
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the chemical space of enol silyl ethers: catalytic dicarbofunctionalization enabled by iron catalysis†
Dinabandhu
Sar
,
Shuai
Yin‡
,
Jacob
Grygus‡
,
Ángel
Rentería-Gómez
,
Melanie
Garcia
and
Osvaldo
Gutierrez
 *
*
Department of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: og.labs@tamu.edu
First published on 20th October 2023
Abstract
Enol silyl ethers are versatile, robust, and readily accessible substrates widely used in chemical synthesis. However, the conventional reactivity of these motifs has been limited to classical two electron (2-e) enolate-type chemistry with electrophilic partners or as radical acceptors in one electron (1-e) reactivity leading, in both cases, to exclusive α-monofunctionalization of carbonyls. Herein we describe a mild, fast, and operationally simple one-step protocol that combines readily available fluoroalkyl halides, silyl enol ethers, and, for the first time, hetero(aryl) Grignard reagents to promote selective dicarbofunctionalization of enol silyl ethers. From a broader perspective, this work expands the synthetic utility of enol silyl ethers and establishes bisphosphine–iron catalysis as enabling technology capable of orchestrating selective C–C bond formations with short-lived α-silyloxy radicals with practical implications towards sustainable chemical synthesis.
Introduction
Enol silyl ethers are versatile, robust, and readily accessible substrates widely used in organic synthesis.1–6 However, in general, the reactivity of these motifs has been limited to classical two-electron (2-e) partners7 and severely restricted to electrophiles that can undergo SN2-type chemistry (Fig. 1A; top).8–19 Recently, multicomponent transition-metal radical C–C cross-couplings have emerged as powerful tools that utilize radical acceptors including those bearing alpha-heteroatoms as lynchpins to facilitate the merger of two open-shell coupling partners via the formation and relay of alkyl radicals followed by C–C bond formation with a catalytic transition metal species. Unfortunately, despite long history of enol silyl ethers as radical acceptors to furnish transient α-silyloxy radicals (i.e., via one electron process),20–35 the installation of a second (and potentially enantioenriched) C–C bond via a one-step selective radical/transition metal cross-coupling procedure remains elusive (Fig. 1A; bottom).36 The lack of catalytic methods to selectively undergo radical/transition metal cross-couplings with these α-silyloxy radicals is likely attributed to the high propensity of these species to undergo β-hydride elimination or exceedingly fast single electron transfer (SET) events under (metalla)photoredox catalysis.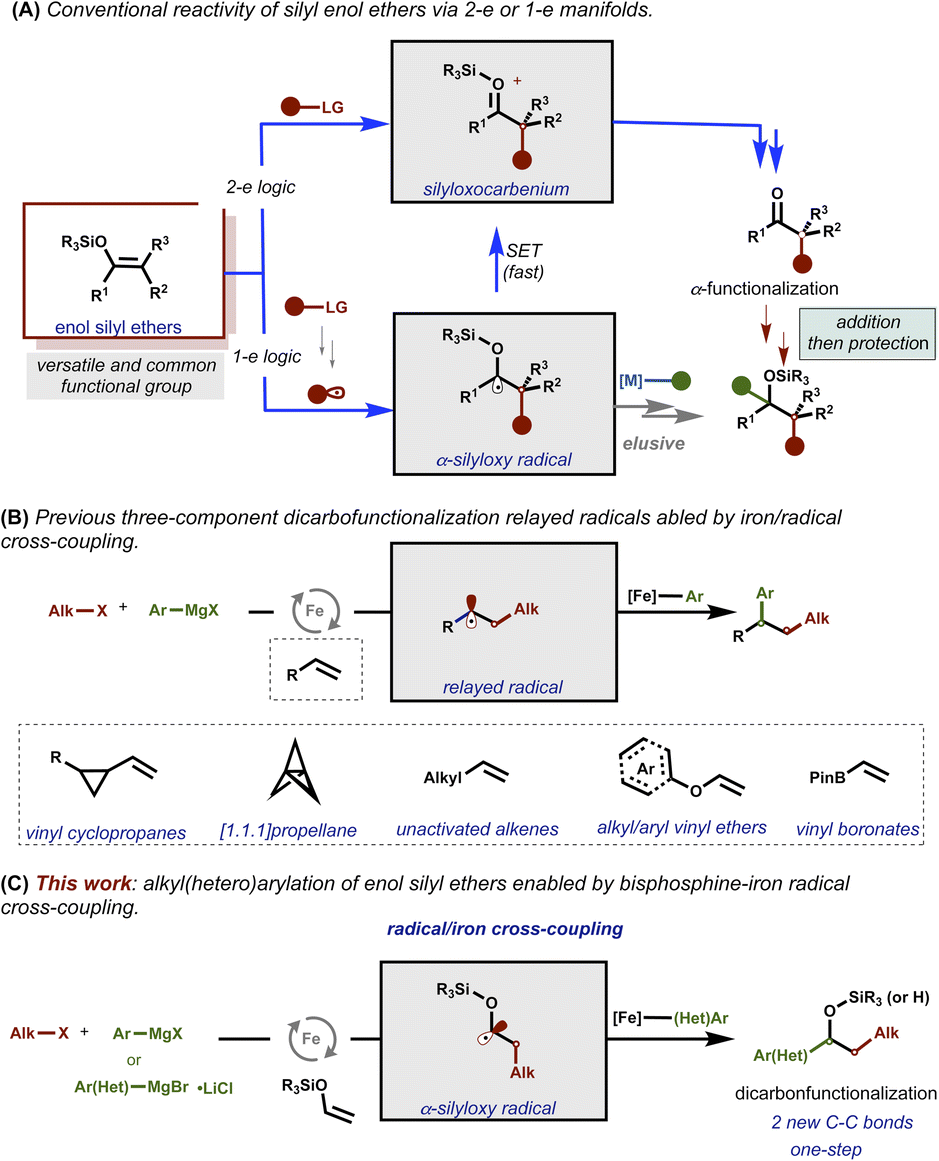 | ||
| Fig. 1 (A) Traditional strategies based on monocarbofunctionalization of silyl enol ethers via 1-e and 2-e enolate-type chemistry. (B) Representative examples of iron-catalyzed dicarbofunctionalization. (C) This work: bisphosphine–iron-catalyzed radical cross-coupling of α-silyloxy radicals. | ||
Recently, our laboratory reported the ability to modulate the speciation of organoiron species to enable multicomponent radical cross-coupling reactions of alkyl halides and sp2-hybridized Grignard reagents with a range of radical acceptors including strain release vinyl cyclopropanes and [1.1.1]propellanes, unactivated alkenes, electron-deficient vinyl boronates, and electron-rich alkyl- and aryl vinyl ethers as effective lynchpins (Fig. 1B).37–41
Based on these reports, we hypothesized that this bisphosphine–iron mechanistic manifold is uniquely positioned to orchestrate a series of events that could allow for unprecedented42–47 recombination of α-silyloxy radicals with organoiron species to selectively form two new C–C bonds with enol silyl ethers (Fig. 1C). Herein, we report the successful one-step multicomponent cross-coupling of enol silyl ethers, di- and tetrafluoroalkyl halides, and hetero/aryl Grignard reagents under bisphosphine–iron catalysis. In contrast to prior methods that need additional synthetic steps, including protecting/deprotecting strategies and purification steps, to add a second C–C bond, this method permits direct formation of valuable and structurally diverse di- and trifluoro alkyl silyl ethers and 2° benzylic alcohols from readily available starting materials.
