
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparative overview of MXenes and metal oxides as cocatalysts in clean energy production through photocatalysis
Mahesh M.
Nair
a,
Alexandra C.
Iacoban
ab,
Florentina
Neaţu
 a,
Mihaela
Florea
*a and
Ştefan
Neaţu
*a
a,
Mihaela
Florea
*a and
Ştefan
Neaţu
*a
aNational Institute of Materials Physics, 405A Atomistilor Street, 077125 Magurele, Romania. E-mail: mihaela.florea@infim.ro; stefan.neatu@infim.ro
bInterdisciplinary School of Doctoral Studies, University of Bucharest, Mihai Kogalniceanu Street 36-46, 050067 Bucharest, Romania
First published on 16th January 2023
Abstract
Photocatalytic conversion of H2O, CO2 and N2 represents one promising approach to harvest and store solar energy, for which efficient visible light responsive semiconductors are inevitable. Often, the presence of a small amount of an additional component called a “cocatalyst”, is required to synergistically enhance the performance of the photocatalyst. Tremendous efforts were made in the past to identify inexpensive materials to be used as cocatalysts, for which metal oxides (MOs) are one of the traditional choices. Among alternative categories of materials investigated, the recently discovered MXenes display enormous potential owing to their unique 2D layered structure, tuneable composition, abundant surface functionalities and superior electronic conductivity. Specifically, MOs and MXenes encompass a variety of distinct as well as analogous characteristics that allows them to be tailored to different extents. Unfortunately, a comprehensive overview covering the synthetic, structural and photocatalytic aspects of MOs and MXenes is not available as of now. Herein, we intend to summarize the progress achieved so far in these two families of materials to be used as cocatalysts for the photoconversion of H2O, CO2 and N2. Followed by a general introduction, we briefly outline the fundamental principles and the role of cocatalysts in photocatalytic reactions. A discussion regarding the use of MOs and MXenes as cocatalysts for the conversion of H2O, CO2 and N2 is then provided in separate sections. Critical assessment regarding structure and morphology control, surface properties and stability concerns can not only help to recognize the challenges that limit further advancement, but can also highlight the future research directions of these materials for the effective transformation of H2O, CO2 and N2.
 Back row, left to right: Ştefan Neaţu, Mahesh M. Nair, and Florentina Neaţu and lower row, left to right: Mihaela Florea, and Alexandra C. Iacoban | Integrated in the Catalytic Material and Catalysis group, the research team is part of the National Institute of Materials Physics, Romania (NIMP) (https://infim.ro/). Back row, left to right: Dr Ştefan Neaţu (senior researcher), Dr Mahesh Nair (Postdoc), and Dr Florentina Neaţu (senior researcher) and lower row, left to right: Dr Mihaela Florea (senior researcher) and Alexandra Iacoban (PhD student). The design, characterisation, and investigation of the physical properties of novel materials are the main goals of the NIMP team's research. Their primary areas of applications implies heterogeneous catalysis, energy conversion, electrochemistry, fuel cells, and materials surface science. |
10th anniversary statementJ. Mater. Chem. A is dedicated to publish original contributions to research at the interface of materials science and chemistry. To celebrate the 10th anniversary of J. Mater. Chem. A, we devoted a special review discussing, what else than materials, new developed materials such as MXenes and old, but still unfashionable materials such as metal oxides. As you will be able to see while reading this review, the use of the two types of materials as cocatalysts leads to obtaining truly extraordinary hybrid composites with applicability in various photocatalytic reactions. Speaking about photocatalysis in general, and in particular about finding new materials with direct applications in the photocatalysis field, J. Mater. Chem. A was, is and it will be a landm ark for both specialized and non-specialized readers passionate about novelty. Therefore, what binds us to the J. Mater. Chem. A is this very passion for novelty in the field of materials chemistry. Now reaching its 10th anniversary, we wish J. Mater. Chem. A to continue to surround us with the joy of reading outstanding papers that arouse our attention and appetite for new discoveries. Happy Anniversary, J. Mater. Chem. A! |
1. Introduction
Being the major contributor to global warming, the emission of large quantities of CO2 by the burning of fossil fuels demands immediate attention. The harmful effects arising from the increasing concentration of atmospheric CO2, coupled with the progressive depletion of the fossil fuel reserves drives the quest for alternative energy sources. Therefore, energy harvesting in a clean and sustainable manner remains the most crucial challenge the world is currently facing. Solar energy is promising in this regard and represents a readily available and exploitable substitute to fossil fuels.1–3 Solar energy needs to be captured and converted into useful forms, especially considering the intermittency (day/night, seasons, etc.). One such existing and established technology is electricity generation using solar cells.4,5 Solar electricity needs to be converted into chemical fuels, since these are the primary energy feedstock for industrial and transportation sectors. However, such transitions are difficult, demand additional costs and are time consuming. Moreover, there are technological challenges involving storage and transportation in a cost-effective and convenient manner. Therefore, direct production of fuels and chemicals using solar energy is considered a more attractive option.Inspired by the natural process of photosynthesis, efforts were made in the recent past to create solar fuels using semiconductor photocatalysts, called ‘artificial leaves’, as shown in Fig. 1. In this case, photon energy from the sun is used to generate electron–hole pairs to drive redox reactions.6–10
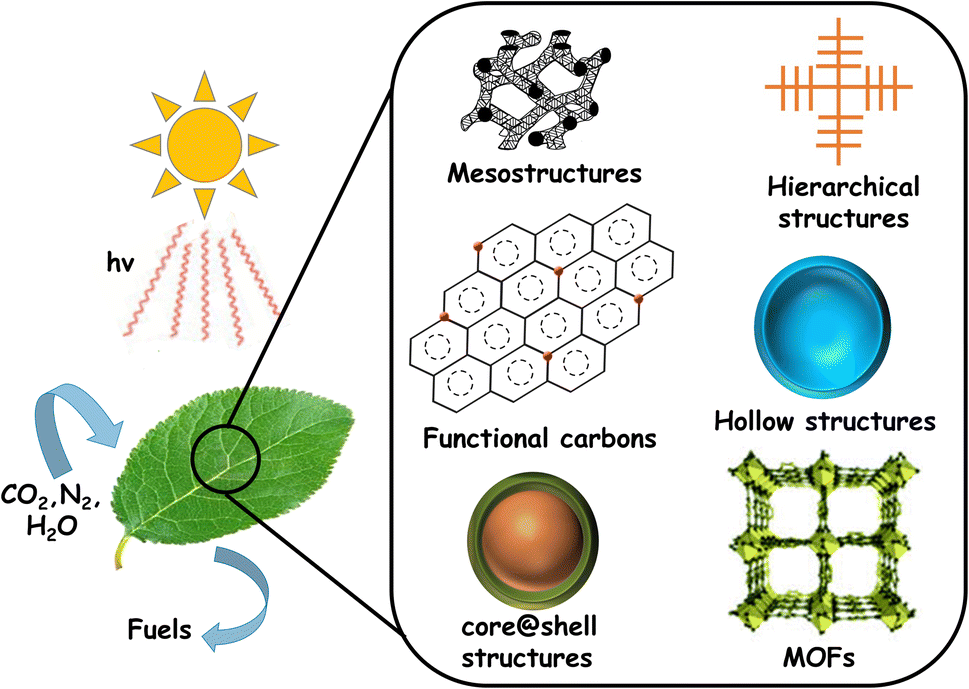 | ||
| Fig. 1 Schematic representation of solar fuel production from CO2, H2O and N2, along with the photocatalytic structural variants commonly studied. | ||
Most of the initial studies focused on the production of H2 by splitting H2O using photocatalysts.11,12 Apart from being an energy carrier itself, H2 can be used to generate hydrocarbons by photoreduction of CO2, thereby creating a significant environmental impact by converting a greenhouse gas into a useful commodity or for N2 photofixation toward a better solar-to-chemical energy conversion.
In short, the production and development of a technology that utilizes sunlight to efficiently convert H2O and CO2 into H2, hydrocarbons, or syngas (CO + H2) can revolutionize the concept of fuel production. Here, functional materials that encompass desired photocatalytic properties such as enhanced solar light absorption, high charge separation, appropriate band gaps, etc., form a major requirement.13–16 Compositional optimization of these materials was the earliest and most straightforward approach followed to achieve this. A variety of oxides, oxynitrides, metal organic frameworks (MOFs), carbonaceous materials, etc., were studied to identify the best candidates for producing solar fuels from H2O and CO2.17–22 Unfortunately, materials that satisfy all the practical requirements have not yet been found. On the bright side, several of these studies confirmed the dependence of photocatalytic properties on the structural features and morphology of the materials.13,15,17
One proven strategy to enhance the photoactivity of a semiconductor is to integrate a small amount of a secondary component. This secondary component called a “cocatalyst” facilitates the separation of charge carriers by forming an interface with the photocatalyst. Also, it can serve as the active site for the surface reaction to take place.23–26 Preliminary studies on the use of noble metals (i.e., Pt, Pd, Ag, and Au) as cocatalysts were promising, however, with limited commercial applicability owing to their high costs.27–29 Numerous non-noble metal alternatives were therefore investigated as cost-effective photocatalysts. So far, transition metal-based materials including oxides, hydroxides, sulfides, etc., have been widely explored.30–34 More recently, the introduction of 2D transition metal carbides, called MXenes, as cocatalysts was found to significantly promote photocatalytic H2O splitting and CO2 reduction.35–40
In general, performance optimization primarily relies on structural and compositional aspects of the materials employed. Metal oxides (MOs) can accommodate a large number of cationic components in varying amounts in their structure, thereby allowing the long-range tailoring of their physicochemical properties. In conjugation, the noteworthy progress achieved so far in inorganic synthesis makes it possible to prepare these materials in a variety of nanostructures and morphologies. Similarly, MXenes, regardless of their short period of evolution, exhibited significant potential to host a number of metallic components in varying proportions in their structure.41–44 Several recent reports also claim the possibility of preparing morphological variants of these materials.38 An additional advantage exclusive to MXenes is the presence of inherent surface functionalities that can be useful for catalytic applications.
Several reviews exist regarding the synthesis and applications of MXenes, including photocatalysis.35–45 Moreover, theoretical results and insights on how MXenes and/or MOs contribute to photocatalysis as cocatalysts exist already in the literature.46–52 The present review summarizes the major milestones achieved regarding the use of MOs and MXenes as cocatalysts for hydrogen production through the water splitting reaction and photocatalytic CO2 and N2 reduction reactions. To maximize the synergistic interactions, it is crucial to maintain optimum contact between the semiconductor and cocatalyst, which depends primarily on the synthetic procedures and nanoscale structural arrangement. Therefore, we separately discuss the general advantages of nanostructures in photocatalysis, the progress so far on the development of synthetic approaches and the resultant structure evolution for MXenes and MOs. Furthermore, applications of these two categories of materials for photocatalytic H2O, CO2 and N2 conversion will be summarized. Comparisons will be made highlighting the advantages and disadvantages, where appropriate. In addition, challenges and future directions for MOs and MXenes regarding the photocatalytic conversion of H2O, CO2 and N2 will be provided from the perspective of the authors.
2. Fundamentals of photocatalysis and the relevance of nanostructures
In general, photocatalysis involves light absorption, charge separation (electrons and holes) and surface reactions initiated by these photogenerated charge carriers (see Fig. 2). Photon energy corresponding to the band gap of the semiconductor will be absorbed resulting in the generation of electrons (e−) in the conduction band and holes (h+) in the valence band. The reactants will be adsorbed at the active sites on the surface of the photocatalyst. The photogenerated charge carriers (e− and h+) can get trapped, recombine, or migrate towards the surface, to drive the desired transformation of the adsorbed reactant species. The products thus formed will then be desorbed from the surface.53,54 During water splitting, the photogenerated holes will interact with H2O, producing O2 and H+. The H+ ions will consume the e− residing at the conduction band forming H2.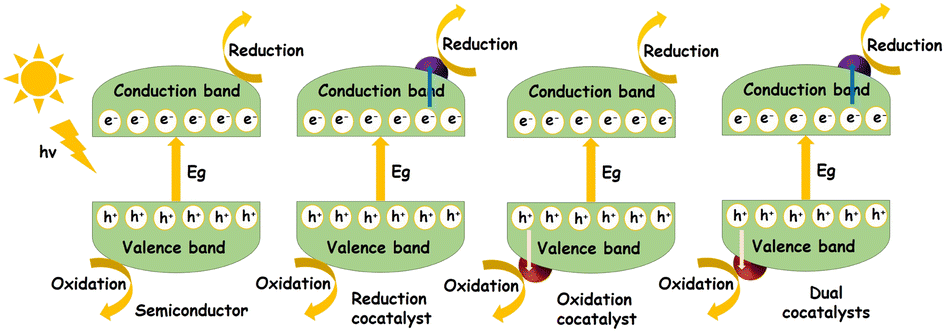 | ||
| Fig. 2 Fundamental principles in photocatalysis including light absorption, charge separation, and surface reactions, along with the influence of cocatalysts. | ||
On the other hand, photoreduction of CO2 is more complex involving multiple electrons that can end up in a series of low molecular weight organic products. Therefore, in this later case, reaction selectivity is also a major concern.55 The N2 photofixation process involves the use of holes generated by light irradiation to oxidize the H2O (as in the case of water splitting), while the e− are used to reduce the N2 to ammonia.56 The photocatalytic water splitting, CO2 reduction and N2 photofixation reactions are shown in eqn (1)–(2), (3)–(7) and (8), respectively. It is to be noted here that apart from electrons and holes, other reactive intermediates are formed during photocatalysis. These reactive oxygen species are particularly useful for the photodegradation of organic pollutants and hence will not be discussed here.57
| H2O + 2h+ → ½O2 + 2H+ | (1) |
| 2H+ + 2e− → H2 | (2) |
| CO2 + 2H+ + 2e− → HCOOH | (3) |
| CO2 + 2H+ + 2e− → CO + H2O | (4) |
| CO2 + 4H+ + 4e− → HCHO + H2O | (5) |
| CO2 + 6H+ + 6e− → CH3OH + H2O | (6) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | (7) |
| N2 + 6H+ + 6e− → 2NH3 | (8) |
Thermodynamically, a reduction reaction will take place if the conduction band is located at a more negative potential and an oxidation reaction requires a more positive valence band than the oxidation potential. Unfortunately, this requirement is often satisfied by materials with much wider band gaps, prohibiting their functioning in the visible region, where the major portion of solar energy is distributed. In addition, prolonged charge separation is strongly desired to minimize the electron–hole recombination that can occur during their migration towards the surface.53,54
Systematic tailoring of the structure and morphology at the nanoscale can have a profound effect on the overall photocatalytic properties of materials.13,15,58,59 Performance enhancement in nanostructures can be attributed to an increased surface area, reduced diffusion barriers and shortened charge transport pathways. Nanomaterials in general exhibit enhanced values of specific surface area. Since only surface atoms participate in photocatalytic reactions, a high surface area implies that more atoms are available as adsorption sites for the reactants. A high surface-to-volume ratio results in an abundant and uniform distribution of accessible active sites and often promotes charge transfer ac ross interfaces. Several studies considered nanostructuring as a way to improve the light harvesting efficiency of photocatalysts.13,15,60,61 A few possible effects of nanostructures on photocatalysis are schematically shown in Fig. 3.
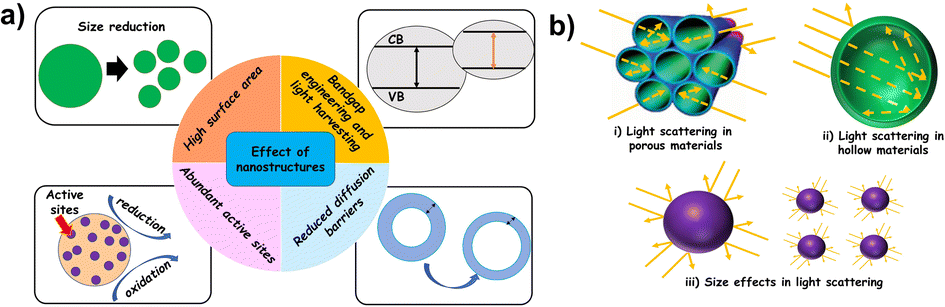 | ||
| Fig. 3 (a) General influence of nanostructures in photocatalysis and (b) light scattering effects under confinement. | ||
Nanostructures will provide an increased number of transport paths resulting in a prolonged interaction time of the photocatalyst with incoming light. This results in an enhanced surface distribution, thereby improving the light absorption effectiveness. For instance, light scattering effects observed in some nanostructures can influence the light harvesting efficiency of photocatalysts. In a conventional bulk material, surface reflection is predominant whereas in nanostructures, the incident light is scattered resulting in secondary absorptions, leading to enhanced light harvesting.61–63
The effect of secondary scattering can be more pronounced where a high degree of confined spaces is available (e.g., nanoporous materials). Thin films or hollow nanostructures with thin walls could promote facile diffusion of charge carriers towards the surface adsorbed reactant species.64 Nanoporous materials generally have particle size distribution in the micrometer regime and can prevent stability issues related to agglomeration prevalent in conventional nanosized particles. Apart from the enhancement of light absorption and transport kinetics, the quantum confinement effects inherent in nanostructures can often facilitate appropriate band gap engineering. Quantum size effects can modify the position of the valence and conduction bands in the photocatalyst. Also, confined nanoparticles can result in the generation of multiple e−/h+ pairs (excitons). Overall, the improved photocatalytic performance of nanostructured materials stems from a combination of their structural features, specific surface area and presence of accessible active sites.65 In reality, photocatalytic conversion of H2O, CO2 and N2 depends on the physicochemical and optical properties of the photocatalyst. Even though nanostructure optimization can improve some of these properties, it can be unfavourable to the others. Therefore, a rational approach for the design of nanostructured photocatalysts must take the disadvantages also into consideration. For instance, an increased surface energy inherent in nanomaterials can influence its thermodynamic stability. Also, high interfacial areas and short diffusion lengths in nanoscale materials can promote undesired and easy recombination of the charge carriers, influencing the reaction rates.
As mentioned above, the presence of a cocatalyst is necessary to improve the photocatalytic properties of semiconductors. In other words, the overall performance stems from the synergistic functioning of both these components in the form of a hybrid. Therefore, any discussion regarding the fundamental mechanism of photocatalysts cannot exclude the role of cocatalysts, improving a photocatalytic reaction by providing reaction sites and by promoting charge separation. Reduction cocatalysts are used to trap electrons whereas oxidation cocatalysts are used for trapping holes. The presence of a cocatalyst should not hinder the active sites from light absorption or accessibility of the reactants and should improve the corresponding half reaction whereas dual cocatalyst-semiconductor photocatalytic systems contribute towards the advancement of the overall redox reaction.23–26
3. MXenes
3.1. Short overview
The presence of a cocatalyst synergistically modifies the performance of a semiconductor photocatalyst and to maximize such interactions, it is important to maintain optimum contact. Therefore, several approaches have been developed to integrate the cocatalyst with the semiconductor. This includes the synthesis of the cocatalyst separately and then combining it with the semiconductor in a post-synthetic manner, or synthesizing the cocatalyst and the semiconductor in one pot. For instance, the synthetic procedure for MXenes is not compatible with routinely used semiconductor photocatalysts. Moreover, the thermal stability of MXenes is limited under oxidizing conditions. Therefore, post-synthetic approaches are always necessary for integrating MXenes as a cocatalyst.3.2. Synthesis and structure evolution
Naguib et al.,66 in 2011, reported a transition metal containing graphene analogue (Ti3C2Tx), called MXene, obtained by selectively extracting Al from the parent MAX phase (Ti3AlC2) using HF.66 In the strategies developed so far, the synthesis of MXenes involves the selective etching of an A-layer from the MAX phase, by exploiting the differences between the metal–metal (M–A) and metal–carbon (M–X) bond strengths.40–42,66 The use of aqueous HF as the etchant resulted in the formation of a multi-layered material with abundant –F, –O and –OH surface terminations (see Fig. 4).67 These materials are represented by the general formula Mn+1XnTx, where M denotes early transition metals, X can be carbon or nitrogen, T indicates the surface functionalities such as hydroxyl groups, halides, etc. and n determines the number of layers, usually between 1 and 4. Recently, MXenes with n = 5 were discovered.68 It is to be noted here that the etching conditions were influenced by the variations in M–Al bond strength and hence, minor disparities in etching conditions are inevitable. The presence of surface functionalities enables the post-synthetic modifications through intercalation or grafting, thereby tailoring the physicochemical properties of these materials as desired.69,70 With regard to the safety issues in handling HF, LiF/HCl mixture, ammonium bifluoride (NH4HF2), etc. were also found to be useful as alternatives.71,72 Other approaches such as electrochemical etching in HCl, hydrothermal treatment (HTT) in NaOH, iodine dissolved in anhydrous acetonitrile, molten salt such as ZnCl2 or CuCl2 at enhanced temperatures were also reported.73–77 However, in some cases, issues such as incomplete etching and the presence of impurities, could not be avoided.74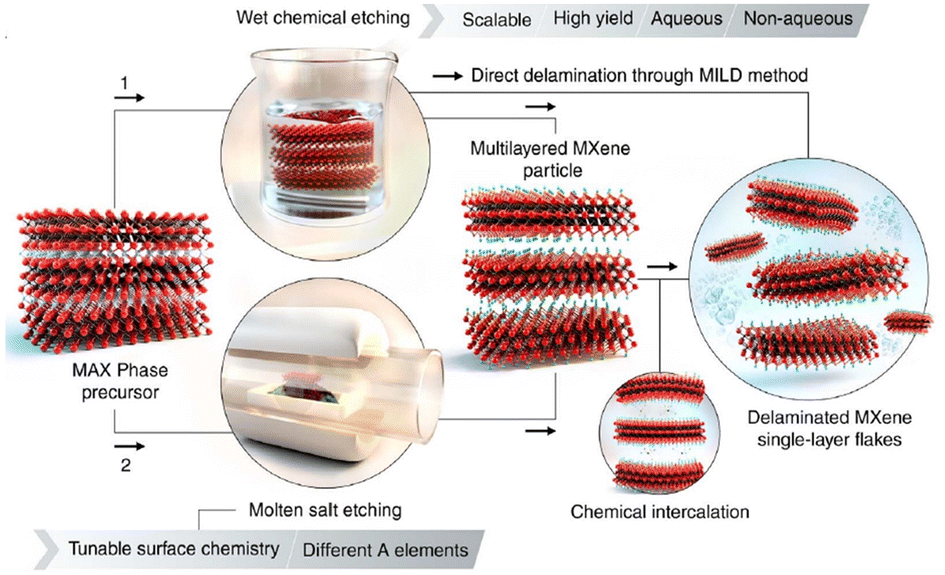 | ||
| Fig. 4 Schematic representation of two etching strategies to remove the A layers from MAX phases to produce multi-layered MXenes and delaminated single layer flakes. Reproduced with permission from ref. 67, Copyright 2021 Science. | ||
In short, the composition of MXenes primarily depends on the parent MAX phase while the structure and surface properties are influenced by the etching protocols. The simplest version of MXenes or conventional MXenes incorporates an early transition metal attached to carbon.66 Afterwards, more complex structural and compositional variants were discovered.78–83 For this, MAX phases with more than one transition metal were used as precursors. o-MXenes consist of multiple transition metal components with an inner layer formed by one component and the outer layer by the other. On the other hand, non-stoichiometric i-MXenes display an ordered array of vacancies in the metal in-plane layer.
Tremendous progress has been achieved so far in controlling the synthesis parameters to obtain 2D MXene layers. Liquid phase exfoliation of the accordion structure resulted in the expansion of interlayer space, which weakens interlayer interactions, resulting in nanosized sheets. Usually, the concept of etching was used to remove the A layer from the precursor MAX phase.
Interestingly, Ren et al.85 extended this etching concept for the synthesis of porous 2D Ti3C2Tx open structures with large surface area.85 Delaminated colloidal solutions were partially oxidized with O2 dissolved in the water, in the presence of Cu2+ as a catalyst. Selectively removal of TiO2 nanoparticles thus formed resulted in porous Ti3C2Tx flakes. Stacking of 2D MXene sheets can limit their applicability and hence strategies are to be developed to hinder their aggregation.
