
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dramatic acceleration by visible light and mechanism of AuPd@ZIF-8-catalyzed ammonia borane methanolysis for efficient hydrogen production†
Naixin
Kang
a,
Ruofan
Shen
b,
Baojun
Li
 *b,
Fangyu
Fu
c,
Bruno
Espuche
de,
Sergio
Moya
d,
Lionel
Salmon
f,
Jean-Luc
Pozzo
*a and
Didier
Astruc
*a
*b,
Fangyu
Fu
c,
Bruno
Espuche
de,
Sergio
Moya
d,
Lionel
Salmon
f,
Jean-Luc
Pozzo
*a and
Didier
Astruc
*a
aISM, UMR CNRS No. 5255, Univ. Bordeaux, 33405 Talence Cedex, France. E-mail: didier.astruc@u-bordeaux.fr
bResearch Center of Green Catalysis, College of Chemistry, Zhengzhou University, 100 Science Road, Zhengzhou 450001, P. R. China
cMOE Key Laboratory of Cluster Science, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 102488, P. R. China
dSoft Matter Nanotechnology Lab, CIC biomaGUNE, Paseo Miramón 182. 20014, Donostia-San Sebastián, Gipuzkoa, Spain
ePOLYMAT, Applied Chemistry Department, Faculty of Chemistry, University of the Basque Country, UPV/EHU, Paseo Manuel de Lardizabal 3, Donostia-San Sebastián, 20018, Spain
fLCC, CNRS & University of Toulouse, 31077 Toulouse Cedex, France
First published on 31st January 2023
Abstract
The generation of H2 from materials with a high content of H atoms is attractive for both sustainable energy and convenient hydrogenation. We report that the novel synthesized AuPd@ZIF-8-alloyed nanoparticles (ZIF = zeolitic imidazolate framework) in which the AuPd nanoparticles (NPs) have a size of 2.43 nm and are shown by Brunauer–Emmett–Teller (BET) surface to be encapsulated into ZIF-8. The AuPd and ZIF-8 nanoparticle is an excellent nanocatalyst for the evolution of H2 in the methanolysis reaction of aminoborane (AB) under visible light irradiation. Visible-light-induced acceleration is due to the Au plasmonic excitation provoking hot electron transfer from Au to Pd-substrate ensemble, whereas the reactions catalyzed by monometallic Au@ZIF-8 or Pd@ZIF-8 undergo only few minutes or no acceleration (respectively) under visible light irradiation. Three mol H2 per mol AB are produced in 6 min at 25 °C (TOF: 86.8 mol H2 molatom−1 min−1) with AuPd@ZIF-8 under visible light compared with incomplete H2 formation in 30 min in the dark. A comparison of various heterogeneous supports shows that the ZIF-8 encapsulation of the nanoalloy is by far the best support for this reaction. The large primary kinetic isotope effect (KIE) kH/kD = 3.4 with visible-light irradiation, the high turnover frequency (TOF) under light illumination (3.7 times higher than in the dark), and density functional theory (DFT) calculations confirm the mechanism and illustrate the more difficult O–H oxidative addition on Pd in methanol than in water in this process. This is due to the weaker acidity of methanol compared to that of water. Coupling labeling and tandem reactions with styrene hydrogenation showed that one H atom of H2 formed is provided by AB, while the other one is from methanol. The cleanness of the H2 generated and the recyclability of the NH4B(OMe)4 product render AB methanolysis attractive for low-temperature H2 production devices and tandem reactions.
Introduction
Hydrogen (H2) generation is promising to provide a source of green energy and to avoid hydrogen storage in laboratories.1–3 Ammonia borane (AB) is among the best H2 sources and the most studied one because of its high hydrogen gravimetric capacity (19.6 wt%), excellent solubility in alcohols, high stability, lack of toxicity and accessible formation of H2 on the surface of catalysts.4–8Small nanoparticles (NPs) are outstanding candidates for nanocatalysts owing to their high ratio between surface and volume and performances for the catalysis of many reactions.9–11 The alloys between Pt or Pd and other transition metals can be tuned in terms of their stereo-electronic properties to optimize the catalysis of many reactions.12–15 For instance, NiPt and PtCo alloyed NPs are known to accelerate the hydrolytic dehydrogenation of AB and to facilitate reactions conducted in tandem with hydrogenation reactions.16,17
AB methanolysis (eqn (1)) is promising although it is much less used than AB hydrolysis as a means of facile H2 generation:
 | (1) |
However, because of the seminal reports by Ramachandran and Gagare of optimized ammonia borane preparation and methanolysis18 and by Jagirdar's group of Co, Ni and Cu-based nanocatalysts19,20 of AB methanolysis, several publications have appeared on this method of hydrogen generation, and the area was reviewed in 2016 by Xu's group21 and in 2022 by Li's group.22 This H2 generation method possesses several significant advantages towards current applications:21,22 (i) AB is highly methanol soluble at room temperature (23% wt);23 (ii) it is highly stable in methanol without a catalyst; (iii) H2 produced by AB methanolysis is pure from ammonia impurities that are poisoning fuel cells; (iv) this reaction generates H2 below 0 °C upon catalysis, which might be useful for portable devices under cold temperature conditions; (v) the AB methanolysis product formed, NH4B(OCH3)4, can regenerate AB upon reaction with LiAlH4 and NH4Cl under ambient conditions.24 Although first-row late transition-metal-based nanocatalysts are privileged,18,24–26 there are also reports using more efficient noble-metal catalysts.23,27
Recently, the plasmon-induced enhancement of catalytic reactivity has received attention for AB dehydrogenation under visible-light irradiation25,26 given the excitation of plasmon that accelerates reactions.26–30 The dissipation of energy proceeds by the scattering of radiative photons or the induction of charge transfer, provoking reaction rate enhancement.31 Local collective heating,30 heterogeneity of charges at the surface,32 and hot electrons injected from the plasmon are known to accelerate hydrogen production upon AB hydrolysis.31–33
Herein, we report the synthesis and characterization of a novel catalyst AuPd@zeolitic imidazolate framework (ZIF)-8 for AB methanolysis, the dramatic visible-light-induced acceleration of this reaction with alloyed AuPd NPs supported by ZIF-8, the high selectivity and superiority of this support and a proposed mechanism based on isotopic labeling upon tandem styrene hydrogenation reaction, high primary kinetic isotope effect (KIE), and density functional theory (DFT) calculations confirming the reaction mechanism. The impressive visible light-induced acceleration involves nanogold plasmon excitation owing to the local field with contributions of hot electron transfer, and the very slight effect of temperature increase is revealed and discussed.
Materials and methods
Materials
Potassium tetrachloroaurate, sodium tetrachloropalladate, zirconium tetrachloride sodium borohydride, acetic acid, zinc nitrate hexahydrate, p-phthalic acid, cobalt nitrate hexahydrate, 2-methylimidazole, methanol (>99.9% HPLC), and methanol-D4 (>99.95%, NMR) were purchased from Sigma-Aldrich. Graphene oxide (GO) was synthesized using the Hummers' method.34 All reaction vessels were washed with aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1) and acetone. All reagents and solutions were prepared using ultra-pure water provided by the Barnstead nanowater purification system (Thermo Fisher Scientific Inc., USA).
:HNO3 = 3:1) and acetone. All reagents and solutions were prepared using ultra-pure water provided by the Barnstead nanowater purification system (Thermo Fisher Scientific Inc., USA).
Syntheses of the nanocatalysts
In general, 100 mg ZIF-8 NPs, dispersed in a mixture of 5 mL water and 5 mL methanol, were introduced into a 50 mL Schlenk flask under argon. The contents of the AuPd alloyed NPs were adjusted based on variations in the precursor molar ratios. The reaction mixture was continuously stirred at ambient temperature for 2 h. The solution was degassed for 10 min. Then, 1 mL of an aqueous solution of recently prepared NaBH4 (10 equiv. per equiv. metal) was rapidly added. The reaction mixture was stirred for an additional 30 min. Then, the nanocatalyst was obtained by centrifugation and washed with deoxygenated methanol. Finally, it was dried overnight at 60 °C in vacuo. The amount of metal content was determined using inductively coupled plasma atomic emission spectroscopy (ICP-AES). For the preparation of AuPd NPs stabilized by other supports, the steps were similar to those used for AuPd@ZIF-8.Catalysis in the dark of AB methanolysis by AuPd @ZIF-8
The methanolysis of AB in the dark was achieved at 25 ± 0.2 °C in a 50 mL Schlenk tube. 50 mg of the AuPd@ZIF-8 catalyst was introduced into a reaction vessel with 4 mL of water. Subsequently, 1 mL of the AB methanol solution was introduced into the solution. In the standard treatment, the content of metal nanoparticles (MNPs) was 1.25% mmol per mmol AB, the ratio between Au and Pd was 1 to 1, and the concentration of AB was 1 mmol L−1. The reaction time and determination of the H2 volume of the produced H2 volume began when the AB solution was imported using a needle (Fig. S1†).Catalysis of AB methanolysis by AuPd@ZIF-8 under visible light
The reactions in the presence of visible light were performed by utilizing visible light-emitting diode (LED) strips (7 W) from Sunshine Lighting Limited, France, at a light intensity of 1540lx (each) with a wavelength of 550 nm. The LED lamps were turned on for 15min before irradiation to ensure that the light intensity was stable. All visible-light reactions were carried out using a condensate circulation system so that the temperature remains at 25 ± 0.2 °C. The thermometer was located inside the reaction vessel. The concentrations of AB and catalyst were identical to those conducted in the dark. The other reaction steps were also conducted identically (Fig. S2†).
