 145, 1475–1496 CrossRef CAS PubMed.
- H. Yazdani, S. E. Hooshmand and R. S. Varma, Org. Chem. Front., 2022, 9, 4178–4191 RSC.
- Y. Yusran, X. Guan, H. Li, Q. Fang and S. Qiu, Natl. Sci. Rev., 2020, 7, 170–190 CrossRef CAS PubMed.
- K. S. Song, P. W. Fritz and A. Coskun, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- X. Wang, H. Liu, J. Zhang and S. Chen, Polym. Chem., 2023, 14, 1293–1317 RSC.
- H. Li, A. Dilipkumar, S. Abubakar and D. Zhao, Chem. Soc. Rev., 2023, 52, 6294–6329 RSC.
- B. Chen, H. Xie, L. Shen, Y. Xu, M. Zhang, M. Zhou, B. Li, R. Li and H. Lin, Small, 2023, 19, 2207313 CrossRef CAS PubMed.
- Y. Yuan, K. T. Bang, R. Wang and Y. Kim, Adv. Mater., 2023, 35, 1–26 Search PubMed.
- C. Yuan, Z. Wang, W. Xiong, Z. Huang, Y. Lai, S. Fu, J. Dong, A. Duan, X. Hou, L. Yuan and Y. Cui, J. Am. Chem. Soc., 2023, 145, 18956–18967 CrossRef CAS PubMed.
- L. Liu, Y. Hu, S. Huang, Y. Jin, J. Cui, W. Gong and W. Zhang, Chem. Sci., 2021, 12, 13316–13320 RSC.
- L. Wei, T. Sun, Z. Shi, Z. Xu, W. Wen, S. Jiang, Y. Zhao, Y. Ma and Y. B. Zhang, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Z. Meng and K. A. Mirica, Chem. Soc. Rev., 2021, 50, 13498–13558 RSC.
- T. Skorjanc, D. Shetty and M. Valant, ACS Sens., 2021, 6, 1461–1481 CrossRef CAS PubMed.
- Y. Qian, J. Li, M. Ji, J. Li, D. Ma, A. Liu, Y. Zhao and C. Yang, Front. Chem., 2022, 10, 1–7 Search PubMed.
- Y. Wei, W. Yang and H. Guo, ChemistrySelect, 2023, 8, e202301828 CrossRef CAS.
- J. Yu, L. Luo, H. Shang and B. Sun, Biosensors, 2023, 13, 636 CrossRef CAS PubMed.
- R. Xue, Y.-S. Liu, S.-L. Huang and G.-Y. Yang, ACS Sens., 2023, 8, 2124–2148 CrossRef CAS PubMed.
- E. Martínez-periñán, M. Martínez-fernández, J. L. Segura and E. Lorenzo, Sensors, 2022, 22, 1–29 CrossRef PubMed.
- X. Li and K. P. Loh, ACS Mater. Lett., 2019, 1, 327–335 CrossRef CAS.
- H. Salemi, M. Debruyne, V. Van Speybroeck, P. Van Der Voort, M. D’hooghe and C. V. Stevens, J. Mater. Chem. A, 2022, 10, 20707–20729 RSC.
- E. Nikkhoo, S. Mallakpour and C. M. Hussain, New J. Chem., 2023, 47, 6765–6788 RSC.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- H. Chen, H. S. Jena, X. Feng, K. Leus and P. van der Voort, Angew. Chem., Int. Ed., 2022, 61, e202204938 CrossRef CAS PubMed.
- T. He and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef CAS PubMed.
- K. Prakash, B. Mishra, D. D. Díaz, C. M. Nagaraja and P. Pachfule, J. Mater. Chem. A, 2023, 14489–14538 RSC.
- S. Bommakanti, A. Puthukkudi, M. Samal, P. Sahu and B. P. Biswal, Cryst. Growth Des., 2023, 23, 6172–6200 CrossRef CAS.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- H. L. Nguyen, Adv. Mater., 2023, 35, 1–7 Search PubMed.
- C. Sun, D. Sheng, B. Wang and X. Feng, Angew. Chem., Int. Ed., 2023, 62, 202303378 CrossRef PubMed.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- I. Janica, V. Patroniak, P. Samorì and A. Ciesielski, Chem. – Asian J., 2018, 13, 465–481 CrossRef CAS PubMed.
- L.-J. Wang, P.-Y. Dong, G. Zhang and F.-M. Zhang, Energy Fuels, 2023, 37, 6323–6347 CrossRef CAS.
