
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Control of nanoparticle dispersion, SEI composition, and electrode morphology enables long cycle life in high silicon content nanoparticle-based composite anodes for lithium-ion batteries†
Maxwell C.
Schulze
 ,
Fernando
Urias
,
Nikita S.
Dutta
,
Zoey
Huey
,
Jaclyn
Coyle
,
Glenn
Teeter
,
Ryan
Doeren
,
Bertrand J.
Tremolet de Villers
,
Sang-Don
Han
,
Nathan R.
Neale
and
G. Michael
Carroll
*
,
Fernando
Urias
,
Nikita S.
Dutta
,
Zoey
Huey
,
Jaclyn
Coyle
,
Glenn
Teeter
,
Ryan
Doeren
,
Bertrand J.
Tremolet de Villers
,
Sang-Don
Han
,
Nathan R.
Neale
and
G. Michael
Carroll
*
National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, Colorado 80401, USA. E-mail: mike.carroll@nrel.gov
First published on 3rd February 2023
Abstract
Striking a balance between high theoretical capacity, earth abundance, and compatibility with existing manufacturing infrastructure, silicon is one of the few materials that meet the requirements for a next-generation anode for rechargeable lithium-ion batteries. Due to complications with extreme volume changes during charging/discharging and reactive interfacial chemistries, the cycle life of silicon-based composite anodes is unacceptable for broad use. Developing a majority silicon composite anode formulation that overcomes these challenges and is compatible with current industrial manufacturing practices requires materials and chemical engineering solutions that account for both electrode morphology and interfacial chemistry. Here, we synthesize surface-functionalized silicon nanocrystals that enable a highly dispersed and homogeneous slurry that can easily be integrated into a standard electrode fabrication process. We use this formulation to fabricate a 76 wt% silicon composite electrode. We show that the contents and the morphology of the silicon electrolyte interphase – a determining factor for the cycle life of silicon-based anodes – can be controlled with a post-synthetic thermal curing procedure. The curing process removes the organic surface functional groups used initially to enable dispersion in the slurry. Removing the organic surface coating reduces the cell impedance, improves the silicon utilization for lithium storage, and boosts the coulombic efficiency to values > 99.9% when electrochemically cycled. When paired with a capacity-matched lithium nickel manganese cobalt oxide LiNi0.8Mn0.1Co0.1O2 cathode, the cell retains 72% of its capacity after 1000 charge/discharge cycles while delivering an initial anode specific capacity of nearly 1000 mA h g−1 and an areal capacity of 2.55 mA h cm−2.
Introduction
The US Department of Energy's Vehicle Technologies Office has identified a cell stack cost goal of $100 kW h−1 for rechargeable lithium-ion battery (LIB) technology to achieve cost parity with internal combustion engines and enable widespread electrification of the transportation sector.1 To achieve this goal, the active materials must be earth-abundant and store high stoichiometric quantities of Li ions at a high cell voltage. The gravimetric energy density of the incumbent anode material, graphite, is too low to meet this goal. With a theoretical capacity of 3500 mA h g−1, Li storage potentials between 0.6 and 0.01 V vs. Li/Li+, and high earth abundance, silicon (Si) is one of only a few potential anode materials that can potentially reach this high bar.2To facilitate the technological transition and minimize disruption to industrial anode production, Si would simply replace graphite in composite-based anodes. However, the requirements to replace graphite in LIB anodes are daunting. The electrode must be able to deliver several thousand charge/discharge cycles with coulombic efficiencies (CE) > 99.99%, greatly exceed the electrode specific gravimetric and volumetric energy densities of graphite anodes, have a decades long shelf-life (calendar life), charge/discharge at high rates, be scalable and adaptable to composite anode manufacturing practices, and be cost competitive. In principle, Si can achieve all these feats.
The potential of Si in LIB anodes has catalyzed significant interest and investment throughout the last several decades.2–7 However, despite these efforts, high-content silicon anodes have not yet overtaken graphite as the state-of-the-art LIB anode material. Anodes consisting of >70% Si with industrially relevant areal capacities (>2.5 mA h cm−2) and realistically scalable fabrication processes have unacceptably low CE leading to cycle lifetimes (80% capacity retention) of only a few hundred cycles.2,8–10 Moreover, reports of majority silicon composite anodes paired with high-voltage NMC cathodes and using industry standard electrolytes (1.2 M LiPF6 in 0.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7 ethylene carbonate:ethylmethyl carbonate with fluoroethylene carbonate (GenF)) typically display useful lifetimes less than 100 cycles.4,11 Indeed, challenges associated with repeatedly charging and discharging silicon continue to hinder Si anode-based cycle lifetimes.
:0.7 ethylene carbonate:ethylmethyl carbonate with fluoroethylene carbonate (GenF)) typically display useful lifetimes less than 100 cycles.4,11 Indeed, challenges associated with repeatedly charging and discharging silicon continue to hinder Si anode-based cycle lifetimes.
Silicon's volume expands and contracts by up to 350% between charging (lithiation) and discharging (delithiation), creating severe mechanical strain on both individual Si particles and across the composite electrode, which leads to rapid capacity loss.2 Reducing the Si particle dimensions to the nanoscale can eliminate particle pulverization,7,12 but long-range electrode-level mechanical strain persists regardless of the Si size. The volumetric changes also affect the electrode's morphology by increasing the solid volume fraction, which reduces porosity and hinders Li-ion transport.13,14 Strategies to mitigate the mechanical stress of lithiating and delithiating silicon range from single particle to electrode-level architecture engineering.2,7,8,15–17 While these efforts usually mark an improvement in the anodes' capacity retention and provide valuable scientific insight, the gains are met with an inability to scale the particle or electrode fabrication procedure. Both improved cycle life and scalability must be realized for a solution to be viable.
The interfacial chemistry between silicon and the electrolyte plays a critical role in Si LIB stability as well.18–30 The interfaces of both Si and lithiated silicon (LixSi) are chemically reactive towards the components of carbonate electrolytes.31–33 Upon cycling, electrolyte components irreversibly reduce and deposit on the Si surface to form a solid-electrolyte-interphase (SEI).33–37 Once formed, the SEI must passivate the LixSi interface against further parasitic reactions in order to promote long cycle lifetimes in full-cells with competitive energy densities, where lean electrolyte conditions are used and Li+ inventories are limited.38–40 Effective passivation requires the SEI to be elastic enough to expand and contract with repetitive cycling without tearing or fracturing, all the while conducting Li+ and allowing the Si to remain electrically connected to the current collector.17,41 Achieving passivation is so difficult, in fact, that even at open circuit, parasitic chemistry occurs that reduces the calendar lifetime and, as such, is one of the major limitations to commercial deployment of Si-based anodes.42 Intentional electrode design involving simple scalable steps that enable control of both the electrode structure and the Si SEI is needed if Si's remarkable energy storage capacities are to be realized in LIB technologies.
Here, we present two innovations for Si-based anodes for LIB technology. First, we functionalize the surface of d = 5.9 nm Si nanoparticles (NPs, Fig. 1a) with a hydrophilic allyloxy (diethyleneoxide methylether) (PEO) coating to form Si@PEO. This functionalization provides the necessary interparticle repulsive forces to prevent particle flocculation and enables precise control of the electrode morphology. When added to N-methyl pyrrolidone (NMP), Si@PEO forms a colloidal suspension which enables homogeneous and reproducible slurries – a feat not possible with hydrogen-terminated SiNPs – that are simply blade coated onto copper current collectors. We find that PEO creates large barriers for ionic transport in the composite electrode and poor interfacial passivation at the silicon surface leading to high cell impedance, low coulombic efficiency, and poor silicon utilization. To remedy this, we introduce thermal curing to remove the PEO from the finished electrode architecture, which enables a thin, well-defined, and mostly inorganic SEI to form on the Si surface. Upon curing, the cell impedance decreases by several orders of magnitude and the CE increases by an order of magnitude. These improvements are derived from facilitated Li+ transport (relative to the PEO coated silicon) and improved interfacial passivation. Our high Si content electrodes deliver areal capacities > 4 mA h cm−2, and when paired with capacity-matched lithium nickel manganese cobalt oxide (NMC) cathodes, retain upwards of 72% of their capacity after 1000 lithiation/delithiation cycles while delivering initial anode specific capacities of nearly 1000 mA h g−1 at an areal capacity of 2.55 mA h cm−2.
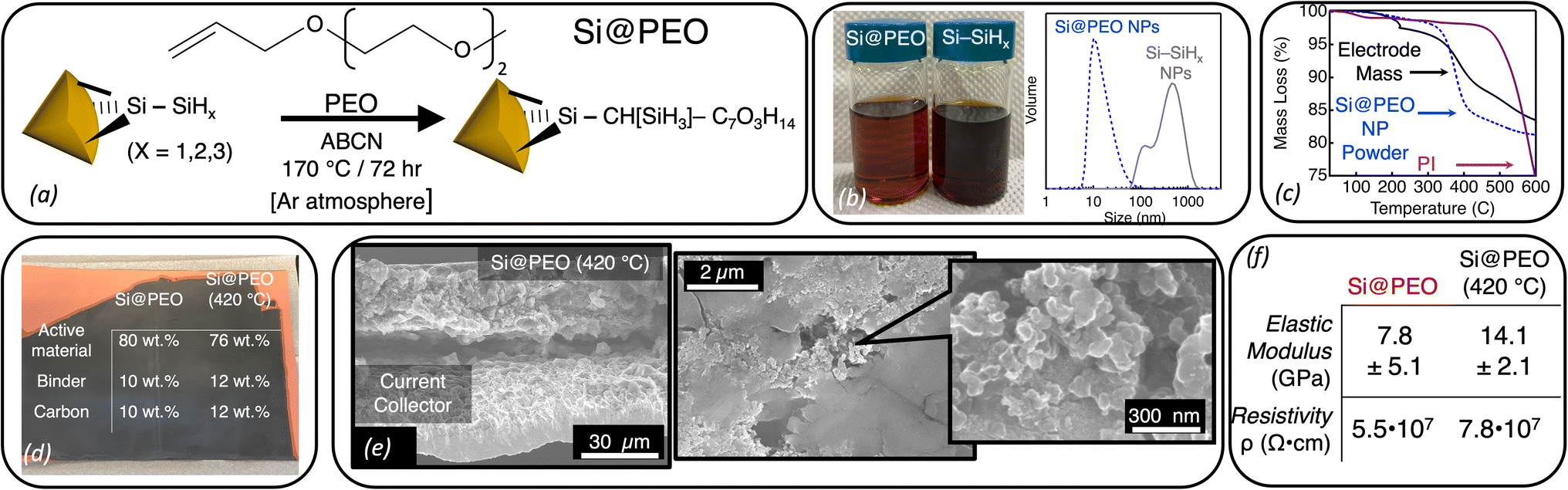 | ||
| Fig. 1 (a) Schematic illustration of the radical-initiated surface functionalization of Si–SiHx NPs to form Si@PEO NPs. (b) (Left) Photograph of Si@PEO NPs and Si–SiHx NPs suspended in NMP. (Right) Dynamic light scattering data for the same Si@PEO (blue dashed) and Si–SiHx (gray) NP suspensions in NMP. (c) Thermal gravimetric analysis data for the Si@PEO NP powder (blue dashed), polyimide (PI) binder (purple), and the dried electrode mass (black) performed under a N2 atmosphere. (d) Photograph of a blade coated Si@PEO electrode with the mass fractions of Si@PEO and Si@PEO (420 °C). (e) (Left) Cross-sectional and (right) plan-view SEM images of a Si@PEO (420 °C) electrode. For clarity, the current collector and the electrode mass are labeled on the left image. Additional SEM images before and after cycling are shown in ESI, Fig. S2–S4.† (f) Averaged elastic modulus and electronic resistivity values for Si@PEO and Si@PEO (420 °C) electrodes. | ||
Results and discussion
Radical-initiated chemistry is a robust and useful technique for derivatizing silicon interfaces with a variety of covalent bonds and functionalities.43–47 Here, we employ radical chemistry to induce a reaction between the alkene functional group on PEO and the hydride-terminated surface (Si–SiHx) of d = 5.9 nm Si NPs to form surface functionalized Si@PEO NPs (Fig. 1a).47,48 Following purification (see the Experimental section), our isolated Si@PEO NPs disperse into polar solvents like water48 and NMP. Fig. 1b shows a photograph of a Si@PEO NP suspension compared to Si–SiHx NPs in NMP. While the Si–SiHx NP suspension scatters visible light and appears opaque, the Si@PEO NP sample is translucent and homogeneous. From dynamic light scattering measurements (Fig. 1b), Si–SiHx NP suspension forms a bimodal distribution of NP flocs with mean diameters of 90 nm and 800 nm. The 5.9 nm Si@PEO NPs, however, are singly distributed with a hydrodynamic radius peak centered at 11 nm, indicating that PEO at the surface of Si eliminates particle flocculation at these concentrations. Compared to the unfunctionalized Si–SiHx NPs, the Si@PEO NP colloid provides an ideal platform for producing homogeneous slurries that result in volumetrically dense composite electrodes with optimal electronic and ionic percolation networks.49Si@PEO electrodes were fabricated by blade coating a slurry mixture containing Si@PEO NPs, Timcal C45 as a conductive additive, and polyimide (PI) binder dissolved in NMP at a weight ratio of 8:1:1 (Si@PEO:C45:PI) onto a copper current collector at six different thicknesses (see the Experimental section for additional details). While typical composite electrodes contain heat-sensitive polymeric binders like (poly)acrylic acid which decompose under aggressive thermal conditions,50 our use of a thermally robust polyimide binder allows for high temperature treatments to remove the electrochemically inactive PEO surface coating. Following a vacuum drying procedure at 150 °C, our electrodes are cured at 420 °C for 4 h under a continuous N2 flow (Si@PEO (420 °C)) to remove the surface PEO. Thermogravimetric analysis (TGA) data of the Si@PEO NP powder shown in Fig. 1c display thermally induced mass loss beginning at 330 °C and continuing until 400 °C. This mass loss is attributed to volatilization and loss of the PEO. The total mass loss of the Si@PEO NP powder is 18%, which corresponds to a surface density of ∼2 PEO per nm2, and is in agreement with prior reports.47,48 TGA of the dried composite electrode displays a similar mass-loss shape which, given that PI does not lose significant mass until T > 500 °C (Fig. 1c), we attribute to the volatilization of PEO within the electrode. This PEO mass accounts for 14% of the total electrode mass (excluding residual NMP at T < 200 °C) which matches the expected mass of PEO in the electrode (14.4%). In other words, when the diffusion of PEO is not impeded, most of the PEO mass is lost upon heating. Given that these electrodes are several microns thick and highly tortuous, it is possible that some PEO becomes trapped in the electrode structure. Nevertheless, from these data we surmise that the thermal curing removes most of the PEO from the electrode leaving a mass ratio of 76% Si, 12% PI, and 12% C45, as shown in Fig. 1d.
The scanning electron microscopy (SEM) images of the Si@PEO (420 °C) electrode in Fig. 1e show a densely packed structure with a thickness of 15 μm and feature dimensions on the order of 100 nm (attributed to Timcal C45).51 At the single micron and hundreds of nm scales, the morphology of the Si@PEO electrode is identical to that of Si@PEO (420 °C) (ESI, Fig. S2–S4†). The transmission electron microscopy images (TEM) (ESI, Fig. S5†) of the Si@PEO (420 °C) electrode indicate that, as expected, the Si NPs retain their 6 nm diameter, as well as their crystallinity, after thermal curing. Scanning spreading resistance microscopy (SSRM) measurements show no change in the silicon-specific resistivity (Fig. 1f and ESI, Fig. S6†), though some evidence for segregation between silicon and C45 is apparent after thermal curing. Fourier transform infrared (FTIR) measurements (ESI, Fig. S7†) on electrodes before and after thermal curing display little to no difference in the identities of the chemical species, just their relative abundance. These spectroscopic, microscopic, and electronic measurements suggest that the morphology and chemistry of the electrode are largely unaffected by thermal curing. Interestingly, however, we do note that the elastic modulus of the top 1 μm of the composite electrode nearly doubles upon curing, which may be related to the minor phase segregation observed in SSRM measurements (Fig. 1f and S8†).
The beneficial effect of thermal curing becomes apparent upon electrochemical analysis. Fig. 2a displays a comparison of the voltage profiles for the first three lithiation/delithiation cycles of Si@PEO and Si@PEO (420 °C) in a half-cell configuration against Li metal in 1.2 M LiPF6 in a 0.29:0.68:0.03 ethylene carbonate:ethylmethyl carbonate:fluoroethylene carbonate (GenF) electrolyte. The charge that passed during the first lithiation (negative scan direction) is 2.50 A h g−1 for both Si@PEO and Si@PEO (420 °C), but upon delithiation, the recovered charge of Si@PEO is only 1.05 A h g−1, whereas that of Si@PEO (420 °C) is 1.83 A h g−1, resulting in 1st cycle coulombic efficiencies (CE) of 42% and 73%, respectively. Since Si@PEO and Si@PEO (420 °C) lithiate to the same degree in the first cycle with nearly identical voltage profiles, but do not delithiate to the same degree, we surmise that much of the loss associated with the first cycle of Si@PEO may be due to Li-trapping or active material loss rather than parasitic side chemistry. In agreement, the delithiation capacities of the second and third cycles for Si@PEO and Si@PEO (420 °C) are nearly the same as the first at 1.04 and 1.93 mA h g−1, respectively. If parasitic chemistry was the root cause of the low 1st cycle CE, we would expect much lower second and third cycle CEs. Because of Li metal's reactivity toward the electrolyte,52,53 continued cycling in a half-cell configuration is prone to artifacts.54,55 For this reason we do not place a high-level of confidence in extended half-cell cycling. For completeness, however, extended half-cell cycling is provided in the ESI† (Fig. S9†) with an associated discussion.
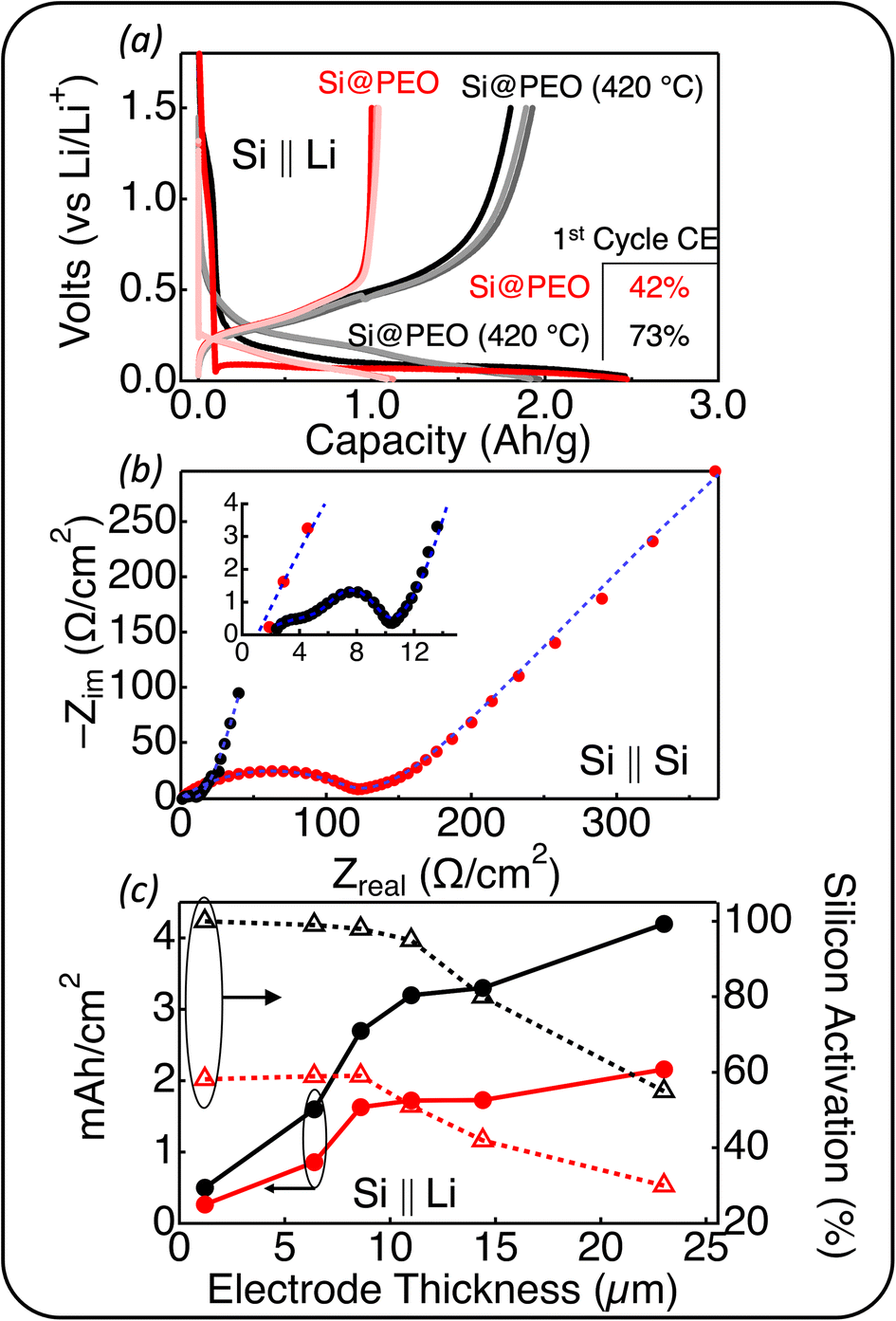 | ||
| Fig. 2 (a) Voltage profiles of Si@PEO (red) and Si@PEO (420 °C) (black) in a half-cell configuration against Li metal for the first three electrochemical lithiation and delithiation cycles. The shade of the color indicates the cycle number where the light colors correspond to later cycles. The inset displays the first cycle coulombic efficiency for each electrode. (b) Nyquist plot of Si@PEO and Si@PEO (420 °C). The electrodes were cycled in a half-cell format against Li metal in a GenF electrolyte, disassembled, and reassembled in a symmetric cell configuration. The frequency range is 500 kHz to 2 mHz. The blue dashed lines are equivalent circuit fits to the data. (c) Areal capacity (solid circles and solid lines) and the silicon activation (open triangles and dashed lines) for Si@PEO (red) and Si@PEO (420 °C) (black) half-cells in the third cycle for electrodes with identical compositions at different thicknesses. | ||
To probe the transport properties in these electrodes, we perform electrochemical impedance spectroscopy (EIS) measurements on symmetric Si@PEO ‖ Si@PEO and Si@PEO (420 °C) ‖ Si@PEO (420 °C) cells. The EIS data shown in Fig. 2b indicate a significant reduction in area specific impedance (ASI, defined by the area of the current collector) for Si@PEO (420 °C) compared to Si@PEO. Although we have reservations about the accuracy of the fit values for the Si@PEO impedance (as outlined in ESI, Fig. S10† and the accompanying discussion), upon fitting the data to an equivalent circuit, the ASI related to the SEI is 2200 Ω cm−2 for Si@PEO and 4.7 Ω cm−2 for Si@PEO (420 °C). The SEI-related impedance semicircle occurs in the frequency regime between 100 Hz and 1 Hz, indicating that the impedance is related to Li+ transport (as opposed to electron transport (MHz) or diffusion of Li in Si (mHz)). Because the ionic conductivity of LiPF6 in amorphous PEO is between one to two orders of magnitude lower than that of 3:7 EC:EMC at 25 °C,56,57 such a disparity in impedance is expected for a Li+ transport mechanism dictated by amorphous PEO indicating that, as expected, PEO is a substantial portion of the SEI which inhibits Li+ transport.
To quantify the energy charge storage performance of these electrodes at practical loadings, we fabricated Si@PEO and Si@PEO (420 °C) electrodes with six different thicknesses. The areal capacity (mA h cm−2) and Si activation for each thickness are shown in Fig. 2c. Here, Si activation is defined as the measured delithiation capacity in the third cycle divided by the electrode's theoretical delithiation capacity (assuming a Si-specific theoretical delithiation capacity of 3.5 A h g−1). Importantly, to account for the mass loss of PEO during thermal curing and obtain an accurate electrode theoretical capacity, the mass fraction due to PEO was subtracted from the Si@PEO (420 °C) electrode. If this correction is not performed, values for Si activation exceeded 1.0, which is clearly not physically possible.
As seen in Fig. 2c, both the Si@PEO and Si@PEO (420 °C) electrodes display a similar trend with increasing electrode thickness: an increasing areal capacity, but a decline in the Si activation beyond an electrode thickness of ∼8 μm. The values of the areal capacity and the Si activation, however, are quite different between Si@PEO and Si@PEO (420 °C). The areal capacity of Si@PEO (420 °C) is nearly double that of Si@PEO at a given electrode thickness. The Si activation for electrodes < 10 μm is near unity for Si@PEO (420 °C) and only 60% for Si@PEO. For the thickest electrodes, 55% of the Si in Si@PEO (420 °C) participates in reversible lithiation delivering 4.2 mA cm−2 while only 30% of the Si is active in Si@PEO, delivering 2.1 mA cm−2. From our EIS analysis, we attribute the declining Si activation with increasing electrode thickness to a reduction in porosity; organic SEI build-up in the first cycle increases the solid volume fraction of the electrode, which inhibits electrolyte mass transport throughout the porous network and increases ionic resistance. Interestingly, upon disassembling the coin cells, the deformation of the Si@PEO (420 °C) electrode is apparent—the degree to which seems to depend on the areal capacity (ESI, Fig. S8†). This deformation may be related to the increase in the elastic modulus with thermal curing. However, no delamination or other major mechanical failure was observed after cycling even the highest loading electrodes. Indeed, these high Si content, high loading Si@PEO (420 °C) electrodes exhibit promising electrochemical properties in half-cell configurations with traditional carbonate-based electrolytes.
The contents of the SEI and the chemical environment at the surface of silicon play a major role in the cycle stability and ionic and electronic resistance. To probe the compositional environment at the surface of these electrodes, we performed X-ray photoelectron spectroscopy measurements before and after cycling on both Si@PEO and Si@PEO (420 °C) (Fig. 3). Because we are interested in the surface coatings of organic-based PEO and it's evolution with thermal curing and cycling, we only present the O 1s and C 1s spectra in the main text. Core-level spectra for Li 1s, P 2p, and Si 2p are available in the ESI (Fig. S11).†
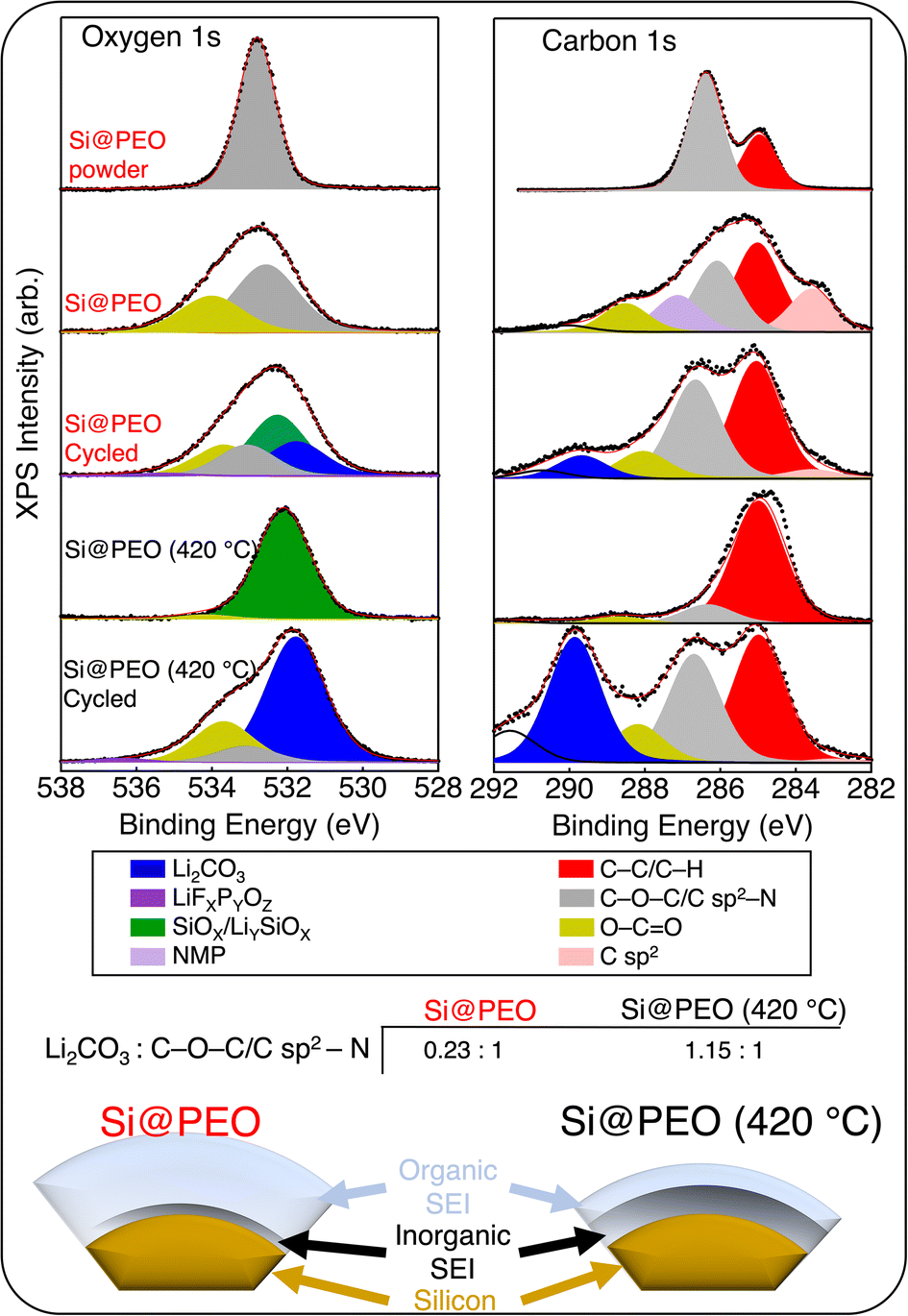 | ||
| Fig. 3 Background subtracted oxygen 1s (left) and carbon 1s (right) X-ray photoelectron spectroscopy data (black dots) for the as-synthesized Si@PEO powder, Si@PEO before and after three cycles in a half cell, and Si@PEO (420 °C) before and after three cycles in a half cell. The data were fit to multiple peaks to deconvolute the spectra. The fitting envelopes are shown as red lines and the individual peaks are shown in different colors. The color key below links the chemical identities to their corresponding peaks. The table at the bottom plots the ratio of inorganic (Li2CO3) to organic (C–O–C/C sp2–N). The scheme below summarizes the ratiometric analysis of the SEI for Si@PEO and Si@PEO (420 °C). | ||
As seen in Fig. 3, the overwhelming signal for the Si@PEO powder in the O 1s and C 1s spectra arises from PEO (the organic functional group O–C–O) and C–C/C–H carbon, as expected. Upon electrode fabrication, signatures from the PI binder (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and conductive carbon (C sp2) are detectable. We note that some NMP remained in the Si@PEO electrode after the drying procedure. Following 420 °C treatment, oxidation at the surface of the silicon is seen with the appearance of SiOx, and very little signal from PEO is present, consistent with our TGA data presented in Fig. 1. Curiously, the abundance of PI binder and conductive carbon decreases upon heating which may be related to the appearance of carbon domains and phase segregation (ESI, Fig. S6†). Upon cycling three times in a half cell, the inorganic species Li2CO3 and LiFxPyOz appear in the O 1s and C 1s spectra in addition to the organic functional groups for both Si@PEO and Si@PEO (420 °C). Because Li2CO3 and LiFxPyOz deposit on top of the Si/SiOx surface and XPS is a surface sensitive technique, the apparent intensity is dominated by Li2CO3 and LiFxPyOz. Importantly, however, the relative abundances of the inorganic and organic species for Si@PEO and Si@PEO (420 °C) are quite different. While the inorganic and organic signals are nearly equal in the spectral composition of Si@PEO (420 °C) (1.15:1 inorganic: organic), the organic components dominate Si@PEO at 0.23:1. These results indicate that curing Si@PEO at 420 °C reduces the organic content of the SEI, producing an SEI more commonly found in cells with uncoated silicon surfaces,58 which is summarized in the scheme in Fig. 3.
O) and conductive carbon (C sp2) are detectable. We note that some NMP remained in the Si@PEO electrode after the drying procedure. Following 420 °C treatment, oxidation at the surface of the silicon is seen with the appearance of SiOx, and very little signal from PEO is present, consistent with our TGA data presented in Fig. 1. Curiously, the abundance of PI binder and conductive carbon decreases upon heating which may be related to the appearance of carbon domains and phase segregation (ESI, Fig. S6†). Upon cycling three times in a half cell, the inorganic species Li2CO3 and LiFxPyOz appear in the O 1s and C 1s spectra in addition to the organic functional groups for both Si@PEO and Si@PEO (420 °C). Because Li2CO3 and LiFxPyOz deposit on top of the Si/SiOx surface and XPS is a surface sensitive technique, the apparent intensity is dominated by Li2CO3 and LiFxPyOz. Importantly, however, the relative abundances of the inorganic and organic species for Si@PEO and Si@PEO (420 °C) are quite different. While the inorganic and organic signals are nearly equal in the spectral composition of Si@PEO (420 °C) (1.15:1 inorganic: organic), the organic components dominate Si@PEO at 0.23:1. These results indicate that curing Si@PEO at 420 °C reduces the organic content of the SEI, producing an SEI more commonly found in cells with uncoated silicon surfaces,58 which is summarized in the scheme in Fig. 3.
The morphology and thickness of the SEI around the Si NPs define the electrode porosity after cycling, which dictates important properties like mass transport and electronic percolation. To understand how the SEI changes with thermal curing, we performed transmission electron microscopy (TEM) measurements on electrodes after three lithiation/delithiation cycles in a half-cell configuration. Representative TEM images for Si@PEO and Si@PEO (420 °C) and particle size statistics are shown in Fig. 4. As seen in Fig. 4, the Si@PEO electrode morphology is a broadly distributed aggregation of particles encapsulated within a large-volume, low-contrast SEI matrix. The Si@PEO (420 °C) morphology, however, is composed of discrete and homogeneously distributed particles. The apparent size of Si@PEO (420 °C) is 13 nm implying an SEI thickness of 3.5 nm. In contrast, the apparent average size of the cycled Si@PEO particle remains nearly unchanged at 6.3 nm compared to the as-prepared Si@PEO at 5.9 nm. Likely, this is the result of the low contrast PEO surface coating inhibiting the growth of higher contrast inorganic SEI components like Li2CO3 or LiF (vide supra). The distribution of particle size is also impacted by thermal curing. One standard deviation from the mean particle size (σ) for Si@PEO (420 °C) is 2 nm, close to the σ = 1.1 nm for uncycled Si@PEO. For cycled Si@PEO, however, the distribution broadens significantly to σ = 4.1 nm. This increase in size distribution may be due to trapped Li within the silicon nanoparticles — consistent with the low first cycle CE — or highly inhomogeneous SEI contents. In either case, these images clearly show that the Si@PEO SEI is large and poorly defined, but once thermally cured, the SEI is homogeneous and conforms to the silicon surface.
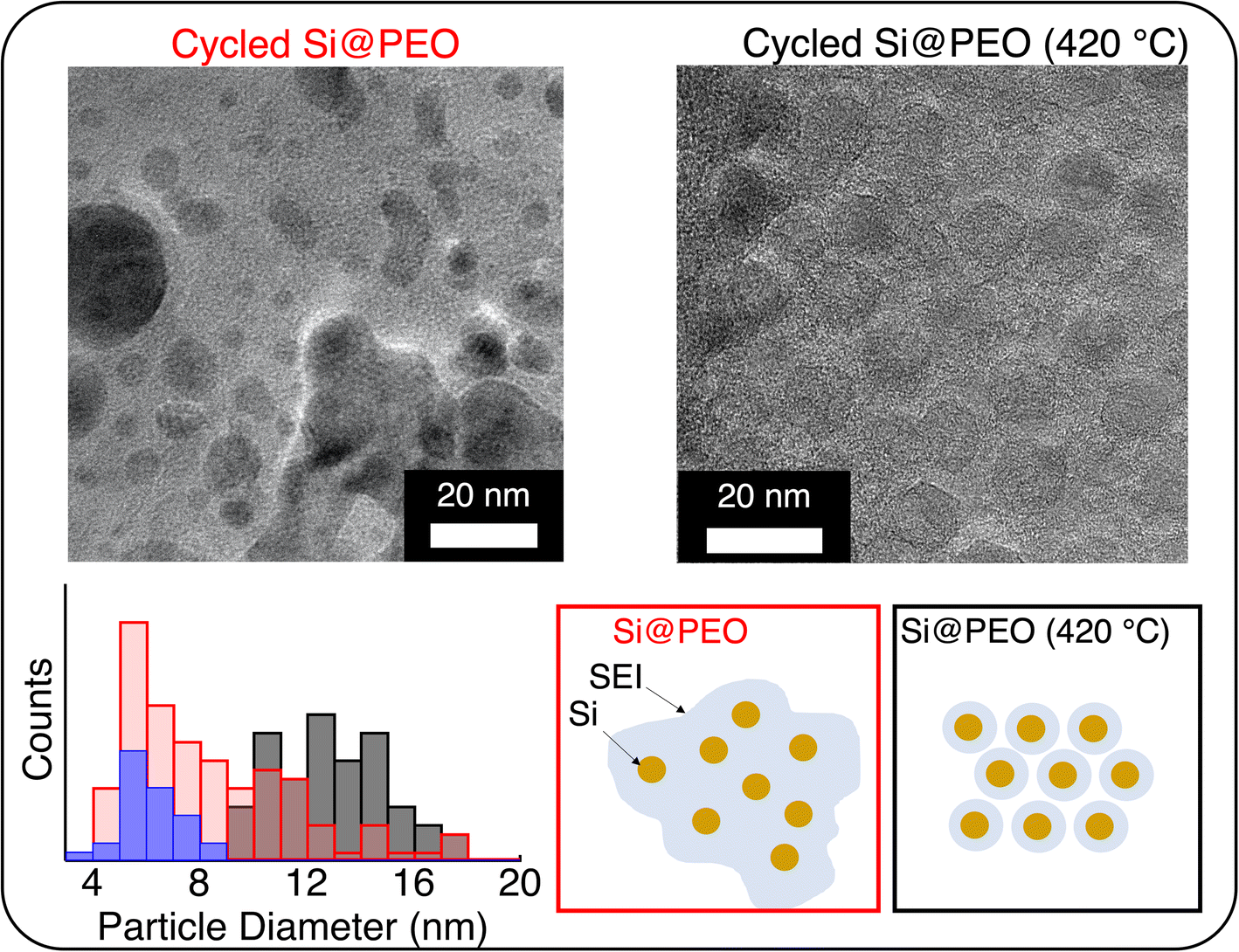 | ||
| Fig. 4 Transmission electron microscopy images of (left) Si@PEO and (right) Si@PEO (420 °C) electrodes after three lithiation/delithiation cycles in a half cell. (Bottom) Histogram of the apparent particle size (including SEI thickness) for the Si@PEO (420 °C) electrode prior to cycling (blue), and cycled Si@PEO (red) and Si@PEO (420 °C) (black) electrodes. An example of size determination is shown in Fig. S5.† The scheme summarizes the different SEI morphologies for each of the samples based on TEM measurements and particle size analysis. | ||
Coupling Si-based anodes with capacity-matched high-voltage NMC-based cathodes provides a configuration for obtaining high energy density Li-ion batteries. To this end, we fabricated Si@PEO and Si@PEO (420 °C) batteries using NMC622 composite electrodes as cathodes (Si@PEO ‖ NMC622 and Si@PEO (420 °C) ‖ NMC622). To match the capacity of our Si@PEO electrodes with that of standardized 1.5 mA h cm−2 NMC622 cathodes provided by the Cell Analysis, Modeling, and Prototyping (CAMP) facility at Argonne National Laboratories, Si@PEO and Si@PEO (420 °C) anodes with a measured areal capacity of approximately 1.75 mA h cm−2 (accounting for 18% SOC electrochemical prelithiation) were chosen (see the Full-Cell Electrode Balancing section in the ESI†). The electrode mass loading for Si@PEO (420 °C) was 1.12 mg cm−2 and 1.75 mg cm−2 for Si@PEO. The additional mass of the Si@PEO electrode accounts for the mass of the PEO surface coating as well as the inactive fraction of silicon. The cycling data for 1000 charge/discharge cycles are shown in Fig. 5.
 | ||
| Fig. 5 Cathode and areal capacities (solid circles) and coulombic efficiencies (hash marks) of 1000 cycles for prelithiated Si@PEO (red) and prelithiated Si@PEO (420 °C) (black) in a full-cell configuration against NMC622. These cells were cycled between 3.0 and 4.2 V at a rate of C/3 99 times followed by a cycle at a rate of C/10 cycle every 100th cycle and this total cycling process was repeated 10 times. The C-rate was calculated based on the limiting capacity of the NMC622 electrode. The silicon electrodes were first cycled in a half-cell configuration against Li metal and electrochemically prelithiated by stopping delithiation at 0.6 V. The n:p ratio was calculated based on the measured capacity of the silicon electrode between 0.6 and 0.01 V in the half-cell. All electrochemical measurements were carried out in a GenF electrolyte. | ||
With a slightly oversized anode capacity (1.05:1 n:p) and electrochemically prelithiated to ∼18% SOC, both Si@PEO ‖ NMC622 and Si@PEO (420 °C) ‖ NMC622 provide an initial cathode specific capacity of 160 mA h g−1, where Si@PEO (420 °C) ‖ NMC622 delivers an anode specific capacity of nearly 1000 mA h g−1 and a total cell stack energy density of 100 W h kg−1. The capacity retention of Si@PEO (420 °C) ‖ NMC622 is nearly 100% after the first 180 cycles, with only 26% capacity loss after 1000 cycles. The inflection point in cycle 180 is likely due to the consumption of the excess Li+ inventory in the prelithiated Si@PEO (420 °C) by parasitic processes. Reflected in the excellent capacity retention, the initial CE of Si@PEO (420 °C) ‖ NMC622 is 99.5% and rises to > 99.9% within 10 cycles, where it remains throughout the course of the measurement. Indeed, the cycle stability of Si@PEO (420 °C) ‖ NMC622 is remarkable for a high-content Si-based anode.
In stark contrast, Si@PEO‖NMC622 loses 50% of its reversible capacity within 20 cycles and only retains 5.5% of its capacity after 1000 cycles. The initial CE of Si@PEO ‖ NMC622 is only 95.8%, which gradually rises to 99% after 200 cycles, the cumulative effect of which results in rapid capacity fade.40 The intermittent C/10 cycles indicate that the capacity loss is not simply slow, reversible Li+ transport (which would be evident from large capacity spikes in slower cycles), but is in fact either loss of active material or loss of Li+ inventory (see the Full-Cell Procedure section in the ESI† for additional discussion). For reference, rate capability data are provided in the ESI (Fig. S12).† Differential capacity and voltage profile plots indicate increasing lithiation overpotential with continued cycling on Si@PEO ‖ NMC622, where little additional overpotential is observed for Si@PEO (420 °C) ‖ NMC622 (ESI, Fig. S13†). Due to their low-lying oxidation potentials, polyether moieties like PEO or glyme-based electrolytes undergo oxidation and rapid irreversible Li+ inventory consumption at the NMC622 interface during electrochemical cycling.12,13,36,59 Given that our Si@PEO electrodes contain 14.4 wt% PEO, it is possible that PEO crosses over from Si@PEO to the NMC622 electrode and contributes to the low CE of Si@PEO ‖ NMC622. The ATR-FTIR spectra of the cycled NMC622 cathode of Si@PEO ‖ NMC622 show distinct differences relative to the same electrode cycled against Si@PEO (420 °C) or Li metal (Fig. S14†), though the overlapping spectral features from other electrode components complicate the analysis and preclude a definitive identification of PEO at the cathode. No apparent changes in the NMC electrode morphology could be detected (ESI, Fig. S15†). Nevertheless, these data highlight the importance of removing surface coatings before assembly into electrochemical cells.
Finally, to push the cell stack energy density capability of our batteries towards more commercially competitive values, we paired 0.64 mgSi cm−2 (thin) and 1.37 mgSi cm−2 (thick) prelithiated Si@PEO (420 °C) electrodes against high-voltage NMC 811 cathodes that have limiting capacities of 1.66 mA h cm−2 (thin Si@PEO (420 °C)‖ NMC811) and 2.55 mA h cm−2 (thick Si@PEO (420 °C)‖NMC811), respectively. The data for 1000 electrochemical cycles of these cells are plotted in Fig. 6. As seen in Fig. 6, the initial cell stack energy density for the two electrode thicknesses is 212 and 105 W h kg−1. If only the electrode mass is considered in these energy density calculations (i.e. neglecting the current collectors, separator, and electrolyte) the energy density increases to > 400 W h kg−1 for each battery. While the evolution of energy density with the cycle number is slightly different for the two thicknesses during the first 100 cycles, the slope of the capacity fade is nearly identical for the remaining 900 cycles. The capacity retention after 1000 cycles for thin Si@PEO (420 °C) ‖ NMC811 is 79% and for thick Si@PEO (420 °C)‖ NMC811 is 72%. These values are consistent with the small differences in the average CE (99.96% and 99.94%). Overall, these data show that these high silicon content electrodes operate well at loadings above 1 mgSi/cm2 when paired with nickel-rich high voltage cathodes in industry standard carbonate electrolytes.
 | ||
| Fig. 6 Energy density (left axis, solid dots) and coulombic efficiency (right axis, hash marks) for prelithiated Si@PEO (420 °C) ‖ NMC811full-cells for two different NMC 811 areal capacity electrodes: 1.66 mA h cm−2 (gray) and 2.55 mA h cm−2 (black). The table below summarizes the battery configuration parameters as well as the average CE and capacity retention. The Si@PEO electrodes were first electrochemically cycled and prelithiated as described in the main text and the Experimental section. No pretreatment was performed on any cathode. All electrochemical experiments were performed in a GenF electrolyte. The energy density reported here considers the mass of both electrode materials (anode + cathode), the current collector for each electrode, the separator, and the electrolyte. Additional details for the calculation are provided in the ESI.† | ||
These results provide valuable insight into the principles of solid-electrolyte interphase engineering for lithium-ion composite electrodes. Specifically, we show that Li+ transport is hindered when Si is surrounded by a PEO matrix. This is evident from the large difference in impedance and silicon utilization when compared to electrodes whose pores are infiltrated with a carbonate electrolyte, as seen in Fig. 2. These observations mirror the challenges faced by solid polymer electrolytes in LIB technology.60 So, while it is known that oligomeric PEO surface coatings do conduct Li+, liquid carbonate electrolytes provide much more ideal transport properties. In addition, we find that after extended cycling, the SEI resulting from PEO coated silicon is much less passivating than an SEI derived from the GenF electrolyte as the coulombic efficiency of a PEO-infiltrated electrode is over an order of magnitude lower than that of an electrode infiltrated with a carbonate electrolyte (99% vs. >99.9%). These findings indicate that a highly organic and poorly defined SEI, as shown in Fig. 3 and 4, is a poor SEI design choice for a densely packed, majority silicon composite electrode in a LIB configuration.
Conclusion
In summary, we developed a method for fabricating densely packed, high Si-content composite anodes from surface-modified 5.9 nm Si NPs. We functionalize the surface of these Si NPs with a molecular oligomer – PEO – to provide enough steric repulsion to limit Si flocculation and generate homogeneous slurries amenable to conventional slurry processing and electrode fabrication. By utilizing a temperature-stable polyimide binder, we show that these electrodes can be thermally cured at 420 °C with minimal impact on the overall electrode structure. The effect of thermal curing removes most of the PEO surface coating and allows a 3.5 nm thick well-defined, mostly inorganic, SEI to be formed at the Si surface, which enables ideal electrochemical lithiation/delithiation properties up to an areal capacity of 4.2 mA h cm−2. If the PEO is not removed, we find that the SEI is large, induces high interfacial impedance, contains mostly organic components, and results in significantly reduced silicon utilization and cycle lifetime.When paired with a capacity-matched NMC622 cathode using common carbonate-based electrolytes, Si@PEO (420 °C) retains 76% of its initial capacity after 1000 cycles while Si@PEO retains only 5% of its capacity in the same cycle period. When pushed to higher limits by increasing the Si loading above 1 mg cm−2 and pairing the electrode with NMC 811 cathodes, Si@PEO (420 °C) retains at least 72% of its capacity after 1000 cycles. This promising electrode formulation offers a plausible path toward achieving the US Department of Energy's Vehicle Technologies Office's goal of <$100 kW h−1 Li-ion battery packs with a simple drop-in substitution to replace graphite with surface-modified silicon.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory (NREL), operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. This research was supported by the U.S. Department of Energy's Vehicle Technologies Office under the Silicon Consortium Project, directed by Brian Cunningham, and managed by Anthony Burrell and by the Laboratory Directed Research and Development (LDRD) Program at NREL. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- B. Cunningham, Silicon and Intermetallic Anode Portfolio Strategy Overview. Annual Merit Review, US Department of Energy 2020 Search PubMed.
- M. N. Obrovac and V. L. Chevrier, Alloy negative electrodes for Li-ion batteries, Chem. Rev., 2014, 114(23), 11444–11502 CrossRef CAS PubMed.
- P. Limthongkul, Y.-I. Jang, N. J. Dudney and Y.-M. Chiang, Electrochemically-driven solid-state amorphization in lithium-silicon alloys and implications for lithium storage, Acta Mater., 2003, 51(4), 1103–1113 CrossRef CAS.
- K. Feng, M. Li, W. Liu, A. G. Kashkooli, X. Xiao, M. Cai and Z. Chen, Silicon-Based Anodes for Lithium-Ion Batteries: From Fundamentals to Practical Applications, Small, 2018, 14(8), 1702737 CrossRef PubMed.
- M. Gu, Y. He, J. Zheng and C. Wang, Nanoscale silicon as anode for Li-ion batteries: The fundamentals, promises, and challenges, Nano Energy, 2015, 17, 366–383 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, 25th anniversary article: Understanding the lithiation of silicon and other alloying anodes for lithium-ion batteries, Adv. Mater., 2013, 25(36), 4966–4985 CrossRef CAS PubMed.
- X. Su, Q. Wu, J. Li, X. Xiao, A. Lott, W. Lu, B. W. Sheldon and J. Wu, Silicon-Based Nanomaterials for Lithium-Ion Batteries: A Review, Adv. Energy Mater., 2014, 4(1), 1300882 CrossRef.
- A. Du, H. Li, X. Chen, Y. Han, Z. Zhu and C. Chu, Recent Research Progress of Silicon-Based Anode Materials for Lithium-Ion Batteries, ChemistrySelect, 2022, 7(19), e202201269 CrossRef CAS.
- Y.-M. Zhao, F.-S. Yue, S.-C. Li, Y. Zhang, Z.-R. Tian, Q. Xu, S. Xin and Y.-G. Guo, Advances of polymer binders for silicon-based anodes in high energy density lithium-ion batteries, InfoMat, 2021, 3(5), 460–501 CrossRef CAS.
- X. Zuo, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Silicon based lithium-ion battery anodes: A chronicle perspective review, Nano Energy, 2017, 31, 113–143 CrossRef CAS.
- J. Guo, D. Dong, J. Wang, D. Liu, X. Yu, Y. Zheng, Z. Wen, W. Lei, Y. Deng, J. Wang, G. Hong and H. Shao, Silicon-Based Lithium Ion Battery Systems: State-of-the-Art from Half and Full Cell Viewpoint, Adv. Funct. Mater., 2021, 31(34), 2102546 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, Size-dependent fracture of silicon nanoparticles during lithiation, ACS Nano, 2012, 6(2), 1522–1531 CrossRef CAS PubMed.
- B. Suthar, P. W. C. Northrop, D. Rife and V. R. Subramanian, Effect of Porosity, Thickness and Tortuosity on Capacity Fade of Anode, J. Electrochem. Soc., 2015, 162(9), A1708–A1717 CrossRef CAS.
- I. Profatilova, E. De Vito, S. Genies, C. Vincens, E. Gutel, O. Fanget, A. Martin, M. Chandesris, M. Tulodziecki and W. Porcher, Impact of Silicon/Graphite Composite Electrode Porosity on the Cycle Life of 18650 Lithium-Ion Cell, ACS Appl. Energy Mater., 2020, 3(12), 11873–11885 CrossRef CAS.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Challenges and Recent Progress in the Development of Si Anodes for Lithium-Ion Battery, Adv. Energy Mater., 2017, 7(23), 1700715 CrossRef.
- J. Wang, T. Xu, X. Huang, H. Li and T. Ma, Recent progress of silicon composites as anode materials for secondary batteries, RSC Adv., 2016, 6(90), 87778–87790 RSC.
- H. Wang, S.-H. Lu, X. Wang, S. Xia and H. Beng Chew, A review of the multiscale mechanics of silicon electrodes in high-capacity lithium-ion batteries, J. Phys. D: Appl. Phys., 2021, 55(6), 063001 CrossRef.
- J. E. Coyle, M. T. Brumbach, G. M. Veith and C. A. Apblett, Investigating the Chemical Reactivity of Lithium Silicate Model SEI Layers, J. Phys. Chem. C, 2020, 124(15), 8153–8161 CrossRef CAS.
- G. Yang, R. L. Sacci, I. N. Ivanov, R. E. Ruther, K. A. Hays, Y. Zhang, P.-F. Cao, G. M. Veith, N. J. Dudney, T. Saito, D. T. Hallinan and J. Nanda, Probing Electrolyte Solvents at Solid/Liquid Interface Using Gap-Mode Surface-Enhanced Raman Spectroscopy, J. Electrochem. Soc., 2019, 166(2), A178 CrossRef CAS.
- C. Stetson, Y. Yin, A. Norman, S. P. Harvey, M. Schnabel, C. Ban, C.-S. Jiang, S. C. DeCaluwe and M. Al-Jassim, Evolution of solid electrolyte interphase and active material in the silicon wafer model system, J. Power Sources, 2021, 482, 228946 CrossRef CAS.
- C. Stetson, M. Schnabel, Z. Li, S. P. Harvey, C.-S. Jiang, A. Norman, S. C. DeCaluwe, M. Al-Jassim and A. Burrell, Microscopic Observation of Solid Electrolyte Interphase Bilayer Inversion on Silicon Oxide, ACS Energy Lett., 2020, 5(12), 3657–3662 CrossRef CAS.
- Y. Ha, C. Stetson, S. P. Harvey, G. Teeter, B. J. Tremolet de Villers, C.-S. Jiang, M. Schnabel, P. Stradins, A. Burrell and S.-D. Han, Effect of Water Concentration in LiPF6-Based Electrolytes on the Formation, Evolution, and Properties of the Solid Electrolyte Interphase on Si Anodes, ACS Appl. Mater. Interfaces, 2020, 12(44), 49563–49573 CrossRef CAS PubMed.
- M. Schnabel, E. Arca, Y. Ha, C. Stetson, G. Teeter, S.-D. Han and P. Stradins, Enhanced Interfacial Stability of Si Anodes for Li-Ion Batteries via Surface SiO2 Coating, ACS Appl. Energy Mater., 2020, 3(9), 8842–8849 CrossRef CAS.
- Y. Yin, E. Arca, L. Wang, G. Yang, M. Schnabel, L. Cao, C. Xiao, H. Zhou, P. Liu, J. Nanda, G. Teeter, B. Eichhorn, K. Xu, A. Burrell and C. Ban, Nonpassivated Silicon Anode Surface, ACS Appl. Mater. Interfaces, 2020, 12(23), 26593–26600 CrossRef CAS PubMed.
- Y. Ha, B. J. Tremolet de Villers, Z. Li, Y. Xu, P. Stradins, A. Zakutayev, A. Burrell and S.-D. Han, Probing the Evolution of Surface Chemistry at the Silicon–Electrolyte Interphase via In Situ Surface-Enhanced Raman Spectroscopy, J. Phys. Chem. Lett., 2020, 11(1), 286–291 CrossRef CAS PubMed.
- C. Stetson, Y. Yin, C.-S. Jiang, S. C. DeCaluwe, M. Al-Jassim, N. R. Neale, C. Ban and A. Burrell, Temperature-Dependent Solubility of Solid Electrolyte Interphase on Silicon Electrodes, ACS Energy Lett., 2019, 4(12), 2770–2775 CrossRef CAS.
- Y. Xu, K. Wood, J. Coyle, C. Engtrakul, G. Teeter, C. Stoldt, A. Burrell and A. Zakutayev, Chemistry of Electrolyte Reduction on Lithium Silicide, J. Phys. Chem. C, 2019, 123(21), 13219–13224 CrossRef CAS.
- F. Wang, L. Wu, B. Key, X.-Q. Yang, C. P. Grey, Y. Zhu and J. Graetz, Electrochemical Reaction of Lithium with Nanostructured Silicon Anodes: A Study by In-Situ Synchrotron X-Ray Diffraction and Electron Energy-Loss Spectroscopy, Adv. Energy Mater., 2013, 3(10), 1324–1331 CrossRef CAS.
- Y. Jin, N.-J. H. Kneusels, P. C. M. M. Magusin, G. Kim, E. Castillo-Martínez, L. E. Marbella, R. N. Kerber, D. J. Howe, S. Paul, T. Liu and C. P. Grey, Identifying the Structural Basis for the Increased Stability of the Solid Electrolyte Interphase Formed on Silicon with the Additive Fluoroethylene Carbonate, J. Am. Chem. Soc., 2017, 139(42), 14992–15004 CrossRef CAS PubMed.
- T. F. Malkowski, Z. Yang, R. L. Sacci, S. E. Trask, M.-T. F. Rodrigues, I. D. Bloom and G. M. Veith, Evaluating the roles of electrolyte components on the passivation of silicon anodes, J. Power Sources, 2022, 523, 231021 CrossRef CAS.