Styrene hydrogenation catalyzed in tandem reaction by AuPd@ZIF-8
A sealed system was prepared, comprising two flasks with a stirring bar in each flask. AB (1.0 mmol, 30.87 mg), AuPd@ZIF-8 (17 mg, 1.25 mmol% per AB) and 2 mL CD3OD were introduced into the left flask. AuPd@ZIF-8 catalyst (15 mg, 2 mmol% per styrene) and styrene (0.5 mmol, 52 mg) in 2 ml CD3OD were introduced into the right flask. Air was removed in vacuo; then, the reaction mixture was stirred for 12 h at room temperature. The solution in the right flask was then centrifugated for analysis by 1H NMR and gas chromatography-mass spectroscopy (GC-MS).DFT calculations
The three nanocatalysts Au@ZIF-8, Pd@ZIF-8 and AuPd@ZIF-8 were simplified as ZIF-8 and 38-atom metal clusters. All the spin-polarized calculations with density functional theory (DFT) were conducted using the DMol3 package.35 The generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof form and Semicore Pseudopotential method (DSPP) with the double numerical basis sets in addition to the polarization functional (DNP) were included.36,37 The density of states, electron density, orbitals and population analysis in dye-free form were examined. The double numerical basis set DNP was utilized for predicting dye electronic properties. To simplify the calculation, the electrons of the structure are excited to consider light irradiation. Thus, the structure of the excited state was involved to simulate the effect of visible light. The energy of the photon at the light wavelength utilized for nanocatalysis was sufficient to arrive at the first Pd and Au excited states. The singlet option using the first excited state was selected. Furthermore, a DFT-D correction using the Grimme scheme was used to consider the interaction of the dispersion.7 The convergence of SCF for each electronic energy was fixed at 1.0 × 10−5 Ha, and the convergence criteria for the geometry optimization were fixed as follows: 1.0 × 10−5 Ha for energy, 0.001 Ha Å−1 for force, and 0.01 Å for displacement. The energy barriers were determined using the methods of linear and quadratic synchronous transit combined with conjugated gradient (CG) refinement. The free energies were G = Etotal + EZPE − TS, in which Etotal, EZPE and TS are the ground-state energy, zero-point energies, and entropy terms, respectively. The two latter terms use frequencies of vibration from DFT. The energies of reaction (G) of various intermediates were defined by ΔG = Gi − Greactant, where the energy of intermediates is Gi and the total energy of the reactants is Greactant.Results and discussion
Characterizations of the catalysts
The AuPd NPs stabilized by ZIF-8 were prepared based on the method of deposition/precipitation using the reducing agent NaBH4.38,39 In particular, these nanocatalysts were synthesized through the co-reduction of KAuCl4 and NaPdCl4 in water and methanol (volume ratio: 1:1) by NaBH4 under ambient conditions (Fig. 1a), then collected by centrifugation and dried. The loading of metal in the samples was investigated by inductively coupled plasma-optical emission spectroscopy (ICP-AES, Table S1†). Fig. 1b and c show that some AuPd NPs are encapsulated in ZIF-8, and Fig. S3† shows the record by transmission electron microscopy (TEM). The average size of the AuPd NPs is 2.43 nm. A distance of 0.22 nm was found in the (111) plane lattice of AuPd NPs.40 The X-ray diffraction of the powder (XRD, Fig. 1d) indicates a crystal pattern that is typical of ZIF-8 but without noticeable peaks corresponding to metallic Au or Pd from similar crystallinity of AuPd@ZIF-8 including ZIF-8 support.41 This is considered by low metal loading and location of some NPs inside ZIF-8. Fig. 1e shows AuPd@ZIF-8 exhibiting apparent isotherms of I N2 type with the uptake of N2 mostly found at a relatively low range of pressure (P/P0 < 0.03), showing that micropores are present inside the structures. Moreover, Fig. 1e shows the distribution of the corresponding size of pores estimated using the DFT non-local model.
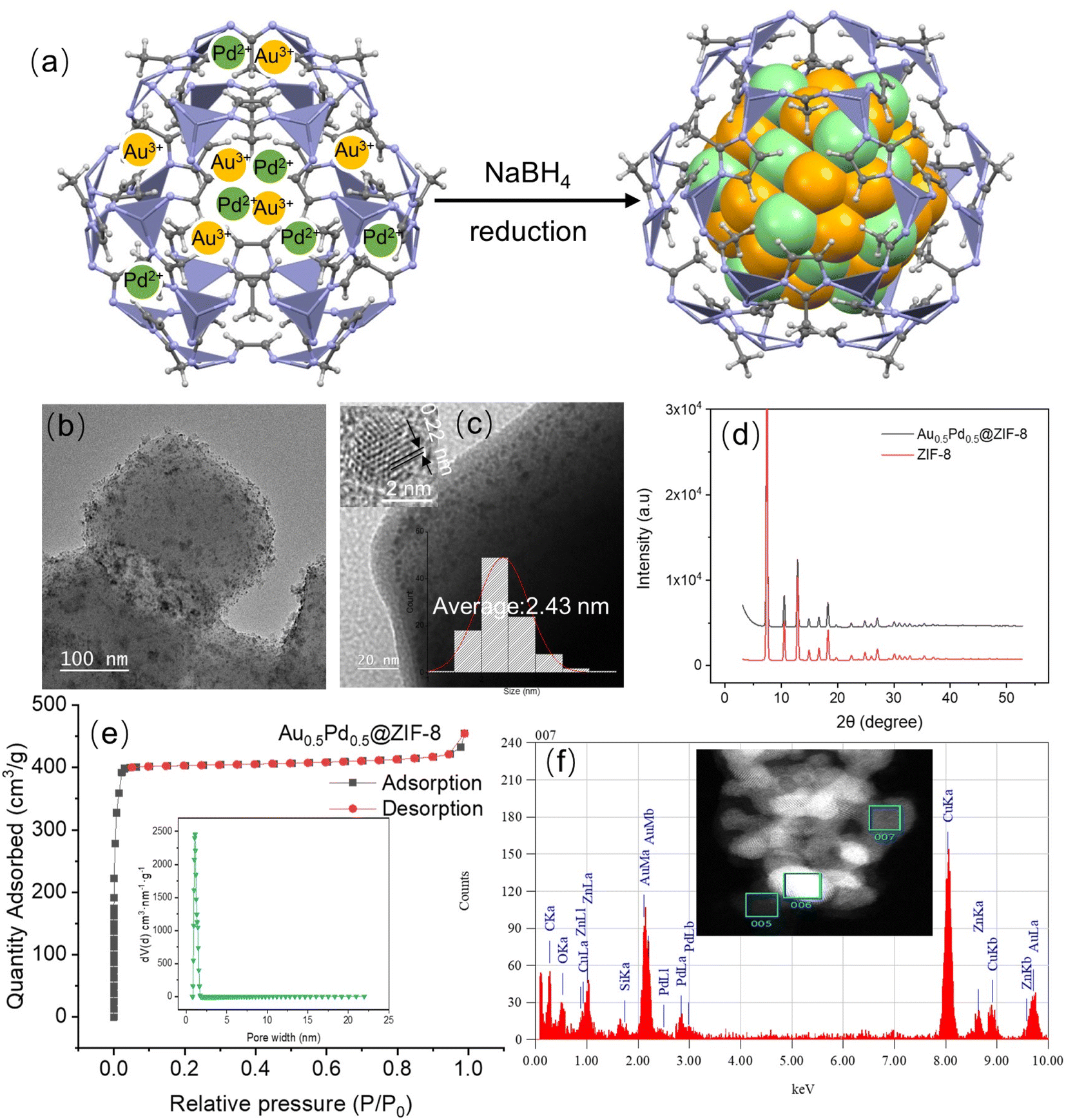 | ||
| Fig. 1 (a) Preparation of AuPd@ZIF-8; (b) image by TEM of AuPd@ZIF-8; (c) image by TEM of AuPd@ZIF-8 with size distribution and HRTEM image inserted; (d) PXRD patterns of ZIF-8 and AuPd@ZIF-8; (e) nitrogen adsorption–desorption isotherms of the alloyed NP on ZIF-8 with the insertion of the corresponding pore distribution of AuPd@ZIF-8; and (f) distributions of Au and Pd on the 007 particles with the insertion of the image by scanning transmission electronic microscopy (STEM). | ||
The size of the pore is 1.5 nm on average, i. e. significantly smaller than the size of the AuPd NP (2.43 nm). This observation is explained as follows. First, salts containing hydrophilic metal cations are introduced inside the hydrophilic ZIF-8 cavity, where they become coordinated by ZIF-8 N atoms. Subsequently, NaBH4 reduces them42 to AuPd NPs. Once these NPs are formed by atom aggregation, they cannot escape from the interior of the ZIF-8 cavity because they are larger than the pores. Consequently, the surface area, measured by applying Brunauer–Emmett–Teller (BET), becomes smaller than before the AuPd NP encapsulation, decreasing from 1787 m2 g−1 to 1269 m2 g−1 after their introduction (Fig. S4 and Table S2†), as reported previously upon related treatments.16,26 The high-angle annular dark field coupling with scan transmission electron microscopy (HAADF-STEM) images were utilized to show the nanostructure of such an alloy and the representative NP. As shown in Fig. 1f and S5,† they indicate that Au and Pd are both located in the alloyed NP, and their distribution along the cross-section of the AuPd NPs confirms the AuPd alloyed nanostructure.
X-ray Photoelectron Spectroscopy (XPS) analysis was applied to search the composition and electronic interaction in the prepared nanocatalysts. The XPS results show binding energies of 83.4 eV and 87.1 eV for Au (0) 4f7/2 and Au (0) 4f5/2, respectively, and binding energies of 334.1 and 339.6 eV corresponding to Pd 3d3/2 and 3d1/2, confirming that Pd(0) is present in the AuPd@ZIF-8 NPs43 (Fig. S6 and Table S3†). The signals representing the binding energies of Au 4f and Pd 3d demonstrate that Au(I) and Pd(II) ions are present, which is preceded by the oxidation of the AuPdNPs by air between the time of synthesis and that of measurement.26 By comparing bimetallic alloys and monometallic NPs, the electronic influence between Au and Pd is analyzed. Here, the binding energies of Au 4f (83.4 eV and 87.1 eV) of the AuPd alloy undergo a shift to values lower than those of AuNPs (84.0 eV and 87.9 eV) (Table S3†). In addition, the binding energies of Pd 3d (334.1 eV and 339.6 eV) of the AuPd alloy also shifted to lower values compared to those (334.9 eV and 340.2 eV) of the PdNPs (Table S3†). The direction of charge transfer is the same, indicating that the charge rearrangement between the two metals is not strong.44
Rate enhancement by visible light of the catalyzed AB methanolysis
The H2 gas obtained in the AB methanolysis reaction corresponds to about three equivalents of H2 per mol AB. As shown in Fig. 2a, in the dark, the efficiency of AB methanolysis catalyzed by Pd@ZIF-8 is higher than that catalyzed by Au@ZIF-8. The catalytic activity of Au0.5Pd0.5@ZIF-8 upon AB methanolysis is intermediate at the beginning of the reactions between those of the two monometallic catalysts, but the hydrogen production catalyzed by these three catalysts is nonquantitative. Here, a strongly positive effect of synergy in the AuPd alloy was obtained although AB methanolysis derived by AuPd alloy is slightly faster than that by Pd NPs after 15 min. There is no significant positive synergy when Au and Pd are alloyed in the dark following the fact that a strong electronic interaction between Pd and Au in the AuPd alloy is not found in the XPS results.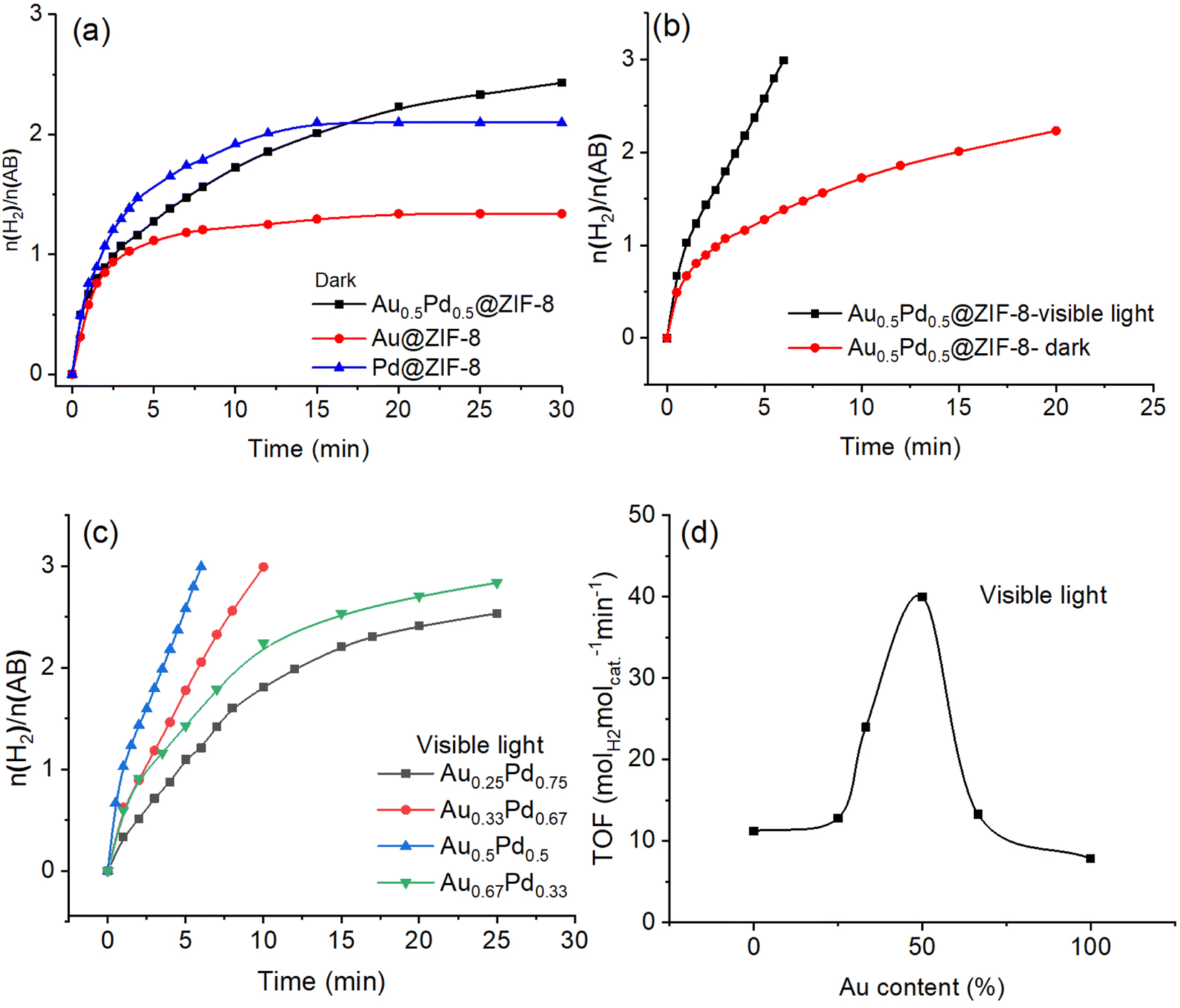 | ||
| Fig. 2 (a) Plot of the amount of mol H2 released during AB methanolysis vs. time catalyzed by AuNP@ZIF-8, PdNP@ZIF-8 and Au0.5Pd0.5NP@ZIF-8. (b) Compared efficiencies of the nanocatalysts among Au0.5Pd0.5@ZIF-8 without illumination by visible light. (c) Plot of the amount of mol H2 released upon methanolysis of AB vs. time catalyzed by Au0.5Pd0.5@ZIF-8 with different ratios of Au and Pd with illumination using visible light. (d) Values of TOFs with various ratios of Au/Pd upon illumination with visible light. In all experiments, the conditions are as follows: 1 mmol of AB; 1.25% mmol MNP; and temperature: 25 ± 0.5 °C. | ||
Remarkably, when the AB methanolysis reactions were moved from dark to irradiation with visible light, the efficiency of AB methanolysis catalyzed by Au0.5Pd0.5@ZIF-8 was considerably enhanced, and the hydrogen generation was complete (Fig. 2).
Under visible light, the TOF value increased by more than 3.7 folds compared to the reaction conducted in the dark, which implies that visible light played a major role in AB methanolysis catalyzed by Au0.5Pd0.5@ZIF-8. Moreover, given the crucial influence of visible light exerted on Au0.5Pd0.5@ZIF-8 catalysis, the light effect was also evaluated for comparison with the Au@ZIF-8 and Pd@ZIF-8 catalysts containing a single metal NP in the ZIF (Fig. S7 and S8†). Upon irradiation using visible light, the rates of AB methanolysis catalyzed by Au@ZIF-8 and Pd@ZIF-8 were somewhat enhanced compared to that in the dark, but these reactions catalyzed by either Au@ZIF-8 or Pd@ZIF-8 were incomplete. Although visible-light irradiation of AuNPs involves a strong plasmon, hot plasmonic electron transfer generated on the surface of AuNPs upon illumination is inefficient because AuNPs are not effective catalysts for AB methanolysis. Consequently, only a very modest enhancement of AB methanolysis efficiency for Au@ZIF-8 was observed with light. For Pd@ZIF-8, no improvement in catalytic H2 evolution activity was noted upon visible-light illumination compared to the reaction in the dark. This is because the plasmon resonance of Pd cannot be excited under visible light at 550 nm (Fig. S9†).
Because the AuPd alloyed nanocatalysts showed excellent performance for AB methanolysis upon illumination of visible light, it was necessary to optimize the proportion of Au and Pd in the alloy (Fig. 2c). Under visible-light irradiation, the best ratio of Au to Pd is 1 to 1, with a high TOF value of 40 molH2 molcat.−1·min−1 (86.7 molH2 molatom−1 min−1). As the Au content increases from Pd NP to AuPd alloy, the reaction rate of AB methanolysis increases until the proportion of 50% Au is reached in the AuPd alloy. Then, the catalytic performance continually decreases upon raising the Au proportion above 50%. Because we know that Pd(0) at the NP surface is, contrary to Au(0), an excellent metal for the oxidative addition of the CH3O–H bond leading to the product and that Au is the surface metal yielding plasmonic hot electron transfer towards Pd to boost the reaction, it is of interest to experimentally evaluate the relative importance of these two effects. The finding that the AuPd catalyst is optimized for equivalent amounts of these two metals in the alloy shows that the importance of these two metals in these two effects is of the same order.
Methanolytic AB dehydrogenation catalyzed by AuPd on various heterogeneous supports
The loading amount of the nanocatalyst AuPd in @ZIF-8 was varied, and the different effects were investigated to improve the catalyst. Thus, AuPd NPs with a series of amounts of ZIF-8 (2.6 wt%, 4.2 wt%, 5.7 wt% and 7.1%) were prepared, and the catalytic efficiencies upon AB methanolysis were evaluated. The best catalytic performance was obtained when the MNP load was 5.7 wt% (Fig. S10†). The catalysts stabilized by other supports (ZIF-67, UIO-66, GO) were synthesized and evaluated in the AB methanolysis reaction using the same method with 5.7% loading of MNPs for Au0.5Pd0.5@ZIF-8, and the catalytic performances were compared. The catalytic activities measured by the TOF values for AuPd NPs supported by ZIF-8, ZIF-67, UIO-66, and GO are compared, showing that ZIF-8 is by far the best support obtained with AuPd under dark or visible light irradiation conditions (Fig. 3).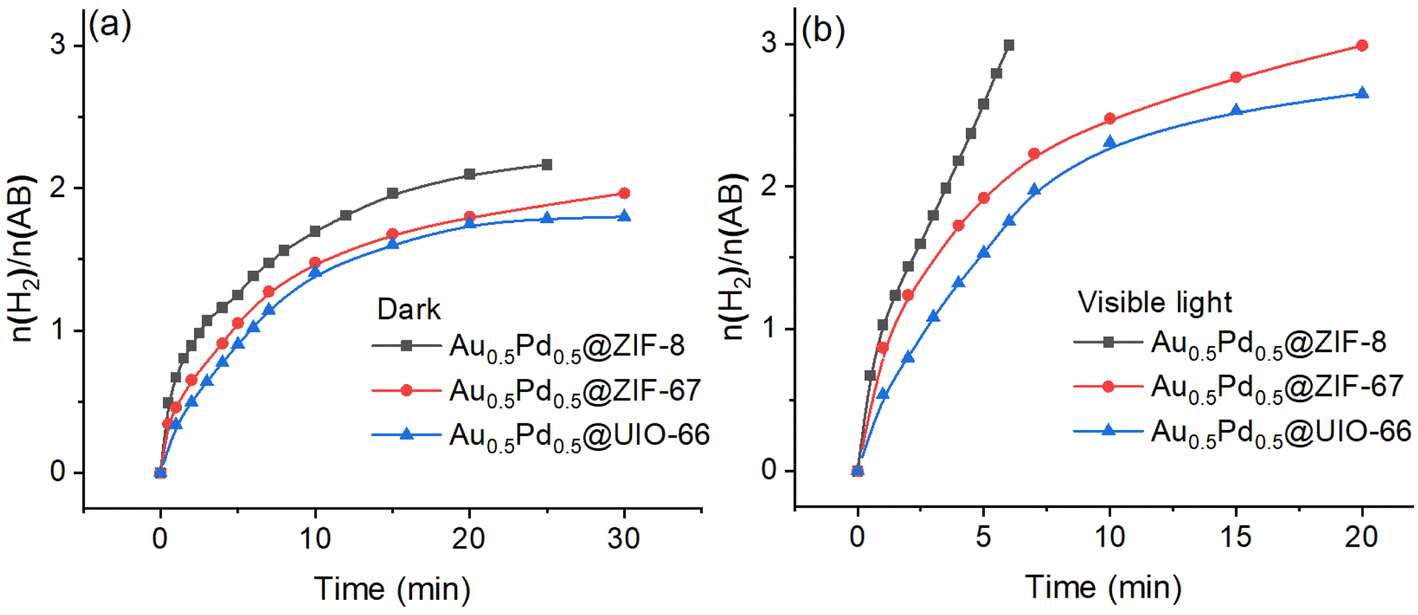 | ||
| Fig. 3 AB methanolysis catalyzed by Au0.5Pd0.5 on different supports: (a) in the absence of light and (b) in the presence of visible light. Reaction conditions: 1 mmol AB; 1.25% mmol MNP; temperature: 25 ± 0.5 °C. | ||
AuPd NPs tend to quickly aggregate when they are supported by GO, which results in incomplete methanolysis, contrasting with catalysis using the other supports. Therefore, the characterization of Au0.5Pd0.5@GO was not considered. The specific support performances without light irradiation exhibit the following order: Au0.5Pd0.5@GO > Au0.5Pd0.5@ZIF-8 > Au0.5Pd0.5@ZIF-67 > Au0.5Pd0.5@UIO-66. The differences in performances find their origin in the various particle sizes and BET surfaces. The sizes of the nanoalloys between the four catalysts exhibit the following order: Au0.5Pd0.5@ZIF-8 < Au0.5Pd0.5@ZIF-67 = Au0.5Pd0.5@UIO-66 (Fig. S11 and S12†). Moreover, the BET surfaces of the three other catalysts exhibit the following order: Au0.5Pd0.5@ZIF-67 > Au0.5Pd0.5@ZIF-8 > Au0.5Pd0.5@UIO-66 (Fig. S13 and Table S4†). To further confirm the excellence of the catalysts with a metal organic framework (MOF) as support, GO was investigated for AB methanolysis (Fig. S14†). Under dark conditions, the MOF-embedded AuPd NP nanocatalysts do not perform better than the GO-supported AuPd nanocatalyst in the initial reaction. This is because when an NP is encapsulated inside a MOF, as confirmed by BET, its surface is protected. Under visible-light illumination, the performances are significantly enhanced for the three MOF-supported nanocatalysts Au0.5Pd0.5@ZIF-8, Au0.5Pd0.5@ZIF-67 and Au0.5Pd0.5@UIO-66 (Fig. 3b), and this reactivity order remains the same for the reactions in the dark. Their rate multiplication factors are 3.7, 2.9 and 2.1, respectively.
Concerning the MOF-supported AuPd NPs (Au0.5Pd0.5@ZIF-8, Au0.5Pd0.5@ZIF-67, and Au0.5Pd0.5@UIO-66), plasmonic bands were observed (Fig. S15†), correspondingly showing significant light-induced enhancement upon photocatalysis. This indicates that ZIF-8 involves high selectivity for AuPd NPs. Therefore, there is an impressive light effect on the Au0.5Pd0.5@ZIF-8 nanocatalyst for AB methanolysis, with ZIF-8 serving as the optimized support for the catalytic activation of the substrates AB and methanol.
Among the four supported reactions, only the reaction catalyzed by Au0.5Pd0.5@GO maintains a similar rate under dark and visible-light irradiation. Moreover, the reaction with this nanocatalyst, Au0.5Pd0.5@GO, generates insufficient hydrogen. This is because GO does not protect the NPs well, as these NPs quickly aggregate at the beginning of the reaction.
The selectively unique ZIF-8 effect on the catalytic performance of the AuPd alloy is best considered by geometrical constraint to the methanol substrate between the ZIF-8 capsule inner surface and the outer Pd surface atoms of the alloy. Indeed, a basic imidazolate N atom of the ZIF activates the acidic methanol H atom, facilitating further CH3O–H bond oxidative addition cleavage at the Pd surface atom in the RDS of the reaction.
Concerning plasmonic metal-based catalysts, the critical feature is that their performance is wavelength dependent because the wavelength of light determines the incoming photon energy. Therefore, to check the effects produced by the plasmon-enhanced catalysis, the catalytic efficiency of AB methanolysis was evaluated upon light irradiation at wavelengths of 450 nm, 520 nm and 620 nm (Fig. S16†). It was found that irradiation at 550 nm was the most effective of the three visible light wavelengths. It is known that, compared to the light at 550 nm, the 450 nm light possesses more photons, but the performance of AB methanolysis at 550 nm is better than that at 450 nm. This shows that illumination with an increased number of photons does not lead to better efficiency because only light wavelengths in the range of the surface plasmon band of Au0.5Pd0.5@ZIF-8 leads to enhancement of catalytic efficiency. As shown in Fig. S9,† the plasmon band of Au0.5Pd0.5@ZIF-8 exhibits a maximum at 560 nm.
Photoinduced rate enhancement of Au0.5Pd0.5@ZIF-8-catalyzed AB methanolysis
It has been reported that enhancement introduced by photons comprises photo-thermalization, local photo field and hot-electron transfer.45 The photoinduced enhancement of AB methanolysis efficiency was analyzed from these three aspects. First, low-energy electrons coupled to phonon modes after thermalization increase the NP temperature, and this effect then dissipates into the medium.46 Thus, to precisely show the effect of heating the reactor itself subsequently to continuous illumination, a temperature probe was located inside the vessel containing the reaction mixture to observe the change in temperature during AB methanolysis catalyzed by Au0.5Pd0.5@ZIF-8 (25 °C, 7 mW cm−1). Because the maximum reaction time is approximately 6 min, the contribution of heating by visible light should be considered. The flask was covered with tin paper to avoid direct visible-light irradiation. Consequently, the variation in temperature during the reaction was approximately 1.2 °C, and the reaction rates under dark or heating by visible light showed only a small difference (Fig. S17†). Thus, the contribution from photo-thermalization is not mainly responsible for light enhancement. This result is related to previous reports.47,48The plasmon-induced electric field is dominated by localized surface electronic states. The plasmon field at the NP surface favors the provision of energy for chemical bond activation.49 Furthermore, the local fields decay nearly exponentially away from the surface of the NP and go along with the formation of gradient forces.50 Stronger electric field-gradient forces are essential to trap reactant molecules and polarize capture targets.50 Finite difference time domain (FDTD) simulations were used to perform the calculation of the local field associated with the SPR (Fig. S18†). Alloying Au with Pd improves the distribution in the space of the electric field strength in NPs. The addition of Au to the Pd NPs increases |E/E0|2 from 6.4 to 13.1. Notably, the NP dimer exhibits a near-field distribution that is stronger than that of individual NPs, with |E/E0|2 reaching 49.7. The enhanced field provokes increased generation of hot electrons and adsorption of the molecular substrate to Au0.5Pd0.5 alloy NPs, leading to stronger adsorption of the metal-adsorbent complex.51 Simultaneously, favorable reactant adsorption onto the metal NPs enhances catalytic activity. Lu et al. observed that the local electric field formed by Ag NPs enhanced adsorption and subsequent photodehydrogenation of ammonia borane.52 Similarly, here, the increased electric field of the AuPd alloy NPs enhanced the adsorption of methanol (MeOH) and AB forming metal-AB-MeOH complexes. This results in a more efficient hot electron transfer, further favoring catalytic activity.
The structural modification of the nanocatalyst during the reaction accelerated by light was characterized by TEM and XPS. The nanoparticle size increases from 2.43 nm before reaction to 2.77 nm after reaction (Fig. S19†), indicating the excellent property of ZIF-8 as support. Thus, the recycling of the Au0.5Pd0.5@ZIF-8 nanocatalyst was tested. The Au0.5Pd0.5@ZIF-8 catalyst remained highly catalytically active upon AB methanolysis during three cycles (Fig. S20†). The XPS data indicate oxidation states and shifts in binding energies. After AB methanolysis, XPS shows that the binding energies are 83.3 and 86.9 eV, which corresponds to Au (0) 4f. The binding energies 334.2 and 339.7 eV demonstrate that Pd remains in the 0-valent state (Fig. 4). Comparing the Au 4f and Pd 3d regions before and after the reaction, binding energy shifts were noticed. The binding energies of Au 4f are 83.4 and 87.1 eV before the reaction, and they shift to lower values (83.3 and 86.9 eV, respectively) (Fig. 4a and c), indicating that there is electronic stimulation on the Au surface. Moreover, the binding energy of Pd 3d (339.6 and 334.1 eV) shifts to higher values (339.7 and 334.2 eV) after the reaction (Fig. 4b and d), demonstrating that the Pd surface became more positively charged after the photoreaction.
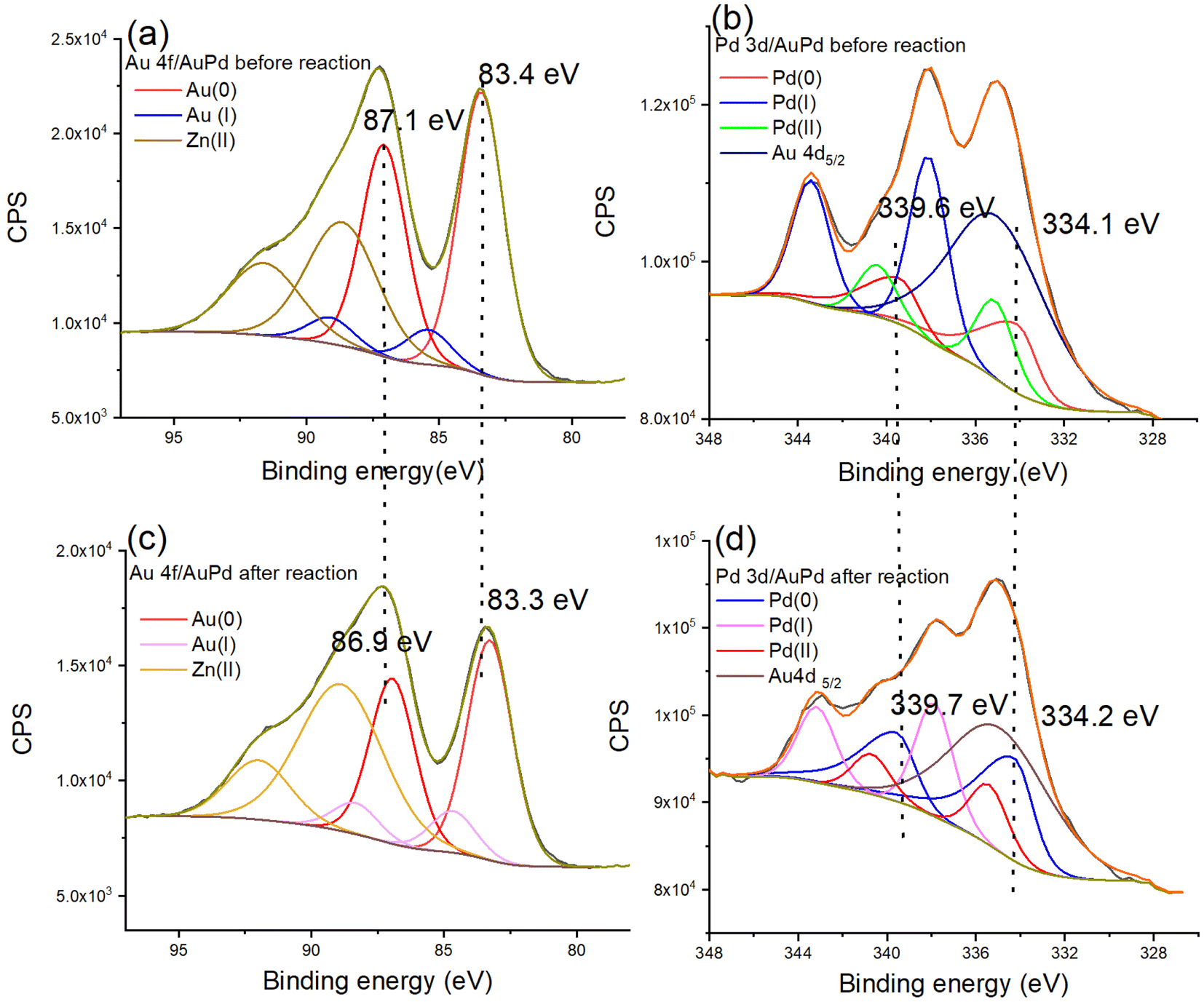 | ||
| Fig. 4 XPS spectra for AuPd@ZIF-8, (a) region of Au 4f for AuPd@ZIF-8 before the light-induced reaction, (b) region of Pd 3d for AuPd@ZIF-8 before the light-induced reaction, (c) region of Au 4f of AuPd@ZIF-8 after the light-induced reaction, and (d) region of Pd 3d for AuPd@ZIF-8 after the light-induced reaction. | ||
During the photo-acceleration reaction, there is electron density transfer from Pd to Au, and Au atoms are consequently electronically enriched. This suggests that, although the time scale of photon-induced electron transfer is of only the order of femtoseconds, some electron density transfer is irreversible following the photo-reaction that involves restructuration to some extent.25 Usually, electron density transfer between NPs and support should be considered at the heterogeneous catalyst interface upon reaction involving illumination with visible light. However, ZIF-8 functions as an insulator, and it has been proven that it neither transfers nor separates electrons induced by the Au NP plasmon. Thus, ZIF-8 is not excited by the light because its LUMO is located at an energy level that is too high.26,53,54
Mechanism of H2 formation upon nanocatalyzed AB methanolysis
In the reaction mechanism of H2 formation upon AB methanolysis, it has been reported that one of the H atoms of generated H2 was provided by AB while the other was by methanol.48,55,56 The kinetic isotope effect (KIE: kH/kD) determination conducted with d4 methanol is an efficient method of identifying the reaction rate-determining step (RDS). In the present reaction, the KIE value is 2.2 under dark conditions and 3.4 under irradiation with visible light (Fig. S21†). These values clearly show that methanol O–H bond cleavage is the rate-determining reaction step in both cases. The higher KIE value under light compared to that in the dark may be due to the additional necessary adsorption rearrangement of methanol in the course of plasmonic electron transfer.57The tandem reaction was conducted to demonstrate that one H atom of H2 is provided by methanol in the reaction of AB methanolysis while the other one is by AB. Typically, AB methanolysis catalyzed by Au0.5Pd0.5@ZIF-8 was carried out in a sealed reactor comprising two connected flasks for styrene hydrogenation with the same catalyst in both flasks (Fig. S22†). The hydrogen gas produced from AB methanolysis catalyzed by Au0.5Pd0.5@ZIF-8 with CD3OD in the left flask was transferred into the right flask in which styrene hydrogenation proceeds in CD3OD, and the deuteration of this hydrogen gas was investigated. Coupled gas chromatography-mass spectrometry and 1H NMR spectroscopy were utilized to characterize the product of this hydrogenation reaction without any treatment. The nanocatalyzed styrene hydrogenation that generated quantitative hydrogen after 12 h at 25 °C produced ethylbenzene that was analyzed using its ethyl signals in the 1H NMR spectrum. The number of hydrogen atoms found for this ethyl group is 4 out of the 5 hydrogen atoms of this group. We conclude that the missing 5th hydrogen atom has been substituted by a deuterium atom (Fig. S23†). Furthermore, the molecular peak in the mass spectrum at 106, 107 (major), and 108 m/z corresponding to CH8H10, C8H9D, and C8H8D2, respectively, statistically shows that one deuterium atom was transferred to styrene, forming mono-deuterated ethylbenzene (Fig. S24 and S25†). These data show that one of the H atoms of H2 is provided by AB and the other hydrogen from methanol. In addition, this experiment demonstrates that Au0.5Pd0.5@ZIF-8 is an excellent catalyst for AB methanolysis.
To explain the difference in catalytic activity among Au@ZIF-8, Pd@ZIF-8 and AuPd@ZIF-8, the d-orbital density of states (DOS) of different NPs was studied to evaluate the d-band center position. The d-band centers of Au (111) and Pd (111) show −3.2 and −2.4 eV, respectively (Fig. 5a). Upon Au and Pd alloying, the d-band center of AuPd (111) is −1.5 eV, which is at a higher energy than that of both Au (111) and Pd (111). The upgrading of the d-band center energy level of AuPd indicates that the electron in the d-orbital of the metal is closer to the energy level of the antibonding orbital of the adsorbate species. The closing tendency between the d-orbital and antibonding orbital induces easier adsorption of reactant molecules on the catalyst surface, which signifies that the energy of adsorption between AuPd and AB is higher than that with either Pd or Au.58,59
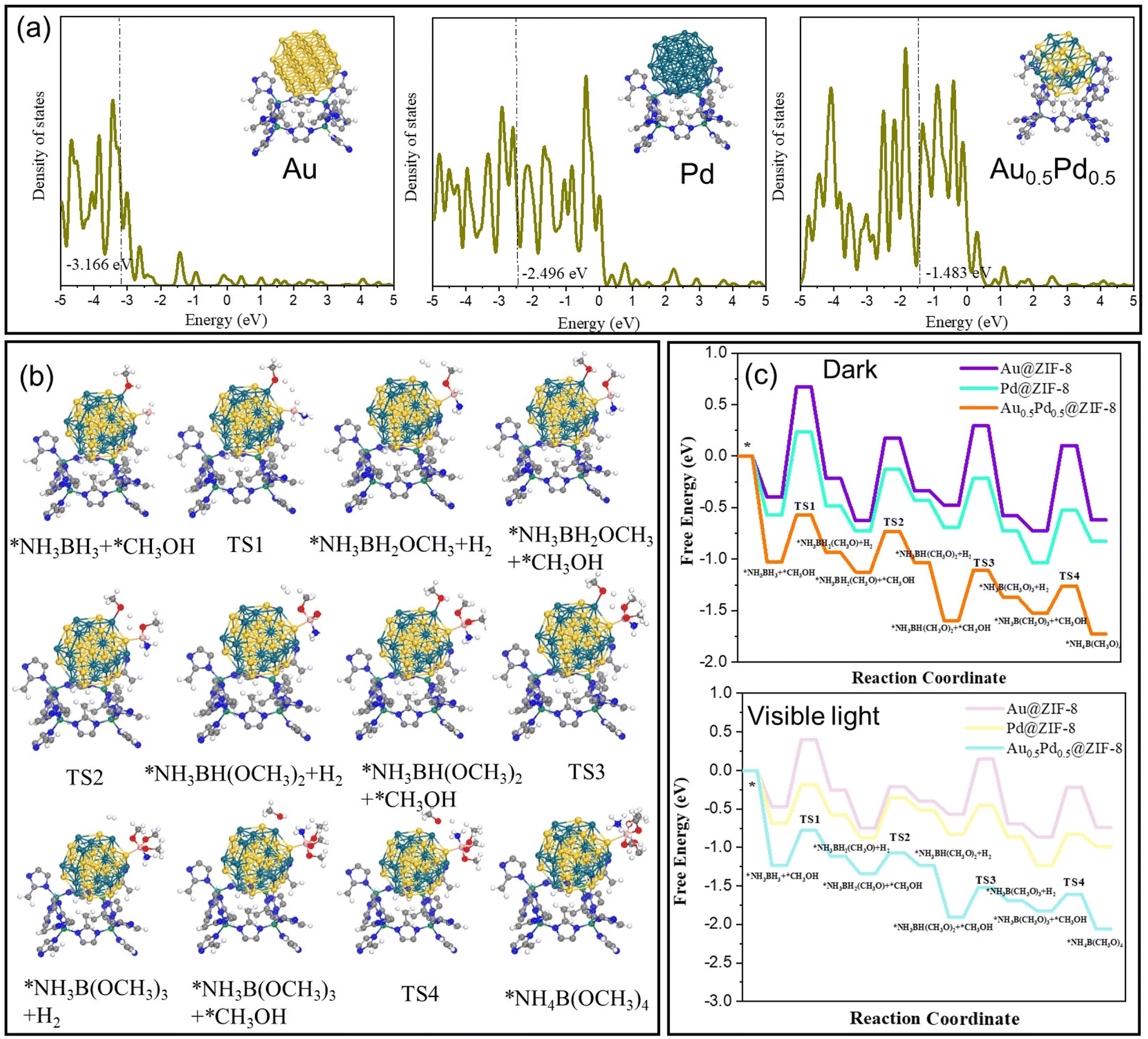 | ||
| Fig. 5 DFT simulation data of NH3BH3 and MeOH in the course of AB methanolysis catalyzed by Au@ZIF-8, Pd@ZIF-8, and Au0.5Pd0.5@ZIF-8. (a) The d-band center of Au (111), Pd (111) and AuPd (111) in catalysts. (b) Path of NH3BH3 and MeOH on Au@ZIF-8, Pd@ZIF-8, and Au0.5Pd0.5@ZIF-8. (c) Gibbs free-energy diagrams of adsorption, activation, and dissociation for NH3BH3 and MeOH on Au@ZIF-8, Pd@ZIF-8, and Au0.5Pd0.5@ZIF-8, respectively, under dark conditions (top) and visible-light illumination (bottom). | ||
DFT calculations were carried out to confirm the mechanism of H2 formation upon nanocatalyzed AB methanolysis. The real model optimization based on the nanocatalysts is delicate, but it was most representative to form the calculation model based on AB methanolysis occurring at the contact between Au0.5Pd0.5 and ZIF-8. As shown in the DFT results, the formation of the NP-coordinated O-CH3 ligand demands the highest energy barrier of reaction in the dark and under visible-light conditions for the 3 nanocatalysts Au@ZIF-8, Pd@ZIF-8 and Au0.5Pd0.5@ZIF-8. The data obtained (Fig. 5) confirm the validity of the catalytic mechanism.
The energy barriers for AB methanolysis under dark conditions on the surfaces of Au@ZIF-8, Pd@ZIF-8 and Au0.5Pd0.5@ZIF-8 show the following respective tendency: 1.07 eV > 0.81 eV > 0.46 eV (Fig. 5c). The above results confirm that the cleavage of the H-OCH3 bond is the only rate-determining step of the AB methanolysis reaction with this alloyed nanocatalyst (TS1 > TS2). The optimized catalyst also provided the active site for the increase in the reaction rate via a reduction in the energy barrier for the rate-limiting step under conditions of illumination with light. Under these conditions, the charge excitation/transfer at the interface of plasmonic NPs and adsorbed substrates proceeded, bringing CH3OH into vibrationally or electronically excited states and subsequently increasing substrate reactivity. A hot electron from the plasmon is injected into the excited state of the Pd-adsorbate entity, enhancing the overall electron density of the states of the Pd-adsorbate sites and provoking faster cleavage of the methanol H⋯O bond by oxidative addition to Pd in PdH⋯H⋯OCH3.
The energy barriers for AB methanolysis under light conditions on the surfaces of Au@ZIF-8, Pd@ZIF-8 and Au0.5Pd0.5@ZIF-8 convey a tendency of 0.88 eV > 0.51 eV > 0.45 eV, respectively (Fig. 5c). Comparison of the dark and visible-light conditions from the viewpoint of the Gibbs free energy yielded the following result: TS1light < TS1dark.
In sum, a proposed mechanism for Au0.5Pd0.5@ZIF-8-catalyzed AB methanolysis accelerated by visible light is shown in Scheme 1.
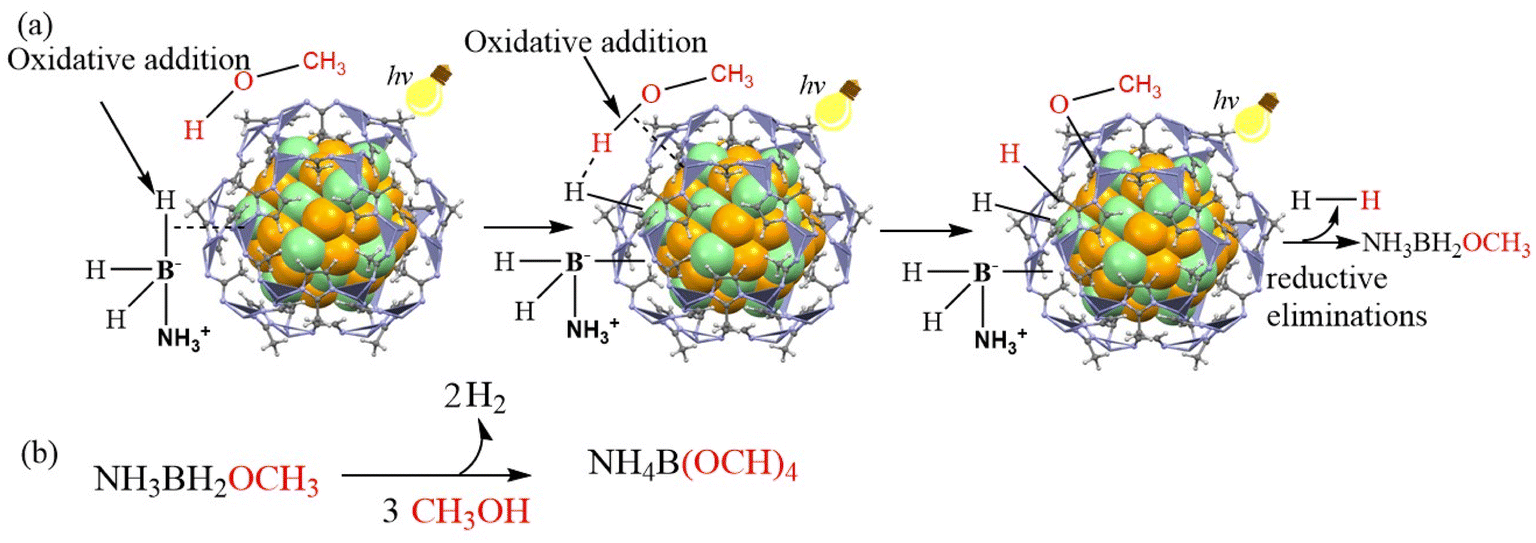 | ||
| Scheme 1 Proposed mechanism for the AB methanolysis generating H2 catalyzed by Au0.5Pd0.5@ZIF-8 and accelerated by illumination with visible light. | ||
The injection of hot electrons into the Pd-adsorbate ensemble comprising AB and methanol and the photoinduced enhancement of the local field contributes to the enhanced catalysis by AuPd@ZIF-8 under illumination with visible light. AB and CH3OH are absorbed on the NP surface to form an NP/substrate ensemble. The transfer onto the unoccupied state of the metal–CH3OH–AB complex of a hot electron induced by the plasmon excitation by light and a hydrogen bond [H3NBH2H]⋯H–OCH3 formed between the hydridic B–H bond of AB and the acidic H atom of methanol constitutes the reaction acceleration driving forces. This activation results in the formation of metal-BH2NH3, metal-OCH3, and two metal-hydride intermediates. Reductive elimination of these metal–ligand species subsequently releases H2 and H3NBH2(OCH3). The latter compound undergoes two additional similar processes to finally produce three equivalents of H2 and NH3B(OCH3)3, whose methanolysis produces NH4B(OCH3)4 and satisfies the stoichiometry of eqn (1).
A comparison of the nanocatalyzed dehydrogenative hydrolysis and methanolysis of AB shows that methanolysis is considerably more boosted by visible light illumination compared to the analogous reaction in the dark (by a factor of 3.7) than hydrolysis (by a factor of 1.4). For instance, the KIE increased from 2.2 in the dark to 3.4 under visible light for AB methanolysis and only from 4 to 4.3 upon AB hydrolysis, emphasizing the much more dramatic positive influence of visible light on AB methanolysis than on AB hydrolysis. This can be understood by looking at the hydrogen bonding between the hydridic Pd–H after the initial hydride transfer from AB and the acidic O–H bond of methanol. This hydrogen bond is stronger in the case of AB hydrolysis than in the case of AB methanolysis because water is more acidic than methanol. This hydrogen bonding weakens the O–H bond and then becomes easier to break by oxidative addition onto the metal surface. Because hydrogen bonding is weaker with methanol than with water, O–H cleavage becomes more difficult with methanol than with water. Thus, with methanol, more activation by visible light is necessary to overcome the rate-limiting O–H oxidative addition step than with water.
Conclusion
Although Au@ZIF-8 does not catalyze AB methanolytic dehydrogenation, alloying Au with various metals allows partial H2 evolution using this reaction in the dark, best conducted with AuPt and AuPd alloys. Upon light irradiation, neither Au@ZIF-8 nor Pd@ZIF-8-catalyzed reaction is significantly improved compared to reactions in the dark, but the rate of AuPd@ZIF-8-catalyzed H2 evolution is considerably boosted upon visible-light illumination, the best-alloyed nanocatalyst being, after optimization, Au0.5Pd0.5@ZIF-8. These equal amounts of Au and Pd in the most effective alloy are considered because the Au plasmonic hot electron transfer and the Pd-induced CH3O–H oxidative addition appear to have equivalent importance in the light-boosted reaction. Comparison between various supports including ZIF-8, ZIF-67, UIO-66, and GO shows rate superiority but the instability of GO (incomplete H2 evolution due to aggregation), whereas ZIF-8 largely overtakes the other supports under visible light with complete evolution of 3 mol H2. This can be rationalized by the geometric constraint inside the ZIF-8 cavity optimizing imidazole activation of the methanol acidity towards CH3O–H bond cleavage in the RDS of the reaction. By varying the proportions of Au and Pd, Au0.5Pd0.5 alloy was found to exhibit maximum rate enhancement (more than 3.7 folds) and excellent catalytic activity involving a reaction rate of 500 μmol min−1 at 22 ± 1 °C. Excitation of the nanogold plasmon upon irradiation with visible light inducing transfer of a hot electron from Au to the Pd-AB-methanol adsorbate along with the hydrogen bond [H3NBH2H]⋯H–OCH3 provokes the oxidative addition of the O–H bond in methanol, the rate-limiting reaction step. This mechanism is ascertained by isotope labeling in a tandem hydrogenation reaction, kinetic isotope effect and DFT calculations. The dramatic reaction rate enhancement provoked by visible-light irradiation due to the plasmonic gold alloy excitation is taken into account by the difficulty of methanol O–H bond activation in the RDS. Upon AB methanolysis, this O–H bond activation by oxidative addition onto the nanocatalyst surface requires a larger driving force than the related water O–H bond activation upon AB hydrolysis. However, the cleanness of the hydrogen produced in this system and the recyclability of the product NH4B(OCH3)4 offer a good choice for the potential design of low-temperature devices.Author contributions
Naixing Kang: investigation, verification, formal analysis, visualization, writing original draft, writing review, formal analysis. Ruofan Shen: investigation (DFT calculations), formal analysis, software. Baojun Li: formal analysis (DFT calculations), software, validation, resources. Fangyu Fu: investigation. Bruno Espuche: investigation, formal analysis, resources. Sergio Moya: investigation, formal analysis, resources, validation. Lionel Salmon: investigation, formal analysis, validation. Jean-Luc Pozzo: supervision, writing review and editing. Didier Astruc: formal analysis, resources, supervision, conceptualization, project administration, writing review and editing.Conflicts of interest
The authors declare that they have no known competing financial interests.Acknowledgements
Helpful comments from Dr Desire di Silvio, (CIC biomaGUNE, San Sebastian) and Dr Tong Wu, (Institute d’Optique, Bordeaux) and financial support from the China Scholarship Council (CSC, PhD grant to Naixin Kang), the MAT2017-88752-R Retos Project from the Ministerio de Economía, Industria y Competitividad, the Gobierno de España (Dr Sergio Moya), the National Natural Science Foundation of China (no. 22279118, Prof. Baojun Li), the University of Bordeaux and the Centre National de la Recherche Scientifique (CNRS) are gratefully acknowledged.References
- T. He, P. Pachfule, H. Wu, Q. Xu and P. Chen, Nat. Rev. Mater., 2016, 1, 16059 CrossRef CAS.
- M. Yue, H. Lambert, E. Pahon, R. Roche, S. Jemei and D. Hissel, Renewable Sustainable Energy Rev., 2021, 146, 111180 CrossRef.
- M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698–9725 RSC.
- Z. Huang and T. Autrey, Energy Environ. Sci., 2012, 5, 9257–9268 RSC.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2021, 50, 3437–3484 RSC.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- C. Wang, Q. Wang, F. Fu and D. Astruc, Acc. Chem. Res., 2020, 53, 2483–2493 CrossRef CAS PubMed.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- D. Astruc, Chem. Rev., 2020, 120, 461–463 CrossRef CAS PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Q. Sun, N. Wang, Q. Xu and J. Yu, Adv. Mater., 2020, 32, 2001818 CrossRef CAS PubMed.
- M. Rakap, Appl. Catal., B, 2015, 163, 129–134 CrossRef CAS.
- Y.-H. Zhou, X. Cao, J. Ning, C. Ji, Y. Cheng and J. Gu, Int. J. Hydrogen Energy, 2020, 45, 31440–31451 CrossRef CAS.
- X. Zhao, H. Hu, G. Li, J. Cai, Y. Wang and G. Fan, J. Alloys Compd., 2022, 912, DOI:10.1016/j.jallcom.2022.165076.
- C. Wang, X. Liu, C. Wu and D. Astruc, J. Mater. Chem. A., 2022, 10, 17614–17623 RSC.
- F. Fu, C. Wang, Q. Wang, A. M. Martinez-Villacorta, A. Escobar, H. Chong, X. Wang, S. Moya, L. Salmon, E. Fouquet, J. Ruiz and D. Astruc, J. Am. Chem. Soc., 2018, 140, 10034–10042 CrossRef CAS PubMed.
- Q. Wang, F. Fu, S. Yang, M. Martinez Moro, M. de los A. Ramirez, S. Moya, L. Salmon, J. Ruiz and D. Astruc, ACS Catal., 2019, 9, 1110–1119 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810–7817 CrossRef CAS PubMed.
- S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870–5874 RSC.
- S. B. Kalidindi, A. A. Vernekar and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2009, 11, 770–775 RSC.
- W.-W. Zhan, Q.-L. Zhu and Q. Xu, ACS Catal., 2016, 6, 6892–6905 CrossRef CAS.
- H. Li, Z. Yao, X. Wang, Y. Zhu and Y. Chen, Energy Fuels, 2022, 36, 11745–11759 CrossRef CAS.
- N. Tunc and M. Rakap, Renewable Energy, 2020, 155, 1222–1230 CrossRef CAS.
- M. Yurderi, A. Bulut, İ. E. Ertas, M. Zahmakiran and M. Kaya, Appl. Catal., B, 2015, 165, 169–175 CrossRef CAS.
- N. Kang, Q. Wang, R. Djeda, W. Wang, F. Fu, M. M. Moro, M. de los A. Ramirez, S. Moya, E. Coy, L. Salmon, J.-L. Pozzo and D. Astruc, ACS Appl. Mater. Interfaces, 2020, 12, 53816–53826 CrossRef CAS PubMed.
- N. Kang, X. Wei, R. Shen, B. Li, E. G. Cal, S. Moya, L. Salmon, C. Wang, E. Coy, M. Berlande, J.-L. Pozzo and D. Astruc, Appl. Catal., B, 2022, 310, 121300 CrossRef.
- S. Zhang, M. Li, L. Li, F. Dushimimana, J. Zhao, S. Wang, J. Han, X. Zhu, X. Liu, Q. Ge and H. Wang, ACS Catal., 2020, 10, 14903–14915 CrossRef CAS.
- Z. Hu, D. Meng, F. Lin, X. Zhu, Z. Fang and X. Wu, Adv. Opt. Mater., 2019, 7, 1801590 CrossRef.
- Q. Zhang, Y. Zhang, K. Xiao, Z. Meng, W. Tong, H. Huang and Q. An, Chem. Eng. J., 2019, 358, 389–397 CrossRef CAS.
- J. R. McLaughlan, D. M. J. Cowell and S. Freear, Phys. Med. Biol., 2018, 63, 015004 CrossRef PubMed.
- Y. Liu, Z. Zhang, Y. Fang, B. Liu, J. Huang, F. Miao, Y. Bao and B. Dong, Appl. Catal., B, 2019, 252, 164–173 CrossRef CAS.
- S. Jo, P. Verma, Y. Kuwahara, K. Mori, W. Choi and H. Yamashita, J. Mater. Chem. A., 2017, 5, 21883–21892 RSC.
- P. Verma, Y. Kuwahara, K. Mori and H. Yamashita, Bull. Chem. Soc. Jpn., 2019, 92, 19–29 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol 3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- Q. Wang and D. Astruc, Chem. Rev., 2020, 120, 1438–1511 CrossRef CAS PubMed.
- P. Michaud, D. Astruc and J. H. Ammeter, J. Am. Chem. Soc., 1982, 104, 3755–3757 CrossRef CAS.
- L. Wang, Y. Zhou, J. Timoshenko, S. Liu, Q. Qiao, K. Kisslinger, M. Cuiffo, Y.-C. Chuang, X. Zuo, Y. Xue, Y. Guo, C. Pan, H. Li, C.-Y. Nam, S. Bliznakov, P. Liu, A. I. Frenkel, Y. Zhu and M. H. Rafailovich, ACS Catal., 2019, 9, 1446–1456 CrossRef CAS.
- G. Xu, A. Yu, Y. Xu and C. Sun, Catal. Commun., 2021, 158, 106338 CrossRef CAS.
- D. Astruc, Acc. Chem. Res., 1991, 24, 36–42 CrossRef CAS.
- L. Zhang, Z. Xie and J. Gong, Chem. Soc. Rev., 2016, 45, 3916–3934 RSC.
- S. K. Das, B. Mohanty, S. C. Sahu, T. J. Sruthi, B. Chakraborty, S. Basu and B. K. Jena, Electrochim. Acta, 2020, 356, 136820 CrossRef CAS.
- H. Kang, J. T. Buchman, R. S. Rodriguez, H. L. Ring, J. He, K. C. Bantz and C. L. Haynes, Chem. Rev., 2019, 119, 664–699 CrossRef CAS PubMed.
- Y. Dubi, I. W. Un and Y. Sivan, Chem. Sci., 2020, 11, 5017–5027 RSC.
- A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed.
- F. Tong, X. Liang, Z. Wang, Y. Liu, P. Wang, H. Cheng, Y. Dai, Z. Zheng and B. Huang, ACS Catal., 2021, 11, 10814–10823 CrossRef CAS.
- H. S. Kojori, J.-H. Yun, Y. Paik, J. Kim, W. A. Anderson and S. J. Kim, Nano Lett., 2016, 16, 250–254 CrossRef PubMed.
- S. Wu, Z. Xu, S. Tian, T. Luo and G. Xiong, J. Mol. Liq., 2022, 360, 119410 CrossRef CAS.
- Milon, M. K. Hossain, D. Roy and F. Ahmed, J. Mol. Struct., 2021, 1237, 130302 CrossRef CAS.
- P. Xu, W. Lu, J. Zhang and L. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 12366–12377 CrossRef CAS.
- Y. Zhang, Q. Li, C. Liu, X. Shan, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2018, 224, 283–294 CrossRef CAS.
- M. Wen, S. Song, Q. Liu, H. Yin, K. Mori, Y. Kuwahara, G. Li, T. An and H. Yamashit, Appl. Catal., B, 2021, 282, 119511 CrossRef CAS.
- X. Su and S. Li, Int. J. Hydrogen Energy, 2021, 46, 14384–14394 CrossRef CAS.
- Y. Feng, X. Zhang, Y. Shao, X. Chen, H. Wang, J. Li, M. Wu, H. Dong, Q. Liu and H. Li, ACS Appl. Mater. Interfaces, 2022, 14, 27979–27993 CrossRef CAS PubMed.
- D. Astruc, Organometallic Chemistry and Catalysis, Springer, Berlin, 2007, p. 87–107 Search PubMed.
- X. Zhang, Y. Zhao, X. Jia, Y. Zhao, L. Shang, Q. Wang, G. I. N. Waterhouse, L. Wu, C. Tung and T. Zhang, Adv. Energy Mater., 2018, 8, 1702780 CrossRef.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu and J. Zhu, Angew. Chem., Int. Ed., 2018, 23, 5076–5080 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08396e |
| This journal is © The Royal Society of Chemistry 2023 |