
Protein redox by a piezoelectric acousto-nanodevice†
Sophia
Selvarajan
ab,
Hyunji
Shim
a,
Eunjeong
Byun
a,
Albert
Kim
 *b and
Seung Hyun
Song
*a
*b and
Seung Hyun
Song
*a
aDepartment of Electronics Engineering, Sookmyung Women's University, Seoul 04310, Republic of Korea. E-mail: shsong.ee@sookmyung.ac.kr
bDepartment of Medical Engineering, University of South Florida, Tampa, FL 33602, USA. E-mail: akim1@usf.edu
First published on 12th July 2023
Abstract
Protein redox is responsible for many crucial biological processes; thus, the ability to modulate the redox proteins through external stimuli presents a unique opportunity to tune the system. In this work, we present an acousto-nanodevice that is capable of oxidizing redox protein under ultrasonic irradiation via surface-engineered barium titanate (BTO) nanoparticles with a gold half-coating. Using cytochrome c as the model protein, we demonstrate nanodevice-mediated protein oxidation. BINased on our experimental observations, we reveal that the electron transfer occurs in one direction due to the alternating electrical polarization of BTO under ultrasound. Such unique unidirectional electron transfer is enabled by modulating the work function of the gold surface with respect to the redox center. The new class of ultrasonically powered nano-sized protein redox agents could be a modulator for biological processes with high selectivity and deeper treatment sites.
Redox reactions in proteins, governed by the electron transfer,1–3 are at the center of the protein functionality and stability as they govern many crucial life-sustaining functions, including respiration,4 photosynthesis,5 metabolism,6 and cell apoptosis.7,8 Moreover, the conformation and aggregation behaviors of proteins that determine the protein stability and influence the development of protein misfolding diseases, such as Alzheimer's or Parkinson's diseases, are closely related to the protein redox states.9,10 Thus, the ability to externally control the protein redox states has great clinical implications as it may enable the modulation of biological processes or protein conformation.11 For example, it has been shown that the oxidation of cytochrome c, which functions as an electron carrier in the respiratory chain, can trigger cell apoptosis.12 Also, the oxidation of amyloid beta aggregates, a hallmark of Alzheimer's disease, has been shown to destabilize the aggregates both experimentally and theoretically.13,14
Modulation and the characterization of the redox proteins have traditionally been achieved by electrochemical methods,15 which have shown that the protein redox process is governed by the electron transfer between the redox sites of the proteins and the electrode.1–3 However, the need for wiring of the electrodes makes the clinical translation using electrochemical techniques challenging. For this reason, researchers have looked for alternative methods without the use of traditional electrodes. A successful example is the optical interrogation of the redox state of cytochrome c based on the electron transfer of the plasmonically excited electrons to the heme site of the cytochrome c under light illumination.16,17 On the other hand, the use of acoustics in protein redox has been relatively under-explored, despite the fact that acoustics can readily address a key limitation of the optical methods in biological systems, that is the limited penetration depth.
In this work, we present an ultrasonically excitable acousto-nanodevice capable of modulating the protein redox states. Ultrasound, as the external stimulus for redox modulation, offers a unique advantage of deep penetration depth in tissues without the need for wiring. The acousto-nanodevice comprises asymmetrically metalized barium titanate (BTO) nanoparticles (i.e., with a half-surface metal coating), which allow the modulation of the electron energy under acoustic waves. Our experimental results demonstrate that cytochrome c (cyt c) oxidation can be mediated by the acousto-nanodevice under ultrasonic irradiation. The oxidation mechanism is tentatively linked to the unidirectional direct electron transfer between the piezoelectric acousto-nanodevice and the heme site of cytochrome c (cyt c).