Results and discussion
Development of a dicarbofunctionalization reaction
Based on recent mechanistic studies in bisphosphine–iron catalysis,48,49 we envisioned that alkyl halide 1.1 could undergo halogen-atom abstraction to form electrophilic radical int-1˙. In turn, regioselective20–33,50–52 Giese addition with electron-rich enol silyl ether 2.1 would result in the formation of α-silyloxy radical int-2˙. Given the unfavourable energies for bisphosphine–iron species to undergo SET with α-silyloxy radicals (see Fig. S10† for calculated redox potentials) and in contrast to prior methods including palladium catalysis34,35 or photocatalysis,20–33 we hypothesized that α-silyloxy radical int-2˙ could undergo selective radical addition to monoaryl bisphosphine–iron(II) species (to form int-3˙) followed by a spin-selective reductive elimination to forge the second C–C bond.41,53 To our delight, using only 2 equiv. of enol silyl ether, we identified Fe(acac)3 and a bisphosphine ligand bearing a thiophene bridge (3,4-bis(dicyclohexylphosphino)thiophene; dcypt) L1 as a suitable iron/ligand combination to enable the formation of the desired three-component coupling product 4, after judicious in situ desilylation with HCl, in 97% 1H NMR yield (Table 1, entry 1). Notably, the use of saturated NH4Cl as a mild quenching strategy also allows for selective dicarbofunctionalization without O–Si bond cleavage (vide infra). The more flexible but sterically and electronically related (1,2-bis(dicyclohexylphosphino)ethane; dcype) L2 ligand was slightly less effective (entry 2) while significantly inferior results were found upon changing the length of the tether and/or P-cyclohexyl substituents (entries 3–5). Presumably, the sterically demanding and electron-rich alkyl substituents in these ligands (L1 and L2) are crucial to control the formation of mono-ligated Fe(II) species involved in radical formation and recombination.41 Finally, with the exception of iron trifluoride (entry 7), other Fe(II) and Fe(III) salts were also effective in this transformation (entries 6–9) albeit lower yields were observed. Furthermore, mixing iron dibromide and L1 led to suitable crystals for X-ray analysis (Table S10†). The crystal showed that Fe(II) salt complexes to only one phosphine ligand to form the corresponding tetrahedral dihalide Fe(II) species, presumably a crucial feature to promote multicomponent iron-catalyzed cross-couplings.41 Subsequent control experiments confirmed that both the iron salt and the ligand are essential to promote the three-component cross-coupling reaction (entries 10–12). Notably, under the standard optimized reaction conditions, the product from atom transfer radical addition (ATRA) to an enol silyl ether was not observed and thus unlikely to proceed via consecutive ATRA followed by Fe-catalyzed cross-coupling. For full optimization studies, including the rate of addition, substrate loadings, solvents, etc., see the ESI (Tables S1–S3†).| a Reactions were carried out on a 0.1 mmol scale. Standard reaction conditions are as follows: 1-(2-bromo-1,1,2,2-tetrafluoroethoxy)-2-methoxybenzene 1.1 (0.1 mmol, 1.0 equiv.), vinyloxy-trimethylsilane 2.1 (0.2 mmol, 2.0 equiv.), p-tolylmagnesium bromide 3.1 (0.2 mmol, 2.0 equiv.) and THF (0.1 mL). Aryl Grignard 3.1 was added dropwise via a syringe pump over 1 h at 0 °C. b Yields were determined by 1H NMR using dibromomethane as an internal standard of the associated deprotected alcohol. Isolated yield (parenthesis). N.D.: not detected. |
|---|
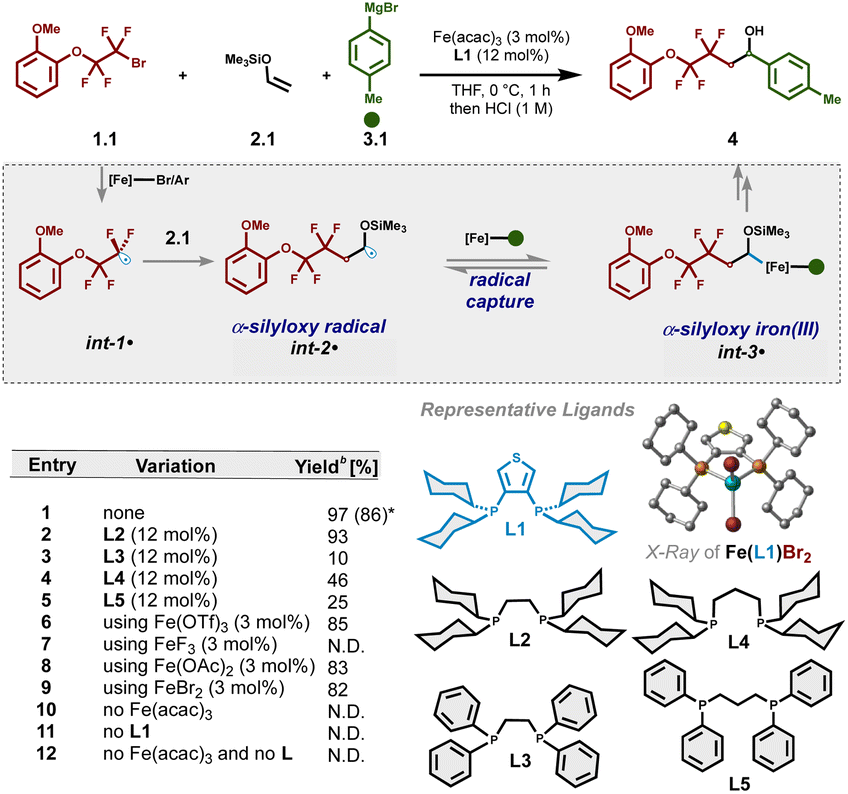
|
Reaction scope
We next evaluated the scope of each coupling partner in this three-component radical cross-coupling reaction involving α-silyloxy radical/organoiron cross-coupling.As shown in Fig. 2, to study the scope and limitations in terms of nucleophiles, a series of heteroaryl- and aryl Grignard reagents, including heteroaryl turbo Grignard reagents, featuring diverse functional groups at varied ring positions were evaluated. Overall, this method tolerated a broad range of electron-rich aryl Grignard nucleophiles bearing substituents at the meta- and para-positions of the aryl ring leading to (after in situ desilylation) the corresponding secondary benzyl alcohol products (4, 8, 10, 12, 14, 18 and 24) or the silyl ether products (5–7, 9, 11, 13, 15, 19 and 25) using sat. NH4Cl as a mild quenching agent in good to excellent yields (39–91%). Similarly, electronically neutral aryl Grignard nucleophiles could also be efficiently engaged in this cross-coupling reaction, as exemplified by secondary alcohol and silyl ether products 16/17 and 20/21. Furthermore, electron-deficient aryl Grignard nucleophiles, including those bearing versatile carbon–halogen bonds for further manipulations via traditional cross-coupling protocols, yielded the targeted alcohols and silyl ether products (22–33) in good to excellent yields.