- C. Qian, L. Feng, W. L. Teo, J. Liu, W. Zhou, D. Wang and Y. Zhao, Nat. Rev. Chem, 2022, 6, 881–898 CrossRef CAS PubMed.
- B. P. Biswal, S. Chandra, S. Kandambeth, B. Lukose, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2013, 135, 5328–5331 CrossRef CAS PubMed.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS PubMed.
- B. Díaz de Greñu, J. Torres, J. García-González, S. Muñoz-Pina, R. de los Reyes, A. M. Costero, P. Amorós and J. V. Ros-Lis, ChemSusChem, 2021, 14, 208–233 CrossRef PubMed.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS PubMed.
- D. W. Burke, C. Sun, I. Castano, N. C. Flanders, A. M. Evans, E. Vitaku, D. C. McLeod, R. H. Lambeth, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Angew. Chem., Int. Ed., 2020, 132, 5203–5209 CrossRef.
- L. Cusin, H. Peng, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2021, 60, 14236–14250 CrossRef CAS PubMed.
- X. Guan, Q. Fang, Y. Yan and S. Qiu, Acc. Chem. Res., 2022, 55, 1912–1927 CrossRef CAS PubMed.
- L. Cusin, H. Peng, A. Ciesielski and P. Samorì, Angew. Chem., Int. Ed., 2021, 60, 14236–14250 CrossRef CAS PubMed; G. Jiang, W. Zou, Z. Ou, W. Zhang, Z. Liang and L. Du, Chem. –Eur. J., 2023, 29, e202203610 CrossRef PubMed.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS PubMed.
- C. Feriante, A. M. Evans, S. Jhulki, I. Castano, M. J. Strauss, S. Barlow, W. R. Dichtel and S. R. Marder, J. Am. Chem. Soc., 2020, 142, 18637–18644 CrossRef CAS PubMed.
- I. Castano, A. M. Evans, H. Li, E. Vitaku, M. J. Strauss, J. L. Brédas, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2019, 5, 1892–1899 CrossRef CAS PubMed.
- H. Li, A. M. Evans, I. Castano, M. J. Strauss, W. R. Dichtel and J. L. Bredas, J. Am. Chem. Soc., 2020, 142, 1367–1374 CrossRef CAS PubMed.
- L. Deng, X. Kang, T. Quan, L. Yang, S. Liu, K. Zhang, M. Gao, Z. Xia and D. Gao, ACS Appl. Mater. Interfaces, 2021, 13, 33449–33463 CrossRef CAS PubMed.
- S. T. Emmerling, L. S. Germann, P. A. Julien, I. Moudrakovski, M. Etter, T. Friščić, R. E. Dinnebier and B. V. Lotsch, Chem, 2021, 7, 1639–1652 CAS.
- Q. Guan, D. D. Fu, Y. A. Li, X. M. Kong, Z. Y. Wei, W. Y. Li, S. J. Zhang and Y. Bin Dong, iScience, 2019, 14, 180–198 CrossRef CAS PubMed.
- Z. Guo, H. Wu, Y. Chen, S. Zhu, H. Jiang, S. Song, Y. Ren, Y. Wang, X. Liang, G. He, Y. Li and Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202210466 CrossRef CAS PubMed.
- Z. Li, Z. W. Liu, Z. Li, T. X. Wang, F. Zhao, X. Ding, W. Feng and B. H. Han, Adv. Funct. Mater., 2020, 30, 1–9 Search PubMed.
- K. Kaya, D. Ditz, A. Jaworski, J. Chen, S. Monti, G. Barcaro, S. Budnyk, A. Slabon and R. Palkovits, Adv. Sustainable Syst., 2023, 7, 2300071 CrossRef CAS.
- N. Huang, R. Krishna and D. Jiang, J. Am. Chem. Soc., 2015, 137, 7079–7082 CrossRef CAS PubMed.
- M. S. Lohse, T. Stassin, G. Naudin, S. Wuttke, R. Ameloot, D. De Vos, D. D. Medina and T. Bein, Chem. Mater., 2016, 28, 626–631 CrossRef CAS.
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2018, 140, 12677–12681 CrossRef CAS PubMed.
- X. Guan, H. Li, Y. Ma, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, Nat. Chem., 2019, 11, 587–594 CrossRef CAS PubMed.
- H. L. Qian, Y. Li and X. P. Yan, J. Mater. Chem. A, 2018, 6, 17307–17311 RSC.
- E. Dautzenberg, G. Li and L. C. P. M. de Smet, ACS Appl. Mater. Interfaces, 2023, 15, 5118–5127 CrossRef CAS PubMed.