Zhao et al.84 proposed a hard templating method to derive hollow spheres and 3D architectures of MXene flakes and the corresponding images are provided in Fig. 5. Hollow spheres and 3D microporous films were made by using polymethyl methacrylate (PMMA) spheres as the template.86 In a similar manner, Xiu et al.87 used an ultrasound assisted aerosol spray drying method, to overcome the possible aggregation of MXene nanosheets.87 These authors successfully assembled Ti3C2Tx into hierarchical 3D architectures by evaporating aerosol droplets produced by ultrasonication, exhibiting geometry-based resistance to aggregation. It was also shown that by introducing polyvinyl pyrrolidine into the MXene colloidal solution, sphere like 3D architectures can be produced. Free standing 3D network structures were developed by assembling MXenes in the presence of templates.88–90 MXene quantum dots (QDs) were fabricated by dissociating their layered structure using HTT, an ultrasonic method, etc.91–94
 | ||
| Fig. 5 (a) SEM images showing the morphology of the as-produced PMMA spheres. (b) TEM image of Ti3C2Tx flakes. SEM image of (c) Ti3C2Tx/PMMA hybrids and (d) hollow Ti3C2Tx spheres after removal of PMMA, showing that the spherical shape is retained after PMMA's thermal evaporation at 450 °C. TEM images showing (e) a hollow Ti3C2Tx sphere and (f) its wall MXene layers. Reproduced with permission from ref. 84, Copyright 2017 Wiley Publications. | ||
For photocatalytic applications, post-synthetic procedures are almost always necessary for MXenes. Simple oxidation treatment sometimes results in partial oxidation of the metallic species in MXenes, resulting in the formation of the corresponding MXene-oxide composites.95,96 In addition, other wet-chemical approaches including HTT, ultrasonication, etc. are frequently employed.97–99
3.3. Photocatalytic applications for water splitting
In spite of MXenes being metallic, the surface functionalities can be effectively tailored so as to directly employ them as semiconductor photocatalysts.100 Studies utilizing deformation potential theory (DPT), density functional theory (DFT) and ab initio molecular dynamics (AIMD) calculations clearly predicted several MXene compositions that can be used for photocatalytic applications.101–104 However, experimental validation of the synthesis of such compositions and the evaluation of their photocatalytic performance are yet to be made.For instance, by performing controlled oxidation of Nb2CTx using CO2 as a mild oxidant, Su et al.95 successfully synthesized Nb2O5/C/Nb2C composites. The materials obtained after an oxidation time of 1 h exhibited hydrogen evolution rates 4 times higher than that of pristine Nb2O5, with an AQY of 0.11%. Hydrogen yield remain consistent during four consecutive cycles. The Schottky layer formed at the interface acted as an electron sink from which backward diffusion to the Nb2O5 photocatalyst was hindered. The presence of an amorphous carbon layer at the Nb2O5/C/Nb2C interface also contributed towards the separation of photogenerated charge carriers.95 This work confirmed that MXenes can act as precursors for photocatalysts. In another study, it was observed that carbon doped TiO2 can be produced from Ti3C2Tx nanosheets under HTT.105 This material exhibited a 9.7 times higher hydrogen production rate compared to commercial TiO2 (P25) using triethanolamine (TEA) as a hole scavenger and in the absence of a cocatalyst. The authors carried out XRD analysis of the material recovered after the reaction and the pattern confirmed the structural stability of this material. This work further confirms the role of carbon in facilitating the separation of charge carriers and the use of MXene as a precursor for photocatalysts.105
Li et al.96 effectively controlled the surface terminations in Ti3C2Tx and synthesized TiO2/Ti3C2Tx hybrids by performing calcination of F- terminated and OH- terminated Ti3C2Tx, separately.96 OH- terminated Ti3C2Tx was obtained by treating Ti3C2Tx with NaOH. Uniformly truncated octahedral bipyramidal TiO2 particles with exposed [001] facets were preferentially formed on the surface of F- terminated MXenes after calcination. This material exhibited two times enhanced hydrogen production compared to TiO2/Ti3C2Tx produced from OH–terminated counterparts.96
| Photocatalyst/cocatalyst | Synthetic method | Hole scavenger | Light region | Optimum H2 production rate [μmol g−1 h−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|
| a ∼ Data taken from a graphical representation of the corresponding reference; NP: value not provided; TEA = triethanolamine. | ||||||
| TiO2/Ti2CTx | HTT (95 °C for 4 h) | Methanol (25%) | Visible | ∼32 | 0.27 | 97 |
| TiO2/Ti3C2Tx | 17.8 | 0.15 | ||||
| TiO2/Nb2CTx | ∼46 | 0.39 | ||||
| Nb2O5/C/Nb2C | Oxidation under CO2 (850 °C for 0.5–1.5 h) | Methanol (25%) | Visible | 7.81 | 0.11 | 95 |
| TiO2/Ti3C2 nanoflowers | HTT (140 °C for 12 h), ion exchange (HCl, 24 h) and calcination in air (300–500 °C for 3 h) | Methanol (20%) | UV-visible | 783.11 | 5.86 (350 nm) | 108 |
| TiO2/Ti3C2Tx composites | Sonication | Methanol (25%) | UV | 2650 | 15.8 (305 nm) | 98 |
| C-doped TiO2 derived from Ti3C2Tx | HTT (160 °C for 9 h) | TEA (10%) | Full spectrum | 33.04 | NP | 105 |
| Truncated octahedral bipyrmidal TiO2/Ti3C2 hybrids | Calcination in air (550 °C for 4 h) | Glycerol (10%) | Full spectrum | ∼130 | 0.9 (365 nm) | 96 |
| 0D/1D/2D Ag/Nb2O5@Nb2CTX nanohybrids | HTT (180 °C for 0.5–10 h) and in situ photodeposition | Methanol (7%) | Full spectrum | 682 | 1.6 (313 nm) | 109 |
| Glycerol (7%) | 824 | |||||
| La2Ti2O7/Ti3C2 composites | HTT (200 °C for 24 h) | Methanol (40%) | UV | 1594 | NP | 110 |
| CdS/Ti3C2 composites | HTT (180 °C for 12 h) | Lactic acid (22%) | Visible | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 342 |
40.1 (420 nm) | 111 |
| 1D/2D CdS/Ti3C2 nanocomposites | HTT (160 °C for 48 h) | Lactic acid (10%) | Visible | 2407 | 35.6 (420 nm) | 112 |
| 1D/2D CdS/Ti3C2 composites | HTT (200 °C for 5 h) | TEA (20%) | Simulated solar light | 63.53 | 2.28 | 113 |
| 2D/2D CdS/Ti3C2Tx composites | HTT (80 °C for 48 h) | Lactic acid (10%) | Visible | 3226 | 47 (420 nm) | 114 |
| CdS/TiO2–C (derived from Ti3C2Tx) | γ-ray irradiation | Lactic acid (20%) | Visible | 1480 | NP | 115 |
| CdS/Mo2C | HTT 180 °C for 24 h) | Lactic acid (17%) | Visible | 17964 |
48.06 (420 nm) | 116 |
| 2D/2D ZnIn2S4/2D Ti3C2Tx sandwich like hierarchical heterostructures | Reflux (80 °C for 2 h) | TEA (10%) | Visible | 3475 | 11.14 (420 nm) | 117 |
| BiVO4@ZnIn2S4/Ti3C2 QD | Ultrasonication | None | Visible | 6.16 | 2.9 (460 nm) | 118 |
| Ti3C2@TiO2/ZnIn2S4 ternary composites | HTT (180 °C for 3 h) | Na2S/Na2SO3 | Simulated solar light | 1185.8 | NP | 119 |
| CdS–MoS2–Ti3C2 composites | HTT (160 °C for 24 h) | Na2S/Na2SO3 | Visible | 9679 | 26.7 (420 nm) | 120 |
| MoxS@TiO2@Ti3C2 composites | HTT (160 °C for 12 h) | TEA | UV-visible-near IR | 10505.8 |
7.535 | 121 |
| CdS@Au/Ti3–xC2Ty ternary composites | Solvothermal treatment (180 °C for 12 h) | Lactic acid (10%) | Visible | 5371 | 16.7 (420 nm) | 122 |
| 2D/2D g-C3N4/Ti3C2 nanosheets | Electrostatic self-assembly | TEA (10%) | Visible | 72.3 | 0.81 (400 nm) | 99 |
| 2D/0D g-C3N4/Ti3C2 composites | Freeze drying | TEA (15%) | Simulated solar light | 5111.8 | 3.654 | 123 |
| p-g-C3N4/Ti3C2Tx hollow spheres | Electrostatic self-assembly with a sacrificial template | TEA (10%) | Visible | 982.8 | NP | 124 |
| 2D/3D g-C3N4/Ti3C2 hybrid | Calcination under N2 (550 °C for 2 h) | TEA (10%) | Visible | 116.2 | NP | 125 |
| 0D black phosphorous/2D Ti3C2/2D g-C3N4 | Grinding and sonication | TEA (10%) | Visible | 18420 |
17.6 (420 nm) | 126 |
| Chl/Ti3C2Tx | Wet chemical | Ascorbic acid | Visible | 1010 | NP | 127 |
| Chl-1@ChI-2/0Ti3C2Tx | Wet chemical | Ascorbic acid | Visible | 143 | NP | 128 |
Wang et al.97 synthesized TiO2/Ti3C2Tx composites in different weight ratios (5–50% Ti3C2Tx) under HTT. Photocatalytic hydrogen evolution from water using methanol as a hole scavenger was monitored for these materials under visible light irradiation. Inclusion of 5 wt% Ti3C2Tx resulted in a 400% enhancement in hydrogen evolution compared to pure rutile TiO2. Photocatalytic activity was further found to be improved by replacing Ti3C2Tx with Ti2CTx or Nb2CTx, among which 5% Nb2CTx exhibited the best performance among the series, with an apparent quantum yield (AQY) of 0.39%. Stability tests performed for three cycles using TiO2/Ti3C2Tx exhibited co nsistent results.
According to the authors, the hydrothermal synthesis method allowed an intimate contact between the semiconductor and the cocatalyst surfaces resulting in a flow of photogenerated electrons. A Schottky barrier thus formed at the interface prevented the electrons from diffusing back, thereby preventing charge recombination. In addition, the highest value of the work function for Nb2CTx (4 eV) among those of the series reflected a larger Schottky barrier explaining its best performance among the series (see Fig. 6).97
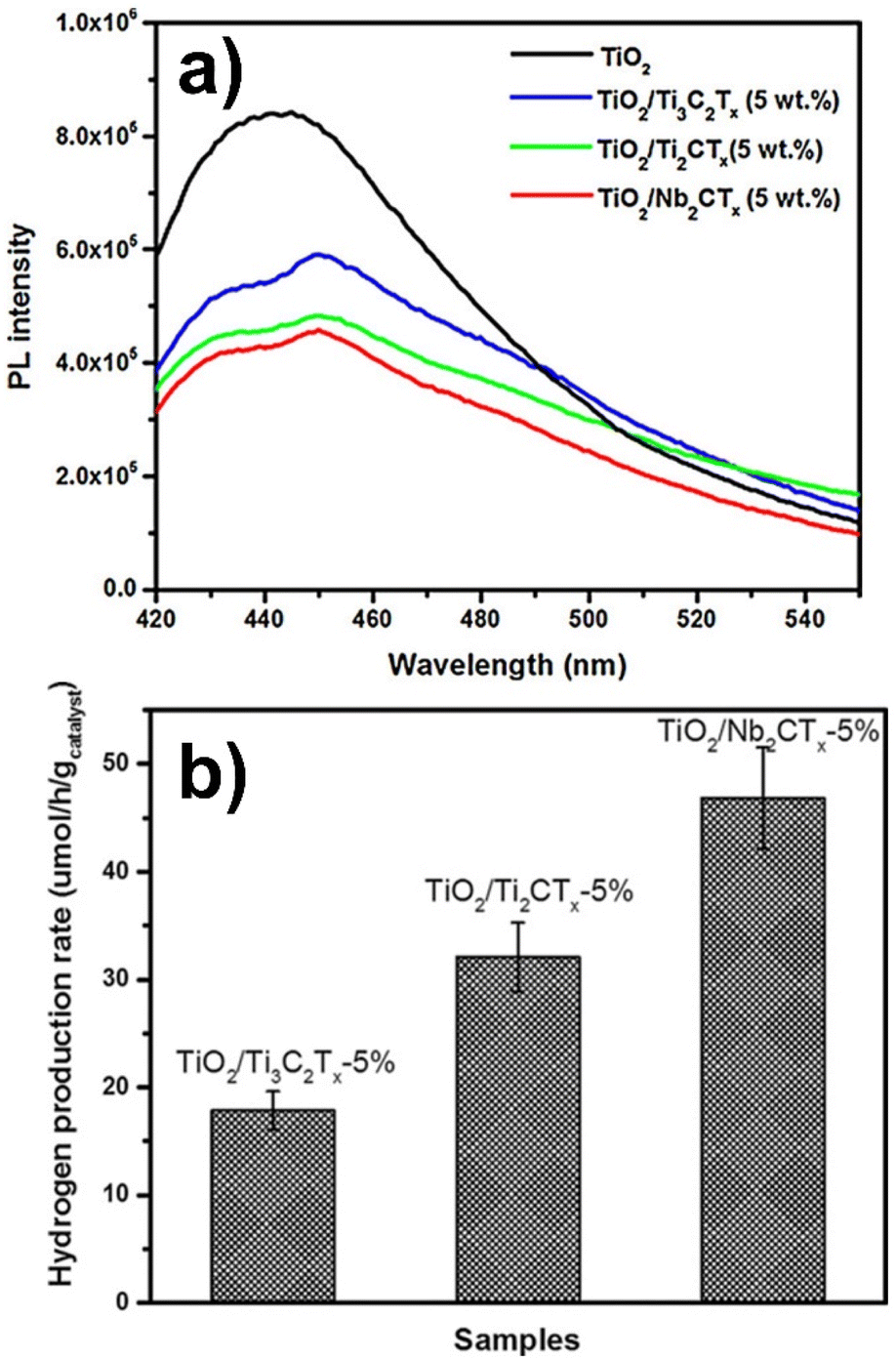 | ||
| Fig. 6 (a) Photoluminescence spectra of pure TiO2 along with those of the materials with different 5 wt% metal carbide cocatalysts. The excitation wavelength was 400 nm. (b) Photocatalytic hydrogen production rates obtained for different metal carbide cocatalysts. Reproduced with permission from ref. 97, Copyright 2016 European Chemical Societies Publishing. | ||
In a similar manner, Li et al.108 synthesized TiO2/Ti3C2Tx nanoflowers from Ti3C2 under HTT followed by ion exchange and calcination.108 Initially, Na2Ti3O7–Ti3C2 composites were synthesized hydrothermally, and were then treated with HCl to replace Na+ with H+ producing H2Ti3O7–Ti3C2. These materials were then calcined at 300–500 °C to produce TiO2/Ti3C2Tx. Optimum materials obtained after calcination at 500 °C displayed overall water splitting in the absence of a sacrificial agent, with an AQY of 5.86%. The better performance was attributed to the ability of 3D porous nanoflower-like morphology to provide more active sites, and its greater ability to reflect and scatter light and reduce the diffusion length of photogenerated charge carriers. The Schottky junction formed at the interface hindered charge recombination resulting in more electrons participating in H2 evolution and more holes participating in O2 evolution.108
Su et al.98 synthesized a series of Ti3C2Tx/TiO2 composite photocatalysts by adding an aqueous dispersion of stacked monolayer Ti3C2Tx in different weight ratios to commercial TiO2 (Degussa P25), under sonication.98 Photocatalytic tests performed in the presence of methanol as a hole scavenger indicated a 9 times higher hydrogen evolution rate (AQY = 15.8%) for the composite with 5 wt% MXene, compared to P25. Interestingly, this composite material exhibited a 2.5 times better performance than when the multi-layer MXene counterpart was used as the cocatalyst. The significantly enhanced activity is attributed to the superior electron conductivity of monolayer Ti3C2Tx and enhanced charge separation at the interface. It is to be noted here that after four cycles, the hydrogen production rate decreased from 2650 μmol g−1 h−1 to 2300 μmol g−1 h−1. Since the structural and surface properties of the photocatalyst recovered after the reaction remained unchanged, the observed decrease in the rate was attributed to the probable detachment between TiO2 and Ti3C2Tx.98
In an attempt to study the charge carrier dynamics, Debow et al.129 observed that band bending resulted in the formation of a Schottky barrier at the contact point between TiO2 and Ti3C2Tx. The hot electrons generated from MXenes are rapidly (180 fs) transferred into the conduction band of TiO2, indicating strong electronic coupling.129
Peng et al.109 fabricated 1D Nb2O5 nanorod arrays with a [001] orientation on 2D Nb2CTx under HTT followed by the photodeposition of Ag nanoparticles. While the low work function of –OH terminated Nb2CTx promoted the trapping of photogenerated holes from Nb2O5 nanorods, Ag nanoparticles acted both as an electron reservoir and active sites for hydrogen evolution, resulting in an effective spatial separation of charge carriers. Under solar light irradiation, these nanohybrids produced 824.2 μmol g−1 h−1 using glycerol as the sacrificial agent.109 Studies regarding stability and reusability were also carried out by these authors. It was found that hydrogen production remained unchanged after three sequential cycles. Moreover, the morphology, phase composition and surface functional groups of Ag/Nb2O5@Nb2CTx recovered after the reaction, was not altered significantly.
In another interesting study, Wang et al.110 hydrothermally synthesized La2Ti2O7/Ti3C2Tx composites. The introduction of 20 wt% Ti3C2Tx resulted in a 16 times higher hydrogen production rate than that of pristine La2Ti2O7, in the presence of methanol. Solid-state NMR and Raman spectroscopy clearly indicated the formation of graphene quantum dots in Ti3C2Tx. In contrast to the vast majority of reports on MXene-based photocatalysts, these authors proposed that graphene quantum dots produced in situ during the HF etching process, rather than Ti3C2Tx itself, played the role of a co-catalyst during the photocatalytic reaction, by suppressing the charge recombination.110 However, in our understanding, no further reports have appeared in this regard.
Metal sulfides are another category of semiconductors widely investigated for photocatalytic applications. In comparison to MOs, these materials comprise a narrow band gap, enabling a visible light response, and hence, several metal sulfides were also coupled with MXenes in photocatalytic water splitting.
Ran et al.111 integrated Ti3C2Tx nanoparticles with a series of metal sulfides in different proportions. Combination with CdS resulted in the best hydrogen yields with an apparent quantum efficiency of 40.1%. Similarly, 386% and 217% enhancement of the photocatalytic performance was respectively observed for Zn0.8Cd0.2S and ZnS by incorporating 1% of Ti3C2Tx. The authors attributed this to a favourable Fermi level position and superior electronic conductivity of MXenes. Significant variations in the performance of CdS/Ti3C2Tx were not observed during seven successive cycling tests. A comparison between the fresh and used materials also indicated consistency in the crystalline phase, morphology and size.111
Hydrothermally synthesized 1D CdS nanorod/2D Ti3C2TX nanosheet heterojunctions displayed 7 times increased hydrogen production compared to pristine CdS nanorods with an AQY of 35.6%, under visible light irradiation in the presence of lactic acid. In this study also, no obvious decrease in hydrogen production was observed for five consecutive cycles. The enhanced performance was attributed to the accelerated charge separation facilitated by the Schottky heterojunction formed, as well as to the synergistic interactions between the components.112
Another study highlighting the effectiveness of combining CdS with Ti3C2Tx under HTT, reported hydrogen evolution rates higher than that of pure CdS and CdS@Pt, with an AQY of 2.28%. The authors also noted 87.61% retention of performance after three cycles. Apart from a slightly weakened intensity of the XRD peaks, no significant changes were observed for the material recovered after the reaction.113 CdS/Ti3C2Tx composites forming 2D/2D Schottky heterojunctions exhibited a 5-fold enhancement of photocatalytic performance of pure CdS, with an AQY of 47%. Stable performance was observed for five consecutive cycles. In this case also, the superior performance was attributed to an intimate interface promoting rapid charge transfer and hindering their recombination, which was confirmed by DFT calculations.114
In another interesting study, Ti3C2Tx/CdS nanocomposites prepared under γ-ray irradiation, upon NaOH treatment resulted in the formation of TiO2–C/CdS nanocomposites. This material exhibited hydrogen evolution rates 6.4 and 7.0 times greater than that of Ti3C2Tx/CdS and CdS, respectively. In this case, oxygen vacancies in amorphous TiO2 also acted as electron trapping sites to facilitate the transfer of photoexcited electrons. No significant loss in photocatalytic performance was observed for five cycles.115
Jin et al.116 synthesized hybrid structures of 2D Mo2C sheets covered with CdS nanocrystals. This hybrid photocatalyst with 2.5 wt% of Mo2C exhibited a 11% higher hydrogen production rate than noble-metal catalysts with an apparent quantum yield of 48.06%.116
Zuo et al.117 used Ti3C2Tx nanosheets to support ultrathin ZnIn2S4 nanosheets, forming sandwich-like hierarchical heterostructures. The resultant materials exhibited improved specific surface area, pore diameter, and hydrophilicity.
A 6.6 times improvement in photocatalytic hydrogen evolution was observed for pure ZnIn2S4 with an AQY of 11.14%, which was attributed to facile charge transfer and inhibition of their recombination (see Fig. 7). The values of hydrogen evolved remained nearly constant after six cycles.117
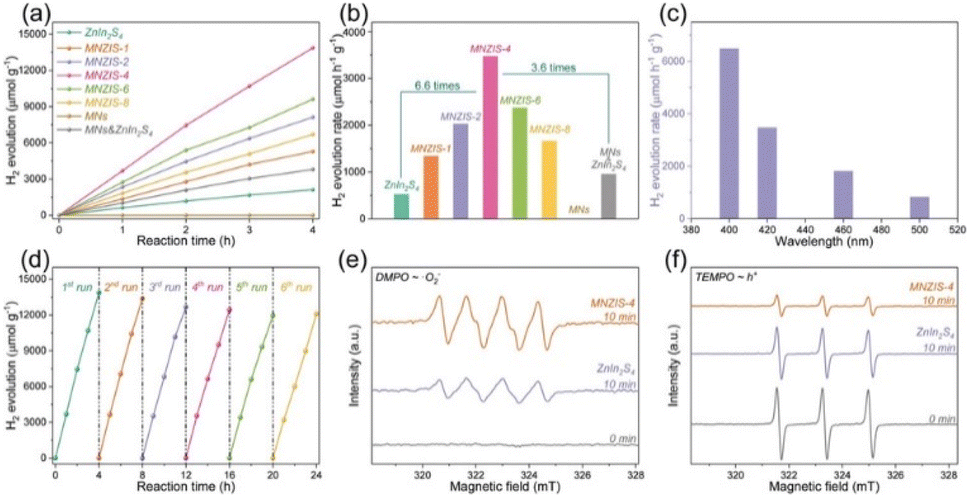 | ||
| Fig. 7 (a) Time–yield plots and (b) comparison of photocatalytic H2 evolution for ZnIn2S4, Ti3C2Tx- ZnIn2S4 (MNZIS-X), MNs, and a physical mixture of MNs & ZnIn2S4, (c) H2 evolution rates for MNZIS-4 with different long-pass cut-off filters, (d) cyclic experiments of H2 evolution using MNZIS-4, and EPR spectra of (e) DMPO ˙O2− and (f) TEMPO h+ for MNZIS-4 and ZnIn2S4. Reproduced with permission from ref. 117, Copyright 2020 Wiley Publications. | ||
A hierarchical core@shell structure consisting of BiVO4@ZnIn2S4 over which Ti3C2 quantum dots were uniformly distributed, was obtained by following a multi-step hydrothermal approach. This material exhibited overall water splitting under visible light irradiation. Stable photocatalytic performance was observed during five cycles. The addition of Ti3C2 significantly improved the light absorption efficiency and range, extending towards the near infrared region. The close contact between BiVO4 and ZnIn2S4 resulted in effective charge separation. MXenes captured electrons from the conduction band of ZnIn2S4 and thereby suppressed charge recombination. The inherent confinement effects of Ti3C2 in the form of quantum dots is also found to contribute.118
Similarly, Huang et al.119 hydrothermally generated TiO2 nanoparticles on the surface of Ti3C2 nanosheets, which were then subjected to an additional HTT to obtain hierarchical Ti3C2@TiO2/ZnIn2S4 structures. Under simulated solar light irradiation, hydrogen generation rates 9.1 times higher than that of Ti3C2@TiO2 and 4.6 times higher than that of pure ZnIn2S4 were obtained. This was attributed to the superior light harvesting, availability of sufficient active sites, intimate interfacial contact and efficient charge separation resulting from the formed heterojunction. A minor decrease in H2 amounts was observed by the authors after four cycles and was attributed to the mass loss that occurred during photocatalyst separation and recovery.119 The CdS–MoS2–Ti3C2Tx composite exhibited a 251.3% enhancement of the hydrogen generation rate of CdS–MoS2, under visible light irradiation, with an AQY of 26.7%. Here, the authors identified three mechanisms for the migration of photogenerated electrons from the conduction band of CdS directly to MoS2 or Ti3C2Tx and to MoS2 through Ti3C2Tx. The adsorbed H+ will get converted into hydrogen either on the surface of Ti3C2Tx or MoS2 edges, implying the significance of the synergy between the components. In this study, the authors noted that adding MXenes above 2% can induce agglomeration and can act as recombination sites for the charge carriers.120 Hydrothermally enabled in situ growth of TiO2 on Ti3C2 nanosheets followed by the distribution of MoS2 on the surface resulted in the formation of MoxS@TiO2@Ti3C2 with molybdenum vacancies and double cocatalysts. This unique composite displayed a 193 times higher hydrogen production rate compared to pure TiO2. The authors observed no significant decrease in hydrogen evolution rates for 3 cycles. Molybdenum vacancies acted as active sites and suppressed charge carrier recombination, whereas MXenes enhanced the electron conductivity.121
Li et al.122 synthesized a ternary CdS@Au/Ti3−xC2Ty composite by a solvothermal method. In particular, core−shell structured CdS@Au nanojunctions were supported on MXenes. The composite with 1 wt% Ti3−xC2Ty and 0.1 wt% Au resulted in 26.6 times enhancement of hydrogen production compared to pristine CdS. The dual Schottky barriers formed at the interface of CdS/Au and CdS/Ti3−xC2Ty facilitated photogenerated electron migration from CdS while limiting back transfer, as evident from XPS analysis, Kelvin probe measurements and DFT calculations. Nanoparticles on the surface of Ti3−xC2Ty nanosheets exhibited signs of aggregation after 4 h which became more pronounced after 29 h, resulting in the decline of photoactivity.122
Polymeric compounds are also investigated for photocatalytic reactions among which graphitic carbon nitrides (g-C3N4) are the most promising. A proper band edge position and narrow bandgap (≈2.9 eV) permits this material to function under visible light. Su et al.99 adapted an electrostatic self-assembly approach to synthesize 2D/2D Ti3C2/g-C3N4 composites by introducing varying amounts of monolayer Ti3C2. Incorporating 3 wt% Ti3C2 resulted in a 10-fold increase in photocatalytic hydrogen evolution performance of pure g-C3N4, under visible light irradiation in the presence of TEA as a hole scavenger. The hydrogen evolution rates were consistent for 3 consecutive cycles of photocatalytic reaction. Moreover, no changes were observed in the composition and structure after the reaction. A slight decrease in light absorption was attributed to the probable separation of Ti3C2 flakes from the g-C3N4 nanosheets. The Schottky junction formed at the interface with the maximum contact surface greatly enhanced electron transport and inhibited their recombination with holes.99
Introduction of 5.5 wt% of Ti3C2 QDs in g-C3N4 by the freeze-drying method resulted in 26 times higher than that of pure g-C3N4, in the presence of TEA (see Fig. 8). Interestingly, these materials also outperformed Pt/g-C3N4. The authors attributed the better performance of these materials to the close contact between Ti3C2 and g-C3N4, large specific surface area and small particle size of Ti3C2 quantum dots. The authors also examined the recyclability and stability of these materials. No significant decrease in the amount of H2 produced was observed during three consecutive eight hour cycles.123
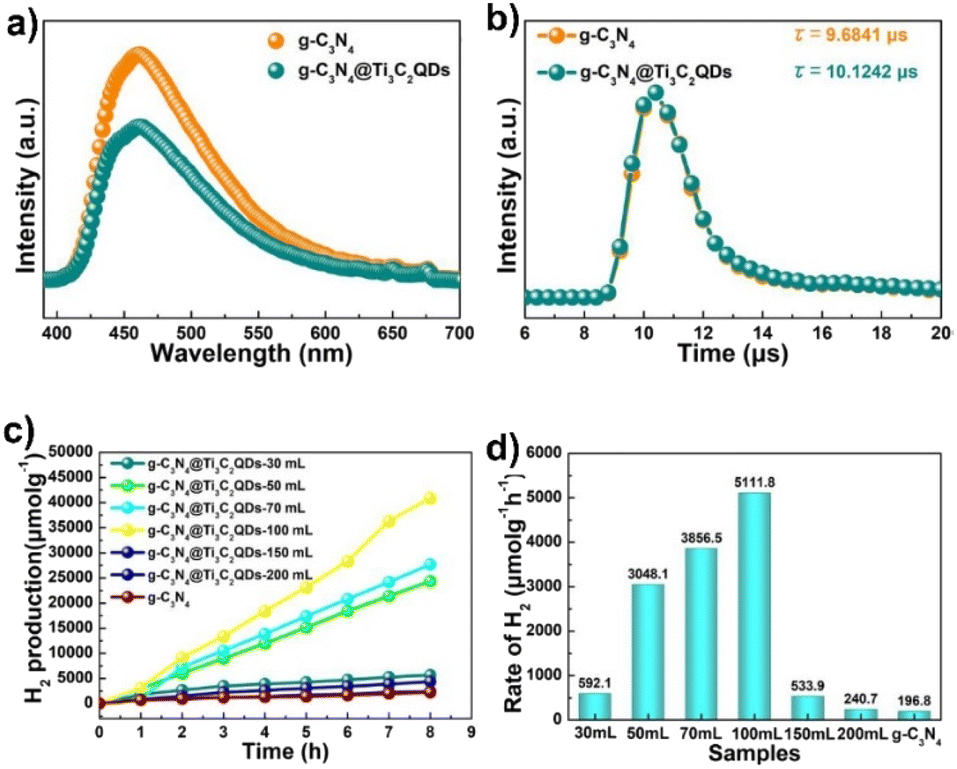 | ||
| Fig. 8 (a) Steady photoluminescence (PL) and (b) time-resolved fluorescence decay spectra of g-C3N4 and g-C3N4@Ti3C2 QDs-100 mL composites, λex = 325 nm. (c) Photocatalytic H2 evolution and (d) rate of g-C3N4 NSs, Pt/g-C3N4, Ti3C2 MXene sheet/g-C3N4 and g-C3N4@Ti3C2 QDs. Reproduced with permission from ref. 123, Copyright 2019 The Royal Society of Chemistry. | ||
Kang et al.124 synthesized g-C3N4/Ti3C2Tx hollow spheres, by electrostatic layer-by-layer assembly, in the presence of amino-functionalized polystyrene beads as sacrificial templates. Protonation of g-C3N4 sheets was carried out using HCl. The resultant hybrid materials exhibited a 3.5 times better hydrogen production rate compared to pure protonated g-C3N4, and a 1.22-fold higher hydrogen production rate than its non-spherical counterpart. The rate of hydrogen production remained approximately the same during three repeated cycles. The 3D hollow structure enabled enhanced light absorption, while the 2D heterostructure between the components shortens the electron migration distance and high surface area providing sufficient active sites.124
2D/3D g-C3N4/Ti3C2 hybrids were fabricated by calcination of melamine together with pre-formed MXenes. This unique material displayed a 6 times higher hydrogen production rate compared to pristine g-C3N4 under visible light irradiation, which was attributed to the Schottky junction formed, preventing charge recombination, together with high electron conductivity of Ti3C2 and an intimate interface.125 The performance remained stable for five cycles. Also, the crystal structure of fresh and recycled samples remained the same, indicating better stability.
Introduction of Ti3C2 into a heterojunction formed between black phosphorus quantum dots and g-C3N4 resulted in hydrogen evolution rates 47.2 and 19.4 times, respectively, higher than that of bulk g-C3N4 and ultrathin g-C3N4. Stable photocatalytic hydrogen evolution was observed for six consecutive cycles. With the apparent quantum yield reaching 17.6%, the composite photocatalysts with an optimum mass ratio exhibited enhanced light absorption and strong interfacial contact. Here, Ti3C2 acted as a bridge to accelerate charge transfer between black phosphorous and g-C3N4.126
Li et al.127 fabricated organic–inorganic composites by depositing supramolecular aggregates of a chlorophyll derivative (zinc methyl 3-devinyl-3-hydroxymethyl-pyropheophorbide a (Chl)) on the surface of Ti3C2Tx. In this case, Chl served as an organic semiconductor and MXene, a co-catalyst. For 2 wt% ChI, 52 ± 5 μmol h−1 g−1 hydrogen evolution was observed under visible light irradiation in the presence of ascorbic acid. Efficient light harvesting followed by exciton transfer in Chl aggregates and the resultant charge separation at the interface were responsible for the observed performance.127 In a similar study, Li et al.128 grafted chlorophyll derivatives, Chl-1 and Chl-2, on the surface of 2D Ti3C2Tx forming a Chl-1@Chl-2@Ti3C2Tx composite. Under visible light irradiation, the hydrogen evolution rate was found to be 143 μmol h−1 g−1 which was substantially higher than the values obtained individually for Chl-1@Ti3C2Tx or Chl-2@Ti3C2Tx.128
3.4. Photocatalytic applications for CO2 reduction
Photocatalytic conversion of CO2 into consumable chemicals and fuels is an ideal approach to harvest solar energy and at the same time counter the challenges related to increasing CO2 emissions. A summary of frequently used reactants, catalysts and products is schematically shown in Fig. 9. Apart from the undesired charge carrier recombination generally encountered in photocatalysis, poor surface adsorption and activation of CO2 molecules must also be addressed in this case. Even though addition of cocatalysts is found to be effective, noble metal free compositions are still scarce in this regard. In comparison to water splitting, fewer studies have been carried out so far using Ti3C2Tx for the photocatalytic conversion of CO2. It needs to be noted here that Ti3C2Tx (or MXenes in general) itself is a source of carbon, and therefore, additional care should be taken while interpreting the results.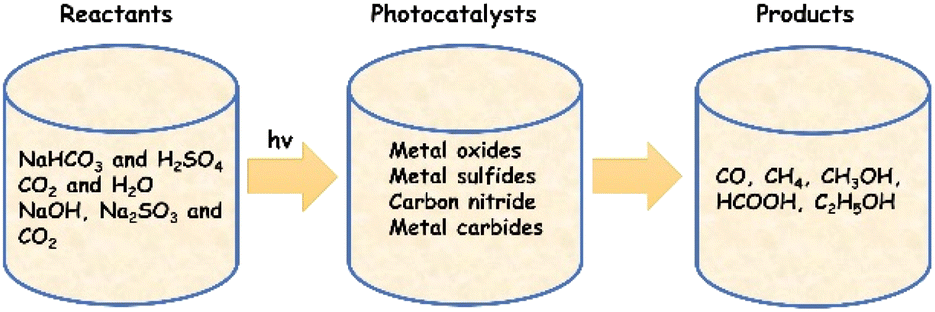 | ||
| Fig. 9 Schematic representation of photocatalytic CO2 conversion highlighting frequently used reactants, photocatalysts and possible products that can be formed. | ||
Table 2 summarizes several examples of MXene-based photocatalysts used in the most recent years for CO2 reduction. The synthetic approaches to combine different semiconductor photocatalysts with MXenes and several reaction parameters are also presented. These reports demonstrated that the use of MXenes as a cocatalyst greatly improves the photocatalytic activity compared to the use of solely the semiconductor photocatalyst. As shown in Table 2, Ti3C2Tx, as a co-catalyst, promotes the separation of charge carriers, leading to the formation of mainly C1 products (i.e., CO, formaldehyde, formic acid, methanol and CH4). The absence of C2 product formation (e.g., ethanol, ethylene or oxalic acid) indicates that Ti3C2Tx cannot change the energy barrier of the photocatalyst.
| Photocatalyst composite | Synthetic method | Reactants | Light region | Products [μmol g−1 h−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|
| a ∼ Data taken from a graphical representation of the corresponding reference; NP: value not provided; MeCN: acetonitrile; TEA: triethanolamine. | ||||||
| TiO2/Ti3C2 | Calcination in air (350–650 °C) | NaHCO3 and HCl | Simulated solar light | CH4 (0.22) | NP | 130 |
| CH3OH (NP) | ||||||
| C2H5OH (NP) | ||||||
| Alkalinized TiO2/Ti3C2 | KOH treatment and physical mixing | CO2 and H2O | Simulated solar light | CH4 (16.61) | 1.61 | 131 |
| CO (11.74) | 0.32 | |||||
| 3D hierarchical TiO2/Ti3C2Tx heterojunction | Calcination in air (300–700 °C for 1 h) | NaHCO3 and H2SO4 | Simulated solar light | CH4 (4.41) | NP | 132 |
| CO (NP) | ||||||
| Ru–Ti3CN–TiO2 | HTT (180 °C for 4 h) | CO2 and H2O | Simulated solar light | CO (99.58 μmol g−1 for 5 h) | NP | 133 |
| CH4 (8.97 μmol g−1 for 5 h) | ||||||
| Core@shell meso- TiO2@ZnIn2S4/Ti3C2 | Electrostatic self-assembly | CO2 and H2O | Visible | CO (30.5 μmol g−1 for 3 h) | NP | 134 |
| CH4 (34 μmol g−1 for 3 h) | ||||||
| Ti3C2 QDs/Cu2O NWs/Cu mesh | Dip coating and calcination under Ar (200 °C for 1 h) | CO2 and H2O | Simulated solar light | CH3OH (78.50) | NP | 135 |
| Ti3C2 sheets/Cu2O NWs/Cu | Dip coating and calcination under Ar (200 °C for 1 h) | CO2 and H2O | Simulated solar light | CH3OH (36.51) | NP | 135 |
| Cu2O/Ti3C2 heterojunction composites | HTT (90 °C for 5 h) | CO2 | Simulated solar light | CO (17.55) | NP | 136 |
| CH4 (0.96) | ||||||
| 2D/2D Ti3C2/Bi2WO6 nanosheets | HTT (120 °C for 24 h) | NaHCO3 and H2SO4 | Simulated solar light | CH4 (1.78) | NP | 137 |
| CH3OH (0.44) | ||||||
| Alkalinized Ti3C2/ZnO | KOH treatment followed by electrostatic self-assembly | CO2 and H2O | Simulated solar light | CO (30.30) | 0.32 | 138 |
| CH4 (20.33) | 0.46 | |||||
| 2D/2D Ti3C2/g-C3N4 nanosheets | Calcination under N2 (550 °C for 2 h) | NaHCO3 and H2SO4 | Visible | CO (5.19) | NP | 139 |
| CH4 (0.044) | ||||||
| 2D/2D/0D TiO2/C3N4/Ti3C2 | Electrostatic self-assembly | NaHCO3 and H2SO4 | Simulated solar light | CO (4.39) | NP | 140 |
| CH4 (1.20) | ||||||
| Alkalinized Ti3C2/g-C3N4 | Wet chemical mixing | CO2 and H2O | Visible | CO (11.21 μmol g−1 for 5 h) | NP | 141 |
| CH4 (0.203 μmol g−1 for 5 h) | ||||||
| C2H4; CH3CHO | ||||||
| Few layer Ti3C2/boron-doped g-C3N4 | Sonication and freeze drying | CO2 and H2O | Visible | CO (14.4 μmol g−1 for 5 h) | NP | 142 |
| CH4 (0.8 μmol g−1 for 5 h) | ||||||
| H2 (3.35 μmol g−1 for 5 h) | ||||||
| g-C3N4/BiOIO3/Ti3C2 ternary composites | Electrostatic self-assembly | CO2 and H2O | Visible | CO (5.88) | NP | 143 |
| CH4 (1.55) | ||||||
| 3D hierarchical Co–Co LDH/Ti3C2Tx nanosheets | HTT (120 °C for 1 h) | MeCN, H2O, TEA, [Ru(bpy)3]Cl2 and CO2 | Visible | CO (12500) |
0.92 | 144 |
| H2 (NP) | ||||||
| 3D hierarchical NiAl LDH/Ti3C2 | HTT (120 °C for 24 h) | CO2 and H2O | Simulated solar light | CO (11.82) | NP | 145 |
| CH4 (1.02) | ||||||
| 0D/2D FAPbBr3/Ti3C2 | Wet chemical synthesis | CO2 and H2O | Simulated solar light | CO (283.41) | NP | 146 |
| CH4 (17.67) | ||||||
| H2 (4.51) | ||||||
| 2D/2D FAPbBr3/Ti3C2 | Wet chemical synthesis | CO2 and H2O | Simulated solar light | CO (93.82) | NP | 147 |
| H2 (∼0.9) | ||||||
| Cs2AgBiBr6 nanocrystals/Ti3C2Tx nanosheets | Ultrasonication | CO2 and H2O | Visible | CO (11.1 μmol g−1) | 0.083 | 148 |
| CH4 (1.3 μmol g−1) | ||||||
| H2 (8.9 μmol g−1) | ||||||
In the same direction, Song et al.132 synthesized 3D hierarchical Ti3C2Tx and performed calcination in air to generate TiO2 on the surface of nanosheets resulting in the formation of a heterostructure with bimodal macro-mesoporosity. These materials were used for photocatalytic conversion of gas phase CO2 produced from the reaction between NaHCO3 and H2SO4 under simulated solar light. In the absence of CO2, product formation was not observed. Also in this case, the composites obtained after calcination at 500 °C, exhibited a 2-fold increase in the CH4 evolution rate compared to commercial TiO2. In this case, the 3D hierarchical structure promoted enhanced light absorption and efficient electron–hole separation whereas strong adsorption of CO2 on Ti3C2 ensured the availability of electrons for the reaction.132
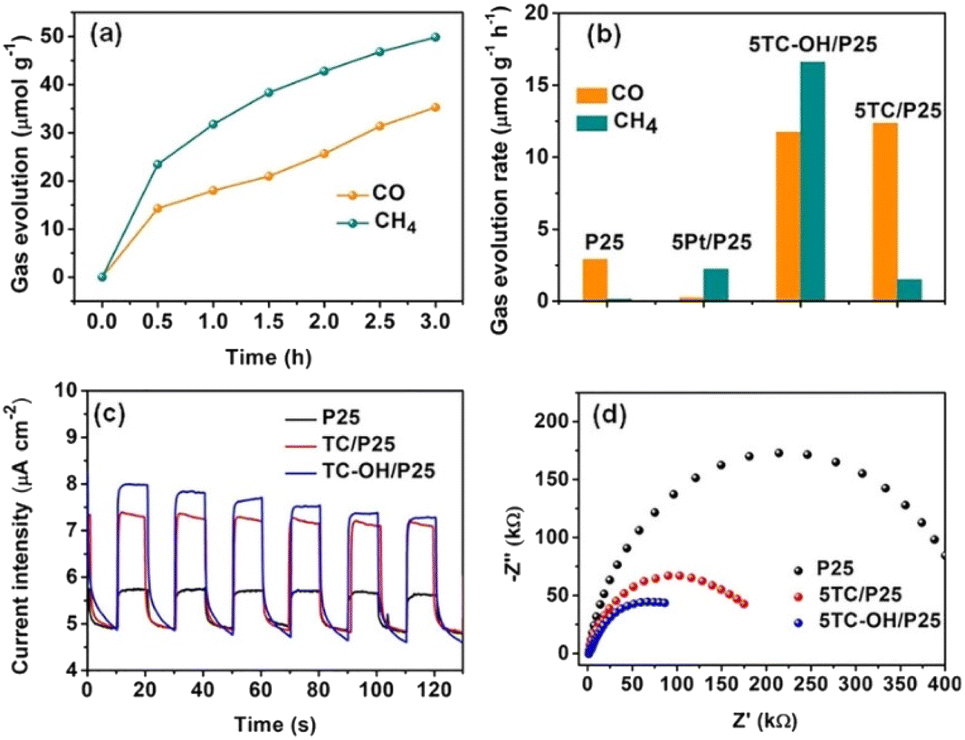 | ||
| Fig. 10 (a) Gas evolution (CO and CH4) over 5TC-OH/P25 as a function of irradiation time under a 300 W Xe lamp. (b) Evolution rates of CO and CH4 over P25, 5Pt/P25, 5TC/P25, and 5TC-OH/P25 under irradiation of a 300 W Xe lamp. (c) Transient photocurrent responses and (d) EIS Nyquist plots of P25, 5TC/P25, and 5TC-OH/P25. Reproduced with permission from ref. 131, Copyright 2018 European Chemical Societies Publishing. | ||
The presence of Ru along with TiO2 on the surface of Ti3CN MXenes produced a mixture of CO, CH4 and other hydrocarbons from CO2 using water as the hydrogen source. With 0.5 wt% Ru, CO and CH4 production rates increased by 20.5 and 9.3 times, respectively, compared to that of P25. Isotopic labelling studies confirmed CO2 as the origin of the reaction products. Gas production rates exhibited a 13.4% decrease during the first three cycles. This was attributed to the accumulation of intermediate products on the active sites, hindering further reaction. To confirm this, the photocatalyst was recovered and washed with deionized water. This resulted in a performance similar to that observed during the first cycle.133
Wang et al.134 prepared a core@shell structured meso-TiO2@ZnIn2S4 and different amounts of Ti3C2 were deposited on the ZnIn2S4 shell. This ternary photocatalyst, under UV-visible light irradiation, resulted in CO and CH4 evolution with slightly better CH4 selectivity. The better performance in comparison with that of the individual components was attributed to accelerated electron transfer to Ti3C2 as well as the enhanced CO2 adsorption capacity of the 3D mesostructure.134
Zeng et al.135 developed a hierarchical heterostructure by decorating Cu2O nanowires with Ti3C2 quantum dots (QDs) on a Cu mesh, adapting an electrostatic self-assembly strategy. Photocatalytic transformation of a CO2 saturated H2O medium was carried out under solar light irradiation, and selective formation of CH3OH was observed. Product yields were found to be 8.25 times higher compared to that of Cu2O nanowires on a Cu mesh. Interestingly, the presence of MXenes as QDs resulted in a 2.15 times higher methanol yield compared to MXene sheets with the same composition. 89% retention of methanol yield was observed after six cycles of photocatalytic reaction. The authors reported a multi-fold influence of Ti3C2Tx on the performance of Cu2O NWs by enhancing charge transport, carrier density, and light adsorption, as well as by decreasing the band bending edge and limiting charge carrier recombination.135 Taking into account the good synergy with Cu2O, a series of Cu2O/Ti3C2Tx heterostructures with varying amounts of Ti3C2Tx, synthesized under hydrothermal conditions were studied for the photoconversion of CO2 to CO in the absence of a hydrogen source.136 Addition of 10 mg of Ti3C2Tx to the initial precursor solution for Cu2O synthesis, exhibited 3 times higher CO production rates compared to that of pristine Cu2O. After three cycles, the amount of CO produced remains 85.8% in comparison with the first cycle. The slight decrease was attributed to the loss of the sample during recovery. Surprisingly, the authors noticed the formation of minor amounts of CH4. However, since the reaction took place under a pure CO2 atmosphere, the detection of CH4 clearly indicates the presence of impurities. Here also, the performance enhancement was attributed to the separation and concentration of electrons on Ti3C2, facilitating CO2 conversion.136
The 2D/2D heterojunctions were found to be very active in photocatalytic transformation of CO2. Therefore, Cao et al.137 fabricated a 2D/2D heterojunction comprising Ti3C2Tx/Bi2WO6 nanosheets under hydrothermal conditions. H2SO4 and NaHCO3 were reacted for the in situ production of CO2 which under simulated solar light irradiation produced CH4 as the major product along with CH3OH. The results indicate a 4.6 times increment in CH4 and CH3OH yields compared to that of pristine Bi2WO6 ultrathin nanosheets. The large interfacial surface area resulting from the 2D/2D heterojunction and lower charge transport distance ensured efficient electron transfer from Bi2WO6 to Ti3C2. This effect coupled with enhanced CO2 adsorption capacity resulted in the observed performance of this study.137
In the same line, Yang et al.139 synthesized a series of 2D/2D Ti3C2/g-C3N4 ultra-thin heterojunctions by performing calcination of varying amounts of Ti3C2Tx and urea. Here, apart from being the precursor for C3N4, the presence of urea facilitated the exfoliation of MXene sheets. Photocatalytic conversion of in situ generated CO2 was carried out under visible light irradiation. In the absence of Ti3C2Tx, CO was the major product observed beside small traces of CH4. The optimized composite material (with 10 mg of Ti3C2) added to the urea precursor, exhibited better yields of CO and CH4 with 8.1 times higher CO2 conversion, compared to pure C3N4. The performance, structure and morphology of the photocatalyst remained reasonably unchanged during five cycles. The detrimental effect of excess Ti3C2Tx was observed in this study also, as previously reported by Low et al.130 The spatial separation of charge carriers facilitated by the formation of an ultra-thin heterojunction, along with an improved CO2 adsorption and activation was responsible for the improvement of performance.139
Integration of Ti3C2 quantum dots into a TiO2/C3N4 core@shell structure resulted in a 2D/2D/0D dual heterojunction photocatalyst. This material exhibited superior photoconversion of in situ generated CO2 into CO and CH4 along with lower amounts of methanol and ethanol. Product yields remained consistent during four cycles of photocatalytic reactions. The formation of dual heterojunctions was found to be responsible for an improved separation and utilization of charge carriers, resulting in the enhanced performance.140
The effect of alkalinized Ti3C2 was also observed by coupling it with g-C3N4. The composite prepared by mixing 5 wt% alkalinized Ti3C2 with g-C3N4 exhibited better CO evolution rates during photocatalytic conversion of a CO2 saturated H2O medium under visible light irradiation, compared to pure g-C3N4. A slight decrease in performance was observed during the first three cycles. However, after washing the catalyst, the activity increased and remained unchanged during the fourth and fifth cycles. A large Fermi level difference between the components contributed towards the enhanced separation of photogenerated charge carriers.141
Wang et al.142 synthesized a few-layer Ti3C2/B-doped C3N4 composite material by adding different volumes of an aqueous solution of Ti3C2Tx (1 mg mL−1) to a fixed amount of B-doped g-C3N4 (0.4 g). Because of the intimate interfacial contact, the optimal (12 mL) MXene suspension added to the composite exhibited 3.2 times and 8.9 times higher CO and CH4 yields, respectively, compared to pristine g-C3N4 under visible light irradiation. A slight decrease (∼14%) in CH4 and CO production was observed during the first three cycles which was then increased after washing with water.142
A BiOIO3/g-C3N4Z-scheme heterojunction modified with Ti3C2 nanosheets was prepared by Hong et al.143 following an electrostatic self-assembly method. Photocatalytic CO2 conversion in an aqueous medium under visible light irradiation indicated a 6.6 times higher CO production rate for heterojunction photocatalysts with 4 wt% Ti3C2, in comparison with pure g-C3N4. No significant reduction in CH4 and CO yields was observed during three cycles.143
Chen et al.144 synthesized a 3D hierarchical Co–Co layered double hydroxide/Ti3C2TX nanoarray under HTT. These materials exhibited a significant enhancement in CO evolution rates, with an apparent quantum efficiency of 0.92%. The high electron conductivity of Ti3C2Tx, coupled with the nanoarray architecture synergistically contributed towards the performance enhancement. For these materials, the product yield remained 90% of its initial value after five cycles.144
Shi et al.145 synthesized a 3D hierarchical NiAl layered double hydroxide/Ti3C2 nanocomposite under HTT by adding different volumes of an aqueous solution of Ti3C2 (15 mg mL−1) to the NiAl-LDH precursor solution. Photoconversion of CO2 was carried out using H2O as the hydrogen source, producing CO with 92% selectivity, along with CH4. Up to 3 times enhancement of CO production was observed and the 2D/2D interface interaction was found to promote charge separation and to facilitate CO2 capture and activation. The CO yield remained constant for four cycles.145
Organic–inorganic hybrid perovskites are another interesting category of materials coupled with MXenes for photocatalytic applications. Que et al.146 anchored FAPbBr3 QDs on Ti3C2 nanosheets resulting in a composite 0D/2D heterojunction photocatalyst for CO2 reduction. The amount of Ti3C2 in the composite was varied between 0 and 1 mg. Under simulated solar light irradiation, CO was formed as the major product along with minor quantities of CH4 and H2 as shown in Fig. 11. In the presence of 0.2 mg of Ti3C2, CO yields were found to be 1.84 times higher compared that of to pure FAPbBr3. No significant changes in product yields were observed during three reaction cycles. Ti3C2 QDs served as the electron acceptor as well as provided more active sites.146
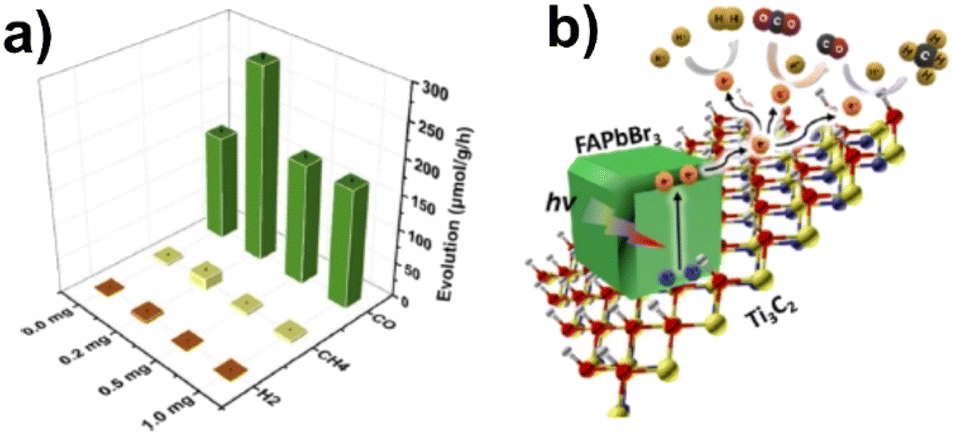 | ||
| Fig. 11 (a) Photocatalytic CO2 reduction performances of FAPbBr3/x-Ti3C2 (x = 0.0, 0.2, 0.5, and 1.0) in deionized water under AM 1.5 G (150 mW cm−2). (b) Schottky heterojunction photocatalytic system for CO2 reduction in FAPbBr3/x-Ti3C2. Reproduced with permission from ref. 146, Copyright 2021 American Chemical Society. | ||
In another study, FAPbBr3 nanoplates were combined with Ti3C2 by Que et al.147 resulting in a 2D/2D Schottky heterojunction. A 1.5-fold enhancement in average CO production was observed for the composite, compared to pure halide perovskite.147 Zhang et al.148 assembled whole inorganic halide double perovskite Cs2AgBiBr6 nanocrystals (NCs) on Ti3C2Tx nanosheets. Superior product yields with 71% selectivity to CO, were obtained for the heterostructures, compared to the individual components. The authors used the electron consumption yield to determine the stability of the material. It remained almost unchanged after 3 cycles. Structural or morphological changes were also not observed during the reaction. The presence of Ti3C2Tx reduced the large exciton binding energy of Cs2AgBiBr6 and promoted the formation of free charge carriers with prolonged life times. Charge carrier recombination was therefore suppressed and this together with enhanced CO2 adsorption, resulted in better performance.148
3.5. Photocatalytic applications for N2 photofixation
In addition to the well-established photocatalytic applications presented above, the MXenes are used as a cocatalyst in photocatalytic systems for the N2 reduction reaction (NRR) to ammonia. In contrast to modern Haber-Bosch technology, which directly utilizes fossil fuels through intensive energy processes at high pressure and temperature and with high CO2 emissions,149,150 the NRR is regarded as an environmentally friendly, sustainable and green NH3 synthesis that utilises N2 and water molecules under mild conditions.151 Obtaining high reaction efficiencies is primarily constrained by electron–hole recombination and limited interfacial charge transfer, but it is also difficult to activate and break the strong and stable NN bond.152 The effectiveness of the NRR can be increased by using MXenes as a cocatalyst to create Schottky junctions with semiconductor materials. Table 3 highlights a few examples of MXene-based photocatalysts that have been applied recently for the NRR.| Photocatalyst | Preparation methods | Reaction conditions | Light source | Sacrificial reagent | NH3 production [μmol g−1 h−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|---|
| a NP: value not provided. b White light (300–780 nm). c Visible light (420–780 nm). | |||||||
| 0D/2D AgInS2/Ti3C2 | HTT (150 °C for 5 h) | 20 mg of sample dispersed in 100 mL 20% methanol solution, RT | 300 W xenon lamp (>400 nm) | Methanol | 38.8 | 0.07 (420 nm) | 153 |
| r-Ti3C2/Au | Solvent-driven | 80 mg of the photocatalyst and 50 mL of water, RT | 300 W xenon lamp | None | 22.6b 12.4c | 0.697 (520 nm) | 154 |
| 200 mg of the photocatalyst and 50 mL of water, RT | Simulated solar light | 21.26 | |||||
| RuO2@TiO2-MXene | HTT (150 °C for 10 h) | 50 mg of photocatalyst with 100 mL distilled water | 300 W xenon lamp, full spectrum (100 mW cm−2) | None | 4.37 | NP | 155 |
| MXene/TiO2/Co-0.5% | Two-step calcination- under N2 (400 °C for 1 h) | 30 mg was dispersed in 100 mL of ultrapure water | 300 W xenon lamp (UV-vis light) | None | 106.7 | NP | 156 |
| CdS@Ti3C2 | HTT (200 °C for 5 h) | 20 mg photocatalyst dispersed in 100 mL aqueous solution containing 20 vol% methanol | 300 W xenon lamp | Methanol | 293.06 μmol L−1 h−1 | 7.88 | 113 |
| Nb2O5/C/Nb2C/g−1-C3N4 | Calcination under CO2 (850 °C) and co-sintering (550 °C for 2 hours) | 50 mg of photocatalyst in 50 mL of double distilled water with 20 vol% methanol | Visible light (λ > 420 nm) | Methanol | 365 | NP | 157 |
| TiO2@C g−1-C3N4:10% | One-step calcination in air (550 °C for 2 hours) | 50 mg of photocatalyst suspended in 100 mL of DD water containing 20 vol% methanol | 300 W xenon lamp | Methanol | 250.6 | 0.14 (420 nm) | 158 |
| OV-C/TiO2-600 | One-step calcination in air (600 °C for 2 hours) | 50 mg of catalyst dispersed in 50 mL of ultrapure water, RT | 300 W xenon lamp (200–800 nm) | H2O | 41 | 0.04 (400 nm) | 159 |
| Methanol | 81 | ||||||
| Ti3C2Tx/TiO2-400 | Calcination in air (400 °C for 15 min) | 10 mg of photocatalysts and 20 mL of deionized water, RT | Xenon lamp (320–780 nm), full spectrum (250 mW cm−2) | None | 422 | NP | 160 |
| W/Ti3C2Tx-U | Ball milling | 20 mL of ultrapure water, 10 mg of purified photocatalysts, RT | Xenon lamp, full-spectrum (250 mW cm−2) | None | 227.5 | 0.05–0.15 in UV-vis and NIR | 161 |
| C3N4/r-Ti3C2 QDs-2 | Electrostatical assembling | 20 mg photocatalyst and 50 mL of 20 vol% methanol, RT | 300 W xenon lamp | Methanol | 328.9b | 0.92 (380 nm) | 162 |
| 197c | |||||||
The 0D/2D AgInS2/Ti3C2Z-scheme heterostructures constructed by HTT, with various mass ratios of AgInS2, showed a notable improvement in photocatalytic activity for the NRR, according to Qin et al.153 For the mass ratio of 30% AgInS2 in N2 saturated solution and under visible light, the greatest ammonia yield rate was 38.8 μmol g−1 h−1, with an AQY = 0.07% at 420 nm. The utilization of Ti3C2 MXenes as a cocatalyst and Z-scheme heterojunction, allowed the effective charge separation and migration of photogenerated electrons and holes, which are responsible for the photocatalytic activity for the NRR.
According to Chang et al.,154 the usage of partly reduced Ti3C2 (r-Ti3C2) with Au nanospheres interlaminated in a layered r-Ti3C2 structure helped to expose several low-valence Ti sites that act as active sites. Injection of photogenerated hot electrons from Au nanospheres under illumination reduces N2 molecules, which are then captured and activated at the active site. In N2 saturated water, the NRR of r-Ti3C2/Au generated NH4+ at a rate of 12.4 μmol g−1 h−1 under visible light, which is 5.9 and 10.3 times higher than those of Ti3C2/Au and r-Ti3C2, respectively, while under white light the rate was 22.6 μmol g−1 h−1, which is 5.8 and 10.2 times higher than those of Ti3C2/Au and r-Ti3C2. By measuring the quantity of ammonia generated in pure water under monochromatic light irradiation, the apparent quantum efficiencies (AQE) of r-T3C2/Au were found to reach 0.697% at 520 nm. The NH3 production rate increased to 21.26 μmol g−1 h−1 when the lighting source was changed to simulated sunlight irradiation (by using an AM 1.5G filter).
The material exhibits high stability, with ammonia generation remaining close to 95%, according to the results of five successive stability test cycles. This may be due to the electrons that are constantly produced and transferred from the Au nanospheres to Ti3C2 under illumination, which can prevent oxidation of MXenes.
Hao et al.155 demonstrated outstanding activity for the NRR in water at room temperature using a RuO2-loaded TiO2-MXene hybrid nanostructure with varied RuO2 ratios. After 450 minutes of Xe-lamp irradiation, the as-prepared RuO2@TiO2-MXene exhibits good N2 reduction performance, reaching a production rate of 4.37 μmol g−1 h−1. The results show that RuO2@TiO2-MXene is really a viable material for the NRR. Since TiO2 creates electrons and holes, MXenes offer efficient electron transport due to metallic conductivity, and RuO2 presents activation sites, and combining the three components in the hybrid structure has a significant impact on the material's photofixation performance.
In a two-step calcination procedure, Gao et al.156 created MXene/TiO2/Co composite materials for the NRR. The production rate of MXene/TiO2/Co-0.5% increased 1.4 times by the addition of Co (50 μmol g−1 h−1) compared to that of MXene/TiO2, and 2.4 times compared with that of TiO2 (69 μmol g−1 h−1). It is significant to note that the ammonia concentration reached 107 μmol g−1 h−1 under UV-vis light. More than this, the photocatalysts exhibit structural stability throughout the reaction as the XRD diffractograms after five cycles of the NRR show no change at all before and after 10 hours of irradiation. Electron paramagnetic resonance (EPR) spectroscopy was used to examine the reaction mechanism and the results suggested that MXene/TiO2/Co-0.5% had a higher oxygen vacancy concentration which is beneficial for photocatalytic nitrogen fixation,163 as shown in Fig. 12.
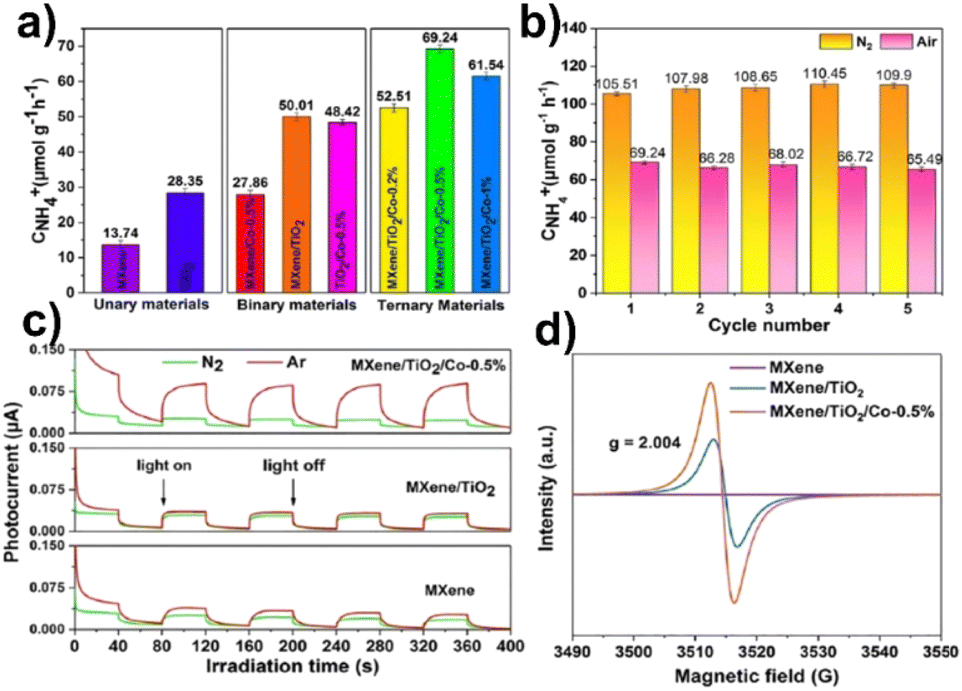 | ||
| Fig. 12 (a) NH4+ production rate in ambient air; (b) the stability of N2 photofixation activity of MXene/TiO2/Co-0.5% under N2 and an air atmosphere; (c) photocurrent spectra of MXene, MXene/TiO2 and MXene/TiO2/Co-0.5%; (d) EPR spectra. Reproduced with permission from ref. 163, Copyright 2015 American Chemical Society. | ||
Sun et al.113 investigated the effects of Ti3C2 as a co-catalyst generated by HTT for the NRR, as well as the photocatalytic performance of CdS@Ti3C2 composites. Pure CdS NRs (nanorods) had a low rate of photocatalytic NH4+ formation (156.62 μmol L−1 h−1), while Ti3C2 does not produce any ammonia. To create the CdS@Ti3C2 composites, several amounts of Ti3C2 ranging from 15 to 100 mg, were tested. Of these, in the presence of a scavenger, CdS@Ti3C2-15 with 15 mg of MXenes has shown the best NRR activity at a rate of 293.06 μmol L−1 h−1, which is 1.9 and 1.4 times higher than that of pure CdS NRs and CdS@Pt, respectively, with an AQY = 7.88%. As a result, Ti3C2 MXenes may be able to replace and outperform Pt in terms of enhancing CdS's capacity for the NRR. Photocatalytic nitrogen fixation on a CdS Ti3C2-15 composite was conducted in the absence of a sacrificial agent at a nitrogen fixation rate of 27 μmol L−1 h−1. The stability test revealed that the production rate is 99.35% of the initial rate after three cycles. Due to their affordability and stability, these composites are expected to play a significant role in the production of ammonia in the future.
Jiang et al.157 reported a 2D MXene-derived Nb2O5/C/Nb2C/g-C3N4 heterojunction which was created by uniformly growing Nb2O5 on Nb2C and then forming g-C3N4 nanosheets in situ on Nb2O5/C/Nb2C. With methanol acting as a scavenger and an optimum Nb2O5/C/Nb2C:g-C3N4 ratio of 1:1, the heterojunctions exhibited a 9.1 times higher nitrogen reduction rate (365 μmol g−1 h−1) in water under visible light, compared with an MXene derived Nb2O5/g-C3N4 composite. Additionally, a pH adjustment from 3 to 9 would enhance the efficiency of the NRR from 180 to 927 μmol L−1 h−1 by changing the energy barrier of proton reduction. The improved nitrogen reduction photocatalytic performance may be due to both the Schottky junction produced at the Nb2O5/Nb2C interface as well as the increased electron and hole separation due to the close contact between the conductive Nb2C and the Nb2O5. The NRR efficiency decreased as the pH of the catalytic system is higher than 9, due to extra negative charges created on the photocatalyst surface that prevent the reaction substrate from effectively making contact with the catalytically active site164 (Fig. 13). Nb2O5/C/Nb2C/g−1-C3N4 heterojunctions are stable materials, with no noticeable NH3 production rate loss observed after 5 cycles.
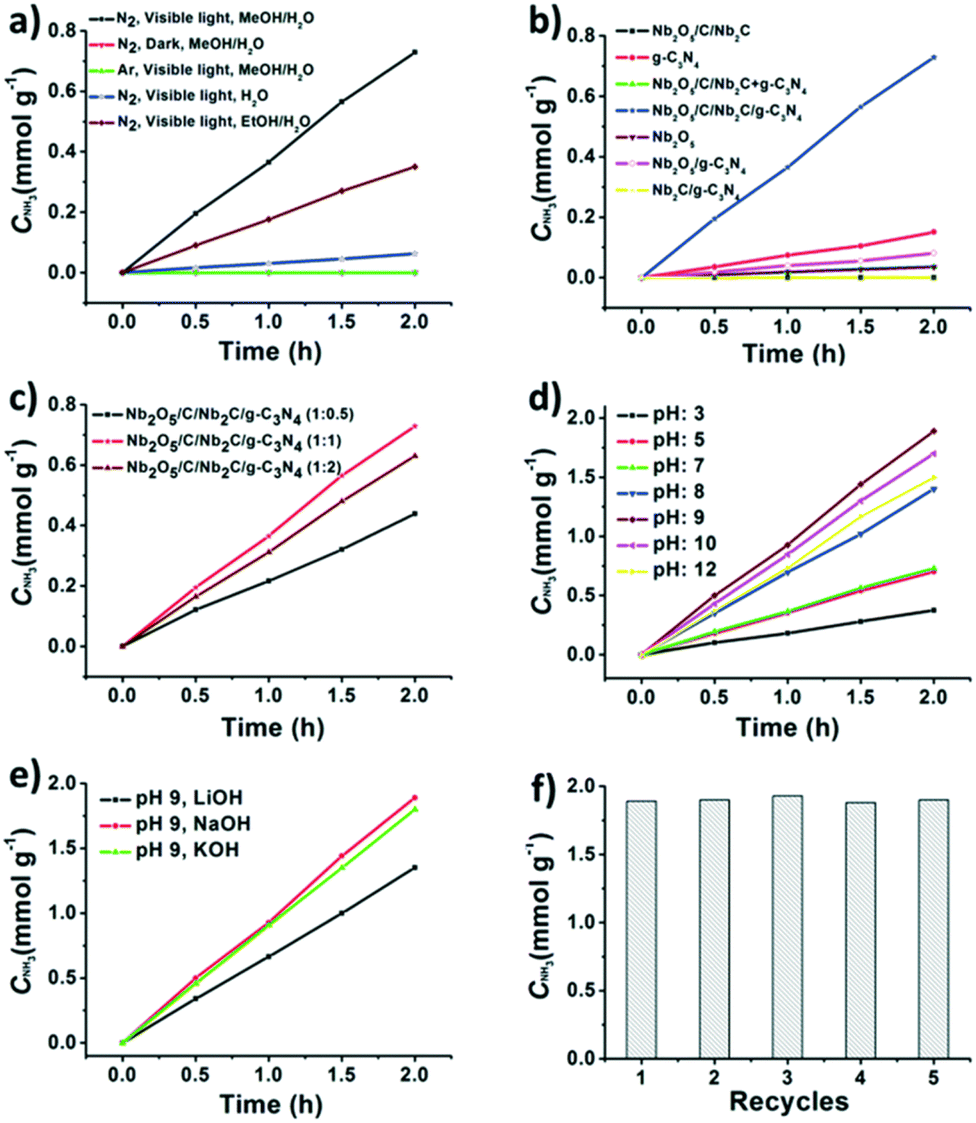 | ||
| Fig. 13 (a) Control experiments on photocatalytic NH3 production with the MXene-derived Nb2O5/C/Nb2C/g-C3N4 heterojunction under different conditions. (b) Photocatalytic activity of the Nb2O5/C/Nb2C, g-C3N4, Nb2O5/C/Nb2C + g-C3N4, Nb2O5/C/Nb2C/g-C3N4, Nb2O5, Nb2O5/g-C3N4 and Nb2C g−1-C3N4 for NH3 production under visible light irradiation (λ > 420 nm). (b) Photocatalytic NH3 production activity of Nb2O5/C/Nb2C/g−1-C3N4 with the ratio variation of Nb2O5/C/Nb2C: melamine from 0.5 to 1 and 2 during catalyst preparation; (c) photocatalytic NH3 production activity of Nb2O5/C/Nb2C/g-C3N4 with different pH values adjusted by using HCl or NaOH solution. Reproduced with permission from ref. 157, Copyright 2020 The Royal Society of Chemistry. | ||
Through one-step calcination in air of a mixture of Ti3C2 and melamine with various ratios of TiO2@C to g-C3N4, Liu et al.158 fabricated a TiO2@C/g-C3N4 heterojunction for the NRR. This approach produces a large number of Ti3+ species which act as active sites. The composite material involves the formation of TiO2 by thermal treatment of Ti3C2Tx MXenes, which is further tightly wrapped by in situ formed g-C3N4 nanosheets. The remarkable performance of the TiO2@C/g-C3N4 heterojunction for photocatalytic nitrogen reduction to ammonia is due to its extensive surface defects, high electron-donating ability, improved light absorption zone, efficient charge transport and reduced recombination, and also strong nitrogen activation ability. The TiO2@C/g-C3N4:10% sample had the highest photocatalytic activity of all the samples, with an ammonia production rate of 250.6 μmol g−1 h−1, with an AQE of 0.14% (420 nm) under visible light irradiation. The performance of TiO2@C/g-C3N4:10% is found to be 18 and 10 times higher than that of g-C3N4 and TiO@C, respectively. A recycling NH3 test on TiO2@C/g-C3N4 revealed no detectable decrease in the NH3 production rate following five cycles in six hours, demonstrating the good stability of the sample.
Using a one-step calcination method at varied temperatures, Qian et al.159 used Ti3C2 as a precursor to produce an oxygen vacancy (OV)-rich C/TiO2 (OV–C/TiO2) material to convert N2 into NH3 under sunlight irradiation. Among the samples, OV-C/TiO2-600 produced NH3 at a rate of 41 μmol g−1 h−1 using water as a proton source (AQY of 0.04% for 400 nm light and 0.01% for of 420 nm light, respectively) which is 26.5 times higher than that of TiO2 Degussa P25. When using methanol as a proton source the NH3 rate increased to 84 μmol g−1 h−1. The addition of oxygen vacancies on the TiO2 surface and carbon doping increased electron–hole pair formation and decreased electron–hole pair recombination. Moreover, there was an increase in the photocarrier lifetime, as well as chemisorption and activation of N2 molecules. The NH3 production yields essentially stayed constant during four photocatalytic cycling trials in 2 h, showing the OV-C/TiO2-600 sample's good stability.
Hou et al.160 developed plasmonic Ti3C2Tx and TiO2 hybrid structures (Ti3C2Tx/TiO2) at various temperatures, with Ti3C2Tx/TiO2-400 exhibiting higher activity for the NRR. At room temperature, the Ti3C2Tx/TiO2-400 hybrid structure yielded 422 μmol g−1 h−1 of NH3 without the use of any sacrificial agents. After being exposed to monochromatic light at 630 nm, Ti3C2Tx/TiO2-400 generated 50 μmol g−1 h−1 of NH3, with an AQE of 0.05%. Once monochromatic light was changed to 740 nm light, the NH3 generation rate for Ti3C2Tx/TiO2-400 improved to 82 μmol g−1 h−1, while the AQE became 0.07%. Additionally, the stability test of Ti3C2Tx/TiO2-400 shows that more than 90% of the initial reaction activity was conserved in ten consecutive rounds of reaction, demonstrating the high stability of Ti3C2Tx/TiO2-400. The activity of Ti3C2Tx/TiO2-400 hybrid structures was attributed to the coexistence of Ti3C2Tx and defective TiO2, which considerably improved charge carrier separation efficiency. The plasmonic Ti3C2Tx phase may collect visible and near-infrared light to generate hot electrons that are transported to the TiO2 conduction band and then trapped by oxygen vacancies.
Under full-spectrum irradiation at room temperature without the use of sacrificial materials, Qi et al.161 demonstrated a remarkable ammonia production rate (227.5 μmol g−1 h−1) utilizing Ti3C2Tx doped with a W atom. Further research on the catalytic performance was conducted using monochromatic light of various wavelengths. At 630 nm, the production rate was 43.6 μmol g−1 h−1, but it dropped to 28 μmol g−1 h−1 when the light source was centered at 740 nm. In particular, the AQEs of W/Ti3C2Tx-U were 0.05–0.15% in UV-vis and NIR wavelengths. In addition, W/Ti3C2Tx-U are stable materials preserving >90% activity after ten subsequent rounds of reaction. Doping with W atoms enhances the physical and chemical adsorption of N2, and they also serve as the active sites for reducing reaction energy during the NRR.
According to Chang et al.,162 mesoporous hollow g-C3N4 spheres were improved with partly reduced Ti3C2 quantum dots (r-Ti3C2 QDs) on the surface for effective NRR. This resulted in the formation of a C3N4/r-Ti3C2 QD Schottky junction via electrostatic self-assembly. The C3N4/r-Ti3C2 QDs' nitrogen photofixation activity was better when exposed to white light than it was when exposed to visible light since the photocatalysts had a limited capacity to absorb visible light. The ammonia production rate of C3N4/r-Ti3C2 QDs reduced from 328.9 μmol g−1 h−1 to 197 μmol g−1 h−1 when the light source was switched from white to visible light (Fig. 14), which, under the same circumstances, are 1.6 and 1.9 times as high as that of C3N4, respectively. The AQE value of C3N4/r-Ti3C2 QDs is 0.92% at 380 nm. When C3N4 is exposed to light, photoinduced electrons and holes are produced, and r-Ti3C2 QDs enable effective migration and separation for these particles. Numerous OV-defect state sites and Ti3+ sites found in r-Ti3C2 QDs serve as active sites for collecting and activating N2 molecules. Quickly moving from C3N4 to r-Ti3C2 QDs via the close contact interface, photogenerated electrons from C3N4 may next be reduced to NH3 by trapping and accumulating photoexcited electrons from g-C3N4.
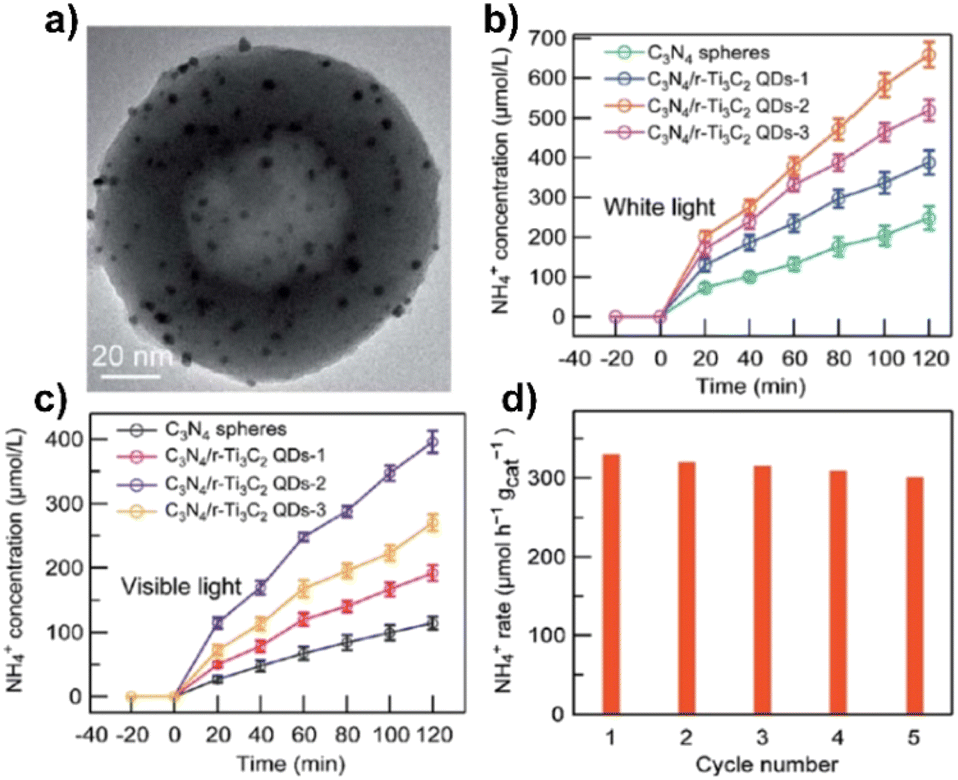 | ||
| Fig. 14 (a) TEM image of C3N4/r-Ti3C2 QDs-2; (b and c) N2 photofixation over the different as-obtained photocatalysts. Time courses of the ammonia concentrations measured under white light illumination (light intensity: 300 mW cm−2 and λ = 300–780 nm) and visible light illumination (light intensity: 300 mW cm−2 and λ = 420–780 nm); (d) stability tests of C3N4/r-Ti3C2 QDs-2 for N2 photofixation under white light. Reproduced with permission from ref. 162, Copyright 2022 The Royal Society of Chemistry. | ||
4. Metal oxides
4.1. Synthesis and structure evolution
The most facile strategy to tailor the physicochemical properties of MOs is by varying their composition. In binary oxides such as TiO2, ZnO, Fe2O3, etc., a small quantity of an additional metallic component can be introduced into the lattice, while maintaining the overall crystal structure.165–167 This approach is more straightforward in mixed oxides such as perovskites, brownmillerites, etc., since such materials include more than one metal in their pristine form.However, more complex compositions with fascinating properties can be made by doping with additional cationic components.168–171 In all these cases, excellent control of desired properties, including photocatalysis, can be achieved. On the other hand, photocatalytic performance also significantly relies on the nanoscale structural features and morphology of the materials involved. Since fabricating nanostructures depends on synthesis methodology, the choice of the synthesis method is important. The conventional approach for making MOs is the solid-state method where thermal energy drives the formation of the desired phase with a high degree of crystallinity. Here, agglomerated large grains are often produced with lower values of specific surface area. Mechanochemical approaches such as ball milling replaced thermal energy with mechanical energy, with limited extent of applicability and poor control over morphology and nanostructure. It is to be noted that mixed oxide compositions more or less require higher temperatures for the formation of the desired single-phase materials. Also, it is difficult to ensure a homogeneous mixing of the precursors in solid state and mechanochemical methods.168–171 Therefore, several soft chemical approaches were developed, incorporating a variety of precursors, templates and reaction media by following a number of complexation, precipitation, hydrothermal and templating routes.172–175 Wet chemical approaches such as the polyol method, sol–gel method, etc., allowed the synthesis of semiconductor photocatalysts with a uniformly distributed particle size and morphology, at comparatively lower temperatures. These methods generally involve the presence of organic molecules such as citric acid, ethylene glycol, etc. The interactions between these organic components with their metallic counterparts along with the mechanism of their cross linking and/or decomposition can exert better control on particle size and morphology.172
One step synthesis of binary and ternary oxides can be achieved by carrying out the reaction in a sealed reaction vessel above the boiling point of the solvent. This method is designated as the hydrothermal method when water is used as the solvent or the solvothermal method when an organic solvent is used instead.173,176 A homogeneous solution of the precursor in an appropriate solvent is subjected to thermal treatment at a temperature higher than the boiling point of the solvent. This promotes an increase in pressure inside the reaction chamber. The combined effect of enhanced temperature and autogenous pressure will drive the formation of the desired product. Excellent control of morphology and nanostructure formation is observed in this method. Simple synthesis parameters such as reactant concentration, pH and temperature were found to significantly influence the formation of the desired phase and morphology in most of the cases. Additional control of microstructure and porosity can be achieved by introducing appropriate amounts of organic templates during synthesis. Even though an additional calcination step is not necessary in most of the cases, high temperature treatments are always required to remove the organic structure directing agents and for the structure stabilization of the materials for applications involving enhanced temperatures.177 To date, a variety of binary and ternary oxide compositions with varied nanostructures and morphologies have been synthesized by following the hydrothermal method.178–180
Followed by the successful synthesis of MCM-41 silica with ordered mesopore channels, templating methods are widely used to produce MOs with controlled porosity and textural parameters.181 The initial focus was on the use of surfactants as soft templates to direct the synthesis of porous oxides.182 Apart from ordered mesoporous silica and carbon materials, limited success was obtained for the soft template method for oxides.183–185 This was primarily due to the non-availability of precursors that undergo hydrolysis in a controlled manner with organic templates. Also, organic template removal is generally carried out at high temperatures. Since the crystallization temperature for most of the MO phases was higher than the decomposition temperature of the template, the quality of the resultant materials was compromised. On the other hand, the nanocasting method was also being developed in parallel, for which an inert porous oxide was used as the hard template.182–187 This method was initially used for the synthesis of ordered mesoporous carbon and was then extended to oxides, utilizing ordered mesoporous silica or carbon as hard templates. A wide variety of binary oxides with extremely high values of surface area and well-ordered pore structure were obtained by this method. Nanocasting was found to be highly versatile and was then used to synthesize complex ternary oxides and non-oxide compositions such as sulfides.188,189 Unfortunately, this multi-step method was time consuming, costly and non-scalable.
Uniform integration of oxide cocatalysts with semiconductors is necessary to develop highly efficient photocatalysts. Several studies reported single step calcination treatments under oxidizing or inert atmospheres to combine oxide cocatalysts with photocatalysts including photocatalysts such as TiO2, g-C3N4, etc.190–192 In addition, other approaches including simple impregnation of the cocatalyst species, HTT, photodeposition, etc., are also followed to develop cocatalyst-photocatalyst combinations.193–195
4.2. Photocatalytic applications for water splitting
MOs are considered as low cost and easily available cocatalysts for photocatalytic applications including H2 evolution by splitting water. Fundamentally, the photogenerated electrons from the semiconductor photocatalyst move towards the MO, acting as a cocatalyst, through the interface, resulting in electron–hole separation. Table 4 summarizes several examples of such co-catalyst/photocatalyst systems used in the most recent years for hydrogen production through water splitting. The synthetic approaches to combine different semiconductor photocatalysts and several reaction parameters are also presented.| Cocatalyst/photocatalyst | Synthetic method | Hole scavenger | Light region | Optimum H2 production rate [μmol h−1 g−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|
| a ∼ Data taken from a graphical representation of the corresponding reference; NP: value not provided; TEA = triethanolamine. | ||||||
| NiOx/CdS | In situ photodeposition | Methanol (30%) | Visible | 445.6 | NP | 196 |
| NiOx/CdS | In situ photodeposition | Methanol (30%) | Visible | 590.8 | 8.6 (400 nm) | 193 |
| NiO/Zn0.8Cd0.2S | In situ photodeposition | None | Visible | 783.11 | 5.86 (350 nm) | 197 |
| NiO/TiO2 nanofibers | Electrospinning and calcination in air (500 °C for 3 h) | Methanol (25%) | Full spectrum | 377 | 1.7 (365 nm) | 200 |
| Amorphous NiO/g-C3N4 | Calcination in air (100–500 °C for 3 h) | TEA (10%) | Visible | 68.8 | ∼0.12 (370 nm) | 190 |
| NiO/g-C3N4 | Impregnation, HTT (125 °C for 10 h) and calcination in air (200–400 °C for 1 h) | None | Visible | 1.41 | NP | 198 |
| NiO/TiO2 | Impregnation and calcination in static air (450 °C for 3 h) | Methanol (5%) | Simulated solar light | ∼130 | NP | 199 |
| NiO/TiO2 | Impregnation and calcination in static air (450 °C for 3 h) | Methanol (5%) | UV | ∼70 | NP | 199 |
| NiO/red phosphorous | Calcination in air (300 °C for 1 h) | Methanol (10%) | Visible | 57.27 | 0.37 (420 nm) | 201 |
| Amorphous MoOx/CdS nanorods | Sonication | Lactic acid (10%) | Simulated solar light | 19300 |
21.3 (420 nm) | 202 |
| Cu2O/g-C3N4 | Calcination in air (520 °C for 4 h) | TEA (10%) | Visible | 241.3 | NP | 203 |
| CuO/TiO2 composite nanofibers | Electrospinning and calcination in air (500 °C for 2 h) | Methanol (10%) | Simulated solar light | 1146.9 | NP | 204 |
| CuO nanodots on TiO2 nanocrystals | Precipitation using ammonium hydroxide followed by calcination in air (200 °C for 1 h) | Methanol (10%) | UV-visible | ∼16000 |
NP | 191 |
| CuO@NiO core@shell nanoparticles | Chemical reduction (using N2H4.H2O) followed by calcination under an inert atmosphere (400 °C for 2 h) | Glycerol (5%) | Solar light | 26100 |
NP | 192 |
| CuO quantum dots/TiO2 nanosheets | Impregnation followed by calcination in air (300 °C for 2 h) | Methanol (20%) | Full spectrum | 18390 |
26.93 (313 nm) | 205 |
| CoO/g-C3N4 | Calcination under Ar (400 °C for 4 h) | TEA (10%) | Visible | 651.3 | NP | 206 |
| CoO/C3N4 nanotubes | Calcination under vacuum (400 °C for 4 h) | TEA (10%) | Visible | 788.6 | 4.9 (420 nm) | 207 |
| Co3O4/C3N4 nanotubes | Calcination in static air (400 °C for 4 h) | TEA (10%) | Visible | 725.7 | 4.07 (420 nm) | 207 |
Thus, Chen et al.196 photodeposited Ni2O3 on the surface of hydrothermally synthesized CdS, and H2 evolution was monitored under visible light irradiation. The presence of Ni2O3 resulted in the enhancement of the hydrogen evolution rate up to 41 times that of pure CdS and up to 1.9 times that of Pt loaded CdS. The authors attributed this improvement to the ability of Ni2O3 to act as electron traps, thereby promoting charge separation. Stability tests were performed for five cycles and the activity was found to decrease to 86.8% of the initial activity. The authors attributed this to the catalyst loss during filtration and washing.196 To study the effect of the chemical environment of photodeposition, nickel oxides were loaded onto CdS under neutral as well as alkaline conditions. Interestingly, the materials synthesized under alkaline conditions resulted in a 117 times enhancement of the hydrogen production rate compared to that of pure CdS. It was proposed that the presence of OH− ions contributed to charge separation by facilitating hole transfer towards the sacrificial agent, methanol.193
Lin et al.30 combined 1D Cd1−xZnxS nanocrystals with oxygen enriched MoS2 resulting in Cd1−xZnxS@O–MoS2 nanohybrids. NiOx species were deposited by following an in situ photodeposition method. The optimized composition of these nanohybrids exhibited 25 times higher hydrogen evolution rates compared to Pt containing CdZnS. Under visible light irradiation using Na2S/Na2SO3 as a sacrificial agent, an AQY of 64.1% was obtained for these materials. Repeated reactions during five cycles indicated minor activity loss. Also, the structure and composition of the recovered material remained unchanged after the photocatalytic reaction.30
Liu et al.190 synthesized a series of amorphous NiO modified g-C3N4 under different temperature treatments. The heterojunctions formed at the interface between amorphous NiO and g-C3N4 created an inner electric field allowing the transfer of photogenerated electrons from C3N4 to NiO. Under visible light irradiation, NiO/g-C3N4 exhibited 430 times higher hydrogen evolution rates compared to pristine g-C3N4 and was consistent during five cycles. Also, the structure and chemical state did not change after the reaction. The authors concluded that the presence of amorphous NiO enhanced the visible light response, promoted the separation and transfer of charge carriers and provided active sites for hydrogen evolution.190
Ning et al.197 synthesized a series of Zn1−xCdxS and an ultrathin layer of NiO was assembled over it by photodeposition. Under visible light irradiation and in the absence of a sacrificial agent, these materials showed significantly higher photocatalytic activity with an AQY of 0.66% for H2 evolution, compared to Pt/ZnCdS, as shown in Fig. 15. The NiO layer suppressed the photocorrosion of ZnCdS and the strong electronic coupling between the NiO layer and Zn1−xCdxS enabled efficient charge separation and transport.197
 | ||
| Fig. 15 (a) Photocatalytic H2 evolution over Zn1−xCdxS. (b) Rate of H2 evolution over the x% NiO/Zn0.8Cd0.2S composite samples. (c) A comparison of the H2 evolution rate over 10% NiO/Zn0.8Cd0.2S, 2% Pt/Zn0.8Cd0.2S, and NiO + Zn0.8Cd0.2S (simple mechanical mixture of NiO and Zn0.8Cd0.2S). (d) Cycling runs for photocatalytic H2 evolution in pure water with the Zn0.8Cd0.2S and 10 wt% NiO/Zn0.8Cd0.2S composite samples under visible light irradiation. (e) Cd2+ leakage profiles under visible light irradiation. Reproduced with permission from ref. 197, Copyright 2019 European Chemical Societies Publishing. | ||
Fu et al.198 synthesized a series of NiO-g-C3N4 by following a successive hydrothermal and calcination procedure. Overall water splitting was achieved for these materials among which the best performance was observed for the materials calcined at 300 °C. In this case, a H2 evolution rate of 1.41 μmol g−1 h−1 and an O2 evolution rate of 0.71 μmol g−1 h−1 were obtained. Experiments carried out for seven cycles along with characterization performed using the recovered catalyst confirmed the stability of these materials. Photocatalytic efficiency decreased for the materials calcined at higher temperatures. Here also, the presence of NiO aided in visible light absorption, separated the charge carriers and provided active sites.198 These results are in agreement with the report by Liu et al.190
Banic et al.199 synthesized a series of NiO/TiO2 materials with varying mass ratios, by wet impregnation. The photocatalytic water splitting reaction was performed in the presence of methanol under simulated solar irradiation and artificial UV irradiation. The materials with 7 wt% NiO enhanced hydrogen production rates up to 7.6 times compared to that of pure TiO2 and 112.8 times compared to that of pure NiO. Under optimum conditions, a 1.8 times higher hydrogen production rate was achieved under simulated solar light compared to that under the influence of artificial UV radiation.199
Since the preparation method is very important, Li et al.200 synthesized mesoporous NiO/TiO2 nanofibers by the electrospinning method. The presence of NiO inhibited the crystal growth of TiO2 resulting in an increase in surface area, pore volume and pore size. Under simulated solar irradiation, an optimum NiO content (0.25 wt%) exhibited a 7 times higher H2 evolution rate with an AQY of 1.7%, compared to pure TiO2.200 Chen et al.201 synthesized a NiO/RP (red phosphorous) composite by HTT followed by high temperature calcination. In this study, the 3 wt% NiO/RP composite exhibited the highest photocatalytic activity for hydrogen production which is 68.56 times higher than that of pure RP under visible light irradiation. The rates did not change significantly for 10 cycles and the physicochemical properties of the recovered materials were in agreement with those of the fresh ones. In the presence of light, electrons are excited towards the conduction band of RP leaving behind holes in the valence band, which simultaneously combines with the electrons from NiO. The holes in NiO are consumed by methanol and photogenerated electrons accumulated on the conduction band of RP which participate in H2 generation. This material also exhibited H2 evolution activity in the near IR region.201
Yan et al.202 prepared nanocomposites consisting of CdS and amorphous MoOx and studied photocatalytic hydrogen production under visible light irradiation. A 16 times enhancement in hydrogen evolution rates was observed compared to that of pristine CdS. The amounts of hydrogen evolved remained stable for five cycles. The photogenerated electrons from CdS resulted in an in situ reduction of Mo6+ in amorphous MoOx creating electronic structure reconstruction in the nanocomposite.202
Chen et al.203 synthesized g-C3N4 modified with different amounts of Cu2O nanoparticles by a simple calcination method. In the presence of Cu2O, up to a 70% increase in visible light induced photocatalytic H2 evolution was observed. Cu2O nanoparticles acted as visible light sensitizers, induced sufficient charge separation and exhibited a negative shading effect so that their content above an optimum level reduced the number of active sites. The rate of hydrogen evolution remained stable for five cycles and the chemical state of Cu in the recovered catalyst was the same as that in the fresh one.203
Zhu et al.204 reported the use of wheat grain like textured TiO2/CuO composite nanofibers synthesized by electrospinning for photocatalytic water splitting. CuO incorporation was found to enhance H2 generation by about 16.8 times in comparison with that of pure TiO2. Significant loss of photocatalytic performance was not observed for four cycles.204
Moon et al.191 reported that size tunable Cu2O nanodots decorated on the surface of TiO2 nanocrystals got reduced to Cu under UV irradiation. The resultant metal–semiconductor Schottky junction enhanced the hydrogen production rate significantly in comparison with P25 and reached approximately 50% of the one achieved with Pt/TiO2 counterparts, as shown in Fig. 16.191
 | ||
| Fig. 16 (A) Hydrogen production vs. illumination time in the presence of CuO-decorated TiO2 catalysts. (B) H2 production rate vs. loading amount of CuO. The inset of (B) is a bar graph showing the activity of CuO-decorated TiO2 in comparison to that of a Pt-decorated system. Reproduced with permission from ref. 191, Copyright 2014 The Royal Society of Chemistry. | ||
Sadanandam et al.205 hydrothermally synthesized TiO2 nanosheets with [001] facets exposed and impregnated with CuO quantum dots. It was observed that during the reaction CuO changed to Cu2O and then to Cu. The energy band alignment of the double heterojunction TiO2/Cu2O/Cu provided effective charge separation, increasing the H2 production efficiency. Under visible light irradiation, the hydrogen production rate of the optimized material was approximately 10 times higher than that of pure TiO2, with an AQY of 26.93%. Cycling tests were carried out for the optimum catalyst and a H2 production retention ratio of 96% is obtained after four cycles. The system was then purged with O2 for 2 h to regenerate the used catalyst. After this, the retention ratio increased to 98%, indicating good cycling stability and photo corrosion resistance.205 The synergy between CuO and NiO, the most studied oxides as cocatalysts, was verified by Ravi et al.192 who synthesized CuO@NiO core@shell nanoparticles deposited on anatase TiO2 and studied them for photocatalytic H2 evolution under direct solar light irradiation. The rate of hydrogen evolution was found to be influenced by the thickness of the NiO shell and cocatalyst loading. 6-fold higher values were obtained in comparison with those of pure TiO2. The performance of this material was examined for few days. The setup was kept under dark conditions after 4 h solar light illumination and was purged with N2 gas before the experiments on the next day. The rate of H2 evolution remained similar for the first 2 days while a minor decrease was noticed on the third day. The authors attributed this to the decrease in the concentration of the sacrificial agent.192
Another MO that was studied as a cocatalyst for water splitting is CoO. Mao et al.206 studied the photocatalytic behaviour of g-C3N4/CoO nanocomposites with varying amounts of CoO, under visible light irradiation This material with 0.5 wt% CoO content exhibited 3 times superior H2 evolution rates compared to pure g-C3N4, owing to enhanced light absorption and separation of charge carriers. Stable performance was observed during three cycling experiments. Also, the recycled material exhibited no sign of aggregation.206
Zhu et al.207 prepared CoOx confined in g-C3N4. The heterojunction formed primarily depended on the conditions of temperature treatment. The type II heterojunction formed between CoO nanoparticles and C3N4 nanotubes, under vacuum, exhibited the highest H2 yield, due to the rapid electron transfer between CoO and C3N4. CoO/C3N4 NTs were found to exhibit 6% activity loss while for Co3O4/C3N4 NTs it was almost 18.0%. A slight decrease in the intensity of the main peaks indexed to Co3O4 NPs was detected by XRD. This indicates the loss of a portion of anchored Co3O4 nanoparticles on C3N4 NTs during the recycling process. Also, some extent of agglomeration was observed for this material.207
4.3. Photocatalytic applications for CO2 reduction
For photocatalytic conversion of CO2, enhancing surface adsorption of CO2 and suppressing charge carrier recombination represent one of the major focuses. As in the case of water splitting, transition MOs based on Cu, Ni, Co and Fe were most widely investigated as cocatalysts, along with CaO, MgO and In2O3 (see Table 5).| Cocatalyst/photocatalyst | Synthetic method | Reactants | Light region | Products [μmol g−1 h−1] | AQY [%] | Ref. |
|---|---|---|---|---|---|---|
| a ∼ Data taken from a graphical representation of the corresponding reference; NP: value not provided. | ||||||
| Cu2O/TiO2 nanosheets | Photodeposition and calcination in air (200 °C, 2 h) | CO2 and H2O | Simulated solar light | CH4 (8.68) | NP | 209 |
| Cu2O/SiC | Precipitation using NaOH followed by chemical reduction using N2H4·H2O | NaOH, Na2SO3 and CO2 | Visible | CH3OH (38) | NP | 210 |
| Octahedral Cu2O/TiO2 nanotube arrays | Electrochemical deposition | CO2 and H2O | Visible | CH4 (∼400 ppm for 4 h) | NP | 211 |
| CuO–TiO2 hollow microspheres | HTT (180 °C for 20 h) | CO2 and H2O | UV | CO (14.5) | 1.285 | 194 |
| CH4 (2.1) | ||||||
| H2 (2.8) | 0.747 | |||||
| FeOx-MWCNT@TiO2 | Impregnation and calcination under N2 (400 °C for 2 h) | CO2 and H2O | Visible | CH4 (0.85 μmol g−1 for 8 h) | NP | 195 |
| CuO-MWCNT@TiO2 | Impregnation and calcination under N2 (400 °C for 2 h) | CO2 and H2O | Visible | CH4 (0.93 μmol g−1 for 8 h) | NP | 195 |
| NiO-MWCNT@TiO2 | Impregnation and calcination under N2 (400 °C for 2 h) | CO2 and H2O | Visible | CH4 (0.44 μmol g−1 for 8 h) | NP | 195 |
| CoO-MWCNT@TiO2 | Impregnation and calcination under N2 (400 °C for 2 h) | CO2 and H2O | Visible | CH4 (0.38 μmol g−1 for 8 h) | NP | 195 |
| ZnO-MWCNT@TiO2 | Impregnation and calcination under N2 (400 °C for 2 h) | CO2 and H2O | Visible | CH4 (0.27 μmol g−1 for 8 h) | NP | 195 |
| Cu–TiO2 in a cordierite honeycomb monolith | Dip coating and calcination in air (500 °C for 5 h) | CO2 and H2 | Simulated solar light | CO (763) | 0.0826 | 212 |
| CH4 (4.20) | NP | |||||
| CuxO/CoPi-SrTiO3 nanorod thin films | Impregnation or photoelectrochemical deposition | CO2 and H2O | UV | CO (0.4 μmol cm−2) | NP | 213 |
| Cu2O on TiO2-pillared K2Ti4O9 | Calcination in air (800 °C for 20 h), ion-exchange (using HCl), exfoliation using (TBAOH), impregnation and reflux (160 °C for 2 h) | CO2 and H2O | Simulated solar light | CH3OH (2.93 μmol g−1 for 5 h) | NP | 214 |
| CuO/Na(1−x)LaxTaO(3+x) | Impregnation and calcination in air (300 °C for 2 h) | NaOH, CO2 and H2O | UV-visible | CH3OH (60.5) | 0.0105 | 215 |
| C2H5OH (15.9) | ||||||
| C2H4O (0.9) | ||||||
| C3H6 (0.15) | ||||||
| C2H6 (0.04) | ||||||
| C2H4 (0.03) | ||||||
| CH4 (0.01) | ||||||
| H2 (0.2) | ||||||
| NiO/Na(1−x)LaxTaO(3+x) | Impregnation and calcination in air (270 °C for 2 h) | NaOH, CO2 and H2O | UV-visible | CH3OH (59.6) | 0.0112 | 215 |
| C2H5OH (18.8) | ||||||
| C2H4O (2.15) | ||||||
| C3H6 (0.4) | ||||||
| C2H6 (0.01) | ||||||
| C2H4 (0.03) | ||||||
| CH4 (0.02) | ||||||
| H2 (0.2) | ||||||
| Co3O4/K2Ti6O13 | Calcination in air (800 °C for 12 h) | CO2 and H2O | Visible | CH3OH (36.4 μmol g−1 for 3 h) | NP | 216 |
| HCHO (453.2 μmol g−1 for 3 h) | ||||||
| CH4 (1.3 μmol g−1 for 3 h) | ||||||
| H2 (8 μmol g−1 for 3 h) | ||||||
| NiO/K2Ti6O13 | Calcination in air (800 °C for 12 h) | CO2 and H2O | Visible | CH3OH (31.2 μmol g−1 for 3 h) HCHO (99.7 μmol g−1 for 3 h) | NP | 216 |
| CH4 (1.1 μmol g−1 for 3 h) | ||||||
| H2 (201 μmol g−1 for 3 h) | ||||||
| CuO/K2Ti6O13 | Calcination in air (800 °C for 12 h) | CO2 and H2O | Visible | CH3OH (159.7 μmol g−1 for 3 h) HCHO (41.4 μmol g−1 for 3 h) | NP | 216 |
| CH4 (0.8 μmol g−1 for 3 h) | ||||||
| H2 (282 μmol g−1 for 3 h) | ||||||
| Cu–Bi2O3/TiO2 | Calcination under H2 (550 °C for 5 h) | CO2 and H2O | UV-visible | CO (5.74) | NP | 217 |
| CH4 (11.90) | ||||||
| NiO/InTaO4 | Sol–gel method and calcination in air (1100 °C for 12 h) | NaOH, CO2 and H2O | Sunlight | CH3OH (11.3) | 0.063 | 218 |
| NiO–InNbO4 | Incipient wetness impregnation and calcination in air (350 °C for 1 h) | CO2 and H2O | Simulated solar light | CH3OH (1.577) | NP | 219 |
| Co3O4–InNbO4 | Incipient wetness impregnation and calcination in air (350 °C for 1 h) | CO2 and H2O | Simulated solar light | CH3OH (1.503) | NP | 219 |
| NiO–KTaO3 | HTT (180 °C for 12 h) and calcination in air (600 °C for 1 h) | CO2 and isopropanol | UV | CH3OH (1523) | NP | 220 |
| NiO–InVO4 | Impregnation and calcination in air (350 °C for 2 h) | CO2, H2O and KHCO3 | Visible | CH3OH (1.526) | NP | 221 |
| Ni@NiO/InTaO4–N | Impregnation, chemical reduction (using NaBH4) and calcination in air (200 °C for 1 h) | CO2 and H2O | Visible | CH3OH (∼300 μmol g−1) | NP | 222 |
| Macro/microporous MgO modified TiO2 | Biotemplating and calcination in air (500 °C for 3 h) | CO2 and H2O | Simulated solar light | CH4 (1.87) | NP | 223 |
| TiO2/MgO | HTT (200 °C for 12 h) | CO2, H2O and NaOH | UV | CO (70.7 μmol g−1 L−1 for 24 h) | NP | 224 |
| CH4 (34.5 μmol g−1 L−1 for 24 h) | ||||||
| HCOOH (19.5 μmol g−1 L−1 for 24 h) | ||||||
| CH3COOH (24.9 μmol g−1 L−1 for 24 h) | ||||||
| Ca/TixSiMCM-41 | HTT (100 °C for 12 h) and calcination (500 °C for 2 h) | CO2 and H2O | UV | CH4 (82 μmol g−1 L−1 for 8 h) | NP | 225 |
Uzunova et al.208 used DFT to deduce the mechanism of CO2 photoconversion to methanol, using H2O as the hydrogen source. The presence of MOs results in bending of a CO2 molecule prior to hydrogenation and the bridging hydroxyl groups which resulted from water dissociation provided H2. Methanol is formed by following a step-wise mechanism via the formation of carboxyl groups, formic acid and formaldehyde intermediates. A comparatively stronger interaction between CO2 and Cu2O compared to that between metallic catalysts was reported.208
Zhu et al.209 deposited Cu2O clusters on TiO2 nanosheets by photodeposition. The authors noticed the formation of surface oxygen vacancies on TiO2 which was attributed to the presence of Cu2O. Under simulated solar light irradiation and in the presence of water as the hydrogen source, methane was the only product formed. The presence of 1 wt% Cu2O resulted in a 30 times enhancement of the photoactivity of pure TiO2. Cu2O clusters increased the surface alkalinity and enhanced CO2 adsorption ability. High concentration of surface oxygen vacancies has also played the role of active sites for CO2 conversion.209
Cu2O loaded SiC photocatalysts were found to be selective towards methanol under visible light irradiation, using water as the hydrogen source. The CH3OH yields were comparatively higher than that by the individual components.210 In agreement with the report by Zhu et al.,209 Cu2O nanoparticles electrochemically deposited on TiO2 nanotubes selectively produced CH4. Under visible light irradiation in the presence of water vapour, an optimum amount of octahedral Cu2O nanoparticles exhibited high efficiency and selectivity towards methane.
The nanotube structure of TiO2 provided active sites for the adsorption of reactants and facilitated the transport of photogenerated charge carriers. Synergistically, Cu2O participated in light absorption and prevented oxidation of the reaction products.211
Fang et al.194 synthesized CuO incorporated TiO2 hollow microspheres with a hollow core and mesoporous shell structure, by a template free one pot HTT, followed by calcination. Photo-reduction of CO2 was carried out over this material using H2O as the hydrogen source under UV irradiation, producing CO and CH4, as shown in Fig. 17. For an optimum CuO loading (3 wt%), 5.8- and 2.7-times higher CO and CH4 yields, respectively, were obtained compared with those of pristine TiO2 hollow microspheres, and 7.3- and 9.0-times higher yields were obtained compared to those of commercial TiO2, respectively. The used catalysts were recovered from the reactor and stored under ambient conditions for 24 h. When this material was tested again, a minimum decrease in product yield was observed indicating regeneration of the catalyst due to desorption of the gas products from its surface during storage.194
 | ||
| Fig. 17 (a) UV-vis DRS for various TiO2 based nano-materials. The CuO content in the CuO–TiO2 catalyst is 3 wt%. (b) Yields of the gas products (CO and CH4) produced during 24 h of photo-driven CO2 reduction on the TiO2 hollow microsphere catalysts with various Cu contents. Reproduced with permission from ref. 194, Copyright 2015 American Chemical Society. | ||
Gui et al.195 prepared MWCNT@TiO2 core@shell nanocomposites, impregnated with a series of binary oxides (FeOx, CuOx, NiO, CoOx and ZnO). The efficiency of these materials for CO2 photoreduction was studied under visible light irradiation in the presence of water vapour. Methane was selectively produced and CuO modified MWCNT@TiO2 exhibited superior performance among the series. The better performance was attributed to the MO band position with respect to that of TiO2.195
Tahir et al.212 used cordierite honeycomb monoliths loaded with N/TiO2 and Cu/TiO2 nanocatalysts using a sol–gel dip coating method for dynamic photocatalytic CO2 reduction with H2 selectively to CO under UV-light irradiation. Both Cu and N doping resulted in a shift of band gap energy towards the visible region and also in an efficient transport and trapping of electrons, hindering carrier recombination. The presence of Cu resulted in a 14 times higher CO production than N/TiO2 and 64 times higher CO production than that of pure TiO2. Here, the CO production decreased after 4 h of irradiation time, indicating deterioration of photoactivity.212
Shoji et al.213 loaded CuxO as a reduction cocatalyst and cobalt phosphate (CoPi) as an oxidation cocatalyst on SrTiO3 nanorod thin films. Photocatalytic experiments under UV irradiation were conducted by immersing the film in an aqueous solution bubbled with CO2. Selective evolution of CO was observed with the highest rate exhibited by the materials with dual cocatalysts. The CO generation rate was found to increase linearly in accordance with the irradiation time. Here, CuxO nanoclusters facilitated CO2 reduction whereas CoPi facilitated surface oxidation to promote oxygen generation. The dual cocatalyst incorporated materials exhibited a 2.5 times superior CO generation rate compared to pure SrTiO3.213
Junior et al.214 synthesized layered semiconductor K2Ti4O9 by the solid-state method and pillared it with TiO2. The resultant mesoporous composite was then loaded with Cu2O nanoparticles as the cocatalyst. CO2 photoreduction was carried out using water as the hydrogen source under simulated solar light. Improved performance was observed for the pillared material TiO2/Ti4O9 than the pristine layered potassium tetratitanate. Here, electron transfer between the guest TiO2 and the host Ti4O92− layers could have played a role, along with the presence of additional photoactive sites. Loading of Cu2O nanoparticles resulted in further improvement of photo activity due to enhanced visible light harvest promoting electron transfer with TiO2 and Ti4O92−. It is to be noted here that since the temperature in the system reached 70 °C during the reaction, the contribution of photothermal effects cannot be ignored.214
Jeyalakshmi et al.215 hydrothermally synthesized Na(1−x)LaxTaO(3+x) and Pt, Ag, Au, CuO, NiO and RuO2 were introduced as cocatalysts by following the wet impregnation method. CO2 photoconversion was carried out in an alkaline medium under UV-visible light. Methanol and ethanol were produced as the major products along with traces of methane, ethane, ethylene, etc. The amounts of methanol produced remained consistent for three cycles over NiO/Na(1−x)LaxTaO(3+x). High AQYs among the series were obtained for 0.2 wt% NiO (11.29%) and 1 wt% CuO (10.50%) as cocatalysts. These oxides reduced the band gap energy of NaTaO3 and at the same time the light absorption onset edge was found to be extended to the visible region.215
Garay-Rodriguez et al.216 used an ultrasound assisted sol–gel method for synthesizing MO (Co3O4, NiO, and CuO) incorporated K2Ti6O13. Interestingly while some extent of Co2+ cations were found to selectively substitute Ti4+ cations, Ni2+ and Cu2+ were incorporated in both Ti4+ and K+ sites. The metal-cation introduction between the tunnels favoured the growth of potassium-poor phases as impurities, especially in the CuO loaded K2Ti6O13 samples. Photoconversion of CO2 using H2O as the hydrogen source under UV-visible light, resulted in different products depending on the loaded MO. It was observed that while the presence of Co3O4 favoured the formation of formaldehyde, NiO favoured hydrogen evolution due to additional photo-reforming of the organic compounds and CuO enhanced the selectivity towards methanol and hydrogen. The product yields remained consistent for three consecutive reaction cycles for Co3O4, NiO, and CuO incorporated K2Ti6O13. Moreover, no structural changes were observed by comparing the XRD patterns of the fresh and used catalysts.216
Jeong et al.217 explored the use of Bi2O3 as a promoter for Cu cocatalysts anchored on the surface of TiO2 nanoparticles for the photocatalytic conversion of CO2 to CH4, under UV-visible light irradiation. The presence of Bi2O3 increased the product selectivity towards methane (see Fig. 18). In the absence of Bi2O3, the photogenerated electrons reduce the CO2 adsorbed on the cocatalyst surface to CO, which has a low binding strength and will get detached as the product. Bi2O3 provided additional binding sites for CO where it undergoes further transformation, producing CH4.217
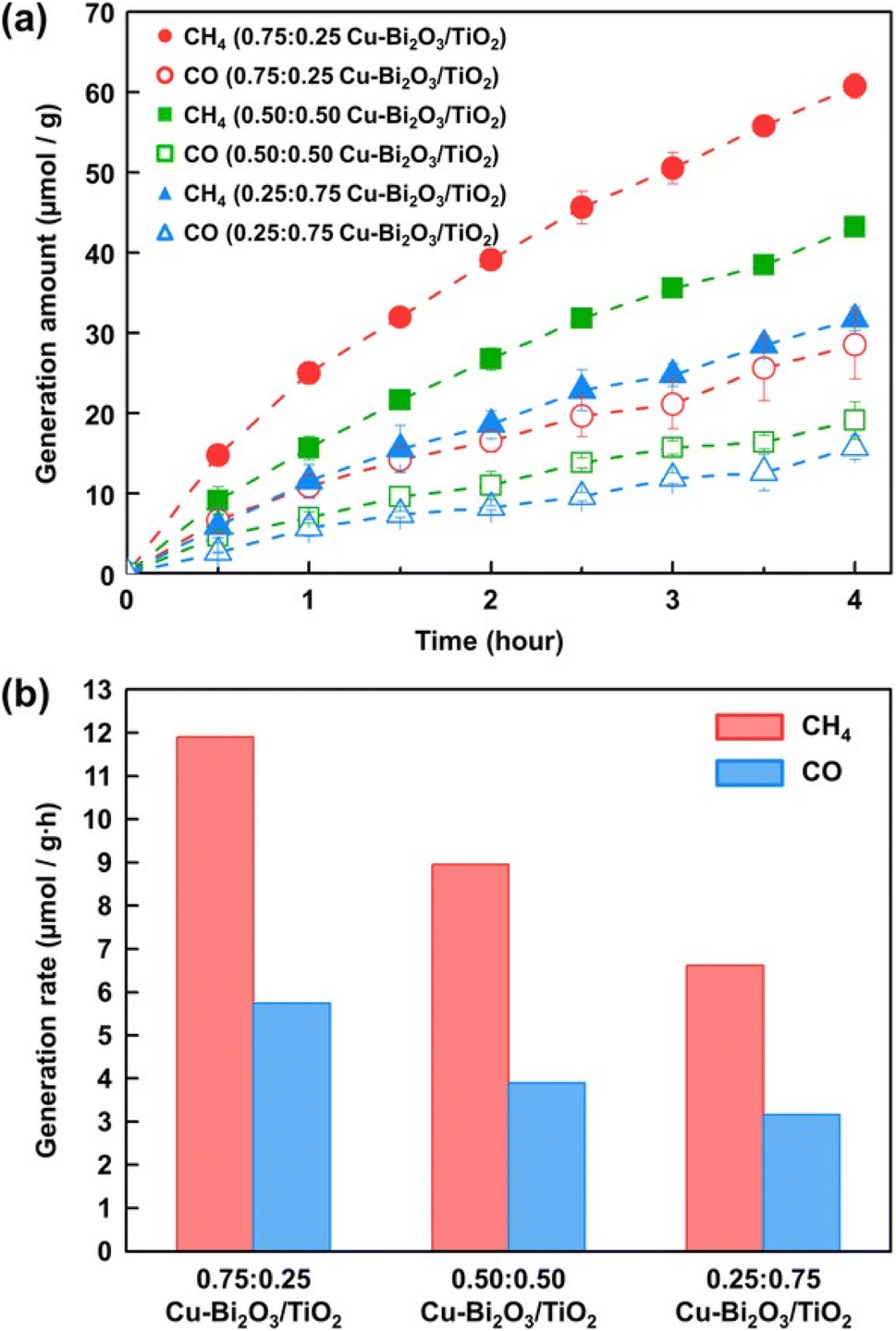 | ||
| Fig. 18 (a) Amount of CH4 and CO generated upon using 0.75:0.25, 0.50:0.50, and 0.25:0.75 Cu–Bi2O3/TiO2 photocatalysts. (b) Rates of CH4 and CO generation in the first 4 h of irradiation. Reproduced with permission from ref. 217, Copyright 2016 European Chemical Societies Publishing. | ||
Wang et al.218 used a NiO loaded InTaO4 photocatalyst to carry out both aqueous phase and vapor phase photoconversion of CO2 using visible light. The rate of methanol production was 11.1 μmol g−1 h−1 which was slightly increased up to 11.30 μmol g−1 h−1 by utilizing concentrated sunlight. The quantum efficiencies were estimated to be 0.0045% and 0.063% in aqueous-phase and gas phase reactors, respectively.218
Lee et al.219 synthesized InNbO4 from the corresponding binary oxides, and cocatalysts (NiO and Co3O4) were impregnated by following the incipient-wetness method. NiO–InNbO4 after a reduction–oxidation pre-treatment exhibited the highest activity for producing methanol under visible light which was attributed to the presence of core–shell type Ni0/NiO on the surface and the presence of a small amount of Nb2O5 as a promoter.219
Comparative studies were carried out between hydrothermally synthesized perovskite-type and pyrochlore-type powders of potassium tantalate (KTaO3). The pyrochlore form exhibited better photocatalytic activities for producing methanol under UV light irradiation which was further increased by adding NiO as a co-catalyst.220 Lee et al.221 synthesized InVO4 by the solid-state reaction method using In2O3 and V3O4 as the starting materials and NiO was added as the cocatalyst by incipient wetness impregnation. The photocatalytic reduction of CO2 with water was carried out under visible light illumination, producing methanol. Introduction of NiO resulted in lowering of the bandgap due to the formation of sub bands in the valence band by creating additional defect sites on the surface of InVO4, resulting in a higher methanol yield with an AQY of 2.8%.221
Tsai et al.222 synthesized N-doped InTaO4 photocatalysts and introduced Ni@NiO core@shell nanostructures as a cocatalyst for transforming CO2 into methanol. Under visible light irradiation, a 2-fold increment in methanol yield was observed for the N-doped samples which was further increased by the presence of Ni@NiO cocatalysts, compared to that of pristine InTaO4. Due to a narrow band gap, Ni@NiO/InTaO4–N absorbed more light, and the Ni@NiO core@shell structures promoted more efficient electron transport.222
Wang et al.223 synthesized porous MgO modified TiO2 using water convolvulus as the biotemplate. This material photoreduces CO2 in the presence of water vapour, selectively producing methane. 0.2 wt% MgO resulted in a 3.5 times increase in the CH4 evolution rate in comparison with pure TiO2. The authors attributed this to various factors including CO2 adsorption capacity, charge carrier transport, and enhanced number of active sites, which depend on the amount of added MgO.223
Torres et al.224 mixed MgO with TiO2 in different proportions and the resultant nanocomposites were studied in the photoconversion of CO2. Enhanced selectivity was observed for CO, while CH4, HCOOH and CH3COOH were also produced in lower amounts. Addition of 1 wt% of MgO enhanced CH4 production compared to that with pure TiO2, however, a further increase in the MgO content significantly deteriorated the performance. The authors attributed this effect to the insulating behaviour of MgO. It is also likely that higher amounts of MgO are blocking the active sites, thereby lowering the light absorption properties.224
Jo et al.225 used a series of MCM-41 mesoporous silica modified with Ca and Ti for the photoreduction of CO2 to CH4. The optimum material among the series exhibited extremely high yields of methane under solar light irradiation. Optimum Ca doping contributed towards photoreduction by providing additional sites for CO2 adsorption and by promoting charge carrier separation. It is to be noted that excess metal sites diminish the performance by providing charge recombination centers.225
4.4. Photocatalytic applications for N2 photofixation
In addition to the well-known photocatalytic applications previously mentioned, MOs are also utilized as a cocatalyst in photocatalytic systems for the photoreduction of N2 to produce NH3. Cocatalysts as MOs are used to broaden the range of absorbed light, as well as to facilitate the electron transport throughout the system and stop electron–hole recombination. Table 6 summarizes several examples of MO-based photocatalysts used in recent years for the NRR.| Cocatalyst/photocatalyst | Preparation methods | Reaction conditions | Light source | Sacrificial reagent | NH3 productiona | AQY [%] | Ref. |
|---|---|---|---|---|---|---|---|
| a The production rates were expressed as how they appear in the references. | |||||||
| Bi2MoO6/OV-BiOBr | HTT (120 °C for 8 h) | 30 mg of the photocatalyst in 60 mL of ultrapure water, RT | 300 W xenon lamp | None | 90.7 μmol g−1 h−1 | NP | 226 |
| 30 wt% SV-1T-MoS2/CdS | HTT (200 °C for 24 h) | 20 mg of photocatalyst in 100 mL 20 vol% methanol solution | Simulated solar light | Methanol | 8220.83 μmol L−1 g−1 h−1 | 4.424 | 227 |
| 18% Sb2O3/W18O49 | Solvothermal (180 °C for 20 h) | 5 mg photocatalyst in the aqueous solution (10 mL) containing 20 vol% methanol, RT | 300 W xenon lamp | Methanol | 35.3 μmol g−1 h−1 | 0.063 | 228 |
| g-C3N4/40 wt% Fe2O3 | Ternary deep eutectic solvents and calcination (550 °C for 4 h) | 10 mg of photocatalyst in 100 mL of distilled water, RT | 300 W xenon lamp | Methanol | 4380 μmol L−1 h−1 | NP | 229 |
For enhanced photocatalytic performance for N2 conversion to ammonia without any noble metal and sacrificial agents under ambient conditions, Xue et al.226 produced heterojunctions based on n-type Bi2MoO6 nanorods and oxygen-vacancy-rich p-type BiOBr nanosheets (Bi2MoO6/OV-BiOBr). The average ammonia generation rate of the Bi2MoO6/OV-BiOBr heterojunctions is 90.7 μmol g−1 h−1, which is about 30 times higher than that of pristine Bi2MoO6 nanorods (3.0 μmol g−1 h−1) and 3 times higher than that of OV-BiOBr nanosheets (31.2 μmol g−1 h−1). This study highlights the significantly increased photocatalytic N2 fixation activity of the Bi2MoO6/OV-BiOBr heterojunctions, without the use of any sacrificial agents.
Sun et al.227 reported sulfur vacancy (SV)-rich O-doped 1T-MoS2 nanosheets (denoted as SV-1T-MoS2), prepared by HTT, as cocatalysts to assist in the photocatalytic nitrogen fixation of CdS nanorods, as a hybrid material SV-1T-MoS2/CdS. Different quantities of the cocatalyst, ranging from 5 to 100 mg, representing 5 to 100 wt%, were used to produce the SV-1T-MoS2/CdS composites. A remarkable photocatalytic N2 reduction rate of 8220 μmol L−1 h−1 g−1 is demonstrated by the optimized 30 wt% SV-1T-MoS2/CdS composites, under simulated solar light irradiation (AM 1.5 G), with methanol as a scavenger, obtaining an AQE of 4.424%. This rate is 2.4 and 4.6 times greater than that of CdSPt (0.1 wt%) and CdS nanorods, respectively, suggesting that SV-1T-MoS2 may exceed Pt in the CdS's photocatalytic nitrogen fixation performance. However, the rate of NH4+ will decrease as the amount of SV-1T-MoS2 in the composites increased from 30 to 100 wt%. This is probably because excessive MoS2 prevents the light from being used. Meanwhile, under visible light (780 nm > λ > 420 nm), the photocatalytic nitrogen fixation rate was 3168 μmol L−1 h−1 g−1.
Three cycles were performed to evaluate the stability over 30 wt% SV-1T-MoS2/CdS, and after 9 hours of testing, 80% of the incipient activity was still present, demonstrating the materials' good cycle stability.
Sb2O3/W18O49, a composite made of 2D Sb2O3 nanosheets and 1D W18O49 nanowires, was created by Hui et al.228via the solvothermal process, by varying the Sb2O3 weight, ranging from 1.8 to 70%. The highest NH3 production was obtained for 18% Sb2O3/W18O49 with a production rate of 35.3 μmol g−1 h−1, with methanol as a scavenger under visible light illumination, which is 3.4 and 5.5 times higher than that of W18O49 and Sb2O3, respectively, with an AQE of 0.063%. However, as the Sb2O3 concentration was increased above 18 wt%, the rate of NH3 synthesis decreased. This is probably due to Sb2O3 aggregation and the low Sb2O3/W18O49 interface active regions, which inhibited N2 adsorption and activation. The heterostructures demonstrated good repeatability with approximately consistent NH3 production rates. Additionally, cycling studies showed that even after five consecutive cycles, the catalytic activity was mainly the same, with a certain decrease of activity due to aggregation.
Mou et al.229 created g-C3N4/MO composites utilizing a simple one-step calcination procedure and a ternary deep eutectic solvent (DES) technique, in which g-C3N4/Fe2O3 was of interest due to its photocatalytic response. Melamine was added in various amounts to control the loading of α-Fe2O3, ranging from 0.1582 g to 2.291 g for the production of 50%-DES, 40%-DES, 35%-DES, and 30%-DES, respectively. At 40 wt%, the best activity was attained (g-C3N4/40 wt% Fe2O3) with an average rate of 4380 μmol L−1 h−1 for producing NH3, with methanol acting as a scavenger. The effective interfacial contact between 2D g-C3N4 nanosheets and α-Fe2O3 nanoparticles, which improves the transfer and separation efficiency of the charge carriers, may be the reason for the high photocatalytic activity.
5. Comparison between MXenes and metal oxides
MXenes and MOs have gained a lot of interest and have been investigated for photocatalytic applications due to their unique properties, such as electronic band structures, catalytic edge effects, planar morphology, etc.230 This section is dedicated to a comparison as succinct as possible between these two categories of materials when they are used as cocatalysts in photocatalytic reactions. The most important criteria taken into account was the employed synthesis methods, their role in promoting photogenerated charge separation, environmental issues, the cost/benefit ratio, the number of combinations in which they can be used and their photocatalytic stability.Thus, in terms of synthesis conditions, both categories of materials can be obtained by following relatively simple approaches. While chemical liquid-phase etching of the MAX phase results in the corresponding MXenes, MOs in their amorphous, polycrystalline or single crystal form can be synthesized from their precursors (nitrates, chlorides, etc.) under thermal or hydrothermal conditions.231 In the previous sections, a summary of individual synthesis methods of the two categories of materials is provided. While making a comparison, we need to take into account the fact that MXenes are still in their infancy, whereas MOs have a long history of evolution. A vast majority of photocatalytic studies using MXenes were carried out using Ti3C2Tx while for oxides several material options were available. For a fair and realistic assessment, the strategies frequently used to integrate these cocatalysts (MXenes and MOs) with the photocatalysts are to be considered. Thus, the choice of a method relies not only on maintaining the structural, compositional and morphological integrity of the materials employed, but also on establishing a homogeneous and intimate contact between them. In addition, the amount of cocatalyst used is important since an excess can be detrimental for the overall performance.
Broadly, the methods used to develop cocatalyst-photocatalyst composites can be classified into two categories: (i) multi-step synthesis – the components can be synthesized separately and can be combined in desired proportions in a post-synthetic manner; and (ii) “one-pot” approaches – in which the cocatalyst and the photocatalyst are formed simultaneously under the same conditions. As mentioned in the previous sections, post-synthetic approaches are always required for MXenes, since the etching conditions (usually HF) are not compatible with the semiconductor photocatalysts. Also, in cases where the desired phase formation and thermal stabilization of the composites under oxidizing conditions are required, total destruction of MXenes can occur, forming the corresponding MOs. It is to be noted here that a few studies reported controlled oxidation of MXenes (as a precursor) producing cocatalyst-photocatalyst composites. However, it is difficult to control the cocatalyst-photocatalyst ratio in this way and to date, it is not sure whether this approach can be generalized. In our understanding, HTT is the most optimum method that can be adapted to both MXenes and MOs to produce photocatalyst composites. An advantage here for MXenes is their rich surface chemistry that enables them to bind appropriately with the semiconductors. Physical mixing approaches such as grinding are also compatible with both these families of materials; however, it is difficult to establish an intimate contact between the cocatalyst and photocatalyst as required to maintain their synergistic functioning.
In photocatalytic reactions, the main role of cocatalysts is to promote the separation of photogenerated charge carriers by acting either as electron mediators or hole mediators. In this regard, starting from their discovery, due to their excellent electrical conductivity (possessing mostly metallic or semi-metallic properties and thus mainly forming Schottky junctions in contact with the semiconductor photocatalysts),232 MXenes have been mainly promoted as a promising reservoir to trap and shuttle the photogenerated electrons from semiconductors. However, as was demonstrated in many studies, the MXene's role of electron- or hole-mediator depends very much on the type of surface functional groups37,102,233–236 and/or the work function compatibility between the MXene and the semiconductor forming the composites.237 This latter situation is also applicable in the case of photocatalytic composites with MOs as cocatalysts. Consequently, across the interfaces between the components of the composite photocatalysts, photogenerated holes will flow from the high-work-function component (the semiconductor acting as a photocatalyst) to the low-work-function component (the MO acting as a cocatalyst), while the photogenerated electrons will follow the opposite direction. In other words, the MO cocatalyst, which is actually a semiconductor material, might work either as an electron mediator or a hole mediator depending on the type of junction it makes with the semiconductor photocatalyst (e.g., p-type/n-type, n-type/n-type junction or even Z or S scheme). To summarize, in terms of the type of junction formed within the composite photocatalyst, the main difference between the MXenes and the MOs is given by their intrinsic properties. MXenes generally possessing a metallic character form Schottky junctions with the semiconductor photocatalyst depending on the terminal functional groups (e.g., –OH, –O, and –F). On the other hand, MOs generally form either p-type/n-type or n-type/n-type junctions as they have semiconductor properties.
Regarding the environmental concerns associated with the use of MXenes or MOs as cocatalysts that might arise, it should be noted that MXene synthesis is much more harmful to the environment than that of the MOs, mainly due to the use of the HF etchant. However, the use of proper handling and disposal measures can minimize the potential hazards, as well as the use of alternative synthetic methods that are currently being developed.
We will reach the same conclusion if we compare the production costs (the production costs for MXenes are comparatively higher, considering the synthesis of parent MAX phases), but perhaps the fairest cost-benefit comparison would be to follow the gain resulting from their use in the three types of reactions discussed in this review.
Regarding the photocatalytic water splitting reaction, multiple reactions occur on the surface of the composite photocatalyst as represented in eqn (9)–(11):
| H2O + e− → H* + OH− | (9) |
| H2O + H* + e− → H2 + OH− | (10) |
| H* + H* + 2e− → H2 | (11) |
In the case of both photocatalytic CO2 reduction and N2 photofixation reactions, the common step is the activation of the reactants (i.e., CO2 and N2). As both reactants are acid molecules (with CO2 possessing much higher acidity than N2), the presence of basic sites, such as the hydroxyl groups, is a prerequisite to perform the activation of these molecules. In view of that, in the case of MXenes, surface alkalinisation is needed in order to create more surface hydroxyl groups as active sites131 to adsorb and activate both CO2 and N2 molecules. This post-synthetic procedure is not necessary in the case of MOs, which already possess large amounts of such hydroxyl groups on the surface.
To summarize, in terms of the best activities reached so far, studies on water splitting indicate that a combination of MXenes (Ti3C2Tx) with black phosphorous and g-C3N4 exhibited the best performance for hydrogen evolution. Also, superior performances were observed for MXenes combined with CdS. In addition, TEA and lactic acid were reported to be the most appropriate hole scavengers under visible light irradiation. In the case of MOs, the highest H2 production rates were observed for CuO as the cocatalyst in the presence of methanol or glycerol as hole scavengers. It is clear from the literature data that extremely high values for AQY were obtained when MXenes were used as cocatalysts. On the other hand, for photocatalytic CO2 conversion, selectivity is a major parameter to consider. Using MXenes as the cocatalyst, CO and CH4 were found to be the main products in most of the cases. Even though a few studies reported the formation of methanol, the product window was narrow. Instead, as a general trend, MOs were found to be more selective for hydrocarbons (other than CH4) and oxygenated products like methanol. There are no big differences in the photocatalytic activity when MOs or MXenes are used as the cocatalyst for N2 photofixation, because the product formation and selectivity are related to the cocatalyst-photocatalyst composite, rather than the individual components.
Regarding the number of possible combinations, by comparing Tables 1–3 with Tables 4–6 one can observe that in the case of MXenes there are limited combinations used, mainly Ti-based MXenes, while for the MOs many more possibilities were explored.
As the last comparison criterion, the durability of the composites based on MXenes and metal oxides represents an important aspect that has to be considered for practical applications. Thus, it has to be specified that leaving a photocatalyst for a longer period of time under the harsh conditions imposed by the photocatalytic reactions will finally lead to photocorrosion phenomena.242 In this view, in comparison with the MOs, one of the biggest advantages of using MXenes as cocatalysts is to improve the photostability of the composites that contain it by shuttling photogenerated carriers from the unwanted side reaction with semiconductors.243,244
To conclude, regardless of the comparison criteria used here, both categories of materials present advantages that should further guide research in the field of photocatalysis towards finding the most efficient cocatalyst/photocatalyst combinations (see Fig. 19).
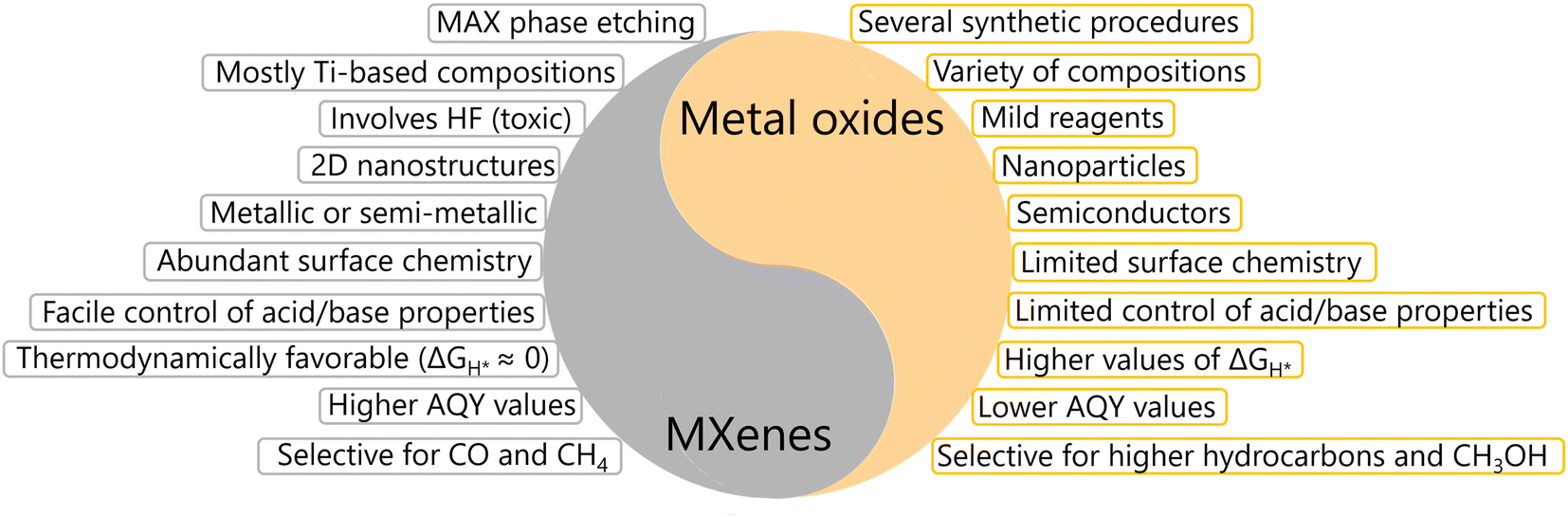 | ||
| Fig. 19 Scheme summarising the advantages and disadvantages of MXenes and metal oxides to be used as cocatalysts in photocatalytic reactions. | ||
6. Summary and future perspectives
This review underlined how important is to optimize the amount and type of the cocatalyst, and its choice should be carefully addressed and controlled. Moreover, the electronic properties, composition, loading, and morphology, among other factors, play an important role, affecting the overall photocatalytic activity of the hybrid materials. The literature survey presented in this review demonstrated that the use of MXenes and MOs as cocatalysts could greatly improve the photoactivity of a photocatalyst. This is mainly due to their capacity to promote the separation of photogenerated charge carriers. Overall, the photocatalytic processes presented in this review appear to be very attractive; however, there are challenges, such as activity and stability or high cost, that need to be elucidated.Since the photocatalytic process takes place on the surface of the photocatalysts, the screening of the surface and electronic properties of the photocatalytic composites should be more systematic. This is mandatory especially in the case of MXenes as their surface is terminated with different functional groups, the type of which not only depending on the etching procedure but also altering under reaction conditions, thus changing the photocatalytic behaviour of the MXene-based composites. In this view, a single characterization approach (which in most cases is carried out before performing the photocatalytic reaction) is insufficient to support a comprehensive understanding of the surface properties of MXenes and thus, a combination of complementary characterization techniques is required.
With regard to the stability issues of the currently used photocatalytic systems, it should be underlined that the effects of the reaction environment on the composite's surface demand more research attention. On this subject, as compared with MOs, MXenes have not been as extensively investigated as cocatalysts in photocatalytic processes. As already mentioned, MXenes are unstable in aqueous environments and in the presence of O2 and UV irradiation, because under such conditions MXenes are transformed into their corresponding oxides.45 However, as shown within this review in subSections 3.3.1 and 3.4.1, this can result in a positive approach for obtaining materials with properties of both type of materials, carbides and oxides, paving the way for new materials for photocatalysis. Moreover, there are studies that confirm the role of carbon in facilitating the separation of charge carriers and the use of MXenes as a precursor for photocatalysts.105,158,159 To sum up, in regard to the stability of MXenes and MOs acting as cocatalysts, probing approaches at the molecular level, such as different in situ/operando spectroscopies, are required in order to investigate the reaction mechanisms happening on the surface of these composites under specific chemical environment and photocatalytic conditions. Nevertheless, additional theoretical and rigorous experimental studies will be helpful for the development of the next generation of both MXene- and MO-based composites with target applications in the area of photocatalysis.
Last but not least, with the focus on the abundant tunability of MXene chemical compositions, but unfortunately with a high production cost, the majority of the photocatalytic studies are still concentrating on the use of Ti-based MXenes and less studies focus on the synthesis of other MXene materials. Therefore, utilization of other MXene-based composites requires further attention in both theoretical and experimental aspects.
To finish, although MXene and MO cocatalysts are a promising and attractive alternative to replace expensive noble-metal cocatalysts in the targeted photocatalytic applications presented here, significant research efforts are still needed. Following the literature displayed in this review, it is clear that there is room for improvement especially in the design and synthesis of MXene-based composites, and in this regard, several approaches can be envisaged, such as developing more environment-friendly methods for the existing ones and extending the preparation protocols to other MXenes besides Ti-based ones.
Author contributions
M. N. and A. I. contributed to Sections 1–4, while F. N., M. F. and S. N. contributed to Sections 5 and 6, all with scientific writing, illustrations and table drafting. M. F. and S. N. supervised the research work and contributed to funding acquisition, outline drafting, reviewing, and editing the overall text, illustrations and tables of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants of the Ministry of Research, Innovation and Digitization, CNCS – UEFISCDI, project number PN–III–P4-PCE-2021-1461 and PN–III–P4-ID-ERC-2021-0007, all within PNCDI III. Financial support from Romanian Ministry of Research, Innovation and Digitization through the Core Program 2023–2026 (contract PC3-PN23080303) is gratefully acknowledged.References
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- R. Schäppi, D. Rutz, F. Dähler, A. Muroyama, P. Haueter, J. Lilliestam, A. Patt, P. Furler and A. Steinfeld, Nature, 2022, 601, 63–68 CrossRef PubMed.
- Q. Wang, C. Pornrungroj, S. Linley and E. Reisner, Nat. Energy, 2022, 7, 13–24 CrossRef.
- R. Wang, T. Huang, J. Xue, J. Tong, K. Zhu and Y. Yang, Nat. Photonics, 2021, 15, 411–425 CrossRef CAS.
- I. Massiot, A. Cattoni and S. Collin, Nat. Energy, 2020, 5, 959–972 CrossRef.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Nørskov, Nat. Mater., 2017, 16, 70–81 CrossRef PubMed.
- B. Qiu, M. Du, Y. Ma, Q. Zhu, M. Xing and J. Zhang, Energy Environ. Sci., 2021, 14, 5260–5288 RSC.
- Z. Wang, Y. Hu, S. Zhang and Y. Sun, Chem. Soc. Rev., 2022, 51, 6704–6737 RSC.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L. Wu, C. Tung and T. Zhang, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- R. M. Navarro Yerga, M. C. Álvarez Galván, F. del Valle, J. A. Villoria de la Mano and J. L. G. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef CAS PubMed.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- X. Sun, S. Jiang, H. Huang, H. Li, B. Jia and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e2022048 Search PubMed.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- L. Jing, W. Zhou, G. Tian and H. Fu, Chem. Soc. Rev., 2013, 42, 9509 RSC.
- M. Ahmed and G. Xinxin, Inorg. Chem. Front., 2016, 3, 578–590 RSC.
- M. Z. Rahman, M. G. Kibria and C. B. Mullins, Chem. Soc. Rev., 2020, 49, 1887–1931 RSC.
- J. Yoon, J. Kim, C. Kim, H. W. Jang and J. Lee, Adv. Energy Mater., 2021, 11, 2003052 CrossRef CAS.
- Q. Xiang, B. Cheng and J. Yu, Angew. Chem., Int. Ed., 2015, 54, 11350–11366 CrossRef CAS PubMed.
- S. Bai, W. Yin, L. Wang, Z. Li and Y. Xiong, RSC Adv., 2016, 6, 57446–57463 RSC.
- J. Ran, M. Jaroniec and S. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef.
- G. Zhao and X. Xu, Nanoscale, 2021, 13, 10649–10667 RSC.
- H. Zhao, L. Jian, M. Gong, M. Jing, H. Li, Q. Mao, T. Lu, Y. Guo, R. Ji, W. Chi, Y. Dong and Y. Zhu, Small Struct., 2022, 3, 2100229 CrossRef CAS.
- D. Wang and X.-Q. Gong, Nat. Commun., 2021, 12, 158 CrossRef CAS PubMed.
- R. Reichert, Z. Jusys and R. J. Behm, J. Phys. Chem. C, 2015, 119, 24750–24759 CrossRef CAS.
- X. Wang, Y. Ruan, S. Feng, S. Chen and K. Su, ACS Sustainable Chem. Eng., 2018, 6, 11424–11432 CrossRef CAS.
- S. Bai, L. Yang, C. Wang, Y. Lin, J. Lu, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2015, 54, 14810–14814 CrossRef CAS PubMed.
- H. Lin, B. Sun, H. Wang, Q. Ruan, Y. Geng, Y. Li, J. Wu, W. Wang, J. Liu and X. Wang, Small, 2019, 15, 1804115 CrossRef PubMed.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha, A. L. Rogach, F. Jäckel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS PubMed.
- G. Ge, M. Liu, C. Liu, W. Zhou, D. Wang, L. Liu and J. Ye, J. Mater. Chem. A, 2019, 7, 9222–9229 RSC.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- R. Tang, S. Xiong, D. Gong, Y. Deng, Y. Wang, L. Su, C. Ding, L. Yang and C. Liao, ACS Appl. Mater. Interfaces, 2020, 12, 56663–56680 CrossRef CAS PubMed.
- L. Cheng, X. Li, H. Zhang and Q. Xiang, J. Phys. Chem. Lett., 2019, 10, 3488–3494 CrossRef CAS.
- Y. Zhao, M. Que, J. Chen and C. Yang, J. Mater. Chem. C, 2020, 8, 16258–16281 RSC.
- X. Xie and N. Zhang, Adv. Funct. Mater., 2020, 30, 2002528 CrossRef CAS.
- K. Huang, C. Li, H. Li, G. Ren, L. Wang, W. Wang and X. Meng, ACS Appl. Nano Mater., 2020, 3, 9581–9603 CrossRef CAS.
- X. Li, Y. Bai, X. Shi, N. Su, G. Nie, R. Zhang, H. Nie and L. Ye, Mater. Adv., 2021, 2, 1570–1594 RSC.
- M. Naguib, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2021, 33, 2103393 CrossRef CAS PubMed.
- K. R. G. Lim, M. Shekhirev, B. C. Wyatt, B. Anasori, Y. Gogotsi and Z. W. Seh, Nat. Synth., 2022, 1, 601–614 CrossRef.
- Á. Morales-García, F. Calle-Vallejo and F. Illas, ACS Catal., 2020, 10, 13487–13503 CrossRef.
- R. Wang, M. Li, K. Sun, Y. Zhang, J. Li and W. Bao, Small, 2022, 18, 2201740 CrossRef CAS.
- F. Bu, M. M. Zagho, Y. Ibrahim, B. Ma, A. Elzatahry and D. Zhao, Nano Today, 2020, 30, 100803 CrossRef CAS.
- I. M. Chirica, A. G. Mirea, Ş. Neaţu, M. Florea, M. W. Barsoum and F. Neaţu, J. Mater. Chem. A, 2021, 9, 19589–19612 RSC.
- Y. Zhao, S. Zhang, R. Shi, G. I. N. Waterhouse, J. Tang and T. Zhang, Mater. Today, 2020, 34, 78–91 CrossRef CAS.
- X. Meng, N. Yun and Z. Zhang, Can. J. Chem. Eng., 2019, 97, 1982–1998 CrossRef CAS.
- A. K. Singh, K. Mathew, H. L. Zhuang and R. G. Hennig, J. Phys. Chem. Lett., 2015, 6, 1087–1098 CrossRef CAS PubMed.
- X. Zhou and H. Dong, ChemCatChem, 2019, 11, 3688–3715 CrossRef CAS.
- P. Liao and E. A. Carter, Chem. Soc. Rev., 2013, 42, 2401–2422 RSC.
- C. Zhang, G. Chen, Y. Si and M. Liu, Phys. Chem. Chem. Phys., 2022, 24, 1237–1261 RSC.
- Y. Dong, P. Duchesne, A. Mohan, K. K. Ghuman, P. Kant, L. Hurtado, U. Ulmer, J. Y. Y. Loh, A. A. Tountas, L. Wang, A. Jelle, M. Xia, R. Dittmeyer and G. A. Ozin, Chem. Soc. Rev., 2020, 49, 5648–5663 RSC.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS.
- M. Humayun, C. Wang and W. Luo, Small Methods, 2022, 6, 2101395 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- R. Huang, X. Li, W. Gao, X. Zhang, S. Liang and M. Luo, RSC Adv., 2021, 11, 14844–14861 RSC.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- C. C. Nguyen, N. N. Vu and T.-O. Do, J. Mater. Chem. A, 2015, 3, 18345–18359 RSC.
- M. Xiao, Z. Wang, M. Lyu, B. Luo, S. Wang, G. Liu, H. Cheng and L. Wang, Adv. Mater., 2019, 31, 1801369 CrossRef.
- D. Chen, X. Zhang and A. F. Lee, J. Mater. Chem. A, 2015, 3, 14487–14516 RSC.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406–8407 CrossRef CAS PubMed.
- X. Wang, M. Liao, Y. Zhong, J. Y. Zheng, W. Tian, T. Zhai, C. Zhi, Y. Ma, J. Yao, Y. Bando and D. Golberg, Adv. Mater., 2012, 24, 3421–3425 CrossRef CAS PubMed.
- T. Xiong, F. Dong and Z. Wu, RSC Adv., 2014, 4, 56307–56312 RSC.
- G. Prieto, H. Tüysüz, N. Duyckaerts, J. Knossalla, G.-H. Wang and F. Schüth, Chem. Rev., 2016, 116, 14056–14119 CrossRef CAS PubMed.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS.
- A. VahidMohammadi, J. Rosen and Y. Gogotsi, Science, 2021, 372, eabf1581 CrossRef CAS PubMed.
- G. Deysher, C. E. Shuck, K. Hantanasirisakul, N. C. Frey, A. C. Foucher, K. Maleski, A. Sarycheva, V. B. Shenoy, E. A. Stach, B. Anasori and Y. Gogotsi, ACS Nano, 2020, 14, 204–217 CrossRef CAS.
- O. Mashtalir, M. Naguib, V. N. Mochalin, Y. Dall'Agnese, M. Heon, M. W. Barsoum and Y. Gogotsi, Nat. Commun., 2013, 4, 1716 CrossRef PubMed.
- M. Naguib, R. R. Unocic, B. L. Armstrong and J. Nanda, Dalton Trans., 2015, 44, 9353–9358 RSC.
- M. Ghidiu, M. R. Lukatskaya, M.-Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 CrossRef CAS PubMed.
- J. Halim, M. R. Lukatskaya, K. M. Cook, J. Lu, C. R. Smith, L.-Å. Näslund, S. J. May, L. Hultman, Y. Gogotsi, P. Eklund and M. W. Barsoum, Chem. Mater., 2014, 26, 2374–2381 CrossRef CAS PubMed.
- W. Sun, S. A. Shah, Y. Chen, Z. Tan, H. Gao, T. Habib, M. Radovic and M. J. Green, J. Mater. Chem. A, 2017, 5, 21663–21668 RSC.
- T. Li, L. Yao, Q. Liu, J. Gu, R. Luo, J. Li, X. Yan, W. Wang, P. Liu, B. Chen, W. Zhang, W. Abbas, R. Naz and D. Zhang, Angew. Chem., Int. Ed., 2018, 57, 6115–6119 CrossRef CAS PubMed.
- H. Shi, P. Zhang, Z. Liu, S. W. Park, M. R. Lohe, Y. Wu, A. Shaygan Nia, S. Yang and X. Feng, Angew. Chem., Int. Ed., 2021, 60, 8689–8693 CrossRef CAS PubMed.
- Y. Li, H. Shao, Z. Lin, J. Lu, L. Liu, B. Duployer, P. O. Å. Persson, P. Eklund, L. Hultman, M. Li, K. Chen, X.-H. Zha, S. Du, P. Rozier, Z. Chai, E. Raymundo-Piñero, P.-L. Taberna, P. Simon and Q. Huang, Nat. Mater., 2020, 19, 894–899 CrossRef CAS PubMed.
- M. Li, J. Lu, K. Luo, Y. Li, K. Chang, K. Chen, J. Zhou, J. Rosen, L. Hultman, P. Eklund, P. O. Å. Persson, S. Du, Z. Chai, Z. Huang and Q. Huang, J. Am. Chem. Soc., 2019, 141, 4730–4737 CrossRef CAS PubMed.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- Q. Tao, M. Dahlqvist, J. Lu, S. Kota, R. Meshkian, J. Halim, J. Palisaitis, L. Hultman, M. W. Barsoum, P. O. Å. Persson and J. Rosen, Nat. Commun., 2017, 8, 14949 CrossRef PubMed.
- L. Dong, H. Kumar, B. Anasori, Y. Gogotsi and V. B. Shenoy, J. Phys. Chem. Lett., 2017, 8, 422–428 CrossRef CAS.
- N. M. Caffrey, J. Phys. Chem. C, 2020, 124, 18797–18804 CrossRef CAS.
- J. Halim, A. S. Etman, A. Elsukova, P. Polcik, J. Palisaitis, M. W. Barsoum, P. O. Å. Persson and J. Rosen, Nanoscale, 2021, 13, 311–319 RSC.
- R. Meshkian, H. Lind, J. Halim, A. El Ghazaly, J. Thörnberg, Q. Tao, M. Dahlqvist, J. Palisaitis, P. O. Å. Persson and J. Rosen, ACS Appl. Nano Mater., 2019, 2, 6209–6219 CrossRef CAS.
- M. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef.
- C. E. Ren, M. Zhao, T. Makaryan, J. Halim, M. Boota, S. Kota, B. Anasori, M. W. Barsoum and Y. Gogotsi, ChemElectroChem, 2016, 3, 689–693 CrossRef CAS.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- L. Xiu, Z. Wang, M. Yu, X. Wu and J. Qiu, ACS Nano, 2018, 12, 8017–8028 CrossRef CAS PubMed.
- Y. Xia, T. S. Mathis, M.-Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- X. Xie, K. Kretschmer, B. Anasori, B. Sun, G. Wang and Y. Gogotsi, ACS Appl. Nano Mater., 2018, 1, 505–511 CrossRef CAS.
