
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUsing molecular straps to engineer conjugated porous polymer growth, chemical doping, and conductivity†
Manikandan
Mohanan
ab,
Humayun
Ahmad
c,
Pooja
Ajayan
d,
Prashant K.
Pandey
a,
Benjamin M.
Calvert
ab,
Xinran
Zhang
be,
Fu
Chen
f,
Sung J.
Kim
 g,
Santanu
Kundu
c and
Nagarjuna
Gavvalapalli
*ab
g,
Santanu
Kundu
c and
Nagarjuna
Gavvalapalli
*ab
aDepartment of Chemistry, Georgetown University, Washington, D.C., USA. E-mail: ng554@georgetown.edu
bInstitute for Soft Matter Synthesis and Metrology, Georgetown University, Washington, D.C., USA
cDepartment of Physics, Georgetown University, Washington, D.C., USA
dDave C. Swalm School of Chemical Engineering, Mississippi State University, Mississippi, USA
eDepartment of Chemistry, University of California, Riverside, California, USA
fDepartment of Chemistry, University of Maryland, College Park, Maryland, USA
gDepartment of Chemistry, Howard University, Washington D.C., USA
First published on 4th May 2023
Abstract
Controlling network growth and architecture of 3D-conjugated porous polymers (CPPs) is challenging and therefore has limited the ability to systematically tune the network architecture and study its impact on doping efficiency and conductivity. We have proposed that π-face masking straps mask the π-face of the polymer backbone and therefore help to control π–π interchain interactions in higher dimensional π-conjugated materials unlike the conventional linear alkyl pendant solubilizing chains that are incapable of masking the π-face. Herein, we used cycloaraliphane-based π-face masking strapped monomers and show that the strapped repeat units, unlike the conventional monomers, help to overcome the strong interchain π–π interactions, extend network residence time, tune network growth, and increase chemical doping and conductivity in 3D-conjugated porous polymers. The straps doubled the network crosslinking density, which resulted in 18 times higher chemical doping efficiency compared to the control non-strapped-CPP. The straps also provided synthetic tunability and generated CPPs of varying network size, crosslinking density, dispersibility limit, and chemical doping efficiency by changing the knot to strut ratio. For the first time, we have shown that the processability issue of CPPs can be overcome by blending them with insulating commodity polymers. The blending of CPPs with poly(methylmethacrylate) (PMMA) has enabled them to be processed into thin films for conductivity measurements. The conductivity of strapped-CPPs is three orders of magnitude higher than that of the poly(phenyleneethynylene) porous network.
Introduction
π-Conjugated porous polymers (CPPs) are porous and have extended π-conjugated networks in 3 dimensions, which makes them attractive for sensing, storage, separation, and optoelectronic applications.1–15 CPPs have several interesting features that make them better suited than 1D-π-conjugated polymers for electronic and energy-related applications.16–20 For example, CPPs provide a porous and morphologically stable architecture for acceptors to interact with the polymer and facilitate charge transfer.21–24 3D-network architectures provide quick and efficient pathways to move charges and energy within the network.25,26 Also, the rate of doping and stability of the charge transfer complex are higher for CPPs than the linear polymers because of the suppression of π–π stacking interactions and higher free surface area.23,24 Despite these interesting features, CPPs are relatively underexplored for energy harvesting applications due to poor processability and lower electrical conductivities.1,4,27 Systematic studies focused towards understanding the effect of the CPP structure and network architecture on doping efficiency and conductivity are essential to explore and harness the advantages of the CPPs porous architecture for energy harvesting applications. However, to date, there are no reports in this direction because controlling the growth and architecture of 3D-porous polymer network is challenging.Controlled synthesis of π-conjugated materials beyond 1 dimension is still in its infancy because linear alkyl pendant chains, which are successful in rendering soluble linear 1D-π-conjugated polymers, do not help to overcome the strong interchain π–π interactions in higher dimensional π-conjugated materials. Typically, in the absence of pendant solubilizing chains, a π-conjugated polymer chain of molecular weight higher than ca. 2 kDa will phase separate, even if it is a linear chain, due to solubility issues. Therefore, during the CPP growth, the rigid three-dimensional network coupled with strong π–π intermolecular interactions make the growing network insoluble in typical organic solvents.28–31 Strong interchain interactions within the aggregates limit the diffusion of monomers into it, leading to incomplete conversion of knots and struts resulting in a lack of control over the network growth and crosslinking density.31,32 The network growth predominantly depends on the heterogeneous reaction between insoluble aggregates themselves or aggregates with soluble monomers/oligomers. This makes it challenging to tune the network growth and crosslinking density.
We have developed strapped aryl monomers33 that can directly mask the π-face and overcome interchain π–π interactions. The strapped aryl monomers (viz. cycloaraliphanes) consist of an aryl face and a cycloalkyl face; the aryl group generates a π-conjugated polymer upon polymerization whereas the cycloalkyl group positioned either above or below the π-conjugation plane masks the π-surface and hinders interchain π–π interactions.33 We have shown that the strapped monomers generate soluble linear 1D-π-conjugated polymers of higher molecular weight (Mn: 23 kDa)33,34 without the need for pendant solubilizing chains, and 2D-oligomers without pendant solubilizing chains.35 This is mainly because the straps reduce interchain interactions, allowing the polymer chains to grow in solution.
In this work, we have investigated the efficacy of the straps in hindering interchain interactions during the CPP network growth and their ability to provide control over network growth and crosslinking density. We hypothesize that the straps reduce interchain π–π interactions in CPPs and generate a swollen network, which enables the monomer to diffuse-in, react, and provide enhanced reaction time for the network to grow compared to the non-strapped units. Thus, the straps provide control over the network growth and enabled us to study the impact of network structural parameters on size, dispersibility, chemical doping, and electrical conductivity. Herein, we have synthesized a series of strapped tris(arylethynyl)phosphane-conjugated microporous polymers (S-CPP-1–3) by varying the knot (A3) to strut (B2) ratio, a control non-strapped CPP (NS-CPP), corresponding trimers (S-trimer and NS-trimer), and a strapped linear polymer (S-LP) as shown in Fig. 1. The impact of the knot to strut ratio on the network size, dispersibility limit, chemical doping efficiency, and conductivity is studied within the S-CPP series and is compared with NS-CPP and S-LP. We have also shown that the physical blend of S-CPPs with insulating commodity polymer (PMMA) helps to overcome the CPPs poor processability, which has been identified as a critical challenge for the widespread use of CPPs, and to process CPPs into thin films for electrical conductivity measurements.
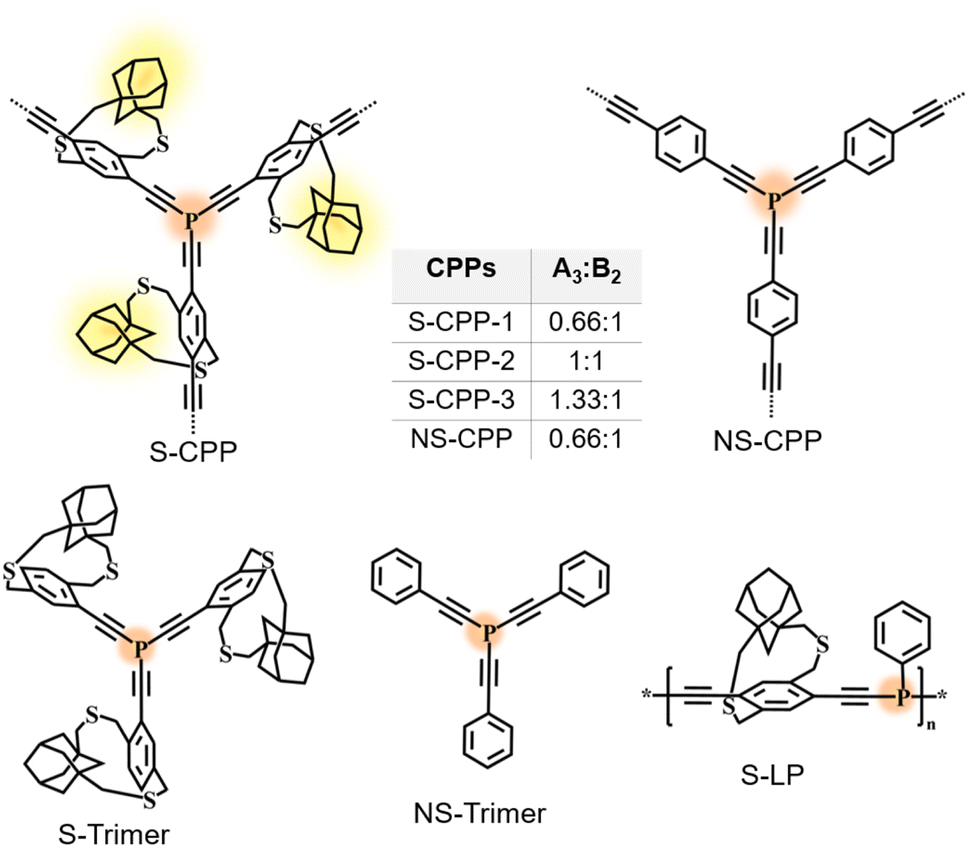 | ||
| Fig. 1 Chemical structures of the series of strapped CPPs (S-CPP-1–3), non-strapped CPP (NS-CPP), model trimers (S-trimer and NS-trimer), and strapped linear polymer (S-LP) studied in this work. | ||
Results and discussion
The strapped tris(arylethynyl)phosphane porous polymer networks (S-CPP-1–3) were synthesized by reacting the phosphorous trichloride knot (A3) with the adamantano cyclophane diacetylene (1) strut (B2) in the presence of a Ni(II) catalyst in a mixture of toluene and triethylamine (Scheme 1). Adamantanocyclophane diacetylene (1) was synthesized following our previous reports.33 PCl3 was selected as the trifunctional A3 monomer for two reasons. First, phosphorous extends the π-conjugation across the network unlike the meta-conjugated 1,3,5-phenylene trifunctional core. For example, the Gates group has shown that phosphorous extends π-conjugation along the polymer backbone and the Lucht group has used the lone pair of electrons on phosphorous to generate polymeric turn-on sensors for gold cations.36–38 Also, tri, di, and mono-reacted phosphorous with aryl acetylenes result in crosslinked (dendritic), linear, and terminal groups respectively, and are relatively easy to differentiate and identify using 31P-NMR compared to a completely carbon-based network. In order to tune the network crosslinking density, the molar ratio of the knot was systematically increased from 0.66 to 1.33 with respect to the strut. S-CPP-1 has the equal stoichiometry of reactive functional groups, which translates to a knot to strut molar ratio of 0.66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 whereas both S-CPP-2 and -3 have a higher mole ratio of the knot than the required and the knot to strut ratio is 1:1 and 1.33:1 for S-CPP-2 and S-CPP-3, respectively.
:1 whereas both S-CPP-2 and -3 have a higher mole ratio of the knot than the required and the knot to strut ratio is 1:1 and 1.33:1 for S-CPP-2 and S-CPP-3, respectively.
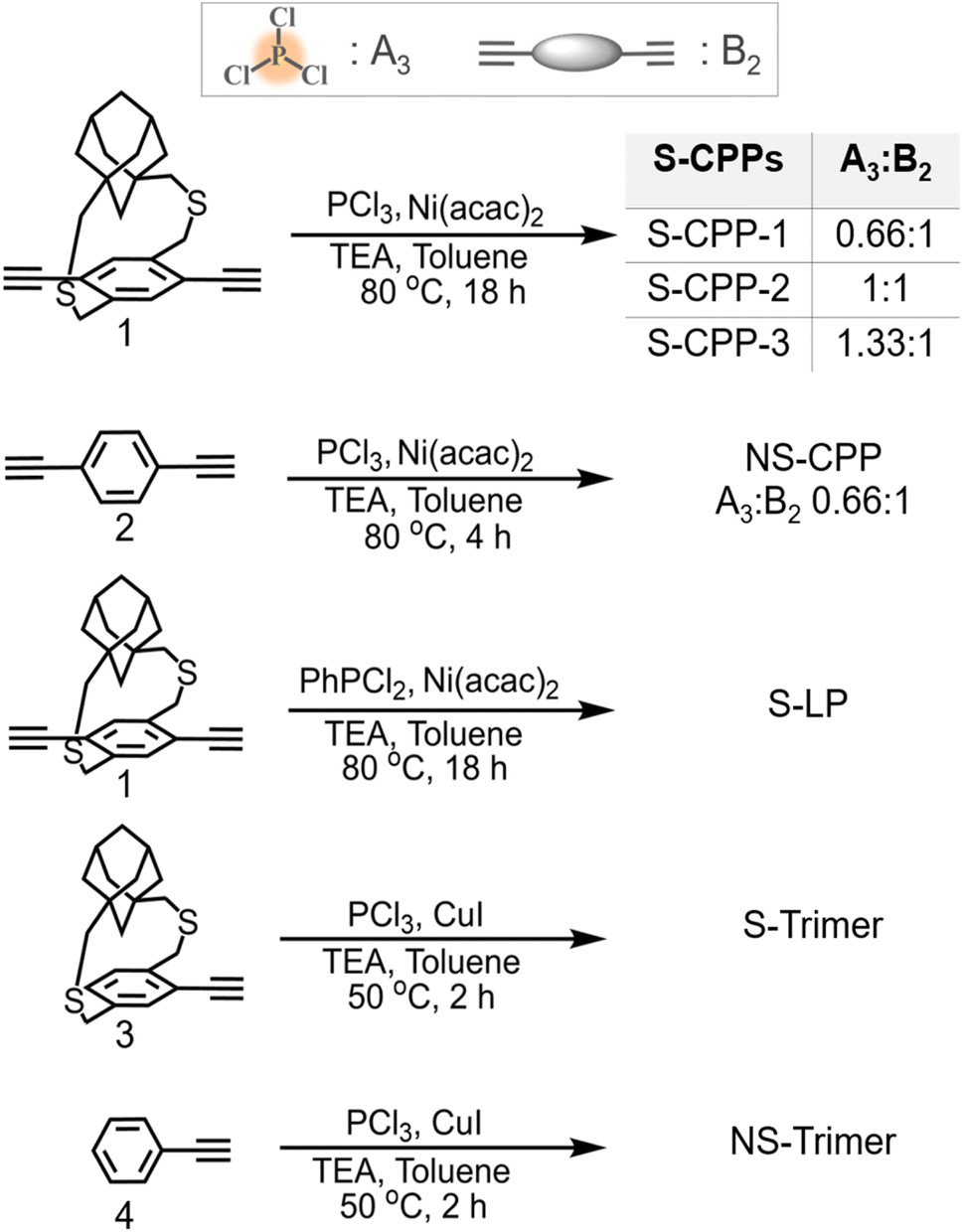 | ||
| Scheme 1 Synthesis of strapped CPPs (S-CPP-1–3), non-strapped CPP (NS-CPP), strapped linear polymer (S-LP), and model trimers (S-trimer & NS-trimer). | ||
As a control CPP, the tris(arylethynyl)phosphane-conjugated porous polymer without straps (NS-CPP) was synthesized by reacting the phosphorous trichloride knot (A3) with the non-strapped 1,4-diethynylbenzene (2) strut (B2). The feed mole ratio between the knot to non-strapped strut is 0.66:1, so NS-CPP is a control network for S-CPP-1. All the porous polymer reaction mixtures were precipitated in methanol. Unreacted monomers and oligomers were extracted using methanol Soxhlet, and the leftover porous polymer was dried under vacuum and used for characterization. S-Trimer and NS-trimer, the trimers representing the branching points of S-CPPs and NS-CPP were synthesized by reacting corresponding arylmonoacetylenes (3 and 4) with phosphorous trichloride. The trimers are helpful in the structural analysis and characterization of the S-CPPs and NS-CPP.