- S. Y. Zhang, X. H. Tang, Y. L. Yan, S. Q. Li, S. Zheng, J. Fan, X. Li, W. G. Zhang and S. Cai, ACS Macro Lett., 2021, 10, 1590–1596 CrossRef CAS PubMed.
- Z. Dong, Y. Yang, X. Cai, X. Tang, Y. Yan, S. Zheng, W. Zhang, S. Cai and J. Fan, J. Solid State Chem., 2022, 316, 123644 CrossRef CAS.
- Y. Zhao, X. Xu, C. Xu, D. Meng, X. Liang and J. Qiu, New J. Chem., 2022, 46, 11980–11984 RSC.
- X. Tang, Y. Yang, X. Li, X. Wang, D. Guo, S. Zhang, K. Zhang, J. Wu, J. Zheng, S. Zheng, J. Fan, W. Zhang and S. Cai, ACS Appl. Mater. Interfaces, 2023, 15(20), 24836–24845 CrossRef CAS PubMed.
- Y. Y. Wen, Z. Y. Dong, X. H. Tang, W. G. Zhang, S. L. Cai and J. Fan, New J. Chem., 2023, 47, 19039–19046 RSC.
- X. Li, Z. Jia, J. Zhang, Y. Zou, Y. Zhang, K. Shu, W. Liu, N. Liu, Y. Li and L. Ma, Small, 2023, 19, 2303775 CrossRef CAS PubMed.
- Y. Liang, L. Feng, X. Liu, Y. Zhao, Q. Chen, Z. Sui and N. Wang, Chem. Eng. J., 2021, 404, 127095 CrossRef CAS.
- R. Liu, L. Huang, H. Tao, X. Lei and Q. Shuai, J. Environ. Chem. Eng., 2022, 10, 107300 CrossRef CAS.
- Q. Gao, X. Li, G. Ning, H. Xu, C. Liu, B. Tian, W. Tang and K. P. Loh, Chem. Mater., 2018, 30, 1762–1768 CrossRef CAS.
- B. Zhang, H. Mao, R. Matheu, J. A. Reimer, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11420–11424 CrossRef CAS PubMed.
- L. Chen, C. Gong, X. Wang, F. Dai, M. Huang, X. Wu, C. Z. Lu and Y. Peng, J. Am. Chem. Soc., 2021, 143, 10243–10249 CrossRef CAS PubMed.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque, Y. Li and B. Chen, Science, 2018, 359, 206–210 CrossRef CAS PubMed.
- H. Li, J. Ding, X. Guan, F. Chen, C. Li, L. Zhu, M. Xue, D. Yuan, V. Valtchev, Y. Yan, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2020, 142, 13334–13338 CrossRef CAS PubMed.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- T. Liu, Y. Zhao, M. Song, X. Pang, X. Shi, J. Jia, L. Chi and G. Lu, J. Am. Chem. Soc., 2023, 145, 2544–2552 CrossRef CAS PubMed.
- S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed.
- Y. Y. Liu, X. C. Li, S. Wang, T. Cheng, H. Yang, C. Liu, Y. Gong, W. Y. Lai and W. Huang, Nat. Commun., 2020, 11, 1–8 CrossRef CAS PubMed.
- H. Hong, J. Liu, H. Huang, C. Atangana Etogo, X. Yang, B. Guan and L. Zhang, J. Am. Chem. Soc., 2019, 141, 14764–14771 CrossRef CAS PubMed.
- A. K. Mohammed, S. Usgaonkar, F. Kanheerampockil, S. Karak, A. Halder, M. Tharkar, M. Addicoat, T. G. Ajithkumar and R. Banerjee, J. Am. Chem. Soc., 2020, 142, 8252–8261 CrossRef CAS PubMed.
- S. Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- M. X. Wu and Y. W. Yang, Chin. Chem. Lett., 2017, 28, 1135–1143 CrossRef CAS.
- G. Barin, V. Krungleviciute, O. Gutov, J. T. Hupp, T. Yildirim and O. K. Farha, Inorg. Chem., 2014, 53, 6914–6919 CrossRef CAS PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- J. Ren, M. Ledwaba, N. M. Musyoka, H. W. Langmi, M. Mathe, S. Liao and W. Pang, Coord. Chem. Rev., 2017, 349, 169–197 CrossRef CAS.
- Y. Liu, W. K. Han, W. Chi, Y. Mao, Y. Jiang, X. Yan and Z. G. Gu, Appl. Catal., B, 2023, 331, 122691 CrossRef CAS.
- N. He, B. Liu, B. Jiang, X. Li, Z. Jia, J. Zhang, H. Long, Y. Zhang, Y. Zou, Y. Yang, S. Xiong, K. Cao, Y. Li and L. Ma, ACS Appl. Mater. Interfaces, 2023, 15, 16975–16983 CrossRef CAS PubMed.
- K. Gottschling, L. Stegbauer, G. Savasci, N. A. Prisco, Z. J. Berkson, C. Ochsenfeld, B. F. Chmelka and B. V. Lotsch, Chem. Mater., 2019, 31, 1946–1955 CrossRef CAS PubMed.
- M. Yin, L. Wang and S. Tang, ACS Appl. Mater. Interfaces, 2022, 14, 55674–55685 CrossRef CAS PubMed.
- H. Lyu, H. Li, N. Hanikel, K. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 12989–12995 CrossRef CAS PubMed.
- X. Yang, Z. Xie, T. Zhang, G. Zhang, Z. Zhao, Y. Wang, G. Xing and L. Chen, Sci. China: Chem., 2022, 65, 190–196 CrossRef CAS.
- H. Veldhuizen, S. A. Butt, A. Van Leuken, B. Van Der Linden, W. Rook, S. Van Der Zwaag and M. A. Van Der Veen, ACS Appl. Mater. Interfaces, 2023, 15, 29186–29194 CrossRef CAS PubMed.
- A. K. Sekizkardes, P. Wang, J. Hoffman, S. Budhathoki and D. Hopkinson, Mater. Adv., 2022, 3, 6668–6686 RSC.
- H. L. Nguyen, C. Gropp, N. Hanikel, A. Möckel, A. Lund and O. M. Yaghi, ACS Cent. Sci., 2022, 8, 926–932 CrossRef CAS PubMed.
- L. Chen, W. Han, X. Yan, J. Zhang, Y. Jiang and Z. Gu, ChemSusChem, 2022, 15, 202201824 CrossRef PubMed.
- C. Sun, Y. Zhu, P. Shao, L. Chen, X. Huang, S. Zhao, D. Ma, X. Jing, B. Wang and X. Feng, Angew. Chem., Int. Ed., 2023, 62, e2022171 Search PubMed.
- L. Grunenberg, G. Savasci, S. T. Emmerling, F. Heck, S. Bette, A. C. Bergesch, C. Ochsenfeld and B. V Lotsch, J. Am. Chem. Soc., 2023, 145, 13241–13248 CrossRef CAS PubMed.
- Y. Liu, W. K. Han, W. Chi, J. X. Fu, Y. Mao, X. Yan, J. X. Shao, Y. Jiang and Z. G. Gu, Appl. Catal., B, 2023, 338, 123074 CrossRef CAS.
- P. W. Fritz and A. Coskun, ACS Cent. Sci., 2022, 8, 871–873 CrossRef CAS PubMed.
- X. Yuan, N. Wu, Z. Guo and H. Zhan, Microporous Mesoporous Mater., 2023, 355, 112573 CrossRef CAS.
- S. Maiti, J. K. Sharma, J. Ling, D. Tietje-Mckinney, M. P. Heaney, T. Runčevski, M. A. Addicoat, F. D'Souza, P. J. Milner and A. Das, Macromol. Rapid Commun., 2023, 44, 2200751 CrossRef CAS PubMed.
- C. Wang, S. Jiang, W. Ma, Z. Liu, L. Liu, Y. Zou, B. Xu and W. Tian, Molecules, 2023, 28, 449 CrossRef CAS PubMed.
- Q. Liao, W. Xu, X. Huang, C. Ke, Q. Zhang, K. Xi and J. Xie, Sci. China: Chem., 2020, 63, 707–714 CrossRef CAS.
- Y. Yang, W. Zhao, H. Niu and Y. Cai, ACS Appl. Mater. Interfaces, 2021, 13, 42035–42043 CrossRef CAS PubMed.
- C. Gao, X. Zhang, M. Zhang, X. Wu, R. Chen, C. Zhang, C. Sun, Y. Du, Q.-H. Xu and B. Hu, ACS Appl. Energy Mater., 2023, 6, 427–9433 Search PubMed.
- L. Zhang, C. Sun, S.-J. Xiao, Q.-G. Tan, G.-P. Yang, J.-Q. Fan, Y.-T. Luo, R.-P. Liang and J.-D. Qiu, ACS Appl. Nano Mater., 2023, 6, 17083–17091 CrossRef CAS.
- C. Arqueros, F. Zamora and C. Montoro, Nanomaterials, 2021, 11, 1–31 CrossRef.
- Z. Xia, Y. Zhao and S. B. Darling, Adv. Mater. Interfaces, 2021, 8, 1–17 Search PubMed.
- A. K. Mohammed and D. Shetty, Environ. Sci.: Water Res. Technol., 2021, 7, 1895–1927 RSC.
- S. Kim, H. Wang and Y. M. Lee, Angew. Chem., Int. Ed., 2019, 58, 17512–17527 CrossRef CAS.
- J. Li, Y. Li, Y. Lu, Y. Wang, Y. Guo and W. Shi, Biomimetics, 2023, 8, 35 CrossRef CAS PubMed.
- L. Feng, C. Qian and Y. Zhao, ACS Mater. Lett., 2020, 2, 1074–1092 CrossRef CAS.
- J. Ma, T. Shu, Y. Sun, X. Zhou, C. Ren, L. Su and X. Zhang, Small, 2022, 18, 1–30 Search PubMed.
- A. Mal, H. Ding, M. Li, W. Li and C. Wang, ACS Appl. Nano Mater., 2022, 5, 13972–13984 CrossRef CAS.
- F. Mohajer, G. Mohammadi Ziarani, A. Badiei, S. Iravani and R. S. Varma, RSC Adv., 2023, 13, 8136–8152 RSC.
- P. Mohan, B. Sasikumar, S. A. G. Krishnan and G. Arthanareeswaran, J. Taiwan Inst. Chem. Eng., 2023, 105067 CrossRef.
- H. H. Zhang, C. Gu, M. S. Yao and S. Kitagawa, Adv. Energy Mater., 2022, 12, 2100321 CrossRef CAS.
- J. Tang, C. Su and Z. Shao, Small Methods, 2021, 5, 1–29 Search PubMed.
- Q. Wang, D. Wang, Q. Liao, C. Ke, Y. Zhang, Q. Han, Y. Zhang, D. Jiang and K. Xi, J. Colloid Interface Sci., 2022, 612, 608–616 CrossRef CAS PubMed.
- Q. Wang, K. Tang, Q. Liao, Y. Xu, H. Xu, Y. Wang, P. Wang, Z. Meng and K. Xi, Adv. Funct. Mater., 2023, 33, 2211356 CrossRef CAS.
- Y. Xiong, Q. Liao, Z. Huang, X. Huang, C. Ke, H. Zhu, C. Dong, H. Wang, K. Xi, P. Zhan, F. Xu and Y. Lu, Adv. Mater., 2020, 32, 1907242 CrossRef CAS PubMed.
- X. Wu, X. Zhang, Y. Li, B. Wang, Y. Li and L. Chen, J. Mater. Sci., 2021, 56, 2717–2724 CrossRef CAS.
- J. Hu, S. K. Gupta, J. Ozdemir and H. Beyzavi, ACS Appl. Nano Mater., 2020, 3, 6239–6269 CrossRef CAS PubMed.
- H. L. Nguyen, Chem. Sci., 2021, 12, 8632–8647 RSC.
- B. Gui, J. Xin, Y. Cheng, Y. Zhang, G. Lin, P. Chen, J.-X. Ma, X. Zhou, J. Sun and C. Wang, J. Am. Chem. Soc., 2023, 145, 11276–11281 CrossRef CAS PubMed.
- Y. Liu, J. Li, J. Lv, Z. Wang, J. Suo, J. Ren, J. Liu, D. Liu, Y. Wang, V. Valtchev, S. Qiu, D. Zhang and Q. Fang, J. Am. Chem. Soc., 2023, 145, 9679–9685 CrossRef CAS PubMed.
- X. Liu, C. Wang, J. Li, Z. Zhang, A. M. Al-enizi, A. Nafady, F. Shui, Z. You, B. Li, Y. Wen and S. Ma, Nat. Commun, 2023, 14, 7022 CrossRef CAS PubMed.
- P. Martinez-Bulit, A. Sorrenti, D. R. S. Miguel, M. Mattera, Y. Belce, Y. Xia, S. Ma, M.-H. Huang, S. Pané and J. Puigmartí-Luis, Chem. Engg. J., 2022, 435, 135117 CrossRef CAS.
- W. Zhang, L. Chen, S. Dai, C. Zhao, C. Ma, L. Wei, M. Zhu, S. Y. Chong, H. Yang, L. Liu, Y. Bai, M. Yu, Y. Xu, X. W. Zhu, Q. Zhu, S. An, R. S. Sprick, M. A. Little, X. Wu, S. Jiang, Y. Wu, Y. B. Zhang, H. Tian, W. H. Zhu and A. I. Cooper, Nature, 2022, 604, 72–79 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |