
Integrating RuO2@TiO2 catalyzed electrochemical chlorine evolution with a NO oxidation reaction for nitrate synthesis†
Longcheng
Zhang
 ab,
Jie
Liang
c,
Xun
He
c,
Qin
Yang
c,
Yongsong
Luo
c,
Dongdong
Zheng
c,
Shengjun
Sun
d,
Jing
Zhang
e,
Hong
Yan
f,
Binwu
Ying
*a,
Xiaodong
Guo
*b and
Xuping
Sun
*cd
ab,
Jie
Liang
c,
Xun
He
c,
Qin
Yang
c,
Yongsong
Luo
c,
Dongdong
Zheng
c,
Shengjun
Sun
d,
Jing
Zhang
e,
Hong
Yan
f,
Binwu
Ying
*a,
Xiaodong
Guo
*b and
Xuping
Sun
*cd
aDepartment of Laboratory Medicine, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail: yingbinwu@scu.edu.cn
bSchool of Chemical Engineering, Sichuan University, Chengdu 610065, Sichuan, China. E-mail: xiaodong2009@scu.edu.cn
cInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, China. E-mail: xpsun@uestc.edu.cn; xpsun@sdnu.edu.cn
dCollege of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University, Jinan 250014, Shandong, China
eInterdisciplinary Materials Research Center, Institute for Advanced Study, Chengdu University, Chengdu 610106, Sichuan, China
fLaboratory Medicine Center, The Second Affiliated Hospital of Nanjing Medical University, Nanjing 210011, Jiangsu, China
First published on 28th February 2023
Abstract
We report a cascade chlorine evolution–nitric oxide (NO) oxidation system for the homogeneous synthesis of nitrate, in which NO can be efficiently converted to nitrate by reacting with the active chlorine species. This highly efficient nitrate synthesis system was demonstrated by using a RuO2 nanoparticle-decorated TiO2 nanobelt array on a titanium plate (RuO2@TiO2/TP) under ambient conditions, capable of attaining a high nitrate yield of 95.22 mg cm−2 h−1 at 2.1 V versus the reversible hydrogen electrode in NO-saturated 0.5 M NaCl + 0.01 M HClO4. Electrochemical in situ Raman spectroscopy studies reveal the adsorption of Cl–O species on the electrode surface and the key role of high-valence Rux+ (x > 4) at high potentials. The electron paramagnetic resonance results confirmed the existence of chlorine radicals and hydroxyl radicals generated by RuO2@TiO2/TP in the electrolyte.
Nitrate, as one of the most crucial forms of oxidized nitrogen, which is essential for the production of fertilizers, explosives, and gunpowder, plays a significant role in our modern society.1–3 The traditional route to nitrate production in industry involves catalytic oxidation of NH3 through the Ostwald process, which results in a high rate of pollution to the environment.4 Moreover, NH3 is mainly produced by the Haber–Bosch process operating at a high temperature and pressure, which is energy-intensive and emits large amounts of greenhouse gases.5 Electrocatalytic nitrogen oxidation is considered an attractive alternative for direct production of nitrate under ambient conditions but suffers from low nitrate yield and faradaic efficiency (FE).6–8
Nitric oxide (NO), as one of the major air pollutants, has a lower bonding energy than N2 (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O: 607 kJ mol−1 and N
O: 607 kJ mol−1 and N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N: 941 kJ mol−1 at 298 K),9–15 suggesting its use as an alternative nitrogen source to N2 for nitrate electrosynthesis via an NO oxidation reaction (NOOR). Wang et al. proposed a plasma-engraved commercial carbon cloth to synthesize nitrate by the electrochemical NOOR, which only occurs at the electrode surface resulting in low nitrate yield.10 The chlor-alkali process is one of the most critical chemical industry processes in the global economy,16,17 and the generated Cl2via an anodic chlorine evolution reaction (CER) is disproportionate in aqueous environments to form oxidative chlorine species.18–21 Our recent work suggests that using active chlorine species generated by the CER can improve the efficiency of NO electro-oxidation22 and may be an alternative path to drive efficient and sustainable nitrate synthesis.
N: 941 kJ mol−1 at 298 K),9–15 suggesting its use as an alternative nitrogen source to N2 for nitrate electrosynthesis via an NO oxidation reaction (NOOR). Wang et al. proposed a plasma-engraved commercial carbon cloth to synthesize nitrate by the electrochemical NOOR, which only occurs at the electrode surface resulting in low nitrate yield.10 The chlor-alkali process is one of the most critical chemical industry processes in the global economy,16,17 and the generated Cl2via an anodic chlorine evolution reaction (CER) is disproportionate in aqueous environments to form oxidative chlorine species.18–21 Our recent work suggests that using active chlorine species generated by the CER can improve the efficiency of NO electro-oxidation22 and may be an alternative path to drive efficient and sustainable nitrate synthesis.
Here, we put forward a cascade catalytic system for efficient nitrate synthesis enabled by a RuO2 nanoparticle-decorated TiO2 nanobelt array on a titanium plate (RuO2@TiO2/TP) as a CER electrocatalyst producing active chlorine species to directly convert NO into nitrate under ambient conditions. Such a RuO2@TiO2/TP catalyst exhibits a low potential of 1.78 V to reach a current density of 20 mA cm−2 and a high active chlorine yield of 83.3 mg cm−2 h−1 at 2.1 V versus the reversible hydrogen electrode (RHE) in 0.5 M NaCl + 0.01 M HClO4. Owing to the generation of active chlorine, the reaction sites for the oxidation of NO to nitrate are not restricted to the electrode surface, but extend throughout the electrolyte, achieving homogeneous nitrate production. RuO2@TiO2/TP affords an outstanding nitrate yield of 95.22 mg cm−2 h−1 at 2.1 V vs. the RHE in NO-saturated 0.5 M NaCl + 0.01 M HClO4. It does not show performance decay for at least 12 h of continuous operation, with a remarkable nitrate accumulation of 1222.44 mg. Electrochemical in situ Raman and quasi in situ electron paramagnetic resonance (EPR) experiments revealed the Rux+ (x > 4) active sites and Cl–O species adsorbed on the electrode surface as well as the presence of chlorine radicals and hydroxyl radicals in the electrolyte catalytically produced by RuO2@TiO2/TP.
The preparation of RuO2@TiO2/TP involves the hydrothermal growth of Na-titanate on the TP, a cation-exchange reaction between Na-titanate and Ru3+, and an air calcination treatment of Ru-titanate/TP (Fig. S1†). Comparison of the X-ray diffraction (XRD) patterns of Na-titanate/TP and Ru-titanate/TP reveals that Ru-titanate/TP has an almost similar crystal structure to that of Na-titanate/TP (Fig. S2a†). In addition, compared with Na-titanate/TP, the diffraction peak of Ru-titanate/TP at the (200) plane has a slight shift to a lower scattering angle (Fig. S2b†). The XRD patterns of both TiO2/TP and RuO2@TiO2/TP (Fig. 1a) show diffraction peaks for anatase TiO2 (JCPDS no. 43-1027) with mainly characteristic peaks at 25.28°, 38.57°, 48.04°, 53.89°, and 55.06° indexed to the (101), (112), (200), (105), and (211) planes, respectively. Meanwhile, a characteristic peak at ca. 28.01° is detected, which is well indexed to the (110) plane of RuO2 (JCPDS no. 43-1027). The scanning electron microscopy (SEM) images reveal the full coverage of the TP substrate (Fig. S3†) by the Na-titanate nanobelt array (Fig. S4a†), whose morphology is well preserved following the subsequent cation-exchange reaction (Fig. S4b†). The SEM images of TiO2/TP (Fig. 1b) and RuO2@TiO2/TP (Fig. 1c) show that the morphologies of both materials are nanobelt arrays. Energy dispersive X-ray spectroscopy (EDX) elemental mapping images acquired from RuO2@TiO2/TP (Fig. 1d) identified the co-existence of Ru, Ti, and O elements with a uniform distribution. More details of RuO2@TiO2 were observed by transmission electron microscopy (TEM). As exhibited in Fig. 1e, some small nanoparticles can be seen in the nanobelt. The high-resolution TEM (HRTEM) image of RuO2@TiO2 shows lattice fringes with distances of 0.318 and 0.352 nm attributed to the (110) and (101) planes of RuO2 and TiO2, respectively (Fig. 1f). The chemical state of RuO2@TiO2/TP can be revealed by X-ray photoelectron spectroscopy (XPS). As depicted in Fig. 1g, the two strong peaks of Ru 3d3/2 at 284.9 eV and Ru 3d5/2 at 280.9 eV are attributed to Ru4+.23,24 The Ti 2p spectrum (Fig. 1h) shows two dominant peaks located at 464.2 eV (Ti 2p1/2) and 458.4 eV (Ti 2p3/2).25 In the O 1s region (Fig. 1i), two peaks located at 531.9 eV and 530.1 eV are regarded as the absorbed water and lattice of oxides, respectively, while the other peak at 529.4 eV refers to the metal–O bond.26,27
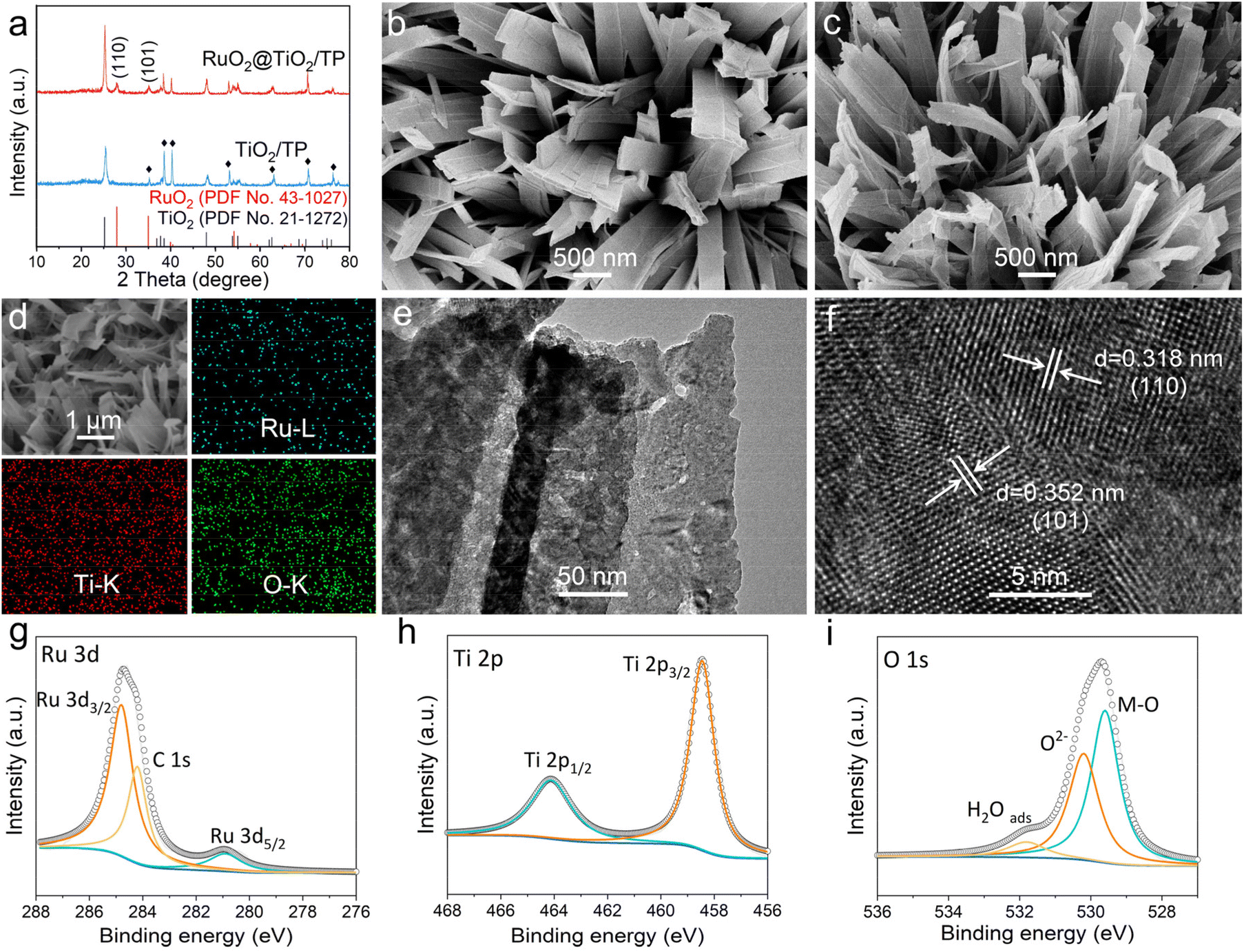 | ||
| Fig. 1 (a) XRD patterns of RuO2@TiO2/TP and TiO2/TP. SEM images of (b) TiO2/TP and (c) RuO2@TiO2/TP. (d) The SEM image and corresponding EDX elemental mapping images of RuO2@TiO2/TP. (e) TEM and (f) HRTEM images of RuO2@TiO2. High-resolution XPS spectra of RuO2@TiO2/TP in (g) the Ru 3d, (h) the Ti 2p, and (i) the O 1s regions. | ||
The electrocatalytic activities of RuO2@TiO2/TP for CER were tested in a gas-tight H-type cell. Fig. 2a shows the linear sweep voltammetry (LSV) curves of RuO2@TiO2/TP, the benchmark RuO2 on TP (RuO2/TP), TiO2/TP, and the bare TP in 0.5 M NaCl + 0.01 M HClO4. This clearly shows a higher current density for RuO2@TiO2/TP compared to RuO2/TP, TiO2/TP, and bare TP at the potential range of 1.7 V–2.1 V. RuO2@TiO2/TP shows a lower potential of 1.78 V compared to RuO2/TP (2.0 V) at a current density of 20 mA cm−2, which is superior to that of most recently reported CER electrocatalysts listed in Table S1.† RuO2@TiO2/TP also achieves a low Tafel slope of 169.1 mV dec−1 (Fig. 2b), which is comparable to RuO2/TP (157.5 mV dec−1) and smaller than those of TiO2/TP (300.4 mV dec−1) and bare TP (328.4 mV dec−1). Active chlorine was evaluated with colorimetric measurements using N,N-diethyl-p-phenylenediamine (DPD), and the ultraviolet-visible (UV-vis) calibration curves are shown in Fig. S5.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28 Additionally, the chronoamperometry curves and UV-vis absorption spectra are illustrated in Fig. S6.† The average FE of active chlorine for RuO2@TiO2/TP reaches a maximum value of 83.4% at 2.1 V. Of note, a high average active chlorine yield of 83.3 mg cm−2 h−1 was also delivered at the same potential, namely 2.1 V (Fig. 2c). To confirm the origin of the active chlorine in the CER, several control experiments were conducted. As shown in Fig. 2d and Fig. S7,† RuO2@TiO2/TP affords a high active chlorine yield of up to 83.3 mg cm−2 h−1 at 2.1 V, while only 1.11 mg cm−2 h−1 and 0.24 mg cm−2 h−1 are measured at the open circuit potential (OCP) and in a blank electrolyte, respectively, indicating that the facilities and medicines were less polluted. In addition, the yields of active chlorine generated by RuO2@TiO2/TP, TiO2/TP, and bare TP during the CER process were compared at 2.1 V (Fig. 2d and Fig. S8†). It can be seen distinctly that TiO2/TP and bare TP produce little active chlorine, suggesting that RuO2 possesses a wonderful catalytic activity toward the CER. The stability of RuO2@TiO2/TP was examined by a long-term stability test at a constant current density of 50 mA cm−2 in 0.5 M NaCl + 0.01 M HClO4. As shown in Fig. 2e, the fluctuation of potential at a current density of 50 mA cm−2 is almost negligible. The good electrochemical stability of our RuO2@TiO2/TP was further verified by the LSV curve after electrolysis, which shows a slight decline compared with the initial curve (Fig. S9†).
28 Additionally, the chronoamperometry curves and UV-vis absorption spectra are illustrated in Fig. S6.† The average FE of active chlorine for RuO2@TiO2/TP reaches a maximum value of 83.4% at 2.1 V. Of note, a high average active chlorine yield of 83.3 mg cm−2 h−1 was also delivered at the same potential, namely 2.1 V (Fig. 2c). To confirm the origin of the active chlorine in the CER, several control experiments were conducted. As shown in Fig. 2d and Fig. S7,† RuO2@TiO2/TP affords a high active chlorine yield of up to 83.3 mg cm−2 h−1 at 2.1 V, while only 1.11 mg cm−2 h−1 and 0.24 mg cm−2 h−1 are measured at the open circuit potential (OCP) and in a blank electrolyte, respectively, indicating that the facilities and medicines were less polluted. In addition, the yields of active chlorine generated by RuO2@TiO2/TP, TiO2/TP, and bare TP during the CER process were compared at 2.1 V (Fig. 2d and Fig. S8†). It can be seen distinctly that TiO2/TP and bare TP produce little active chlorine, suggesting that RuO2 possesses a wonderful catalytic activity toward the CER. The stability of RuO2@TiO2/TP was examined by a long-term stability test at a constant current density of 50 mA cm−2 in 0.5 M NaCl + 0.01 M HClO4. As shown in Fig. 2e, the fluctuation of potential at a current density of 50 mA cm−2 is almost negligible. The good electrochemical stability of our RuO2@TiO2/TP was further verified by the LSV curve after electrolysis, which shows a slight decline compared with the initial curve (Fig. S9†).
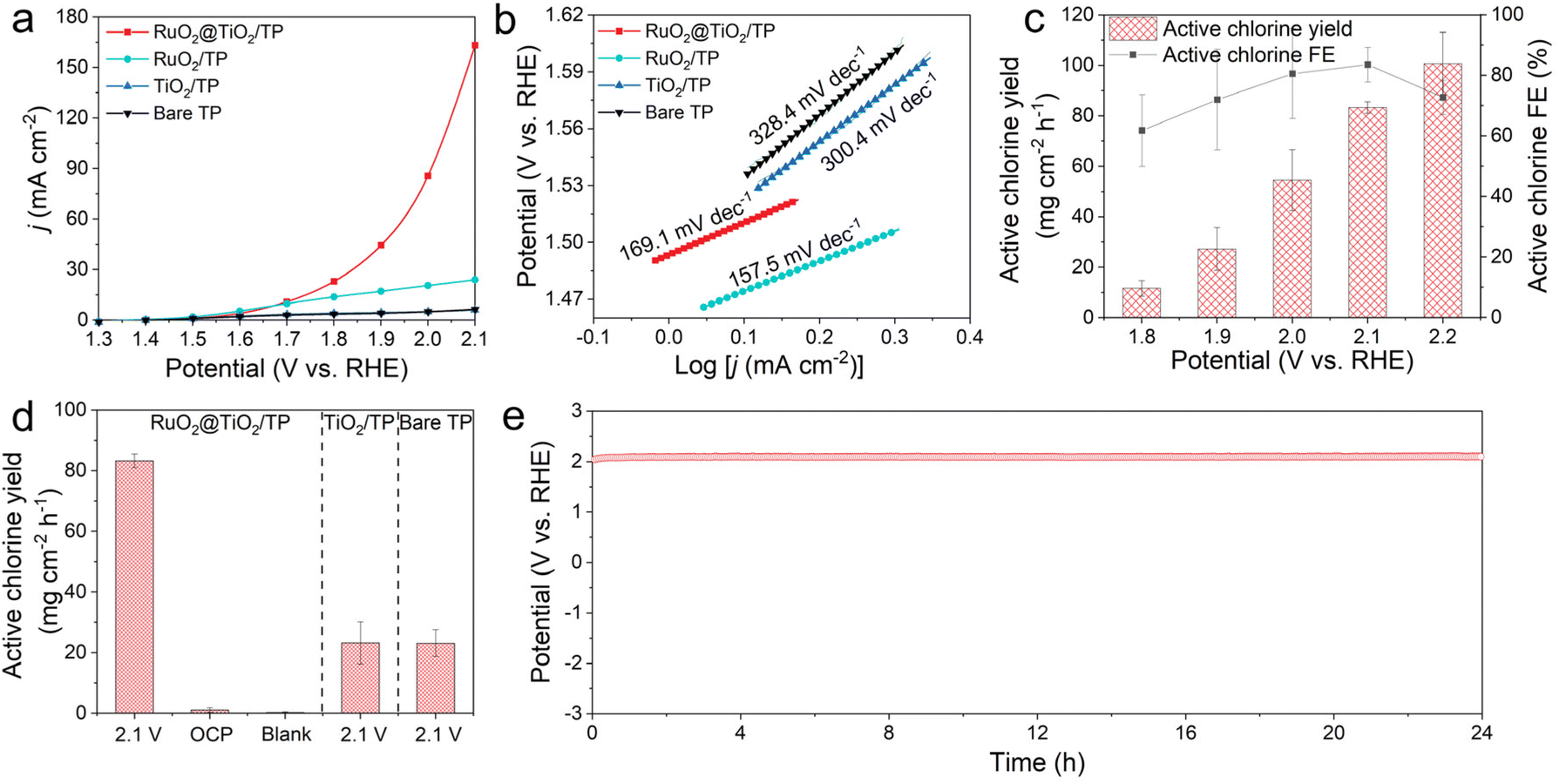 | ||
| Fig. 2 (a) LSV curves of RuO2@TiO2/TP, RuO2/TP, TiO2/TP, and bare TP at a scan rate of 5 mV s−1 in 0.5 M NaCl + 0.01 M HClO4 and (b) the corresponding Tafel plots of different catalysts. (c) Active chlorine yields and FEs of RuO2@TiO2/TP at different potentials. (d) Comparison of the active chlorine yields for RuO2@TiO2/TP under different electrochemical conditions, and for TiO2/TP and bare TP at 2.1 V in 0.5 M NaCl + 0.01 M HClO4. (e) Long-term stability test of RuO2@TiO2/TP at a constant current density of 50 mA cm−2 in 0.5 M NaCl + 0.01 M HClO4. | ||
In situ electrochemical Raman spectroscopy was employed to probe the dynamic evolution of the surface structure of RuO2@TiO2/TP during the CER (Fig. S10†). As shown in Fig. 3a, the peaks for the Ti–O and Ru–O bands appear at 399 cm−1 and 524 cm−1 in the potential window from the OCP to 2.015 V, respectively.29,30 With gradually increasing the potential to 1.915 V, the typical Raman peaks at 820 cm−1 and 933 cm−1 appeared, which were attributed to the Rux+ (x > 4) and Cl–O stretching bands, respectively,31–33 and the intensity gradually became larger when the potential was increased. These results illustrate that the surface of RuO2 was oxidized into Rux+ (x > 4) and adsorbed by the Cl–O species. Electrochemical impedance spectroscopy is a classical method for probing the electrochemical behavior of the electrode/electrolyte interfaces.34,35 A series of impedance spectra for RuO2@TiO2/TP are exhibited in Nyquist (Fig. 3b) and Bode (Fig. 3c) formats over a range of potentials (from the OCP to 2.26 V). These Nyquist plots were fitted with an equivalent circuit (Fig. S11†). Of note, the impedance responses of RuO2@TiO2/TP at the low-frequency region (below 102 Hz) were remarkable once the potentials were increased above 1.71 V. The impedance responses at these frequencies could be related to the metal oxyhydroxides on the RuO2 electrode surface, and the newly formed metal oxyhydroxides at high potentials may be the active phase of the CER.36,37 Inspired by the formation of various radicals in the oxidation system,20 we speculated that there may be free radicals in the electrolyte. Quasi in situ EPR spectroscopy was applied to investigate the Cl-related intermediate. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) as a spin trapping agent was added into 0.5 M NaCl + 0.01 M HClO4 to improve the lifetimes of the intermediate38 and to facilitate the EPR tests (Fig. 3d). Notably, three peaks at 3494, 3509, and 3524 G indexed to DMPO-Cl39 can be detected at a potential of 2.1 V in 0.5 M NaCl + 0.01 M HClO4, implying the generation of chlorine radicals40 catalyzed by RuO2@TiO2/TP. The quartet of peaks at 3487, 3502, 3517, and 3532 G with an intensity ratio of 1:2:2:1 corresponded to DMPO-OH. The control experiment showed that no relevant DMPO-Cl and DMPO-OH signals were detected in the blank electrolyte.
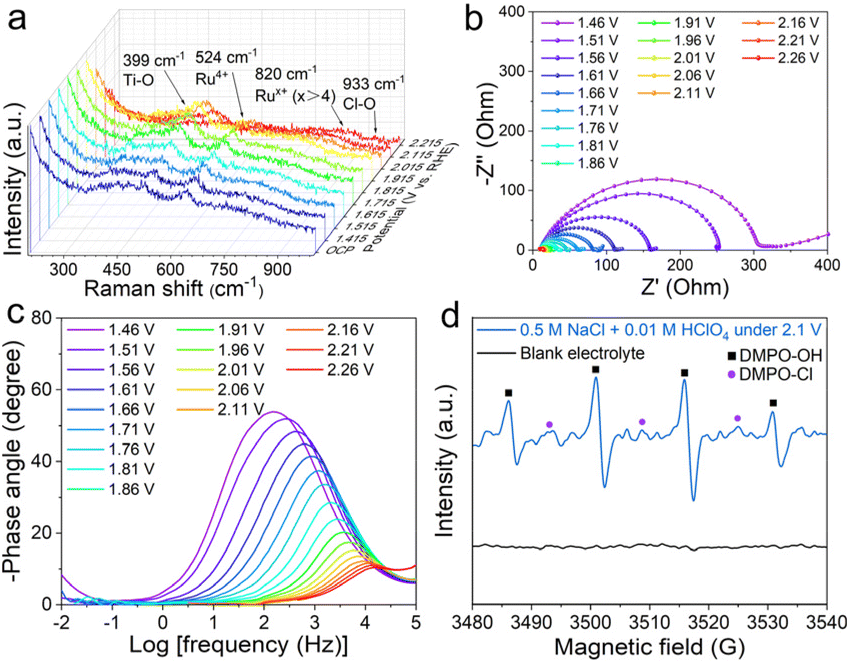 | ||
| Fig. 3 (a) In situ Raman spectra recorded for RuO2@TiO2/TP, shown as a function of potential, during the CER in 6 M NaCl + 0.01 M HClO4. (b) Nyquist plots and (c) Bode phase plots for RuO2@TiO2/TP at different applied potentials in 0.5 M NaCl + 0.01 M HClO4. (d) Quasi in situ EPR trapping of the radicals. | ||
Fig. 4a exhibits the LSV curves of RuO2@TiO2/TP in Ar- and NO-saturated 0.5 M NaCl + 0.01 M HClO4 in a gas-tight H-cell under ambient conditions. As the potential exceeds 1.9 V, the oxidation current density of RuO2@TiO2/TP slightly decreases in the NO-saturated electrolyte, indicating that NO can reduce the current density of the CER. This observation is worthy of study, and we further discuss this point in the later product analysis part. The produced nitrate and nitrite are quantified based on the standard method by using UV-vis spectrophotometry (Fig. S12 and Fig. S13,† respectively). The influence of the different potentials on the yields of nitrate and nitrite was investigated (Fig. 4b and c, respectively). It can be seen that the nitrate yields increase with more positive potential, and achieve a high yield of 95.22 mg cm−2 h−1 at 2.1 V, considerably outperforming the previously reported electrocatalysts for NOR under ambient conditions (Table S2†). Of note, yields of the nitrite are essentially the same and negligible at each potential, with the highest value of 0.27 mg cm−2 h−1 at 1.9 V. The corresponding chronoamperometry curves and UV-vis absorption spectra are displayed in Fig. S14.† NO reacts with O2 in air easily to produce NO2 chemically, which can also easily dissolve in water to form nitrate.10 The control experiment of NO and O2 co-saturated electrolyte without electricity was conducted. As shown in Fig. 4d and Fig. S15,† in 0.5 M NaCl + 0.01 M HClO4, the nitrate yield from the reaction between NO and O2 is very low relative to our system, being about 15.6 mg (Fig. 4d). Subsequently, we systematically investigated the NOOR performance of RuO2@TiO2/TP. It can be seen distinctly that RuO2@TiO2/TP produces trace nitrate, and the maximum nitrate yield of 2.76 mg cm−2 h−1 was achieved at 1.9 V in NO-saturated 0.5 M Na2SO4 + 0.01 M HClO4 (Fig. 4e and Fig. S16†). Of note, RuO2@TiO2/TP produced much more nitrite than nitrate at each potential for the NOOR. A high nitrite yield of 14.31 mg cm−2 h−1 was obtained at 2.2 V, which was significantly higher than that when NO reacts with active chlorine generated by the CER at all potentials (Fig. 4f). The cascade catalytic system consisting of the CER and NO oxidation exhibiting an enhanced nitrate yield can be explained as follows. Due to the low solubility of NO in water, a conventional NOOR occurs at the electrode surface and is quickly limited by mass transport at high current densities, resulting in poor nitrate yields. In our cascade catalysis system, chloride is oxidized by RuO2@TiO2/TP to active chlorine species in the electrolyte, which can rapidly oxidize NO to nitrate. Chloride also acts as a redox mediator between the electrode and NO, and the active chlorine species donate electrons to NO in solution and return to the initial chloride state (Fig. 4g). Moreover, the total nitrate yields increase almost linearly from 78.01 mg at 1 h to 1222.44 mg at 12 h, which reveals the good stability of our RuO2@TiO2/TP catalyst in the cascade catalytic system with the CER and NO oxidation (Fig. 4h and Fig. S17†). Notably, the XRD pattern and SEM image of post-electrolysis RuO2@TiO2/TP confirm that the crystal structure and nanobelt array are maintained, respectively (Fig. S18 and S19†). Moreover, the Ru 3d region of post-electrolysis RuO2@TiO2/TP shows a high-valence Ru oxidation state, which is consistent with previous reports (Fig. S20†).24,41
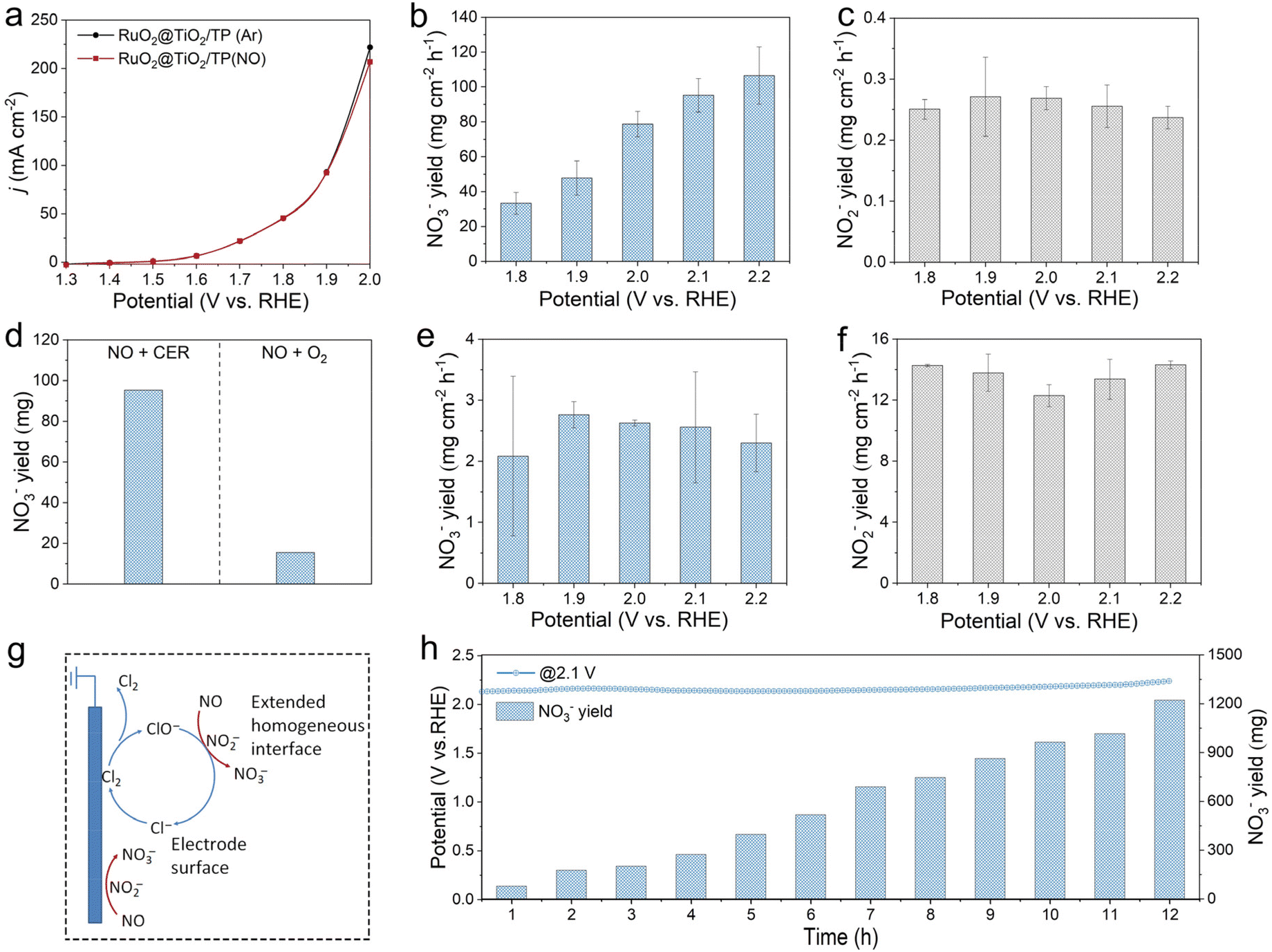 | ||
| Fig. 4 (a) LSV curves of RuO2@TiO2/TP in Ar- and NO-saturated 0.5 M NaCl + 0.01 M HClO4. (b) Nitrate and (c) nitrite yields over RuO2@TiO2/TP at each given potential in NO-saturated 0.5 M NaCl + 0.01 M HClO4. (d) Nitrate yields of RuO2@TiO2/TP for 1 h of electrolysis in gas-tight (in an NO-saturated electrolyte) and open electrolytic cells (under an NO + O2 atmosphere). (e) Nitrate and (f) nitrite yields over RuO2@TiO2/TP at each given potential in NO-saturated 0.5 M Na2SO4 + 0.01 M HClO4. (g) Possible reaction pathways for nitrate production. (h) Long-term stability tests for continuous generation of nitrate over RuO2@TiO2/TP in NO-saturated 0.5 M NaCl + 0.01 M HClO4. | ||
In the present study, we demonstrate an efficient production process of nitrate under ambient conditions by using RuO2@TiO2/TP as the electrode to produce active chlorine to oxidize NO in aqueous solution. Such a RuO2@TiO2/TP catalyst exhibits a low potential of 1.78 V to reach a current density of 20 mA cm−2 and a high active chlorine yield of 83.3 mg cm−2 h−1 at 2.1 V in 0.5 M NaCl + 0.01 M HClO4. Owing to the high active chlorine yield, our system enables a superior nitrate yield of 95.22 mg cm−2 h−1 in NO-saturated 0.5 M NaCl + 0.01 M HClO4 at 2.1 V, and trace amounts of unwanted nitrite (0.25 mg cm−2 h−1) are generated. Moreover, a 12 h stability test with steady nitrate build-up is performed. The electrochemical in situ Raman analysis revealed the active site role of Rux+ (x > 4) in catalyzing the CER and the interaction between the Cl atom and O atom at high potentials (1.915 V to 2.215 V). The quasi in situ EPR results confirmed the presence of chlorine radicals and hydroxyl radicals catalytically produced by RuO2@TiO2/TP in the electrolyte. Our work highlights a promising strategy for highly efficient conversion of NO into nitrate, and provides opportunities for the synthesis of high-value chemicals from other small molecules with low solubility via this cascade system.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22072015).References
- J. Liang, Q. Liu, A. A. Alshehri and X. Sun, Recent advances in nanostructured heterogeneous catalysts for N-cycle electrocatalysis, Nano Res. Energy, 2022, 1, e9120010 CrossRef.
- A. J. Medford and M. C. Hatzell, Photon-driven nitrogen fixation: current progress, thermodynamic considerations, and future outlook, ACS Catal., 2017, 7, 2624–2643 CrossRef CAS.
- J. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Beyond fossil fuel-driven nitrogen transformations, Science, 2018, 360, eaar6611 CrossRef PubMed.
- Y. Wang, Y. Yu, R. Jia, C. Zhang and B. Zhang, Electrochemical synthesis of nitric acid from air and ammonia through waste utilization, Natl. Sci. Rev., 2019, 6, 730–738 CrossRef CAS PubMed.
- I. Dybkjaer, in Ammonia, Catalysis and Manufacture, ed. A. Nielsen, Springer, Heidelberg, 1995, 199–308 Search PubMed.
- C. Dai, Y. Sun, G. Chen, A. C. Fisher and Z. J. Xu, Electrochemical oxidation of nitrogen towards direct nitrate production on spinel oxides, Angew. Chem., Int. Ed., 2020, 59, 9418–9422 CrossRef CAS PubMed.
- Y. Wang, T. Li, Y. Yu and B. Zhang, Electrochemical synthesis of nitric acid from nitrogen oxidation, Angew. Chem., Int. Ed., 2022, 61, e202115409 CAS.
- S. Han, C. Wang, Y. Wang, Y. Yu and B. Zhang, Electrosynthesis of nitrate via the oxidation of nitrogen on tensile-strained palladium porous nanosheets, Angew. Chem., Int. Ed., 2021, 60, 4474–4478 CrossRef CAS PubMed.
- Y. Ji, L. Li, W. Cheng, Y. Xiao, C. Li and X. Liu, A CeP nanoparticle-reduced graphene oxide hybrid: an efficient electrocatalyst for the NH3 synthesis under ambient conditions, Inorg. Chem. Front., 2021, 8, 2103–2106 RSC.
- D. Wang, N. He, L. Xiao, F. Dong, W. Chen, Y. Zhou, C. Chen and S. Wang, Coupling electrocatalytic nitric oxide oxidation over carbon cloth with hydrogen evolution reaction for nitrate synthesis, Angew. Chem., Int. Ed., 2021, 60, 24605–24611 CrossRef CAS PubMed.
- C. Liu, Y. Wang and B. Zhang, Synthesis of ammonia via an electroreduction removal of NO from exhausted gas: an upgrading to N2 fixation, Sci. China: Chem., 2020, 63, 1173–1174 CrossRef CAS.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, High-efficiency electrocatalytic NO reduction to NH3 by nanoporous VN, Nano Res. Energy, 2022, 1, e9120022 CrossRef.
- L. Zhang, J. Liang, Y. Wang, T. Mou, Y. Lin, L. Yue, T. Li, Q. Liu, Y. Luo, N. Li, B. Tang, Y. Liu, S. Gao, A. A. Alshehri, X. Guo, D. Ma and X. Sun, High-performance electrochemical NO reduction into NH3 by MoS2 nanosheet, Angew. Chem., Int. Ed., 2021, 60, 25263–25268 CrossRef CAS PubMed.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S.-Z. Qiao, Nitrogen vacancies on 2D layered W2N3: a stable and efficient active site for nitrogen reduction reaction, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- J. Liang, W.-F. Hu, B. Song, T. Mou, L. Zhang, Y. Luo, Q. Liu, A. A. Alshehri, M. S. Hamdy, L.-M. Yang and X. Sun, Efficient nitric oxide electroreduction toward ambient ammonia synthesis catalyzed by a CoP nanoarray, Inorg. Chem. Front., 2022, 9, 1366–1372 RSC.
- R. K. B. Karlsson and A. Cornell, Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes, Chem. Rev., 2016, 116, 2982–3028 CrossRef CAS PubMed.
- M. Hou, L. Chen, Z. Guo, X. Dong, Y. Wang and Y. Xia, A clean and membrane-free chlor-alkali process with decoupled Cl2 and H2/NaOH production, Nat. Commun., 2018, 9, 438 CrossRef PubMed.
- D. Shao, W. Yan, L. Cao, X. Li and H. Xu, High-performance Ti/Sb-SnO2/Pb3O4 electrodes for chlorine evolution: preparation and characteristics, J. Hazard. Mater., 2014, 267, 238–244 CrossRef CAS PubMed.
- Y. Wang, Y. Liu, D. Wiley, S. Zhao and Z. Tang, Recent advances in electrocatalytic chloride oxidation for chlorine gas production, J. Mater. Chem. A, 2021, 9, 18974–18993 RSC.
- H. Park, C. D. Vecitis and M. R. Hoffmann, Electrochemical water splitting coupled with organic compound oxidation: the role of active chlorine species, J. Phys. Chem. C, 2009, 113, 7935–7945 CrossRef CAS.
- Q. Wang, T. Li, C. Yang, M. Chen, A. Guan, L. Yang, S. Li, X. Lv, Y. Wang and G. Zheng, Electrocatalytic methane oxidation greatly promoted by chlorine intermediates, Angew. Chem., Int. Ed., 2021, 60, 17398–17403 CrossRef CAS PubMed.
- J. Liang, L. Zhang, X. He, Y. Wang, Y. Luo, D. Zheng, S. Sun, Z. Cai, J. Zhang, K. Ma, Y. Zheng, X. Sun and C. Tang, Redox mediators promote electrochemical oxidation of nitric oxide toward ambient nitrate synthesis, J. Mater. Chem. A, 2023, 11, 1098–1107 RSC.
- D. Mateo, J. Albero and H. García, Titanium-perovskite-supported RuO2 nanoparticles for photocatalytic CO2 methanation, Joule, 2019, 3, 1949–1962 CrossRef CAS.
- D. J. Morgan, Resolving ruthenium: XPS studies of common ruthenium materials, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- D. Zhao, J. Liang, J. Li, L. Zhang, K. Dong, L. Yue, Y. Luo, Y. Ren, Q. Liu, M. S. Hamdy, Q. Li, Q. Kong and X. Sun, A TiO2–x nanobelt array with oxygen vacancies: an efficient electrocatalyst toward nitrite conversion to ammonia, Chem. Commun., 2022, 58, 3669–3672 RSC.
- J. Augustynski, L. Balsenc and J. Hinden, X–Ray photoelectron spectroscopic studies of RuO2-based film electrodes, J. Electrochem. Soc., 1978, 125, 1093–1097 CrossRef CAS.
- H. Chen, T. Wu, X. Li, S. Lu, F. Zhang, Y. Wang, H. Zhao, Q. Liu, Y. Luo, A. M. Asiri, Z. Feng, Y. Zhang and X. Sun, Modulating oxygen vacancies of TiO2 nanospheres by Mn-doping to boost electrocatalytic N2 reduction, ACS Sustainable Chem. Eng., 2021, 9, 1512–1517 CrossRef CAS.
- J. G. Vos and M. T. M. Koper, Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry, J. Electroanal. Chem., 2018, 819, 260–268 CrossRef CAS.
- X. Wang, X. Wan, X. Qin, C. Chen, X. Qian, Y. Guo, Q. Xu, W.-B. Cai, H. Yang and K. Jiang, Electronic structure modulation of RuO2 by TiO2 enriched with oxygen vacancies to boost acidic O2 evolution, ACS Catal., 2022, 12, 9437–9445 CrossRef CAS.
- H. Li, S. Zha, Z.-J. Zhao, H. Tian, S. Chen, Z. Gong, W. Cai, Y. Wang, Y. Cui, L. Zeng, R. Mu and J. Gong, The nature of loading-dependent reaction barriers over mixed RuO2/TiO2 catalysts, ACS Catal., 2018, 8, 5526–5532 CrossRef CAS.
- A. Satsuma, M. Yanagihara, J. Ohyama and K. Shimizu, Oxidation of CO over Ru/Ceria prepared by self-dispersion of Ru metal powder into nano-sized particle, Catal. Today, 2013, 201, 62–67 CrossRef CAS.
- Y. Zhao, M. Xi, Y. Qi, X. Sheng, P. Tian, Y. Zhu, X. Yang, C. Li and H. Jiang, Redirecting dynamic structural evolution of nickel-contained RuO2 catalyst during electrochemical oxygen evolution reaction, J. Energy Chem., 2022, 69, 330–337 CrossRef CAS.
- K. Nishiyama, A. Ueda, S. Tanoue, T. Koga and I. Taniguchi, Direct observation of perchlorate incorporation induced by redox reaction of ferrocene terminated self-assembled monolayer studied by in situ FT-surface enhanced raman spectroscopy, Chem. Lett., 2000, 29, 930–931 CrossRef.
- M. E. G. Lyons and M. P. Brandon, The significance of electrochemical impedance spectra recorded during active oxygen evolution for oxide covered Ni, Co and Fe electrodes in alkaline solution, J. Electroanal. Chem., 2009, 631, 62–70 CrossRef CAS.
- C.-S. Hsu, N.-T. Suen, Y.-Y. Hsu, H.-Y. Lin, C.-W. Tung, Y.-F. Liao, T.-S. Chan, H.-S. Sheu, S.-Y. Chen and H. M. Chen, Valence- and element-dependent water oxidation behaviors: in situ X-ray diffraction, absorption and electrochemical impedance spectroscopies, Phys. Chem. Chem. Phys., 2017, 19, 8681–8693 RSC.
- V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Tailoring the selectivity for electrocatalytic oxygen evolution on ruthenium oxides by zinc substitution, Angew. Chem., Int. Ed., 2010, 49, 4813–4815 CrossRef CAS PubMed.
- K. S. Exner, J. Anton, T. Jacob and H. Over, Chlorine evolution reaction on RuO2(110): Ab initio atomistic thermodynamics study-pourbaix diagrams, Electrochim. Acta, 2014, 120, 460–466 CrossRef CAS.
- G. R. Buettner, Spin trapping: ESR parameters of spin adducts, Free Radicals Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed.
- Z. Shen, J. Li, Y. Zhang, J. Bai, X. Tan, X. Li, L. Qiao, Q. Xu and B. Zhou, Highly efficient total nitrogen and simultaneous total organic carbon removal for urine based on the photoelectrochemical cycle reaction of chlorine and hydroxyl radicals, Electrochim. Acta, 2019, 297, 1–9 CrossRef CAS.
- K. Guo, Z. Wu, C. Chen and J. Fang, UV/chlorine process: an efficient advanced oxidation process with multiple radicals and functions in water treatment, Acc. Chem. Res., 2022, 55, 286–297 CrossRef CAS PubMed.
- A. Goryachev, M. Etzi Coller Pascuzzi, F. Carlà, T. Weber, H. Over, E. J. M. Hensen and J. P. Hofmann, Electrochemical stability of RuO2(110)/Ru(0001) model electrodes in the oxygen and chlorine evolution reactions, Electrochim. Acta, 2020, 336, 135713 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and ESI figures. See DOI: https://doi.org/10.1039/d3qi00209h |
| This journal is © the Partner Organisations 2023 |