
Crafting core–shell heterostructures of carbon nanotubes and N,O-coordinated cobalt site-impregnated conjugated porous polymers for highly efficient oxygen reduction†
Rui
Ma‡
b,
Zhengjiao
Xie‡
b,
Xiaoxue
Xu
a,
Chenglong
Tang
b,
Xueqin
Liu
a,
Yunhai
Zhu
 a,
Xun
Cui
*a and
Yingkui
Yang
*ab
a,
Xun
Cui
*a and
Yingkui
Yang
*ab
aState Key Laboratory of New Textile Materials and Advanced Processing Technologies, Wuhan Textile University, Wuhan 430200, China. E-mail: xcui@wtu.edu.cn; ykyang@wtu.edu.cn
bSchool of Chemistry and Materials Science, South-Central Minzu University, Wuhan 430074, China
First published on 20th June 2023
Abstract
The ability to develop high-efficiency carbon-based single-metal-atom catalysts (SMACs) via facile routes represents an important endeavor toward high-performance oxygen reduction reaction (ORR). Herein, we report a simple yet viable one-step pyrolysis-free approach via cobalt ion-mediated in situ Schiff-base condensation of π-conjugated monomers in the presence of highly conductive carbon nanotubes (CNTs) for crafting core–shell coaxially heterostructured CNTs/N,O-coordinated cobalt site-impregnated conjugated porous polymers (denoted as CNTs/Co–N,O-CPs) as a superior electrocatalyst for highly efficient ORR. Notably, the CNTs/Co–N,O-CPs possess an advantageous 1D heterostructure with abundant hierarchical porosity, thereby greatly favoring the efficient exposure of active sites and fast electron and mass transport. More importantly, theoretical calculations reveal that the strong orbital interaction between central Co and coordinated O atoms considerably optimizes the adsorption strengths of ORR-relevant intermediates, accounting for the markedly enhanced intrinsic activity of CNTs/Co–N,O-CPs. Consequently, the CNTs/Co–N,O-CPs manifest superior performance for the ORR (i.e., onset potential of 1.004 V and half-wave potential of 0.868 V) and zinc–air batteries (i.e., maximal power density of 181 mW cm−2 and specific capacity of 814 mA h gZn−1) in an alkaline medium, outperforming most previously reported carbon-based SMACs and even surpassing the state-of-the-art Pt/C catalyst. As such, the rationally crafted pyrolysis-free CNTs/Co–N,O-CPs may stand out as a robust ORR electrocatalyst for high-performance energy conversion devices.
 Xun Cui | Dr Xun Cui is currently a Professor at the State Key Laboratory of New Textile Materials and Advanced Processing Technologies, Wuhan Textile University. He received his PhD degree in Chemical Engineering and Technology from Chongqing University in 2018, and then worked as an assistant professor at South-Central Minzu University. After being a postdoctoral research fellow in Prof. Zhiqun Lin's group at the Georgia Institute of Technology from 2018 to 2020, he joined the Faculty of Materials Science and Chemistry at the China University of Geosciences (Wuhan) as a research professor in 2021. He then moved to Wuhan Textile University as a professor in 2022. His research interests include advanced materials synthesis, electrocatalysis, photoelectrocatalysis, and renewable energy conversion and storage devices. He has published 60+ peer-reviewed journal articles with more than 3900 citations. He was recognized by the editors as an outstanding reviewer for the Journal of Materials Chemistry A in 2019. |
Introduction
The sluggish kinetics of oxygen electrocatalysis represent one of the key challenges in various oxygen-related sustainable energy conversion technologies, such as metal–air batteries, fuel cells, and water electrolysis.1–3 Therefore, the ability to develop highly efficient electrocatalysts is of practical interest for enabling high-performance electrochemical energy conversion.4–6 To date, Pt-based metals are widely considered to be state-of-the-art electrocatalysts for the oxygen reduction reaction (ORR).7,8 However, the scarce reserves have greatly impeded their practical utilization.9,10 As such, it is highly desirable to exploit noble-metal-free alternatives with high electrocatalytic ORR performance.In this context, over the past several years, single-metal-atom catalysts (SMACs) have garnered extensive interest due to their high atomic utilization, unique electronic structure, and considerable electrocatalytic activity.11–13 In particular, carbon-based SMACs (M–N–C, M = Fe, Co, Ni, Cu, etc.) exhibit great potential in the field of the electrocatalytic ORR because of their favourable oxygen absorption, electrical conductivity, and structural robustness.14–16 However, despite great progress, the design and synthesis of highly efficient carbon-based SMACs remain challenging. On the one hand, the central metal sites of most carbon-based SMACs reported to date are limited to the porphyrin-like M–N4 configuration in which the coordination microenvironment has not been optimized, thereby greatly hindering the further enhancement of intrinsic electrocatalytic ORR activity.17,18 On the other hand, carbon-based SMACs are currently widely prepared by a high-temperature pyrolysis (usually >700 °C) of the pre-synthesized precursors.19–21 Nevertheless, the pyrolysis approach often tends to cause the aggregation of metal sites and a change in the pre-designed coordination microenvironment, giving rise to extremely complex and poorly defined active sites and consequently undesirable electrocatalytic ORR activity. Moreover, the implementation of pyrolysis also leads to the collapse of the pre-crafted carbon matrix and poor reproducibility of the resultant electrocatalysts. To this end, it is highly desirable to invoke a pyrolysis-free strategy for producing carbon-based SMACs, particularly with a rationally tailored metal atomic centre and coordination microenvironment.
Recently, pyrolysis-free conjugated porous polymers (CPs) have been explored as a promising platform for electrocatalysis, owing to their high charge-carrier mobility, small band gap, good thermodynamic stability, as well as precisely controllable capabilities regarding active site modulation and porosity regulation.22–27 In sharp contrast to conventional pyrolyzed carbon-based SMACs, the absence of high-temperature pyrolysis in the preparation process endows the resultant CPs with well-preserved active sites and microstructures, hence ensuring a clear structure–property relationship. Previous studies have shown that the orbital hybridization between the π-conjugated ligands and the d-block transition metal sites is capable of effectively facilitating the delocalization of electrons and prominently enhancing the stability.28 To date, despite some metallophthalocyanine- and metalloporphyrin-containing CP-based SMACs with well-defined M–N4 active sites being reported in the literature, their catalytic performances are still inferior to that of the benchmark Pt/C catalyst.29,30 Notably, crystal and ligand field theory suggests that the splitting and binding energies of a given four-coordinated central metal site depend primarily on the surrounding ligands (i.e., coordination atoms).31 Accordingly, rationally tailoring the coordination atoms may give rise to a unique d orbital electronic configuration of central metal sites, hence further enhancing the intrinsic electrocatalytic ORR activity of pyrolysis-free CP-based SMACs. This, however, remains challenging and is yet to be largely explored.
Herein, we report a simple yet viable one-step pyrolysis-free route for crafting core–shell coaxially heterostructured carbon nanotubes/N,O-coordinated cobalt site-impregnated conjugated porous polymers (i.e., CNTs/Co–N,O-CPs) that function as a superior electrocatalyst for high-efficiency ORR. Specifically, the cobalt ion-mediated in situ Schiff-base condensation of π-conjugated monomers in the presence of highly conductive CNTs directly yields the CNTs/Co–N,O-CP electrocatalyst. Notably, well-defined Co–N2O2 sites are supported by a polymeric matrix as a Co–N,O-CP shell, which wraps on the surface of the 1D CNT core, forming core–shell coaxially heterostructured CNTs/Co–N,O-CPs. Intriguingly, the highly conjugated structure and large specific surface area of the CNT core impart CNTs/Co–N,O-CPs with an advantageous 1D heterostructure with abundant hierarchical porosity and high charge mobility, thereby greatly favouring the efficient exposure of active sites and fast electron and mass transport. Moreover, theoretical calculations signify that the strong orbital interaction between central Co and coordinated O atoms in CNTs/Co–N,O-CPs could optimize the adsorption strengths of the ORR-relevant intermediates, thereby lowering the reaction energy barrier of the ORR. Consequently, the CNTs/Co–N,O-CPs deliver superior electrocatalytic ORR performance with onset and half-wave potentials of 1.004 V and 0.868 V, respectively, and long-term stability under alkaline conditions. Finally, CNTs/Co–N,O-CPs are implemented in zinc–air batteries as an air cathode, manifesting a maximal power density of 181 mW cm−2, a superior specific capacity of 814 mA h gZn−1, and outstanding durability. As such, crafting core–shell coaxially heterostructured CNTs/Co–N,O-CPs via pyrolysis-free synthesis represents a facile strategy for highly efficient ORR electrocatalysts, and in turn, high-efficiency air cathodes for zinc–air batteries.
Results and discussion
As depicted in Fig. 1a, the synthetic route to the core–shell coaxially heterostructured CNTs/Co–N,O-CP electrocatalyst involves a cobalt ion-mediated one-step in situ Schiff-base condensation between 1,3,5-triformylphloroglucinol (TP) and 1,2-diaminobenzene (DB) building blocks in the presence of well-dispersed CNTs. During the synthesis, both π-conjugated TP and DB molecules can be well attached to the surface of CNTs through the van der Waals interaction, thereby enabling the uniform wrapping of the in situ polymerized Co–N,O-CP shell on the CNT core. The Fourier transform infrared (FTIR) spectrum of the resulting CNTs/Co–N,O-CPs (Fig. S1a†) exhibits the disappearance of characteristic peaks at 3386 cm−1 and 3363 cm−1 (–NH2 group) and 1643 cm−1 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group) and the appearance of new characteristic signals at 1625 cm−1 (CO), 1577 cm−1 (CC), and 1253 cm−1 (C–N), signifying the complete transformation of the aldehyde and amine moieties and the successful polymerization of CPs. Moreover, the peaks in the wave number range of 550–670 cm−1 and 400–490 cm−1 can be assigned to the Co–N and Co–O bonds, respectively, suggesting the existence of N/O-coordinated Co species in CNTs/Co–N,O-CPs (Fig. S1b†).32,33 Note that the CNTs/N,O-CPs (control sample) that are synthesized in the absence of cobalt ions show similar FTIR results except for the absence of characteristic peaks corresponding to Co–N and Co–O bonds (Fig. S2†).
O group) and the appearance of new characteristic signals at 1625 cm−1 (CO), 1577 cm−1 (CC), and 1253 cm−1 (C–N), signifying the complete transformation of the aldehyde and amine moieties and the successful polymerization of CPs. Moreover, the peaks in the wave number range of 550–670 cm−1 and 400–490 cm−1 can be assigned to the Co–N and Co–O bonds, respectively, suggesting the existence of N/O-coordinated Co species in CNTs/Co–N,O-CPs (Fig. S1b†).32,33 Note that the CNTs/N,O-CPs (control sample) that are synthesized in the absence of cobalt ions show similar FTIR results except for the absence of characteristic peaks corresponding to Co–N and Co–O bonds (Fig. S2†).
 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of CNTs/Co–N,O-CPs. (b) SEM, (c) TEM, SAED, (d) HAADF-STEM, and (e) HAADF-STEM and EDX elemental mapping images of CNTs/Co–N,O-CPs. | ||
The morphology and microstructure of CNTs/Co–N,O-CPs were first examined by scanning and transmission electron microscopy (SEM and TEM). Unlike the pristine CNTs with a smooth surface (Fig. S3 and S4†), the CNTs/Co–N,O-CPs show that a seamless Co–N,O-CP shell with a thickness of approximately 4 nm is uniformly wrapped around the CNT core (Fig. 1b, c and S5†). Meanwhile, no visible Co-related nanoparticles can be detected, which may indicate the atomic dispersion of Co species. Besides, the X-ray diffraction (XRD) pattern of CNTs/Co–N,O-CPs only exhibits two peaks at around 24° and 44° (Fig. S6†), corresponding to the (002) and (100) planes of CNTs, respectively, which is in good accordance with the selected-area electron diffraction (SAED) pattern (inset of Fig. 1c).34,35 The absence of diffraction signals corresponding to Co-related crystalline structures in both XRD and SAED patterns is well in line with the SEM and TEM observations. Furthermore, the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of CNTs/Co–N,O-CPs confirms that the Co species are atomically dispersed throughout the entire polymeric matrix (Fig. 1d). The actual loading of Co is determined to be 5.6 wt% by inductively coupled plasma mass spectrometry (ICP-MS). Moreover, the combined energy-dispersive X-ray spectroscopy (EDX) area-scan and line-scan profiles (Fig. 1e and S7†) and electron energy loss spectroscopy (EELS) analysis (Fig. S8†) further support the core–shell heterostructure of CNTs/Co–N,O-CPs and the homogeneous dispersion of C, N, O, and Co.