Our acousto-nanodevice system for protein redox modulation is composed of two parts: the ultrasound as the external control stimulus and the engineered piezoelectric nanoparticles that directly interface with the protein. We chose ultrasound as the controlling external stimulus for the protein redox because it offers a deep penetration depth (on the order of centimeters) and can be administered safely.18–22
We first describe the experimental results of cyt c oxidation by the acousto-nanodevice under external acoustic waves. We chose cyt c as the model protein23 in this proof-of-concept study because its structure and functionality are well established in the literature.24–26 The redox states of cyt c were monitored using UV-vis absorption measurements, which reflect the oxidation/reduction state of heme (Fe2+ ↔ Fe3+) (refer to the ESI† for the UV-vis sample preparation and measurement conditions).27 We prepared the acousto-nanodevice with a gold half-coating for the cyt c oxidation experiment, as the gold surface provides an adsorption site for cyt c (direct chemisorption), retaining its native electron transfer properties.28–30 (For the detailed fabrication procedure of the acousto-nanodevice, refer to the ESI Fig. S1.†)
Following the acousto-nanodevice preparation, we added a low concentration of the acousto-nanodevice (on the order of 1 nM) in a cuvette with 5 μM of reduced cyt c; the cuvet was then placed in a large water tank, and a low-intensity ultrasound (∼150 mW cm−2) was applied for 30 minutes (Fig. 1a.). After the ultrasound irradiation, we observed the color change of cyt c from light pink (reduced cyt c) to a light orange (oxidized cyt c), as shown in Fig. 1b inset. The UV-vis absorption spectrum confirmed the oxidation of cyt c. A blueshift of ∼5 nm around the soret region (415–410 nm; Fig. 1c inset) was observed due to the oxidation of heme (Fe2+ to Fe3+), suggesting an overall net electron transfer from the reduced cyt c to the acousto-nanodevice (Fig. 1d). These results also agree with the reported absorption peaks of oxidized (409 nm) and reduced cyt c (415 nm).27 In contrast, there were no observable changes to the UV-vis spectrum when the same system (i.e., an acousto-nanodevice under ultrasound) was applied to the oxidized cyt c (Fig. 1e). The spectrum remained largely unchanged from the reference oxidized cyt c (Fig. 1e), suggesting that there is little electron transfer between oxidized cyt c and the acousto-nanodevice (i.e., unidirectional electron transfer) (Fig. 1f).
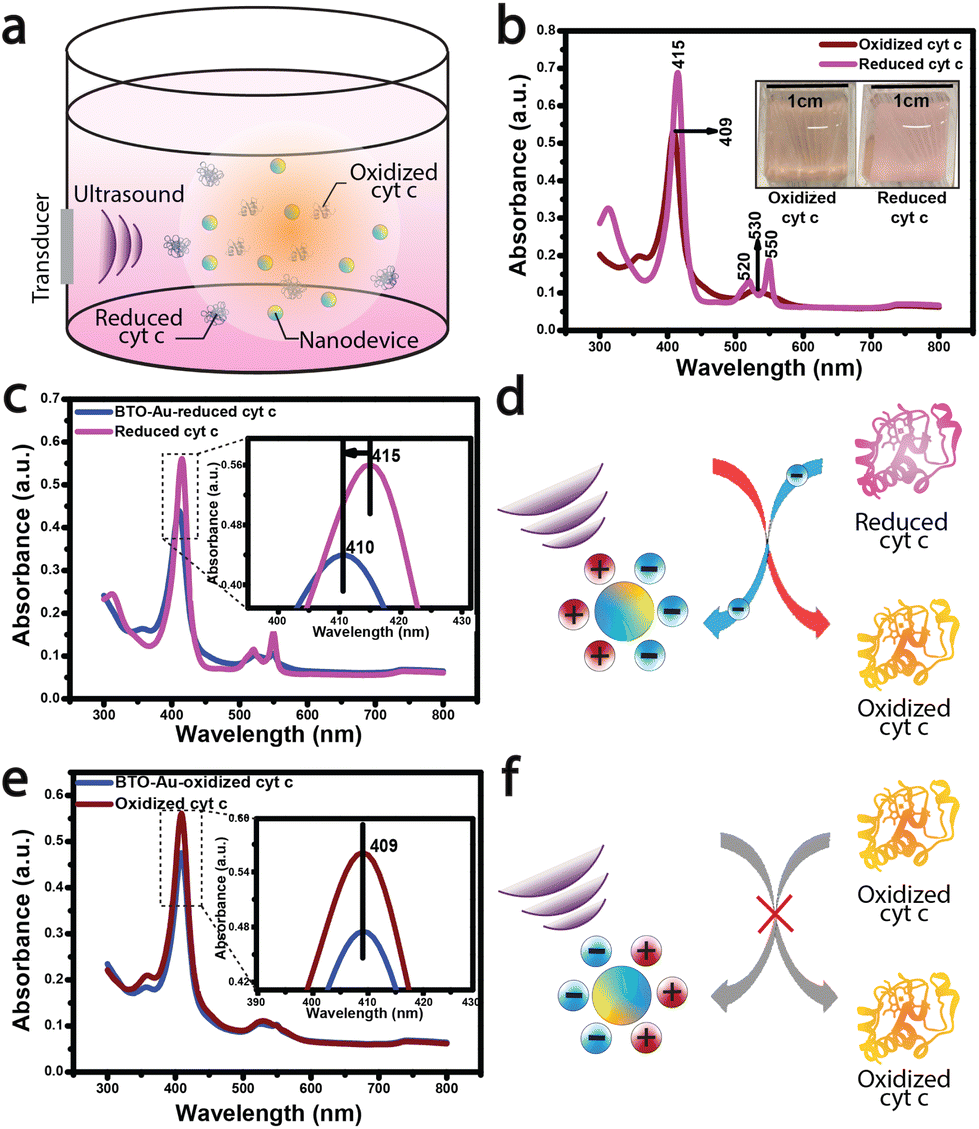 | ||
| Fig. 1 Cytochrome c oxidation by an acousto-nanodevice: (a) schematic illustration of cyt c oxidation by a nanodevice. (b) UV-vis spectrum of oxidized and reduced cyt c (baseline) (inset shows the visual color change due to oxidation of the reduced cyt c upon its interaction with BTO-Au nanoparticles in the presence of ultrasound). (c and d) UV-vis spectrum showing cyt c oxidation mediated by acoustically triggered BTO-Au nanoparticles and schematics depicting uni-directional charge transfer from protein to BTO-Au, causing its oxidation. (e and f) UV-vis spectrum showing the absence of a reducing effect in cyt c by acoustically triggered BTO-Au nanoparticles and its corresponding schematics depicting no charge transfer from BTO-Au to protein. | ||
To find out if the oxidation of reduced cyt c is indeed due to the acousto-nanodevice under ultrasonic irradiation, we performed three different control experiments, which illuminate the role of the surface metal coating and the effect of ultrasonic irradiation. The reduced cyt c was treated with (1) pristine BTO nanoparticles (i.e., no surface metal coating) without sonication, (2) pristine BTO nanoparticles with sonication, and (3) acousto-nanodevice without sonication. The degree of oxidation was quantified by the peak fitting of the UV-vis spectra. We decomposed the peak according to the Beer–Lambert law: the overall UV-vis absorbance of the sample, A, was determined as below:
| A = αox·cox·l + αred·cred·l, |
Fig. 2a–d as well as Fig. S3† (detailed description given in supplementary information) shows the representative UV-vis spectra of all experimental groups (three controls and one test). While the spectra of the three control groups (Fig. 2a–c) were largely the same as the spectrum of the reduced cyt c reference, the spectrum of the test group (Fig. 2d) showed significant oxidation of cyt c; the intensity of the soret peak at 415 nm decreased while the peak at 409 nm increased due to the blueshift of ∼5 nm around the soret region as shown in Fig. S3. For quantitative analysis of the oxidation effect caused by each group, we characterized the oxidation effects of each group by tracking the concentration of the oxidized and reduced cyt c.
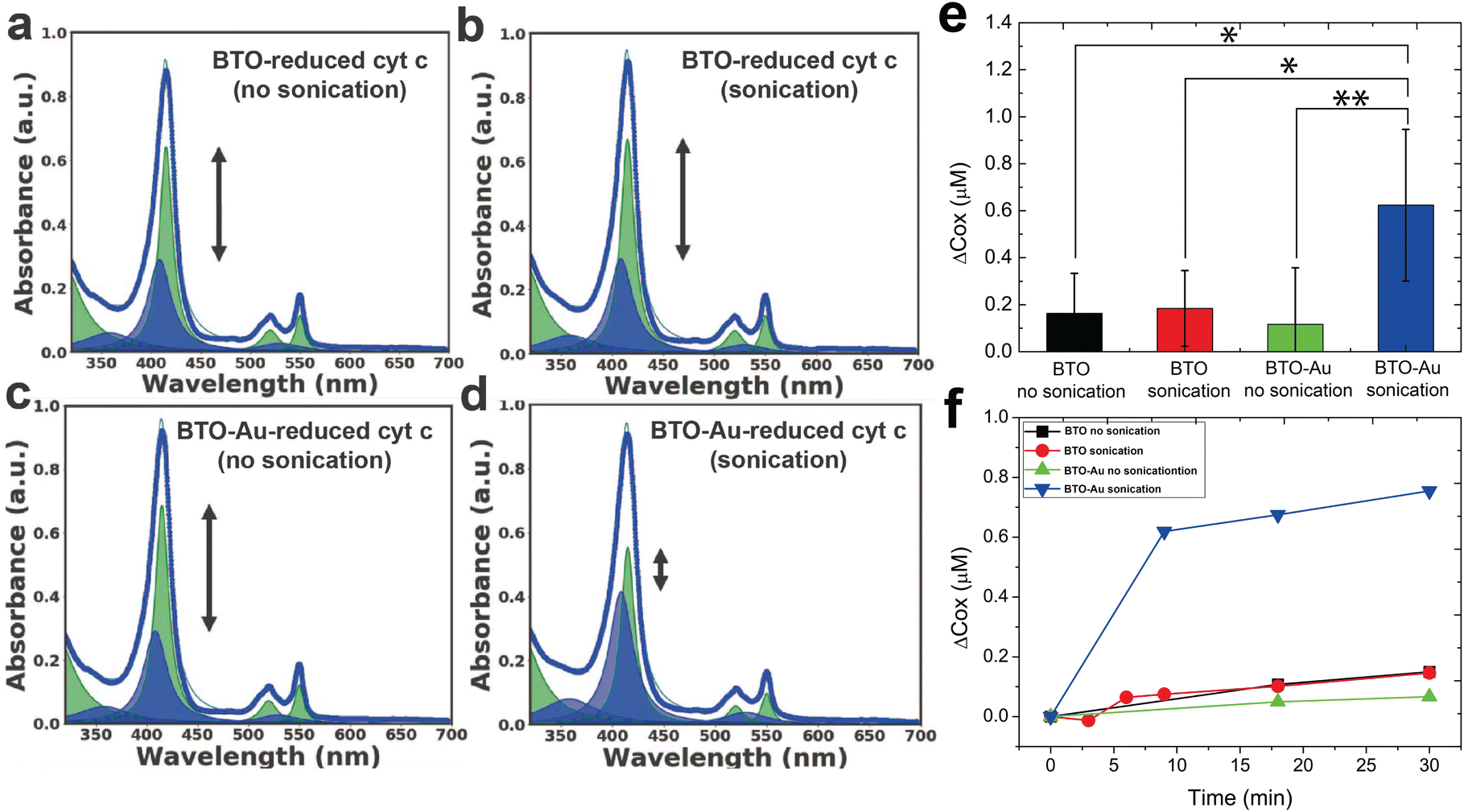 | ||
| Fig. 2 Acoustically triggered protein redox: the underlying mechanism. Lorentzian fit for the UV-vis spectrum of (a) BTO-reduced cyt c (no sonication), (b) BTO-reduced cyt c (sonication), (c) BTO-Au-reduced cyt c (no sonication), and (d) BTO-Au-reduced cyt c (sonication). Except for BTO-Au-reduced cyt c (sonication), there is no significant change in the degree of oxidation for other conjugates. (e) The average change in oxidation following 30 minutes of sonication. (f) 2D graph showing the degree of oxidation with respect to time (obtained from Lorentzian fitting) between the sonicated and non-sonicated conjugates of reduced cyt c. Error bars represent SD; n ≥ 4; p-value was determined using a two-tailed t-test. NS: not significant, (*) p < 0.05, (**) p < 0.01. | ||
The average change in the concentration of oxidized cyt c (Δcox [μM]) following a 30-minute sonication of four independent experiments is summarized in Fig. 2e. We tentatively assign the large variation in the oxidation of the experimental group (i.e., BTO-Au sonication in Fig. 2e) to the variation in the acousto-nanodevice concentrations of the different batches. Note that the collection efficiency of the acousto-nanodevice varied from batch to batch, even though we fabricated the acousto-nanodevice with the same procedure. Nevertheless, the experimental group (acousto-nanodevice and sonication) showed statistical significance compared to all three sham control groups: the p-value was 0.009 for the acousto-nanodevice without sonication, 0.016 for the pristine BTO without sonication, and 0.020 for the pristine BTO with sonication (refer to the ESI for a detailed statistical analysis†). The time evolution of the change in the oxidized cyt c concentration is summarized in Fig. 2f; progressive oxidation of the cyt c by the acousto-nanodevice occurred within 10 min of sonication. In addition, we note that the overall concentration of the cyt c (cox + cred) remained constant throughout the experiment.
In addition, we examined the stability of reduced cyt c in both the presence and absence of ultrasound (Fig. S4†). We observed no detectable changes in the UV-vis spectra in the presence/absence of sonication for an extended time. All experiments also maintained constant pH and temperature, both of which might affect the stability and redox of proteins. We used phosphate-buffered saline (Sigma-Aldrich) to maintain a pH of 7.4. We also utilized a large volume of the water tank (20 L) and a sufficiently low ultrasound intensity (150 mW cm−2) to ensure that the thermal effects due to continuous ultrasound irradiation were minimal; we monitored the temperature during all of the experiments (ΔT < 0.1 °C). Furthermore, we could exclude the surface plasmonic effect at the metallic surface as a contributing factor to the overall oxidation of cyt c since the ultrasonic excitation frequency was 2.3 MHz, far below the typical plasmonic frequency on the order of 1014 Hz.31 We also performed an additional control experiment by changing the surface metal to chromium, for which we observed no oxidation effect (Fig. S5†). Thus, we conclude that the plasmonic effect of the metallic surface is not a dominant factor. Moreover, the fact that there is no modulation of the oxidation state of cyt c by BTO-Cr is indicative of the importance of the metal's work function in determining the redox control through the acousto-nanodevice.
Based on the results presented in Fig. 2, we now discuss the mechanism of oxidation. Firstly, the fact that significant oxidation was observed only using the acousto-nanodevice under ultrasonic irradiation indicates that both the externally applied ultrasound and the presence of the gold half-coating are jointly responsible for the observed oxidation effect. Based on the observation, we tentatively propose the following as the principle of acousto-nanodevice-driven protein redox (Fig. 3a).
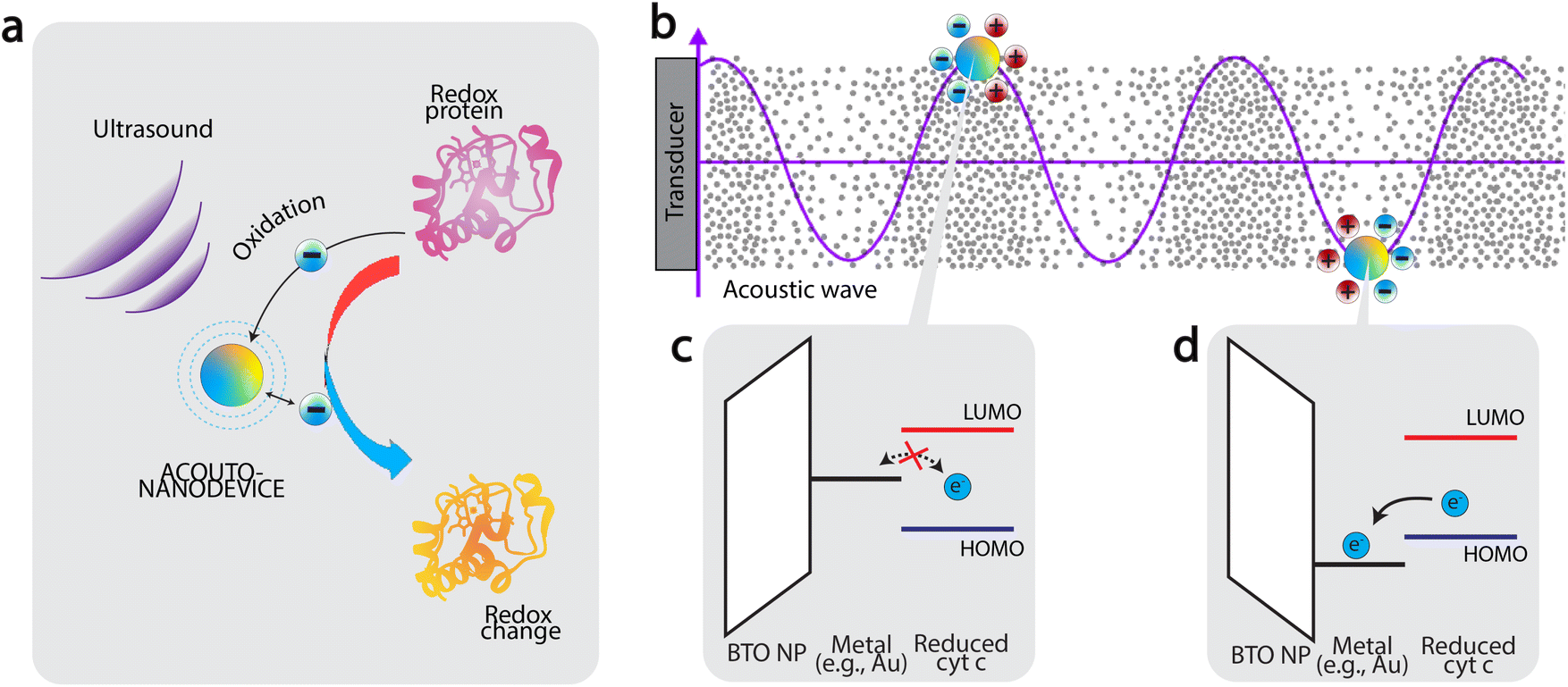 | ||
| Fig. 3 Protein oxidation by acousto-nanodevice driven under external acoustics. (a) principle of acousto-nanodevice-driven protein redox (b) The work function of the surface metal of the acousto-nanodevice oscillates under incoming acoustic waves, (c–d) a unidirectional electron transfer from protein to acousto-nanodevice is achieved because of the band alignment between the surface metal and the HOMO/LUMO levels of the protein redox center. | ||
In our proposed model, the alternating electrical polarization of BTO nanoparticles under the external ultrasound modulates the work function of the half-coated metal (Fig. 3b). With an appropriate selection of the metal work function, the electron transfer between the acousto-nanodevice and the protein redox center can only occur at a certain polarization, effectively achieving unidirectional electron transfer.
Application of this principle to induce protein oxidation by the acousto-nanodevice is schematically illustrated in Fig. 3c and d. The precondition to the protein oxidation by the acousto-nanodevice is that the surface metal work function should oscillate above and below the highest occupied molecular orbital (HOMO) level of the protein redox center. In more detail, we expect that when the polarization points toward the metal surface, the metal work function is increased; if the resulting band alignment is that the work function sits below the lowest unoccupied molecular orbital (LUMO) of the protein redox site, but above HOMO level, there is no electron transfer (Fig. 3c). When the polarization points toward the non-metalized surface, the metal work function can be lowered below the HOMO level, allowing the acousto-nanodevice to extract an electron from the protein. As a result, the band alignment is proposed to be responsible for the unidirectional electron transfer from the protein to the acousto-nanodevice (Fig. 3d). Thus, under external acoustic waves, the net effect of the oscillating work function due to alternating electrical polarization results in a net oxidation effect on proteins. Similarly, protein reduction may also be possible by selecting a metal whose work function is sufficiently low (e.g., Al).
We presented an acousto-nanodevice capable of modulating the redox state of a redox protein (i.e., reduced cyt c) under ultrasonic irradiation. The results clearly demonstrated that protein oxidation occurred only when a half-surface gold-coated acousto-nanodevice was activated by ultrasound. We also experimentally confirmed that the acousto-nanodevice may be engineered to be either an oxidation or a reduction agent, depending on the type of the surface metal. Based on the various sham controls, we postulate the working mechanism of the acousto-nanodevice is that the alternating electrical polarization of BTO nanoparticles can tentatively modulate the work function of the metal, thereby inducing unidirectional electron transfer from the protein redox centers to the acousto-nanodevice. To the best of our knowledge, the present work is the first demonstration of modulating the redox state of proteins selectively by an ultrasonic-mediated acousto-nanodevice. Given the importance of protein redox in biological processes, we look forward to the further development of the acousto-nanodevice for targeting specific redox proteins for external modulations, including both reduction and oxidation.
Author contributions
S. H. S. and A. K. conceived the idea and designed the experiment. S. H. S. and S. S. performed the sample preparation and characterization. S. H. S. and S. S. evaluated and interpreted the data. S. S. wrote the manuscript under the guidance of A. K. and S. H. S. as primary investigators. H. S. and E. B. assisted with manuscript preparation and experiments. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) No. 2018R1C1B5086311 (Song) and the National Science Foundation (NSF) No. ECCS-2245090 (Kim).References
- Q. Chi, O. Farver and J. Ulstrup, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16203–16208 CrossRef CAS PubMed.
- A. Lagunas, A. Guerra-Castellano, A. Nin-Hill, I. Díaz-Moreno, A. Miguel, J. Samitier, C. Rovira and P. Gorostiza, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- J. M. Artes, I. Diez-Perez, F. Sanz and P. Gorostiza, ACS Nano, 2011, 5, 2060–2066 CrossRef CAS PubMed.
- X. Liu, C. N. Kim, J. Yang, R. Jemmerson and X. Wang, Cell, 1996, 86, 147–157 CrossRef CAS PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- D. Stock, A. G. Leslie and J. E. Walker, Science, 1999, 286, 1700–1705 CrossRef CAS PubMed.
- J. Yuan and B. A. Yankner, Nature, 2000, 407, 802–809 CrossRef CAS PubMed.
- M. Giorgio, E. Migliaccio, F. Orsini, D. Paolucci, M. Moroni, C. Contursi, G. Pelliccia, L. Luzi, S. Minucci and M. Marcaccio, et al. , Cell, 2005, 122, 221–233 CrossRef CAS PubMed.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- W. M. Carroll, Lancet Neurol., 2019, 18, 418–419 CrossRef PubMed.
- Y. Ding, K. A. Ball, K. J. Webb, Y. Gao, A. D'Alessandro, W. M. Old, M. H. Stowell and X. Ding, Small, 2020, 16, 2003506 CrossRef CAS PubMed.
- C. Garrido, L. Galluzzi, M. Brunet, P. Puig, C. Didelot and G. Kroemer, Cell Death Differ., 2006, 13, 1423–1433 CrossRef CAS PubMed.
- D. Bayliss, J. L. Walsh, G. Shama, F. Iza and M. G. Kong, New J. Phys., 2009, 11, 115024 CrossRef.
- J. Razzokov, M. Yusupov and A. Bogaerts, Sci. Rep., 2019, 9, 5476 CrossRef CAS PubMed.
- J. J. Warren, M. E. Ener, A. Vlček Jr., J. R. Winkler and H. B. Gray, Coord. Chem. Rev., 2012, 256, 2478–2487 CrossRef CAS PubMed.
- H. Xin, W. J. Sim, B. Namgung, Y. Choi, B. Li and L. P. Lee, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- Y. Choi, T. Kang and L. P. Lee, Nano Lett., 2009, 9, 85–90 CrossRef CAS PubMed.
- S. H. Song, A. Kim and B. Ziaie, IEEE Trans. Biomed. Eng., 2015, 62, 2717–2723 CAS.
- J. Lee, K. Ko, H. Shin, S.-J. Oh, C. J. Lee, N. Chou, N. Choi, M. T. Oh, B. C. Lee and S. C. Jun, et al. , Microsyst. Nanoeng., 2019, 5, 28 CrossRef PubMed.
- G. Leinenga, C. Langton, R. Nisbet and J. Götz, Nat. Rev. Neurol., 2016, 12, 161 CrossRef PubMed.
- M. C. Ziskin, Radiographics, 1993, 13, 705–709 CrossRef CAS PubMed.
- A. Abrahao, Y. Meng, M. Llinas, Y. Huang, C. Hamani, T. Mainprize, I. Aubert, C. Heyn, S. E. Black and K. Hynynen, et al. , Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- B. I. Lee, S. Lee, Y. S. Suh, J. S. Lee, A.-k. Kim, O.-Y. Kwon, K. Yu and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 11472–11476 CrossRef CAS PubMed.
- D. S. Wuttke, M. J. Bjerrum, J. R. Winkler and H. B. Gray, Science, 1992, 256, 1007–1009 CrossRef CAS PubMed.
- C. Lange and C. Hunte, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2800–2805 CrossRef CAS PubMed.
- T. d. F. Paulo, T. P. de Sousa, D. S. de Abreu, N. H. Felício, P. V. Bernhardt, L. G. de F. Lopes, E. H. Sousa and I. C. Diógenes, J. Phys. Chem. B, 2013, 117, 8673–8680 CrossRef CAS PubMed.
- M.-E. Aubin-Tam and K. Hamad-Schifferli, Langmuir, 2005, 21, 12080–12084 CrossRef CAS PubMed.
- H. A. Heering, F. G. Wiertz, C. Dekker and S. de Vries, J. Am. Chem. Soc., 2004, 126, 11103–11112 CrossRef CAS PubMed.
- M. E. Siwko and S. Corni, Phys. Chem. Chem. Phys., 2013, 15, 5945–5956 RSC.
- X. Ding, M. Yang, J. Hu, Q. Li and A. McDougall, Microchim. Acta, 2007, 158, 65–71 CrossRef CAS.
- J.-J. Greffet, Plasmonics, Springer, 2012, pp. 105–148 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01523h |
| This journal is © The Royal Society of Chemistry 2023 |