Both the trimer structures were confirmed through ATR-IR (Fig. 2a) and 1H-, 13C- and 31P-NMR, and mass spectrometry (see ESI†). Both the trimers have no terminal ethynyl C–H stretch in the ATR-IR and concurrently a P–C stretch is observed at 1221 cm−1, which confirms the P–C bond formation. The key difference between the S-trimer and NS-trimer is that the former showed both alkyl and aryl C–H stretches whereas the latter showed only aryl C–H stretch in the C–H stretch region. The ATR-IR spectra of S-CPPs and NS-CPP are shown in Fig. 2b. All the CPPs, similar to the trimers, have a broad C–P stretch around 1220 cm−1 confirming the formation of the P–C bond in the CPPs. All the CPPs, unlike the trimers, have relatively lower intensity ethynyl C–H stretch along with the appearance of internal C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C alkyne stretch at 2200 cm−1. The terminal ethynyl C–H stretch is due to unreacted alkynes in the CPPs, which comprise the terminal groups of the system. The network formation was also confirmed from the 13C-CP-MAS NMR spectra (Fig. 2c, S1–S4†). Two sharp peaks corresponding to the ethynyl carbons in 1 (86.4 and 82.5 ppm) appear as broad peaks in the 13C-CPMAS NMR of S-CPPs indicating the network formation. Also, the chemical shift of the internal carbon (δ: 86.4 ppm) of the alkyne in 1 is shifted to a higher value (δ: 103.5 ppm) and the chemical shift of the terminal carbon of the alkyne 1 has slightly changed upon network formation. The changes in chemical shifts of ethynyl carbons upon the P–C bond formation follow the same trend in all the CPPs (S-CPP-1–3, and NS-CPP).
C alkyne stretch at 2200 cm−1. The terminal ethynyl C–H stretch is due to unreacted alkynes in the CPPs, which comprise the terminal groups of the system. The network formation was also confirmed from the 13C-CP-MAS NMR spectra (Fig. 2c, S1–S4†). Two sharp peaks corresponding to the ethynyl carbons in 1 (86.4 and 82.5 ppm) appear as broad peaks in the 13C-CPMAS NMR of S-CPPs indicating the network formation. Also, the chemical shift of the internal carbon (δ: 86.4 ppm) of the alkyne in 1 is shifted to a higher value (δ: 103.5 ppm) and the chemical shift of the terminal carbon of the alkyne 1 has slightly changed upon network formation. The changes in chemical shifts of ethynyl carbons upon the P–C bond formation follow the same trend in all the CPPs (S-CPP-1–3, and NS-CPP).
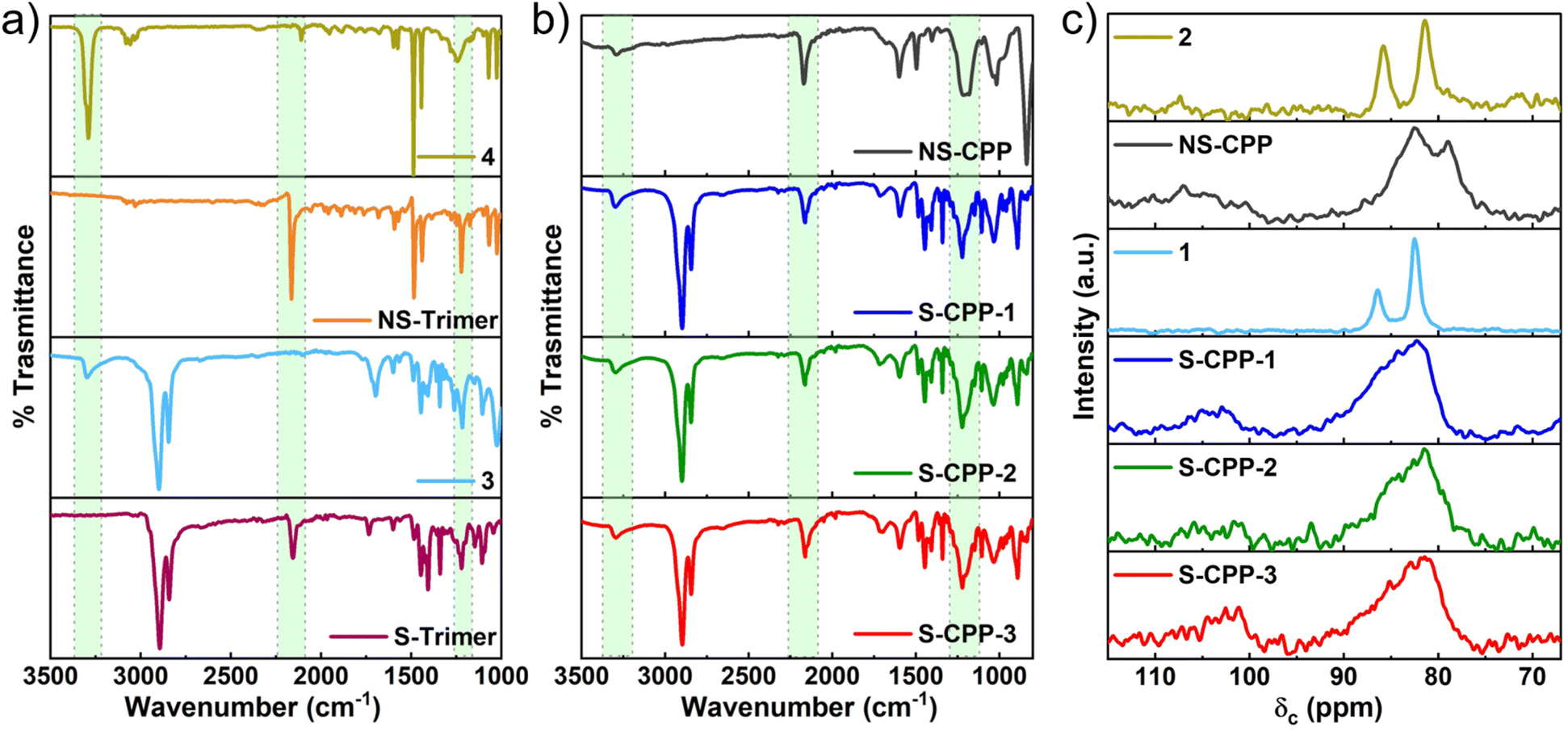 | ||
| Fig. 2 ATR-IR spectra of (a) arylmonoacetylenes (3 & 4) and trimers; (b) S-CPP-1–3 and NS-CPP (highlighted in green from left to right: ethynyl –C–H stretch, alkyne –CC– stretch, and –P–C– stretch); (c) 13C CP-MAS-NMR spectra of monomers (1 & 2) and CPPs are compared to highlight the change in ethynyl carbons chemical shifts and peak widths upon network formation (δ: ca. 86.4 and 82.5 ppm for monomer 1 and δ: ca. 103.5 and 82.1 ppm for S-CPP-1). | ||
In order to determine and quantify the types of phosphorous groups in the CPPs, solid-state 31P– CP-MAS NMR spectra are recorded and are shown in Fig. 3. All the CPPs showed a peak of ca. −90 ppm and a broad peak from 0 to 30 ppm. 31P-NMR of both the trimers in solution shows a peak at −90 ppm (Fig. S31 and S34†), which agrees with the previously reported chemical shift of tris(phenylethynyl)phosphane compounds.39,40 Therefore, the peak at −90 ppm corresponds to the tris(arylethynyl)phosphane. Typically, the phosphorous is more deshielded upon oxidation and shows peaks at a higher chemical shift. For comparison, the NS-trimer was oxidized by reacting with hydrogen peroxide and the 31P-NMR spectrum was recorded. The oxidized trimer showed a peak around −54 ppm (Fig. S37†); however, both the CPPs have no peak in this region indicating that tris(arylethynyl)phosphane is not oxidized in the CPPs. An attempt to determine the oxidized phosphorous stretch from the IR spectrum of the oxidized NS-trimer (Fig. S5†) was futile as the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch (1240 cm−1) overlaps with the P–C stretch (1221 cm−1). Typically, the mono and diaryl oxidized phosphorous peaks appear between 0 and 30 ppm as shown previously in related compounds.41,42 Incompletely reacted phosphorous trichloride can be oxidized by the moisture in the air or during the polymer reprecipitation in methanol and/or soxhlation with methanol. Therefore, the broad peak centered at ca. 30 ppm corresponds to incompletely converted and oxidized phosphorous species. The peak centered at ca. −90 ppm is assigned to the crosslinked or dendritic (D) phosphorous units and the peak centered at ca. 30 ppm is assigned to a combination of di- and mono-ethynylated phosphorous species and corresponds to linear (L) and terminal (T) groups in the CPPs. Thus, by using the area under the deconvoluted peaks from the 31P-NMR spectra, the ratio of D to L + T for S-CPP-1 is determined to be 38%.
O stretch (1240 cm−1) overlaps with the P–C stretch (1221 cm−1). Typically, the mono and diaryl oxidized phosphorous peaks appear between 0 and 30 ppm as shown previously in related compounds.41,42 Incompletely reacted phosphorous trichloride can be oxidized by the moisture in the air or during the polymer reprecipitation in methanol and/or soxhlation with methanol. Therefore, the broad peak centered at ca. 30 ppm corresponds to incompletely converted and oxidized phosphorous species. The peak centered at ca. −90 ppm is assigned to the crosslinked or dendritic (D) phosphorous units and the peak centered at ca. 30 ppm is assigned to a combination of di- and mono-ethynylated phosphorous species and corresponds to linear (L) and terminal (T) groups in the CPPs. Thus, by using the area under the deconvoluted peaks from the 31P-NMR spectra, the ratio of D to L + T for S-CPP-1 is determined to be 38%.
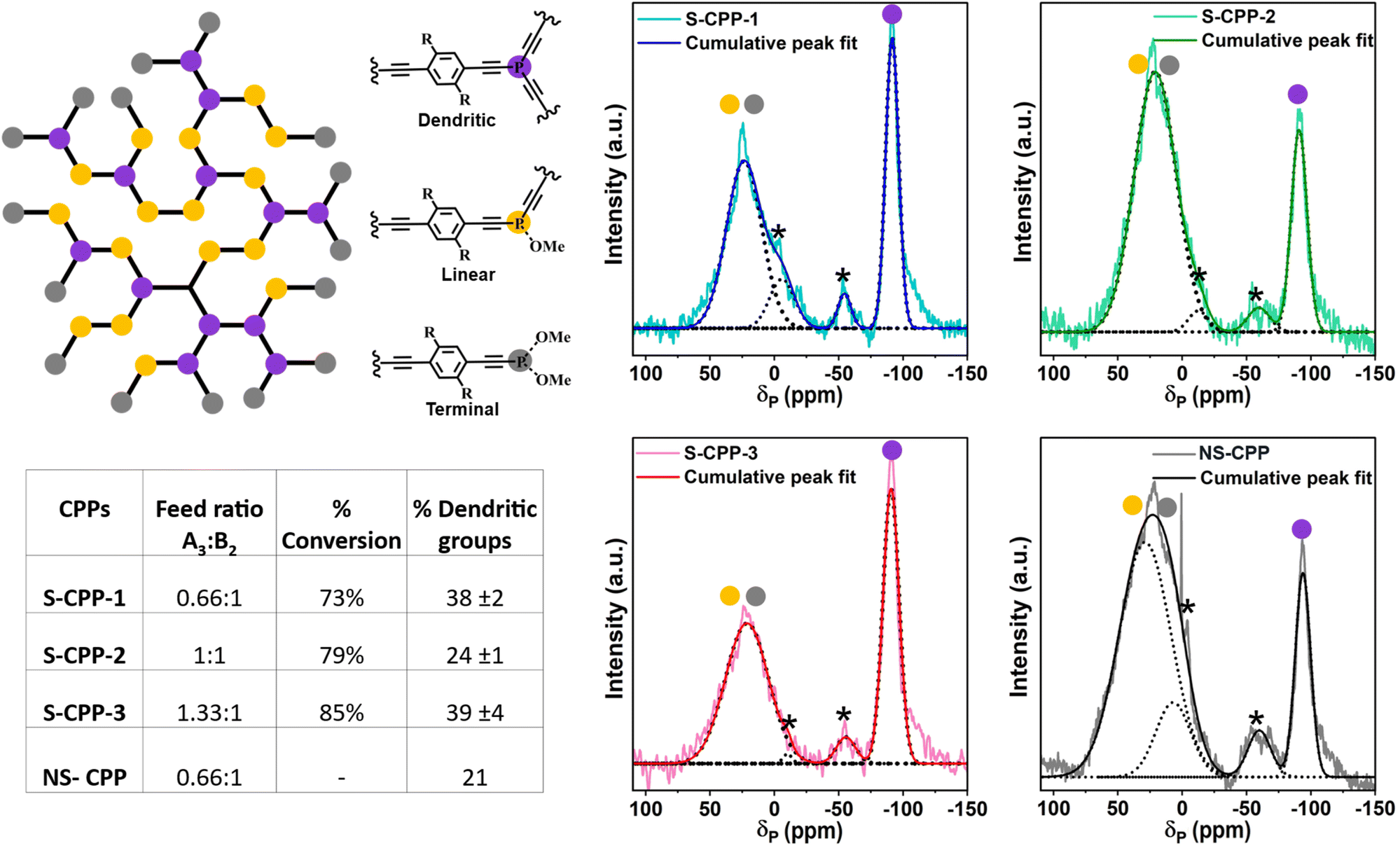 | ||
| Fig. 3 (Top left) Cartoon representation of the CPP network; purple, yellow and grey balls represent dendritic, linear, and terminal groups respectively (R = adamantyl strap for S-CPP-1–3, and –H for NS-CPP); (bottom left) table showing the experimentally determined percentage of strut conversion and percentage dendritic groups for each polymer network; (right) solid-state 31P CP-MAS-NMR spectra of the CPPs, the peaks are deconvoluted [dotted lines] to determine the percentage of dendritic groups in each polymer network (* peaks represent the spinning sidebands). | ||
A similar analysis has been done for NS-CPP and it has about 21% dendritic groups, which is about half of the percentage of crosslinked groups in the S-CPP-1. The higher percentage of dendritic groups in S-CPP-1 indicates that it has a higher crosslinking density than the NS-CPP. Visually, the non-strapped reaction mixture becomes cloudy within an hour followed by the precipitation, whereas for S-CPPs it takes at least 12–15 h for the precipitation to form. This indicates that the straps enhance the network residence time in the reaction mixture and crosslinking density. DLS studies indicate that the straps also generate a swollen network (vide infra). Thus, the straps reduce interchain π–π interactions, enhance the network residence time, and generate a swollen network. This allows the monomer to diffuse into the network and react, hence increasing the crosslinking density compared to NS-CPP.
Similar to S-CPP-1, S-CPP-2, and S-CPP-3 are synthesized by increasing the knot ratio from 0.66 to 1 and 1.33. Both these reaction mixtures contain more knot than the required stoichiometry of reactive functional groups. ATR-IR (Fig. 2b), 13C- and 31P-CP-MAS NMR (Fig. 2c, 3, S3, and S4†) analysis confirm the formation of the network. The area under the peaks from 31P-CP-MAS NMR is used to determine the percentage of dendritic groups. The percentage of dendritic groups in S-CPP-2 and S-CPP-3 is 24% and 39% respectively. Interestingly, the S-CPP solvodynamic radius (Table 1) increased systematically as the knot ratio is increased (vide infra). Thus, a series of S-CPPs with increasing diameter are generated by changing the knot to strut ratio. In the case of non-conjugated hyperbranched polymers, typically, the degree of branching reduces as the knot ratio increases.43 However, there are no reports on how the degree of branching (crosslinking density) of π-conjugated polymer porous networks varies as the knot ratio increases. In this work, the percentage of dendritic groups did not show a clear trend as the knot ratio increased. This could be due to the change in growth kinetics associated with the change in swellability and morphology of the network which alters the percentage of monomer conversion, thus pushing the crosslinking density away from the non-conjugated polymers trend line. This highlights the importance of finding more efficient straps to hinder interchain interactions even at higher knot ratios. The thermal stability of S-CPPs ranges from 170 to 215 °C (Fig. S7 and Table S2†). The BET surface areas of CPPs were determined using nitrogen adsorption isotherms and are relatively low (Fig. S8 and Table S3†).
Straps have been used to hinder interchain interactions and even to generate soluble linear π-conjugated polymers without pendant solubilizing chains. Similarly, here also the straps are expected to hinder interchain interactions within the network and generate a swollen network. As anticipated, S-CPPs are dispersible in typical solvents used to dissolve conjugated polymers including chloroform and o-dichlorobenzene (o-DCB). Since S-CPPs are dispersible in organic solvents, the network size was determined using dynamic light scattering (DLS) (see section 8 in the ESI). In order to determine the role of straps in network swellability, the CPP size was determined in various solvents and is shown in Table 1. The size of S-CPPs in o-dichlorobenzene is about 3 times bigger than that in a bad solvent such as acetonitrile (ACN). THF and chloroform are marginal solvents for S-CPPs because the network size is intermediate to that of good and bad solvents. Thus, the network swells in good solvents and collapses in bad solvents. The size of NS-CPP did not follow any trend in swelling even though it had a similar knot to strut feed ratio as that of S-CPP-1. These results indicate that straps allow the network to easily overcome interchain interactions and swell in a good solvent. Another important observation from this study is that the solvodynamic radii of the S-CPP-1–3 increase as the knot to strut ratio increases. The increase in network size is correlated with the B2 monomer conversion in the reaction mixture. The percent B2 conversion was determined using 1H-NMR (Fig. S9–S12†) and as the knot ratio increases the percent B2 conversion increases resulting in the growth of a larger network.
Dispersibility limits of the CPPs are determined (Fig. 4a) to see if the strapped adamantyl groups help to reduce interchain interactions and stabilize particles against aggregation and sedimentation. CPP dispersions were prepared by sonicating excess CPPs in chloroform followed by centrifugation. The amount of CPP dispersed in the supernatant was determined using the Beer–Lambert law (Fig. S13–S20†) and dispersibility limits are shown in Fig. 4a. The dispersibility limit of S-CPP-1–3 decreases as the knot to strut ratio increases. The percentage of dendritic groups is approximately closer for S-CPP-1 and S-CPP-3; therefore, the higher dispersibility limit for S-CPP-1 is attributed to its smaller network size. S-CPP-2 network dispersibility limit is in between S-CPP-1 and -3, and the trend matches well with that of the network size. The dispersibility limits of S-CPPs are 8–16 times higher than that of NS-CPP. In particular, the dispersibility limit of S-CPP-1 is 16 times higher than that of control NS-CPP with a similar knot to strut ratio. The higher dispersibility limit of the S-CPP-1 compared to that of NS-CPP is due to the presence of adamantyl straps. Thus, the straps shield the π-face of the aryl repeat unit and hinder interparticle π–π interactions against the formation of larger aggregates and sedimentation.
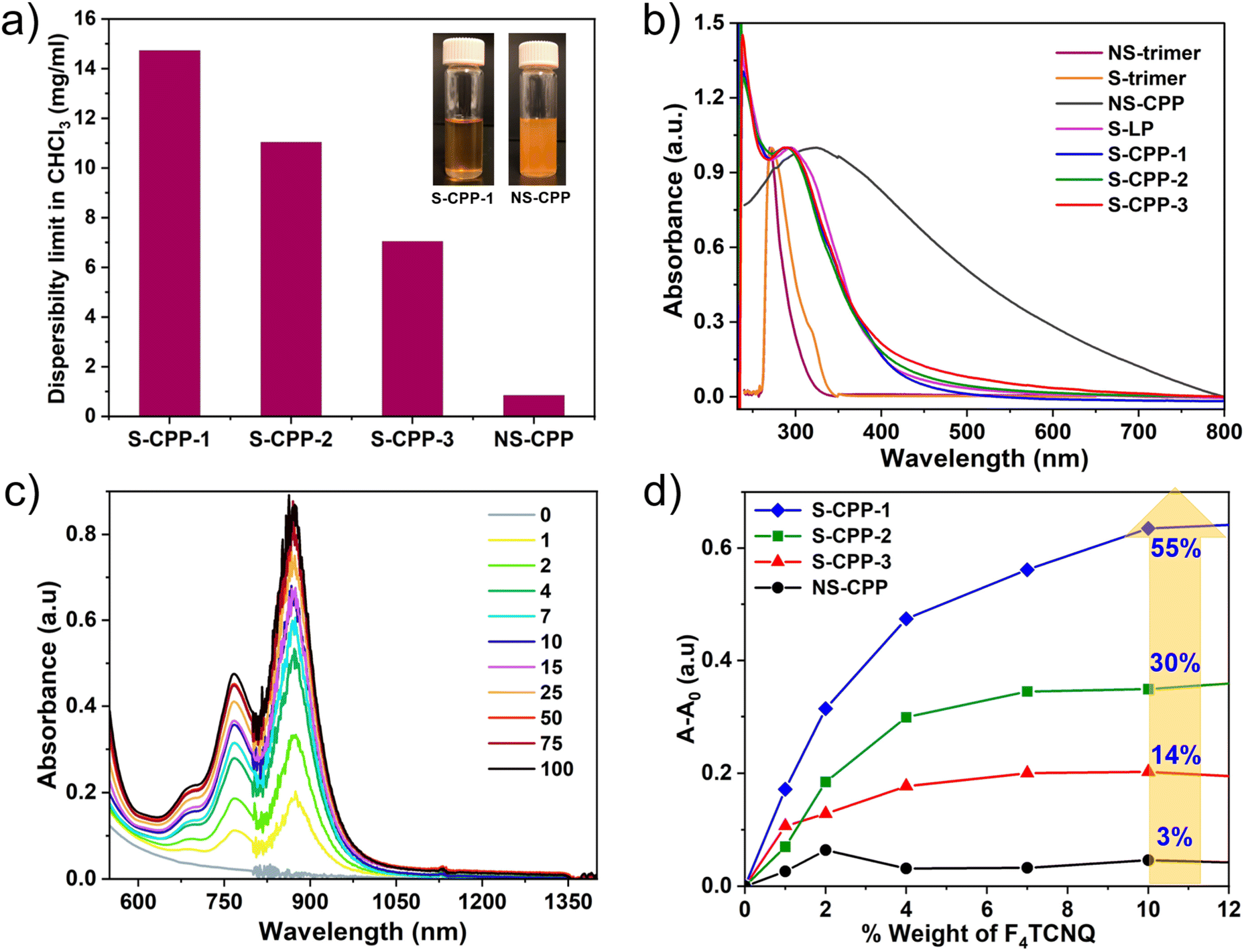 | ||
| Fig. 4 (a) Dispersibility limit of CPPs in CHCl3 (1 mg ml−1 dispersions of S-CPP-1 and NS-CPP are shown for comparison in the inset); (b) UV-vis absorption spectra of CPPs, S-LP, trimers in chloroform; (c) increase in the absorbance of F4TCNQ˙− upon increasing the loading of F4TCNQ in S-CPP-1 solution (legend: weight% of F4TCNQ with respect to S-CPP-1); (d) change in absorbance at 873 nm (corresponding to the absorbance of F4TCNQ˙−) as the weight% of F4TCNQ is increased in solution for S-CPP-1–3 and NS-CPP, doping efficiency of CPPs increases as the porous polymer network solvodynamic size decreases. | ||
UV-vis absorption spectra of all the CPPs along with the trimers in chloroform are shown in Fig. 4b. S-CPPs have a relatively narrow absorption peak that ranges from 240 to 550 nm with an absorption maximum at ca. 241 nm and another intense transition at ca. 290 nm. The low energy transition is red shifted compared to the S-trimer absorption maxima (ca. 272 nm), which is an indication of extended π-conjugation in the CPPs compared to that of trimers. The extension of π-conjugation through phosphorous has been previously reported by multiple groups including the Lucht group.38,44 However, reports on studying the extension of π-conjugation when the phosphorous ethynyl is involved as part of the conjugated polymer backbone are limited.36,37,45 This result shows that triethynyl phosphorous is involved in the extension of π-conjugation in the S-CPPs. The UV-vis absorption maximum and band edge of all the S-CPPs are the same even though the crosslinking densities are different indicating that the extension of π-conjugation across the network is the same for both 24% and 38% crosslinking densities. The negligible effect of network size on UV-vis absorption spectra of S-CPPs indicates that there are no interchain interactions within the network as the particle size increases. On the other hand, NS-CPP does not generate a clear dispersion even after a longer sonication and thus the light scattering from the larger aggregates gives rise to a broad absorption range from 240 to 800 nm with an absorption maximum at ca. 321 nm. Unlike NS-CPP, S-CPP-1–3 have a relatively narrow absorption range because they are well dispersible in chloroform and generate a stable dispersion. Using cyclic voltammetry (Fig. S21†) and UV-vis absorption band edge, frontier energy levels of the S-CPPs were determined (Table 2). There is no considerable difference in the optical band gap and frontier energy levels of the S-CPPs indicating that the network size and crosslinking density have minimal impact on these two properties.
| CPPs | A3:B2 feed ratio |
Sizea (nm) | Dispersibility limitb (mg mL−1) | λ max (nm) | HOMOc (eV) | LUMOd (eV) | Doping efficiency (%) | Conductivity × (10−9) (S cm−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Dispersion | Thin film | |||||||||
| a Solvodynamic radius in o-DCB at 25 °C. b Dispersibility limit in chloroform at room temperature. c Calculated from the oxidation peak onset in CV. d Calculated from Eg − HOMO, where the Eg is the optical band gap obtained from the UV-vis band edge. | ||||||||||
| S-CPP-1 | 0.66:1 |
246 ± 8 | 14.7 | 290 | 365 | −5.19 | −2.11 | 55 | 5.4 ± 0.6 | |
| S-CPP-2 | 1:1 |
310 ± 29 | 11.0 | 288 | 313 | −5.18 | −2.02 | 30 | 5.6 ± 0.7 | |
| S-CPP-3 | 1.33:1 |
385 ± 41 | 7.1 | 289 | 304 | −5.16 | −1.99 | 14 | 6 ± 1.7 | |
| NS-CPP | 0.66:1 |
328 ± 13 | 0.9 | 320 | 322 | — | — | 3 | 4 ± 1.2 | |
Organic semiconductors are doped with small molecules to enhance the conductivity.46–48 The generated series of S-CPPs allows us to study the impact of CPP network size and crosslinking density on chemical doping efficiency. CPPs undergo p-type doping up on the addition of F4TCNQ, resulting in the formation of a reduced F4TCNQ˙− with new UV-vis-NIR absorption peaks centered at 767 nm and 873 nm.49 In order to understand the impact of the knot to strut ratio on the efficiency of chemical doping, the dopant was titrated into the CPP dispersion (Fig. 4c and S22–S26†), and the change in absorbance of the F4TCNQ˙− peak (873 nm peak) was plotted against the weight percentage of the dopant as shown in Fig. 4d. The absorbance of the F4TCNQ˙− peak increased as the dopant weight percentage increased up to 20 wt% and after that the increase was minimal. More importantly, the intensity of the F4TCNQ˙− peak decreased as the knot ratio increased for a given weight percentage of dopant (S-CPP-1 > S-CPP-2 > S-CPP-3). The doped NS-CPP dispersion showed the lowest intensity of the F4TCNQ˙− peak indicating the lowest chemical doping efficiency among all the CPPs studied in this work. As a reference, the linear ethynylphosphine homopolymer (S-LP) was synthesized by reacting phenyl dichlorophoshine (A2) with 1 (B2) (see the ESI†). The relative doping efficiency of CPPs was determined by normalizing the doping efficiency of CPPs with respect to the S-LP and is shown in Table 2.
The chemical doping efficiency of S-CPPs is 4–18 times higher than that of NS-CPP. The chemical doping efficiency of S-CPP-1 is 18 times higher than that of control NS-CPP. Within the strapped series as the knot ratio increases the doping efficiency decreases i.e., larger particles showed lower doping efficiency. As mentioned above the network size and crosslinking density have minimal impact on the energy levels and band gap. Both S-CPP-1 and S-CPP-3 have similar crosslinking densities but different chemical doping efficiency; thus, the crosslinking density also cannot explain the increase in chemical doping efficiency as we reduce the knot feed ratio. Therefore, the increase in chemical doping efficiency is mostly due to the change in the network size. The smaller particles provide a higher outer surface area for F4TCNQ to interact, thereby leading to higher doping efficiency. The higher doping efficiency of S-CPP-1 compared to NS-CPP is also attributed to the maximum outer surface area provided by the smaller S-CPP-1 particles.
Typically, electrical conductivities of doped-CPPs are measured after pressing the powder into a pellet form.50,51 However, for widespread use of CPPs, a simplified processing technique that allows them to process into various device configurations is required. Recently, to overcome the limitations of semiconducting polymers including environmental stability, high modulus, and processability they have been physically blended with insulating commodity polymers.52–55 This technique combines the advantageous properties of each of these materials and in many reported cases, the conductivity, and/or electronic performance of the semiconducting polymer is better than that of pure semiconducting polymer, which highlights the advantage of this approach.55–59 Surprisingly, the blend approach has so far not been used for CPPs.
Herein, to overcome the processability issue of CPPs, the CPPs were blended with poly(methylmethacrylate) (PMMA) as shown in Fig. 5. Under a nitrogen atmosphere, CPPs were dispersed in chlorobenzene by sonicating at 50 °C for 20 minutes followed by mixing with predissolved PMMA solution in chlorobenzene to obtain a homogeneous polymer insulator blend. Then, F4TCNQ solution in chlorobenzene was added into the blend and stirred for a few minutes at room temperature such that the final weight ratio between CPP:PMMA:F4TCNQ is 1:0.25:0.5. The resultant blend was drop cast into thin films and their UV-vis absorption spectra and electrical conductivities were measured. As anticipated, the thin film UV-vis spectra showed the F4TCNQ radical anion peaks similar to what was observed for the dispersions (Fig. S28†). As a control experiment, PMMA alone was mixed with the F4TCNQ, and their absorption spectra were recorded. The F4TCNQ radical anion peak was not observed in the case of the control sample indicating that PMMA is an optimal choice for measuring the conductivity of doped-semiconducting polymers (Fig. S28†). The electrical conductivities of CPPs are shown in Table 2 and are ca. 4–6 × 10−9 S cm−1. The conductivity values are about three orders of magnitude higher than that of poly(phenyleneethynylene) CPP.27 The key difference between CPPs studied in this work and poly(phenyleneethynylene) CPP is that in the former, the struts are connected via phosphorous, which is known to extend the conjugation across the network as shown by the Lucht and Gates whereas in the poly(phenyleneethynylene) CPP the struts are connected via meta phenylene linkages and hence do not extend the conjugation between struts. The conductivity of NS-CPP is similar to that of S-CPP-1–3 indicating that straps do not hinder interchain charge transport.
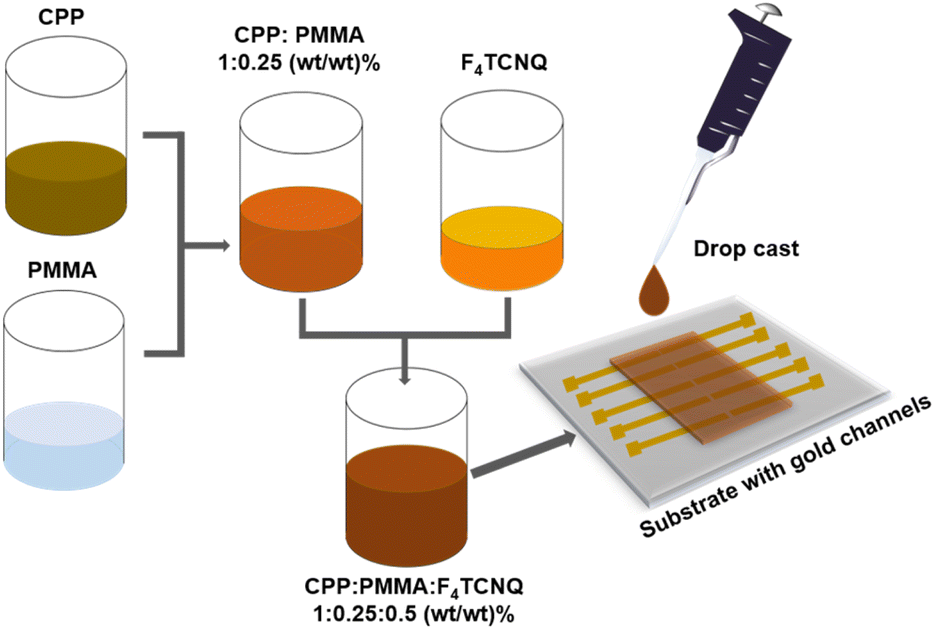 | ||
| Fig. 5 Processability of CPPs is enhanced by blending them with insulating polymers; CPP:PMMA:F4TCNQ (1:0.25:0.5) was blended in chlorobenzene and the resultant solution was drop cast into a thin film on top of a prefabricated chip with gold channels for conductivity measurements. | ||
Conclusions
To summarize, π-face masking straps reduced interchain π–π interactions during the network growth, enhanced the network residence time in the reaction mixture, and generated a swollen network for monomers to diffuse in and react, which allowed us to vary the network size and crosslinking density. Tris(arylethynyl)phosphane CPP networks of varying size and crosslinking density are synthesized by varying the knot to strut ratio. For the strapped CPPs, as the knot to strut ratio decreases, the particle size decreases leading to a higher dispersibility limit and chemical doping efficiency. The dispersibility limit and doping efficiency of the strapped porous network are higher than those of the control non-strapped porous network, which highlights the advantage of straps. For the first time, we have shown that the poor processability of CPPs can be overcome by blending them with insulating commodity polymers. The phosphorous knots efficiently extended conjugation in the CPP networks, which resulted in electrical conductivities three orders of magnitude higher than that of the poly(phenyleneethynylene) network. This work paves the way for employing π-face masking strapped aryl building blocks to control the growth of higher dimensional π-conjugated materials.Author contributions
M. M. performed synthesis, structural and optical characterizations, and was involved in the draft writing. B. M. C. helped with the precursor synthesis. H. A. & S. K. collected and analyzed the conductivity data. F. C., S. J. K., P. K. P., P. A., & X. Z. collected and helped with the analysis of the structural characterization data. N. G. conceived the idea, guided the experimental work, involved in the data analysis, and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation CAREER Grant (NSF-1944184) and Georgetown University.References
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- D. Taylor, S. J. Dalgarno, Z. Xu and F. Vilela, Chem. Soc. Rev., 2020, 49, 3981–4042 RSC.
- M. Goel and M. Thelakkat, Macromolecules, 2020, 53, 3632–3642 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012 RSC.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423–426 CrossRef CAS PubMed.
- G. A. Leith, C. R. Martin, A. Mathur, P. Kittikhunnatham, K. C. Park and N. B. Shustova, Adv. Energy Mater., 2021, 12, 2100441 CrossRef.
- M. G. M. Bai, H. V. Babu, V. Lakshmi and M. R. Rao, Mater. Chem. Front., 2021, 5, 2506–2551 RSC.
- Y. Tian and G. Zhu, Chem. Rev., 2020, 120, 8934–8986 CrossRef CAS PubMed.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed.
- C. Gu, N. Huang, J. Gao, F. Xu, Y. Xu and D. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4850–4855 CrossRef CAS PubMed.
- W. S. Y. Ong, R. A. Smaldone and S. C. Dodani, Chem. Sci., 2020, 11, 7716–7721 RSC.
- C. Wang, C. Li, E. R. C. Rutledge, S. Che, J. Lee, A. J. Kalin, C. Zhang, H.-C. Zhou, Z.-H. Guo and L. Fang, J. Mater. Chem. A, 2020, 8, 15891–15899 RSC.
- Y. Hong, V. Rozyyev and C. T. Yavuz, Small Sci., 2021, 1, 2000078 CrossRef CAS.
- D. Jung, Z. Chen, S. Alayoglu, M. R. Mian, T. A. Goetjen, K. B. Idrees, K. O. Kirlikovali, T. Islamoglu and O. K. Farha, ACS Appl. Mater. Interfaces, 2021, 13, 10409–10415 CrossRef CAS PubMed.
- Y. Gong, H. Hu, S. Huang, S. Lu and W. Zhang, ACS Appl. Nano Mater., 2022, 5, 10090–10096 CrossRef CAS.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- P. Zhang, K. Wu, J. Guo and C. Wang, ACS Macro Lett., 2014, 3, 1139–1144 CrossRef CAS PubMed.
- Y. Song, C. Zhu and S. Ma, EnergyChem, 2022, 4, 100079 CrossRef CAS.
- P. She, Y. Qin, X. Wang and Q. Zhang, Adv. Mater., 2022, 34, 2101175 CrossRef CAS PubMed.
- S. Xu and Q. Zhang, Mater. Today Energy, 2021, 20, 100635 CrossRef CAS.
- J. Lim, M.-S. Kim, W. Jang, D. H. Wang and J. K. Park, Dyes Pigm., 2020, 178, 108332 CrossRef CAS.
- C. Gu, N. Huang, Y. Chen, L. Qin, H. Xu, S. Zhang, F. Li, Y. Ma and D. Jiang, Angew. Chem., Int. Ed., 2015, 54, 13594–13598 CrossRef CAS PubMed.
- H. Veldhuizen, L. Elzen, T. Mahon, R. Abellon and A. Nagai, Macromol. Chem. Phys., 2020, 221, 1900415 CrossRef CAS.
- K. V. Rao, S. Mohapatra, T. K. Maji and S. J. George, Chemistry, 2012, 18, 4505–4509 CrossRef CAS PubMed.
- X. Liu, Y. Xu and D. Jiang, J. Am. Chem. Soc., 2012, 134, 8738–8741 CrossRef CAS PubMed.
- J.-X. Jiang, A. Trewin, D. J. Adams and A. I. Cooper, Chem. Sci., 2011, 2, 1777–1781 RSC.
- J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed. Engl., 2012, 51, 12727–12731 CrossRef CAS PubMed.
- G. Cheng, B. Bonillo, R. S. Sprick, D. J. Adams, T. Hasell and A. I. Cooper, Adv. Funct. Mater., 2014, 24, 5219–5224 CrossRef CAS.
- A. Laybourn, R. Dawson, R. Clowes, T. Hasell, A. I. Cooper, Y. Z. Khimyak and D. J. Adams, Polym. Chem., 2014, 5, 6325–6333 RSC.
- F. S. Jia-Xing Jiang, A. Trewin, C. D. Wood, H. Niu, J. T. A. Jones, Y. Z. Khimyak and A. I. Cooper, J. Am. Chem. Soc., 2008, 130, 7710–7720 CrossRef PubMed.
- L. J. Abbott and C. M. Colina, J. Chem. Eng. Data, 2014, 59, 3177–3182 CrossRef CAS.
- S. Chaudhuri, M. Mohanan, A. V. Willems, J. A. Bertke and N. Gavvalapalli, Chem. Sci., 2019, 10, 5976–5982 RSC.
- R. Lillis, M. R. Thomas, M. Mohanan and N. Gavvalapalli, Macromolecules, 2021, 54, 3112–3119 CrossRef CAS.
- M. K. Das, F. Hameed, R. Lillis and N. Gavvalapalli, Mater. Adv., 2020, 1, 2917–2925 RSC.
- B. W. Rawe, M. R. Scott, C. M. Brown, H. K. MacKenzie and D. P. Gates, Macromolecules, 2017, 50, 8916–8927 CrossRef CAS.
- H. K. Mackenzie, B. W. Rawe, K. Samedov, H. T. G. Walsgrove, A. Uva, Z. Han and D. P. Gates, J. Am. Chem. Soc., 2020, 142, 10319–10324 CrossRef CAS PubMed.
- Z. Jin and B. L. Lucht, J. Am. Chem. Soc., 2005, 127, 5586–5595 CrossRef CAS PubMed.
- I. P. Beletskaya, V. V. Afanasiev, M. A. Kazankova and I. V. Efimova, Org. Lett., 2003, 5, 4309–4310 CrossRef CAS PubMed.
- J. E. Reynolds, K. M. Walsh, B. Li, P. Kunal, B. Chen and S. M. Humphrey, Chem. Commun., 2018, 54, 9937–9940 RSC.
- G. Baba, C. G. Tea, S. A. Touré, M. Lesvier and J. M. Denis, J. Organomet. Chem., 2002, 643–644, 342–349 CrossRef CAS.
- X. Yu, S. Zhang, Z. Jiang, H.-S. Zhang and T. Wang, Eur. J. Org. Chem., 2020, 2020, 3110–3113 CrossRef CAS.
- H. Chen and J. Kong, Polym. Chem., 2016, 7, 3643–3663 RSC.
- B. L. Lucht and N. O. S. Onge, Chem. Commun., 2000, 2097–2098 RSC.
- V. A. Wright, B. O. Patrick, C. Schneider and D. P. Gates, J. Am. Chem. Soc., 2006, 128, 8836–8844 CrossRef CAS PubMed.
- S.-L. Cai, Y.-B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S.-R. Zheng, W.-G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
- J. M. Rotter, R. Guntermann, M. Auth, A. Mahringer, A. Sperlich, V. Dyakonov, D. D. Medina and T. Bein, Chem. Sci., 2020, 11, 12843–12853 RSC.
- Z. Meng, R. M. Stolz and K. A. Mirica, J. Am. Chem. Soc., 2019, 141, 11929–11937 CrossRef CAS PubMed.
- D. A. Dixon, J. C. Calabrese and J. S. Miller, J. Phys. Chem., 1989, 93, 2284–2291 CrossRef CAS.
- Y. Byun, L. S. Xie, P. Fritz, T. Ashirov, M. Dinca and A. Coskun, Angew. Chem., Int. Ed. Engl., 2020, 59, 15166–15170 CrossRef CAS PubMed.
- P. W. Fritz, T. Chen, T. Ashirov, A. D. Nguyen, M. Dinca and A. Coskun, Angew. Chem., Int. Ed. Engl., 2022, 61, e202116527 CAS.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
- J.-Y. Chen, C.-C. Kuo, C.-S. Lai, W.-C. Chen and H.-L. Chen, Macromolecules, 2011, 44, 2883–2892 CrossRef CAS.
- J.-Y. Chen, H.-C. Hsieh, Y.-C. Chiu, W.-Y. Lee, C.-C. Hung, C.-C. Chueh and W.-C. Chen, J. Mater. Chem. C, 2020, 8, 873–882 RSC.
- A. Gumyusenge, X. Luo, H. Zhang, G. M. Pitch, A. L. Ayzner and J. Mei, ACS Appl. Polym. Mater., 2019, 1, 1778–1786 CrossRef CAS.
- C. M. Wolf, L. Guio, S. C. Scheiwiller, R. P. O'Hara, C. K. Luscombe and L. D. Pozzo, Macromolecules, 2021, 54, 2960–2978 CrossRef CAS.
- J. Xu, S. Wang, G.-J. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-Gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y.-H. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B.-H. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- S. Zhang, Y. H. Cheng, L. Galuska, A. Roy, M. Lorenz, B. Chen, S. Luo, Y. T. Li, C. C. Hung, Z. Qian, P. B. J. St. Onge, G. T. Mason, L. Cowen, D. Zhou, S. I. Nazarenko, R. F. Storey, B. C. Schroeder, S. Rondeau-Gagné, Y. C. Chiu and X. Gu, Adv. Funct. Mater., 2020, 30, 2000663 CrossRef CAS.
- H. Ahmad, S. Zhang, C.-T. Liu, G. Ma, J. D. Azoulay, X. Gu, M. K. Gangishetty and S. Kundu, ACS Appl. Polym. Mater., 2022, 4, 8812–8824 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00983a |
| This journal is © The Royal Society of Chemistry 2023 |