- Q. Zhao, Q. Zhu, J. Miao, P. Zhang, P. Wan, L. He and B. Xu, Small, 2019, 15, 1904293 CrossRef CAS PubMed.
- Q. Xue, H. Zhang, M. Zhu, Z. Pei, H. Li, Z. Wang, Y. Huang, Y. Huang, Q. Deng, J. Zhou, S. Du, Q. Huang and C. Zhi, Adv. Mater., 2017, 29, 1604847 CrossRef PubMed.
- Q. Zhang, Y. Sun, M. Liu and Y. Liu, Nanoscale, 2020, 12, 1826–1832 RSC.
- B. Shao, Z. Liu, G. Zeng, H. Wang, Q. Liang, Q. He, M. Cheng, C. Zhou, L. Jiang and B. Song, J. Mater. Chem. A, 2020, 8, 7508–7535 RSC.
- H. Mao, C. Gu, S. Yan, Q. Xin, S. Cheng, P. Tan, X. Wang, F. Xiu, X. Liu, J. Liu, W. Huang and L. Sun, Adv. Electron. Mater., 2020, 6, 1900493 CrossRef CAS.
- T. Su, R. Peng, Z. D. Hood, M. Naguib, I. N. Ivanov, J. K. Keum, Z. Qin, Z. Guo and Z. Wu, ChemSusChem, 2018, 11, 688–699 CrossRef CAS PubMed.
- Y. Li, D. Zhang, X. Feng, Y. Liao, Q. Wen and Q. Xiang, Nanoscale Adv., 2019, 1, 1812–1818 RSC.
- H. Wang, R. Peng, Z. D. Hood, M. Naguib, S. P. Adhikari and Z. Wu, ChemSusChem, 2016, 9, 1490–1497 CrossRef CAS PubMed.
- T. Su, Z. D. Hood, M. Naguib, L. Bai, S. Luo, C. M. Rouleau, I. N. Ivanov, H. Ji, Z. Qin and Z. Wu, ACS Appl. Energy Mater., 2019, 2, 4640–4651 CrossRef CAS.
- T. Su, Z. D. Hood, M. Naguib, L. Bai, S. Luo, C. M. Rouleau, I. N. Ivanov, H. Ji, Z. Qin and Z. Wu, Nanoscale, 2019, 11, 8138–8149 RSC.
- K. Maeda, H. Wakayama, Y. Washio, A. Ishikawa, M. Okazaki, H. Nakata and S. Matsuishi, J. Phys. Chem. C, 2020, 124, 14640–14645 CrossRef CAS.
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC.
- X. Zhang, X. Zhao, D. Wu, Y. Jing and Z. Zhou, Nanoscale, 2015, 7, 16020–16025 RSC.
- G. Gao, A. P. O'Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- K. Xiong, P. Wang, G. Yang, Z. Liu, H. Zhang, S. Jin and X. Xu, Sci. Rep., 2017, 7, 15095 CrossRef PubMed.
- G. Jia, Y. Wang, X. Cui and W. Zheng, ACS Sustainable Chem. Eng., 2018, 6, 13480–13486 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- Y. Li, X. Deng, J. Tian, Z. Liang and H. Cui, Appl. Mater. Today, 2018, 13, 217–227 CrossRef.
- C. Peng, X. Xie, W. Xu, T. Zhou, P. Wei, J. Jia, K. Zhang, Y. Cao, H. Wang, F. Peng, R. Yang, X. Yan, H. Pan and H. Yu, Chem. Eng. J., 2021, 421, 128766 CrossRef CAS.
- B. Wang, M. Wang, F. Liu, Q. Zhang, S. Yao, X. Liu and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 1914–1918 CrossRef CAS PubMed.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- R. Xiao, C. Zhao, Z. Zou, Z. Chen, L. Tian, H. Xu, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B, 2020, 268, 118382 CrossRef CAS.
- B. Sun, P. Qiu, Z. Liang, Y. Xue, X. Zhang, L. Yang, H. Cui and J. Tian, Chem. Eng. J., 2021, 406, 127177 CrossRef CAS.
- M. Ding, R. Xiao, C. Zhao, D. Bukhvalov, Z. Chen, H. Xu, H. Tang, J. Xu and X. Yang, Sol. RRL, 2021, 5, 2000414 CrossRef CAS.
- N. Zhao, Y. Hu, J. Du, G. Liu, B. Dong, Y. Yang, J. Peng, J. Li and M. Zhai, Appl. Surf. Sci., 2020, 530, 147247 CrossRef CAS.
- S. Jin, Z. Shi, H. Jing, L. Wang, Q. Hu, D. Chen, N. Li and A. Zhou, ACS Appl. Energy Mater., 2021, 4, 12754–12766 CrossRef CAS.
- G. Zuo, Y. Wang, W. L. Teo, A. Xie, Y. Guo, Y. Dai, W. Zhou, D. Jana, Q. Xian, W. Dong and Y. Zhao, Angew. Chem., Int. Ed., 2020, 59, 11287–11292 CrossRef CAS PubMed.
- X. Du, T. Zhao, Z. Xiu, Z. Xing, Z. Li, K. Pan, S. Yang and W. Zhou, Appl. Mater. Today, 2020, 20, 100719 CrossRef.
- K. Huang, C. Li and X. Meng, J. Colloid Interface Sci., 2020, 580, 669–680 CrossRef CAS.
- R. Chen, P. Wang, J. Chen, C. Wang and Y. Ao, Appl. Surf. Sci., 2019, 473, 11–19 CrossRef CAS.
- Y. Li, L. Ding, Z. Liang, Y. Xue, H. Cui and J. Tian, Chem. Eng. J., 2020, 383, 123178 CrossRef CAS.
- Z. Li, W. Huang, J. Liu, K. Lv and Q. Li, ACS Catal., 2021, 11, 8510–8520 CrossRef CAS.
- Y. Li, L. Ding, Y. Guo, Z. Liang, H. Cui and J. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 41440–41447 CrossRef CAS.
- J. Kang, S. Byun, S. Kim, J. Lee, M. Jung, H. Hwang, T. W. Kim, S. H. Song and D. Lee, ACS Appl. Energy Mater., 2020, 3, 9226–9233 CrossRef CAS.
- J. Li, L. Zhao, S. Wang, J. Li, G. Wang and J. Wang, Appl. Surf. Sci., 2020, 515, 145922 CrossRef CAS.
- T. Song, L. Hou, B. Long, A. Ali and G. J. Deng, J. Colloid Interface Sci., 2021, 584, 474–483 CrossRef CAS PubMed.
- Y. Li, X. Chen, Y. Sun, X. Meng, Y. Dall'Agnese, G. Chen, C. Dall'Agnese, H. Ren, S. Sasaki, H. Tamiaki and X. Wang, Adv. Mater. Interfaces, 2020, 7, 1902080 CrossRef CAS.
- Y. Li, Y. Sun, T. Zheng, Y. Dall'Agnese, C. Dall'Agnese, X. Meng, S. Sasaki, H. Tamiaki and X. Wang, Chem. – Eur. J., 2021, 27, 5277–5282 CrossRef CAS PubMed.
- S. Debow, T. Zhang, X. Liu, F. Song, Y. Qian, J. Han, K. Maleski, Z. B. Zander, W. R. Creasy, D. L. Kuhn, Y. Gogotsi, B. G. DeLacy and Y. Rao, J. Phys. Chem. C, 2021, 125, 10473–10482 CrossRef CAS.
- J. Low, L. Zhang, T. Tong, B. Shen and J. Yu, J. Catal., 2018, 361, 255–266 CrossRef CAS.
- M. Ye, X. Wang, E. Liu, J. Ye and D. Wang, ChemSusChem, 2018, 11, 1606–1611 CrossRef CAS PubMed.
- Q. Song, B. Shen, J. Yu and S. Cao, ChemNanoMat, 2021, 7, 910–915 CrossRef CAS.
- Q. Tang, P. Xiong, H. Wang and Z. Wu, J. Colloid Interface Sci., 2022, 619, 179–187 CrossRef CAS.
- K. Wang, X. Li, N. Wang, Q. Shen, M. Liu, J. Zhou and N. Li, Ind. Eng. Chem. Res., 2021, 60, 8720–8732 CrossRef CAS.
- Z. Zeng, Y. Yan, J. Chen, P. Zan, Q. Tian and P. Chen, Adv. Funct. Mater., 2019, 29, 1806500 CrossRef.
- J. Zhang, J. Shi, S. Tao, L. Wu and J. Lu, Appl. Surf. Sci., 2021, 542, 148685 CrossRef CAS.
- S. Cao, B. Shen, T. Tong, J. Fu and J. Yu, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- J. Li, Z. Wang, H. Chen, Q. Zhang, H. Hu, L. Liu, J. Ye and D. Wang, Catal. Sci. Technol., 2021, 11, 4953–4961 RSC.
- C. Yang, Q. Tan, Q. Li, J. Zhou, J. Fan, B. Li, J. Sun and K. Lv, Appl. Catal., B, 2020, 268, 118738 CrossRef CAS.
- F. He, B. Zhu, B. Cheng, J. Yu, W. Ho and W. Macyk, Appl. Catal., B, 2020, 272, 119006 CrossRef CAS.
- Q. Tang, Z. Sun, S. Deng, H. Wang and Z. Wu, J. Colloid Interface Sci., 2020, 564, 406–417 CrossRef CAS PubMed.
- H. Wang, Q. Tang and Z. Wu, ACS Sustainable Chem. Eng., 2021, 9, 8425–8434 CrossRef CAS.
- L. Hong, R. Guo, Y. Yuan, X. Ji, Z. Lin, X. Yin and W. Pan, Colloids Surf., A, 2022, 639, 128358 CrossRef CAS.
- W. Chen, B. Han, Y. Xie, S. Liang, H. Deng and Z. Lin, Chem. Eng. J., 2020, 391, 123519 CrossRef CAS.
- Q. Shi, X. Zhang, Y. Yang, J. Huang, X. Fu, T. Wang, X. Liu, A. Sun, J. Ge, J. Shen, Y. Zhou and Z. Liu, J. Energy Chem., 2021, 59, 9–18 CrossRef CAS.
- M. Que, Y. Zhao, Y. Yang, L. Pan, W. Lei, W. Cai, H. Yuan, J. Chen and G. Zhu, ACS Appl. Mater. Interfaces, 2021, 13, 6180–6187 CrossRef CAS.
- M. Que, W. Cai, Y. Zhao, Y. Yang, B. Zhang, S. Yun, J. Chen and G. Zhu, J. Colloid Interface Sci., 2022, 610, 538–545 CrossRef CAS PubMed.
- Z. Zhang, B. Wang, H.-B. Zhao, J.-F. Liao, Z.-C. Zhou, T. Liu, B. He, Q. Wei, S. Chen, H.-Y. Chen, D.-B. Kuang, Y. Li and G. Xing, Appl. Catal., B, 2022, 312, 121358 CrossRef CAS.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- S. D. Minteer, P. Christopher and S. Linic, ACS Energy Lett., 2019, 4, 163–166 CrossRef CAS.
- A. Liu, Y. Yang, X. Ren, Q. Zhao, M. Gao, W. Guan, F. Meng, L. Gao, Q. Yang, T. Ma and X. Liang, ChemSusChem, 2020, 13, 3766–3788 CrossRef CAS PubMed.
- J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed.
- J. Qin, X. Hu, X. Li, Z. Yin, B. Liu and K. Lam, Nano Energy, 2019, 61, 27–35 CrossRef CAS.
- B. Chang, Y. Guo, D. Wu, L. Li, B. Yang and J. Wang, Chem. Sci., 2021, 12, 11213–11224 RSC.
- C. Hao, Y. Liao, Y. Wu, Y. An, J. Lin, Z. Gu, M. Jiang, S. Hu and X. Wang, J. Phys. Chem. Solids, 2020, 136, 109141 CrossRef CAS.
- W. Gao, X. Li, S. Luo, Z. Luo, X. Zhang, R. Huang and M. Luo, J. Colloid Interface Sci., 2021, 585, 20–29 CrossRef CAS.
- H. Jiang, C. Zang, Y. Zhang, W. Wang, C. Yang, B. Sun, Y. Shen and F. Bian, Catal. Sci. Technol., 2020, 10, 5964–5972 RSC.
- Q. Liu, L. Ai and J. Jiang, J. Mater. Chem. A, 2018, 6, 4102–4110 RSC.
- J. Qian, S. Zhao, W. Dang, Y. Liao, W. Zhang, H. Wang, L. Lv, L. Luo, H.-Y. Jiang and J. Tang, Adv. Sustainable Syst., 2021, 5, 2000282 CrossRef CAS.
- T. Hou, Q. Li, Y. Zhang, W. Zhu, K. Yu, S. Wang, Q. Xu, S. Liang and L. Wang, Appl. Catal., B, 2020, 273, 119072 CrossRef CAS.
- L. I. Qi, J. Xu, S. Wang, K. Yu, W. Zhu, T. Hou, L. Zhang, W. Zhang, S. Liang and L. Wang, Trans. Nonferrous Met. Soc. China, 2022, 32, 233–250 CrossRef.
- B. Chang, Y. Guo, H. Liu, L. Li and B. Yang, J. Mater. Chem. A, 2022, 10, 3134–3145 RSC.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- W. Song, E. M. Lopato, S. Bernhard, P. A. Salvador and G. S. Rohrer, Appl. Catal., B, 2020, 269, 118750 CrossRef CAS.
- S. Martha, P. Chandra Sahoo and K. M. Parida, RSC Adv., 2015, 5, 61535–61553 RSC.
- Y. Sun, T. Zhang, C. Li, K. Xu and Y. Li, J. Mater. Chem. A, 2020, 8, 13415–13436 RSC.
- Y. Yang, S. Niu, D. Han, T. Liu, G. Wang and Y. Li, Adv. Energy Mater., 2017, 7, 1700555 CrossRef.
- X. Li, H. Zhao, J. Liang, Y. Luo, G. Chen, X. Shi, S. Lu, S. Gao, J. Hu, Q. Liu and X. Sun, J. Mater. Chem. A, 2021, 9, 6650–6670 RSC.
- Z. Zeng, Y. Xu, Z. Zhang, Z. Gao, M. Luo, Z. Yin, C. Zhang, J. Xu, B. Huang, F. Luo, Y. Du and C. Yan, Chem. Soc. Rev., 2020, 49, 1109–1143 RSC.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- S. Royer, D. Duprez, F. Can, X. Courtois, C. Batiot-Dupeyrat, S. Laassiri and H. Alamdari, Chem. Rev., 2014, 114, 10292–10368 CrossRef CAS PubMed.
- A. E. Danks, S. R. Hall and Z. Schnepp, Mater. Horiz., 2016, 3, 91–112 RSC.
- D. R. Modeshia and R. I. Walton, Chem. Soc. Rev., 2010, 39, 4303 RSC.
- X. Huang, G. Zhao, G. Wang and J. T. S. Irvine, Chem. Sci., 2018, 9, 3623–3637 RSC.
- Y. Jun, J. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS PubMed.
- Y. Mao, T.-J. Park, F. Zhang, H. Zhou and S. S. Wong, Small, 2007, 3, 1122–1139 CrossRef CAS PubMed.
- M.-M. Titirici, M. Antonietti and A. Thomas, Chem. Mater., 2006, 18, 3808–3812 CrossRef CAS.
- A. Querejeta, A. Varela, M. Parras, F. del Monte, M. García-Hernández and J. M. González-Calbet, Chem. Mater., 2009, 21, 1898–1905 CrossRef CAS.
- S. Bie, Y. Zhu, J. Su, C. Jin, S. Liu, R. Yang and J. Wu, J. Mater. Chem. A, 2015, 3, 22448–22453 RSC.
- J. J. Urban, L. Ouyang, M.-H. Jo, D. S. Wang and H. Park, Nano Lett., 2004, 4, 1547–1550 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- D. Gu and F. Schüth, Chem. Soc. Rev., 2014, 43, 313–344 RSC.
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217–1231 CAS.
- A.-H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793–1805 CrossRef CAS.
- F. Schüth, Angew. Chem., Int. Ed., 2003, 42, 3604–3622 CrossRef PubMed.
- M. M. Nair, H. Yen and F. Kleitz, C. R. Chim., 2014, 17, 641–655 CrossRef CAS.
- M. M. Nair, S. Kaliaguine and F. Kleitz, ACS Catal., 2014, 4, 3837–3846 CrossRef CAS.
- Y. Shi, Y. Wan and D. Zhao, Chem. Soc. Rev., 2011, 40, 3854 RSC.
- B. T. Yonemoto, G. S. Hutchings and F. Jiao, J. Am. Chem. Soc., 2014, 136, 8895–8898 CrossRef CAS PubMed.
- J. Liu, Q. Jia, J. Long, X. Wang, Z. Gao and Q. Gu, Appl. Catal., B, 2018, 222, 35–43 CrossRef CAS.
- G. D. Moon, J. B. Joo, I. Lee and Y. Yin, Nanoscale, 2014, 6, 12002–12008 RSC.
- P. Ravi, V. Navakoteswara Rao, M. V. Shankar and M. Sathish, Int. J. Hydrogen Energy, 2020, 45, 7517–7529 CrossRef CAS.
- X. Chen, W. Chen, H. Gao, Y. Yang and W. Shangguan, Appl. Catal., B, 2014, 152–153, 68–72 CrossRef CAS.
- B. Fang, Y. Xing, A. Bonakdarpour, S. Zhang and D. P. Wilkinson, ACS Sustainable Chem. Eng., 2015, 3, 2381–2388 CrossRef CAS.
- M. M. Gui, S.-P. Chai and A. R. Mohamed, Appl. Surf. Sci., 2014, 319, 37–43 CrossRef CAS.
- X. Chen, W. Chen, P. Lin, Y. Yang, H. Gao, J. Yuan and W. Shangguan, Catal. Commun., 2013, 36, 104–108 CrossRef CAS.
- X. Ning, W. Zhen, X. Zhang and G. Lu, ChemSusChem, 2019, 12, 1410–1420 CrossRef CAS PubMed.
- Y. Fu, C. Liu, C. Zhu, H. Wang, Y. Dou, W. Shi, M. Shao, H. Huang, Y. Liu and Z. Kang, Inorg. Chem. Front., 2018, 5, 1646–1652 RSC.
- N. Banic, J. Krstić, S. Stojadinović, A. Brnović, A. Djordjevic and B. Abramović, Int. J. Energy Res., 2020, 44, 8951–8963 CrossRef CAS.
- L. Li, B. Cheng, Y. Wang and J. Yu, J. Colloid Interface Sci., 2015, 449, 115–121 CrossRef CAS PubMed.
- N. Chen, J. Cao, M. Guo, C. Liu, H. Lin and S. Chen, Int. J. Hydrogen Energy, 2021, 46, 19363–19372 CrossRef CAS.
- B. Yan, J. Li, Z. Lin, C. Du and G. Yang, ACS Appl. Nano Mater., 2019, 2, 6783–6792 CrossRef CAS.
- J. Chen, S. Shen, P. Guo, M. Wang, P. Wu, X. Wang and L. Guo, Appl. Catal., B, 2014, 152–153, 335–341 CrossRef CAS.
- L. Zhu, M. Hong and G. W. Ho, Nano Energy, 2015, 11, 28–37 CrossRef CAS.
- G. Sadanandam, X. Luo, X. Chen, Y. Bao, K. P. Homewood and Y. Gao, Appl. Surf. Sci., 2021, 541, 148687 CrossRef CAS.
- Z. Mao, J. Chen, Y. Yang, D. Wang, L. Bie and B. D. Fahlman, ACS Appl. Mater. Interfaces, 2017, 9, 12427–12435 CrossRef CAS PubMed.
- Y. Zhu, T. Wan, X. Wen, D. Chu and Y. Jiang, Appl. Catal., B, 2019, 244, 814–822 CrossRef CAS.
- E. L. Uzunova, N. Seriani and H. Mikosch, Phys. Chem. Chem. Phys., 2015, 17, 11088–11094 RSC.
- S. Zhu, S. Liang, Y. Tong, X. An, J. Long, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2015, 17, 9761–9770 RSC.
- H. Li, Y. Lei, Y. Huang, Y. Fang, Y. Xu, L. Zhu and X. Li, J. Nat. Gas Chem., 2011, 20, 145–150 CrossRef CAS.
- Y. Li, W. Zhang, X. Shen, P. Peng, L. Xiong and Y. Yu, Chin. J. Catal., 2015, 36, 2229–2236 CrossRef CAS.
- M. Tahir and B. Tahir, Appl. Surf. Sci., 2016, 377, 244–252 CrossRef CAS.
- S. Shoji, A. Yamaguchi, E. Sakai and M. Miyauchi, ACS Appl. Mater. Interfaces, 2017, 9, 20613–20619 CrossRef CAS PubMed.
- M. Alves Melo Júnior, A. Morais and A. F. Nogueira, Microporous Mesoporous Mater., 2016, 234, 1–11 CrossRef.
- V. Jeyalakshmi, R. Mahalakshmy, K. R. Krishnamurthy and B. Viswanathan, Catal. Today, 2016, 266, 160–167 CrossRef CAS.
- L. F. Garay-Rodríguez and L. M. Torres-Martínez, J. Mater. Sci.: Mater. Electron., 2020, 31, 19248–19265 CrossRef.
- S. Jeong, W. D. Kim, S. Lee, K. Lee, S. Lee, D. Lee and D. C. Lee, ChemCatChem, 2016, 8, 1641–1645 CrossRef CAS.
- Z.-Y. Wang, H.-C. Chou, J. C. S. Wu, D. P. Tsai and G. Mul, Appl. Catal., A, 2010, 380, 172–177 CrossRef CAS.
- D.-S. Lee, H.-J. Chen and Y.-W. Chen, J. Phys. Chem. Solids, 2012, 73, 661–669 CrossRef CAS.
- X. Shao, X. Yin and J. Wang, J. Colloid Interface Sci., 2018, 512, 466–473 CrossRef CAS PubMed.
- D.-S. Lee and Y.-W. Chen, J. CO2 Util., 2015, 10, 1–6 CrossRef.
- C.-W. Tsai, H. M. Chen, R.-S. Liu, K. Asakura and T.-S. Chan, J. Phys. Chem. C, 2011, 115, 10180–10186 CrossRef CAS.
- F. Wang, Y. Zhou, P. Li, L. Kuai and Z. Zou, Chin. J. Catal., 2016, 37, 863–868 CrossRef CAS.
- J. A. Torres, A. E. Nogueira, G. T. S. T. da Silva, O. F. Lopes, Y. Wang, T. He and C. Ribeiro, J. CO2 Util., 2020, 35, 106–114 CrossRef CAS.
- S. W. Jo, B. S. Kwak, K. M. Kim, J. Y. Do, N.-K. Park, S. O. Ryu, H.-J. Ryu, J.-I. Baek and M. Kang, Appl. Surf. Sci., 2015, 355, 891–901 CrossRef CAS.
- X. Xue, R. Chen, C. Yan, Y. Hu, W. Zhang, S. Yang, L. Ma, G. Zhu and Z. Jin, Nanoscale, 2019, 11, 10439–10445 RSC.
- B. Sun, Z. Liang, Y. Qian, X. Xu, Y. Han and J. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7257–7269 CrossRef CAS PubMed.
- X. Hui, L. Li, Q. Xia, S. Hong, L. Hao, A. W. Robertson and Z. Sun, Chem. Eng. J., 2022, 438, 135485 CrossRef CAS.
- H. Mou, J. Wang, D. Zhang, D. Yu, W. Chen, D. Wang and T. Mu, J. Mater. Chem. A, 2019, 7, 5719–5725 RSC.
- L. Yan, Q. Zhang, W. Deng, Q. Zhang and Y. Wang, Catalytic Valorization of Biomass and Bioplatforms to Chemicals through Deoxygenation, Elsevier Inc., 1st edn, 2020, vol. 66 Search PubMed.
- H. H. B. T.-S, in Transition Metal Oxides,ed. S. S. and C. Kung, Elsevier, 1989, vol. 45, pp. 121–135 Search PubMed.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248 CrossRef CAS PubMed.
- M. Khazaei, M. Arai, T. Sasaki, A. Ranjbar, Y. Liang and S. Yunoki, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 75411 CrossRef.
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CrossRef CAS PubMed.
- C. Peng, H. Wang, H. Yu and F. Peng, Mater. Res. Bull., 2017, 89, 16–25 CrossRef CAS.
- Y. Liu, H. Xiao and W. A. Goddard, J. Am. Chem. Soc., 2016, 138, 15853–15856 CrossRef CAS PubMed.
- C. Peng, P. Wei, X. Li, Y. Liu, Y. Cao, H. Wang, H. Yu, F. Peng, L. Zhang, B. Zhang and K. Lv, Nano Energy, 2018, 53, 97–107 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- J. Bonde, P. G. Moses, T. F. Jaramillo, J. K. Nørskov and I. Chorkendorff, Faraday Discuss., 2009, 140, 219–231 RSC.
- Y. H. Li, C. Peng, S. Yang, H. F. Wang and H. G. Yang, J. Catal., 2015, 330, 120–128 CrossRef CAS.
- B. Weng, M.-Y. Qi, C. Han, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2019, 9, 4642–4687 CrossRef CAS.
- T. Cai, L. Wang, Y. Liu, S. Zhang, W. Dong, H. Chen, X. Yi, J. Yuan, X. Xia, C. Liu and S. Luo, Appl. Catal., B, 2018, 239, 545–554 CrossRef CAS.
- X. Xie, N. Zhang, Z.-R. Tang, M. Anpo and Y.-J. Xu, Appl. Catal., B, 2018, 237, 43–49 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |