
Decoupling the contributions of industrially relevant conditions to the stability of binary and ternary FeNi-based catalysts for alkaline water oxidation†
Yang
Xiao
,
Kamran
Dastafkan
 ,
Zhen
Su
,
Chengli
Rong
and
Chuan
Zhao
*
,
Zhen
Su
,
Chengli
Rong
and
Chuan
Zhao
*
School of Chemistry, University of New South Wales, Sydney, NSW 2052, Australia. E-mail: chuan.zhao@unsw.edu.au
First published on 21st August 2023
Abstract
As a crucial half-reaction in electrochemical water splitting, advancing the stability performance of the oxygen evolution reaction (OER) under industrial conditions is of great importance for practical water electrolysis. Nonprecious FeNi-based binary and ternary catalysts have been developed with improved activity toward the OER in alkaline media. However, these catalysts often exhibit degradation under industrial water electrolysis operation conditions. Here, we demonstrate the impact of industrially relevant conditions on the long-term stability performance of binary NiFe and ternary FeNiCr hydroxide as a showcase of highly active catalysts. Under the simulation of industrial operating conditions in alkaline water electrolysers (AWEs), the ternary FeNiCr catalyst exhibits a faster decline in the OER stability compared to the binary FeNi catalyst. Decoupling of industrial conditions revealed that the concentration of alkaline electrolyte has the biggest effect on stability decline followed by temperature and current density. Results show a different metal dissolution pattern during the OER compared to the binary FeNi catalyst, entailing changes in surface morphology and overpotential increase. The findings in this study reveal the significance of testing conditions in developing high-performance and durable catalysts for practical water electrolysis.
Introduction
Catalyst development for water electrolysis has often overlooked the gap between laboratory and industrial scale testing conditions, which leads to an overestimation of the stability and durability of catalysts. It is crucial to design catalysts that can withstand the harsh industrial operating conditions of water electrolysers. The stability decline under industrially relevant conditions is particularly significant for the OER half-reaction in alkaline media. Compared to the hydrogen evolution reaction (HER), the OER is complex and requires higher overpotentials due to its large thermodynamic potential and sluggish multi-electron transfer kinetics.1,2 Despite the progress in catalyst development for alkaline OER, achieving the desired balance for the structure–performance relationship has remained a significant hurdle due to the lack of long-term stability under industrially relevant conditions.Industrially relevant conditions in traditional AWEs as a well-established technology in water electrolysis in alkaline media include high concentrations of the alkaline electrolyte (5–7 M KOH), elevated cell and electrolyte temperature (up to 80 °C), and large current densities (approaching 500 mA cm−1 and above).3–5 Severe loss of electrocatalytic efficiency, catalyst degradation, and stability decline are often observed at these conditions when testing the majority of nonprecious metal-based electrocatalysts. Strategies to promote the stability performance under industrially relevant conditions have been very few including using intermittent reduction methodology to revivify a degraded electrocatalyst by manipulating reversible phase segregation6 and introducing cationic vacancies on the substrate surface of NiFe double layer hydroxides (LDHs)7 or delaminating them into atomically thin nanosheets to improve stability in the OER process.8 Despite these efforts, most reported catalysts currently lack the required stability to meet the requirements for device level applications.
In recent years, FeNi-based structures have emerged as benchmark catalysts for alkaline OER. However, the development has been focused mostly on the activity enhancement via metal/non-metal doping, structure modulation and defect engineering.9–14 Previously, promising activity improvements were demonstrated for ternary FeNi-based systems, such as FeNiCr, FeNiV, and FeNiMo, under benign alkaline conditions, i.e., 1 M KOH and room temperature.15–19 Regulating the electronic and compositional structure has resulted in the boosted intrinsic activity of Ni/Fe active sites compared to the binary FeNi structure at low and moderate OER current densities.20–22 However, these FeNi-based catalysts remain stable only under standard laboratory conditions and particularly the long-term stability performance falls short of the efficiency required for their practical use in AWE electrolysers.23,24 Consequently, developing high-performance OER catalysts that surpass binary FeNi LDHs, as well as FeNi(O)x and FeNi(OH)x systems, using accelerated degradation testing conditions is crucial to commencing efficient large-scale hydrogen production using alkaline water electrolysers.25–27
Herein, to examine the intrinsic activity and stability performance of FeNi-based model catalysts and to understand the impact of operation testing conditions on their durability, FeNi and FeNiCr hydroxides are employed as highly active catalysts for alkaline OER. The long-term stability performance is investigated under both laboratory and industrially relevant conditions. Although the addition of Cr to the FeNi system improves the intrinsic catalytic activity of Fe/Ni active sites and the ternary FeNiCr catalyst remains stable under laboratory conditions, it shows a faster stability decline under simulated operation conditions in AWEs compared to the FeNi binary system. Furthermore, both the activity and stability of ternary FeNiCr and binary FeNi catalysts are examined as a function of temperature, current density, and alkaline electrolyte concentration, separately, in relation to metal dissolution, morphology change, and phase transformation. The results indicate that the electrolyte concentration is the major factor contributing to stability decline with Cr addition to the FeNi system. This study shows the critical role of accelerated degradation tests and the dire need for considering the industrially relevant conditions when developing efficient FeNi-based catalysts.
Results and discussion
To study the stability performance under industrially relevant conditions, it is important to consider both the initial activity and long-term stability of catalysts, as the rapid decay in the initial period of stability tests and the rate of performance deterioration over time give crucial information about the catalyst structure and efficiency for water electrolysis. In this study, the binary FeNi and ternary FeNiCr hydroxides are firstly prepared over Fe foams (FeNi/FF and FeNiCr/FF) via a corrosion engineering approach reported in our previous study.28 To monitor the catalyst degradation during the stability performance under simulated operation conditions in AWEs, a home-made three-electrode half-cell with independent control over current density, temperature, and electrolyte concentration is used (Fig. 1a). A pump with a flow controller connected to a container with the electrolyte at elevated temperatures and to the half-cell allowed maintaining the alkaline electrolyte at different temperatures and concentrations. The potentiostat workstation was connected to a booster to apply larger current densities to the electrodes. The electrolyte was constantly stirred and circulated under these conditions to mimic the electrolytic operation testing conditions in AWEs. The ternary FeNiCr hydroxide catalyst was evaluated against the benchmark binary FeNi hydroxide for the OER in alkaline electrolyte, and then characterized before and after the long-term stability testing.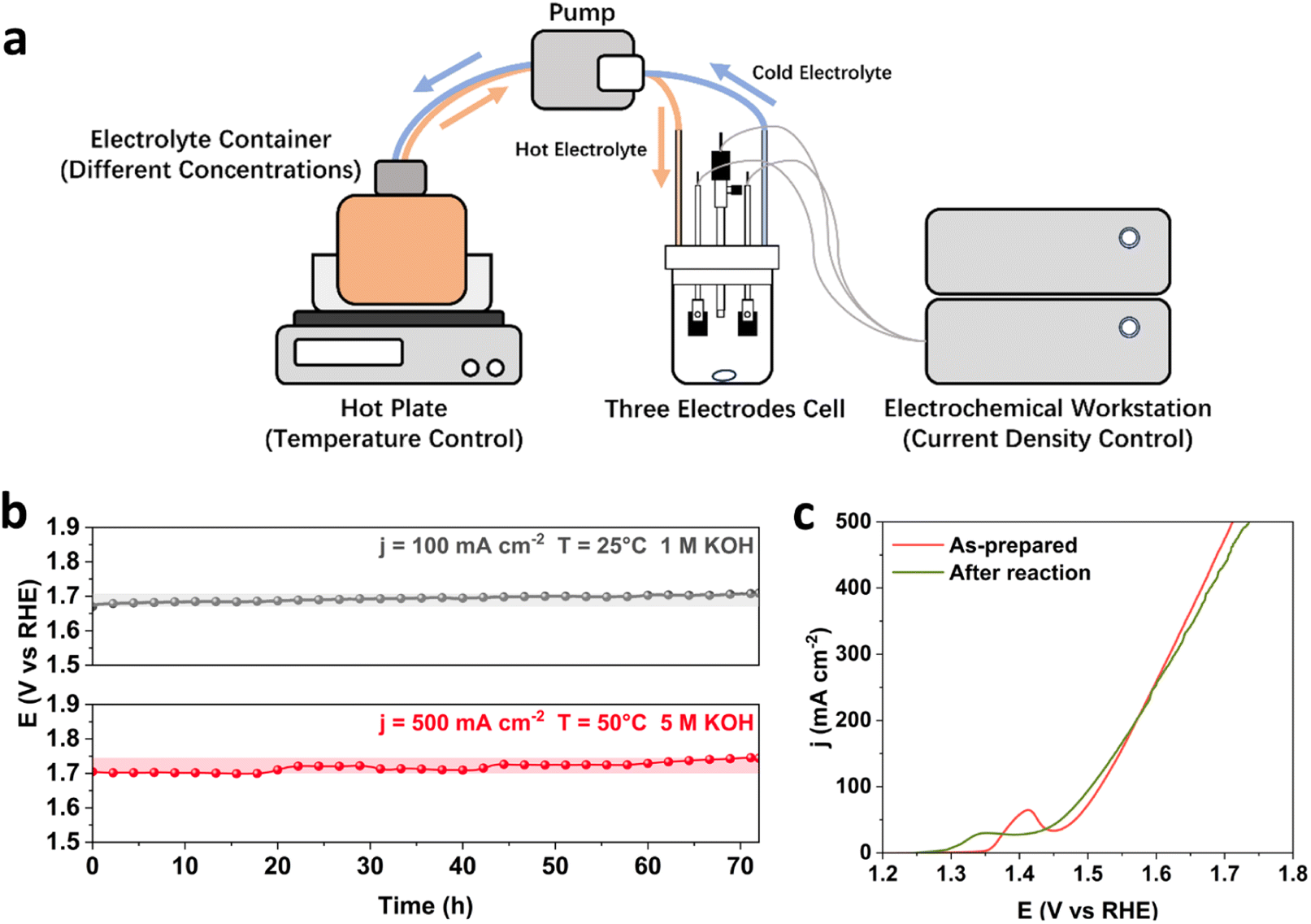 | ||
| Fig. 1 (a) The schematic diagram of the experimental setup. Electrochemical characterization of FeNi/FF: (b) long-term OER stability performance of FeNi/FF under standard and simulated industrial conditions. (c) Polarization curves of FeNi/FF before and after the stability test under simulated industrial conditions. | ||
Stability of binary FeNi under industrially relevant conditions
As the benchmark catalyst for alkaline water oxidation, binary FeNi structures have a relatively stable performance in alkaline media, while degradation worsens for most FeNi-based structures upon varying current density, temperature, and concentration of KOH.4,29 Here, we first subject the FeNi/FF electrode to long-term stability tests under both standard laboratory and industrially relevant conditions. At 25 °C and in 1 M KOH, FeNi/FF exhibits a slow increase in the overpotential at 100 mA cm−2 (Fig. 1b). When testing at 500 mA cm−2 and 50 °C in 5 M KOH, FeNi/FF shows a noticeable increase in overpotential, as confirmed by the polarization curves before and after the reaction. Interestingly, the Ni oxidation peak prior to the OER process shifts to lower potential after the long-term stability tests under industrially relevant conditions (Fig. 1c). To date, the phase transformation in FeNi-based structures has been less understood under the electrolytic conditions of water electrolysers with elevated currents, temperatures and alkaline electrolyte concentrations. Depending on the obtained local electronic structure and the synthesis environment, the binary FeNi structure may form the β-NiOOH phase at the surface at oxidative potentials which emanates from the effect of Fe species on Ni sites.30 Then, under the harsh industrial electrolytic conditions phase transformation to the less reactive γ-NiOOH phase could happen more, which is captured by the shift in the Ni oxidation to the lower potential. The observed stability decline over 72 hours of testing under simulated industrial conditions is consistent with the previously reported stability characteristics.29 This slight increase in overpotential over a period of tens of hours does not necessarily meet the durability demands in alkaline water electrolysers requiring catalyst lifetimes of thousands or even more hours.24 This makes it important to investigate the relationship between OER operation conditions and catalyst degradation mechanisms.The degradation of FeNi/FF involves multiple mechanisms, typically resulting from a combination of factors. These mechanisms encompass the dissolution of Fe and Ni species,8 transformation in the active phase and surface morphology,6 detachment of the FeNi catalyst layer from the substrate,31 and blockage of active sites caused by gas bubbles.32 Among these mechanisms, the dissolution and redeposition of Fe and Ni are considered to be one of the most important contributing factors, despite the general stability of transition metals at alkaline pH. On one hand, the dissolution of Fe and Ni as catalytic active centres to form soluble ions, such as FeO42−, results in the direct loss of active sites.33,34 On the other hand, in most binary FeNi structures, such as FeNi hydroxide,35 spinel oxide,36 and LDHs,25 Fe is more soluble than Ni, which results in an unbalanced element consumption. More Fe species are dissolved from the catalyst surface and redeposited into separate phases, which contributes to structural degradation through phase transformation and phase segregation. It has been shown that the dynamic process of dissolution and redeposition of FeNi-based structures accelerates the segregation of Fe and the formation of secondary phases such as FeOOH, which already contribute to the decrease in the catalyst activity under laboratory OER conditions.37 The above mechanisms for catalyst degradation and stability decline are only expedited at elevated current densities, temperature, and alkaline electrolyte concentration, and accelerate the dissolution process of Fe and Ni active sites. Under testing conditions close to industrial operation parameters in water electrolysers, the long-term OER is reported to cause a structural transformation of FeNi LDHs into a mixture of NiO and NiFe2O4,38 with less intrinsic catalytic activity. The structure of FeNi catalysts has also been reported to transform into a combination of separated phases of Ni(OH)2 and amorphous FeOOH at 80 °C and 7.5 M KOH, which no longer benefit from the catalytic synergy between Fe and Ni in the original binary structural phases.39 The formation of low active phases at the surface breaks the pre-designed catalyst structure and significantly changes the elemental ratios in the transformed structures, significantly contributing to the degradation of binary FeNi-based catalysts.
Stability of ternary FeNiCr under industrially relevant conditions
Under the industrially relevant conditions of 50 °C and 5 M KOH, Cr addition boosts the performance of Fe and Ni active sites (Fig. S1a and b†). FeNiCr/FF shows stability performance at a moderate current density of 100 mA cm−2 at 25 °C and 1 M KOH electrolyte with no significant decay after 72 h (Fig. 2a). However, under industrially relevant conditions, i.e., 50 °C, 5 M KOH, and at 500 mA cm−2, FeNiCr/FF depicts a faster and bigger decline in the catalytic performance compared to FeNi/FF, despite the slight improvement in the initial activity (Fig. S1c†). The OER polarization curves of FeNiCr/FF, obtained with the as-prepared electrode and after a three-day stability test at 500 mA cm−2, 50 °C, and 5 M KOH (Fig. 2b and c), exhibit a significant decrease in the electrocatalytic performance. FeNiCr/FF exhibits a larger double layer capacitance (Cdl) after the OER at the above conditions, reflecting a larger exposed surface area (Fig. 2c), despite a weakened OER activity. These results indicate that the addition of Cr into the NiFe structure is not beneficial for a stable water electrolysis despite improving the intrinsic activity of binary FeNi structures at standard electrolytic conditions. Given the bigger impact of industrial conditions on the long-term stability as well as on morphology and structure variation in the ternary FeNiCr hydroxides, FeNiCr/FF endures a higher active site loss and a different phase transformation than the binary hydroxide in FeNi/FF. This is captured by the slight shift of Ni oxidation peak to a lower potential in Fig. 2b, suggesting the rate of phase transformation for producing more γ-NiOOH is slowed down and the β-NiOOH phase may still exist at the surface.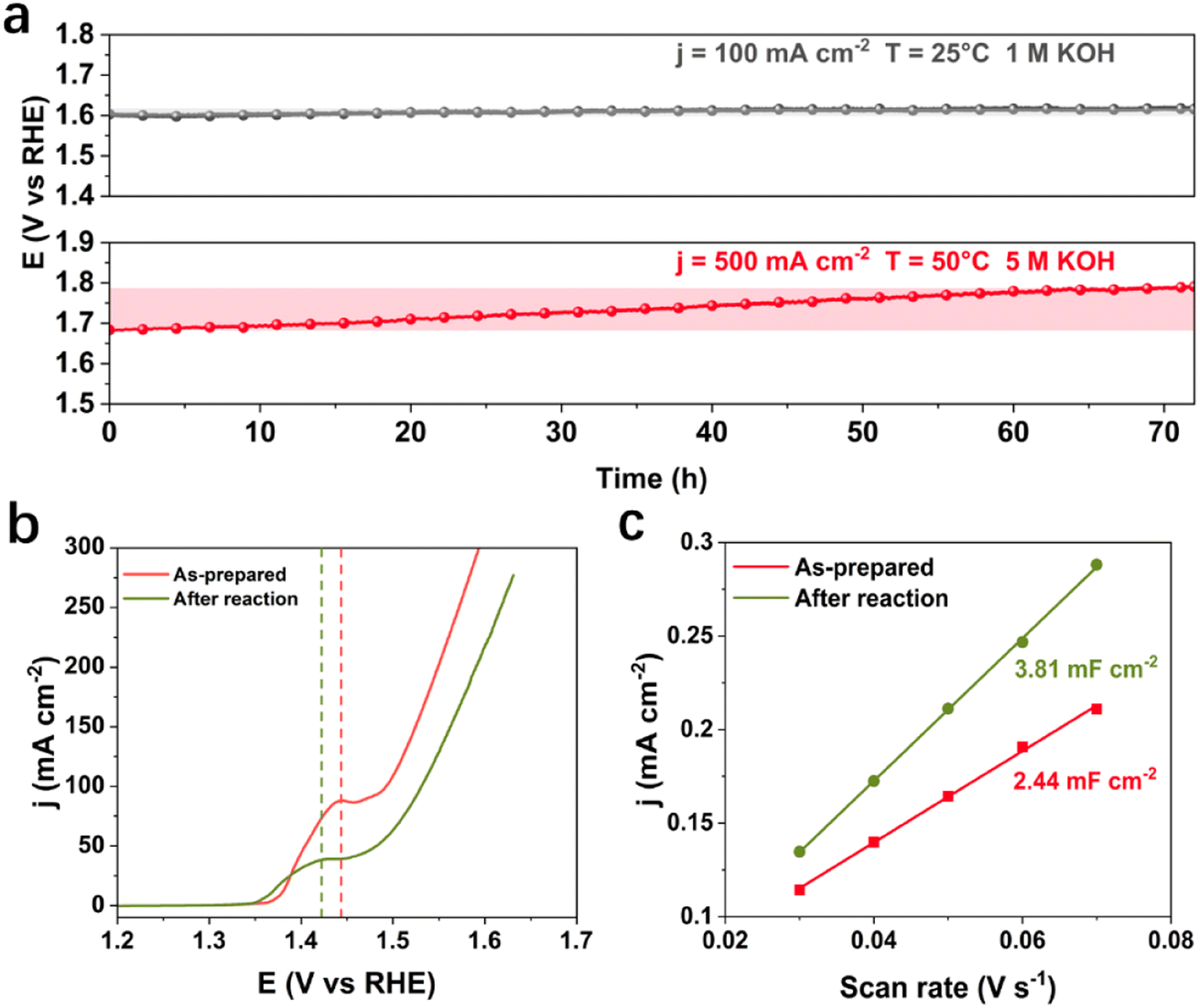 | ||
| Fig. 2 Electrochemical characterization of FeNiCr/FF under industrially relevant conditions. (a) Long-term OER stability performance of FeNiCr/FF under standard and simulated industrial conditions. (b) Polarization curves and (c) double-layer capacitance in comparison with the as-prepared electrode. | ||
The change in morphology and chemical composition of the reacted FeNiCr catalyst during long-term OER under the simulated industrial conditions was investigated. The fine structures over FeNiCr/FF are completely roughened and also the bigger aggregations disappeared after a three-day stability test (Fig. 3a and S2a†). Meanwhile, the FeNiCr structure over the Fe substrate remains amorphous (Fig. 3b and S2b†). The change in the morphology, especially for the aggregated particles, may involve physical detachment from Fe foam. The corresponding elemental EDS mapping verifies a significant decrease for Cr species, caused by Cr dissolution during the stability test, compared to the homogeneous distribution of Fe, Cr, and Ni species in the as-prepared FeNiCr structure (Fig. 3c and S2c†). There is also a slight increase in the Fe content in the catalyst from the corresponding TEM-EDS mapping after three days of OER. This is most likely because the as-prepared FeNiCr film grown onto Fe foam as both a support and an Fe source via a corrosion engineering process has less Fe in the outermost layers due to the limitation in the mass transport of the ions and charges for the electrochemical and chemical reactions to occur at local sites of the Fe foam.40 Instead, the inner regions of the catalyst film which are closer to the Fe foam support have more Fe. With the outermost catalyst surface exposed and degraded in the electrolyte under the simulated industrial conditions, the inner regions of the catalyst film are exposed. The newly exposed FeNiCr catalyst layers with more Fe grown during the corrosion process then reveal more Fe via characterization. However, the enlarged larger surface area, as a result of surface roughening due to Cr dissolution as well as the exposure of more Fe from the FeNiCr structure, is considerably negated by the morphology and physical loss of the FeNiCr catalyst, which together with the change in the elemental distribution in the ternary hydroxide structure, decrease the intrinsic activity of FeNiCr/FF.
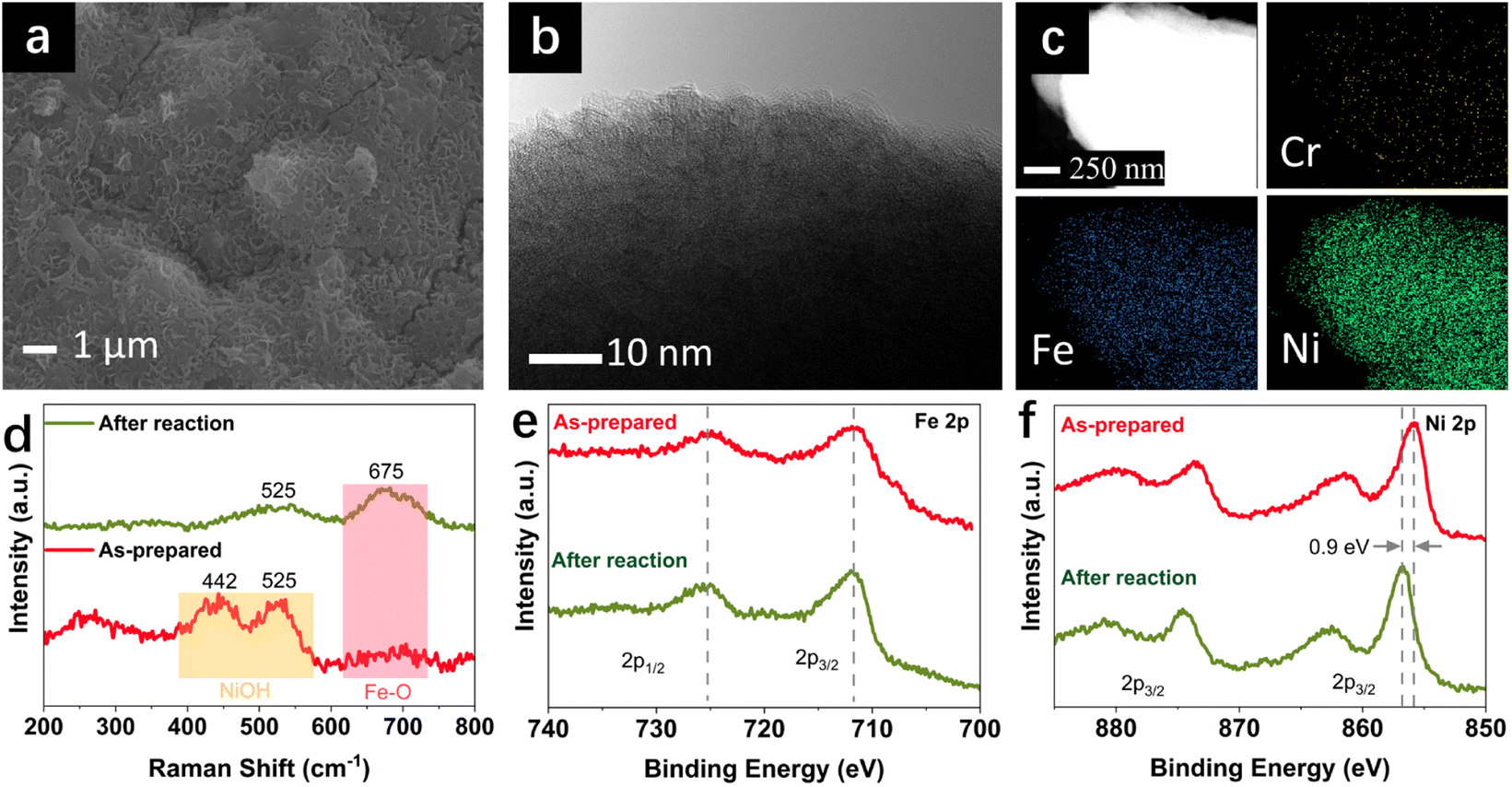 | ||
| Fig. 3 Characterization of FeNiCr/FF after a three-day OER stability test at 500 mA cm−2, 50 °C, and 5 M KOH: (a) SEM, (b) HRTEM, (c) EDS mapping, (d) Raman spectra, and high-resolution XPS spectra for (e) Fe 2p and (f) Ni 2p. | ||
The chemical structure of the FeNiCr catalyst and oxidation states of Fe, Ni, and Cr species in FeNiCr/FF subjected to long-term stability tests under industrially relevant conditions were then investigated. The characteristic Raman peaks for Ni–O bonding vibrations at 442 cm−1 and 525 cm−1 in the as-prepared electrode dampen significantly where a broad peak between 400 cm−1 and 600 cm−1 is observed after the stability test, pointing to the mixed γ- and β-NiOOH phases at the surface of FeNiCr (Fig. 3d). Meanwhile, the Fe–O bond at 675 cm−1 intensifies after the three-day stability test, confirming the higher Fe content in the newly exposed inner regions of the FeNiCr catalyst after the outermost surface layer is lost during the OER.
The X-ray photoelectron spectroscopy (XPS) analysis of FeNiCr/FF after long-term stability testing under the simulated industrial conditions depicts a similar Fe3+ oxidation state without any noticeable change in Fe 2p peaks (Fig. 3e). The Ni 2p3/2 peak at 855.8 eV shifted to higher energy by 0.9 eV after the long-term OER, indicating the typical transition from Ni2+ to Ni3+ states (Fig. 3f). The Cr 2p signals disappeared after the OER stability test, due to the dissolution of Cr in the concentrated KOH electrolyte (Fig. S3a†). The core-level O 1s signal shifted toward a lower energy, indicating more metal–oxygen bonding in the metal (oxy)hydroxide phase (Fig. S3b†). Notably, the surface of the graphite counter electrode was also changed as a result of the re-deposition of Fe species, as confirmed by the corresponding SEM-EDS elemental mapping (Fig. S4†).
According to these observations, several factors negate the synergistic effect of Cr on Fe and Ni active sites and contribute to the inferior performance of the ternary FeNiCr under the simulated industrial electrolytic conditions, i.e., the significant Cr loss within the first 24 h, unfavourable phase transformation to a mixed state of γ- and β-NiOOH at the surface compared to the dominant β-NiOOH phase in FeNiCr under standard conditions,28 and severe morphology change and physical loss of the catalyst. To further understand the impact of industrial conditions on the stability performance of FeNiCr/FF, the contribution of each industrial condition was decoupled, and independent long-term stability tests were carried out to investigate the extent to which varying current densities, temperatures, and electrolyte concentrations affect the degradation of ternary FeNiCr hydroxides. Subsequently, the corresponding changes in surface morphology and chemical composition are analysed.
Effect of current density
The effect of current density on stability performance was investigated at 25 °C and 1 M KOH. The SEM images of FeNiCr/FF collected after long-term stability tests at 100, 500, and 1000 mA cm−2 show a slow detachment of particulate-shaped catalysts on the surface as well as the roughening of fine nanostructures covering the electrode surface (Fig. 4a). With increasing the current density from 100 to 1000 mA cm−2, FeNiCr/FF shows no significant stability decay at lower current densities, and even the increase in overpotential at 1000 mA cm−2 is only 1.22% after 72 h (Fig. 4b). A comparison of the corresponding LSV curves obtained before and after the stability test at these current densities also demonstrates no significant change in the OER activity (Fig. S5†).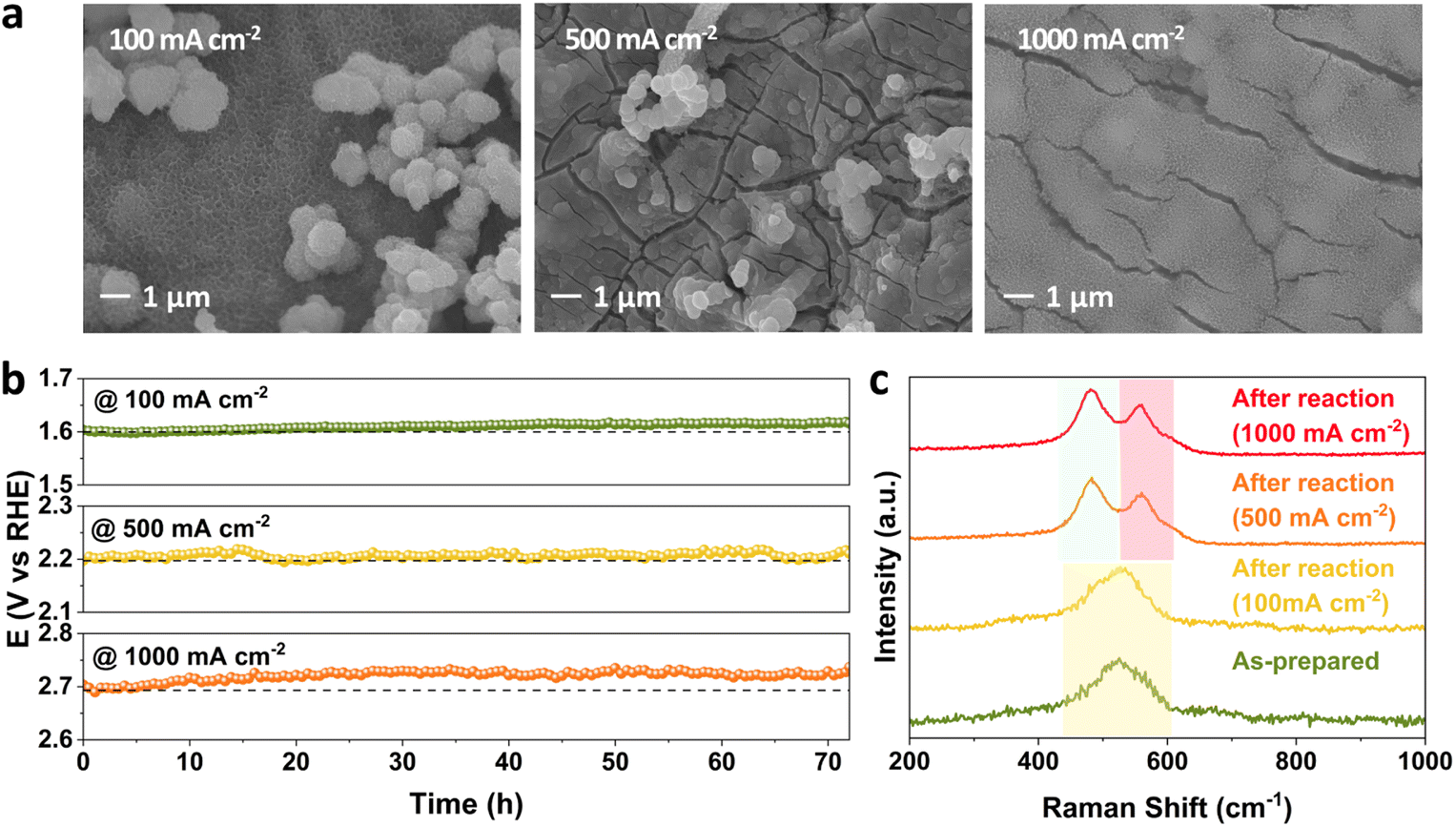 | ||
| Fig. 4 Monitoring the stability, morphology and structure of FeNiCr/FF during the OER with independently controlled current density from 100 mA cm−2 to 1000 mA cm−2. (a) SEM images, (b) chronopotentiometry profiles, and (c) Raman spectra. | ||
Measuring the concentration of Fe, Ni, and Cr species leached out into the KOH electrolyte during three days of stability testing by ICP-MS reveals a significantly different Cr dissolution profile than those of Fe and Ni. A rapid spike in Cr concentration in 1 M KOH in the first 24 h followed by its relative stabilization for the remaining time is recorded at all three current densities (Fig. S6a†). The amount of Cr dissolved at a fixed current density of 1000 mA cm−2 is only slightly higher than that under other current densities. The dissolution of Cr during alkaline water oxidation has been reported in several studies due to the dissolvable species generated at high pH and high oxidation potentials.41,42 Both Fe and Ni dissolution are negligible compared to that of Cr (Fig. S6b and c†).
It should also be noted that the amount of Fe re-deposited onto the counter electrode is only a small fraction of the total Fe lost in the electrolyte, and is mainly controlled by current density. Here, with increasing the current density the amount of the re-deposited Fe is expected to increase. However, still the highest and lowest Fe dissolution into the electrolyte during the three days of stability testing is detected at 1000 and 100 mA cm−2 (Fig. S6b†), respectively, indicating that the observed trends of Fe dissolution by the effect of current density are not impacted by the Fe re-deposition.
Well-resolved Ni–O vibrations pertaining to the oxyhydroxide phase in FeNiCr/FF are formed and shifted to 481 and 558 cm−1 after the long-term stability tests at the elevated current densities of 500 and 1000 mA cm−2 (Fig. 4c). The higher intensity of the Ni–O signal at 481 cm−1 points to the formation of the γ-NiOOH phase.43 Furthermore, the peak positions in the Fe 2p and Ni 2p profiles of the corresponding XPS spectra did not change before and after the stability tests (Fig. S7a and b†). The Cr 2p signal disappeared after the OER at all current densities (Fig. S7c†). The O 1s signal continuously shifted to lower energy with increasing current density with higher metal–oxygen coordination, indicating the phase transformation to the oxyhydroxide phase (Fig. S7d†).
These results suggest that current density has less impact on the OER stability performance, despite the physical changes observed. Addition of Cr induces synergistic and structure regulating effects on the ternary FeNiCr hydroxides, but Cr is not directly involved in the formation of Fe/Ni active sites, hence the stability curves do not show a significant decline despite a large amount of Cr being dissolved during the OER process, in agreement with previous reports.44,45 The change of surface morphology with increasing current density is attributed to a combination of Cr dissolution from the electrode surface and the large amount of gas bubble flushing generated at high current densities. Iron is stable in alkaline environments, however according to Pourbaix diagrams, Fe dissolution can be explained by the formation of soluble Fe species. Nevertheless, the limited amount of dissolved Fe species implies that the surface active sites are still retained at 1000 mA cm−2.
Effect of temperature
The effect of temperature on the performance of FeNiCr/FF is studied at a moderate current density of 100 mA cm−2 in 1 M KOH. Significant changes in the surface morphology of FeNiCr/FF after long-term OER stability testing can also be observed with increasing temperature. Fine nanostructures of FeNiCr hydroxides are preserved at 50 °C and aggregated with further increasing the temperature, forming cracked films on the surface of Fe foam, while the spherical nanoparticles could be detected even at 80 °C (Fig. 5a). The stability slightly decayed at 50 °C but it deteriorated at 80 °C during the OER for 72 h (Fig. 5b). The corresponding LSV curves at 25 °C, 50 °C, and 80 °C verify the stability decline with the increase in temperature (Fig. S8†). Monitoring the concentration of Cr, Fe, and Ni via ICP-MS during the three-day stability performance reveals a similar spike in Cr dissolution on the first day, followed by a relative stabilization (Fig. S9a†). Fe dissolution is relatively enhanced by temperature, while Ni dissolution remains negligible at all temperatures (Fig. S9b and c†).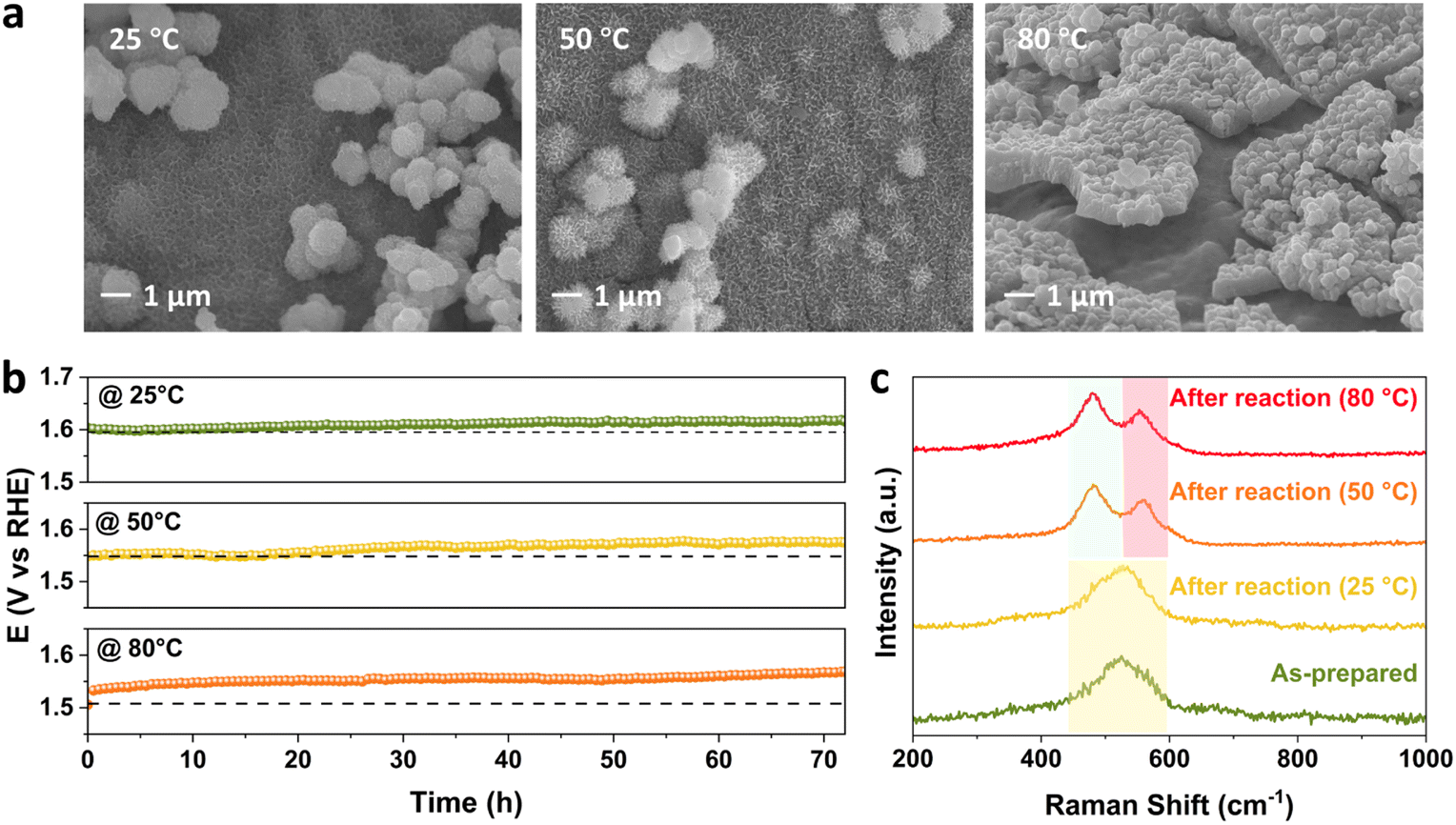 | ||
| Fig. 5 Monitoring the stability, morphology and structure of FeNiCr/FF during the OER with independently controlled temperature from 25 °C to 80 °C. (a) SEM images, (b) chronopotentiometry profiles, and (c) Raman spectra. | ||
The Raman spectra of FeNiCr/FF demonstrate similar shifts with increasing the temperature to the effect of current density. The signal splitting of Ni–O vibration occurs at higher temperatures of 50 °C and 80 °C giving well-resolved Raman shifts centred around 481 cm−1 and 558 cm−1 (Fig. 5c). The appearance of these Raman shifts pertaining to γ-NiOOH and β-NiOOH phases, respectively, follows the trend observed with the effect of current density. No change was observed in Ni 2p XPS peak positions after stability tests at different temperatures, while the intensity of Fe 2p features reduces at 80 °C (Fig. S10a and b†). Similar to the effect of current density, Cr species are completely removed from the electrode structure (Fig. S10c†), and the O 1s signal shifted to lower binding energies (Fig. S10d†) with increasing the temperature. Thus, the surface reconstruction and phase transformation to the FeNi oxyhydroxide phase happen to the same degree as with the effect of temperature.
Compared to the effect of current density, increasing the temperature has a slightly bigger impact on the stability performance of FeNiCr/FF. The dissolution of Cr is slightly lower, however, more Fe is lost into the electrolyte with increasing the temperature to 80 °C, which along with the decreased intensity of Fe 2p XPS features entails that Fe dissolution is impacted more by the effect of temperature. Therefore, in addition to the morphology change and phase transformation, Fe dissolution contributes to the reduced performance observed for the FeNiCr catalyst. In addition, while the increase in temperature does not affect the removal of fine nanostructures and bigger nanoparticles of FeNiCr hydroxides as much as the effect of high current density, more physical detachment and surface cracking occurs at higher temperatures, i.e., 80 °C.
Effect of alkaline electrolyte concentration
To investigate the effect of the concentration of alkaline electrolyte on the stability performance, the KOH concentration was increased from 1 M to 5 M and 10 M at 25 °C and 100 mA cm−2. The change in morphology at the FeNiCr/FF surface is the highest with respect to the effects of current density and temperature. Fig. 6a shows that both particles and surface fine nanostructures are highly aggregated upon increasing the KOH concentration. This explains the following highest decline observed in the performance of FeNiCr/FF in 10 M KOH after the three-day stability test compared to that at 1000 mA cm−2 and 80 °C. A clear stability decay is observed with increasing the KOH concentration after a three-day stability test (Fig. 6b). The corresponding OER LSV curves also demonstrate an increasing activity decline and enhanced overpotentials after stability tests at the fixed KOH concentrations (Fig. S11†). In particular, the intensity of the Ni2+–Ni3+ oxidation peak between 1.3 and 1.4 V vs. RHE is significantly decreased after the stability test in 10 M KOH, alluding to the loss of Ni active sites.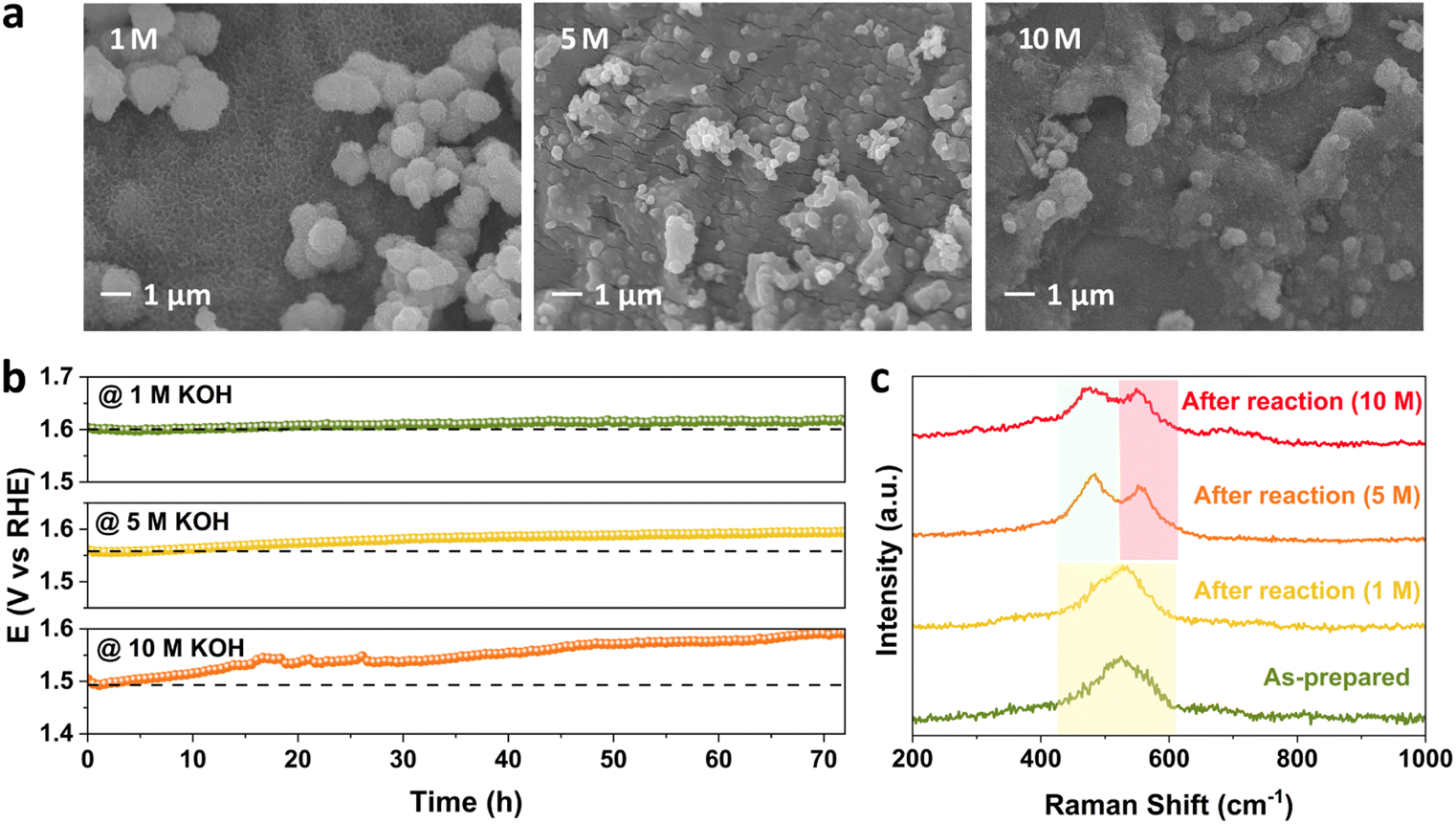 | ||
| Fig. 6 Monitoring the stability, morphology and structure of FeNiCr/FF during the OER with independently controlled concentration of electrolyte from 1 M to 10 M. (a) SEM images, (b) chronopotentiometry profiles, and (c) Raman spectra. | ||
Besides the physical loss of the catalyst film, metal dissolution is significantly higher at high alkaline electrolyte concentrations, resulting in a severe decrease in surface active sites and the active surface area. Relatively similar dissolution profiles are obtained by ICP-MS for Cr, Fe, and Ni metals, however with much higher rates for all metals (Fig. S12†). Also, while the gap between Cr and Ni dissolution profiles is particularly higher upon increasing the KOH concentration, a sharp increase in Fe dissolution is observed in 10 M KOH electrolyte, suggesting that the Fe dissolution is a major issue for Fe-containing and Fe-supported catalysts among other factors like catalyst detachment and phase transformation. This also explains why FeNi is more stable under industrially relevant conditions. With increasing KOH concentration, FeNiCr has more vacancies left due to the high solubility of Cr, resulting in more exposure and dissolution of Fe and Ni sites. In addition to this observation on the significant impact of high alkaline electrolyte concentrations, the amount of re-deposited Fe onto the counter electrode does not change due to the fixed amount of moderate current density applied, and hence does not affect the Fe dissolution trends owing to the effects of temperature and alkaline electrolyte concentration where comparable amounts of dissolved Fe are found. Even the sharp rise of Fe dissolution in 10 M KOH would not be influenced by Fe re-deposition, further indicating the electrolyte concentration plays the key role in the stability decline under industrially relevant conditions.
The trend in phase transformation is similar in 5 M KOH, but in 10 M KOH, the Ni–O vibrations dampen and broaden due to the higher metal dissolution, while the intensity difference between the two peaks decreases, alluding to the retention of the β-NiOOH phase at the surface of FeNiCr besides the formation of the γ-NiOOH phase in concentrated alkaline media (Fig. 6c). Lack of any significant change in the Fe 2p and Ni 2p XPS peak positions indicates the retained electronic structure of the remaining surface active sites (Fig. S13a and b†). Both the Cr loss and variation in O atoms are similar to the effects of current density and temperature (Fig. S13c and d†).
Herein, although adding the third metal Cr has been proven beneficial for the intrinsic activity of binary FeNi catalysts, it does not benefit the performance under simulated industrial conditions. The enhancement of energy input and charge carrier transport upon increasing temperature and electrolyte concentration, respectively, only improves the initial activity of the as-prepared structure which does not address the intrinsic activity of Fe and Ni active sites. Both activity and stability decrease due to the significant Cr loss at the early stage of the OER, enhanced Fe dissolution at high alkaline electrolyte concentrations, phase transformation to a mixed stated of γ- and β-NiOOH phases, and change in surface morphology. As a result, activity decay and stability decline are higher for the ternary FeNiCr hydroxides than the well-established binary FeNi hydroxide catalysts under industrially relevant conditions. This necessitates future strategies for developing multimetallic FeNi-based catalysts to improve the activity while maintaining the long-term stability of alkaline water oxidation. It is recommended to retain an appropriate electrolyte concentration for augmenting the charge carrier and mass transport, while fixing the temperature and current density to meet the operation conditions and activity in water electrolysers, thus protecting the catalyst from degradation and physical loss at high alkaline electrolyte concentrations. Regulating the electronic structure, surface area, morphology and hetero-interface engineering should be practiced with other catalysts not only to demonstrate the higher impact of alkaline electrolyte concentration on the stability decline, but also to mitigate the undesirable phase transformation and metal dissolution for the long term.
Conclusions
We demonstrate the importance of the evaluation of catalysts in conditions close to those in alkaline water electrolysers. Introducing a third metal as a co-active site or dopant has been demonstrated to be useful in recent years. However, improving the performance is limited only to standard lab-scale conditions. Here, we demonstrate that the activity enhancement of FeNi hydroxides as OER catalysts in alkaline media by the addition of Cr is in fact not valid under the industrially relevant conditions met in AWEs, including large current densities, elevated temperature, and concentrated alkaline electrolytes. The dissolution of the added Cr is fast and significant at the beginning of the stability test, and surface morphology transforms severely involving catalyst detachment and aggregation. The simulated industrial conditions of AWEs indicate the phase transformation to γ-NiOOH unlike the formation of β-NiOOH as the more active phase for the OER under standard conditions. Although the remaining Cr species continue to induce the generation of β-NiOOH and particularly at high alkaline electrolyte concentrations result in a mixed phase state, the increased phase transformation to γ-NiOOH by elevating the current density and temperature, along with metal dissolution and surface morphology change contribute more to deactivation and stability decline. By investigating the effects of current density, temperature, and alkaline electrolyte concentration individually, the alkaline electrolyte concentration is shown to have the highest impact on the stability performance and structural change of FeNiCr followed by temperature. The impact of the current density is noticed only at large values. This study sheds light on the importance of electrolytic conditions and instability mechanisms met in traditional alkaline water electrolysers for catalyst development and the required durability in industrial water electrolysis applications.Author contributions
Yang Xiao: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft. Kamran Dastafkan: conceptualization, formal analysis, project administration, writing – review & editing. Zhen Su: formal analysis, investigation. Chengli Rong: formal analysis, investigation. Chuan Zhao: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
The authors declare no competing conflicts of interest.Acknowledgements
The authors thank the UNSW Mark Wainwright Analytical Centre (MWAC) at UNSW for all characterization instruments. This study was financed by the Australian Research Council (DP220103294, IC200100023 and CE230100017).References
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- B. Zhong, P. Kuang, L. Wang and J. Yu, Appl. Catal., B, 2021, 299, 120668 CrossRef CAS.
- X. Liu, R. Guo, K. Ni, F. Xia, C. Niu, B. Wen, J. Meng, P. Wu, J. Wu, X. Wu and L. Mai, Adv. Mater., 2020, 32, e2001136 CrossRef PubMed.
- Q. Wen, K. Yang, D. Huang, G. Cheng, X. Ai, Y. Liu, J. Fang, H. Li, L. Yu and T. Zhai, Adv. Energy Mater., 2021, 11, 2102353 CrossRef CAS.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C.-J. Sun, L. Li, D. Sokaras, C. Dong, S.-Z. Qiao, X.-W. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- L. Peng, N. Yang, Y. Yang, Q. Wang, X. Xie, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Angew. Chem., Int. Ed., 2021, 60, 24612–24619 CrossRef CAS PubMed.
- R. Chen, S.-F. Hung, D. Zhou, J. Gao, C. Yang, H. Tao, H. B. Yang, L. Zhang, L. Zhang, Q. Xiong, H. M. Chen and B. Liu, Adv. Mater., 2019, 31, 1903909 CrossRef CAS PubMed.
- Z. Lu, W. Xu, W. Zhu, Q. Yang, X. Lei, J. Liu, Y. Li, X. Sun and X. Duan, Chem. Commun., 2014, 50, 6479–6482 RSC.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- H. Liang, A. N. Gandi, C. Xia, M. N. Hedhili, D. H. Anjum, U. Schwingenschlögl and H. N. Alshareef, ACS Energy Lett., 2017, 2, 1035–1042 CrossRef CAS.
- K. Dastafkan, S. Wang, C. Rong, Q. Meyer, Y. Li, Q. Zhang and C. Zhao, Adv. Funct. Mater., 2022, 32, 2107342 CrossRef CAS.
- X. Bo, Y. Li, R. K. Hocking and C. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 41239–41245 CrossRef CAS.
- X. Wan, Y. Song, H. Zhou and M. Shao, Energy Mater. Adv., 2022, 2022, 4225–4237 Search PubMed.
- X. Bo, R. K. Hocking, S. Zhou, Y. Li, X. Chen, J. Zhuang, Y. Du and C. Zhao, Energy Environ. Sci., 2020, 13, 4225–4237 RSC.
- Y. Yang, L. Dang, M. J. Shearer, H. Sheng, W. Li, J. Chen, P. Xiao, Y. Zhang, R. J. Hamers and S. Jin, Adv. Energy Mater., 2018, 8, 1703257 Search PubMed.
- K. N. Dinh, P. Zheng, Z. Dai, Y. Zhang, R. Dangol, Y. Zheng, B. Li, Y. Zong and Q. Yan, Small, 2018, 14, 1703257 CrossRef.
- Y. Duan, Z.-Y. Yu, S.-J. Hu, X.-S. Zheng, C.-T. Zhang, H.-H. Ding, B.-C. Hu, Q.-Q. Fu, Z.-L. Yu, X. Zheng, J.-F. Zhu, M.-R. Gao and S.-H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed.
- K. Dastafkan, Q. Meyer, X. Chen and C. Zhao, Small, 2020, 16, 2002412 CrossRef CAS.
- C.-X. Zhao, B.-Q. Li, M. Zhao, J.-N. Liu, L.-D. Zhao, X. Chen and Q. Zhang, Energy Environ. Sci., 2020, 13, 1711–1716 RSC.
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 14, 1800136 CrossRef PubMed.
- C. Xuan, J. Wang, W. Xia, J. Zhu, Z. Peng, K. Xia, W. Xiao, H. L. Xin and D. Wang, J. Mater. Chem. A, 2018, 6, 7062–7069 RSC.
- F. Zeng, C. Mebrahtu, L. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef CAS.
- F.-Y. Chen, Z.-Y. Wu, Z. Adler and H. Wang, Joule, 2021, 5, 1704–1731 CrossRef CAS.
- H. Zhang, X. Li, A. Hähnel, V. Naumann, C. Lin, S. Azimi, S. L. Schweizer, A. W. Maijenburg and R. B. Wehrspohn, Adv. Funct. Mater., 2018, 28, 1706847 CrossRef.
- S. Yang, R. Du, Y. Yu, Z. Zhang and F. Wang, Nano Energy, 2020, 77, 105057 CrossRef CAS.
- S. Niu, W.-J. Jiang, T. Tang, L.-P. Yuan, H. Luo and J.-S. Hu, Adv. Funct. Mater., 2019, 29, 1902180 CrossRef.
- Y. Xiao, K. Dastafkan, Y. Li, T. Zhao, Z. Su, H. Qi and C. Zhao, ACS Sustain. Chem. Eng., 2022, 10, 8597–8604 CrossRef CAS.
- M. Etzi Coller Pascuzzi, A. J. W. Man, A. Goryachev, J. P. Hofmann and E. J. M. Hensen, Catal. Sci. Technol., 2020, 10, 5593–5601 RSC.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- G. F. Swiegers, R. N. L. Terrett, G. Tsekouras, T. Tsuzuki, R. J. Pace and R. Stranger, Sustainable Energy Fuels, 2021, 5, 1280–1310 RSC.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- S. Klaus, L. Trotochaud, M.-J. Cheng, M. Head-Gordon and A. T. Bell, ChemElectroChem, 2016, 3, 66–73 CrossRef CAS.
- F. D. Speck, K. E. Dettelbach, R. S. Sherbo, D. A. Salvatore, A. Huang and C. P. Berlinguette, Chem, 2017, 2, 590–597 CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- M. Li, Y. Xiong, X. Liu, X. Bo, Y. Zhang, C. Han and L. Guo, Nanoscale, 2015, 7, 8920–8930 RSC.
- F. Bao, E. Kemppainen, I. Dorbandt, F. Xi, R. Bors, N. Maticiuc, R. Wenisch, R. Bagacki, C. Schary, U. Michalczik, P. Bogdanoff, I. Lauermann, R. van de Krol, R. Schlatmann and S. Calnan, ACS Catal., 2021, 11, 10537–10552 CrossRef CAS.
- C. Andronescu, S. Barwe, E. Ventosa, J. Masa, E. Vasile, B. Konkena, S. Möller and W. Schuhmann, Angew. Chem., Int. Ed., 2017, 56, 11258–11262 CrossRef CAS PubMed.
- C. Andronescu, S. Seisel, P. Wilde, S. Barwe, J. Masa, Y. T. Chen, E. Ventosa and W. Schuhmann, Chemistry, 2018, 24, 13773–13777 CrossRef CAS PubMed.
- W. Zhao, H. Xu, H. Luan, N. Chen, P. Gong, K. Yao, Y. Shen and Y. Shao, Adv. Energy Mater., 2022, 12, 2102372 CrossRef CAS.
- Y. Duan, J. Y. Lee, S. Xi, Y. Sun, J. Ge, S. J. H. Ong, Y. Chen, S. Dou, F. Meng, C. Diao, A. C. Fisher, X. Wang, G. G. Scherer, A. Grimaud and Z. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 7418–7425 CrossRef CAS PubMed.
- S. Anantharaj, H. Sugime and S. Noda, ACS Appl. Energy Mater., 2020, 3, 12596–12606 CrossRef CAS.
- P. W. Menezes, S. Yao, R. Beltrán-Suito, J. N. Hausmann, P. V. Menezes and M. Driess, Angew. Chem., Int. Ed., 2021, 60, 4640–4647 CrossRef CAS PubMed.
- X. Bo, Y. Li, R. K. Hocking and C. Zhao, ACS Appl. Mater. Interfaces, 2017, 9, 41239–41245 CrossRef CAS PubMed.
- M. H. Wang, Z. X. Lou, X. Wu, Y. Liu, J. Y. Zhao, K. Z. Sun, W. X. Li, J. Chen, H. Y. Yuan, M. Zhu, S. Dai, P. F. Liu and H. G. Yang, Small, 2022, 18, 2200303 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta03905f |
| This journal is © The Royal Society of Chemistry 2023 |