X-ray photoelectron spectroscopy (XPS) was then used to obtain more compositional information on CNTs/Co–N,O-CPs (Fig. 2a, b and S9†). As displayed in the high-resolution Co 2p XPS spectrum (Fig. 2a), two peaks at binding energies of 796.6 eV and 781.3 eV can be assigned to Co 2p1/2 and Co 2p3/2, respectively, which are very close to those of Co2+ (typically at around 796.8 eV and 781.7 eV), suggesting that the Co species in CNTs/Co–N,O-CPs are positively charged and their average valence state is approximately +2.36 It is noteworthy that the absence of signals corresponding to Co0 (typically at around 778.5 eV) rules out the presence of aggregated Co species, which agrees well with the aforementioned XRD, SAED, and electron microscope observations.36 In addition, two deconvoluted peaks at binding energies of 399.2 eV and 400.2 eV in the high-resolution N 1s XPS spectrum of CNTs/Co–N,O-CPs (Fig. 2b) correspond to Co–N and C–N, respectively.37 Meanwhile, the high-resolution O 1s XPS spectrum of CNTs/Co–N,O-CPs (Fig. S10†) exhibits three deconvoluted peaks at 531.4 eV, 532.2 eV, and 533.9 eV, which can be ascribed to Co–O, CO, and C–O bonds, respectively. These results further support the presence of N/O-coordinated Co species, which is in good agreement with the FTIR observation.
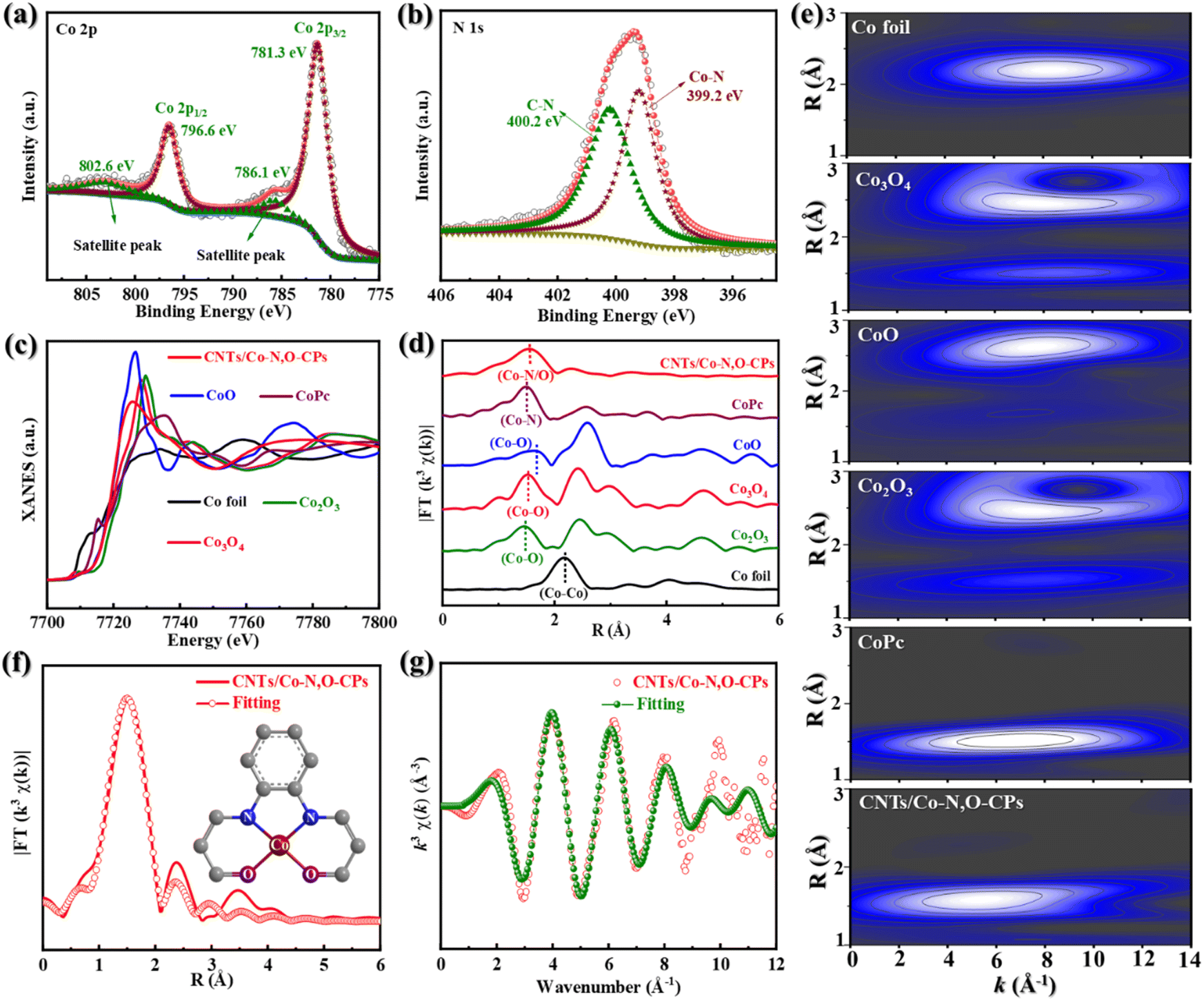 | ||
| Fig. 2 High-resolution XPS spectra of (a) Co 2p and (b) N 1s of CNTs/Co–N,O-CPs. (c) Co K-edge XANES spectra, (d) FT-EXAFS spectra, and (e) WT-EXAFS images of CNTs/Co–N,O-CPs, Co foil, CoO, Co3O4, Co2O3 and CoPc. EXAFS fitting curves of CNTs/Co–N,O-CPs in (f) R space and (g) k space. | ||
Furthermore, Co K-edge extended X-ray absorption fine structure (EXAFS) characterization studies were performed to elucidate detailed structural information on the N/O-coordinated Co species in CNTs/Co–N,O-CPs. As exhibited in Fig. 2c, the X-ray absorption near-edge structure (XANES) spectrum of CNTs/Co–N,O-CPs shows that its absorption edge position is close to that of CoO, revealing that the valence state of positively charged Co species in CNTs/Co–N,O-CPs is around +2. This observation is in good agreement with the XPS results. In addition, the Fourier-transform EXAFS (FT-EXAFS) spectrum of CNTs/Co–N,O-CPs only exhibits one dominant peak at 1.53 Å, which can be assigned to the Co–N/O first-shell coordination (Fig. 2d). Meanwhile, no Co–Co characteristic peaks can be detected for CNTs/Co–N,O-CPs, implying the atomic dispersion of N/O-coordinated Co species. Notably, the FT-EXAFS spectrum of CNTs/Co–N,O-CPs is similar to that of Co phthalocyanine (CoPc), and clearly distinct from that of Co foil, CoO, Co3O4, and Co2O3, demonstrating similar coordination structures to those of N/O-coordinated Co species in CNTs/Co–N,O-CPs and Co–N4 sites in CoPc. The wavelet-transform EXAFS (WT-EXAFS) analysis reveals that both CNTs/Co–N,O-CPs and CoPc show an intensity maximum at a similar R space of ∼1.5 Å and different k spaces of ∼5.5 Å−1 and ∼6.6 Å−1 (Fig. 2e), signifying that their coordination structures are slightly different. Meanwhile, in comparison with Co foil, CoO, Co3O4, and Co2O3, the absence of an intensity maximum corresponding to the Co–Co path (at a k space of ∼8.0 Å−1) in CNTs/Co–N,O-CPs excludes the presence of Co-related crystalline structures. Additionally, the EXAFS fitting of CNTs/Co–N,O-CPs was carried out to obtain detailed coordination information on N/O-coordinated Co species (Fig. 2f, g and Table S1†). The best-fitting curves and parameters clearly indicate that the first-shell peak at 1.53 Å can be attributed to an atomically dispersed Co site coordinating with two N atoms and two O atoms as the Co–N2O2 structure.
We then turn our attention to the electrocatalytic ORR activity of CNTs/Co–N,O-CPs. For comparison, Co site-free CNTs/N,O-CPs, Co–N4 site-containing CNTs/Co–N-CPs, and commercial Pt/C as the references were tested as well. Note that the Co–N4 site-containing CNTs/Co–N-CPs were synthesized using π-conjugated tetraaminophenyl porphyrin (TAPP) and terephthalaldehyde (TPA) as the building blocks (Fig. S11–S13, see the Experimental section in the ESI† for more details). As presented in the cyclic voltammetry (CV) profiles (Fig. 3a), the CNTs/Co–N,O-CPs show an ORR peak at 0.852 V, which is more positive than those of CNTs/N,O-CPs (0.788 V), CNTs/Co–N-CPs (0.815 V) and Pt/C (0.848 V) in O2-saturated 0.1 M KOH, implying a superior electrocatalytic ORR activity of CNTs/Co–N,O-CPs. The linear sweep voltammetry (LSV) curves measured at 1600 rpm using a rotating disk electrode (RDE) are depicted in Fig. 3b. Compared to CNTs/N,O-CPs, CNTs/Co–N-CPs and Pt/C, the ORR activity of the CNTs/Co–N,O-CPs is clearly the highest as evidenced by the largest onset potential (Eonset, 1.004 V) and half-wave potential (E1/2, 0.868 V), although its limited-diffusion current density at 0.2 V (Jd @ 0.2 V, 6.30 mA cm−2) is slightly lower than that of Pt/C (6.36 mA cm−2) (Fig. 3c). Intriguingly, the estimated Tafel slope of CNTs/Co–N,O-CPs (87 mV dec−1) is comparable to that of Pt/C (81 mV dec−1) (Fig. S14†), manifesting its favourable ORR kinetics. To our knowledge, the electrocatalytic ORR activity of CNTs/Co–N,O-CPs is one of the best among various, most advanced electrocatalysts reported in the literature (Table S2†).
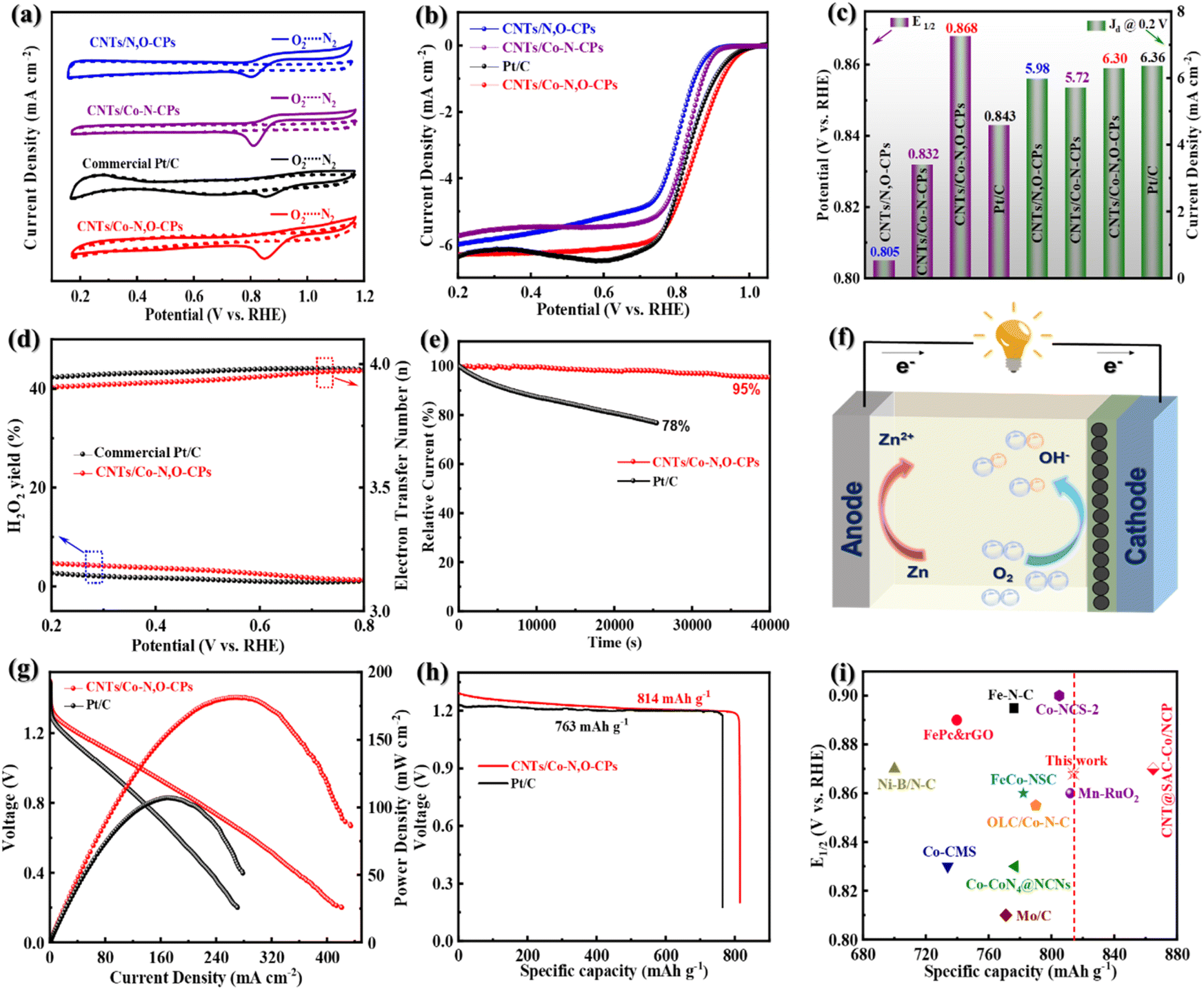 | ||
| Fig. 3 (a) CV curves, (b) LSV curves, and (c) E1/2 and Jd @ 0.2 V of CNTs/Co–N,O-CPs, CNTs/Co–N-CPs, CNTs/N,O-CPs, and Pt/C. (d) Peroxide yields, electron transfer numbers and (e) chronoamperometric responses of CNTs/Co–N,O-CPs and Pt/C. (f) Schematic diagram of a zinc–air battery. (g) Discharge polarization curves, the corresponding power density plots and (h) specific discharging capacities of zinc–air batteries with CNTs/Co–N,O-CPs and Pt/C as the cathode catalysts. (i) Comparison of the E1/2 and specific capacities of CNTs/Co–N,O-CPs and other previously reported state-of-the-art carbon-based electrocatalysts. | ||
Notably, the Co site-free CNTs/N,O-CPs (Fig. S15–S17†) has Brunauer–Emmett–Teller (BET) specific surface area and pore-size distribution comparable to those of CNTs/Co–N,O-CPs (Fig. S17 and S18†), and displays markedly lower electrocatalytic ORR performance (Fig. 3b and c), signifying that the exceptional activity of CNTs/Co–N,O-CPs primarily stems from the Co–N2O2 sites. The slightly lower BET specific surface area of CNTs/N,O-CPs (164 m2 g−1) than that of CNTs/Co–N,O-CPs (197 m2 g−1) (Table S3†) may be due to the influence of the mediating effect of cobalt ions for the in situ Schiff-base condensation process between TP and DB monomers on the microstructure of the resultant conjugated polymer. As revealed in Fig. S17 and S18,† the presence of cobalt ions for CNTs/Co–N,O-CPs in the synthesis process increases the micropore volume and decreases the meso- and macropore volumes, collectively resulting in a slight improvement of the BET specific surface area. It should also be noted that the CNTs/Co–N-CPs (control sample), similar to CNTs/Co–N,O-CPs, features abundant hierarchical porosity and a high BET specific surface area of 201 m2 g−1 (Fig. S19 and Table S3†), further indicating that the Co–N2O2 sites play a more critical role in catalysing oxygen reduction than the Co–N4 sites.
As previously reported in the literature, the pyrolysis-free CPs generally possess electrical conductivity inferior to that of the conventional inorganic electrocatalytic materials.24,30 In addition, the pyrolysis-free CPs usually exhibit a densely stacked architecture with inadequate hierarchical porosity.26,27,36 These characteristics lead to unsatisfactory activity, which limits their applications in electrocatalysis to some extent. Intriguingly, the core–shell coaxially heterostructured CNTs/Co–N,O-CPs in this work exhibit excellent ORR performance, suggesting that CNTs may also play a critical role in boosting the ORR process. To study the effect of CNTs on ORR activity, the control sample of Co–N,O-CPs was synthesized via a similar procedure in the absence of CNTs (Fig. S20–S22, see the Experimental section in the ESI† for more details). In sharp contrast to the 1D core–shell coaxially heterostructured CNTs/Co–N,O-CPs with abundant hierarchical porosity (Fig. 1 and S18†), the Co–N,O-CPs display a morphology of densely stacked powders (Fig. S20†) and thus a significantly lower BET specific surface area (Fig. S22†), thereby preventing the exposure of Co–N2O2 sites and hindering the rapid electron and mass transport. Consequently, the Co–N,O-CPs deliver an extremely poor ORR activity (Fig. S23†). The above results reveal that the construction of interfacial electron transfer channels and hierarchical porosity via compositing pyrolysis-free Co–N,O-CPs with highly conductive CNTs with large specific surface area to form core–shell heterostructured CNTs/Co–N,O-CPs is an effective approach to significantly improve the ORR performance of pyrolysis-free Co–N,O-CPs.
To evaluate the ORR pathway, the LSV curves at various rotating speeds were recorded using a RDE, and the corresponding Koutecky–Levich (K–L) plots were obtained (Fig. S24–S26†). Clearly, all the K–L plots display good linearity in the potential range of 0.2 V to 0.6 V, demonstrating a first-order reaction kinetics for the ORR.38 The average electron transfer number (n) of CNTs/Co–N,O-CPs was determined to be 3.93, fairly close to the theoretical value of benchmark Pt/C (i.e., 4.0), suggesting a nearly four-electron ORR pathway. To further corroborate the four-electron ORR mechanism of CNTs/Co–N,O-CPs, the rotating ring disk electrode (RRDE) measurements were employed to quantify the peroxide yield and the electron transfer number. As exhibited in Fig. 3d, both the CNTs/Co–N,O-CPs and Pt/C display low yields of peroxide (<5%) and high values of n close to 4, which agrees well with the results estimated from the K–L plots, further suggesting an essential four-electron ORR pathway.
Apart from the electrocatalytic ORR activity, durability and resistance of CNTs/Co–N,O-CPs to methanol were further studied by chronoamperometric and accelerated durability measurements. As exhibited in Fig. 3e, the CNTs/Co–N,O-CPs show excellent durability with a 95% retention of current density after 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s, surpassing commercial Pt/C (78% retention after 25000 s). Furthermore, no visible decline in activity (E1/2) is observed after 5000 potential cycles (Fig. S27†), confirming the superior durability of CNTs/Co–N,O-CPs. Notably, no distinct structural and compositional change in CNTs/Co–N,O-CPs is observed after the durability test (Fig. S28–S31†). The superior durability of CNTs/Co–N,O-CPs may result from its robust chemical stability and unique architecture, which prevents the dissolution and aggregation of Co species, promotes the electron and mass transport, and ensures a four-electron ORR pathway. Additionally, a slight current decay is detected for CNTs/Co–N,O-CPs with the injection of methanol into the electrolyte solution (Fig. S32†), while a sharp current decline is observed for Pt/C, reflecting the excellent tolerance of CNTs/Co–N,O-CPs to methanol.
000 s, surpassing commercial Pt/C (78% retention after 25000 s). Furthermore, no visible decline in activity (E1/2) is observed after 5000 potential cycles (Fig. S27†), confirming the superior durability of CNTs/Co–N,O-CPs. Notably, no distinct structural and compositional change in CNTs/Co–N,O-CPs is observed after the durability test (Fig. S28–S31†). The superior durability of CNTs/Co–N,O-CPs may result from its robust chemical stability and unique architecture, which prevents the dissolution and aggregation of Co species, promotes the electron and mass transport, and ensures a four-electron ORR pathway. Additionally, a slight current decay is detected for CNTs/Co–N,O-CPs with the injection of methanol into the electrolyte solution (Fig. S32†), while a sharp current decline is observed for Pt/C, reflecting the excellent tolerance of CNTs/Co–N,O-CPs to methanol.
Motivated by its superior ORR performance, CNTs/Co–N,O-CPs were exploited as the air cathode for zinc–air batteries (Fig. 3f). As displayed in Fig. S33,† the CNTs/Co–N,O-CP-based zinc–air battery exhibits a stable open-circuit voltage of 1.51 V, which is slightly higher than that of its Pt/C-based counterpart, suggesting good electrocatalytic performance of CNTs/Co–N,O-CPs for practical use. Furthermore, the maximal power density and specific capacity of the CNTs/Co–N,O-CP-based zinc–air battery are determined to be 181 mW cm−2 and 814 mA h gZn−1 (at 10 mA cm−2), respectively, outperforming the Pt/C-based battery (107 mW cm−2 and 763 mA h gZn−1, respectively) (Fig. 3g and h). Notably, the performance of CNTs/Co–N,O-CPs as the air cathode is comparable to that of most previously reported state-of-the-art carbon-based ORR electrocatalysts (Fig. 3i and Table S2†).39–49 Meanwhile, the CNTs/Co–N,O-CP-based zinc–air battery exhibits only a negligible voltage drop after 130 h of continuous discharging (at 10 mA cm−2) (Fig. S34†), signifying superior stability. Finally, as a demo, two CNTs/Co–N,O-CP-based zinc–air batteries placed in series can drive a light-emitting diode (LED) for 10 h (Fig. S35†). Taken together the results discussed above, the CNTs/Co–N,O-CPs have great potential as a pyrolysis-free ORR electrocatalyst for high-performance zinc–air batteries.
To further scrutinize the critical role of Co–N2O2 sites in CNTs/Co–N,O-CPs in the enhanced ORR performance, we conducted the density functional theory (DFT)-based computational calculations (see the ESI† for more details). Based on the EXAFS results, two theoretical models (i.e., Co–N2O2 and Co–N4) are computationally established. The optimal configurations of oxygen-related intermediates (*OO, *OOH, *O, and *OH) adsorbing on Co–N2O2 and Co–N4 are displayed in Fig. 4a–d and S36.† The calculated free energy diagram of four-electron oxygen reduction on Co–N4 exhibits the highest uphill step (i.e., from *OOH to *O) with a free energy change of 1.66 eV at U = 1.23 V (Fig. 4e), suggesting very strong adsorption of *OOH and a high activation energy barrier for the reaction proceeding through *OOH to *O. In sharp contrast, Co–N2O2 shows a considerably weakened adsorption strength for *OOH, which is conducive to the continuation of the ORR process, and therefore, the reduction of *OH to OH− with a markedly lowered free-energy change of 1.23 eV becomes the rate-determining step. These results are in good accordance with the experimental results and highlight the critical role of Co–N2O2 sites in facilitating ORR kinetics. To shed more light on the effect of Co–N2O2 sites on the ORR activity, the partial density of states (PDOS) and charge difference analyses were further carried out. Fig. 4f displays the charge density and differential charge density images of Co–N2O2 and Co–N4. Clearly, the central Co atom has a stronger electronic interaction with the coordinated O atom than the coordinated N atom. Furthermore, the d-orbital energy level of the central Co atom in Co–N2O2 exhibits a stronger coupling interaction with the p-orbital energy level of coordinated O atoms (Fig. 4g and h), which matches well with the information reflected by charge difference analysis. Since the adsorption of oxygen-related intermediates to metal active sites depends on the electronic interaction between oxygen and metal atoms, it is reasonable to speculate that the strong coupling effect between central Co and coordinated O atoms would weaken the binding strength between oxygen-related intermediates and active Co sites.50,51 The optimized adsorption could accelerate the desorption process and improve the utilization of the active central Co sites, thus promoting the kinetics of the four-electron ORR process. Taken together the experimental and computational results, the excellent electrocatalytic ORR performance of CNTs/Co–N,O-CPs may likely be a direct consequence of the advantageous 1D heterostructure with abundant hierarchical porosity and the optimized electronic structure of the Co–N2O2 active sites, which ensure the efficient exposure of the highly active sites and rapid electron and mass transport.
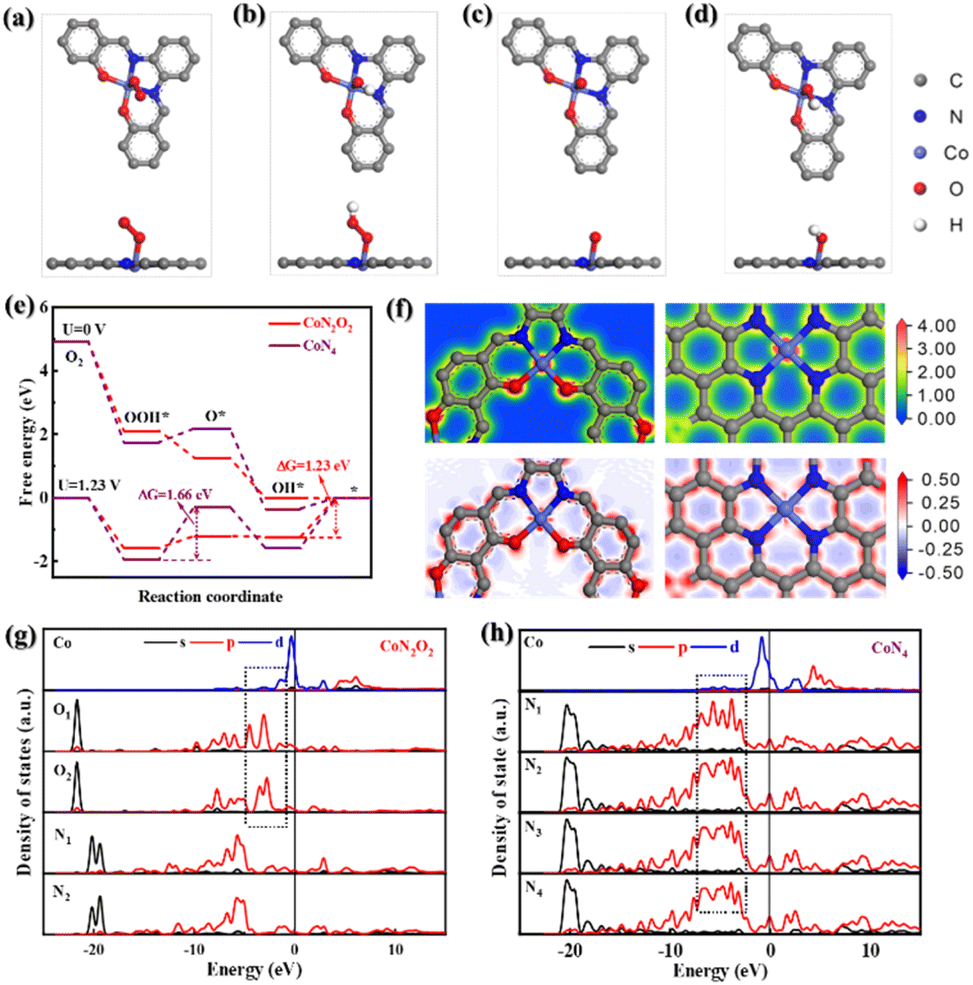 | ||
| Fig. 4 Optimized geometrical structures (top and side views) of (a) *OO, (b) *OOH, (c) *O, and (d) *OH adsorbed on Co–N2O2. (e) The free energy diagrams of Co–N2O2 and Co–N4 at U = 0 V and U = 1.23 V with the four-electron pathway. (f) Charge density images (top) and differential charge density images (bottom) of Co–N2O2 and Co–N4. The colours red and blue represent the charge accumulation and charge depletion, respectively. Partial density of states (PDOS) of (g) Co–N2O2 and (h) Co–N4. | ||
Conclusions
In summary, we developed a viable one-step pyrolysis-free strategy to craft core–shell coaxially heterostructured CNTs/Co–N,O-CPs as a robust ORR electrocatalyst for zinc–air batteries. The CNTs/Co–N,O-CPs manifest excellent electrocatalytic performance in an alkaline medium with Eonset and E1/2 (1.004 V and 0.868 V, respectively) higher than those of the benchmark Pt/C catalyst and superior long-term durability. Furthermore, the CNTs/Co–N,O-CP-based zinc–air battery exhibits outstanding performance with both favourable maximal power density (181 mW cm−2) and specific capacity (814 mA h gZn−1) and excellent durability. Such excellent ORR electrocatalytic performance of CNTs/Co–N,O-CPs can be ascribed to the advantageous 1D heterostructure with rich hierarchical porosity, which facilitates the efficient exposure of more active sites and rapid electron and mass transport. Moreover, DFT-based computational calculations demonstrate that the strong orbital interaction between central Co and coordinated O atoms contributes to the lowered energy barriers of oxygen-related intermediates, thereby giving rise to high intrinsic activity toward four-electron oxygen reduction. This work offers an effective paradigm to improve electrocatalytic ORR kinetics through pyrolysis-free synthesis, which can be easily extended to the design of other high-performance electrocatalysts.Author contributions
Xun Cui and Yingkui Yang conceived the project. Rui Ma, Zhengjiao Xie, XiaoXue Xu, and Chenglong Tang carried out the experimental investigation and wrote the manuscript. Xun Cui, Xueqin Liu, Yunhai Zhu, and Yingkui Yang helped to revise the manuscript. All authors were involved in the analysis and discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52003300, 51973235 and 52173091), Program for Leading Talents of National Ethnic Affairs Commission of China (MZR21001), Hubei Provincial Natural Science Foundation of China (2021CFA022), Wuhan Science and Technology Bureau (2020010601012198) and Hubei Key Laboratory of Polymer Materials, Hubei University.Notes and references
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- X. Cui, L. Gao, S. Lei, S. Liang, J. Zhang, C. D. Sewell, W. Xue, Q. Liu, Z. Lin and Y. Yang, Adv. Funct. Mater., 2021, 31, 2009197 CrossRef CAS.
- L. Gao, X. Cui, Z. Wang, C. D. Sewell, Z. Li, S. Liang, M. Zhang, J. Li, Y. Hu and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023421118 CrossRef CAS PubMed.
- C. D. Sewell, Z. Wang, Y.-W. Harn, S. Liang, L. Gao, X. Cui and Z. Lin, J. Mater. Chem. A, 2021, 9, 20375–20384 RSC.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- Y. Yan, S. Liang, X. Wang, M. Zhang, S.-M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 CrossRef CAS PubMed.
- W. Xue, Q. Zhou, X. Cui, S. Jia, J. Zhang and Z. Lin, Nano Energy, 2021, 86, 106073 CrossRef CAS.
- Y.-W. Harn, S. Liang, S. Liu, Y. Yan, Z. Wang, J. Jiang, J. Zhang, Q. Li, Y. He, Z. Li, L. Zhu, H.-P. Cheng and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2014086118 CrossRef CAS PubMed.
- H. Wang, R. Liu, Y. Li, X. Lu, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, R. Zhang, M. Lei and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- T. Wang, Y. He, Y. Liu, F. Guo, X. Li, H. Chen, H. Li and Z. Lin, Nano Energy, 2020, 79, 105487 CrossRef.
- X. Zheng, P. Li, S. Dou, W. Sun, H. Pan, D. Wang and Y. Li, Energy Environ. Sci., 2021, 14, 2809–2858 RSC.
- W. Zheng, R. Zhu, H. Wu, T. Ma, H. Zhou, M. Zhou, C. He, X. Liu, S. Li and C. Cheng, Angew. Chem., Int. Ed., 2022, 61, e202208667 CAS.
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS PubMed.
- E. Luo, Y. Chu, J. Liu, Z. Shi, S. Zhu, L. Gong, J. Ge, C. H. Choi, C. Liu and W. Xing, Energy Environ. Sci., 2021, 14, 2158–2185 RSC.
- R. Ma, X. Cui, X. Xu, Y. Wang, G. Xiang, L. Gao, Z. Lin and Y. Yang, Nano Energy, 2023, 108, 108179 CrossRef CAS.
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef PubMed.
- A. Kumar, S. Ibraheem, T. A. Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef CAS.
- J. Guo, C.-Y. Lin, Z. Xia and Z. Xiang, Angew. Chem., Int. Ed., 2018, 57, 12567–12572 CrossRef CAS PubMed.
- P. Peng, L. Shi, F. Huo, C. Mi, X. Wu, S. Zhang and Z. Xiang, Sci. Adv., 2019, 5, eaaw2322 CrossRef CAS PubMed.
- T. Wang, Z. Zhang, X. Wang, W. Wang, T. Ma and X. Liu, Appl. Catal., B, 2022, 315, 121590 CrossRef CAS.
- Z. You, B. Wang, Z. Zhao, Q. Zhang, W. Song, C. Zhang, X. Long and Y. Xia, Adv. Mater., 2023, 35, 2209129 CrossRef CAS PubMed.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- X. Cui, S. Lei, A. C. Wang, L. Gao, Q. Zhang, Yi. Yang and Z. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- X. Long, D. Li, B. Wang, Z. Jiang, W. Xu, B. Wang, D. Yang and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 11369–11373 CrossRef CAS PubMed.
- Z. Gan, S. Lu, L. Qiu, H. Zhu, H. Gu and M. Du, Chem. Eng. J., 2021, 415, 127850 CrossRef CAS.
- C. Liu, F. Liu, H. Li, J. Chen, J. Fei, Z. Yu, Z. Yuan, C. Wang, H. Zheng, Z. Liu, M. Xu, G. Henkelman, L. Wei and Y. Chen, ACS Nano, 2021, 15, 3309–3319 CrossRef CAS PubMed.
- K. Fan, C. Zhang, Y. Chen, Y. Wu and C. Wang, Chem, 2021, 7, 1224–1243 CAS.
- B. Huang, L. Li, X. Tang, W. Zhai, Y. Hong, T. Hu, K. Yuan and Y. Chen, Energy Environ. Sci., 2021, 14, 2789–2808 RSC.
- X. Cui, L. Gao, R. Ma, Z. Wei, C.-H. Lu, Z. Li and Y. Yang, J. Mater. Chem. A, 2021, 9, 20985–21004 RSC.
- X. Cui, L. Gao, C.-H. Lu, R. Ma, Y. Yang and Z. Lin, Nano Convergence, 2022, 9, 34 CrossRef CAS PubMed.
- X. Kong, J. Ke, Z. Wang, Y. Liu, Y. Wang, W. Zhou, Z. Yang, W. Yan, Z. Geng and J. Zeng, Appl. Catal., B, 2021, 290, 120067 CrossRef CAS.
- X. Kong, Y. Liu, P. Li, J. Ke, Z. Liu, F. Ahmad, W. Yan, Z. Li, Z. Geng and J. Zeng, Appl. Catal., B, 2020, 268, 118452 CrossRef CAS.
- X. Meng, X. Cui, M. Rager, S. Zhang, Z. Wang, J. Yu, Y. W. Harn, Z. Kang, B. K. Wagner, Y. Liu, C. Yu, J. Qiu and Z. Lin, Nano Energy, 2018, 52, 123–133 CrossRef CAS.
- S. Lei, Y. Lu, X. Zhang, P. Gao, X. Cui and Y. Yang, Chem. Commun., 2019, 55, 2305–2308 RSC.
- R. Ma, X. Cui, Y. Wang, Z. Xiao, R. Luo, L. Gao, Z. Wei and Y. Yang, J. Mater. Chem. A, 2022, 10, 5918–5924 RSC.
- X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Wang, Z. Feng, J. Wang, M. H. Engelhard, H. Zhang, Y. He, Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- C. Shi, Y. Liu, R. Qi, J. Li, J. Zhu, R. Yu, S. Li, X. Hong, J. Wu, S. Xi, L. Zhou and L. Mai, Nano Energy, 2021, 87, 106153 CrossRef CAS.
- X. Lu, H. Xu, P. Yang, L. Xiao, Y. Li, J. Ma, R. Li, L. Liu, A. Liu, V. Kondratiev, O. Levin, J. Zhang and M. An, Appl. Catal., B, 2022, 313, 121454 CrossRef CAS.
- Z. Mei, S. Cai, G. Zhao, Q. Jing, X. Sheng, J. Jiang and H. Guo, Energy Storage Mater., 2022, 50, 12–20 CrossRef.
- F. Wang, R. Zhang, Y. Zhang, Y. Li, J. Zhang, W. Yuan, H. Liu, F. Wang and H. L. Xin, Adv. Funct. Mater., 2023, 2213863 CrossRef CAS.
- Y. Wu, C. Ye, L. Yu, Y. Liu, J. Huang, J. Bi, L. Xue, J. Sun, J. Yang, W. Zhang, X. Wang, P. Xiong and J. Zhu, Energy Storage Mater., 2022, 45, 805–813 CrossRef.
- C. Zhou, X. Chen, S. Liu, Y. Han, H. Meng, Q. Jiang, S. Zhao, F. Wei, J. Sun, T. Tan and R. Zhang, J. Am. Chem. Soc., 2022, 144, 2694–2704 CrossRef CAS PubMed.
- Z. Liang, N. Kong, C. Yang, W. Zhang, H. Zheng, H. Lin and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 12759–12764 CrossRef CAS PubMed.
- S. Liang, L.-C. Zou, L.-J. Zheng, F. Li, X.-X. Wang, L.-N. Song and J.-J. Xu, Adv. Energy Mater., 2022, 12, 2103097 CrossRef CAS.
- R. Yang, D. Yang, M. Wang, F. Zhang, X. Ji, M. Zhang, M. Jia, X. Chen, D. Wu, X. J. Li, Y. Zhang, Z. Shi and C. Shan, Adv. Funct. Mater., 2022, 32, 2207331 CrossRef.
- Y. Zhao, Z. Zhang, L. Liu, Y. Wang, T. Wu, W. Qin, S. Liu, B. Jia, H. Wu, D. Zhang, X. Qu, G. Qi, E. P. Giannelis, M. Qin and S. Guo, J. Am. Chem. Soc., 2022, 144, 20571–20581 CrossRef CAS PubMed.
- J.-C. Li, Y. Meng, L. Zhang, G. Li, Z. Shi, P.-X. Hou, C. Liu, H.-M. Cheng and M. Shao, Adv. Funct. Mater., 2021, 31, 2103360 CrossRef CAS.
- X. Wei, D. Zheng, M. Zhao, H. Chen, X. Fan, B. Gao, L. Gu, Y. Guo, J. Qin, J. Wei, Y. Zhao and G. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14639–14646 CrossRef CAS PubMed.
- Z. Lu, B. Wang, Y. Hu, W. Liu, Y. Zhao, R. Yang, Z. Li, J. Luo, B. Chi, Z. Jiang, M. Li, S. Mu, S. Liao, J. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 2622–2626 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02217j |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |