
Light-gated specific oxidase-like activity of a self-assembled Pt(II) nanozyme for environmental remediation†
Rohit
Kapila
 ,
Bhaskar
Sen
,
Alisha
Kamra
,
Shana
Chandran
and
Subinoy
Rana
*
,
Bhaskar
Sen
,
Alisha
Kamra
,
Shana
Chandran
and
Subinoy
Rana
*
Materials Research Centre, Indian Institute of Science, C. V. Raman Road, Bangalore 560012, Karnataka, India. E-mail: subinoy@iisc.ac.in; Tel: +91 080-22932914
First published on 14th August 2023
Abstract
Artificial enzyme equivalents, also known as nanozymes, are a practical tool for environmental remediation when compared to their natural counterparts due to their high operational stability, efficiency, and cost-effectiveness. Specific oxidase mimicking nanozymes are well suited to degrade toxic chemicals from industrial waste such as phenols and azo dyes. Therefore, photocatalytic nanozymes using visible/sunlight would provide a viable strategy for sustainable environmental remediation. Herein, we introduce an aggregation-induced emissive Pt(II) complex, which self-assembles in water providing NanoPtA nanotapes. These structures exhibit a specific oxidase-like nanozyme activity driven by light. The NanoPtA structure assists in the photogeneration of singlet oxygen in water via a triplet excited 3MMLCT state, leading to a specific oxidase-like activity instead of a peroxidase-like activity. The self-assembled nanozyme showed great stability under harsh environmental conditions and exhibited photo-induced specific oxidase-mimetic activity, which was considerably more efficient than the natural enzyme or other specific nanozymes. We demonstrated efficient NanoPtA-induced photocatalytic degradation of various phenolic compounds and azo dyes within 5–10 minutes of light irradiation. Notably, the system operates under sunlight and exhibits reusability over twenty cycles of catalytic reactions. Another fascinating aspect of NanoPtA is the unaltered catalytic performance for more than 75 days, providing a robust enzyme-equivalent for practical sustainable environmental remediation.
1. Introduction
Natural enzymes as biocatalysts are known for their specificity and efficiency in catalyzing biological reactions in living systems.1 However, they possess certain limitations that hinder their broad applications, including restricted stability, reduced efficacy under harsh conditions, and tedious as well as expensive synthesis and purification procedures. These limitations and their short shelf life impede their practical use and adoption in various industrial sectors.2,3 To address these challenges, a class of nanomaterials that possess enzyme-equivalent activity, known as nanozymes, has been extensively studied.4,5 Nanozymes traditionally refer to nanomaterials that facilitate the conversion of enzyme substrates into products, exhibiting reaction kinetics following the Michaelis–Menten model under physiological conditions, irrespective of their working mechanism compared to the natural enzyme.6 Essentially, nanozymes are artificial synthetic enzymes that display enzyme-like activities. Nanozymes typically consist of catalytically active metal ions and often organic linkers, which contribute to their enzyme mimetic properties. Compared to natural enzymes, nanozymes exhibit superior catalytic performance, selectivity, and stability in various applications including point-of-care diagnostics,7 biotechnology,8 antibacterial agents,9 and biofuel production.10 Diverse natural enzyme-like catalytic activities have been achieved using nanozymes, such as peroxidase,11,12 superoxide dismutase,13,14 esterase,15,16 oxidase,17,18 catalase,19,20 and halo-peroxidase.21,22 A notable feature of nanozymes is the more expansive physical and chemical operational windows, including high salt concentration, temperature, and pH tolerance.2 Due to the advantages of stability under harsh conditions and superior enzymatic performance over natural enzymes, nanozymes would be excellent candidates for environmental monitoring and remediation in challenging and unpredictable outdoor environments.The development of nanozymes with multiple enzymatic activities has mainly focused on designing micro/nanostructures using metals/metal oxides,23,24 carbon-based nanomaterials,25 polymers,26,27 and metal–organic frameworks.28,29 Although these materials have effective enzyme equivalent activities, certain challenges including non-dynamic nature under different stimuli, lack of specificity, low density of active sites, and complex/multi-step synthesis procedures restrict the achievement of “life-like” materials. Using the principles of supramolecular chemistry to mimic enzymes offers great opportunities to achieve such biomimetic materials. This strategy includes the development of nanozymes based on supramolecular structures that share a number of important characteristics similar to natural enzymes such as amphiphilicity, precise control of conformational states, supramolecular interactions driving substrate recognition for augmented activity, and specific hydrophobic microenvironments for catalysis.30 Supramolecular nanozymes provide modular and adaptive platforms that are created by assembling building blocks using noncovalent interactions,31 enabling the design of soft-materials to mimic the coordination environment found in enzyme active sites. Significantly, supramolecular nanozymes present essential features in common with natural enzymes. Consequently, diverse supramolecular nanozymes have been introduced containing different scaffolds such as peptides,32 micelles,33 vesicles,34 nanofibers,35 nanotubes,36 metal coordinated system,37 and hydrogels.38 Metal-based supramolecular systems including metal complexes,39 metallacycles,40 and metallacages41 offer useful spectroscopic properties including aggregation induced emission (AIE), which can be exploited to achieve nanozyme activities. For example, reactive oxidative species (ROS) enable specific oxidase mimetic catalysis,42 unlike traditional nanozymes, which show mixed peroxidase-like activity. Specific oxidase mimetic enzyme-equivalents avoid hydrogen peroxide in the reaction medium and have great implications in practical applications including therapeutics and environmental remediation.
Self-assembled metallo-supramolecular complexes featuring AIE offer promising platforms for generating photo-regulated reactive oxygen species. These supramolecular systems utilize bottom-up assemblies through the unique noncovalent metal–metal and π–π interactions, with advantageous photophysical properties.43 Herein, we developed a supramolecular Pt(II)-based nanotape assembly (NanoPtA), which causes enhanced AIE in aqueous medium as depicted in Scheme 1. This system shows excellent visible light-driven oxidase mimetic activity, precluding any peroxidase-like activity. Mechanistic studies suggest that the photocatalytic generation of singlet oxygen in the aqueous medium bestows the specific oxidase activity. The enzyme equivalent activity allowed us to explore the potential of NanoPtA in degrading organic pollutants and selective azo dye degradation as an environmental remediation approach. The system showed catalytic activity against a range of phenolic and azo-dye substrates. Given the exciting aspect of NanoPtA operating under sunlight/visible light, the present nanozyme is poised to demonstrate practical environmental sustainability. In addition, a practical feature of NanoPtA is its reusability for at least 20 cycles, retaining its catalytic activity for more than 75 days. To the best of our knowledge this is the first report on light-gated specific oxidase-like activity using the Pt(II) complex assembly. This system presents a highly desirable alternative to conventional oxidation methods that often suffer from selectivity and efficacy.
 | ||
| Scheme 1 A schematic illustration of NanoPtA assembly formation in water with variation in chain length resulting in different luminescence. | ||
2. Experimental details
Materials
All the chemicals and solvents used for synthesis and spectroscopic studies were used as received without further purification. Chemicals such as 2,6-pyridinedicarboxylic acid, o-phenylenediamine (OPD), polyphosphoric acid (PPA), dopamine hydrochloride, 4-amino antipyrine (4-AP), 2,4-dichlorophenol (2,4-DP), 2,2′-azino-bis(3-ethylbenzo thiazoline-6-sulfonic acid) (ABTS), 3,3′,5,5′-tetramethylbenzidine (TMB), Congo red, Reactive Red 52, methanol and phenols were obtained from SRL chemicals, India. 4-Methoxyphenol, 4-nitrophenol were purchased from Spectrochem, India. Malachite green oxalate, Evans Blue, Amido Black 10B and Amaranth were obtained from Avra chemicals, India. Adrenaline and Sudan Black were purchased from TCI chemicals. Trypan blue and HEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid) bufferwere obtained from Sigma-Aldrich. All the experiments were performed using Milli-Q water.Synthesis of the ligands and complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; v/v), which afforded the product in a good yield (0.38 g, 75%).
:1; v/v), which afforded the product in a good yield (0.38 g, 75%).
All the reaction schemes for the ligand and complex are shown in the ESI.†
Oxidase-mimicking activity induced by visible light
The catalytic oxidation of TMB, ABTS, and OPD was used to study the NanoPtA oxidase-mimicking activity induced by visible light. In a typical experiment, stock solutions of the catalyst NanoPtA (1.0 mM) in methanol and 5.0 mM chromogenic substrates such as TMB, OPD, or ABTS in DMSO were prepared. Catalytic oxidation studies were then performed under visible light where the final solution contained 10 μL of NanoPtA, 100 μL of chromogenic substrate and 890 μL of sodium acetate buffer (100 mM, pH 4.0). The reaction solution was irradiated for 10 min with visible light (470 nm) at room temperature and the absorbance maximum was monitored at 652 nm, 418 nm, and 450 nm for TMB, ABTS, and OPD, respectively using a SpectraMax® M5e microplate reader.Kinetic assay for TMB oxidation
Kinetics studies were carried out with a TMB substrate by varying the concentrations from 0 to 0.5 mM. After 5 min visible light irradiation (470 nm), the colorless solution turned blue. The absorbance was measured at 652 nm. The acquired data were fitted using the Michaelis–Menten equation, and the kinetic parameters were determined.where KM defines the Michaelis–Menten constant, Vmax represents the maximum velocity, V0 is the initial rate, and S represents the substrate concentration.
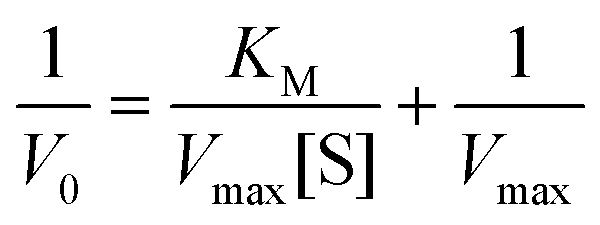
Detection of ROS and the scavenger study
The ROS generation was measured by using UV-vis absorbance spectroscopy. Various known reactants and scavengers were used to study the presence of ROS. To monitor the presence of singlet oxygen (1O2), the photooxidation reaction of 1,3-diphenyl isobenzofuran (DPBF) was utilized. A DPBF (20 μM) solution was added to 10 μM NanoPtA in 100 mM acetate buffer at (pH 4.0). The solution was kept under visible light irradiation for different intervals of time. The estimation of the singlet oxygen was confirmed in the presence of different scavengers such as sodium azide (10 mM), L-histidine (0.4 mM) as a singlet oxygen quencher, isopropanol as a hydroxyl radical quencher (10 mM), and KI as a H+ quencher (10 mM), which were added to the vial containing TMB 0.5 mM and 10 μM of NanoPtA.Terephthalic acid (TA) assay
For the TA assay, we used hydrogen peroxide (5.0 mM), terephthalic acid (0.5 mM) and the catalyst NanoPtA (10 μM). The assay was performed in 100 mM acetate buffer (pH 4.0). Fluorescence spectra ˙OH trapped TA was monitored at 435 nm using 315 nm as the excitation wavelength after 1 h irradiation of visible light (470 nm).3. Results and discussion
Fabrication of the self-assembled Pt(II) system
We synthesized three Pt(II) complexes (Pt-1, Pt-6 and Pt-8) containing conjugated aromatic tridentate donor ligands substituted with C1, C6 and C8 alkyl chains on the benzimidazole rings (Scheme 1 and Schemes S1 and S2†), with good yields through a slight modification of a previously reported protocol.44 First, the BiP ligand was substituted with an alkyl halide in the presence of a base followed by reaction with K2PtCl4 salt to afford the desired Pt-1, Pt-6 and Pt-8 complexes. The detailed synthetic routes and characterization data are presented in the ESI.† All the ligands and complexes were confirmed by 1H and 13C NMR spectroscopy and mass spectrometry (Fig. S1–S11†). Next, we studied the self-assembly of Pt-6 to form NanoPtA with increasing water fractions in methanol and investigated its photophysical properties. The decrease of two high energy absorption bands at 310 and 390 nm in the UV-vis spectra of NanoPtA (100 μM) are attributed to the intra-ligand charge transfer (ILCT)44 originating from the n–π* and π–π* transitions (Fig. 1a). A less intense band at 420–500 nm was assigned to the dz2(Pt)–π* (C6-BiP) metal-to-ligand charge transfer (1MLCT) transition. Notably, a lower energy absorption band at around 550 nm is observed upon introducing water in methanol solution, which is intensified upon increasing the water fraction (Fig. 1a). The plot of absorbance at 550 nm versus water volume fraction showed a steady linear increase (Fig. 1b). This band is ascribed to the metal–metal-to-ligand charge transfer (1MMLCT) transition and has been previously described for the formation of Pt⋯Pt dimers and oligomers.45 These results suggest that the Pt-6 complex tends to undergo self-assembly in water to form NanoPtA due to enhanced intermolecular dz2 interactions between the adjacent Pt(II) centres (Pt⋯Pt).46 Moreover, an aggregation induced emission (AIE) band at 650 nm in the photoluminescence (PL) spectroscopy was observed with an increase in water fraction, which originates from the excited states of the 3MMLCT transition (λexc. 390 nm) (Fig. 1c and S12†) and shows a quantum yield of 12.7% (Fig. S13†). The photophysical properties of the Pt(II) complex were dictated by the aggregation and solubility of the complexes. Notably, Pt-6 exhibits an enhanced emission and luminescence lifetime in its aggregated state compared to the complexes with lower (Pt-1) or higher (Pt-8) alkyl chains (Fig. 1e and Fig. S14†). We conducted further studies on Pt-6 considering these advantageous photophysical properties.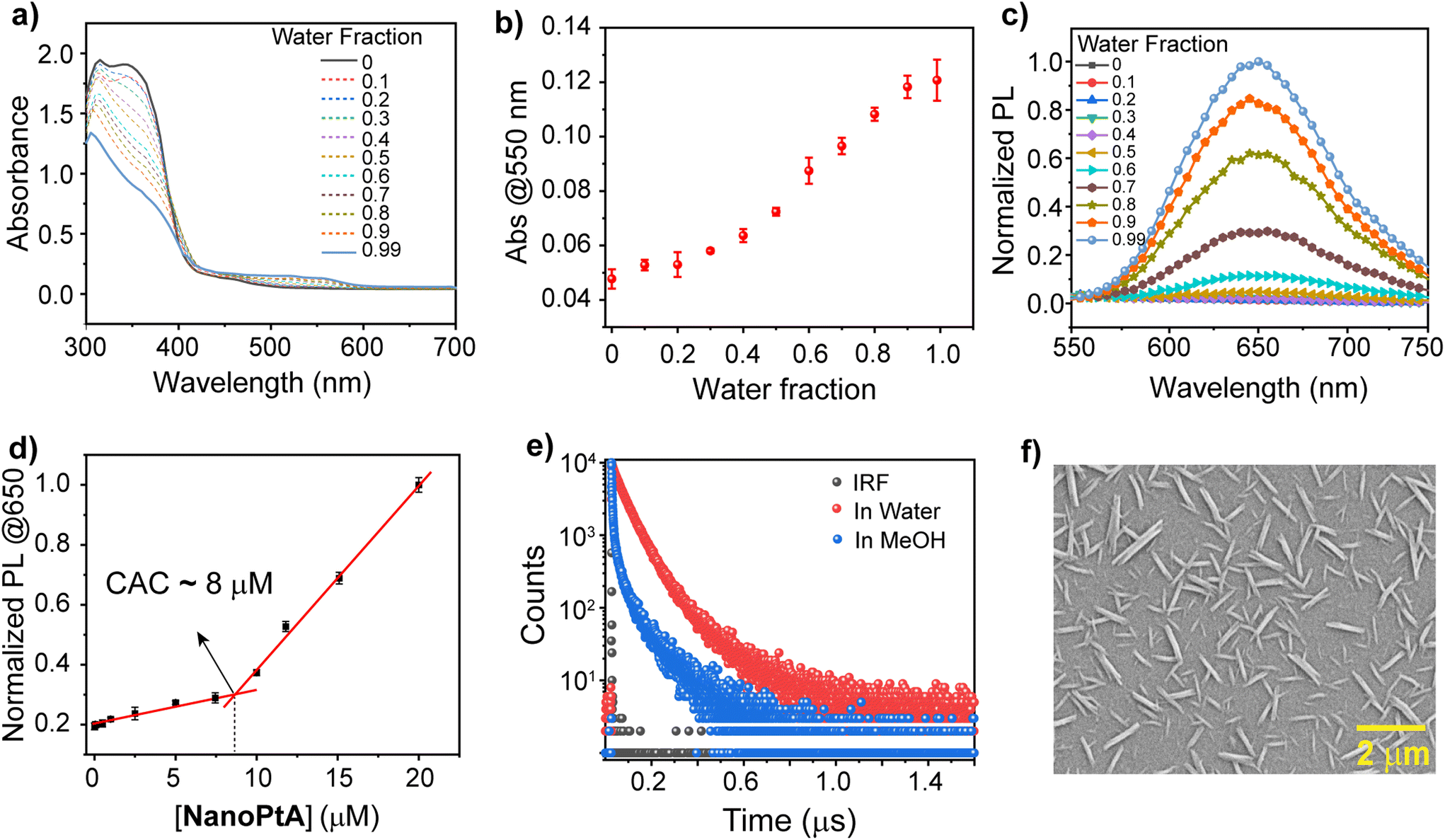 | ||
| Fig. 1 (a) UV-vis spectra of NanoPtA (100 μM) with an increasing volume fraction of water in methanol (0–99%; v/v). (b) Plot of absorbance maxima at 550 nm of NanoPtA with increasing water fraction. (c) PL spectra of NanoPtA (10 μM) with varying water fractions (0–99%; v/v). (d) Determination of the critical aggregation constant (CAC) of NanoPtA by measuring PL intensity at 650 nm. (e) Comparison of the photoluminescence lifetime of NanoPtA in MeOH and in aqueous solution, whereas IRF is the instrument response function. (f) FESEM images of the formed assembly of NanoPtA in 99% water–methanol. | ||
The combined UV-vis and PL spectroscopy results indicate that monomeric Pt-6 in methanol self-assembles into NanoPtA in aqueous medium. Using the self-emissive behaviour of Pt-6 in higher water fractions, we calculated the critical aggregation constant (CAC) of NanoPtA in the 99% water fraction and found the CAC to be as low as ∼8 μM (Fig. 1d). Furthermore, the average luminescence lifetime (τav) of 4.8 ns for Pt-6 in MeOH increased gradually with the increase in water fraction (40%, 60%, 80%, and 99% water–methanol; v/v) to 137 ns (Fig. 1e and Fig. S15†). This further confirms the existence of 3MMLCT transitions in the aqueous medium, which probably originate from the self-assembled state through noncovalent interactions such as π–π, hydrophobic and Pt⋯Pt interactions between the adjacent centers. In addition, we performed field emission scanning electron microscopy (FESEM) analysis to obtain an insight into the morphology of the self-assembled structures. FESEM images revealed that NanoPtA forms nanotape structures with an average length of ∼0.5–1.5 μm and width ∼90–100 nm (Fig. 1f and Fig. S16†). Besides, dynamic light scattering (DLS) studies revealed that the hydrodynamic diameter of Pt-6 increased with the increasing polarity of the solvent (Fig. S17†), with the largest diameter (Zaverage) of ∼1.1 μM at the highest water fraction (99% water–methanol) (Fig. S17b†). Altogether, we fabricated a tape-like self-assembled structure using aggregation of Pt-6 in aqueous medium, leading to significant AIE.
Photocatalytic oxidase-like activity of NanoPtA
The emergence of AIE from the self-assembled structure prompted us to explore the oxidase-like activity, which generally depends on ROS. The oxidase-like activity of NanoPtA was studied above its CAC (≥8.0 μM) using a colorimetric assay that included substrates 3,3′,5,5′-tetramethylbenzidine (TMB) with an apparent colour change from colourless to blue after oxidation (Fig. 2a).47 It was observed that the treatment of NanoPtA (10 μM) with visible light (at 470 nm) for 10 min turns the TMB (0.5 mM) into a blue coloured solution in acetate buffer (100 mM, pH 4.0) (Fig. 2b). The significant absorbance appearance at 652 nm indicates the oxidation of TMB in the presence of the NanoPtA catalyst upon irradiation with visible light. In contrast, TMB did not show any colour change in the absence of the NanoPtA catalyst although the reaction mixture was exposed to visible light. To establish the optimum time span for the complete oxidation of TMB, we studied a time dependent TMB absorbance change by varying the light exposure time in the presence of NanoPtA. The gradual increase in absorbance at 652 nm with different time intervals (Fig. S18†) confirmed a nearly linear increase in oxidase activity with increasing exposure time, before the saturation at 10 min (Fig. 2c). Further, control experiments with the assembly components such as C6-BiP ligand and K2PtCl4 salt even at 5 and 10 equivalents to NanoPtA, respectively, did not show significant oxidase activity (Fig. 2d). In fact, the monomeric form of NanoPtA in MeOH displayed no enzymatic activity (Fig. 2d), indicating that the excellent oxidase-like activity originated from the assembled state. Further, we tested the activity of the three assembled materials, which showed the most efficient oxidase activity by self-assembled Pt-6 (Fig. S19†), followed by Pt-8 and Pt-1.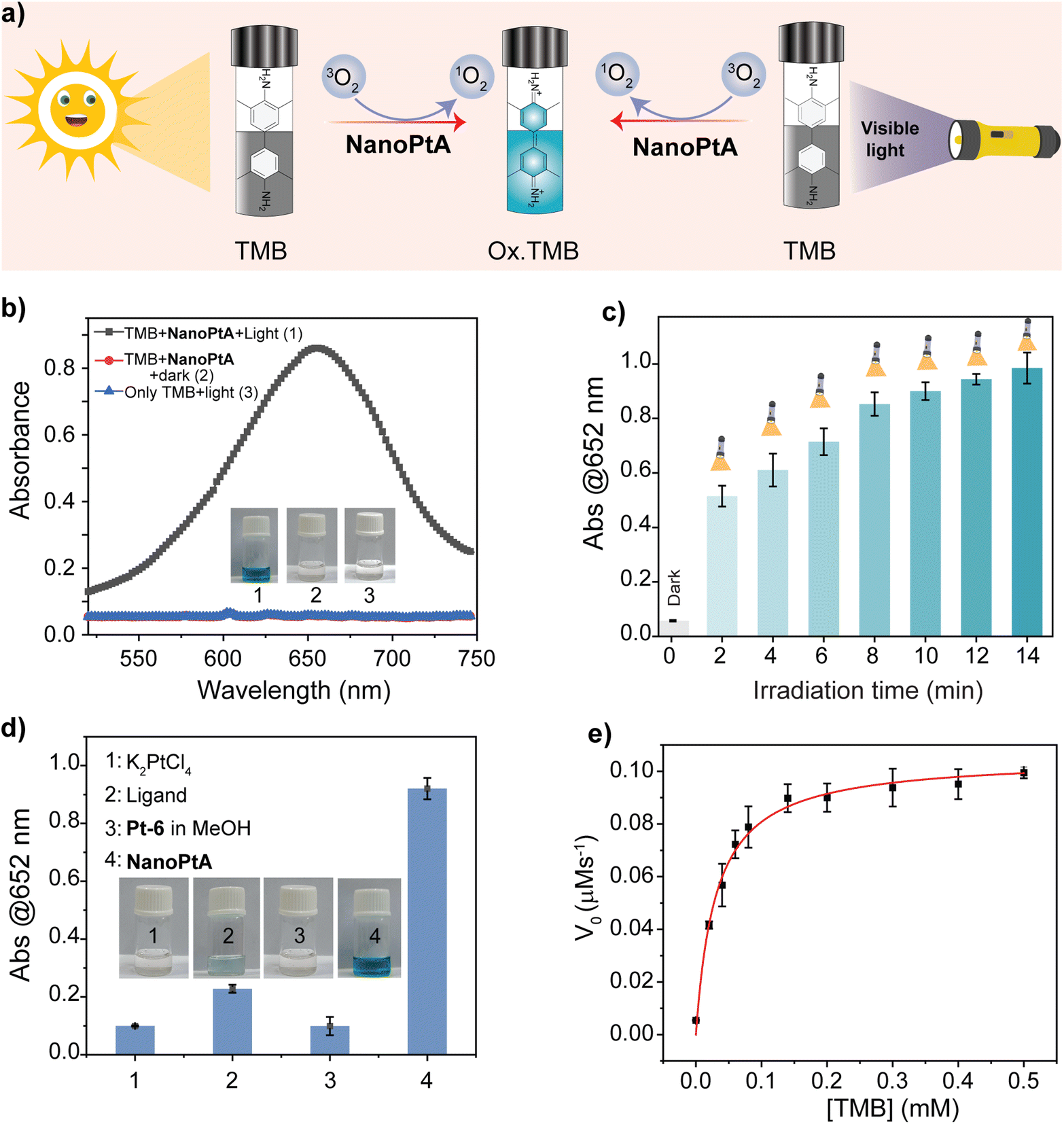 | ||
| Fig. 2 (a) A schematic representation of TMB oxidation both in the presence of visible light and sunlight. (b) UV-vis spectra of TMB (0.5 mM) in the absence and presence of NanoPtA (10 μM) after visible light irradiation (470 nm) in acetate buffer (100 mM, pH 4.0) for 10 min. (c) Absorbance of oxidized TMB (0.5 mM) at 652 nm after visible light irradiation monitored at different time intervals. (d) A comparison of the photo-oxidation of TMB (0.5 mM) by NanoPtA (10 μM) in buffer and in methanol and K2PtCl4 (100 μM) and the ligand C6-BiP (50 μM) in acetate buffer (100 mM, pH 4.0) for 10 min. (Inset) Images of the corresponding solutions of (1) with K2PtCl4, (2) ligand, (3) Pt-6 in MeOH, and (4) NanoPtA. (e) The plot of initial oxidation rate of TMB monitored at 652 nm versus TMB concentration (0–0.5 mM) in the presence of NanoPtA (10 μM) upon 10 min visible light irradiation. Light source = 470 nm visible light. | ||
To evaluate the catalytic performance of the NanoPtA assembly, we performed steady state kinetic assays with increasing concentrations of TMB in the presence of NanoPtA in acetate buffer (100 mM, pH 4.0). Enhanced activity was observed with increasing TMB concentration, which was fitted to the Michaelis–Menten equation. Based on the kinetics plot Fig. 2e, the Michaelis constant (KM) and the maximal reaction velocity (Vmax) were determined to be 0.03 mM and 0.105 μM s−1, respectively. The calculated KM was lower than the previously reported nanozymes, including CeO2 NPs, and Fe–N/C (Table S1†), indicating that NanoPtA possesses a good affinity for TMB. Further, we investigated the practical applicability of NanoPtA for the photooxidation of TMB using sunlight. At first, we measured the extent of TMB oxidation by varying both TMB (0–0.5 mM) and NanoPtA catalyst concentration (0–50 μM) keeping the same experimental conditions under 10 min sunlight exposure. The results depicted in Fig. S20a† indicate that an increase in the concentration of NanoPtA leads to an enhanced yield of oxidized TMB. Also, a substrate concentration-dependent catalytic reaction was observed using a constant catalyst concentration under sunlight (Fig. S20b†). In addition, other substrates like OPD and ABTS were employed to investigate the generalizability of the photocatalytic oxidase-mimicking activity of NanoPtA under visible light (470 nm) irradiation. The production of distinct yellow and green colours with absorption maxima at 417 nm and 448 nm,48 respectively, indicates that NanoPtA can oxidize both OPD and ABTS (Fig. S21†). Therefore, it can be confirmed that NanoPtA possesses photocatalytic oxidase-mimicking activity for a variety of substrates.
Mechanistic studies of the oxidase-like activity of NanoPtA
Fluorophores that contain benzodiazole, benzothiadiazole, benzotriazole, and metal-based luminescent materials are known to efficiently generate ROS upon photoexcitation through a common mechanism,49 as illustrated in Fig. 3a. We tested if the oxidase-like activity of NanoPtA followed a similar mechanism involving ROS. We employed the ROS detection agent 1,3-diphenylisobenzofuran (DPBF), which showed an initial light-yellow colour with an absorbance peak at 418 nm. A gradual decrease in the absorption peak was observed with an increasing duration of visible light irradiation (Fig. 3b). This decrease in absorption intensity can be attributed to the photooxidation of DPBF, which resulted in the formation of 1,2-dibenzoyl benzene (DBB) through ring opening,50 mediated by the singlet oxygen generated (inset of Fig. 3b). However, the DPBF peak did not change in dark conditions even in the presence of NanoPtA. These results validate the generation of singlet oxygen as one of the ROS by the NanoPtA assembly under visible light irradiation.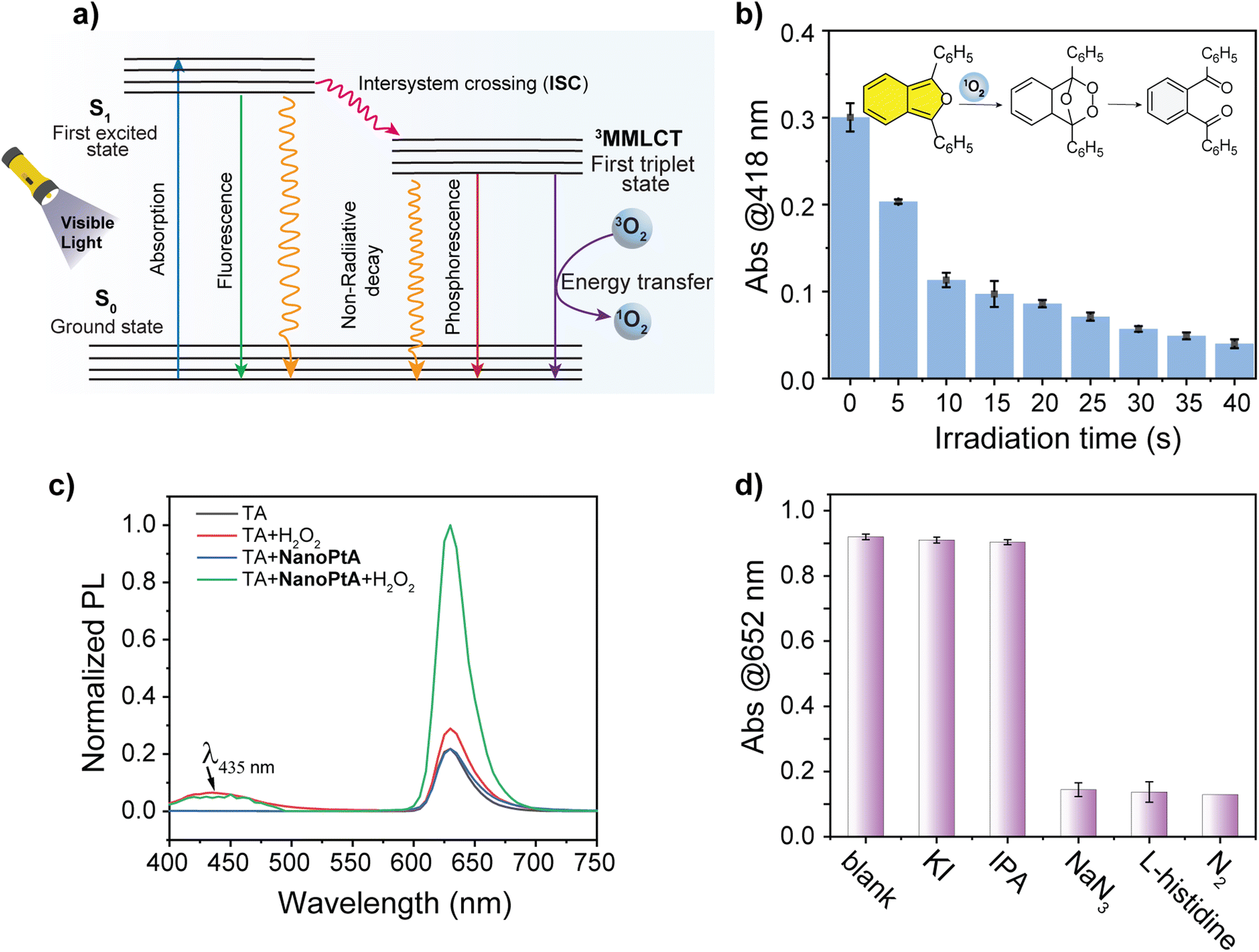 | ||
| Fig. 3 (a) A schematic representation for the generation of singlet oxygen by NanoPtA described by the Jablonski diagram. (b) The extent of photooxidation of 1,3-DPBF monitored at 418 nm with variation of illumination time by visible light (470 nm) in the presence of NanoPtA (10 μM). (c) The fluorescence spectra of the TA assay for the detection of in situ generated hydroxyl radicals (˙OH) using H2O2 (5 mM), NanoPtA (10 μM) and TA (0.5 mM) after 1 h visible light irradiation. (d) Effects of scavengers on the TMB oxidation in the presence of NanoPtA under visible light irradiation. Reaction conditions: 0.5 mM TMB, 10 μM NanoPtA, 20 mM IPA, 5 mM KI, 1.0 mM NaN3, and 0.4 mM L-histidine. Light irradiation time: 10 min. | ||
Given the common existence of non-specific peroxidase and oxidase-like activities in different nanozymes, we examined the specificity of the self-assembled system. To delineate between the two, we used a terephthalic acid (TA) assay using hydrogen peroxide (H2O2). The peroxidase-like activity would decompose H2O2 into a highly reactive hydroxyl radical (˙OH), which reacts with TA to generate 2-hydroxy terephthalic acid (TAOH) (Scheme S3†), resulting in the fluorescence peak at 435 nm (Fig. 3c). The absence of a strong emission band at 435 nm in the presence of NanoPtA and H2O2 under visible light suggests that the self-assembled system does not produce a hydroxyl radical. Further support of this mechanism was provided by quenching of different reactive species, which may be produced during the aerobic catalytic reaction triggered by visible light. We used quenchers to scavenge different reactive species including ˙OH, 1O2, and photogenerated holes (h+). The addition of isopropanol (IPA) as ˙OH scavenger51 and potassium iodide (KI) as an h+ scavenger52,53 did not cause any significant change in the absorbance of the ox.TMB (Fig. 3d), confirming that ˙OH and h+ were not generated in the catalytic reaction. Conversely, the catalytic activity of NanoPtA was significantly suppressed in the presence of NaN3 and histidine, which were used to scavenge 1O2.53,54 This suggests that 1O2 is the primary ROS generated in the photocatalytic reaction. Also, the NanoPtA-induced catalytic activity was significantly reduced in the absence of dissolved oxygen when the reaction was purged with nitrogen to perform TMB oxidation in an oxygen-free environment. Moreover, according to the Jablonski diagram (Fig. 3a), AIEgen molecules can return to their ground state through three pathways after excitation: (i) fluorescence emission, (ii) generation of reactive oxygen species (ROS) through intersystem crossing (ISC) or transfer to oxygen, and (iii) nonradiative heat dissipation, which are useful for photothermal therapy and photoacoustic imaging.55 Upon aggregation, NanoPtA generates singlet 1MMLCT and triplet 3MMLCT states. Normally, triplet transitions are non-emissive due to the spin selection rule, but the heavy Pt(II) metal center and Pt⋯Pt interactions in NanoPtA allow intersystem crossing, enabling the transition from the singlet to the triplet state by relaxing the spin selection rule. In aqueous solution, dissolved oxygen in the ground state exists in the triplet state. When Pt(II) molecules in the triplet 3MMLCT state interact with oxygen, they transmit their energy to ground state oxygen (3O2) due to the feasible spin selection rule to produce singlet oxygen (1O2). Altogether, these studies confirm that the photocatalytic oxidation by NanoPtA is exclusively regulated by singlet oxygen, indicating specific oxidase-like activity without any peroxidase-like mechanism.
Photocatalytic oxidation of phenolic pollutants and neurotransmitters
Toxic phenolic aromatics are commonly found in wastewater and effluents from various industries such as chemicals, petrochemicals, pharmaceuticals, textiles, and steel, which cause significant environmental damage.56 Due to their toxicity, the oxidation and breakdown of phenolic compounds are critical for achieving environmental sustainability. Use of a photocatalytic system would help to produce a viable green technology for environmental remediation. We investigated the oxidation of different phenolic compounds using NanoPtA as a catalyst, in the presence of a chromogenic substrate 4-aminoantipyrine (4-AP) with visible light irradiation for 10 minutes. The oxidized products of phenolic substrates form a red quinone imine adduct with 4-AP (Fig. 4a) with an absorbance at 510 nm. Initially, we performed the catalytic reaction with different concentrations of NanoPtA assembly at a constant substrate concentration (1.0 mM of 2,4-DP and 4-AP each) to investigate the effect of the NanoPtA. Upon irradiation of visible light for 10 minutes, the absorption peak at 510 nm corresponding to the quinone imine product increased with an increase in concentration of NanoPtA (Fig. 4b). Furthermore, we measured the effect of 2,4-DP concentration on the rate of reaction with NanoPtA. It can be observed that the rate of reaction increases linearly as depicted by absorbance at 510 nm (Fig. S22†). Notably, the NanoPtA catalyst efficiently oxidizes all the phenolic substrates in the presence of light (Fig. 4c). It was observed that the phenolic compounds with electron-withdrawing functional groups were oxidized slower than those with electron-donating groups.57 Altogether, our study highlights the promising potential of NanoPtA as a photocatalyst for the oxidation of organic pollutants containing phenolic groups.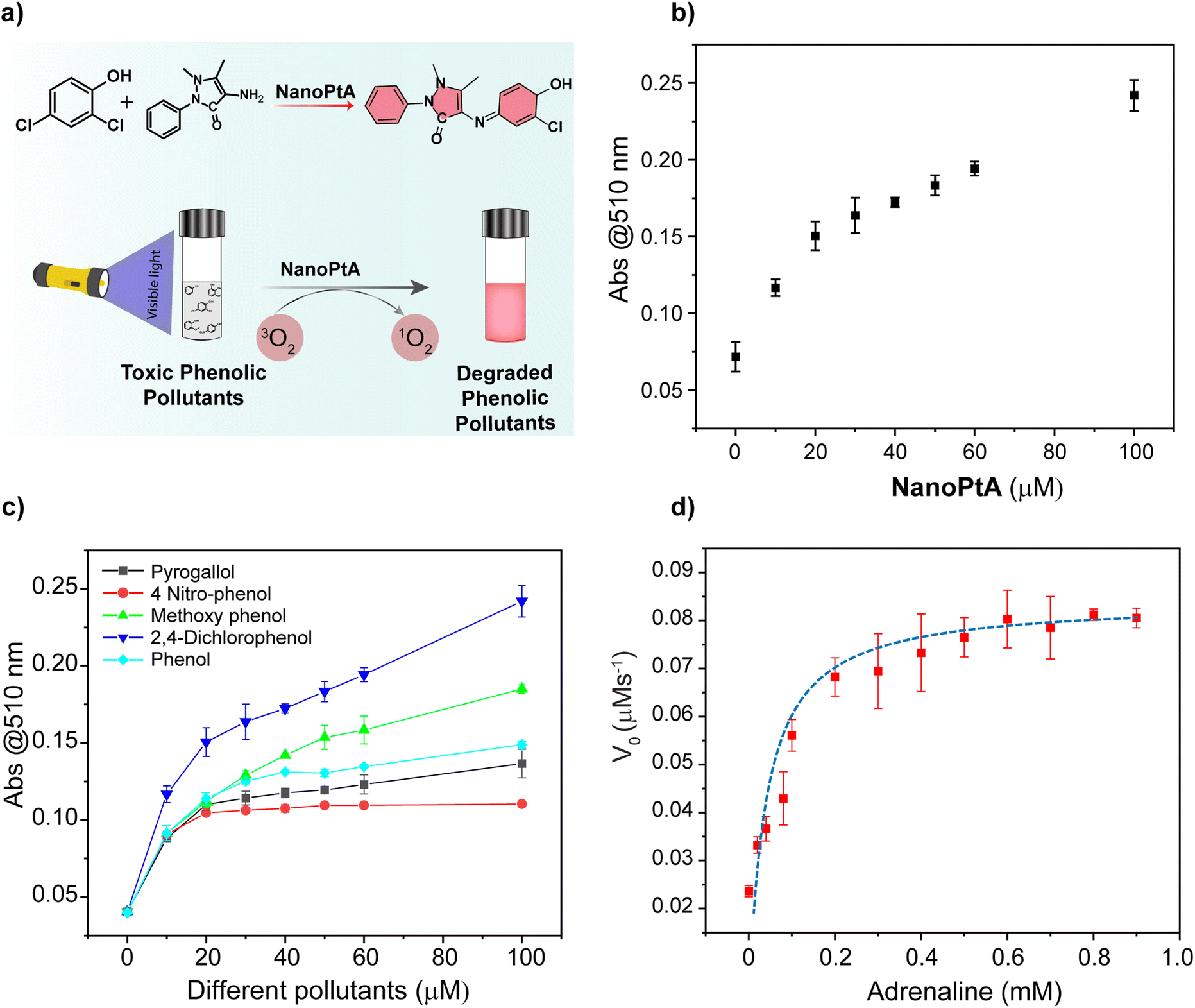 | ||
| Fig. 4 (a) A schematic representation of the visible light mediated phenolic pollutants degradation by the NanoPtA enzyme. (b) UV-vis absorption changes of the chromogenic reaction between 2,4-DP and 4-AP at 510 nm with varying NanoPtA concentrations. (c) Oxidation of different phenolic substrates monitored by quinone imine formation at 510 nm catalyzed by NanoPtA (10 μM) upon 10 min visible light irradiation. Lines are drawn to guide the eye. (d) A Michaelis–Menten plot of adrenaline oxidation catalyzed by NanoPtA (10 μM). Light source: visible light of 470 nm. | ||
Given the effective catalytic activity of NanoPtA in the oxidation of phenolic compounds, we examined the degradation of biologically relevant molecules such as dopamine (DP) and adrenaline (AD), which contain catechol groups. DP is associated with various neurological disorders, including attention deficit hyperactivity disorder, schizophrenia, and neurodegenerative disorders like Alzheimer's and Parkinson's diseases.58 AD is a crucial neurotransmitter used in the treatment of several medical conditions such as bronchial asthma, cardiac arrest, allergies, and superficial bleeding.59 Upon the addition of NanoPtA, the previously colourless solutions of AD and DP turned brown, indicating the oxidation of these biomolecules. To assess the catalytic oxidation of the catecholamines as enzyme substrates, we measured absorbance at 485 nm for AD (Fig. 4d) and 475 nm for DP (Fig. S23†). Interestingly, the oxidation of both AD and DP follow the Michaelis–Menten kinetics, confirming the enzyme-like properties of NanoPtA.6 Given the colorimetric response of the oxidation of the neurotransmitters, this system could be utilized for point-of-care (PoC) detection with a limit of detection (LOD) of 0.7 μM and 4.6 μM for AD and DP, respectively (Fig. S24†). Herein, the LOD was determined from the 3σ/slope of the plots, where σ is the standard deviation of the three blank samples. Also, colorimetric detection of catechol-containing neurotransmitters would enable PoC diagnosis of neurological disorders.
Photocatalytic dye degradation
Organic dyes are a key component of different industries, such as those producing food, leather, textiles, paper, cosmetics, and pharmaceuticals. Even under strict regulations, it has been difficult to restrict the disposal of these dyes into open environments and water bodies, posing a global hazard to human health, aquatic life, and animals.60 We investigated the capability of NanoPtA for dye degradation using the photocatalytic singlet oxygen generation. A 10 min irradiation of Trypan blue (TrB) with visible light (at 470 nm) resulted in immediate decolorization, indicating degradation of the dye. Although 5 μM dye was degraded nearly 100% quantitatively, the 40 μM solution degraded ∼95% within this time (Fig. 5a). The control experiment without NanoPtA did not cause any decolorization and degradation. Moreover, almost no change was observed for the reaction in the dark with various TrB dye concentrations, indicating that the catalytic dye degradation is photo activated. Further, we tested TrB degradation using sunlight as the excitation source, which would provide a better sustainable solution. A 20 μM TrB solution in water faded within 10 min under sunlight in the presence of 10 μM of NanoPtA (Fig. 5b). The absorbance at 590 nm corresponding to the dye showed that ∼95% had degraded in 10 min, indicating the high efficiency of the supramolecular nanozyme. However, the dye without NanoPtA was not degraded even after shining light for the same duration, demonstrating the nanozyme mediated degradation of the dye.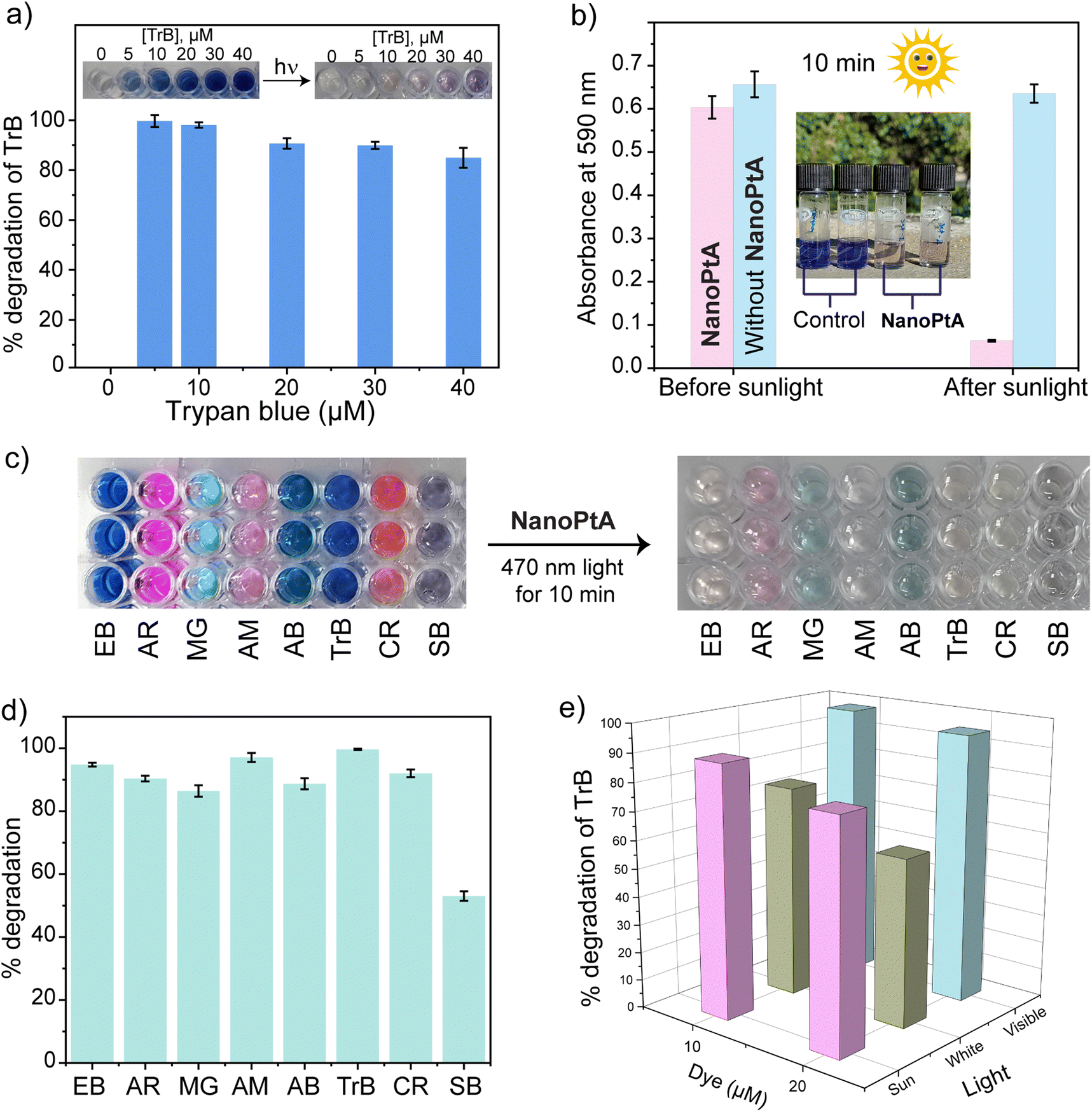 | ||
| Fig. 5 (a) % of TrB degradation monitored by its absorbance at 590 nm with varying concentrations of TrB dye (0–40 μM). (b) The degradation plot of TrB dye at 550 nm in the presence of NanoPtA (10 μM) after 5 min sunlight exposure. (c) Photos of the degradation of different dyes in the presence of NanoPtA (10 μM) under visible light. (d) % Degradation plot of multiple dyes with NanoPtA (10 μM) under visible light. (e) Comparison of % degradation of TrB dye under exposure of different wavelengths. Light source = visible light 470 nm, white light = 40 W LED bulb. | ||
To further examine the generalizability of NanoPtA on more organic azo containing dyes, the photocatalytic degradation of Evans blue (EB), acid red (AR), malachite green (MG), amaranth (AM), amido black (AB), Congo red (CR), and Sudan black (SB) were investigated. The color of the dyes faded rapidly in the presence of NanoPtA following visible light irradiation for 10 min (Fig. 5c). In this time, most of the dyes degraded more than 85% up to 98% (Fig. 5d), whereas SB was the least efficient with ∼50% degradation. As well as studying dye degradation under sunlight, we also compared the efficiency of three different types of light sources: visible light (470 nm), white light (40-W LED bulb) and sunlight. The dye degradation was efficient under all the sources with 98% under visible light, 89% under sunlight and 75% under white light for a 10 μM dye solution (Fig. 5e). Moreover, NanoPtA concentration-dependent dye degradation was noted for all the light conditions (Fig. S25†). Additionally, to confirm the unique oxidase-like activity of NanoPtA, we performed a control experiment with H2O2 for the degradation of TrB dye in the absence of light with or without NanoPtA. As shown in Fig. S26a–S26d,† H2O2 was unable to oxidize the TrB dye in the absence of light or NanoPtA in the dark, indicating 1O2 mediated dye degradation. Further, we tested if NanoPtA exhibits a catalase enzyme-like activity that converts H2O2 into H2O and O2.61 A reaction mixture containing TrB, H2O2 and NanoPtA was degassed with N2 for 10 min to remove the dissolved oxygen, followed by irradiation with 470 nm light for another 10 min. The reaction mixture showed nearly the same absorbance at 590 nm as the control of TrB and NanoPtA without H2O2 (Fig. S27†). These findings showcase the potential of NanoPtA as a specific oxidase-nanozyme in developing sustainable solutions for the degradation of harmful azo dyes in industrial wastewater.
Robustness and reusability of NanoPtA
To make enzyme mimetics practically useful, they need to be robust and stable for longer durations. We studied the stability of the NanoPtA assembly by subjecting samples to harsh conditions such as extreme pH, high salt concentration and high temperature. The photocatalytic activity of NanoPtA was investigated under different ionic strengths varying the NaCl concentrations from 0 to 500 mM. In our earlier studies, we showed a significant decrease in the inherent activity of the natural oxidases such as laccase with an increase in NaCl concentration.34 The decline in activity is mainly due to the salting-out effect, which results in the modulation of the enzyme's active site structure and charge distribution.62 In contrast, the NanoPtA assembly exhibited notable effectiveness in a high salinity environment. At least 90% of activity was found to be retained even at a salt concentration of 500 mM (Fig. 6a). This implies that NanoPtA is a robust oxidase enzyme-equivalent with potential in practical applications, particularly in treating industrial wastewater, where natural enzymes face difficulties due to the high ionic strength.62 Furthermore, we studied the photocatalytic activity of the NanoPtA assembly at varying pH values (4.0, 7.0, and 11.0) in the presence of visible light by observing the degradation of several dyes (Fig. 6b). It is evident that NanoPtA maintained its activity and degraded almost all the dyes by more than 70% except for the SB dye. The SB dye, which lacked ortho-hydroxy groups like the other dyes might have caused less degradation. Also, the degradation activity is largely independent of the pH. Moreover, we evaluated the thermostability of NanoPtA in solution by measuring its catalytic activity at different temperatures. Fig. S28† shows that NanoPtA displayed significant catalytic activity in the temperature range 25–80 °C, retaining at least 85% activity even at 80 °C. In contrast, natural oxidase enzymes such as laccase perform optimally at 50 °C, rapidly losing their activities to only 5% at 80 °C.63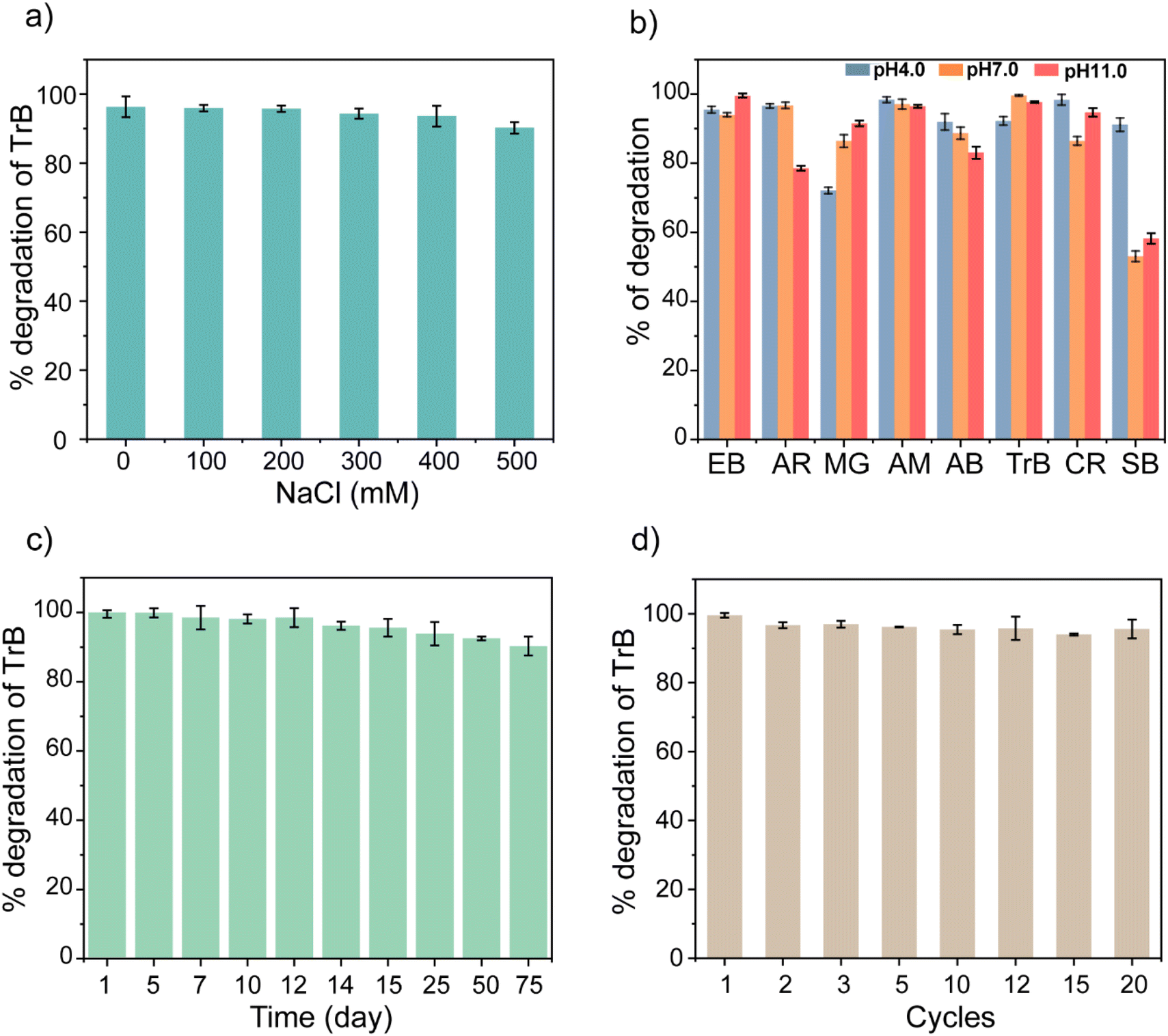 | ||
| Fig. 6 (a) Effect of NaCl salt concentration on degradation of TrB dye by NanoPtA. (b) Comparison of % degradation of different dyes with NanoPtA (10 μM) at variable pH values (4.0, 7.0 and 11.0). (c) The % degradation plot of TrB dye, showing the long-term storage catalytic activity of NanoPtA. (d) The % degradation plot of TrB dye, relative activity of NanoPtA after multiple cycles. Experimental conditions: [TrB] = 20 μM, NanoPtA = 10 μM, light source = visible light 470 nm. | ||
Further, the storage stability of NanoPtA was investigated at room temperature. It is noteworthy that NanoPtA retained >90% of its initial catalytic activity even after 75 days of storage, which was observed from the TrB dye degradation (Fig. 6c). This greater catalytic stability can be attributed to the superior structural stability of the self-assembled system compared to the native enzyme. In general, the self-assembled structure has exceptional operational stability in harsh environments, which enhances its usefulness in real-world applications. Moreover, we evaluated the reusability of NanoPtA in the photocatalytic dye degradation. The process involved the degradation of TrB dye by monitoring the absorbance of the system before and after exposing it to visible light in the presence of NanoPtA. Additional amounts of dye was added to the same system after complete conversion under light irradiation. It is evident that >98% activity is retained up to the 20th cycle of the catalytic reaction, signifying the robustness of the nanozyme (Fig. 6d). These findings validate the utility of NanoPtA for effective environmental remediation in industrial settings.
4. Conclusions
In this work, we introduced a specific enzyme-mimetic activity using a Pt(II)-based supramolecular assembly. While the Pt(II) complex exists as a monomer in methanol, hydrophobic collapse in water leads to aggregated nanotape structures that exhibit AIE. A high luminescence intensity was observed compared to the monomer, leading to the activation of singlet oxygen species upon exposure to visible light. The singlet oxygen-mediated reaction exhibited excellent oxidase-like activity without any peroxidase component, which is a common property of traditional metal-based nanozymes. Another notable feature of the system is the efficient photocatalytic oxidation of phenolic compounds and various azo dyes. These reactions are shown to be effective under visible light sources including sunlight, with quantitative degradation at significantly shorter irradiation times. To address the practical applicability of the supramolecular enzyme mimetic, we demonstrated reusability of NanoPtA for at least 20 cycles. Besides, the notable storage stability at room temperature for more than 75 days and unaltered activity in challenging environmental conditions unlike natural enzymes, makes the system a promising candidate for sustainable wastewater treatment. To the best of our knowledge, NanoPtA is an exceptional example of a discrete supramolecular structure in water, which exhibits remarkable specific oxidase-like activity via photoinduced oxidation. It is anticipated that the extraordinary photocatalytic capabilities of the self-assembled nanozyme will have applications in numerous disciplines, including switchable platforms, biosensing, therapeutics, and environmental protection.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. R. thanks the SERB (grant no. CRG/2022/009021 and ECR/2018/002555) for financial support. The authors thank the Department of Science and Technology (DST-FIST sanction letter no. SR/FST/PSII009/2010) for the instrumental facility at MRC. R. K. acknowledges CSIR for providing a doctoral fellowship. B. S. is thankful for the IISc post-doctoral fellowship under the IOE scheme. A. K. is grateful to MoE for Prime Minister Research Fellowship for doctoral studies.References
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Dis. Primers, 2021, 1, 46 CrossRef CAS.
- Y. Huang, J. Ren and X. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS.
- R. Zhang, X. Yan and K. Fan, Acc. Mater. Res., 2021, 2, 534–547 CrossRef CAS.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- H. Wei, L. Z. Gao, K. L. Fan, J. W. Liu, J. Y. He, X. G. Qu, S. J. Dong, E. K. Wang and X. Y. Yan, Nano Today, 2021, 40, 101269 CrossRef CAS.
- C. P. Kurup and M. U. Ahmed, Biosensors, 2023, 13, 461 CrossRef CAS PubMed.
- Y. Ai, Z.-N. Hu, X. Liang, H.-B. Sun, H. Xin and Q. Liang, Adv. Funct. Mater., 2022, 32, 2110432 CrossRef CAS.
- Y. Tao, E. Ju, J. Ren and X. Qu, Adv. Mater., 2015, 27, 1097–1104 CrossRef CAS PubMed.
- P. G. Le and M. I. Kim, Nanomaterials, 2021, 11, 2116 CrossRef CAS.
- L. He, Y. Li, Q. Wu, D. M. Wang, C. M. Li, C. Z. Huang and Y. F. Li, ACS Appl. Mater. Interfaces, 2019, 11, 29158–29166 CrossRef CAS PubMed.
- W. He, Y. Liu, J. Yuan, J.-J. Yin, X. Wu, X. Hu, K. Zhang, J. Liu, C. Chen, Y. Ji and Y. Guo, Biomaterials, 2011, 32, 1139–1147 CrossRef CAS PubMed.
- N. Singh, S. K. NaveenKumar, M. Geethika and G. Mugesh, Angew. Chem., Int. Ed., 2021, 60, 3121–3130 CrossRef CAS PubMed.
- R. Ragg, A. M. Schilmann, K. Korschelt, C. Wieseotte, M. Kluenker, M. Viel, L. Völker, S. Preiß, J. Herzberger, H. Frey, K. Heinze, P. Blümler, M. N. Tahir, F. Natalio and W. Tremel, J. Mater. Chem. B, 2016, 4, 7423–7428 RSC.
- J. Chen, L. Huang, Q. Wang, W. Wu, H. Zhang, Y. Fang and S. Dong, Nanoscale, 2019, 11, 5960–5966 RSC.
- E. Ghanem and F. M. Raushel, Toxicol. Appl. Pharmacol., 2005, 207, 459–470 CrossRef PubMed.
- S. Jain, B. Sharma, N. Thakur, S. Mishra and T. K. Sarma, ACS Appl. Nano Mater., 2020, 3, 7917–7929 CrossRef CAS.
- J. Chen, Q. Ma, M. Li, D. Chao, L. Huang, W. Wu, Y. Fang and S. Dong, Nat. Commun., 2021, 12, 3375 CrossRef CAS PubMed.
- C. Wang, Y. Li, W. Yang, L. Zhou and S. Wei, Adv. Healthcare Mater., 2021, 10, 2100601 CrossRef CAS PubMed.
- C.-P. Liu, T.-H. Wu, C.-Y. Liu, K.-C. Chen, Y.-X. Chen, G.-S. Chen and S.-Y. Lin, Small, 2017, 13, 1700278 CrossRef PubMed.
- G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469–3478 CrossRef CAS.
- M. Hu, K. Korschelt, M. Viel, N. Wiesmann, M. Kappl, J. Brieger, K. Landfester, H. Thérien-Aubin and W. Tremel, ACS Appl. Mater. Interfaces, 2018, 10, 44722–44730 CrossRef CAS.
- W. Chen, S. Li, J. Wang, K. Sun and Y. Si, Nanoscale, 2019, 11, 15783–15793 RSC.
- N. Alizadeh and A. Salimi, J. Nanobiotechnol., 2021, 19, 26 CrossRef CAS PubMed.
- Z. Wang, R. Zhang, X. Yan and K. Fan, Mater. Today, 2020, 41, 81–119 CrossRef CAS.
- R. Huang, C.-H. Li, R. Cao-Milán, L. D. He, J. M. Makabenta, X. Zhang, E. Yu and V. M. Rotello, J. Am. Chem. Soc., 2020, 142, 10723–10729 CrossRef CAS PubMed.
- D. Velasco, E. Tumarkin and E. Kumacheva, Small, 2012, 8, 1633–1642 CrossRef CAS PubMed.
- M. Li, J. Chen, W. Wu, Y. Fang and S. Dong, J. Am. Chem. Soc., 2020, 142, 15569–15574 CrossRef CAS.
- H. Cheng, Y. Liu, Y. Hu, Y. Ding, S. Lin, W. Cao, Q. Wang, J. Wu, F. Muhammad, X. Zhao, D. Zhao, Z. Li, H. Xing and H. Wei, Anal. Chem., 2017, 89, 11552–11559 CrossRef CAS.
- J. S. Siegel, Science, 1996, 271, 949–949 CrossRef CAS.
- Z. Dong, W. Yongguo, Y. Yin and J. Liu, Curr. Opin. Colloid Interface Sci., 2011, 16, 451–458 CrossRef CAS.
- M. D. Toscano, K. J. Woycechowsky and D. Hilvert, Angew. Chem., Int. Ed., 2007, 46, 3212–3236 CrossRef CAS PubMed.
- X. Huang, Z. Dong, J. Liu, S. Mao, J. Xu, G. Luo and J. Shen, Langmuir, 2007, 23, 1518–1522 CrossRef CAS.
- M. Solra, S. Das, A. Srivastava, B. Sen and S. Rana, ACS Appl. Mater. Interfaces, 2022, 14, 45096–45109 CrossRef CAS.
- M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082–12083 CrossRef CAS PubMed.
- Y. Tang, L. Zhou, J. Li, Q. Luo, X. Huang, P. Wu, Y. Wang, J. Xu, J. Shen and J. Liu, Angew. Chem., Int. Ed., 2010, 49, 3920–3924 CrossRef CAS PubMed.
- P. Bhatt, M. Solra, S. I. Chaudhury and S. Rana, Biosensors, 2023, 13, 277 CrossRef CAS.
- Y. Gao, F. Zhao, Q. Wang, Y. Zhang and B. Xu, Chem. Soc. Rev., 2010, 39, 3425–3433 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- X. Lin, F. Chen, X. Yu, H. Wang, H. Qiu, Y. Li, S. Yin and P. J. Stang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203994119 CrossRef CAS PubMed.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- H. Rao, X. Xue, M. Luo, H. Liu and Z. Xue, Chin. Chem. Lett., 2021, 32, 25–32 CrossRef CAS.
- X. Zheng, M. H.-Y. Chan, A. K.-W. Chan, S. Cao, M. Ng, F. K. Sheong, C. Li, E. C. Goonetilleke, W. W. Y. Lam, T.-C. Lau, X. Huang and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116543119 CrossRef CAS.
- I. Mathew and W. Sun, Dalton Trans., 2010, 39, 5885–5898 RSC.
- C. Po, A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12136–12143 CrossRef CAS PubMed.
- Y.-S. Wong, M. Ng, M. C.-L. Yeung and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 973–982 CrossRef CAS PubMed.
- L. A. Marquez and H. B. Dunford, Biochemistry, 1997, 36, 9349–9355 CrossRef CAS PubMed.
- L. Liu, C. Sun, J. Yang, Y. Shi, Y. Long and H. Zheng, Chem. – Eur. J., 2018, 24, 6148–6154 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- P. P. Neelakandan, A. Jiménez, J. D. Thoburn and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 14378–14382 CrossRef CAS.
- C. An, J. Wang, C. Qin, W. Jiang, S. Wang, Y. Li and Q. Zhang, J. Mater. Chem., 2012, 22, 13153–13158 RSC.
- R. Ma, X. Wang, J. Huang, J. Song, J. Zhang and X. Wang, Vacuum, 2017, 141, 157–165 CrossRef CAS.
- M. Bancirova, Luminescence, 2011, 26, 685–688 CrossRef CAS PubMed.
- I. B. C. Matheson, R. D. Etheridge, N. R. Kratowich and J. Lee, Photochem. Photobiol., 1975, 21, 165–171 CrossRef CAS PubMed.
- G. X. Feng, G. Q. Zhang and D. Ding, Chem. Soc. Rev., 2020, 49, 8179–8234 RSC.
- S. Zhang, F. Lin, Q. Yuan, J. Liu, Y. Li and H. Liang, J. Environ. Sci., 2020, 88, 103–111 CrossRef CAS PubMed.
- X. Xu, J. Wang, R. Huang, W. Qi, R. Su and Z. He, Catal. Sci. Technol., 2021, 11, 3402–3410 RSC.
- H. Juárez Olguín, D. Calderón Guzmán, E. Hernández García and G. Barragán Mejía, Oxid. Med. Cell. Longevity, 2016, 2016, 9730467 Search PubMed.
- K. S. R. Sastry, Y. Karpova, S. Prokopovich, A. J. Smith, B. Essau, A. Gersappe, J. P. Carson, M. J. Weber, T. C. Register, Y. Q. Chen, R. B. Penn and G. Kulik, J. Biol. Chem., 2007, 282, 14094–14100 CrossRef CAS PubMed.
- D. Ayodhya and G. Veerabhadram, Mater. Today Energy, 2018, 9, 83–113 CrossRef.
- P. Campomanes, U. Rothlisberger, M. Alfonso-Prieto and C. Rovira, J. Am. Chem. Soc., 2015, 137, 11170–11178 CrossRef CAS.
- A. M. Hyde, S. L. Zultanski, J. H. Waldman, Y.-L. Zhong, M. Shevlin and F. Peng, Org. Process Res. Dev., 2017, 21, 1355–1370 CrossRef CAS.
- T. Maity, S. Jain, M. Solra, S. Barman and S. Rana, ACS Sustainable Chem. Eng., 2022, 10, 1398–1407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02081a |
| This journal is © The Royal Society of Chemistry 2023 |