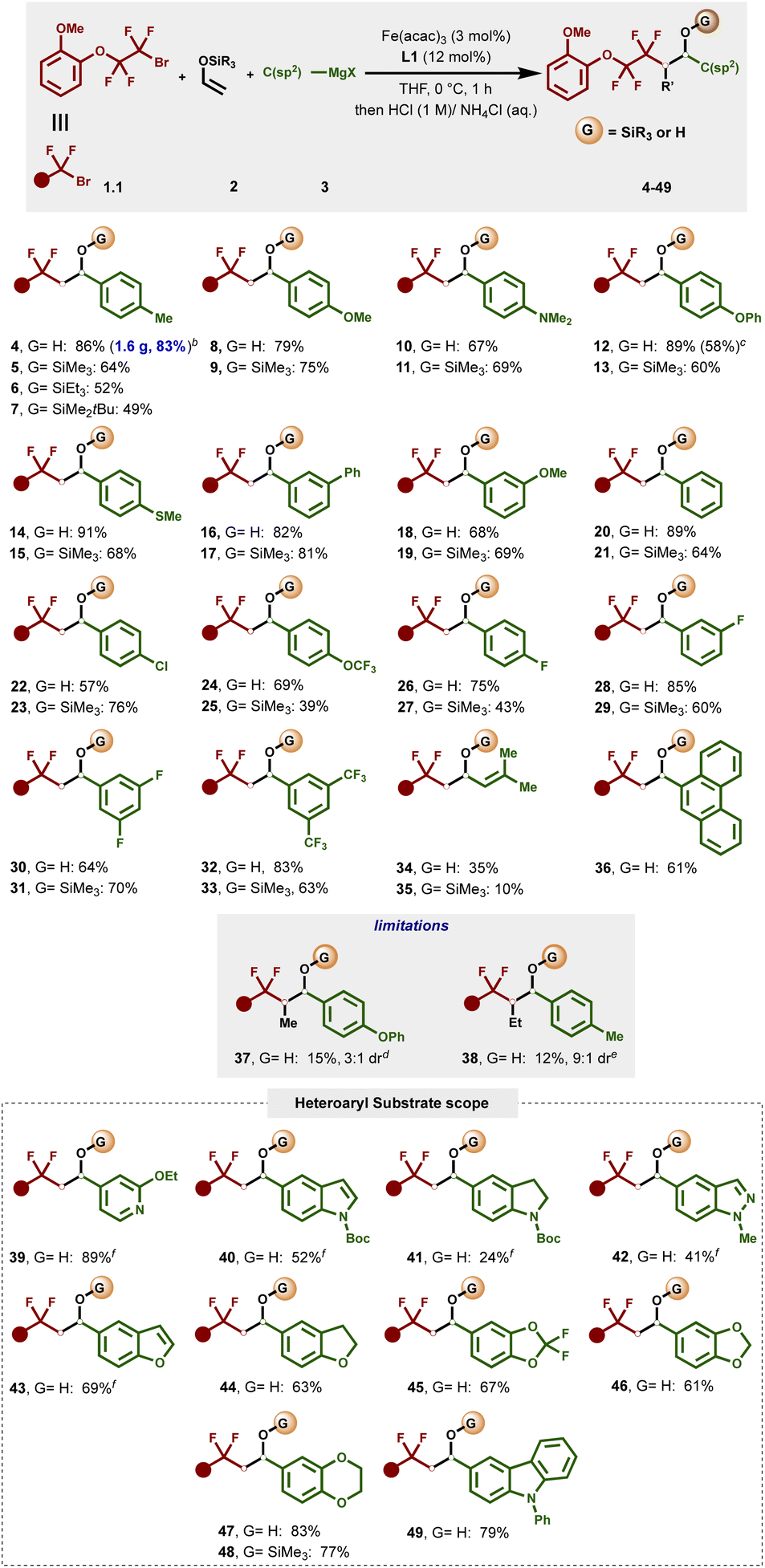 | ||
| Fig. 2 Scope of Grignard reagents. aReaction conditions: reactions were carried out with 1.1 (0.2 mmol), 2 (0.4 mmol), 3 (0.4 mmol), Fe(acac)3 (3 mol%) and L1 (12 mol%) with THF (0.2 mL) at 0 °C. Grignard was added dropwise by using a syringe pump for 1 h. Reaction was performed on a 0.2 mmol scale unless otherwise stated. All reported yields are isolated yields. bUsing a 5.4 mmol scale. cTriethyl(vinyloxysilane) was used as an alkene precursor. d1-Trimethylsilyloxy-1-propene was used as an alkene precursor. e1-Trimethylsilyloxy-1-butene was used as an alkene precursor. fUsing turbo Grignard (Het)ArMgBr·LiCl as a nucleophile. See the ESI† for details. | ||
Unfortunately, the reaction with ortho-substituted aryl Grignard reagents was not successful in this three-component radical cross-coupling transformation presumably due to the high barrier to undergo C–C bond formation with a sterically hindered mono-aryl bisphosphine–iron species (Fig. S6, 3.S6†). Gratifyingly, alkenyl Grignard nucleophile also proved to be a competent partner, affording the desired secondary alcohol (34) and silyl ether product (35), albeit in lower yields. In addition to benzene derivatives, this protocol also tolerates π-extended systems as exemplified using phenanthroline Grignard nucleophile (36). Currently, this method is limited to monosubstituted enol silyl ethers as those bearing additional substituents (i.e., 2.4 and 2.5; Fig. S2†) led to the desired products (37 and 38) in low yields and moderate diastereoselectivity. We next surveyed the scope of this reaction with hetero(aryl) Grignard and turbo Grignard [i.e., (Het)ArMgBr·LiCl] reagents. Gratifyingly, for the first time, we show that these nucleophiles can participate in multicomponent iron-catalyzed cross-coupling reactions. Specifically, the dicarbofunctionalization of enol silyl ethers proved compatible with a wide range of pharmaceutical-relevant heteroaryl nucleophiles including N- and O-containing heteroatoms that can chelate to transition metals and limit catalysis using alternative protocols (Fig. 2; inset). The list of compatible partners include electron-rich 2-ethoxy-4-pyridyl, N-Boc-protected indole and indoline, N-methyl indazole, benzofuran, dihydroxybenzofuran, 2,2-difluorobenzo[d][1,3]dioxole, benzo[d][1,3]dioxole, 2,3-dihydrobenzo[b][1,4]dioxine, and N-phenylcarbazole that deliver the desired cross-coupling products (39–49) in moderate to excellent yields (24–89% yield). Finally, to show the practical utility of the reaction, we executed the reaction on a 5.4 mmol scale, and isolated 4 in 83% yield (parenthesis).
Due to their ability to modulate physical and chemical properties, difluoromethylene and tetrafluoroethylene linkages54–61 are prevalent in the pharmaceutical and agrochemical industries. However, there are no reports on catalytic, one-step dicarbofunctionalization of enol silyl ethers using these valuable motifs.
As shown in Fig. 3, in contrast to tertiary alkyl radicals that required excess enol silyl ether (50), a range of electrophilic radical precursors readily participated in this transformation using 1 equiv. of alkyl halide and 2 equiv. of enol silyl ether and Grignard reagent. Importantly, α,α-difluoro ester bromide engages in the radical cascade/cross-coupling with a range of aryl Grignard reagents leading to challenges in accessing it via traditional routes and pharmaceutically relevant γ-aryl, α,α-difluoro lactone precursors (51–57). Compound 51 produced single crystals by recrystallization in chloroform whose structure was unambiguously determined by X-ray crystallography (Table S7†). Overall, a broad range of di-, tetra- and perfluoro-alkyl radical precursors bearing various functionalities including diethoxyalkyl as protected aldehydes (58), extended alkyl (59 and 60), fluoroalkyl rich (73 and 75) chains, aryl (61) and heteroaryl (74), aryl (thio)ethers (62–64 and 72), esters (65), and diverse amides (66–71) were compatible in this transformation. Notably, a bifunctional reagent bearing two carbon–halogen bonds (i.e., 1-bromo-1,1,2,2-tetrafluoro-4-iodobutane) also reacted with enol silyl ether 2.1 and aryl Grignard nucleophile to give 76 in 55% yield. Unfortunately, unlike their unprotected counterparts (67–69) N-methylated amides failed to give the corresponding product (77). For further substrate limitations see the ESI (Fig. S4–S6†).
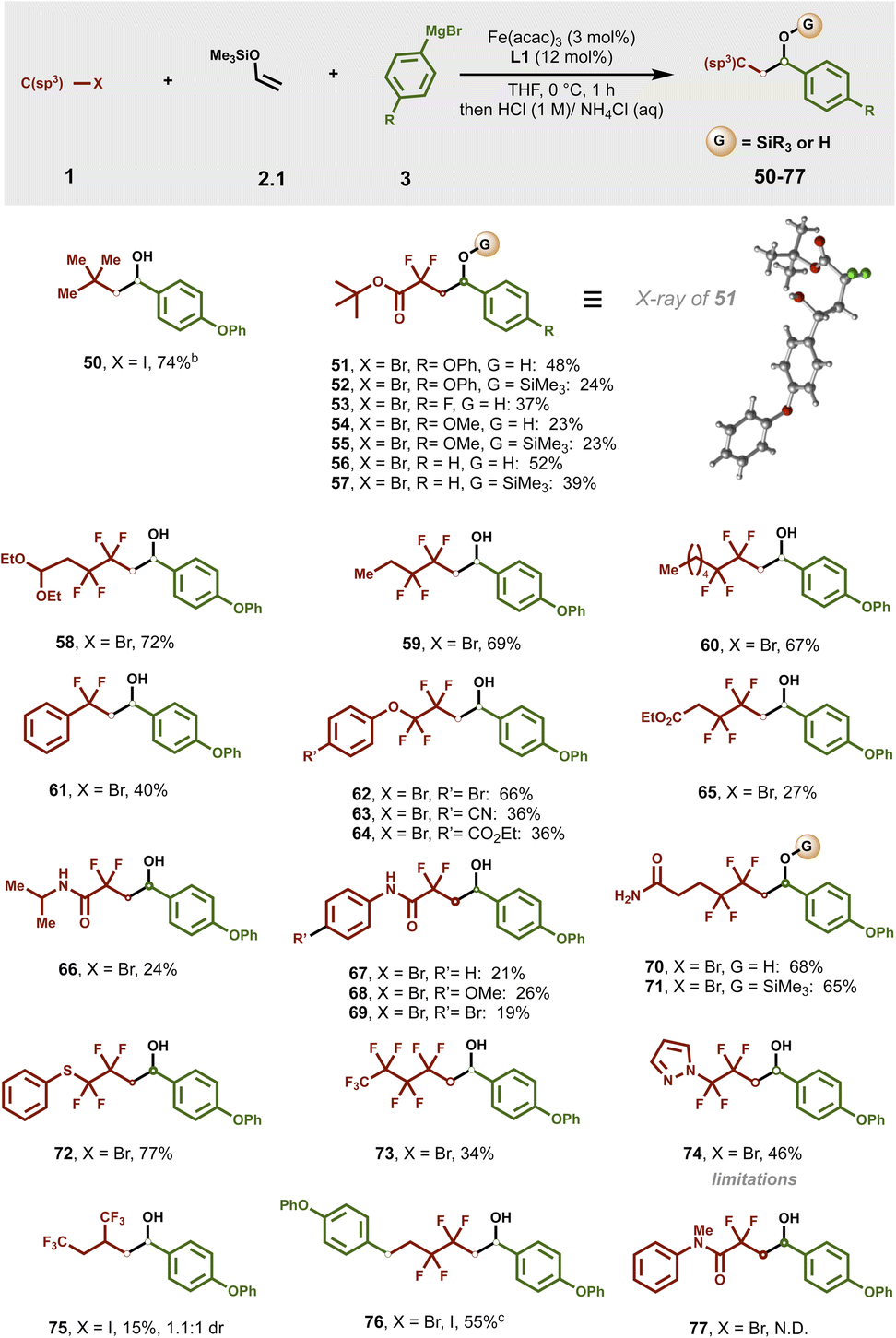 | ||
| Fig. 3 Scope of alkyl halides in dicarbofunctionalization of enol silyl ethers. aReaction conditions: reactions were carried out with 1 (0.2 mmol), 2.1 (0.4 mmol), 3 (0.4 mmol), Fe(acac)3 (3 mol%) and L1 (12 mol%) with THF (0.2 mL) at 0 °C for 1 h. Grignard was added dropwise by using a syringe pump for 1 h. Reaction was performed on a 0.2 mmol scale unless otherwise stated. All cited yields are isolated yields. bUsing 2.8 mmol of 2.1. c1-Bromo-1,1,2,2-tetrafluoro-4-iodobutane was used as a radical precursor. | ||
Synthetic derivations
Derivatization of the secondary alcohol to diverse important synthetic functionalities was further explored to showcase the synthetic utility and practicality of this one-pot, multicomponent iron-catalyzed dicarbofunctionalization of enol silyl ethers (Fig. 4). Selective reduction of the hydroxy group of 4 afforded the corresponding alkane in 62% yield (78). In addition, 4 can be easily converted to the corresponding β-fluoroenones (79) through Swern oxidation followed by dehydrofluorination in good yield.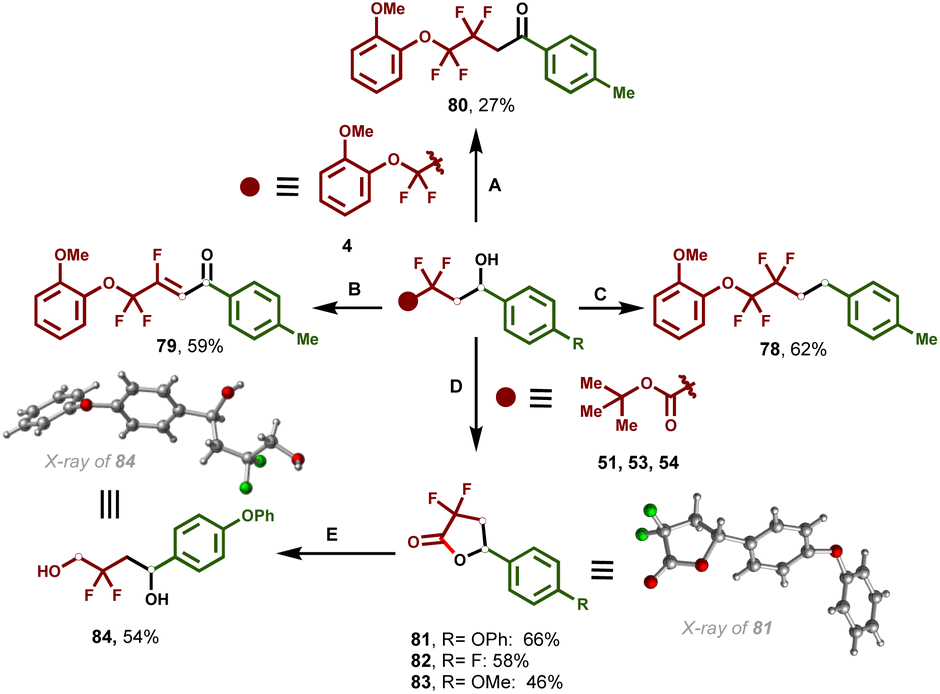 | ||
Fig. 4 Synthetic derivations. Condition A: (NH4)2Ce(NO3)6 (2.5 equiv.) MeCN/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 50 °C, and 16 h. Condition B: (1) (COCl)2 (1 equiv.), DMSO (2 equiv.), CH2Cl2, −78 °C, and 30 min; (2) Et3N (4 equiv.), −78 °C, 30 min, and rt. Condition C: Pd/C (10%), TES (1 equiv.), MeOH, and 30 min. Condition D: TFA, CH2Cl2, 0 °C-rt, and 8 h. Condition E: NaBH4, MeOH, and rt. :1), 50 °C, and 16 h. Condition B: (1) (COCl)2 (1 equiv.), DMSO (2 equiv.), CH2Cl2, −78 °C, and 30 min; (2) Et3N (4 equiv.), −78 °C, 30 min, and rt. Condition C: Pd/C (10%), TES (1 equiv.), MeOH, and 30 min. Condition D: TFA, CH2Cl2, 0 °C-rt, and 8 h. Condition E: NaBH4, MeOH, and rt. | ||
Selective oxidation using ceric ammonium nitrate (CAN) led to the corresponding ketone (80). In addition, hydroxy esters (51–54) synthesized using optimized iron-catalyzed multicomponent cross-coupling were further derivatized to the corresponding medicinally relevant γ-aryl, α,α-difluorolactones (81–83). In turn, lactone 81 was further reduced using sodium borohydride (NaBH4) to the corresponding diol (84). Compounds 81 and 84 produced single crystals whose structure was confirmed by X-ray crystallographic analysis (Tables S8 and S9† respectively).
Mechanistic elucidation
Next, to gain insight into the mechanism by which this bisphosphine–iron catalyst is uniquely suitable for effective α-silyloxy radical/cross-coupling, we undertook a series of experimental and computational studies. First, to probe the formation of α-silyloxy radicals in this transformation, we used 4-bromo,3,3,4,4-tetrafluoro-1-butene (A′) as a radical trap. As shown in Fig. 5A, A′ led to the formation of a cyclic secondary alcohol (85) and silyl ether (86) in good to excellent yield, consistent with a radical relay/cross-coupling cascade. Furthermore, DFT calculations also support the formation of an α-silyloxy radical followed by a rapid 5-exo radical cyclization (barrier ∼ 9 kcal mol−1) and radical/cross-coupling sequence (Fig. S12†). Notably, this multicomponent annulation/cross-coupling (MAC) protocol could be extended to other systems including disubstituted enol silyl ethers leading to the corresponding annulation product (87), albeit in lower yield with 5:1 dr. Cyclic enol silyl ethers (e.g., 1-(trimethylsiloxy)cyclopentene) also formed the corresponding medicinally relevant bicyclo[3.3.0]diquinane ring systems bearing a hydroxyl (88) and silyl ether (89) at the ring juncture,62 both as trans isomers, as revealed by NOESY (Fig. S7 and S8†) with high levels of diastereoselectivity (up to 15:1 dr).
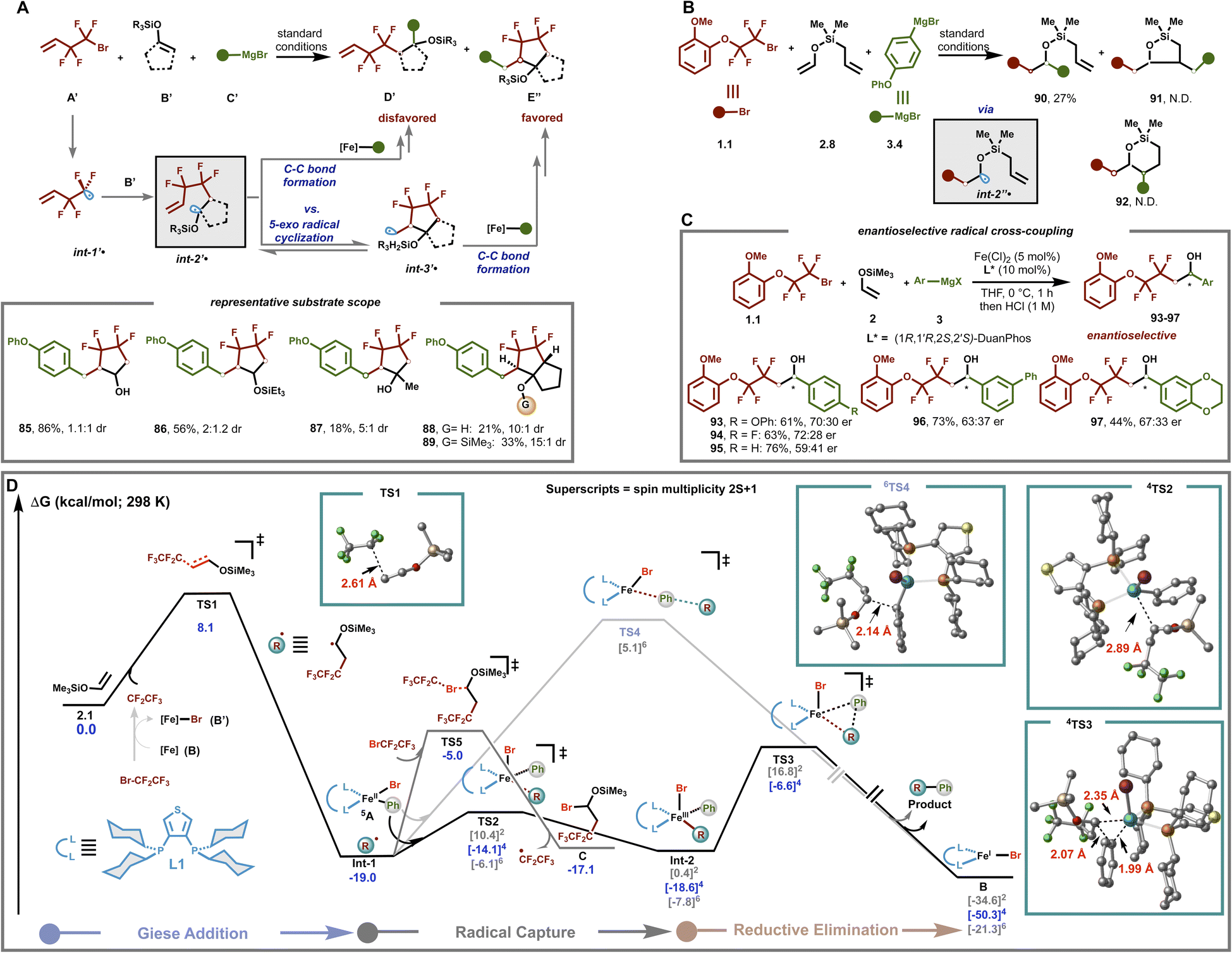 | ||
| Fig. 5 Mechanistic investigation. [A] Radical cascade annulation. [B] Reactivity evaluation between (fluoro)alkyl radical addition to enol silyl ether, and cascade cyclization of silicon-tethered olefin. [C] Enantioselective C–C bond formation with α-silyloxy radicals. [D] DFT calculation for dicarbofunctionalization of enol silyl ether. Calculated Gibbs free energies [uM06L-d3/def2tzvpp-SMD(THF)//uB3LYP-d3/def2-svp-CPCM(THF)] are given in kcal mol−1. All cited yields are isolated yields and er was determined by chiral HPLC. | ||
To gain insights into the chemo- and regio-selectivity, we next proceeded to examine the reactivity using mechanistic probe 2.8 that bears two electronically distinct, but sterically similar, alkene functionalities (Fig. 5B). Interestingly, we only observed the uncyclized (dicarbofunctionalization) product 90 with 27% yield, presumably from direct cross-coupling of organo-iron species with int2′′˙. This result implies that selective addition of the difluoroalkyl radical to the electron-rich enol silyl ether (rather than an alkyl substituted alkene) followed by a radical cross-coupling event with organoiron is faster than competing 5-exo radical cyclization (calculated barrier ∼ 14 kcal mol−1; see Fig. S11†) or potential halogen atom abstraction of alkyl halides by the electron-rich α-silyloxy radical (see the ESI†). Taken together, the results in Fig. 5A and B coupled with the computational studies allow us to “bracket” the C(sp3)–C(sp2) bond formation step at ∼10–14 kcal mol−1. Finally, preliminary results showed that the protocol is also applicable to the asymmetric dicarbofunctionalization of enol silyl ethers leading to the desired chiral alcohols 93–97 (Fig. 5C and Table S4†). These results are more consistent with enantioselective radical/metal cross-coupling between the α-silyloxy radical and chiral mono-aryl organoiron(II) species (vide infra) rather than the formation of TMS–oxonium species followed by non-stereoselective Grignard addition.
