
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical, scalable, and transition metal-free visible light-induced heteroarylation route to substituted oxindoles†
Jadab
Majhi
 ,
Albert
Granados
,
Bianca
Matsuo
,
Vittorio
Ciccone
,
Roshan K.
Dhungana
,
Mohammed
Sharique
and
Gary A.
Molander
*
,
Albert
Granados
,
Bianca
Matsuo
,
Vittorio
Ciccone
,
Roshan K.
Dhungana
,
Mohammed
Sharique
and
Gary A.
Molander
*
Roy and Diana Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104-6323, USA. E-mail: gmolandr@sas.upenn.edu
First published on 3rd January 2023
Abstract
The synthetic application of (hetero)aryl radicals in organic synthesis has been known since the last century. However, their applicability has significantly suffered from ineffective generation protocols. Herein, we present a visible-light-induced transition metal-free (hetero)aryl radical generation from readily available (hetero)aryl halides for the synthesis of 3,3′-disubstituted oxindoles. This transformation is amenable to a wide range of (hetero)aryl halides as well as several easily accessible acrylamides, and it is also scalable to multigram synthesis. Finally, the versatility of the oxindole products is demonstrated through their conversion to a variety of useful intermediates applicable to target-directed synthesis.
Introduction
The oxindole scaffold is a privileged motif that is prevalent in a variety of organic frameworks including biologically active molecules, pharmaceuticals, and natural products.1–7 In this regard, the 3,3′-disubstituted oxindoles have been of special interest because of their numerous applications in drug discovery programs (Fig. 1A).4,8–10 Given the wide range of biological activities and ample synthetic utility of oxindoles, the construction of these scaffolds remains an important goal in synthetic organic chemistry.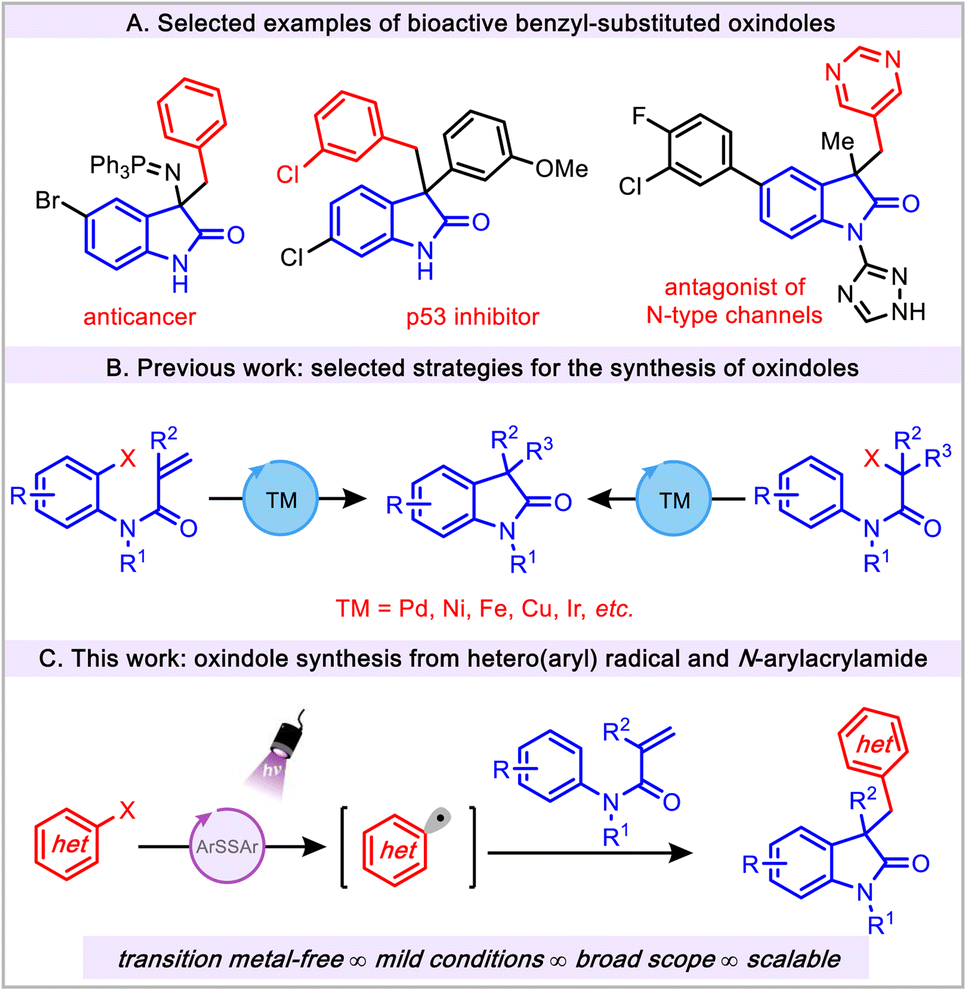 | ||
| Fig. 1 Inspiration and challenges for the preparation of substituted oxindoles. | ||
Existing synthetic methods mainly rely on the use of preexisting oxindole cores to form 3,3′-disubstituted oxindoles via a typical functionalization step.11 In addition, cyclization reactions represent widely used strategies for the construction of oxindole motifs. However, these methods inherently require an ortho-functionalized aryl group and 2-substituted activated anilides (Fig. 1B).12–17 In addition, these transformations not only create difficulties in substrate preparation and are limited in scope, but also require the use of considerable amounts of transition metals (Ni, Cu, and Pd, etc.) and harsh conditions.12 This is a suboptimal scenario because of the significant metal contamination in the production of bioactive oxindole congeners. Recently, N-arylacrylamides have attracted attention for accessing 3-substituted oxindoles via radical cascade reactions.18 Current applications of these methods demand the use of precious transition metals (Pd, Rh and Ir, etc.), ligands, elevated temperature, and/or toxic radical initiators. To circumvent the aforementioned issues, transition metal-free approaches have been utilized in the preparation of oxindole derivatives, but these methods also require a strong oxidant such as elemental iodine.19,20
Visible light photoredox catalysis has emerged as a useful and powerful tool for the construction of oxindoles. In this vein, a large number of visible light photoredox catalysis approaches are based on transition metal catalysts (Ir, Ru and La, etc.).21 Despite the widespread and elegant application of benzyl-substituted oxindoles (Fig. 1A) in medicinal chemistry, it is surprising that only a few synthetic methods have been reported employing aryl radical sources to construct the aforementioned scaffold. These strategies are limited to the use of classical aryl radical sources, namely, aryl diazonium salts (Ered = −0.16 V vs. SCE).22,23 Although these salts are efficient aryl radical precursors, they impose several major drawbacks, such as their chemical instability, limited scope, and challenging manipulation properties, thus reducing their practicality. Despite the widespread synthetic utility of the aryl radicals24,25 in the field of photoredox catalysis, the application of these radical precursors in the preparation of oxindoles has not been explored. Furthermore, given the emerging application of aryl radical chemistry, the use of (hetero)aryl radicals is still in its infancy (vide infra).26–30 Herein, we describe a novel transition metal-free, mild, and operationally simple thiol-mediated photochemical protocol for the construction of substituted oxindoles using readily available (hetero)aryl halides as radical precursors (Fig. 1C). This efficient protocol showcases the successful avoidance of thioether side product formation owing to the potential reactivity of aryl radicals with thiol under visible light conditions.31–33
Discussion
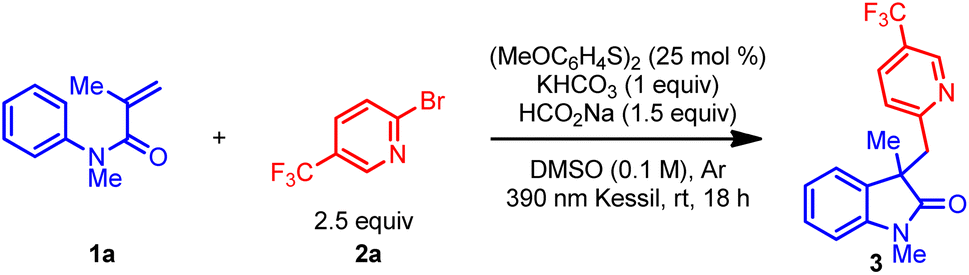
Table 1 outlines the preliminary studies for the development of the proposed transformation using readily available N-phenylacrylamide (1a) and (hetero)aryl bromide 2a as model substrates. For rapid identification of suitable reaction conditions, we used microscale High-Throughput Experimentation (HTE) screening. Treatment of the catalyst bis(4-methoxyphenyl) disulfide, sodium formate, and potassium bicarbonate in DMSO under 390 nm LED array irradiation afforded the highest relative yield in the HTE screen (see ESI†). Performing the reaction on mmol scale delivered the desired oxindole product 3 with a 70% 1H NMR yield (Table 1 entry 1). Next, the effect of light sources in the reaction was investigated (entries 2 and 3). Replacing the purple Kessil lamp (λmax = 390 nm) with a blue Kessil lamp (λmax = 427 nm) delivered the product in a slightly lower yield (entry 2), while no product formed when the lamp was removed (entry 3). Further, the use of the 4-methoxybenzenethiol monomer instead of the dimer also resulted in lower yields (Table 1, entry 4 vs. 1). Hence, we elected to continue the optimization studies with the dimer, owing to its commercial availability as an odorless solid. Additional studies demonstrated that an optimal loading of sodium formate is essential for this transformation (Table 1, entry 1 vs. 5 and 6). Next, changing the reaction environment from an inert (argon) to an open-to-air condition displayed only slight reduction in the yield, suggesting that the method is tolerant of an ambient atmosphere (entry 7). Finally, the impact of the concentration was examined, which demonstrated that concentrated reaction conditions further improved the efficiency of this transformation (entry 8 vs. 1).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||
|---|---|---|
| Entry | Deviation from std conditions | Yield of 3a (%) |
| a Calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. b Isolated yield from 0.5 mmol scale. | ||
| 1 | None | 70 |
| 2 | Blue Kessil (427 nm) | 62 |
| 3 | No light irradiation | 0 |
| 4 | MeOC6H4SH (25 mol%) | 58 |
| 5 | No formate | Traces |
| 6 | 3 equiv. HCO2Na | 60 |
| 7 | Open-to-air | 60 |
| 8 | 0.20 M | 75(69)b |
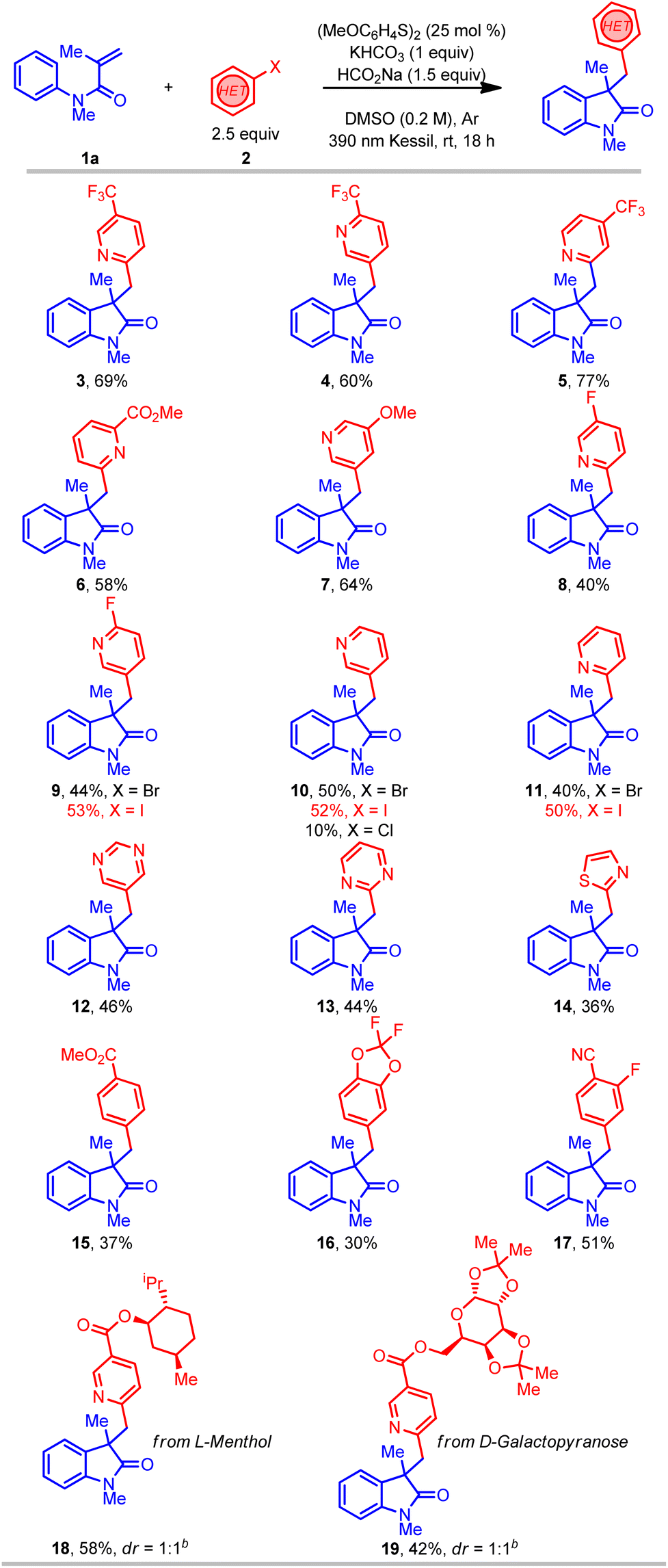
With suitable reaction conditions in hand, the scope of the transformation was explored with a range of (hetero)aryl halides (Table 2). In contrast to previously reported methods for aryl radical generation that solely focused on the use of simple aryl halides (vide supra), we elected to examine comparatively more challenging (hetero)aryl halides. For instance, a wide range of bromopyridine derivatives with different electron-withdrawing and electron-donating groups (CF3, CN, CO2R, F, and OMe) were incorporated at various positions on the aromatic ring, all of which readily afforded the desired benzylated oxindole products in good yield (3–9). More challenging electron-neutral substrates (pyridine, pyrimidine, and thiazole) without any substituents also reacted well and delivered the desired 3,3′-disubstituted oxindoles in moderate yields (10–14). Notably, this transformation was also extended to aryl iodides with even higher yields (9–11), however, the corresponding aryl chloride afforded the desired product (10) in much lower yield. Furthermore, some non-(hetero)aryl substrates were also successfully used in this transformation and afforded comparable yields (15–17) using the same standard reaction conditions. Gratifyingly, more complex molecules such as (hetero)aryl bromide esters derived from L-menthol and D-galactopyranose were also effectively installed in the preparation of oxindole products with good yield (18–19). Overall, with the ability to install the (hetero)aryl groups for the synthesis of the privileged oxindole compounds, this protocol provides a powerful complementary alternative to the more conventional methods.
| a All values indicate the yield of the isolated product. Unless otherwise noted: acrylamide (1 equiv., 0.5 mmol), aryl halide (2.5 equiv., 1.25 mmol), sodium formate (1.5 equiv., 0.75 mmol), potassium bicarbonate (1 equiv., 0.5 mmol), 1,2-bis(4-methoxyphenyl) disulfide (25 mol%, 0.125 mmol), DMSO (0.2 M), 18 h, irradiating with a 390 nm PR160 Kessil lamp. b 2 equiv. of aryl bromide was used. |
|---|
|
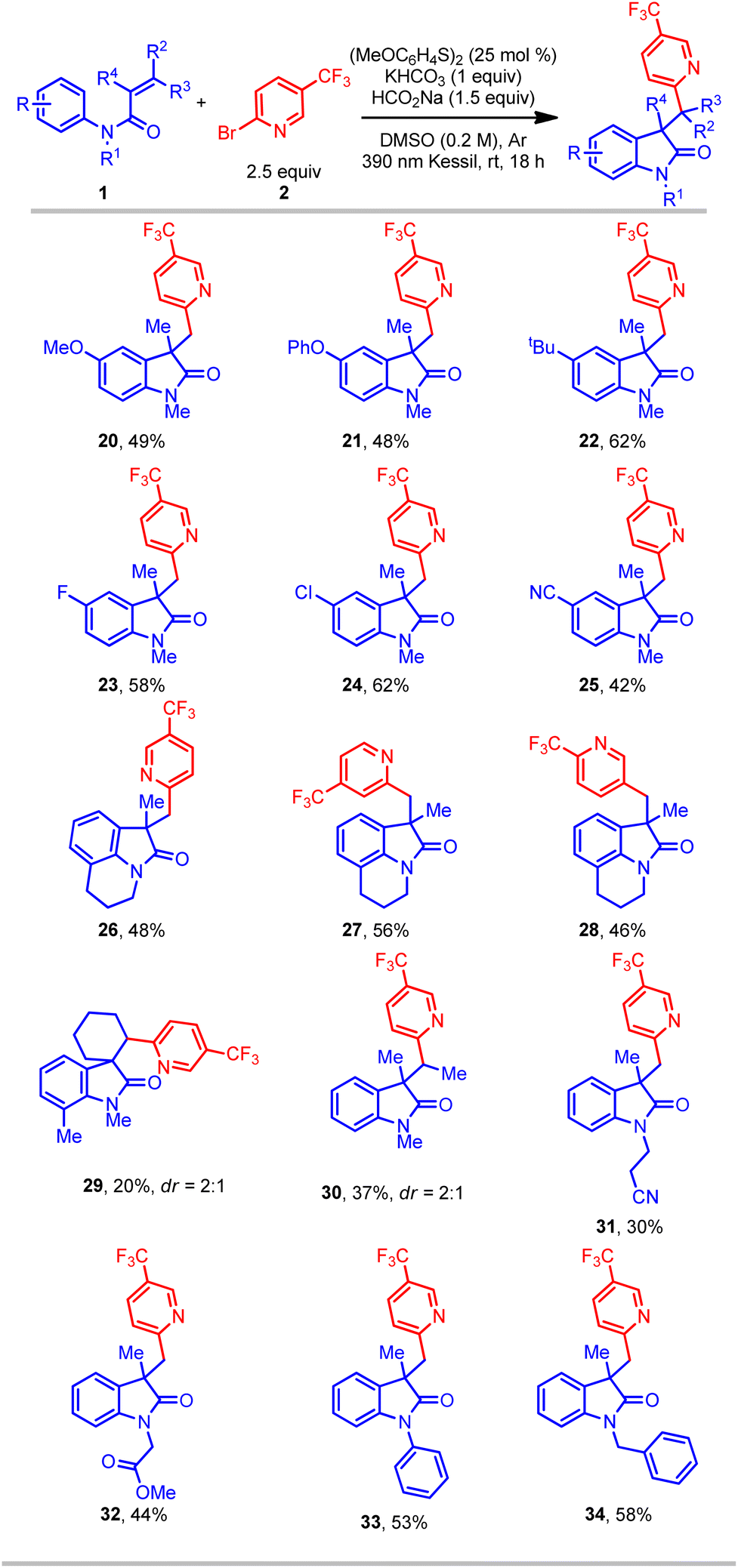
Further studies focused on the scope of N-arylacrylamides in this protocol (Table 3). A wide range of different N-arylacrylamides containing electron-donating (OMe, tBu, etc.) and electron-withdrawing (Cl, CN, etc.) groups afforded the oxindoles with good yields (20–25). Complex fused tricyclic oxindoles are of immense interest because of their unique biological activities.34–36 Gratifyingly, under this protocol fused tricyclic compounds containing a range of (hetero)aryl bromides and tetrahydroisoquinoline were successfully obtained with good yield (26–28). Gratifyingly, a meta-ethyl-substituted acrylamide provided the oxindole product in good yield, albeit with a low (1:1.4) regioselectivity in cyclization onto the aromatic ring (see ESI†). Another motif of relevance, spirocyclic compounds, are important molecules for pharmaceutical applications.4,5 However, such substructures are considered challenging targets in organic synthesis. In this context, a cyclic acrylamide easily afforded the desired spirocyclic compound (29), albeit with a lower yield. Furthermore, an acyclic internal acrylamide also provided oxindole 30 with good yield. The 2-phenyl-substituted acrylamide (R4 = Ph) afforded only trace amount of product, perhaps because of the inherent stability and consequent lack of reactivity of the radical intermediate. Finally, several N-functionalized acrylamides (bearing nitrile, ester, phenyl, and benzyl groups) delivered the corresponding oxindole products (31–34) in good yield. Overall, this transformation provides a simple, efficient, and direct route to access an array of all-carbon-substituted quaternary oxindole compounds.
| a All values indicate the yield of the isolated product. Unless otherwise noted: acrylamide (1 equiv., 0.5 mmol), aryl halide (2.5 equiv., 1.25 mmol), sodium formate (1.5 equiv., 0.75 mmol), potassium bicarbonate (1 equiv., 0.5 mmol), 1,2-bis(4-methoxyphenyl) disulfide (25 mol%, 0.125 mmol), DMSO (0.2 M), 18 h, irradiating with a 390 nm PR160 Kessil lamp. |
|---|
|
Next, attention was turned to the exploration of the synthetic utility of the oxindole product 3. For example, the treatment of oxindole 3 with NBS regioselectively delivered brominated product 35 with excellent yield, which could be a useful intermediate in organic synthesis.37 Remarkably, the benzylic carbon center remains unreactive under the NBS conditions, perhaps because of the deactivating nitrogen and CF3 groups. Additionally, the p-bromo-substituted acrylamide also afforded the same oxindole 35 under the optimized conditions, albeit with moderate yield (Scheme 1, left). Next, treatment of 3 with Lawesson's reagent afforded the indoline-2-thione 36 with excellent yield.37 Finally, a telescoped demethylation process delivered free N–H oxindole 37 with an excellent yield.38 Overall, the ability to convert the oxindole products into an array of important scaffolds without hampering the (hetero)aryl group highlights the potential application of this process to target-directed synthesis.
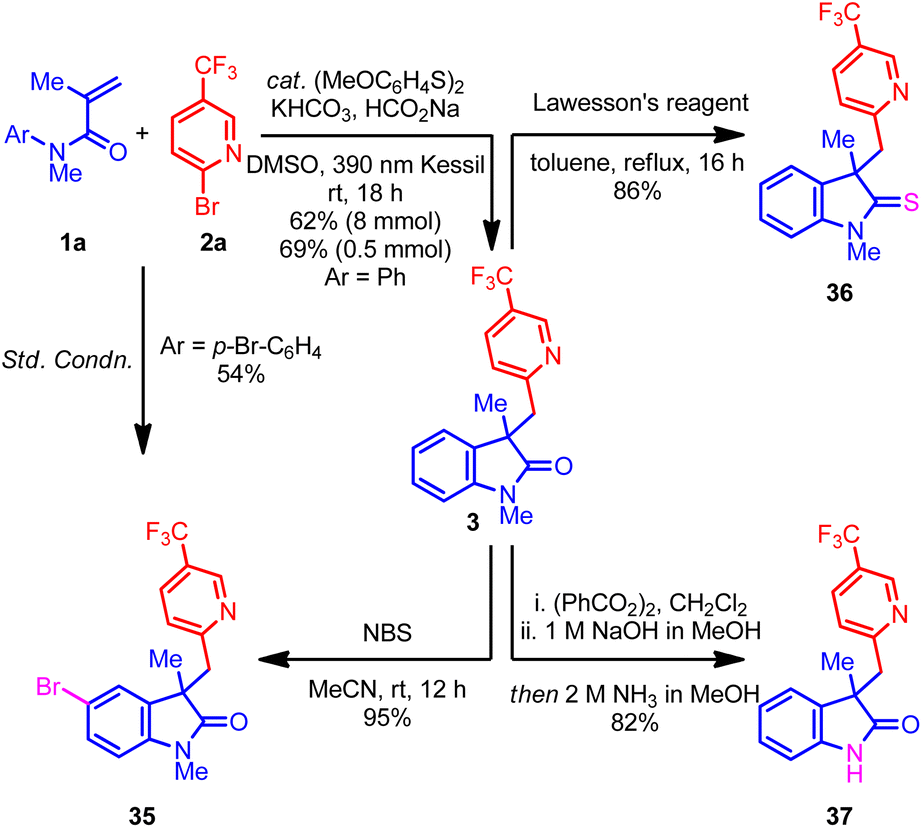 | ||
| Scheme 1 Gram-scale reaction and synthetic application of the (hetero)aryl oxindole 3. | ||
Finally, the operative mechanism of this transformation was explored. To this end, the photochemical quantum yield (Φ) was determined, which revealed a Φ value of 0.5 (see ESI†).39 Radical-trapping experiments were carried out as well. By the addition of TEMPO to the reaction mixture, (hetero)aryl radical generation was indicated (see ESI†). Considering the mechanistic findings depicted herein and the literature,40–42 a plausible mechanism for this transformation is shown in Scheme 2. First, the disulfide catalyst undergoes homolytic fragmentation upon irradiation to the thiyl radical, which abstracts a hydrogen atom from sodium formate, forming the thiophenol and formate radical anion. Subsequently, the thiolate I can be generated after the deprotonation of thiophenol or by a single-electron reduction of the disulfide by the formate radical anion. The excited species II is generated after photoirradiation of I by irradiation with 390 nm light. Next, species II (Ered = −3.31 V vs. SCE)42 reduces the corresponding (hetero)aryl halide (Ered ≈ −1.50 to −2.50 V vs. SCE),43 providing the (hetero)aryl radical III and the thiyl radical. At this point, the generated (hetero)aryl radical III undergoes regioselective and irreversible Giese addition to acrylamide 1, forming the radical species IV. Finally, the thiyl radical undergoes SET with the radical species IV to regenerate I and the oxidized cationic V species, which delivers the final arylated oxindole derivative 3 after deprotonation.
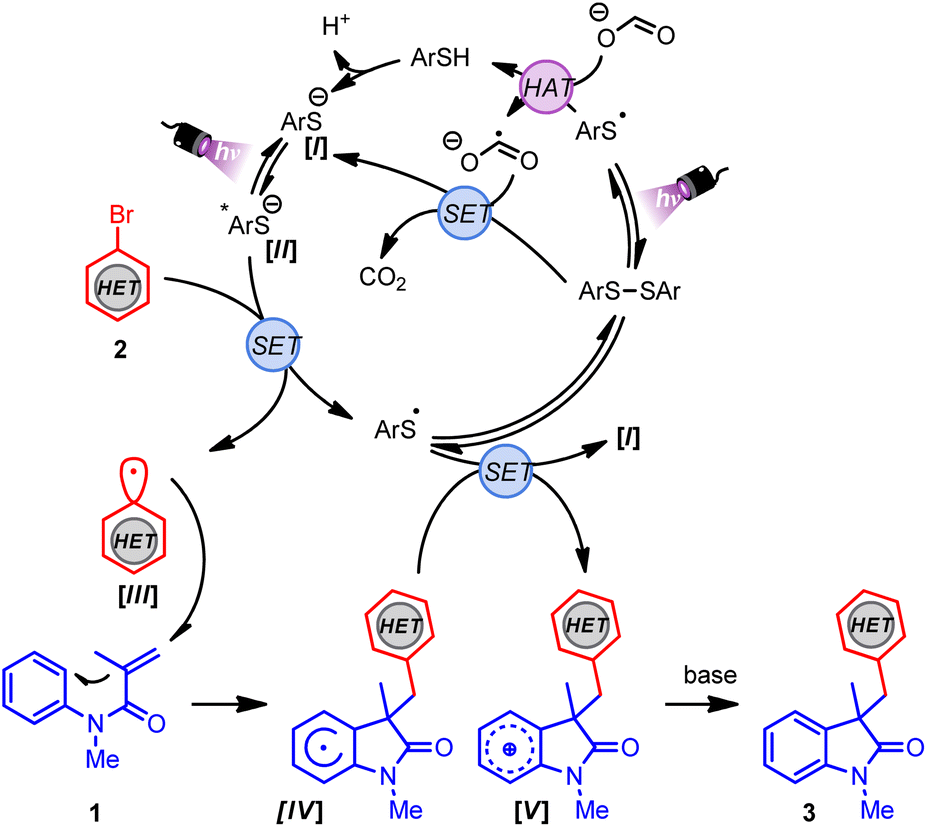 | ||
| Scheme 2 Proposed mechanism for the preparation of benzylated oxindoles. | ||
Conclusions
In conclusion, a transition metal-free visible-light induced (hetero)arylation has been developed to prepare substituted oxindoles from a readily available feedstock. This is a significant development, given that the developed protocol is mild, scalable, and displays a broad substrate scope. In addition, the reaction is amenable to a wide range of functional groups of (hetero)aryl halides and acrylamides. Furthermore, the gram-scale preparation and conversion of the oxindole to several functionalized derivatives illustrates the utility of these intermediates for target-directed synthesis. To the best of our knowledge, this represents the first example of oxindole preparation with aryl radicals derived from aryl halides.Data availability
The ESI† includes all experimental details, including HTE screening results, quantum yield measurements, mechanistics studies, synthesis and characterization of all starting materials and products reported in this study. NMR spectra of all products are included as well.Author contributions
Jadab Majhi, Albert Granados, Bianca Matsuo, Vittorio Ciccone, Roshan K. Dhungana, Mohammed Sharique, and Gary A. Molander designed and conducted experimental work. All the authors contributed to the writing of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support provided by NIGMS (R35 GM 131680 to G. M). The NSF Major Research Instrumentation Program (award NSF CHE-1827457), the NIH supplement awards 3R01GM118510-03S1 and 3R01GM087605-06S1, as well as the Vagelos Institute for Energy Science and Technology supported the purchase of the NMRs used in this study. We thank Dr Charles W. Ross, III (UPenn) for mass spectral data, and Kessil for the donation of lamps.Notes and references
- M. Kaur, M. Singh, N. Chadha and O. Silakari, Eur. J. Med. Chem., 2016, 123, 858–894 CrossRef CAS PubMed.
- Y. M. Khetmalis, M. Shivani, S. Murugesan and K. V. G. Chandra Sekhar, Biomed. Pharmacother., 2021, 141, 111842 CrossRef CAS PubMed.
- M. K. Christensen, K. D. Erichsen, C. Trojel-Hansen, J. Tjørnelund, S. J. Nielsen, K. Frydenvang, T. N. Johansen, B. Nielsen, M. Sehested, P. B. Jensen, M. Ikaunieks, A. Zaichenko, E. Loza, I. Kalvinsh and F. Björkling, J. Med. Chem., 2010, 53, 7140–7145 CrossRef CAS PubMed.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS PubMed.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS PubMed.
- H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Angew. Chem., Int. Ed., 2007, 46, 2254–2256 CrossRef CAS PubMed.
- C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS PubMed.
- M. S. Christodoulou, F. Nicoletti, K. Mangano, M. A. Chiacchio, G. Facchetti, I. Rimoldi, E. M. Beccalli and S. Giofrè, Bioorganic Med. Chem. Lett., 2020, 30, 126845 CrossRef CAS PubMed.
- Y. Shen, N. Lei, C. Lu, D. Xi, X. Geng, P. Tao, Z. Su and K. Zheng, Chem. Sci., 2021, 12, 15399–15406 RSC.
- C. Wang and L. Liu, Org. Chem. Front., 2021, 8, 1454–1460 RSC.
- E. Kókai, G. Simig and B. Volk, Molecules, 2017, 22, 24 CrossRef PubMed.
- A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed.
- E. J. Hennessy and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 12084–12085 CrossRef CAS PubMed.
- E. J. Kiser, J. Magano, R. J. Shine and M. H. Chen, Org. Process Res. Dev., 2012, 16, 255–259 CrossRef CAS.
- C. Theunissen, J. Wang and G. Evano, Chem. Sci., 2017, 8, 3465–3470 RSC.
- Y. Yamane, K. Yoshinaga, M. Sumimoto and T. Nishikata, ACS Catal., 2019, 9, 1757–1762 CrossRef CAS.
- E. M. Larin, J. Masson-Makdissi, Y. J. Jang and M. Lautens, ACS Catal., 2022, 12, 12744–12749 CrossRef CAS.
- C.-C. Li and S.-D. Yang, Org. Biomol. Chem., 2016, 14, 4365–4377 RSC.
- N. Radhoff and A. Studer, Chem. Sci., 2022, 13, 3875–3879 RSC.
- J. R. Donald, R. J. K. Taylor and W. F. Petersen, J. Org. Chem., 2017, 82, 11288–11294 CrossRef CAS PubMed.
- J. Singh and A. Sharma, Adv. Synth. Catal., 2021, 363, 4284–4308 CrossRef CAS.
- W. Fu, F. Xu, Y. Fu, M. Zhu, J. Yu, C. Xu and D. Zou, J. Org. Chem., 2013, 78, 12202–12206 CrossRef CAS PubMed.
- Z. Gonda, F. Béke, O. Tischler, M. Petró, Z. Novák and B. L. Tóth, Eur. J. Org. Chem., 2017, 2017, 2112–2117 CrossRef CAS.
- N. Kvasovs and V. Gevorgyan, Chem. Soc. Rev., 2021, 50, 2244–2259 RSC.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- J. D. Nguyen, E. M. D. Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- R. S. Shaikh, S. J. S. Düsel and B. König, ACS Catal., 2016, 6, 8410–8414 CrossRef CAS.
- T. Constantin, F. Juliá, N. S. Sheikh and D. Leonori, Chem. Sci., 2020, 11, 12822–12828 RSC.
- N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed.
- H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616–13619 CrossRef CAS PubMed.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- H. Li, Y. Liu and S. Chiba, JACS Au, 2021, 1, 2121–2129 CrossRef CAS PubMed.
- M. Li, J.-S. Zhang, Y.-M. Ye and J.-N. Fang, J. Nat. Prod., 2000, 63, 139–141 CrossRef CAS PubMed.
- C. Kikuchi, T. Ando, T. Watanabe, H. Nagaso, M. Okuno, T. Hiranuma and M. Koyama, J. Med. Chem., 2002, 45, 2197–2206 CrossRef CAS PubMed.
- P. V. N. Reddy, K. C. Jensen, A. D. Mesecar, P. E. Fanwick and M. Cushman, J. Med. Chem., 2012, 55, 367–377 CrossRef CAS PubMed.
- H. Li, X. Yan, J. Zhang, W. Guo, J. Jiang and J. Wang, Angew. Chem., Int. Ed., 2019, 58, 6732–6736 CrossRef CAS PubMed.
- W. G. Beyersbergen van Henegouwen, R. M. Fieseler, F. P. J. T. Rutjes and H. Hiemstra, J. Org. Chem., 2000, 65, 8317–8325 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- K. Chen, N. Berg, R. Gschwind and B. König, J. Am. Chem. Soc., 2017, 139, 18444–18447 CrossRef CAS PubMed.
- S. Ghosh, Z.-W. Qu, S. Pradhan, A. Ghosh, S. Grimme and I. Chatterjee, Angew. Chem., Int. Ed., 2022, 61, e202115272 CAS.
- C. Liu, K. Li and R. Shang, ACS Catal., 2022, 12, 4103–4109 CrossRef CAS.
- L. Pause, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 1999, 121, 7158–7159 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and mechanistic studies details, as well as spectral data. See DOI: https://doi.org/10.1039/d2sc05918e |
| This journal is © The Royal Society of Chemistry 2023 |