- R. T. Pekarek, A. Affolter, L. L. Baranowski, J. Coyle, T. Hou, E. Sivonxay, B. A. Smith, R. D. McAuliffe, K. A. Persson, B. Key, C. Apblett, G. M. Veith and N. R. Neale, Intrinsic chemical reactivity of solid-electrolyte interphase components in silicon–lithium alloy anode batteries probed by FTIR spectroscopy, J. Mater. Chem. A, 2020, 8(16), 7897–7906 RSC.
- C. L. Seitzinger, R. L. Sacci, J. E. Coyle, C. A. Apblett, K. A. Hays, R. R. Armstrong, A. M. Rogers, B. L. Armstrong, T. H. Bennet, N. R. Neale and G. M. Veith, Intrinsic Chemical Reactivity of Silicon Electrode Materials: Gas Evolution, Chem. Mater., 2020, 32(7), 3199–3210 CrossRef CAS.
- E. Peled and S. Menkin, Review—SEI: Past, Present and Future, J. Electrochem. Soc., 2017, 164(7), A1703–A1719 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries, Electrochim. Acta, 2010, 55(22), 6332–6341 CrossRef CAS.
- N. Takenaka, A. Bouibes, Y. Yamada, M. Nagaoka and A. Yamada, Frontiers in Theoretical Analysis of Solid Electrolyte Interphase Formation Mechanism, Adv. Mater., 2021, 33(37), 2100574 CrossRef CAS PubMed.
- K. Xu, Electrolytes and Interphases in Li-Ion Batteries and Beyond, Chem. Rev., 2014, 114(23), 11503–11618 CrossRef CAS PubMed.
- J. Shin, T.-H. Kim, Y. Lee and E. Cho, Key functional groups defining the formation of Si anode solid-electrolyte interphase towards high energy density Li-ion batteries, Energy Storage Mater., 2020, 25, 764–781 CrossRef.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries, npj Comput. Mater., 2018, 4(1), 15 CrossRef.
- J. R. Dahn, J. C. Burns and D. A. Stevens, Importance of Coulombic Efficiency Measurements in R&D Efforts to Obtain Long-Lived Li-Ion Batteries, Electrochem. Soc. Interface, 2016, 25(3), 75 CAS.
- M. C. Schulze and N. R. Neale, Half-Cell Cumulative Efficiency Forecasts Full-Cell Capacity Retention in Lithium-Ion Batteries, ACS Energy Lett., 2021, 6(3), 1082–1086 CrossRef CAS.
- J. D. McBrayer, C. A. Apblett, K. L. Harrison, K. R. Fenton and S. D. Minteer, Mechanical studies of the solid electrolyte interphase on anodes in lithium and lithium ion batteries, Nanotechnology, 2021, 32(50), 502005 CrossRef CAS PubMed.
- J. D. McBrayer, M.-T. F. Rodrigues, M. C. Schulze, D. P. Abraham, C. A. Apblett, I. Bloom, G. M. Carroll, A. M. Colclasure, C. Fang, K. L. Harrison, G. Liu, S. D. Minteer, N. R. Neale, G. M. Veith, C. S. Johnson, J. T. Vaughey, A. K. Burrell and B. Cunningham, Calendar aging of silicon-containing batteries, Nat. Energy, 2021, 6(9), 866–872 CrossRef CAS.
- J. M. Buriak, Organometallic chemistry on silicon and germanium surfaces, Chem. Rev., 2002, 102(5), 1271–1308 CrossRef CAS PubMed.
- L. M. Wheeler, N. C. Anderson, P. K. B. Palomaki, J. L. Blackburn, J. C. Johnson and N. R. Neale, Silyl Radical Abstraction in the Functionalization of Plasma-Synthesized Silicon Nanocrystals, Chem. Mater., 2015, 27(19), 6869–6878 CrossRef CAS.
- G. M. Carroll, R. Limpens and N. R. Neale, Tuning Confinement in Colloidal Silicon Nanocrystals with Saturated Surface Ligands, Nano Lett., 2018, 18(5), 3118–3124 CrossRef CAS PubMed.
- G. M. Carroll, R. Limpens, G. F. Pach, Y. Zhai, M. C. Beard, E. M. Miller and N. R. Neale, Suppressing Auger Recombination in Multiply Excited Colloidal Silicon Nanocrystals with Ligand-Induced Hole Traps, J. Phys. Chem. C, 2021, 125(4), 2565–2574 CrossRef CAS.
- M. C. Schulze, G. M. Carroll, T. R. Martin, K. Sanchez-Rivera, F. Urias and N. R. Neale, Hydrophobic versus Hydrophilic Interfacial Coatings on Silicon Nanoparticles Teach Us How to Design the Solid Electrolyte Interphase in Silicon-Based Li-Ion Battery Anodes, ACS Appl. Energy Mater., 2021, 4(2), 1628–1636 CrossRef CAS.
- S. Jiang, B. Hu, R. Sahore, H. Liu, G. F. Pach, G. M. Carroll, L. Zhang, B. Zhao, N. R. Neale and Z. Zhang, Tailoring the Surface of Silicon Nanoparticles for Enhanced Chemical and Electrochemical Stability for Li-Ion Batteries, ACS Appl. Energy Mater., 2019, 2(9), 6176–6183 CrossRef CAS.
- A. Kraytsberg and Y. Ein-Eli, Conveying Advanced Li-ion Battery Materials into Practice The Impact of Electrode Slurry Preparation Skills, Adv. Energy Mater., 2016, 6(21), 1600655 CrossRef.
- J. J. Maurer, D. J. Eustace and C. T. Ratcliffe, Thermal characterization of poly(acrylic acid), Macromolecules, 1987, 20(1), 196–202 CrossRef CAS.
- M. E. Spahr, D. Goers, A. Leone, S. Stallone and E. Grivei, Development of carbon conductive additives for advanced lithium ion batteries, J. Power Sources, 2011, 196(7), 3404–3413 CrossRef CAS.
- D.-H. Liu, Z. Bai, M. Li, A. Yu, D. Luo, W. Liu, L. Yang, J. Lu, K. Amine and Z. Chen, Developing high safety Li-metal anodes for future high-energy Li-metal batteries: strategies and perspectives, Chem. Soc. Rev., 2020, 49(15), 5407–5445 RSC.
- J. Zheng, M. S. Kim, Z. Tu, S. Choudhury, T. Tang and L. A. Archer, Regulating electrodeposition morphology of lithium: towards commercially relevant secondary Li metal batteries, Chem. Soc. Rev., 2020, 49(9), 2701–2750 RSC.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, The interplay between solid electrolyte interface (SEI) and dendritic lithium growth, Nano Energy, 2017, 40, 34–41 CrossRef CAS.
- S. C. Nagpure, T. R. Tanim, E. J. Dufek, V. V. Viswanathan, A. J. Crawford, S. M. Wood, J. Xiao, C. C. Dickerson and B. Liaw, Impacts of lean electrolyte on cycle life for rechargeable Li metal batteries, J. Power Sources, 2018, 407, 53–62 CrossRef CAS.
- N. Zhang, T. Deng, S. Zhang, C. Wang, L. Chen, C. Wang and X. Fan, Critical Review on Low-Temperature Li-Ion/Metal Batteries, Adv. Mater., 2022, 34(15), 2107899 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, Poly(ethylene oxide)-based electrolytes for lithium-ion batteries, J. Mater. Chem. A, 2015, 3(38), 19218–19253 RSC.
- S.-D. Han, K. N. Wood, C. Stetson, A. G. Norman, M. T. Brumbach, J. Coyle, Y. Xu, S. P. Harvey, G. Teeter, A. Zakutayev and A. K. Burrell, Intrinsic Properties of Individual Inorganic Silicon–Electrolyte Interphase Constituents, ACS Appl. Mater. Interfaces, 2019, 11(50), 46993–47002 CrossRef CAS PubMed.
- D. Guyomard and J. M. Tarascon, Rechargeable Li1 + x Mn2O4/Carbon Cells with a New Electrolyte Composition: Potentiostatic Studies and Application to Practical Cells, J. Electrochem. Soc., 1993, 140(11), 3071–3081 CrossRef CAS.
- P. Yao, H. Yu, Z. Ding, Y. Liu, J. Lu, M. Lavorgna, J. Wu and X. Liu, Review on Polymer-Based Composite Electrolytes for Lithium Batteries, Front. Chem., 2019, 7, 522 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta08935a |
| This journal is © The Royal Society of Chemistry 2023 |