Lastly, we carried out dispersion-corrected density functional theory (DFT) calculations to gain insight into the nature of C–C bond formation (Fig. 5D). Based on the related mechanistic studies on the iron catalyzed cross-coupling reaction,41,63 the direct halogen abstraction of the corresponding fluoroalkyl halide by an intermediate-spin distorted square planar bis-aryl bisphosphine–iron species (or by an Fe(I) species) can lead to ˙CF2CF3 radical formation (Fig. S14†). In turn, ˙CF2CF3 could undergo fast and irreversible Giese addition to enol silyl ether 2.1 to form radical Int-1 (barrier is only 8.1 kcal mol−1). Then, Int-1 found adds rapidly and reversibly to the tetrahedral, monoligated mono-aryl Fe(II) species 5A through the selective quartet spin state TS2 leading to the formation of distorted square pyramidal Fe(III) alkyl intermediate Int-2, in the same spin state. Finally, subsequent and irreversible reductive elimination via selective quartet spin state TS3 produces the targeted product and Fe(I) species (4B), which can then restart the catalytic process.65 Overall, in agreement with the “bracketed” 10–14 kcal mol−1 barrier (vide supra), the C(sp2)–C(sp3) bond formation between these two species is predicted to be ∼12 kcal mol−1. Finally, as in the past, calculations rule out the outer-sphere C–C bond formation via high spin 6TS4.41,53,63 Furthermore, we also studied an alternative pathway in which a carbo-bromination product (C) is formed via ATRA64 to an enol silyl ether through TS5, but the calculations ruled out the this pathway due to a higher energy barrier (∼14 kcal mol−1) compared with 4TS2.
Conclusion
In conclusion, we have developed a unique iron–bisphosphine manifold that expands the chemical space and reactivity patterns of enol silyl ethers leading to selective dicarbofunctionalization with alkyl halides and (hetero)aryl Grignard and turbo Grignard reagents as coupling partners. Overall, this method allows selective formation and cross-coupling of α-silyloxy radicals leading to the formation of two new C–C bonds in one step under mild conditions, exceedingly fast reaction times, and with broad functional group tolerance. Experimental and computational studies were used to gain insights into the mechanism, selectivity, and nature of C–C bond formation. From a broader perspective, we anticipate that this method will pave the way towards forming transient carbon-centered radicals with low redox potentials and undergoing enantioselective carbon–carbon bond formation with chiral organoiron species.Data availability
All detailed experimental procedures, additional tables for optimization data, characterization data, computational data (methods, results, and coordinates), NMR spectra, and crystallographic data are available in the ESI.†Author contributions
D. S. developed the method, conducted experiments, and wrote the manuscript. S. Y. and J. G. performed materials preparation and chromatography. M. G. performed material preparation. A. R.-G. carried out DFT calculations. O. G. directed the investigations and wrote the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
O. G. thanks the National Institutes of Health (R35GM137797), National Science Foundation (#2246853), Camille and Henry Dreyfus Foundation, and Welch Foundation (A-2102-20220331) for funding, and Texas A&M University HPRC resources (https://hprc.tamu.edu) for computational resources. We thank Dr Nattamai Bhuvanesh for help with X-ray diffraction and Dr Yohannes H. Rezenom for help with the HRMS.Notes and references
- G. Stork and P. F. Hudrlik, J. Am. Chem. Soc., 1968, 90, 4462–4464 CrossRef CAS.
- J. K. Rasmussen, Synthesis, 1977, 91–110 CrossRef CAS.
- X.-S. Hu, J.-S. Yu and J. Zhou, Chem. Commun., 2019, 55, 13638–13648 RSC.
- I. Kuwajima and E. Nakamura, Acc. Chem. Res., 1985, 18, 181–187 CrossRef CAS.
- M. Parasmam, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef PubMed.
- For a recent example of an alternative method for the synthesis of stereochemical defined silyl enol ethers, see and references therein: S. Guven, G. Kundu, A. Weßels, J. S. Ward, K. Rissanen and F. Schoenebeck, J. Am. Chem. Soc., 2021, 143, 8375–8380 CrossRef CAS PubMed.
- For a recent example that uses enolates as electrophilic partners in 2-e logic, see and references therein: Z.-T. He and J. F. Hartwig, Nat. Chem., 2019, 11, 177–183 CrossRef CAS PubMed.
- T. Song, S. Arseniyadis and J. Cossy, Org. Lett., 2019, 21, 603–607 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, L. H. Lim, G. C. P. Quang, H. Hirao and J. S. Zhou, Angew. Chem., Int. Ed., 2013, 52, 5807–5812 CrossRef CAS PubMed.
- F.-M. Liao, Z.-Y. Cao, J.-S. Yu and J. Zhou, Angew. Chem., Int. Ed., 2017, 56, 2459–2463 CrossRef CAS PubMed.
- A. Bigot, A. E. Williamson and M. J. Gaunt, J. Am. Chem. Soc., 2011, 133, 13778–13781 CrossRef CAS PubMed.
- J. S. Harvey, S. P. Simonovich, C. R. Jamison and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 13782–13785 CrossRef CAS PubMed.
- M. Seto, J. L. Roizen and B. M. Stoltz, Angew. Chem., Int. Ed., 2008, 47, 6873–6876 CrossRef CAS PubMed.
- M. Chen and J. F. Hartwig, Angew. Chem., Int. Ed., 2014, 53, 8691–8695 CrossRef CAS PubMed.
- T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192–17193 CrossRef CAS PubMed.
- E. Belanger, K. Cantin, O. Messe, M. Tremblay and J.-F. Paquin, J. Am. Chem. Soc., 2007, 129, 1034–1035 CrossRef CAS PubMed.
- P.-Y. Wang, I. Massad and I. Marek, Angew. Chem., Int. Ed., 2021, 60, 12765–12769 CrossRef CAS PubMed.
- Q. Zhu, E. C. Gentry and R. R. Knowles, Angew. Chem., Int. Ed., 2016, 55, 9969–9973 CrossRef CAS PubMed.
- T. Iwama, V. B. Birman, S. A. Kozmin and V. H. Rawal, Org. Lett., 1999, 1, 673–676 CrossRef CAS PubMed.
- R. Mao, Z. Yuan, Y. Li and J. Wu, Chem.–Eur. J., 2017, 23, 8176–8179 CrossRef CAS PubMed.
- D. Spinnato, B. Schweitzer-Chaput, G. Goti, M. Oseka and P. Melchiorre, Angew. Chem., Int. Ed., 2020, 59, 9485–9490 CrossRef CAS PubMed.
- P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed.
- W. Kong, C. Yu, H. An and Q. Song, Org. Lett., 2018, 20, 349–352 CrossRef CAS PubMed.
- F. Pettersson, G. Bergonzini, C. Cassani and C.-J. Wallentin, Chem.–Eur. J., 2017, 23, 7444–7447 CrossRef CAS PubMed.
- S.-H. Cai, J.-H. Xie, S. Song, L. Ye, C. Feng and T.-P. Loh, ACS Catal., 2016, 6, 5571–5574 CrossRef CAS.
- E. Selmi-Higashi, J. Zhang, X. C. Cambeiro and S. Arseniyadis, Org. Lett., 2021, 23, 4239–4243 CrossRef CAS PubMed.
- V. I. Supranovich, V. V. Levin and A. D. Dilman, Beilstein J. Org. Chem., 2020, 16, 1550–1553 CrossRef CAS PubMed.
- L. I. Panferova, M. O. Zubkov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Angew. Chem., Int. Ed., 2021, 60, 2849–2854 CrossRef CAS PubMed.
- H. Cao, S. Ma, Y. Feng, Y. Guo and P. Jiao, Chem. Commun., 2022, 58, 1780–1783 RSC.
- H.-B. Yang and N. Selander, Chem.–Eur. J., 2017, 23, 1779–1783 CrossRef CAS PubMed.
- N. Esumi, K. Suzuki, Y. Nishimoto and M. Yasuda, Org. Lett., 2016, 18, 5704–5707 CrossRef CAS PubMed.
- Y.-b. Wu, G.-P. Lu, B.-J. Zhou, M.-J. Bu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 5965–5968 RSC.
- C. Banoun, F. Bourdreux, E. Magnier and G. Dagousset, Org. Lett., 2021, 23, 8926–8930 CrossRef CAS PubMed.
- B. Zhao, R. Shang, G.-Z. Wang, S. Wang, H. Chen and Y. Fu, ACS Catal., 2020, 10, 1334–1343 CrossRef CAS.
- W.-L. Xing, R. Shang, G.-Z. Wang and Y. Fu, Chem. Commun., 2019, 55, 14291–14294 RSC.
- J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutierrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503 CrossRef CAS PubMed.
- L. Liu, W. Lee, M. Yuan, C. Acha, M. B. Geherty, B. Williams and O. Gutierrez, Chem. Sci., 2020, 11, 3146–3155 RSC.
- A. Rentería-Gómez, W. Lee, S. Yin, M. Davis, A. R. Gogoi and O. Gutierrez, ACS Catal., 2022, 12, 11547–11556 CrossRef.
- L. Liu, W. Lee, C. R. Youshaw, M. Yuan, M. B. Geherty, P. Y. Zavalij and O. Gutierrez, Chem. Sci., 2020, 11, 8301–8305 RSC.
- M. E. Rotella, D. Sar, L. Liu and O. Gutierrez, Chem. Commun., 2021, 57, 12508–12511 RSC.
- L. Liu, M. C. Aguilera, W. Lee, C. R. Youshaw, M. L. Neidig and O. Gutierrez, Science, 2021, 374, 432–439 CrossRef CAS PubMed.
- T. Nakashima, H. Fujimori, K. Ohmatsu and T. Ooi, Chem.–Eur. J., 2021, 27, 9253–9256 CrossRef CAS PubMed.
- T. Nakashima, K. Ohmatsu and T. Ooi, Org. Biomol. Chem., 2021, 19, 141–145 RSC.
- K. Ohmatsu, T. Nakashima, M. Sato and T. Ooi, Nat. Commun., 2019, 10, 2706 CrossRef PubMed.
- W. Xie, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 20588–20593 CrossRef CAS PubMed.
- L. Huang, M. Szewczyk, R. Kancherla, B. Maity, C. Zhu, L. Cavallo and M. Rueping, Nat. Commun., 2023, 14, 548 CrossRef CAS PubMed.
- K. Liu and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202206533 CrossRef CAS PubMed.
- M. L. Neidig, S. H. Carpenter, D. J. Curran, J. C. DeMuth, V. E. Fleischauer, T. E. Iannuzzi, P. G. N. Neate, J. D. Sears and N. J. Wolford, Acc. Chem. Res., 2019, 52, 140–150 CrossRef CAS PubMed.
- J. D. Sears, P. G. N. Neate and M. L. Neidig, J. Am. Chem. Soc., 2018, 140, 11872–11883 CrossRef CAS PubMed.
- D. Li, J. Org. Chem., 2021, 86, 609–618 CrossRef CAS PubMed.
- L. Xu, X. Liu, G. R. Alvey, A. Shatskiy, J.-Q. Liu, M. D. Kärkäs and X.-S. Wang, Org. Lett., 2022, 24, 4513–4518 CrossRef CAS PubMed.
- Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2012, 48, 5355–5357 RSC.
- W. Lee, J. Zhou and O. Gutierrez, J. Am. Chem. Soc., 2017, 139, 16126–16133 CrossRef CAS PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed.
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
- M. E. Christy, C. D. Colton, M. Mackay, W. H. Staas, J. B. Wong, E. L. Engelhardt, M. L. Torchiana and C. A. Stone, J. Med. Chem., 1977, 20, 421–430 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. Václavík, I. Klimánková, A. Budinská and P. Beier, Eur. J. Org Chem., 2018, 3554–3593 CrossRef.
- J.-F. Tan, C. T. Bormann, K. Severin and N. Cramer, Chem. Sci., 2022, 13, 3409–3415 RSC.
- C. F. Heinrich, C. P. Laurence, M. P. Geoffroy and M. Miesch, Synthesis, 2016, 48, 1607–1615 CrossRef CAS.
- A. K. Sharma, W. M. C. Sameera, M. Jin, L. Adak, C. Okuzono, T. Iwamoto, M. Kato, M. Nakamura and K. Morokuma, J. Am. Chem. Soc., 2017, 139, 16117–16125 CrossRef CAS PubMed.
- W. Pu, D. Sun, W. Fan, W. Pan, Q. Chai, X. Wang and Y. Lv, Chem. Commun., 2019, 55, 4821 RSC.
- M. C. Aguilera, A. R. Gogoi, W. Lee, L. Liu, W. Brennessel, O. Gutierrez and M. L. Neidig, ACS Catal., 2023, 13, 8987 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2170253, 2211073, 2213284 and 2254575. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc04549h |
| ‡ These authors contributed equally: Shuai Yin and Jacob Grygus. |
| This journal is © The Royal Society of Chemistry 2023 |